
In 2018, FDA published draft guidance on drug development for major depressive disorder, which included advice for evaluating novel antidepressants (FDA, 2018), said Tiffany Farchione, director of the Division of Psychiatry in the Office of Neuroscience at FDA. Although the draft guidance was published before the approvals of esketamine and brexanolone, it was informed by the agency’s regulatory considerations from these drugs and other products under development at the time, she said.
Traditional antidepressants include all antidepressants approved prior to 2019—SSRIs, serotonin and norepinephrine reuptake inhibitors (SNRIs), MAO inhibitors, and other drugs that are taken orally on a daily basis, said Farchione. By contrast, novel antidepressants could include drugs given as a single dose with long-lasting effects, drugs given at fixed intervals, drugs given on an as-needed basis, and drugs given to jumpstart treatment for an initial period before the effects of traditional antidepressants are noticeable, she said. To approve such drugs, Farchione said, FDA needs to see data that enable the agency to write a label that tells prescribers how to use the product. She added that the study design will vary depending on the intended use of the drug. For example, to assess the efficacy of a rapidly acting antidepressant, an early endpoint is useful. In addition to that, later endpoints are also needed to characterize the durability of the effect. The population for whom the drug is intended also must be clearly defined, she said. For example, a drug may be developed for the treatment of treatment-resistant depression, PPD, or other types of depression.
Farchione noted that for traditional antidepressants, FDA has allowed maintenance studies to be conducted after market approval through randomized withdrawal studies because experience has demonstrated that these treatments continue to work. However, for novel antidepressants, data are needed to demonstrate appropriate dosing regimens over the long term for this chronic cyclical illness.
THE APPROVAL OF ESKETAMINE
As noted earlier, both esketamine and brexanolone received breakthrough therapy designation and priority reviews, and both had public advisory committee meetings prior to approval, said Farchione. Both of these drug approvals also include REMS, she said. For example, esketamine must be administered under supervision because it can induce feelings of dissociation. The initial treatment strategy for esketamine, which prescribes two doses per week, was based on preliminary data from previous ketamine studies. Because the drug is indicated for treatment-resistant depression, eligible trial participants must have failed at least two antidepressants of the same or different class, given at an adequate dose and of adequate duration. Farchione said inclusion in the trials was assessed retrospectively so that physicians could determine suitability for the trial based on the patient’s medication history. This approach enabled the trial to align with clinical practice, said Farchione.
There were concerns that patients who had not responded to many treatments, and thus were in need of effective care, might be given a placebo rather than an active drug, Farchione said. To address these concerns, trial participants were started on a new oral antidepressant at the same time that the double-blind intranasal esketamine treatment was initiated. This “in conjunction with” treatment, where an oral antidepressant and intranasal ketamine were in simultaneous use, was another aspect of clinical trials that differed from trials of other conventional drugs in review. It also resulted in esketamine’s FDA labeling including the words “in conjunction with” rather than calling it an adjunctive treatment. Furthermore, FDA obtained commitments that the submitting drug development company would study whether esketamine can be given as a monotherapy, said Farchione.
Because prior experience with traditional antidepressants is not applicable to drugs approved under a novel paradigm, Farchione said, FDA informed the submitting drug development company that it would not be sufficient to demonstrate an acute treatment effect to gain approval, but that they would also need to provide long-term data. In this case, the drug development company opted to conduct a trial supporting its unique dosing approach in which dosing changes over the course of treatment, starting with twice per week in the induction phase followed by once weekly or once every 2 weeks in the maintenance phase, she added.
The agency also addressed the issue of time to onset in a unique way, Farchione continued. Because the definition of “rapidly acting” is not specific, the FDA label includes a figure showing the time course of efficacy (see Figure 6-1). The figure shows that while the two treatment curves separate as early as 24 hours after initial dosing, symptoms continue to improve with time, resulting in a statistically significant treatment effect
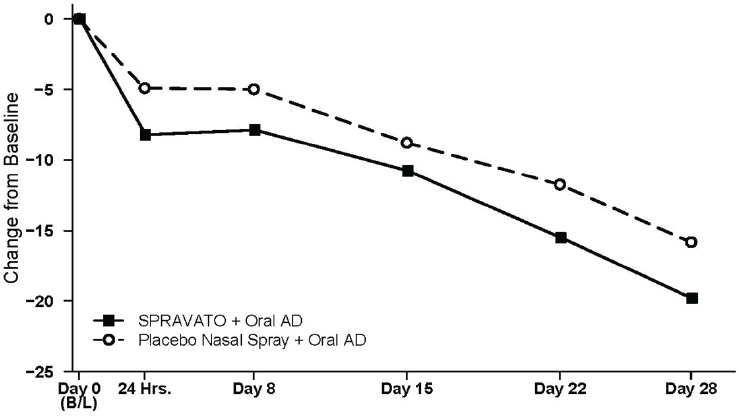
NOTES: In this flexible dose study, dosing was individualized based on efficacy and tolerability. Few subjects (<10 percent) had reduction in SPRAVATO dosage from 84 mg to 56 mg twice weekly.
SOURCES: Presented by Tiffany Farchione, March 9, 2021; esketamine full prescribing information, initial approval, March 5, 2019.
at the day 28 endpoint. Interestingly and unexpectedly, participants who received the intranasal placebo also improved when they switched their oral antidepressants; however, those in the esketamine group improved more, which led to the product approval, she said.
THE APPROVAL OF BREXANOLONE
Prior to the introduction of brexanolone, PPD was treated similarly to an episode of major depressive disorder, said Farchione. However, as noted by Samantha Meltzer-Brody, a more rapidly acting drug was needed to prevent the risk of lasting negative effects on both the mother and child.
Administered as a one-time, 60-hour infusion in a targeted population, the trials for brexanolone were complex, said Farchione. Participants in these studies were no more than 6 months postpartum. To assess the durability of the antidepressant effect while limiting the number of participants who might otherwise drop out of the study, the primary endpoint was the
Hamilton Depression Rating Scale (HDRS), assessed at hour 60 (the end of the infusion) and the secondary endpoint at day 30. Farchione said the rationale for making the second assessment at day 30 was that most participants did not start the trial immediately after giving birth, and if depression recurred more than 30 days later, it was more likely to reflect a traditional depressive episode that could be treated with a traditional antidepressant.
Administration of the drug required continuous monitoring, said Farchione, because in the development program some patients experienced excessive sedation or even loss of consciousness. When this occurred, patients were aroused without incident when the drug infusion was suspended, she said. Because investigators were unable to identify predictors of loss of consciousness, a more stringent monitoring plan was necessary. In addition, the company agreed to a post-marketing commitment to evaluate alternative dosing regimens, such as in an outpatient, non-24-hour facility. As with esketamine, the time to onset efficacy curves were included in the FDA label (see Figure 6-2). Farchione noted that a second study (not shown here) showed no difference from placebo at day 30 because the placebo group continued to improve and eventually caught up with the brexanolone-treated group.
Farchione noted that neither the Montgomery-Asberg Depression Rating Scale (MADRS), which was used in the esketamine trials, nor the HDRS are particularly good for detecting early signals of efficacy because many of the items rated by these measures would not be expected to change quickly. She said FDA accepted letters of intent for two patient-reported clinical outcome measures in development that focus on recall of depression symptoms over hours rather than days. She added that if either esketamine or brexanolone is evaluated for other chronic conditions such as PTSD, maintenance studies will most likely be required.
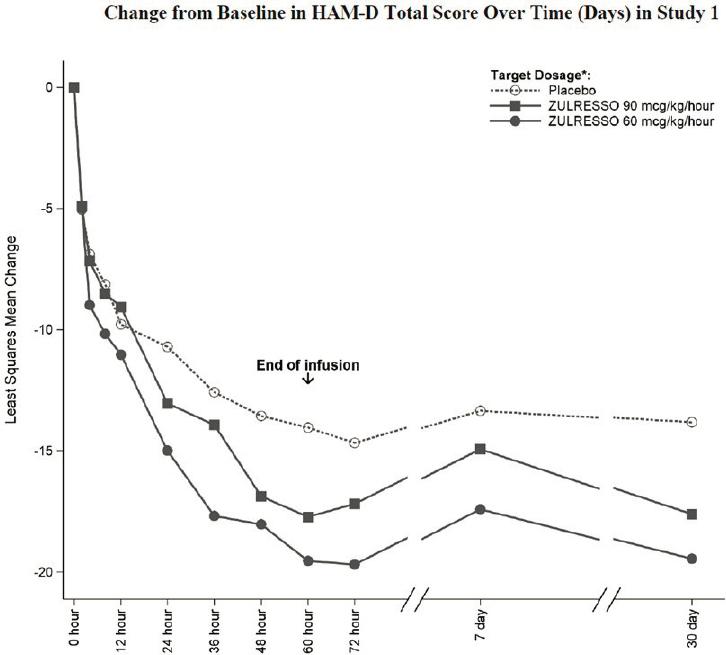
NOTE: ZULRESSO was administered via a 6-hour intravenous infusion as follows: 90 mcg/kg/hour-target dosage: 30 mcg/kg/hour for 4 hours, 60 mcg/kg/hour for 20 hours, 90 mcg/kg/hour for 28 hours, 60 mcg/kg/hour for 4 hours, 30 mcg/kg/hour for 4 hours; 60 mcg/kg/hour-target dosage: 30 mcg/kg/hour for 4 hours, 60 mcg/kg/hour for 52 hours, 30 mcg/kg/hour for 4 hours.
SOURCES: Presented by Tiffany Farchione, March 9, 2021; brexanolone full prescribing information, initial approval, March 19, 2019.






