Appendix B
Epidemiology and Attributes of Specific Childhood Cancers
CONTENTS
SELECTED HEMATOLOGIC MALIGNANCIES
Non-Hodgkin lymphoma (except Burkitt lymphoma)
SELECTED CENTRAL NERVOUS SYSTEM TUMORS
Ependymoma (includes choroid plexus tumor)
Medulloblastoma (intracranial and intraspinal embryonal tumors)
SELECTED NON–CENTRAL NERVOUS SYSTEM SOLID TUMORS
Ewing sarcoma (includes related sarcoma of bone)
Germ cell tumors (includes trophoblastic tumors and neoplasms of gonads)
Neuroblastoma (includes other peripheral nervous cell tumors)
Non-rhabdomyosarcoma soft-tissue sarcomas
Wilms tumor (nephroblastoma and other nonepithelial renal tumors)
This appendix presents summary information on 18 childhood cancers. For each cancer, key data on its occurrence and features are listed, followed by figures on age-specific annual incidence, age-specific annual incidence by sex, age-specific annual incidence by race/ethnicity, survival by age at diagnosis, and survival by year of diagnosis.
All of the data were obtained by the committee from available sources, and the figures were created by the committee. Other characteristics of the data include the following:
- The diagnosis categories correspond to the International Classification of Childhood Cancer presented in Table 2-1 (in Chapter 2).
- Incidence and survival data are from the National Cancer Institute’s Surveillance, Epidemiology, and End Results Program for 1990–2016.
- Estimated numbers of cases diagnosed annually in the United States are based on the incidence rate applied to the 2010 U.S. census for individuals under the age of 18 living in the United States.
- Five-year survival estimates are based on patients diagnosed for 2010–2016.
- Ten-year survival estimates are based on patients diagnosed for 2000–2009.
- Interpretation of incidence and survival data presented by age, sex, race/ethnicity, and year of diagnosis categories should take into consideration the fact that some subgroups may include a relatively small number of cases, which may impact the stability of the estimates.
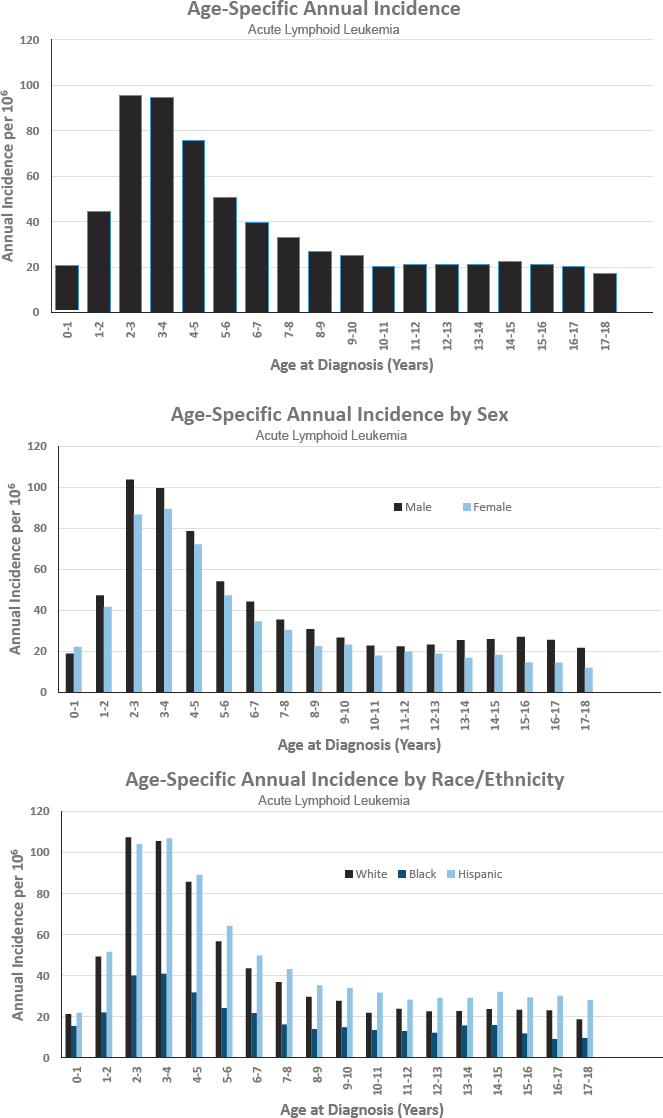
ACUTE LYMPHOID LEUKEMIA
Diagnosed 0–17 Years of Age
- Annual incidence rate is 376 per 1,000,000 population
- Estimated 2,790 cases diagnosed annually
- Represents approximately 21 percent of cancers diagnosed before age 18
- Ratio of males to females is 1.2 to 1.0
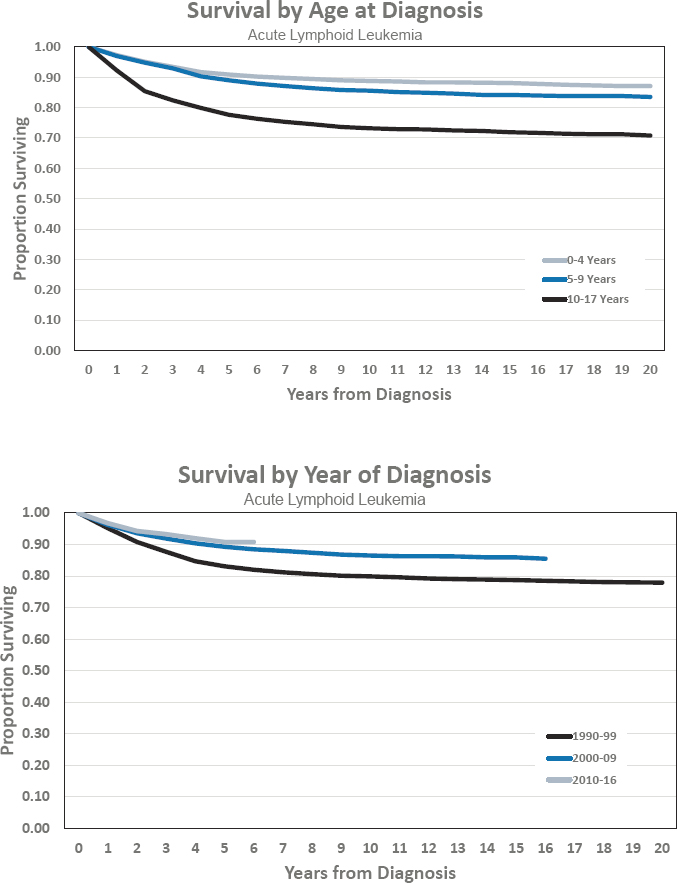
- Estimated 5-year survival of 90.8 percent
- Estimated 10-year survival of 86.5 percent
Features of Acute Lymphoid Leukemia
- Represents numerous subtypes defined by immunophenotype and genetics.
- Prognosis can vary depending on demographic factors such as age at diagnosis, clinical presentation, biologic features, and genetic markers within the leukemia cells.
- Prognostic subgroups can be defined as standard risk, high risk, and very high risk (see Annex Table 5-1 in Chapter 5).
- Risk-based treatment approaches are used to determine treatment agents/modality/intensity.
- Genetic sequencing and genomic profiling of the leukemic cells continue to identify new subtypes and markers of prognosis.
Figures for Acute Lymphoid Leukemia
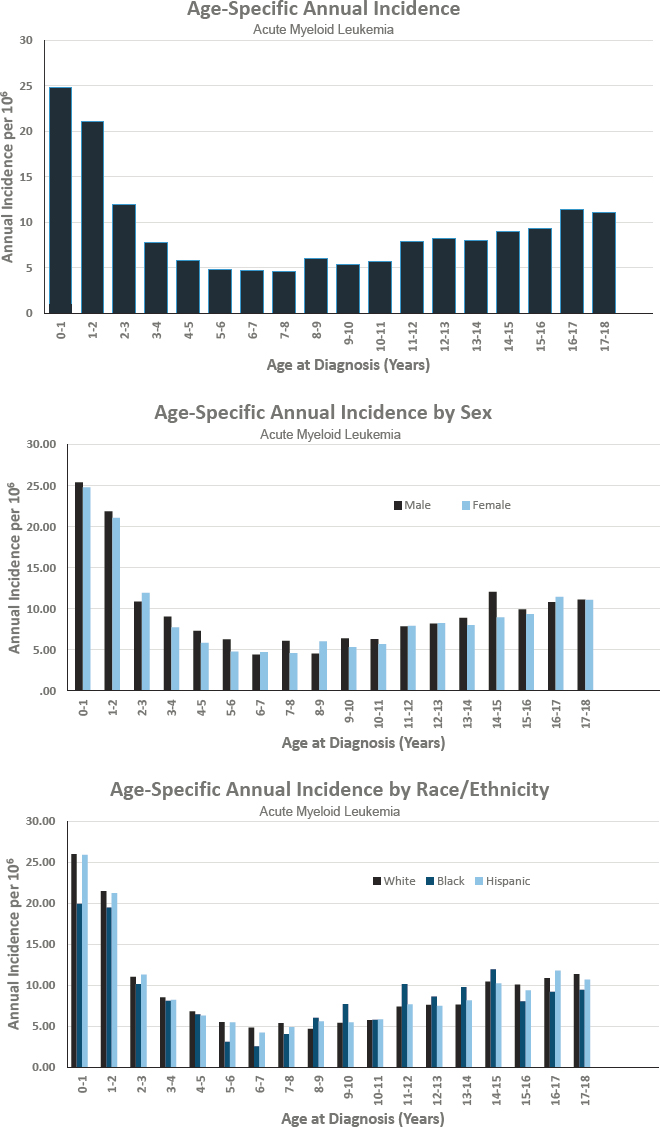
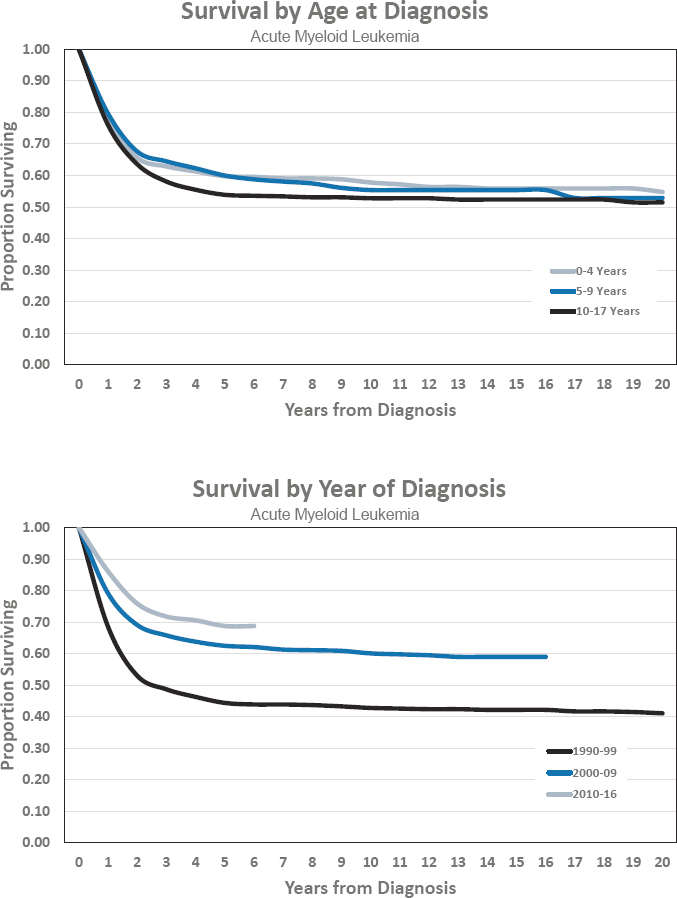
ACUTE MYELOID LEUKEMIA
Diagnosed 0–17 Years of Age
- Annual incidence rate is 81 per 1,000,000 population
- Estimated 600 cases diagnosed annually
- Represents approximately 5 percent of cancers diagnosed before age 18
- Ratio of males to females is 1.1 to 1.0
- Estimated 5-year survival of 68.8 percent
- Estimated 10-year survival of 60.1 percent
Features of Acute Myeloid Leukemia
- Includes seven distinct subtypes (designated M1 to M7 under the FAB [French-American-British] system).
- Prognosis can vary depending on demographic factors such as age at diagnosis, biologic features including white blood cell count at presentation, and genetic markers within the leukemia cells.
- Prognosis may be influenced by genetic and molecular abnormalities, immunophenotype, and clinical features including white blood cell count at diagnosis and cerebrospinal fluid involvement.
- Induction and consolidation therapies include multi-agent chemotherapy. Relapsed or refractory disease may be treated with allogeneic hematopoietic stem cell transplantation.
- Further identification and characterization of molecular abnormalities continue to refine prognostic tools and therapy options.
Figures for Acute Myeloid Leukemia
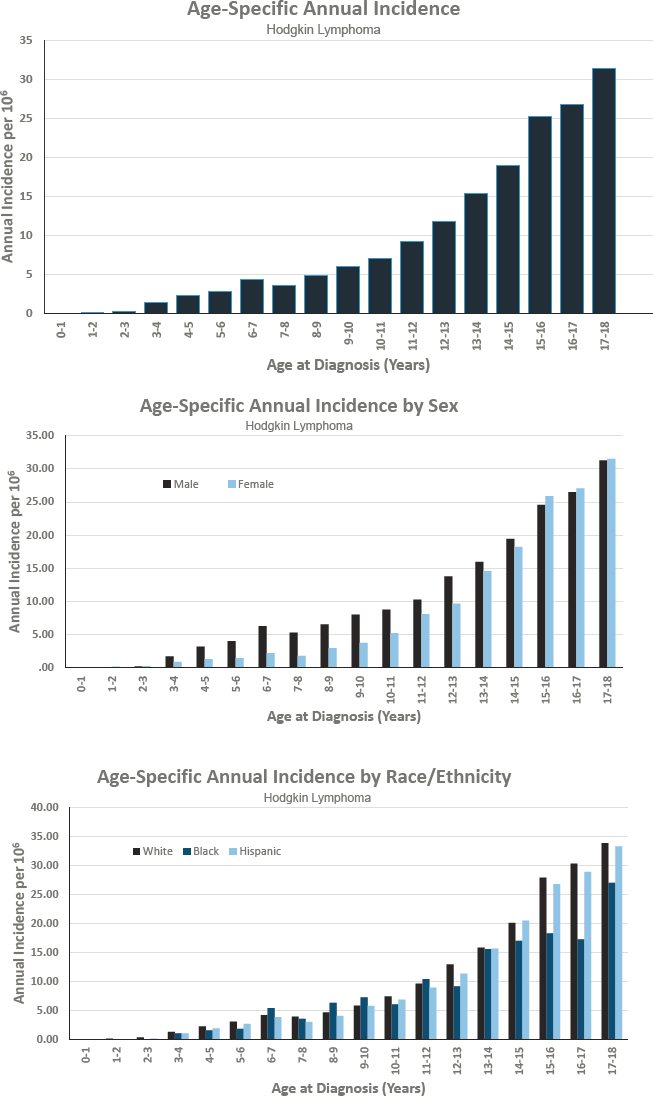
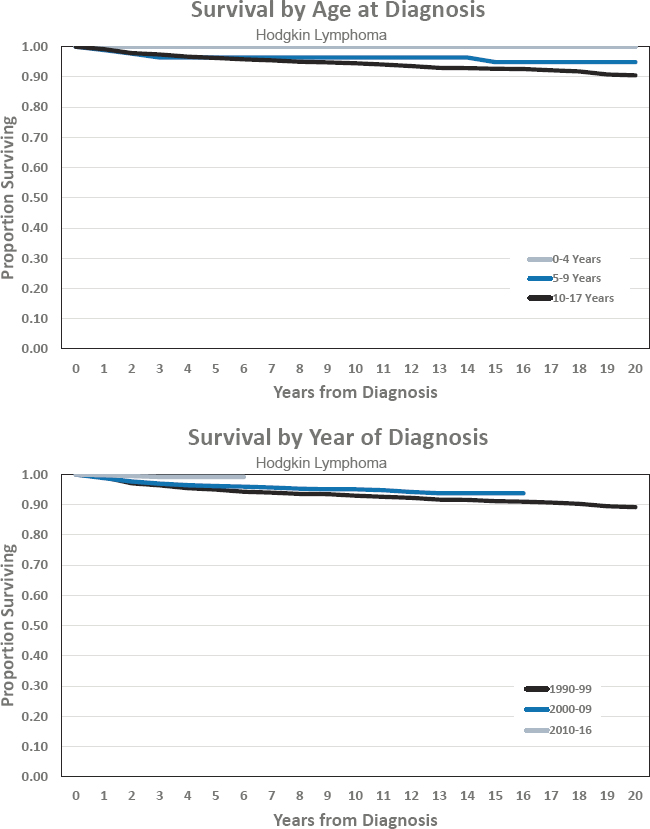
HODGKIN LYMPHOMA
Diagnosed 0–17 Years of Age
- Annual incidence rate is 93 per 1,000,000 population
- Estimated 690 cases diagnosed annually
- Represents approximately 5 percent of cancers diagnosed before age 18
- Ratio of males to females is 1.2 to 1.0
- Estimated 5-year survival of 99.2 percent
- Estimated 10-year survival of 95.1 percent
Features of Hodgkin Lymphoma
- Hodgkin lymphoma is a type of lymphoma that originates from lymphocytes and can occur anywhere in the lymphatic system.
- Prognosis is determined by a combination of stage, presence or absence of bulk disease, and presence or absence of B symptoms. Though Hodgkin lymphoma is highly curable, its therapy is associated with high rates of chronic health problems and subsequent cancers.
- Prognosis is categorized as low, intermediate, or high risk. These risk groups are not entirely consistent across clinical trials (see Annex Table 5-1 in Chapter 5).
- Risk-based and response-adapted approaches involve multi-agent chemotherapy, with or without low-dose involved-field, involved-site, or, more recently, involved-node radiotherapy.
- The very good survival rates for childhood Hodgkin lymphoma have driven therapeutic focus to minimizing long-term morbidity.
Figures for Hodgkin Lymphoma
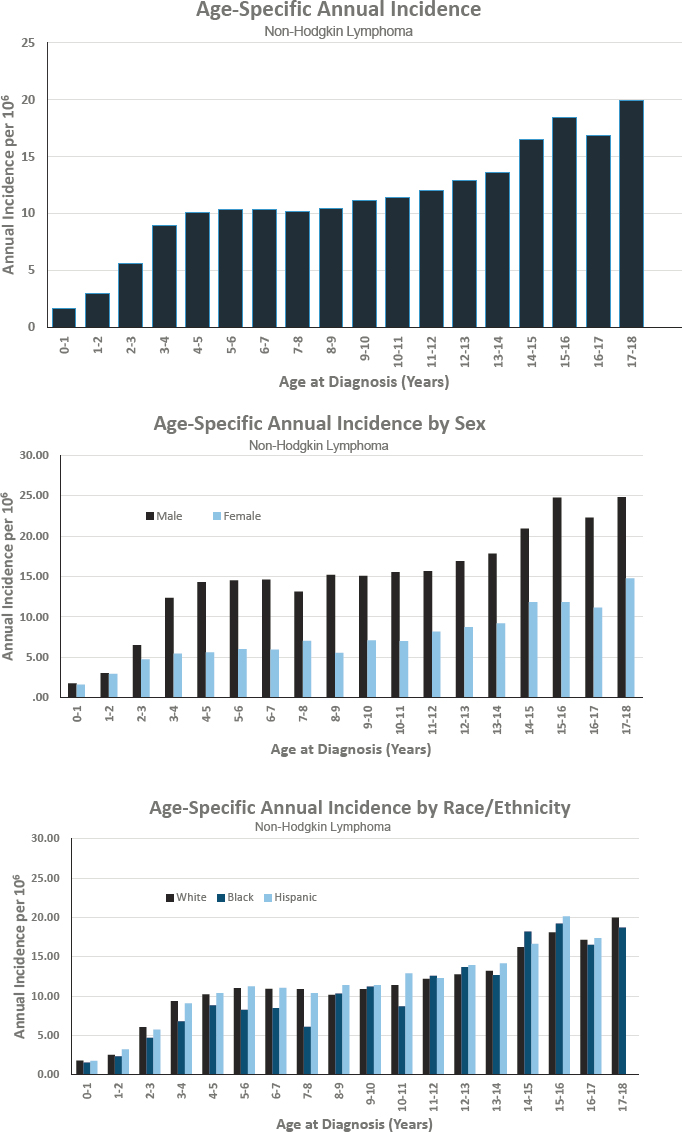
NON-HODGKIN LYMPHOMA
Diagnosed 0–17 Years of Age
- Annual incidence rate is 101 per 1,000,000 population
- Estimated 750 cases diagnosed annually
- Represents approximately 6 percent of cancers diagnosed before age 18
- Ratio of males to females is 2 to 1
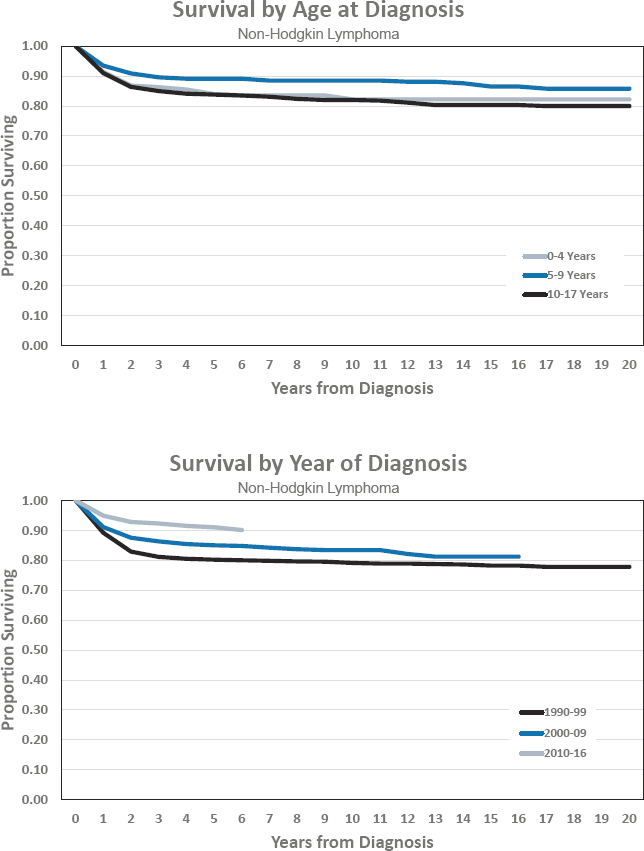
- Estimated 5-year survival of 91.0 percent
- Estimated 10-year survival of 83.4 percent
Features of Non-Hodgkin Lymphoma
- Non-Hodgkin lymphoma includes subtypes of lymphoma beginning in B lymphocytes, T lymphocytes, or natural killer cells.
- Prognosis can vary depending on the subtype of lymphoma. The risk of late effects is associated with the treatment program.
- Risk-determining factors include the pathologic subtype and stage (Murphy classification) (see Annex Table 5-1 in Chapter 5).
- Treatment for non-Hodgkin lymphoma is driven by the subtype and staging. Surgery and chemotherapy are most common, with radiation therapy in limited use.
Figures for Non-Hodgkin Lymphoma
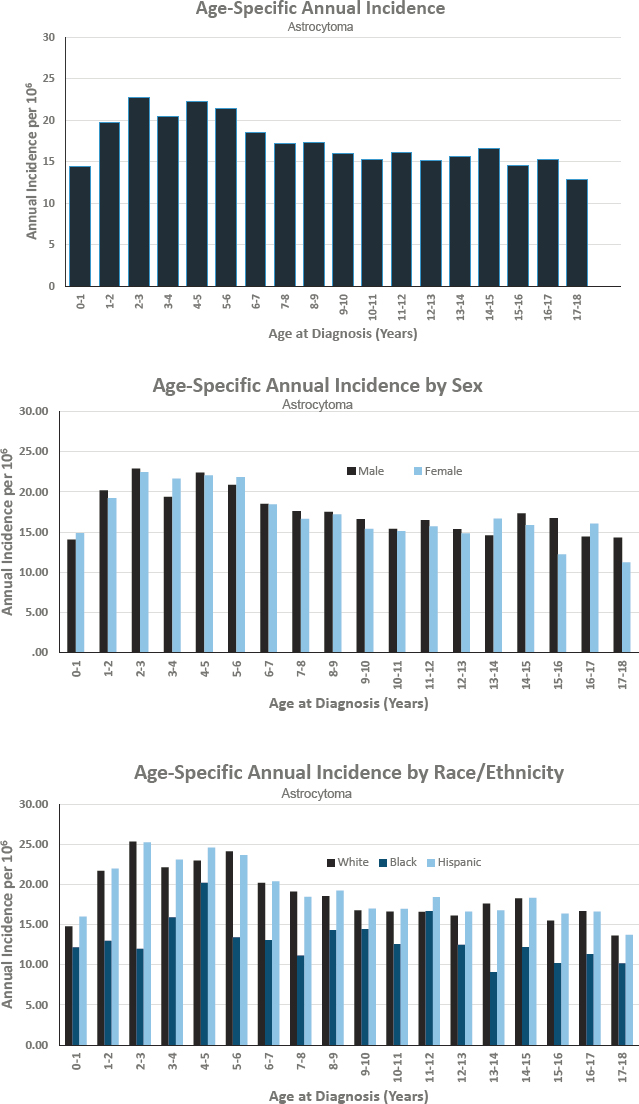
ASTROCYTOMA
Diagnosed 0–17 Years of Age
- Annual incidence rate is 182 per 1,000,000 population
- Estimated 1,360 cases diagnosed annually
- Represents approximately 10 percent of cancers diagnosed before age 18
- Ratio of males to females is 1 to 1
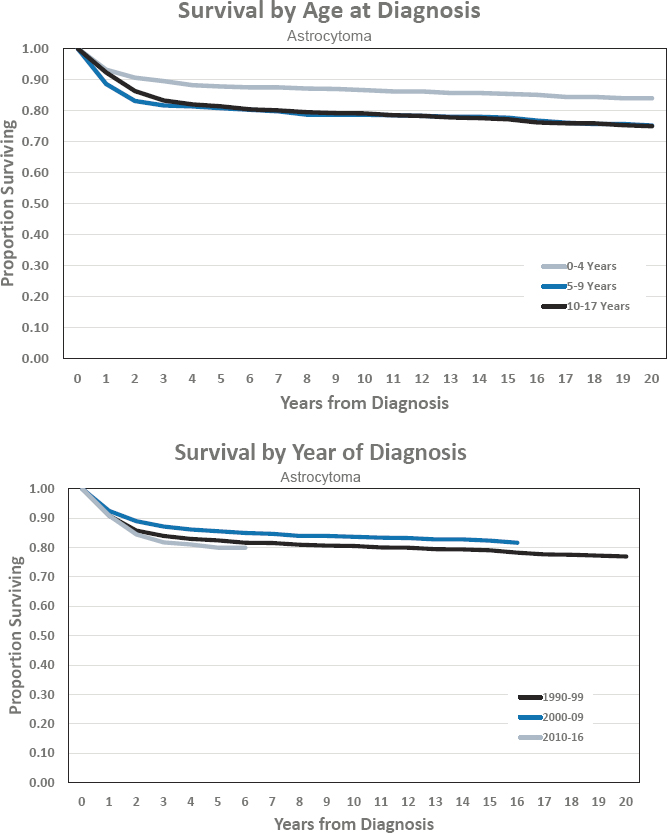
- Estimated 5-year survival of 79.9 percent
- Estimated 10-year survival of 83.6 percent
Features of Astrocytoma
- Astrocytoma is the most common type of pediatric central nervous system (CNS) tumor; it is a broad histologic category of tumors with both low- (World Health Organization [WHO] grade I and II) and high- (WHO grade III and IV) grade types and numerous subtypes defined by histologic, biologic, and genetic characteristics.
- Low-grade astrocytomas are by far the most common type of astrocytoma and have a better prognosis and survival outcomes compared with high-grade astrocytomas, which have a very poor prognosis and overall survival.
- Standard treatment for low-grade glioma after surgery often consists of classic chemotherapy, though promising new molecularly targeted agents are now being tested.
- Children with high-grade glioma are treated with a combination of surgery and radiation + chemotherapy or a molecularly targeted agent on a clinical trial.
- As the molecular and genetic landscape of astrocytoma (both low- and high-grade) continues to advance, a better understanding of more-well defined risk groups and use of specific molecularly targeted agents will progress.
Figures for Astrocytoma
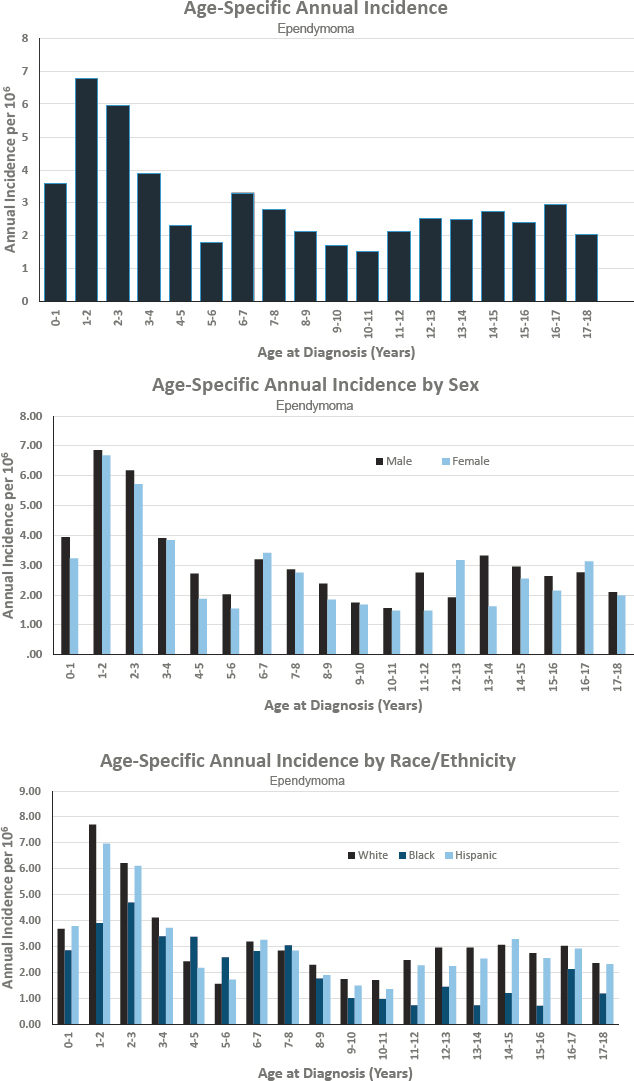
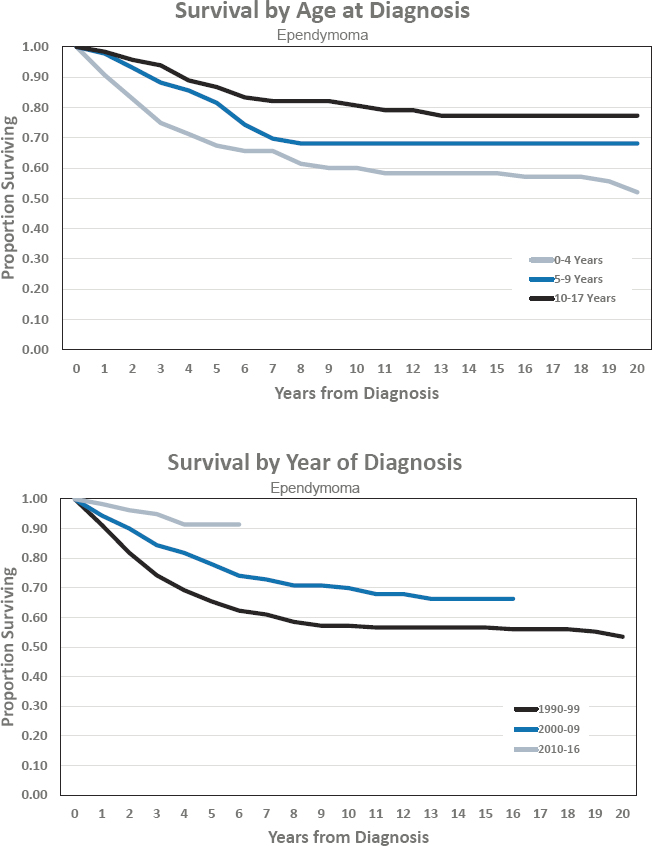
EPENDYMOMA
Diagnosed 0–17 Years of Age
- Annual incidence rate is 40 per 1,000,000 population
- Estimated 300 cases diagnosed annually
- Represents approximately 2 percent of cancers diagnosed before age 18
- Ratio of males to females is 1.1 to 1.0
- Estimated 5-year survival of 91.4 percent
- Estimated 10-year survival of 69.9 percent
Features of Ependymoma
- Ependymomas/ependymal tumors are a class of glial tumors that can occur anywhere in the brain or spine.
- The majority of ependymomas occur in the posterior fossa, although they can occur throughout the CNS and are typically associated with a ventricular surface.
- Metastasis throughout the CNS occurs in fewer than 10 percent of cases and is associated with poor prognosis.
- Risk categories include age, degree of surgical resection, and stage (local or metastatic disease).
- Treatment of ependymomas consists of surgical resection followed by radiation to the tumor area. The prognosis is variable.
- The treatment of subependymomas and myxopapillary ependymomas is typically surgical resection. Both tumors have excellent prognosis.
- The molecular underpinnings of ependymoma are currently yielding promising new risk stratifications that better predict prognosis and may guide future treatment.
Figures for Ependymoma
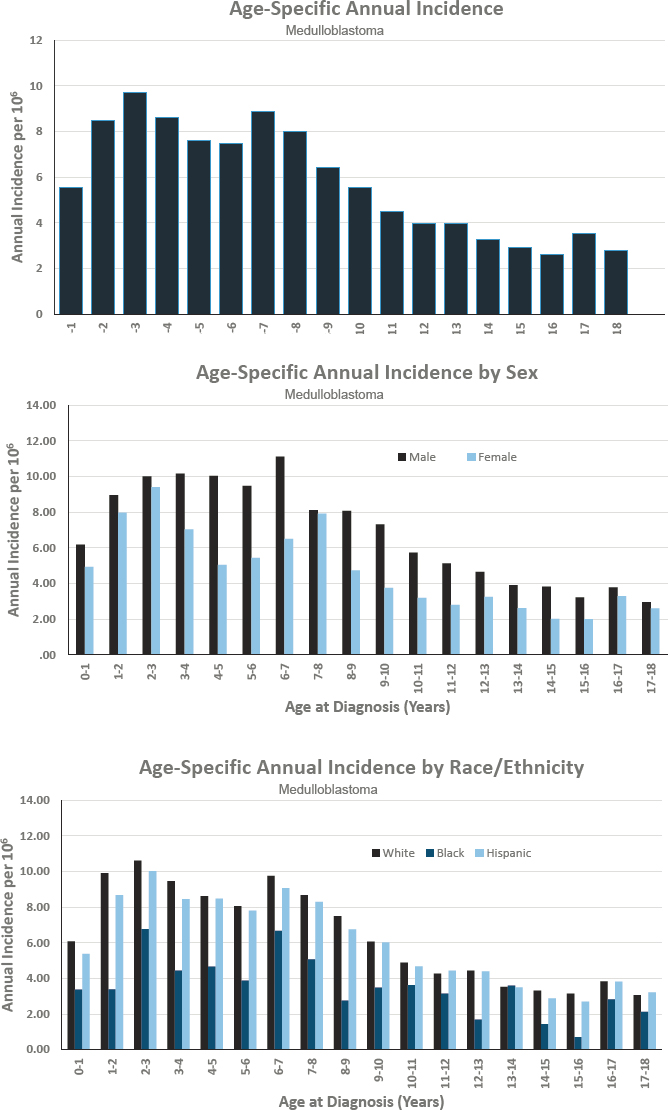
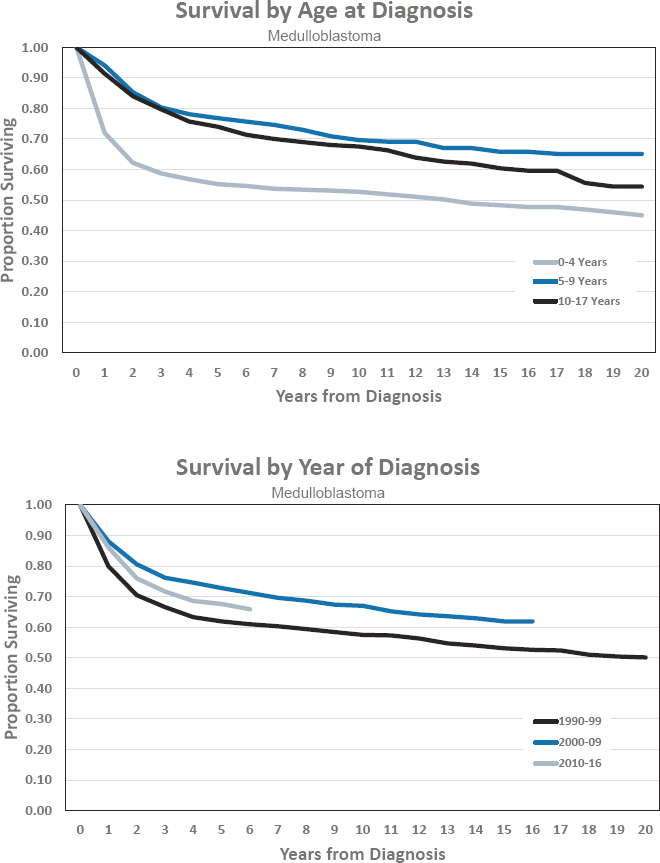
MEDULLOBLASTOMA
Diagnosed 0–17 Years of Age
- Annual incidence rate is 64 per 1,000,000 population
- Estimated 475 cases diagnosed annually
- Represents approximately 4 percent of cancers diagnosed before age 18
- Ratio of males to females is 1.4 to 1.0
- Estimated 5-year survival of 67.6 percent
- Estimated 10-year survival of 67.0 percent
Features of Medulloblastoma
- Medulloblastomas are the most common embryonal tumor of childhood, accounting for 9 percent of all CNS tumors and occurring predominantly in the first two decades of life.
- Approximately one-third are metastatic at diagnosis, which is associated with poor prognosis.
- Children less than 3 years old have a generally poor survival prognosis except for infants with extensively nodular medulloblastomas, who have a good prognosis.
- Risk stratification currently is based on molecular subtypes and clinical criteria and falls into four categories for children greater than 3 years old: (1) low-risk, WNT molecular subtype; (2) standard-risk, minimal residual disease following resection, no metastases, not in a molecular high-risk category; (3) high-risk, presence of significant residual disease after resection or metastases, not in molecular very high-risk category; and (4) very high-risk, SHH (sonic hedgehog) molecular subtype with TP53 mutation or Group 3 molecular subtype with MYC amplification.
- Treatment for children older than 3 years includes surgery, external beam craniospinal radiation with tumor bed boost, and multidrug chemotherapy for 12–18 months.
- Treatment using high-dose chemotherapy with autologous stem cell rescue is included in some experimental protocols, including in children less than 3 years of age.
- Survivors may have extensive physical, neurological, and cognitive deficits that negatively impact quality of life and decrease the probability of living independently as adults, especially when treatment occurs at less than 7 years of age.
- The molecular subtypes have different mechanisms of oncogenesis. Future clinical trials will increasingly rely on molecular targeted therapies, and protocols are predicted to evolve to allow introduction of targeted therapies while minimizing treatment with conventional chemotherapy and radiation.
Figures for Medulloblastoma
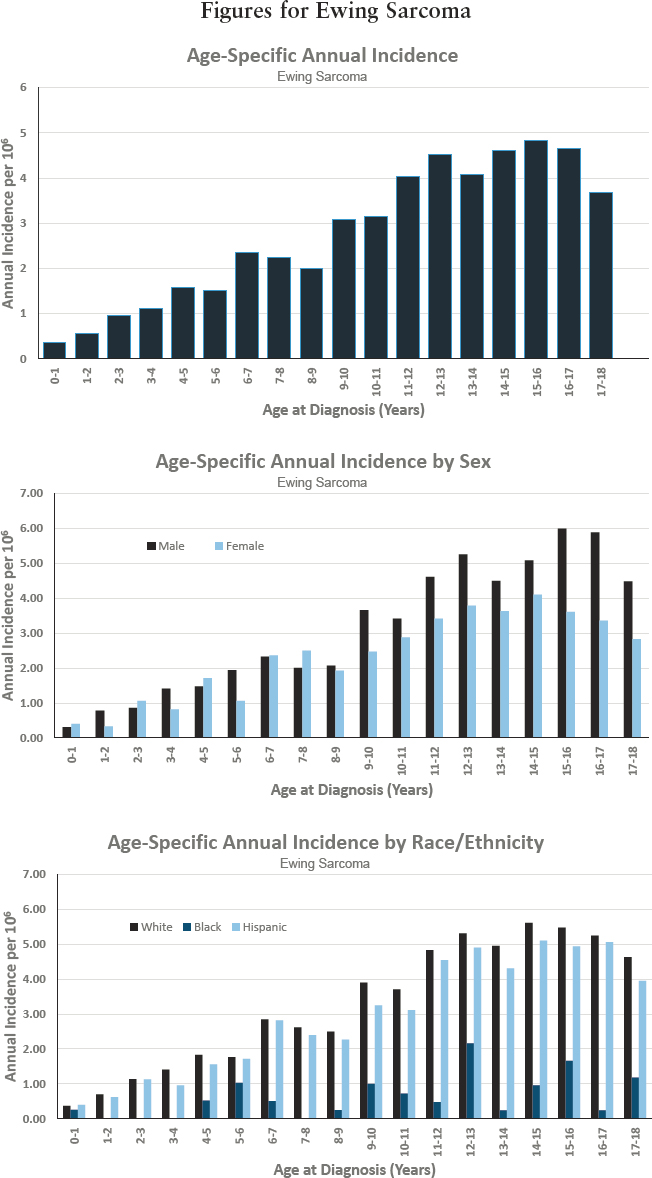
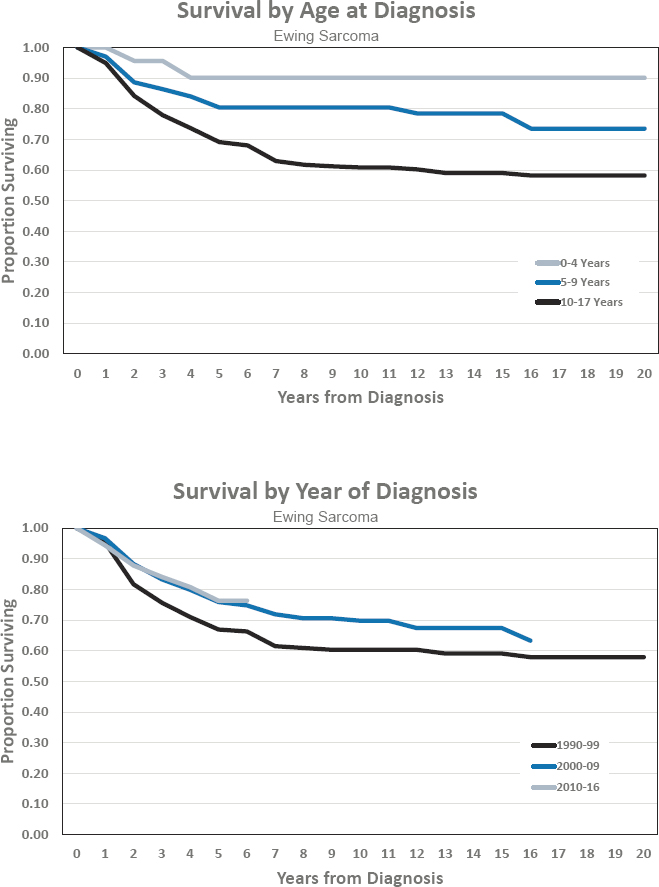
EWING SARCOMA
Diagnosed 0–17 Years of Age
- Annual incidence rate is 32 per 1,000,000 population
- Estimated 240 cases diagnosed annually
- Represents approximately 2 percent of cancers diagnosed before age 18
- Ratio of males to females is 1.3 to 1.0
- Estimated 5-year survival of 76.3 percent
- Estimated 10-year survival of 69.8 percent
Features of Ewing Sarcoma
- Primary bone tumors are responsible for 6 percent of all childhood cancers.
- Affects mainly Whites and is extremely uncommon among Blacks (in both the United States and Africa) and Asians.
- Most often arises in the long bones of the extremities (predominantly the femur, but also the tibia, fibula, and humerus) and has a predilection for the flat bones of the pelvis and shoulder girdle. A minority of Ewing sarcomas arise in soft tissue.
- Ninety percent of cases have the t(11;22)(q24;q12) translocation, which fuses the 5’ portion of the EWSR1 gene on chromosome 22 to the 3’ portion of the FLI1 gene on chromosome 11 and generates an aberrant transcription factor through fusion of the EWSR1 gene with the FLI1 gene. This is detected using fluorescence in situ hybridization.
- Prognosis varies depending on patient age and sex, tumor location and size, presence or absence of metastases, elevated serum lactate dehydrogenase, and a variety of biological factors.
- Patients are stratified by the presence or absence of metastatic disease. There are five distinct prognostic groups based on age, pelvic location, ethnicity, and the presence of metastatic disease.
- Treatment consists of chemotherapy, surgery, and surgery with radiation.
Figures for Ewing Sarcoma
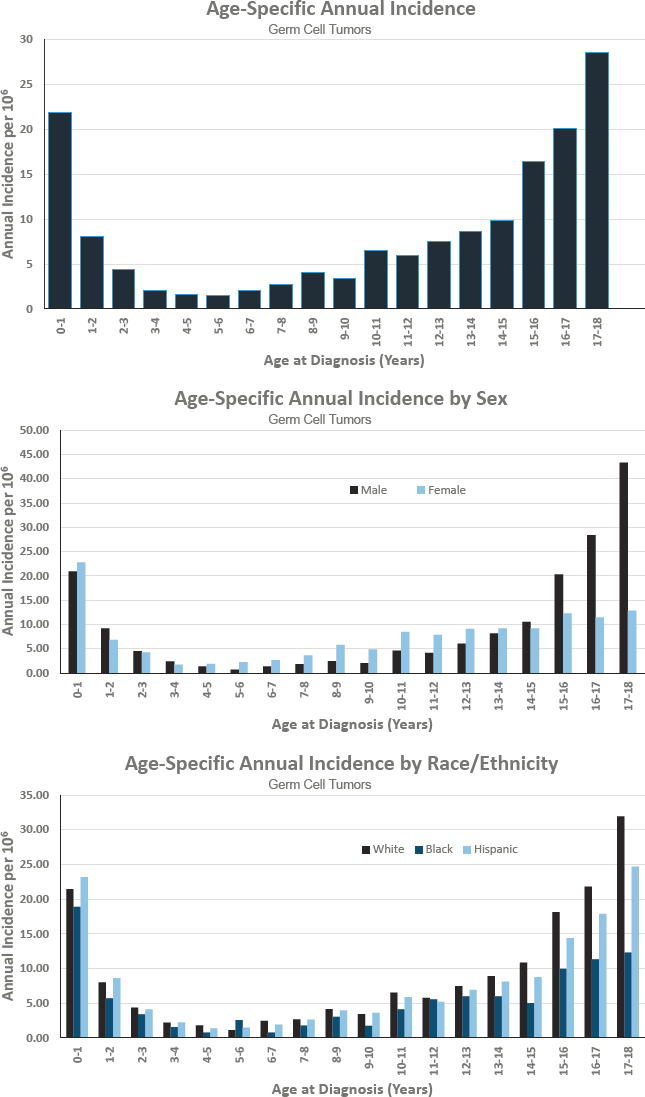
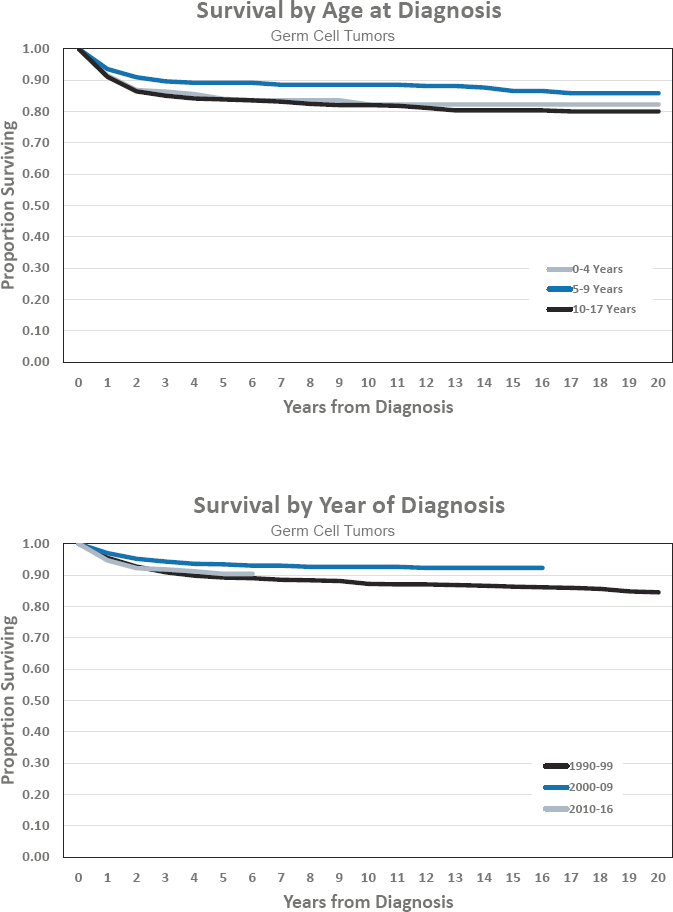
GERM CELL TUMORS
Diagnosed 0–17 Years of Age
- Annual incidence rate is 48 per 1,000,000 population
- Estimated 350 cases diagnosed annually
- Represents approximately 3 percent of cancers diagnosed before age 18
- Ratio of males to females is 1.3 to 1.0
- Estimated 5-year survival of 90.4 percent
- Estimated 10-year survival of 92.6 percent
Features of Germ Cell Tumors
- Germ cells normally give rise to eggs in females or sperm in males, so the tumors that derive from them may be composed of several different cell types of varying maturity, ranging from embryonal to mature tissues. The most common pediatric malignant germ cell tumor (GCT) is the yolk sac tumor, which secretes alpha fetoprotein. Other malignant classes include choriocarcinoma, embryonal carcinoma, dysgerminoma, and seminoma. As a group, GCTs are among the most curable tumors of childhood.
- The most important features driving prognosis are the age of the patient, the cell types present, the height of plasma tumor marker elevation, the location of the tumor in the gonad (testis or ovary) or extragonadal (sacrococcygeal or mediastinal), and the stage of disease.
- Mature teratomas are generally benign. Immature teratomas can be cured with surgery alone. Malignant GCTs can be classified into low, standard, and high risk.
- Surgery is the mainstay of therapy for all GCTs. Standard- and high-risk malignant GCTs also require chemotherapy for cure.
- While GCTs are relatively uncommon in young children, they make up 15 percent of tumors occurring in adolescents and young adults, and efforts are being made to harmonize treatment algorithms between pediatric and medical oncologists. This combined effort will also accelerate understanding of the genetic underpinnings of this family of tumors.
Figures for Germ Cell Tumors
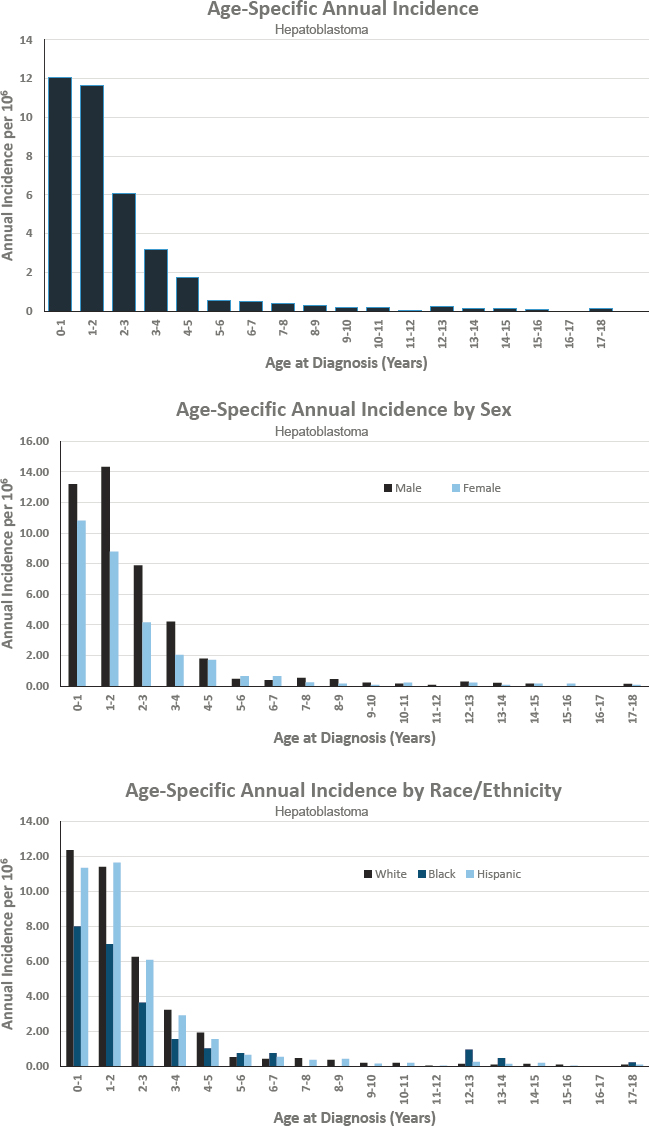
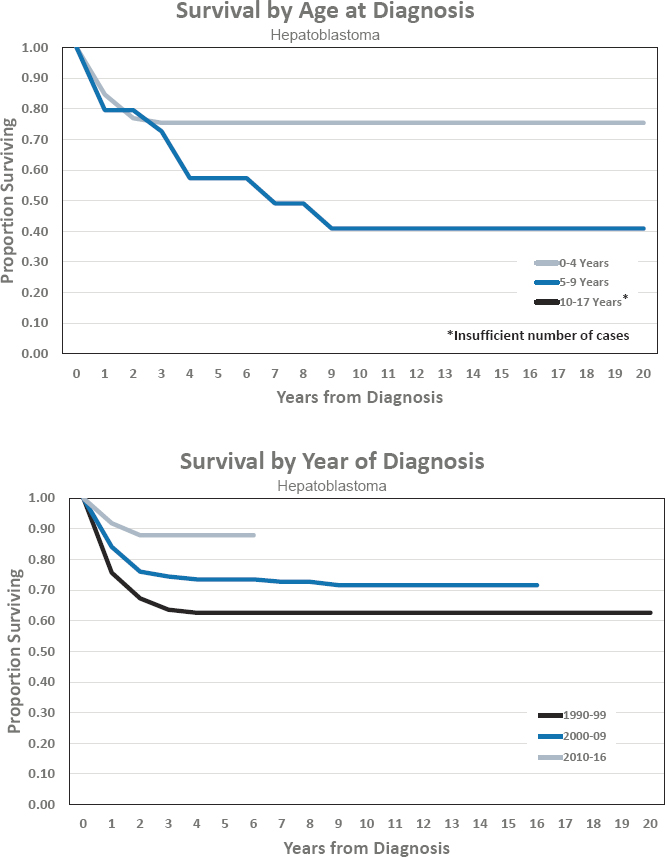
HEPATOBLASTOMA
Diagnosed 0–17 Years of Age
- Annual incidence rate is 23 per 1,000,000 population
- Estimated 175 cases diagnosed annually
- Represents approximately 1 percent of cancers diagnosed before age 18
- Ratio of males to females is 1.5 to 1.0
- Estimated 5-year survival of 87.9 percent
- Estimated 10-year survival of 71.6 percent
Features of Hepatoblastoma
- Hepatoblastoma is typically diagnosed before 4 years of age and is associated with a number of genetic conditions.
- Patients with these conditions should be screened for hepatoblastoma by abdominal ultrasound and determination of alpha-fetoprotein in the blood every 3 months.
- Treatment for hepatoblastoma is guided by local tumor extent prior to therapy (PRETEXT system) and after therapy (POSTTEXT system).
- Complete surgical resection by an expert team is the cornerstone for curative therapy.
- The overall survival for patients with hepatoblastoma has improved as a result of advances in surgical, chemotherapy, and transplant options, but the outcome for unresectable or recurrent hepatoblastoma remains poor.
- The outcome is best for children with resectable disease at diagnosis, but many patients present with unresectable disease.
- Patients whose tumors remain unresectable after chemotherapy should be considered for liver transplant at the earliest possible time point.
Figures for Hepatoblastoma
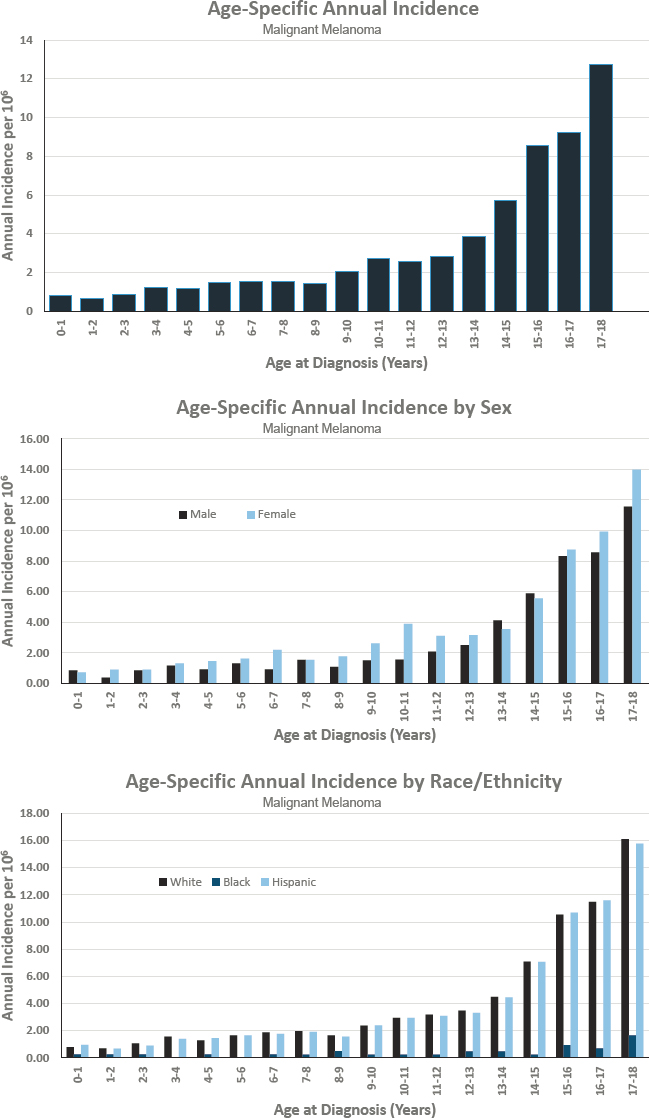
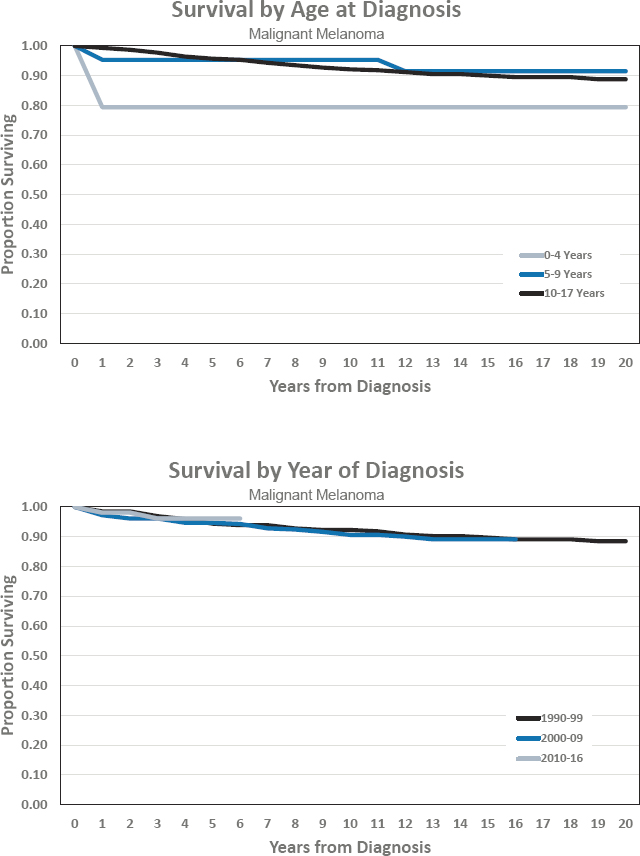
MALIGNANT MELANOMA
Diagnosed 0–17 Years of Age
- Annual incidence rate is 30 per 1,000,000 population
- Estimated 225 cases diagnosed annually
- Represents approximately 2 percent of cancers diagnosed before age 18
- Ratio of males to females is 0.8 to 1.0
- Estimated 5-year survival of 96.1 percent
- Estimated 10-year survival of 90.6 percent
Features of Malignant Melanoma
- Melanoma is the most common skin cancer in children, but overall, it is a rare pediatric malignancy.
- Age, stage, and biology are the most significant determinants of behavior. Congenital metastatic melanoma and melanoma in the context of a congenital skin disorder are usually fatal. In general, sporadic pediatric melanoma has a better prognosis than its adult counterpart, but prepubescent children do better than adolescents.
- Melanoma can be classified into low, standard, and high risk (metastatic disease).
- Complete resection with negative margins is required for all melanoma. Patients with spitzoid melanoma and young patients with malignant melanoma often have positive sentinel node biopsies but remain at low risk for recurrence. Adolescents with malignant melanoma should be treated according to adult algorithms.
- Immunotherapy using immune checkpoint inhibitors that block the PD-1 and CTLA-4 pathways and BRAF and MEK inhibition for BRAF mutated tumors have changed the outlook for adult metastatic melanoma. The congenital melanomas are unlikely to benefit because of unique biology, but too few children with sporadic melanoma have needed to receive these interventions to draw definitive conclusions about their efficacy in pediatric patients.
Figures for Malignant Melanoma
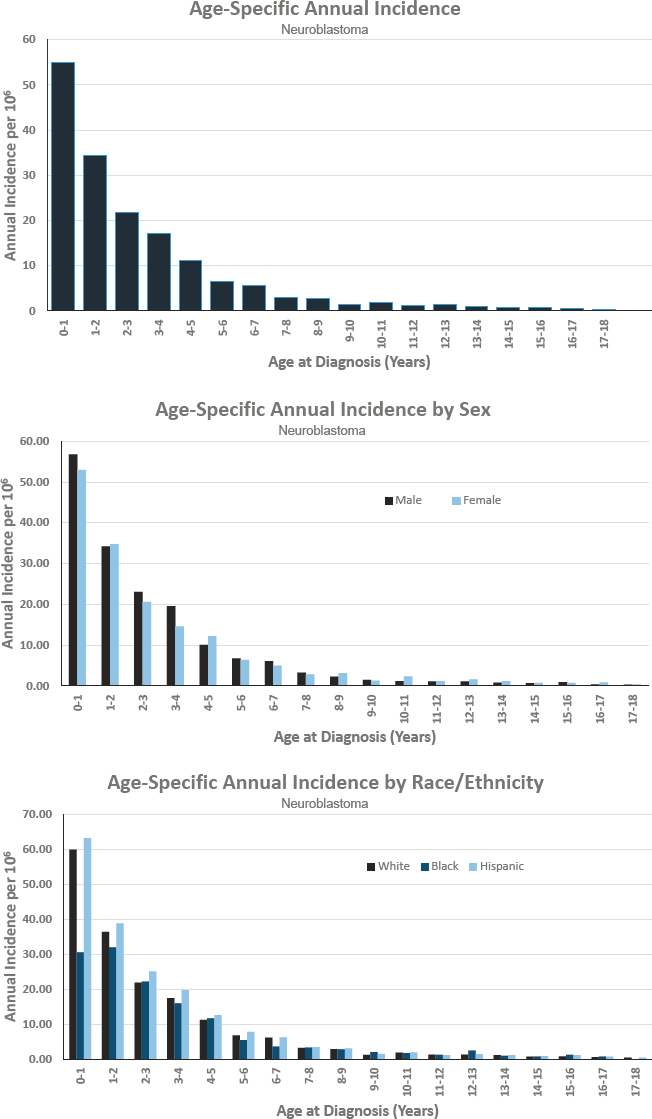
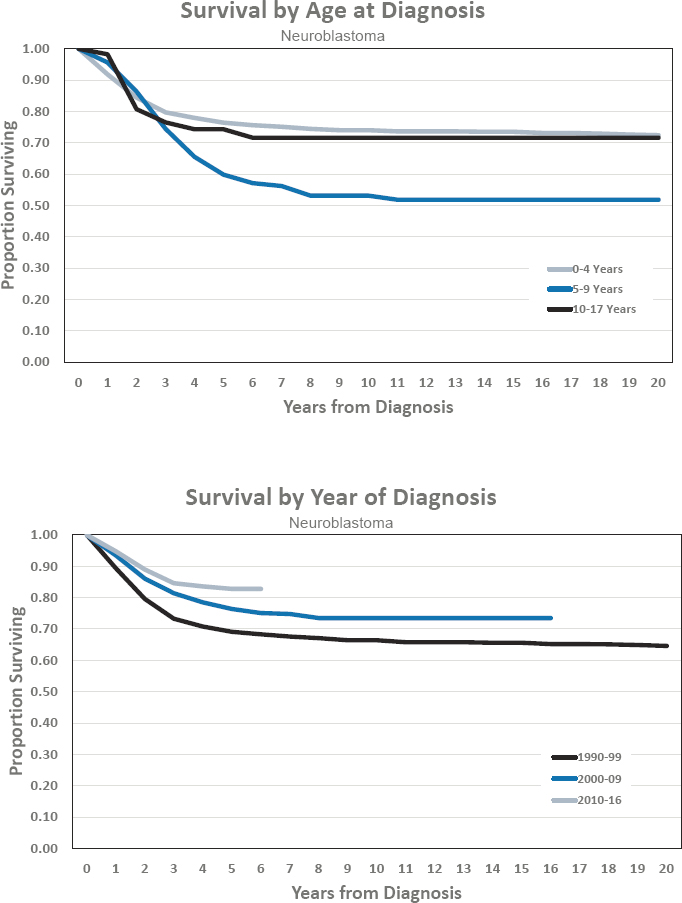
NEUROBLASTOMA
Diagnosed 0–17 Years of Age
- Annual incidence rate is 95 per 1,000,000 population
- Estimated 700 cases diagnosed annually
- Represents approximately 5 percent of cancers diagnosed before age 18
- Ratio of males to females is 1 to 1
- Estimated 5-year survival of 82.8 percent
- Estimated 10-year survival of 73.5 percent
Features of Neuroblastoma
- Neuroblastoma is the most common pediatric solid tumor occurring outside the brain, usually originating in the adrenal gland or sympathetic nerves that run along both sides of the spine from neck to pelvis. Approximately 90 percent of patients present under the age of 5. Neuroblastoma can exhibit a broad spectrum of behavior, from spontaneous regression to aggressive disease requiring intensive multimodality treatment to achieving cure in only half of patients.
- The most important features driving prognosis are age; stage; and certain biological features of the tumor, including unfavorable or favorable histology, the presence of MYCN amplification, the mean number of chromosomes (ploidy), and segmental chromosomal aberrations including loss of 11q. There are two staging systems still in use for neuroblastoma: the International Neuroblastoma Staging System (INSS) and the newer International Neuroblastoma Risk Group Staging System (INRGSS).
- Prognostic subgroups can be defined as very low, low, intermediate, and high risk. There is also a “special” category in both the INSS (4S) and INRGSS (MS) that applies to infants and toddlers with good biology who may have a pattern of metastatic disease limited to the skin, liver, and minimal bone marrow; 4S/MS-stage disease and small perinatal tumors may sometimes undergo spontaneous regression.
- Risk-based treatment approaches are used to determine treatment agents, intensity, and local control modalities, but location remains an important driver of disability regardless of risk categorization.
- Immunotherapy with anti-GD2 antibodies is a recent breakthrough in the prevention of relapse. Genetic sequencing and genomic profiling of tumors may identify new potential targets for drug treatment, including ALK.
Figures for Neuroblastoma
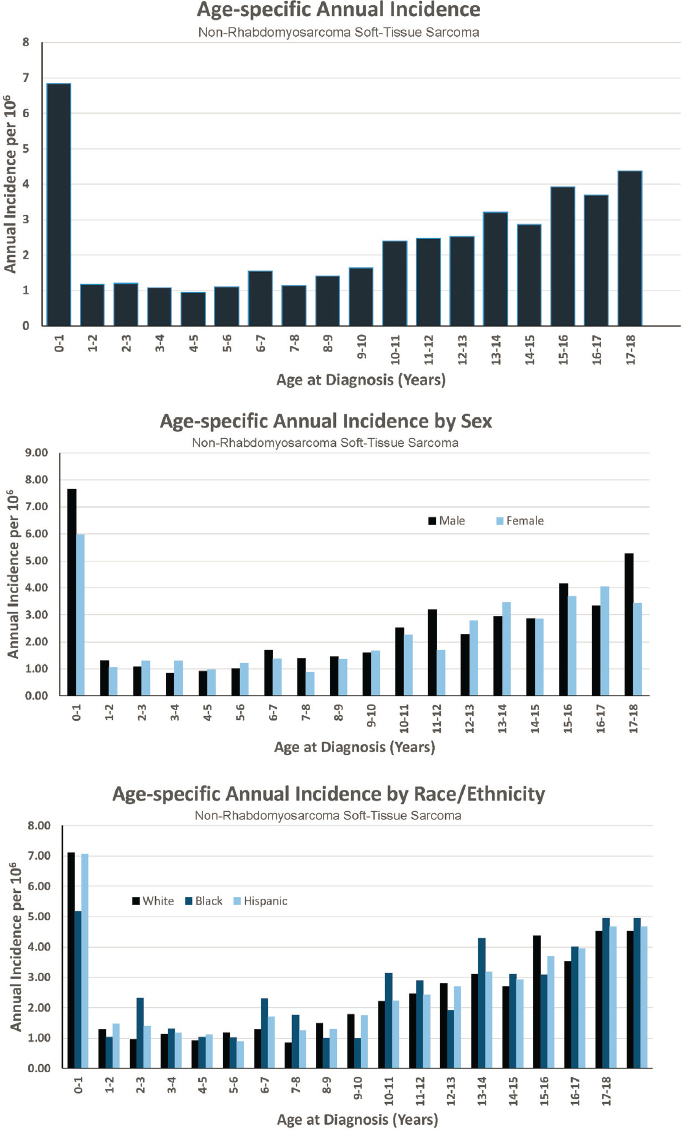
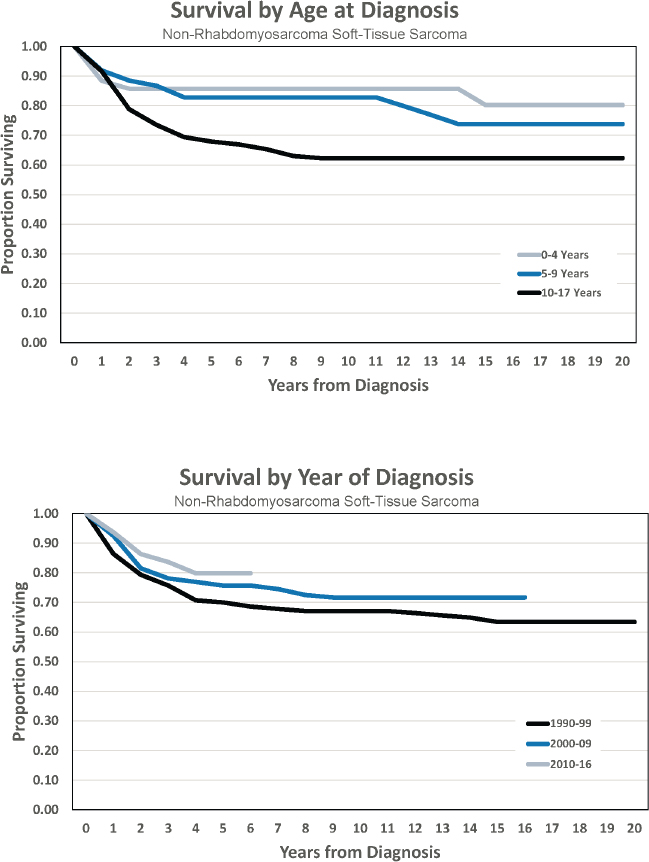
NON-RHABDOMYOSARCOMA SOFT-TISSUE SARCOMAS (NRSTS)
Diagnosed 0–17 Years of Age
- Annual incidence rate is 46 per 1,000,000 population
- Estimated 350 cases diagnosed annually
- Represents approximately 3 percent of cancers diagnosed before age 18
- Ratio of males to females is 1.1 to 1.0
- Estimated 5-year survival of 79.8 percent
- Estimated 10-year survival of 71.7 percent
Features of Non-Rhabdomyosarcoma Soft-Tissue Sarcomas
- NRSTS are a heterogeneous group of malignant tumors that, like rhabdomyosarcomas, arise from primitive mesenchymal tissue. They are more common in adolescents and adults than in younger children.
- Types of NRSTS include synovial sarcoma, fibrosarcomas, peripheral nerve sheath tumors, desmoplastic small round cell tumor (DSRCT), and other fibromatous neoplasms.
- Some types of NRSTS are defined by a characteristic chromosomal translocation or fusion protein, including synovial sarcoma (t(X;18)(p11;q11)) and DSRCT (EWS/WT-1 fusion).
- Genetic and exposure risk factors for NRSTS include, among others, Li-Fraumeni syndrome, familial adenomatous polyposis, germline mutations of the RB1 and SMARCB1 (INI1) genes, neurofibromatosis type 1, and prior radiation exposure.
- Prognosis varies greatly depending on the tumor type, primary site, size, grade, histology, and resectability, as well as depth of tumor invasion, presence and sites of metastases, and use of radiation therapy.
- Treatment is tailored to tumor type, size, and location and can include chemotherapy, surgery, radiation, and targeted therapy.
Figures for Non-Rhabdomyosarcoma Soft-Tissue Sarcomas
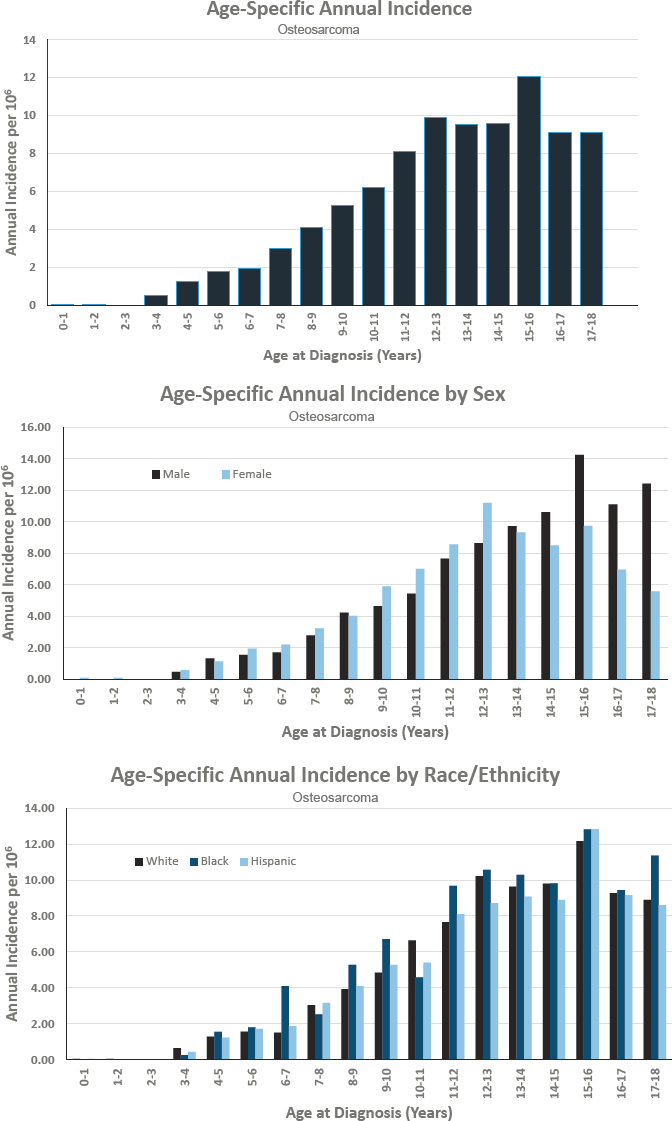
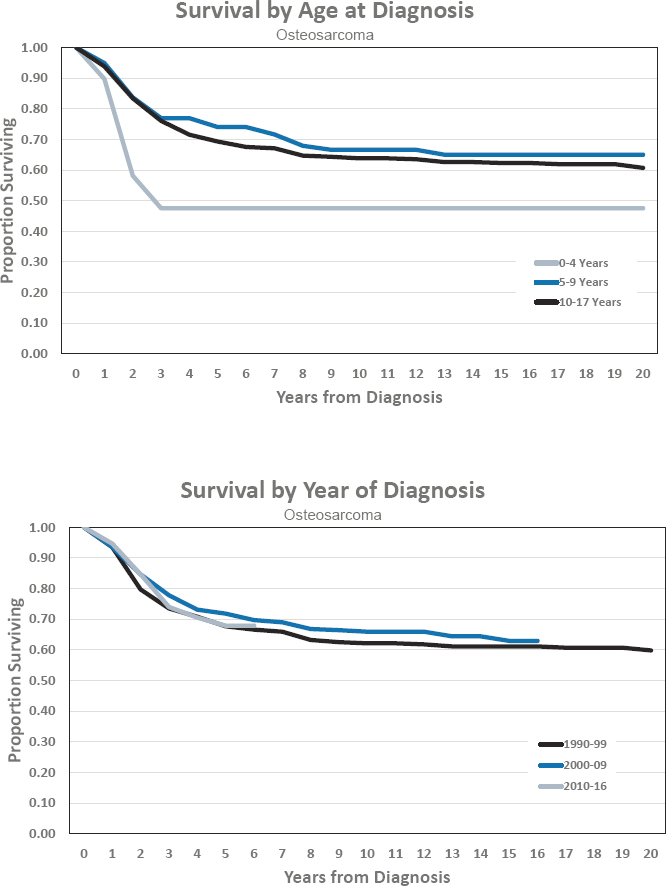
OSTEOSARCOMA
Diagnosed 0–17 Years of Age
- Annual incidence rate is 55 per 1,000,000 population
- Estimated 400 cases diagnosed annually
- Represents approximately 3 percent of cancers diagnosed before age 18
- Ratio of males to females is 1.1 to 1.0
- Estimated 5-year survival of 67.9 percent
- Estimated 10-year survival of 65.9 percent
Features of Osteosarcoma
- Osteosarcoma is a primary malignant bone tumor.
- Prognosis varies depending on the site, size, and initial treatment of the primary tumor; the presence of clinically detectable metastatic disease; the surgical resectability; and degree of necrosis of the primary tumor following preoperative chemotherapy.
- There are two risk categories for patients with metastatic disease: low-risk cases with resectable metastases at a single site that should be managed with a very aggressive chemotherapeutic and surgical approach; and high-risk cases for which new, innovative therapeutic approaches are needed.
- Treatment depends on grade. Treatment for low-grade tumors consists of surgery alone. Treatment for intermediate- and high-grade tumors consists of neoadjuvant chemotherapy, surgery, and adjuvant chemotherapy.
- Treatments and outcomes have not changed over the past 40 years.
Figures for Osteosarcoma
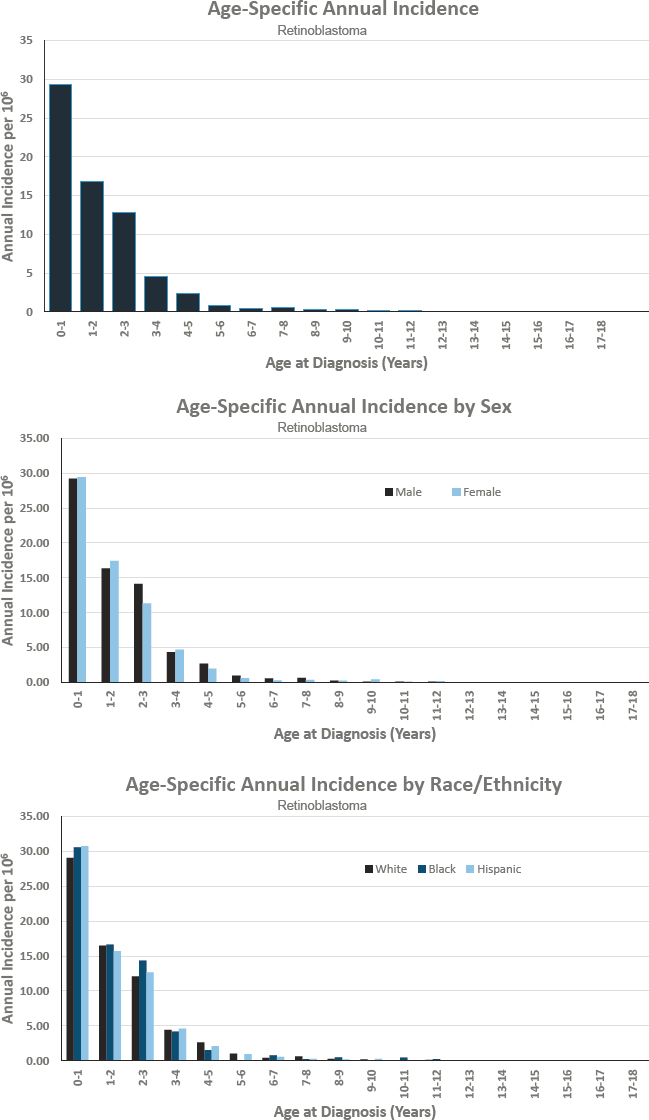
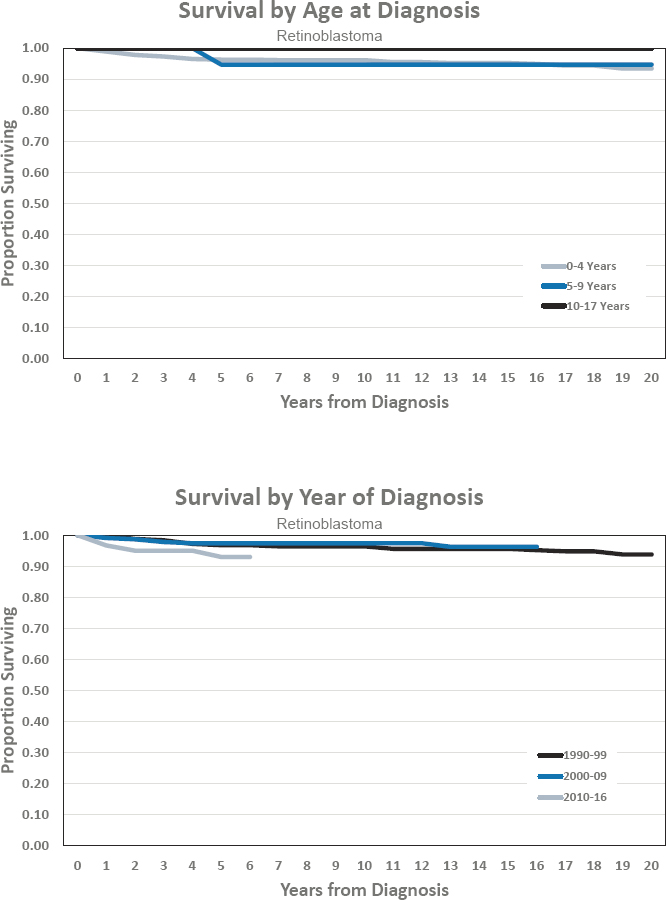
RETINOBLASTOMA
Diagnosed 0–17 Years of Age
- Annual incidence rate is 37 per 1,000,000 population
- Estimated 275 cases diagnosed annually
- Represents approximately 2 percent of cancers diagnosed before age 18
- Ratio of males to females is 1 to 1
- Estimated 5-year survival of 93.0 percent
- Estimated 10-year survival of 97.4 percent
Features of Retinoblastoma
- Retinoblastoma is the most common neoplasm of the eye in childhood, and two-thirds of cases occur in children less than 2 years old. It can occur in one eye (unilateral, 75 percent) or in both eyes (bilateral or multifocal, 25 percent).
- Heritable retinoblastoma (25 percent) is characterized by a germline mutation in the RB1 gene.
- The presence of positive family history or bilateral or multifocal disease is suggestive of heritable disease.
- For children with a family history of retinoblastoma, detailed screening guidelines have been developed. They include a detailed dilated eye exam under anesthesia at regular intervals.
- Several staging systems for retinoblastoma have been developed that take into account whether disease is in one or both eyes or multifocal and the extent of disease within the eye.
- The goal of the treatment of retinoblastoma is to cure the patient but also to preserve vision and reduce long-term toxicities and subsequent neoplasms.
- Retinoblastoma requires treatment by a multidisciplinary team with varied expertise using a risk-adapted individualized approach.
- Treatments for retinoblastoma include enucleation; focal treatments, such as cryotherapy; intra-arterial chemotherapy into the ophthalmic artery; systemic chemotherapy; and radiation therapy.
- Retinoblastomas are sensitive to radiation. However, because of the risk of second malignancies and other late effects, radiation is avoided if at all possible.
- Survivors of retinoblastoma are at high risk of developing subsequent neoplasms and also suffer from other late effects, including diminished orbital growth after enucleation, visual field defects, and chemotherapy-associated hearing loss.
Figures for Retinoblastoma
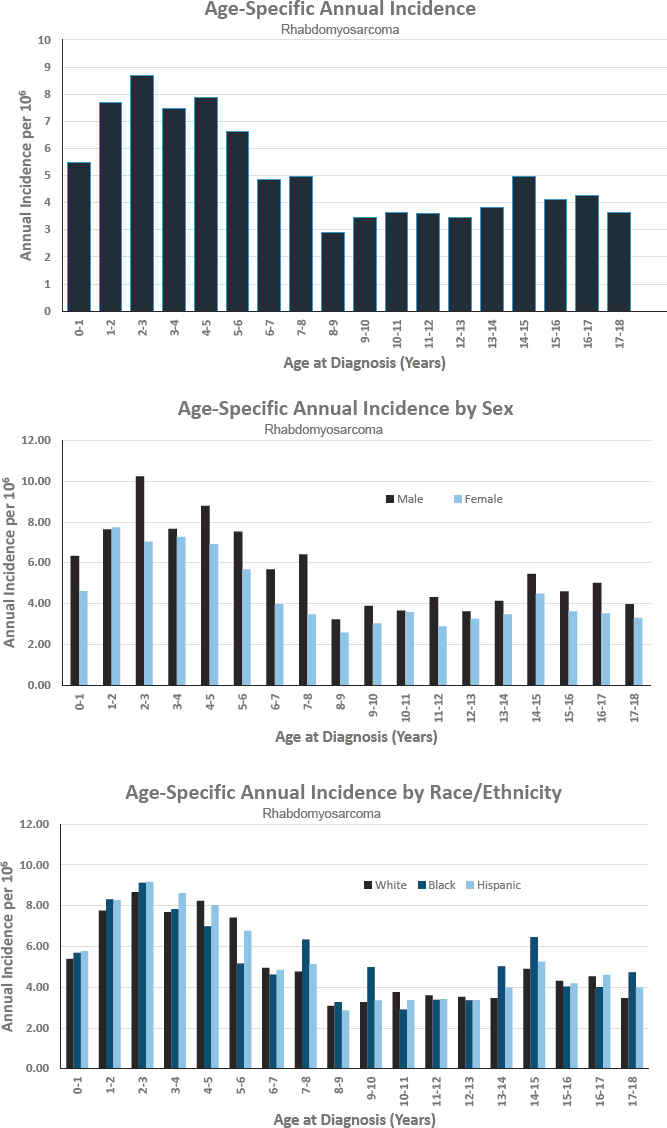
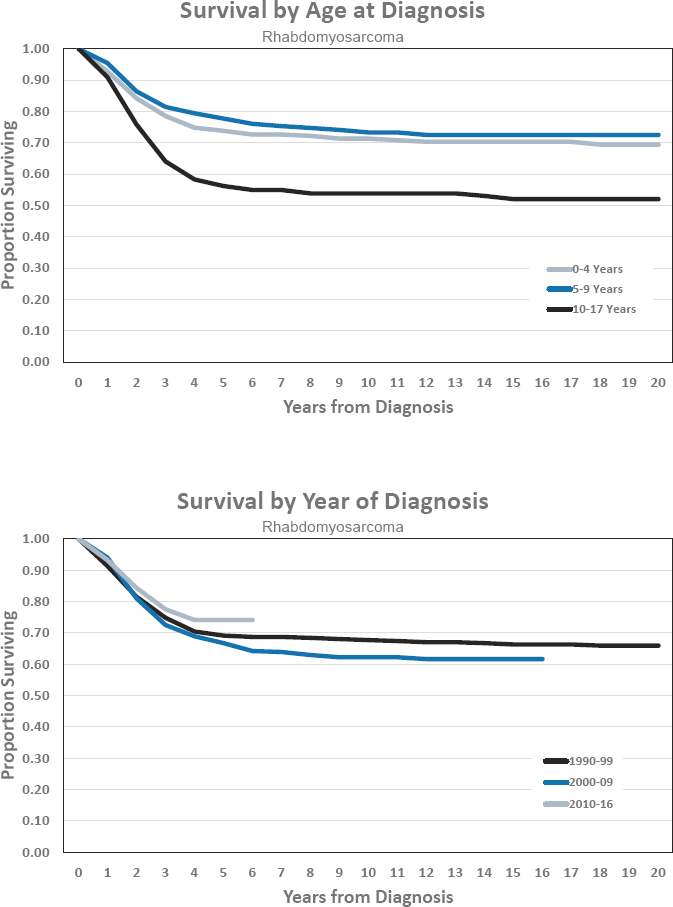
RHABDOMYOSARCOMA (RMS)
Diagnosed 0–17 Years of Age
- Annual incidence rate is 51 per 1,000,000 population
- Estimated 375 cases diagnosed annually
- Represents approximately 3 percent of cancers diagnosed before age 18
- Ratio of males to females is 1.3 to 1.0
- Estimated 5-year survival of 74.1 percent
- Estimated 10-year survival of 62.2 percent
Features of Rhabdomyosarcoma
- Treatment of RMS is complex and based on site, stage, extent of surgical resection at diagnosis, and PAX/FOXO gene status. These factors are assessed at diagnosis, allowing assignment to risk group. Chemotherapy, surgery, and radiation treatment are determined by risk group assignment.
- Prognosis can vary greatly depending on the site of disease, the invasiveness of the tumor (which is a surrogate for resectability), and the efficacy of radiotherapy. Also, PAX/FOXO gene status trumps alveolar or embryonal histology. For example, an embryonal RMS of the orbit has a ~90 percent survival and is curable, whereas an unresectable embryonal RMS of the pelvis has a ~50 percent survival with almost certain relapse.
- Prognostic subgroups can be defined as low risk, intermediate risk, high risk, and relapsed (see Annex Table 7-2 in Chapter 7).
- Risk-based treatment approaches are used to determine chemotherapy dose and duration, and timing of radiation and surgery.
- Specific to RMS are various PAX-FOX01 (PAX3 or PAX7) fusions. Whether an RMS tumor is fusion positive or fusion negative is the first factor that determines risk assignment at diagnosis.
- Other genetic markers of good and poor prognosis are presently being determined to further separate children with RMS into higher- and lower-risk groups at the time of diagnosis. This will allow a more tailored approach to tumors based on expected response to therapy and help identify novel targeted therapies for patients with these genetic aberrations.
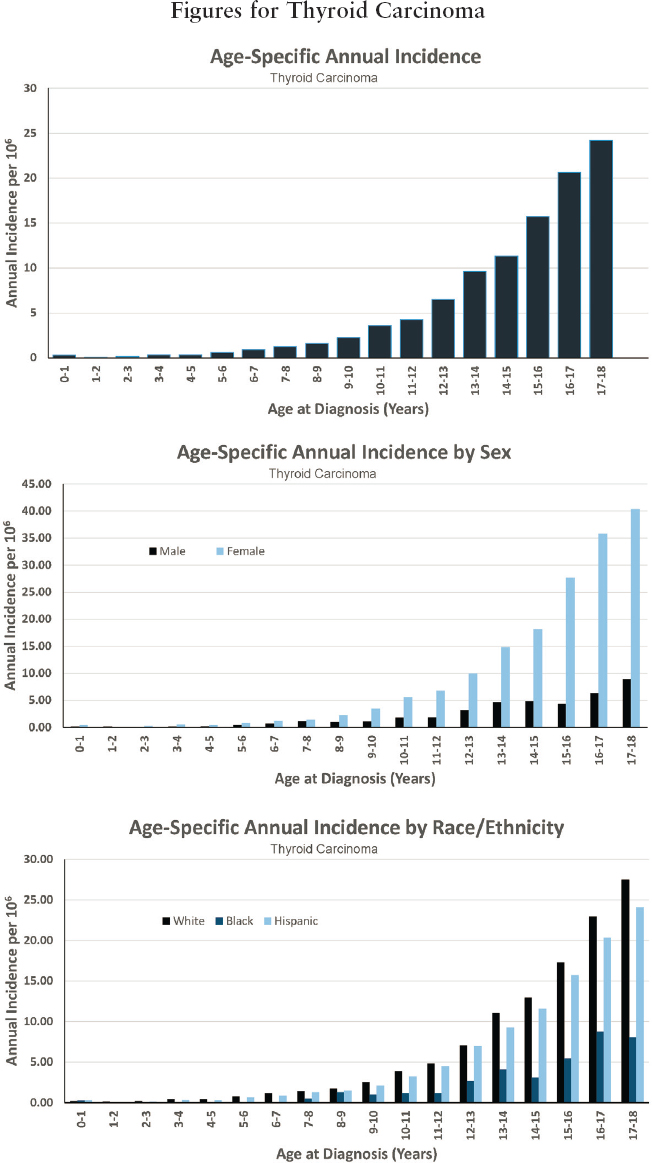
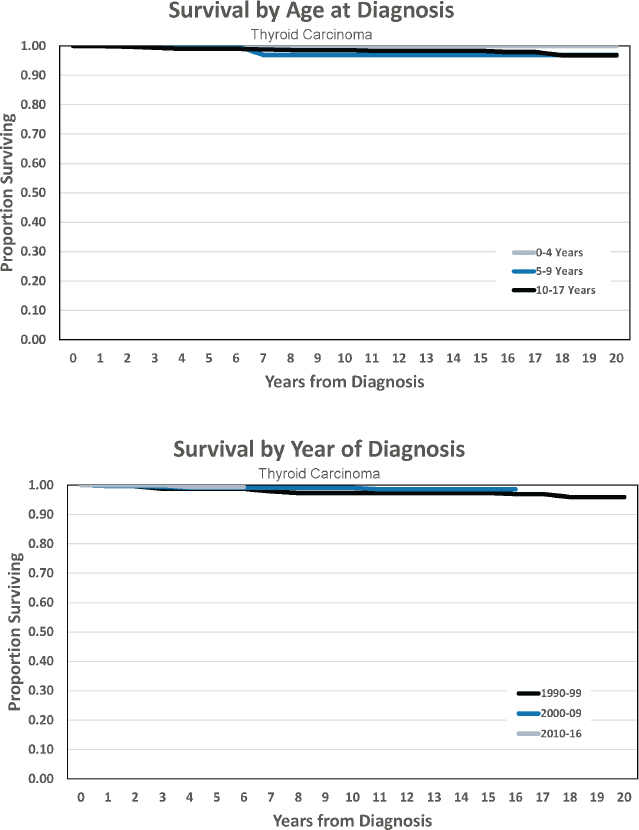
THYROID CARCINOMA
Diagnosed 0–17 Years of Age
- Annual incidence rate is 77 per 1,000,000 population
- Estimated 575 cases diagnosed annually
- Represents approximately 4 percent of cancers diagnosed before age 18
- Ratio of males to females is 0.2 to 1.0
- Estimated 5-year survival of 99.3 percent
- Estimated 10-year survival of 99.0 percent
Features of Thyroid Carcinoma
- Thyroid carcinomas are the most common secondary neoplasm in children and young adults surviving childhood cancers.
- Genetic tumor predisposition syndromes (multiple endocrine neoplasia [MEN], associated with the RET gene) have an important role in a subset of thyroid carcinomas.
- Differentiated thyroid carcinomas (DTCs) account for the majority of thyroid carcinomas, followed by medullary thyroid carcinomas (10 percent), and anaplastic thyroid carcinomas (1 percent).
- The clinical presentation is typically a thyroid mass, and sometimes enlarged lymph nodes. Presentation at younger age and larger tumor size are associated with more aggressive behavior.
- Treatment includes surgery, radioactive iodine ablation, and targeted therapies.
- Total thyroidectomy is the treatment of choice and should be performed by a surgeon with expertise in endocrine surgeries. If thyroid carcinoma has spread to the neck lymph nodes, central or lateral neck dissection is performed.
- 131I (Iodine) therapy is administered to patients with DTC and persistent or recurrent local or nodal disease that cannot be resected or known metastases. Treatment and dosing should be performed by experts in pediatric thyroid carcinoma.
- Targeted therapies may be considered, depending on the molecular profile of the tumor.
- Prophylactic thyroidectomy is recommended for MEN, with the timing dependent on the type of germline mutation.
- Most children will require lifelong thyroid hormone replacement. Long-term potential side effects of 131I treatment include dysfunction of the salivary glands, damage to the lung (pulmonary fibrosis), and secondary malignancies.
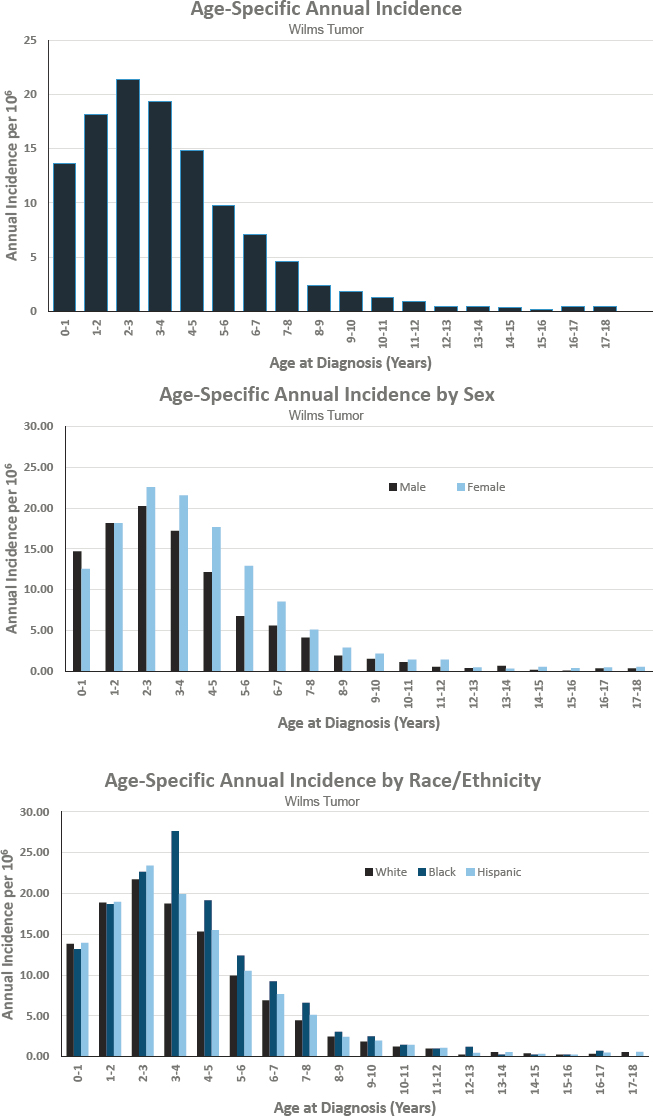
WILMS TUMOR
Diagnosed 0–17 Years of Age
- Annual incidence rate is 69 per 1,000,000 population
- Estimated 500 cases diagnosed annually
- Represents approximately 4 percent of cancers diagnosed before age 18
- Ratio of males to females is 0.8 to 1
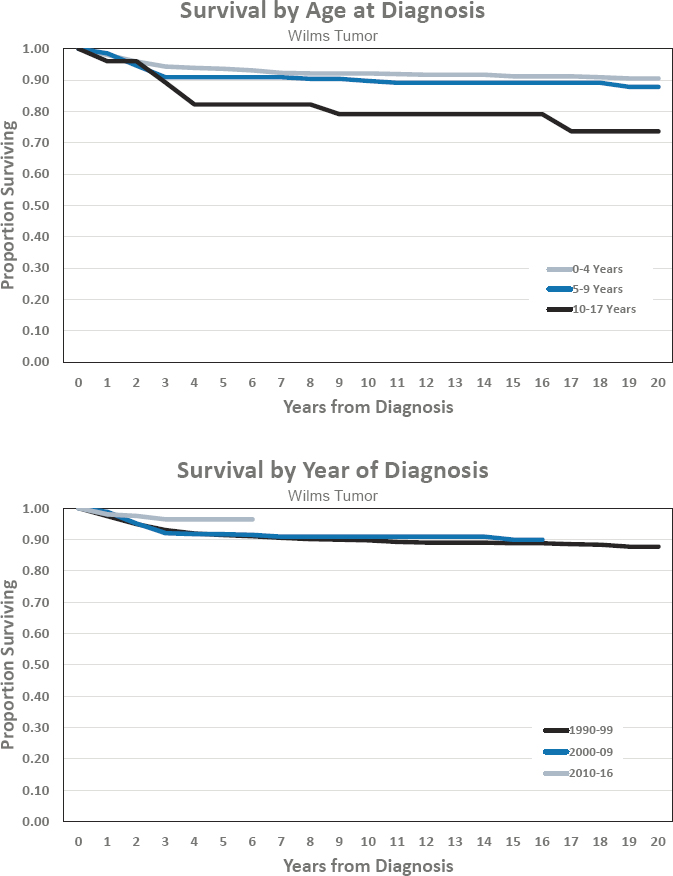
- Estimated 5-year survival of 96.4 percent
- Estimated 10-year survival of 90.9 percent
Features of Wilms Tumor
- Wilms tumor is the most frequent kidney tumor in children. Most cases (75 percent) occur in children less than 5 years of age.
- Wilms tumor is associated with a number of genetic conditions, including WAGR syndrome, Beckwith-Wiedemann syndrome, and Denys-Drash syndrome. In these children with increased risk for Wilms tumor, regular screening for Wilms tumor is mandatory.
- Survival for all patients with Wilms tumor together is greater than 90 percent, and even patients presenting with metastatic disease have high chance for cure.
- A number of prognostic factors have been identified and are used to tailor treatment for Wilms tumor.
- Very low-risk patients undergo surgery only while high-risk patients undergo surgery, multimodality chemotherapy, and radiation therapy.
- Patients with relapsed Wilms tumor may still have a good chance for cure depending on prior treatment and histology. Relapse with favorable histology Wilms tumor has a much better prognosis than relapse with anaplastic histology.
- Late effects from chemotherapy, radiation, and surgery need to be carefully monitored and include renal and cardiac dysfunction.
Figures for Wilms Tumor