
19
Choline: Human Requirements and Effects on Human Performance
Steven H.Zeisel1
INTRODUCTION
Choline is an essential component of the human diet and is important for the normal functioning of all cells (Zeisel and Blusztajn 1994). As such, it has great potential for use as a dietary modulator of human performance. Several mechanisms can explain how these effects are mediated. Acetylcholine synthesis can be influenced by the availability of choline; it is an important neurotransmitter controlling such diverse neural functions as memory and control of muscle function. Choline-phospholipids are extremely important structural elements of cells and are essential for the normal processing of dietary fat. Recently, researchers have begun to understand that choline-phospholipids also are transducers of signals from the exterior of cells to the nucleus. This signaling mechanism is so important and is so widely
distributed that its manipulation by changing the diet is likely to be a powerful tool for improving human performance.
Choline is present in some form in all cells, predominantly as one of the choline-phospholipids phosphatidylcholine, lysophosphatidylcholine, choline plasmalogen, platelet-activating factor, or sphingomyelin. All of these are essential components of all membranes (Zeisel, 1990). The importance of choline as a nutrient was first appreciated during the pioneering work on insulin when the association between a low-choline diet and fatty infiltration of the liver was recognized (Best and Huntsman, 1935). The term lipotropic was coined to describe choline and the other substances that prevent the deposition of fat in the liver.
Until recently, choline was considered a dispensable nutrient for humans because there is an endogenous pathway for the de novo biosynthesis of the choline moiety (Bremer and Greenberg, 1961). In addition, the demand for choline is modified by the rate of growth of an individual and by complex interrelationships between choline and the nutrients methionine, folic acid, and vitamin B12 (lipotropes) (Zeisel, 1988). However, it is known that human cells grown in culture have an absolute requirement for choline (Eagle, 1955), and recent studies have established that choline is indeed an essential nutrient for normal humans when methionine is not available in excess of requirements (Zeisel et al., 1991) (see discussion below).
DIETARY SOURCES OF CHOLINE
Calculations of dietary choline intake are based on estimates of the free choline and phosphatidylcholine contents of foods (Engel, 1943; Food and Nutrition Board, 1973; McIntire et al., 1944; Weihrauch and Son, 1983; Zeisel et al., 1986). Measurements of the lysophosphatidylcholine, glycerophosphocholine, and phosphocholine contents of rat tissues (Pomfret et al., 1989) show that these choline-containing compounds are also present in high concentrations in many tissues (e.g., the concentrations of each of these three esters in rat muscle were approximately 100 nmol/g.) Thus, the foods eaten by humans probably also contain significant amounts of these esters of choline. In addition, the choline concentrations in tissues rise postmortem. For these reasons, choline intake (especially unesterified choline intake) is probably greatly underestimated.
In addition to the naturally occurring choline in foods, significant amounts are added as dietary supplements. Choline chloride and choline bitartrate are listed in the Code of Federal Regulations as nutrients and/or dietary supplements that are generally recognized as safe (Federation of American Societies for Experimental Biology, 1975). More than 5,000 kg of choline chloride and
more than 12,000 kg of choline bitartrate were used in the manufacture of foods in 1970 (Federation of American Societies for Experimental Biology, 1975). Most of the choline added to foods was found in infant formulas (Federation of American Societies for Experimental Biology, 1975).
On the basis of these data and reasonable estimates of food intake, total choline intake in the adult human (as free choline and the choline in phosphatidylcholine and other choline esters) is greater than 700–1,000 mg/day (Federation of American Societies for Experimental Biology, 1981; Zeisel, 1981). When humans were switched from a diet of normal foods to a defined diet containing 500 mg of choline (750 mg of choline chloride), Zeisel and colleagues (1991) observed that plasma choline and phosphatidylcholine concentrations decreased in most subjects. This suggests that normal dietary intake of choline exceeds 500 mg. Consumption of choline is higher in humans who ingest phosphatidylcholine (also called lecithin) as a dietary supplement (the capsules or granules sold over the counter are usually only 35 percent phosphatidylcholine).
Plasma choline concentrations in fasting subjects vary from 7 to 20 µM, with the plasma of most adult human subjects having concentrations of 10 µM, whereas plasma phosphatidylcholine concentrations are approximately 1–1.5 mM (Aquilonius et al., 1975; Sheard et al., 1986; Zeisel et al., 1980, 1991). Liver phosphocholine is the most labile pool of choline and seems to be the most sensitive indicator of choline nutriture (Pomfret et al., 1990). In the adult human, serum choline concentrations fluctuate modestly (increase 1.5-fold) when common choline-containing foods are ingested (Zeisel et al., 1980). Total body stores of choline in humans can be estimated on the basis of measurements of choline pool concentrations in animal tissues; it is estimated that a 70-kg human contains more than 5 mmol of free choline (500 mg) and more than 300 mmol of choline (30 g) in esterified form (Zeisel et al., 1991).
The only source of choline other than from the diet is from the de novo biosynthesis of phosphatidylcholine catalyzed by phosphatidylethanolamine-N-methyltransferase (PeMT). This enzyme synthesizes phosphatidylcholine via sequential methylation of phosphatidylethanolamine by using S-adenosylmethionine as a methyl donor (Blusztajn et al., 1979; Ridgway and Vance, 1987; Zeisel, 1981). Most PeMT activity is found in the liver (Bjornstad and Bremer, 1966).
CHOLINE AND METHYL GROUP METABOLISM
The demand for choline as a methyl donor is probably the major factor that determines how rapidly a diet deficient in choline induces pathology. The pathways of choline and 1-carbon metabolism intersect at the formation of
methionine from homocysteine (Figure 19–1) (Finkelstein et al., 1982; Mudd and Poole, 1975; Wong and Thompson, 1972). Methionine is regenerated from homocysteine in a reaction catalyzed by betaine: homocysteine methyltransferase, in which betaine, a metabolite of choline, serves as the methyl donor (Finkelstein et al., 1982). The only alternative mechanism for regeneration of methionine is via a reaction catalyzed by 5-methyltetra-hydrofolate:-homocysteine methyltransferase, which uses a methyl group generated de novo from the 1-carbon pool (Finkelstein et al., 1982, 1988). Methionine is converted to S-adenosylmethionine in a reaction catalyzed by methionine adenosyl transferase. S-Adenosylmethionine is the active methylating agent for many enzymatic methylations.
A disturbance in folate or methionine metabolism results in changes in choline metabolism and vice versa. During choline deficiency, the hepatic choline concentration decreases rapidly (Zeisel et al., 1989). At the same time, hepatic S-adenosylmethionine concentrations are decreased (Barak et al., 1982; Poirier et al., 1977; Shivapurkar and Poirier, 1983; Zeisel et al., 1989). It has been suggested that the availability of methionine limits S-adenosylmethionine synthesis during choline deficiency because the 5-methyltetrahydrofolate:-homocysteine methyltransferase reaction alone cannot fulfill the total requirement for methionine, and the betaine-dependent remethylation of homo
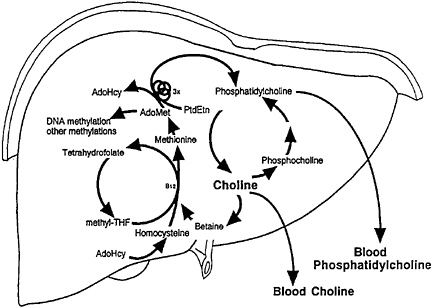
FIGURE 19–1 Choline, folate, and methionine metabolism are closely interrelated. AdoHcy, S-adenosylhomocysteine; AdoMet, S-adenosylmethionine; PtdEtn, phosphatidylethanolamine; THF,
cysteine is limited by the availability of betaine (Finkelstein et al., 1982). Choline deficiency is also associated with inhibition of hepatic glycine-N-methyltransferase activity, which is believed to be important for the removal of excess S-adenosylmethionine from the liver (Cook et al., 1989). Betaine concentrations in the livers of choline-deficient rats are markedly diminished (Barak and Tuma, 1983; Finkelstein et al., 1982; Wong and Thompson, 1972), as are total folate concentrations (Horne et al., 1989).
Methotrexate, which is widely used in the treatment of cancer, psoriasis, and rheumatoid arthritis, limits the availability of methyl groups by competitively inhibiting dihydrofolate reductase, a key enzyme in intracellular folate metabolism. When 1-carbon metabolism is poisoned, the only alternative to choline as a source of methyl groups for regeneration of methionine is lost. Hepatic choline, phosphocholine, S-adenosylmethionine, and betaine concentrations are diminished after treatment with methotrexate (Barak and Kemmy, 1982; Barak et al., 1984; Freeman-Narrod et al., 1977; Pomfret et al., 1990; Svardal et al., 1988). Folate metabolism is also altered in choline-deficient rats (Horne et al., 1989; Selhub et al., 1991), which is reflected by the greater residence time of folate molecules within liver.
REQUIREMENT FOR CHOLINE IN HUMANS
In the rat (Lombardi, 1971), hamster (Handler and Bernheim, 1949), guinea pig (Tani et al., 1967), pig (Blair and Newsome, 1985; Fairbanks and Krider, 1945), dog (Best and Huntsman, 1932; Hershey and Soskin, 1931), monkey (Hoffbauer and Zaki, 1965), trout (Ketola, 1976), quail (Ketola and Young, 1973), and chicken (Ketola and Nesheim, 1974), choline deficiency results in liver dysfunction. Extremely large amounts of lipid (mainly triglycerides) accumulate in the liver during choline deficiency (Blusztajn and Zeisel, 1989; Lombardi, 1971; Lombardi et al., 1968; Yao and Vance, 1988, 1989), beginning within hours to days after rats are started on a choline-deficient diet (daCosta et al., 1993). Triacylglycerol accumulation occurs because it is secreted from liver as very-low density lipoprotein (VLDL), and phosphatidylcholine is a required component of VLDL (Yao and Vance, 1988, 1989).
Healthy humans fed a choline-deficient diet for 3 weeks developed biochemical changes consistent with choline deficiency (Zeisel et al., 1991). Subjects were admitted to the Clinical Research Center at Boston University School of Medicine and were constantly observed for 5 weeks. During the first week all subjects consumed the same choline-containing diet. During the middle 3 weeks of the study the control group continued on the choline-containing diet while the choline-deficient group consumed the same
diet without choline. During the fifth week all subjects consumed the choline-containing diet. Humans ingesting a choline-deficient diet for 3 weeks had diminished plasma choline and phosphatidylcholine concentrations, as well as diminished erythrocyte membrane phosphatidylcholine concentrations. Serum alanine transaminase activity, a measure of hepatocyte damage, increased significantly when a choline-deficient diet was ingested. This experiment establishes a requirement for choline in the diets of normal humans.
Humans with Special Needs for Choline
Choline deficiency may be of clinical importance in several groups of individuals. Humans running a marathon have lower blood choline concentrations after the run than before the run (Conlay et al., 1986). Supplementation with choline before and during a 32-km (20-mile) run prevented this drop in plasma choline and improved the subjects’ run times by 5 min (Sandage et al., 1992). The reasons for this drop in choline are undefined and might not reflect the utilization of choline but the redistribution of choline as fluid pools shift during exertion.
The demand for choline in normal adults is likely to be smaller than the demand for choline in infants, because large amounts of choline must be used to make phospholipids in growing organs (Zeisel, 1990). The observed changes that occurred in choline-deficient adult humans might have been greater if growing children were studied. Malnourished humans, in whom stores of choline, methionine, and folate have been depleted (Chawla et al., 1989; Sheard et al., 1986), are also more likely than healthy adult subjects to need dietary choline. Fatty liver develops in obese rats in which 90 percent of their small intestine was bypassed. Choline supplementation prevented this, and choline-deficient diets in such animals exacerbated the accumulation of fat in the liver (Kaminski et al., 1980). Amino acid-glucose solutions used in the total parenteral nutrition of humans contain no choline (Chawla et al., 1985; Sheard et al., 1986). The lipid emulsions used to deliver extra calories and essential fatty acids during parenteral nutrition contain choline in the form of phosphatidylcholine (a 20 percent emulsion contains 13.2 µmol/ml), and humans treated with parenteral nutrition required 1,000–1,700 µmol (approximately 800–1,360 mg) of choline-containing phospholipid per day during the first week of parenteral nutrition therapy to maintain plasma choline levels (Sheard et al., 1986). Burt et al. (1980) reported that plasma choline concentrations were decreased in patients receiving parenteral nutrition at the same time that liver dysfunction was present. Conditions that enhance hepatic triglyceride synthesis (such as carbohydrate loading) increase the requirement for choline
for the export of triglyceride from liver (Carroll and Williams, 1982). Thus, treatment of malnourished patients with high-calorie parenteral nutrition solutions at a time when choline stores are depleted might enhance the likelihood of hepatic dysfunction. When supplemental choline (in the form of lecithin) was administered during parenteral nutrition in humans, plasma choline levels returned to normal, and the incidence of hepatic dysfunction and steatosis diminished (Buchman et al., 1992). Subjects treated with placebo did not get better. These observations, that humans fed very-high-calorie diets develop fatty liver that can be alleviated by choline treatment, may be of relevance to designers of very-high-calorie rations for soldiers.
OTHER MECHANISMS WHEREBY CHOLINE INFLUENCES CELLULAR FUNCTION
As discussed earlier, choline is required for two types of signaling mechanisms that are fundamental modulators of the functions of many cells. It is a precursor for the widely distributed neurotransmitter acetylcholine, and it is a precursor for choline-phospholipids, which play a vital role in the regulation of transmembrane signaling.
Cholinergic Neurotransmission in the Brain
Only a small fraction of dietary choline is acetylated, catalyzed by the activity of choline acetyltransferase (Haubrich et al., 1975b; White and Cavallito, 1970). Choline acetyltransferase is highly concentrated in the terminals of cholinergic neurons (Malthe and Fonnum, 1972), but it is also present in such non-nervous system tissues as the placenta (Rama Sastry and Henderson, 1972). The availability of choline and acetyl-coenzyme A (acetyl-CoA) influence choline acetyltransferase activity (Cohen and Wurtman, 1975, 1976; Haubrich et al., 1974, 1975a). In the brain it is unlikely that choline acetyltransferase is saturated with either of its substrates, so that choline (and possibly acetyl-CoA) availability determines the rate of acetylcholine synthesis (White and Wu, 1973). Some investigators report that administration of choline or phosphatidylcholine results in the accumulation of acetylcholine within brain neurons (Haubrich et al., 1974, 1975a; Cohen and Wurtman, 1975, 1976), whereas others observe that such acceleration of acetylcholine synthesis by choline administration can be detected only after pretreatments with agents that cause cholinergic neurons to fire rapidly (Miller et al., 1989; Trommer et al., 1982; Wecker, 1986, 1988; Wecker and Dettbarn, 1979; Wecker et al., 1989). Increased brain acetylcholine synthesis is associated with an augmented
release into the synapse of this neurotransmitter. A temporal dissociation between choline administration and effects on brain acetylcholine synthesis and release has been observed (Trommer et al., 1982). The choline taken up by the brain may first enter a storage pool (perhaps the phosphatidylcholine in membranes) before being converted to acetylcholine.
Pharmacological amounts (5–20 g) of supplemental choline characteristically have been administered to alter cholinergic neurotransmission. Choline chloride supplementation (500 mg/kg of body weight in drinking water) increased the number of dendritic spines in the cerebral cortexes of old mice (Bertoni-Freddari et al., 1985; Mervis et al., 1985). In these same animals, memory, as assessed by learning performance, was improved by choline supplementation (Bartus et al., 1980). Choline supplementation during both pre- and postnatal development had long-term enhancing effects on spatial memory in rats, and this improvement was still detected in adults who were treated as infants (Meck et al., 1989). In those studies, effects were detected as late as 8 months after treatment; these changes have now been shown to persist beyond 26 months of age (W.H.Meck, Columbia University, personal communication, 1993). Choline supplementation during the perinatal period led to a significant reduction in both the number of working memory errors and the number of reference memory errors made both during acquisition and at steady-state performance.
The mechanism for this effect of choline on brain function has not been elucidated. Adult rats treated perinatally with choline had increased muscarinic receptor densities as measured by [3H]quinuclidinyl benzilate binding in both the hippocampus and the frontal cortex when compared with those of untreated littermates (Meck et al., 1989). Levels of choline acetyltransferase in the hippocampus were significantly lower in choline-treated rats, although there was no significant change in their levels in the frontal cortex (Meck et al., 1989). Acetylcholine neurotransmission is important for memory in normal humans and rodents (Bartus et al., 1982), and acetylcholine synthesis can be driven by the increased availability of choline (Blusztajn and Wurtman, 1983). Increased choline can enhance activity in the basal forebrain cholinergic system, thereby leading to improved memory function in a variety of species, including rats, mice, mollusks, and humans (Barry and Gelperin, 1982; Bartus et al., 1980; Sahley et al., 1986; Sitaram et al., 1978a,b). Choline may influence brain function via its effects on phospholipid biosynthesis (Schmidt and Wecker, 1981; Wecker, 1986, 1990). These observations suggest that the effects of choline on brain function, mediated by increased cholinergic neurotransmission or by changes in brain membrane and structures, may be a performance-enhancing effect of choline that can be exploited when designing rations for soldiers.
Neuromuscular Junction
Administration of choline increases acetylcholine (ACh) synthesis and release in peripheral neurons (muscle, adrenal, and heart cholinergic neurons) (Wurtman et al., 1980). Choline availability directly influences ACh synthesis and release at the neuromuscular junction. Definitive demonstration of this has been obtained by using the isolated phrenic nerve-diaphragm preparation (Bierkamper and Goldberg, 1980). The phrenic nerve was stimulated at 7 Hz (0.2 ms, 0.6 V), and release of ACh was measured and compared with spontaneous release (no stimulation). Physiological variations in choline in the perfusing medium (0–60 µM) resulted in marked changes in ACh release. In the absence of choline, electrical stimulation of the nerve for 1 h evoked a mean release of 5.13±0.62 pmol/min. In the presence of 30 or 60 µM choline, choline-stimulated release increased to 6.75±0.07 and 9.78±1.67 pmol/min, respectively. These changes were highly significant (P<0.01).
Humans fed a choline-deficient diet for 3 weeks (see earlier discussion) (Zeisel et al., 1991) were tested electromyographically for muscle conduction velocity while exerting a constant force with their anterior tibialis muscle. As fatigue began, conduction velocity diminished. The slope of decay of conduction velocity was more than two times faster in those given a choline-deficient diet than in controls (S.Zeisel, unpublished data, 1993).
The effects of choline on peripheral cholinergic parasympathetic neurotransmission are also significant. Intravenous choline administration (8–32 mg/kg) produces a sharp fall in blood pressure in rats (Singh, 1973) but not in cats (40 mg/kg of body weight intravenously [Kapp et al., 1970]). Oral administration of choline (10 g/day) had a slight hypotensive effect in humans (Boyd et al., 1977). Choline could be acting by increasing vagal tone to the heart or by dilating arterioles. Although added choline increases acetylcholine release in in vitro preparations of heart (Loffelholz, 1981), changes in the cardiac rate have not been observed in healthy humans treated with choline.
The urinary bladder muscle is also controlled by parasympathetic cholinergic neurotransmission. Bladders isolated from choline-deficient animals showed a 46 percent increase in the maximum response to acetylcholine compared with those from animals with normal levels of choline, whereas bladders from animals on choline-enriched diets showed a 15 percent decrease in maximum contractile response. The results are consistent with the hypothesis that muscarinic receptors are down-regulated to compensate for the increased parasympathetic activity associated with choline-enriched diets and up-regulated to compensate for the decreased parasympathetic activity associated with choline-deficient diets (Wallace et al., 1985)
Choline and Signal Transduction
Current Concepts of Signal Transduction
Signals are transmitted across membranes via a highly interactive cascade of molecular events (Figure 19–2). In the simplest form, an extracellular sensor detects a signal and transmits this message into the interior of the cell via a protein phosphorylation cascade to a final component that acts as a transcriptional regulator. Phospholipids act as vital elements in transmembrane signaling. Agonist-induced hydrolysis of phosphatidyl-inositol bisphosphate linositides (PtdIns) has been established as a major mechanism for transmitting messages into the interior of cells via protein phosphorylation cascades, which ultimately regulate gene transcription. There is a growing body of evidence that choline phospholipids (phosphatidylcholine, sphingomyelin, and their metabolites) are also important mediators and modulators of transmembrane signaling. These functions may explain how choline phospholipids influence normal physiological processes as well as a diverse group of pathological processes.
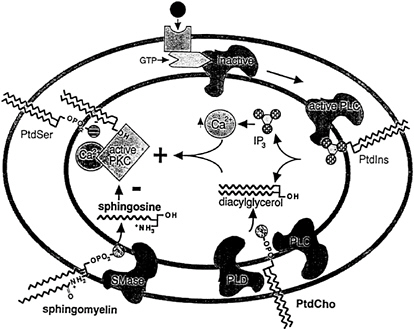
FIGURE 19–2 Choline-phospholipids and signaling. Activation of receptor results in the subsequent activation of phospholipases (PLC, phospholipase C; PLD, phospholipase D), which hydrolyze phosphatidylinositol bisphosphate (PtdIns) and phosphatidylcholine (PtdCho). Diacylglycerol and inositol-1,4,5-trisphosphate (IP3) are generated. These second messengers activate protein kinase C (PKC). Subsequent hydrolysis of sphingomyelin generates sphingosine and ceramide, which are inhibitors of PKC and act to terminate signaling. SMase, sphingomyelinase; PtdSer, phosphatidylserine; OH, hydroxyl group; NH3+, ammonium ion Ca2+, calcium ion; GTP, guanoisne triphosphate; OPO2, phosphate.
Hundreds of messengers whose effects are mediated by hydrolysis of phospholipids have been identified. An incomplete list includes acetylcholine (M1 receptor), norepinephrine (α1), epinephrine (α1), dopamine, histamine (H1 receptor), serotonin, vasopressin, angiotensin II, cholecystokinin, gastrin, pancreozymin, substance P, bradykinin, thromboxane, thrombin, collagen, platelet-activating factor, secretagogues, growth factors, and most mitogens. For many of these messengers, the receptor-ligand interaction leads to altered conformation of the receptor so that it can activate a GTP (guanosine diphosphate)- binding protein (G protein). In the inactive state, GDP (guanosine triphosphate) is bound to the complex. Activation triggers replacement of GDP with GTP. The activated G protein stimulates the next effector protein in the signal cascade. The GTPase activity intrinsic to the α subunit eventually hydrolyzes GTP, ending the signal (Meldrum et al., 1991).
The activation of the G protein results in the subsequent activation of phospholipase C (PLC) activity within the plasma membrane. The PLCs are a family of phosphodiesterases that hydrolyze the glycerophosphate bond of intact phospholipids to generate 1,2-sn-diacylglycerol (DAG) and an aqueous soluble head group. It is believed that specific receptors couple to specific PtdIns-PLC isotypes (Meldrum et al., 1991). In a similar manner, specific receptors appear to be linked to activation of specific phosphatidlycholine-PLCs (Exton, 1990) (see discussion below).
The action of PLC triggers the next event in the signal cascade, which is the activation of protein kinase C (PKC; serine-threonine kinases). The first step in PKC activation is the formation of a enzyme-calcium ion (Ca2+)-phospholipid complex. The products generated by PtdIns-PLC include inositol-1,4,5-trisphosphate (IP3) and DAG. IP3 is a water soluble product that acts to release calcium from stores in the endoplasmic reticulum. This increase in cytosolic calcium makes more calcium available for binding to PKC isotypes that are Ca2+ dependent (PKCsα, ß1/2, and γ; PKCs δ, ε, ζ, θ and η lack the calcium-binding C2- domain of PKC and therefore are not calcium dependent; Stabel and Parker, 1991). Calcium increases the tightness of association of these PKCs with the membrane, thereby increasing membrane occupancy. This facilitates binding of DAG, which, as described earlier, is the other product of PLC activity. The DAG-PKC complex approaches the membrane more closely, placing the kinase in a pocket of negatively charged phosphatidylserine head groups, into which Ca2+ is attracted. Thus, DAG increases the affinity of PKC for calcium. Normally, PKC is folded so that an endogenous pseudosubstrate region on the protein is bound to the catalytic site, thereby inhibiting activity. The combination of DAG and Ca2+ causes a conformational change in PKC, causing flexing at a hinge region so as to withdraw the pseudosubstrate and unblock the PKC catalytic site. The
appearance of diacylglycerol in membranes is usually transient, and therefore, PKC is activated only for a short time after a receptor has been stimulated.
The events that occur downstream from PKC are just beginning to be characterized. Serine-threonine kinases and tyrosine kinases catalyze phosphorylation events distal to PKC. These phosphorylation cascades serve to enhance amplification of the original signal. Clearly, PKC signals impinge on several known intracellular control circuits (Stabel and Parker, 1991). The targets for phosphorylation by PKC include receptors for insulin, epidermal growth factor, and many of the proteins involved in the control of gene expression, cell division, and cell differentiation (Nishizuka, 1986; Weinstein, 1990). It is likely that a plethora of PKC targets will be studied in the next few years.
Choline Phospholipids and Signal Transduction
The hydrolysis of phosphatidylcholine (PtdCho) occurs in response to a range of agonists, some of which activate PtdCho-specific PLCs and phospholipase D (PLD; PLD generates phosphatidic acid and choline) (Exton, 1990). PtdCho hydrolysis can act to sustain a message that was initially transmitted via inositide breakdown. Sustained activation of PKC is essential for triggering cell differentiation and proliferation (Nishizuka, 1992). PtdCho breakdown can generate second messengers independent of PtdIns breakdown. This is important, because several isotypes of PKC are activated by DAG in the absence of an increase in intracellular calcium (see earlier discussion) (Stabel and Parker, 1991). The fatty acid species in PtdCho are different from those in PtdIns; therefore, the diradylglycerols generated from each will differ. PtdCho is made up of different molecular species, including ester-, ether- and vinyl-linked species. Further diversity exists, because the acyl chain structure can vary greatly. Therefore, hydrolysis of PtdCho can generate multiple species of diradylglycerols. The predominant components of diradylglycerol in tissues are the ester-linked 1,2-diacyl species and the 1-alkyl-2-acyl glycerols (ether-linked 1-O-alkyl-2-acyl and vinyl-linked 1-O-alk-1’-enyl-2-acyl species). These subclasses of DRGs may differ in their ability to activate PKC (Bass et al., 1989; Cabot and Jaken, 1984; Daniel et al., 1988; Dawson and Cook, 1987; Ford et al., 1989; Heymans et al., 1987). It is possible that the DRGs generated from PtdCho are recognized by special isotypes of PKC, which act in a different domain than does inositide-stimulated PKC, and this may provide a mechanism to maintain signal specificity.
Other products of PtdCho hydrolysis, such as phosphatidic acid, lysophosphatidylcholine (lysoPtdCho), and free fatty acids also are second messengers (Besterman et al., 1986; Exton, 1990). Phosphatidic acid can act as a mitogen (Wakelam et al., 1991). LysoPtdCho stimulates PKC activity (Nishizuka, 1992), but it is a membrane-lytic detergent with potential toxic
effects. LysoPtdCho generation is important in chemotaxis, relaxation of smooth muscle, and activation of T lymphocytes (Nishizuka, 1992). Phospholipase A2 (PLA2) generates free fatty acids from PtdCho. It is activated by many agonists including tyrosine kinase activators (epidermal growth factor and platelet-derived growth factor) and PKC activators (Nishizuka, 1992). Arachidonic acid, which is generated by PLA2, can be a precursor for lipoxygenase- or cyclo-oxygenase generated products (Stabel and Parker, 1991). Oleic and arachidonic acids are able to activate soluble PKC but not membrane-bound PKC (Khan et al., 1992). This may be important for differential activation of the separate isotypes of PKC.
Although PtdCho hydrolysis generates a series of messengers that sustains the PKC phosphorylation cascade, the choline-phospholipid sphingomyelin (SM) is hydrolyzed to generate messengers that terminate the cascade. The hydrolysis of SM by sphingomyelinase (which produces ceramide and phosphocholine from SM) is activated by multiple agonists, including 1α,25-dihydroxyvitamin D3, tumor necrosis factor, and γ interferon (Merrill, 1992). Ceramide is a potent inhibitor of cell growth as well as a promoter of cell differentiation. Its metabolite, sphingosine, is a potent inhibitor of PKC and acts by blocking DAG activation (Merrill and Stevens, 1989). Lysosphingomyelin is also formed during the hydrolysis of SM (see Figure 19– 2) and inhibits PKC (Hannun and Bell, 1989). The synthesis of SM from ceramide and PtdCho generates DAG (Figure 19–2), but it is not known whether this DAG is delivered to a subcellular location where it is available for PKC activation. At present it is suspected that a major function for receptor-mediated activation of sphingomyelinase is the generation of products that stop the signaling cascade.
Choline and Carcinogenesis
Choline deficiency is an excellent example of how choline can influence PKC signal transduction. Choline is the major dietary source for labile methyl groups, and its metabolism is interrelated with methionine and folate metabolism; choline deficiency depletes all of these methyl donors (Zeisel, 1990). Choline is the only single nutrient for which dietary deficiency is associated with the development of cancer (Newberne and Rogers, 1986).
As discussed before, during choline deficiency, secretion of triglyceride (TG) is inhibited. This causes TG and DRG to accumulate (daCosta et al., 1993), and this accumulation is associated with significant increases in PKC activity in hepatic plasma membranes. There is a stable activation of PKC, an increase in the total PKC pool in the cell, or both (daCosta et al., 1993), with changes in several PKC isotypes (at 6 weeks of choline deficiency, the amounts of PKCs α and δ are increased 2-fold and 10-fold, respectively). The
accumulation of DAG and the subsequent activation of PKC within the liver during choline deficiency may be the critical abnormality that eventually contributes to the development of hepatic cancer in these animals. Several lines of evidence indicate that cancers might develop secondary to abnormalities in PKC-mediated signal transduction (Weinstein, 1990). It is interesting that choline deficient rats not only have a higher incidence of spontaneous hepatocarcinoma but that they are also markedly sensitized to the effects of administered carcinogens (Newberne and Rogers, 1986). Perturbed PKC signal transduction may lower the threshold dose of carcinogen needed to initiate the development of cancers.
CONCLUSIONS AND RECOMMENDATIONS
-
Choline is an essential nutrient for humans and can affect human performance via multiple mechanisms. Of immediate practical application in the design of rations is choline’s use for the handling and packaging of lipid within the liver. Rations must be formulated to be calorically dense (high fat), and the calories administered to troops are much higher than those ingested by the normal population. This high-calorie diet should increase demands for choline to package lipid within liver.
-
In the near term, it appears reasonable to explore choline’s ability to enhance muscle function in individuals performing strenuous exercise. The data indicating efficacy are preliminary and need to be carefully replicated before changes in ration compositions are implemented.
-
Finally, the ubiquitous role that phosphatidylcholine plays in transmembrane signaling suggests that significant enhancement of human performance might be obtained through dietary manipulation of such signaling. Several drug companies are actively pursing the identification of pharmacologic agents that can be used to manipulate transmembrane signaling. A significant investment in basic research on dietary interactions with cell signaling needs to be made before practical applications to human rations can be recommended. The very potent effects already demonstrated for dietary modulation of cell signaling (carcinogenesis) suggest that any investment made on such basic research would be worthwhile.
ACKNOWLEDGMENTS
Some of the work described here was supported by grants from the National Institutes of Health (AG-09525 and RR-00046) and the American Institute for Cancer Research.
REFERENCES
Aquilonius, S.M., G.Ceder, T.U.Lying, H.O.Malmlund, and J.Schuberth 1975 The arteriovenous difference of choline across the brain of man. Brain Res. 99:430–433.
Barak, A.J., and R.J.Kemmy 1982 Methotrexate effects on hepatic betaine levels in choline-supplemented and choline-deficient rats. Drug Nutr. Interact. 1:275–278.
Barak, A.J., and D.J.Tuma 1983 Betaine, metabolic by-product or vital methylating agent? Life Sci. 32:771–744.
Barak, A.J., H.C.Beckenhauer, and D.J.Tuma 1982 Use of S-adenosylmethionine as an index of methionine recycling in rat liver slices. Anal. Biochem. 127:372–375.
Barak, A.J., D.J.Tuma, and H.C.Beckenhauer 1984 Methotrexate hepatotoxicity. J. Am. Coll. Nutr. 3:93–96.
Barry, S.R. and A.Gelperin. 1982 Exogenous choline augments transmission at an identified cholinergic synapse in terrestrial mollusk Limax maximus. J. Neurophysiol. 48:439–450.
Bartus, R.T., R.L.Dean, J.A.Goas, and A.S.Lippa 1980 Age-related changes in passive avoidance retention: Modulation with dietary choline. Science 209:301–303.
Bartus, R.T., R.Dean, B.Beer, and A.S.Lippa. 1982 The cholinergic hypothesis of geriatric memory dysfunction. Science 217:408–414.
Bass, D.A., L.C.McPhail, J.D.Schmitt, S.Morris-Natschke, C.E.McCall and R.L.Wykle 1989 Selective priming of rate and duration of the respiratory burst of neutrophils by 1,2-diacyl and 1-O-alkyl-2-acyl diglycerides. Possible relation to effects on protein kinase C. J. Biol. Chem. 263:19610–19617.
Bertoni-Freddari, C., R.F.Mervis, C.Giuli, and C.Pieri 1985 Chronic dietary choline modulates synaptic plasticity in the cerebellar glomeruli of aging mice. Mech. Aging Dev. 30:1–9.
Best, C.H., and M.E.Huntsman 1932 The effects of the components of lecithin upon the deposition of fat in the liver. J. Physiol. 75:405–412.
1935 Effect of choline on liver fat of rats in various states of nutrition. J. Physiol. 83:255–274.
Besterman, J.M., V.Duronio, and P.Cuatrecasas. 1986 Rapid formation of diacylglycerol from phosphatidylcholine: A pathway for generation of a second messenger. Proc. Natl. Acad. Sci. 83:6785–6789.
Bierkamper, G.G., and A.M.Goldberg 1980 Release of acetylcholine from the vascular perfused rat phrenic nerve hemidiaphragm. Brain Res. 202:234–237.
Bjornstad, P., and J.Bremer 1966 In vivo studies on pathways for the biosynthesis of lecithin in the rat J. Lipid Res. 7:38–45.
Blair, R., and F.Newsome 1985 Involvement of water-soluble vitamins in diseases of swine. J. Animal Sci. 60:1508–1517.
Blusztajn, J.K., and R.J.Wurtman 1983 Choline and cholinergic neurons. Science 221:614–620.
Blusztajn, J.K., and S.H.Zeisel 1989 1,2-sn-Diacylglycerol accumulates in choline-deficient liver. A possible mechanism of hepatic carcinogenesis via alteration in protein kinase C activity? FEBS Lett. 243:267–270.
1979 Synthesis of lecithin (phosphatidylcholine) from phosphatidylethanolamine in bovine brain. Brain Res. 179:319–327.
Blusztajn, J.K., S.H.Zeisel, and R.J.Wurtman 1985 Developmental changes in the activity of phosphatidylethanolamine N-methyltransferases in rat brain. Biochem. J. 232:505–511.
Boyd, W.D., W.J.Graham, G.Blackwood, I.Glen, and J.McQueen 1977 Clinical effects of choline in Alzheimer senile dementia. Letter. Lancet 2:711.
Bremer, J., and D.Greenberg 1961 Methyl transfering enzyme system of microsomes in the biosynthesis of lecithin (phosphatidylcholine) . Biochim. Biophys. Acta 46:205–216.
Buchman, A.L., M.Dubin, D.Jenden, A.Moukarzel, M.H.Roch, K.Rice, J.Gornbein, M.E. Ament, and C.D.Eckhert 1992 Lecithin increases plasma free choline and decreases hepatic steatosis in long-term total parenteral nutrition patients. Gastroenterology 102:1363–1370.
Burt, M.E., I.Hanin, and M.F.Brennan 1980 Choline deficiency associated with total parenteral nutrition. Lancet 2:638–639.
Cabot, M.C., and S.Jaken 1984 Structural and chemical specificity of diradylglycerols for protein kinase C activation. Biochem. Biophys. Res. Commun. 125:163–169.
Carroll, C., and L.Williams 1982 Choline deficiency in rats as influenced by dietary energy somics. Nutr. Rep. Inter. 25:773.
Chawla, R.K., C.J.Berry, M.H.Kutner, and D.Rudman 1985 Plasma concentrations of transsulfuration pathway products during nasoenteral and intravenous hyperalimentation of malnourished patients. Am. J. Clin. Nutr. 42:577–584.
Chawla, R.K., D.C.Wolf, M.H.Kutner, and H.L.Bonkovsky 1989 Choline may be an essential nutrient in malnourished patients with cirrhosis. Gastroenterology 97:1514–1520.
Cohen, E.L., and R.J.Wurtman 1975 Brain acetylcholine: Increase after systemic choline administration. Life Sci. 16:1095–1102.
1976 Brain acetylcholine: Control by dietary choline. Science 191:561–562.
Conlay, L.A., R.J.Wurtman, K.Blusztajn, I.L.Coviella, T.J.Maher and G.E.Evoniuk 1986 Decreased plasma choline concentrations in marathon runners. Letter. N Engl. J. Med. 315:892.
Cook, R.J., D.W.Horne, and C.Wagner 1989 Effect of methyl group deficiency on one-carbon metabolism in rats. J. Nutr. 119:612–617.
daCosta, K.A., E.F.Cochary, J.K.Blusztajn, S.F.Garner, and S.H.Zeisel. 1993 Accumulation of 1,2-sn-diradylglycerol with increased membrane-associated protein kinase C may be the mechanism for spontaneous hepatocarcinogenesis in choline deficient rats. J. Biol. Chem. 268:2100–2105.
Daniel, L.W., G.W.Small, J.D.Schmitt, C.J.Marasco, K.Ishaq, and C.Piantadosi 1988 Alkyl-linked diglycerides inhibit protein kinase C activation by diacylglycerols. Biochem. Biophys. Res. Commun. 151:291–297.
Dawson, W.D. and J.S.Cook 1987 Parallel changes in amino acid transport and protein kinase C localization in LLL-PK1 cells treated with TPA or diradylglycerols. J. Cell. Physiol. 132:104–110.
Eagle, H. 1955 The minimum vitamin requirements of the L and Hela cells in tissue culture, the production of specific vitamin deficiencies, and their cure. J. Exp. Med. 102:595–600.
Engel, R.W. 1943 The choline content of animal and plant products. J. Nutr. 25:441–446.
Exton, J.H. 1990 Signaling through phosphatidylcholine breakdown. J. Biol. Chem. 265:1–4.
Fairbanks, B.W. and J.L.Krider 1945 Significance of B vitamins in swine nutrition. N. Am. Vet. 26:18–23.
Federation of American Societies for Experimental Biology 1975 Evaluation of the Health Aspects of Choline Chloride and Choline Bitartrate as Food Ingredients. PB262–654. Springfield, Va.: National Technical Information Service.
1981 Effects of Consumption of Choline and Lecithin on neurological and Cardiovascular Systems. PB82–133257. Springfield VA, National Technical Information Service.
Finkelstein, J.D., J.J.Martin, B.J.Harris, and W.E.Kyle 1982 Regulation of the betaine content of rat liver. Arch. Biochem. Biophys. 218:169–173.
Finkelstein, J.D., J.J.Martin, and B.J.Harris 1988 Methionine metabolism in mammals. The methionine-sparing effect of cystine. J. Biol. Chem. 263:11750–11754.
Food and Nutrition Board 1973 Comprehensive GRAS Survey, Usage Levels Reported for NAS Appendix A Substances (Group 1) Used in Regular Foods. Washington, D.C.: National Academy of Sciences.
Ford, D.A., R.Miyake, P.E.Glaser, and R.W.Gross 1989 Activation of protein kinase C by naturally occurring ether-linked diglycerides. J. Biol. Chem. 264:13818–13824.
Freeman-Narrod, M., S.A.Narrod, and R.P.Custer 1977 Chronic toxicity of methotrexate in rats: Partial to complete projection of the liver by choline: Brief communication. J. Natl. Cancer Inst. 59:1013–1017.
Handler, P. and F.Bernheim 1949 Choline deficiency in the hamster. Proc. Soc. Exp. Med. 72:569.
Hannun, Y.A. and R.M.Bell 1989 Regulation of protein kinase C by sphingosine and lysosphingolipids. Clin. Chim. Acta 185:333–345.
Haubrich, D.R., P.W.Wedeking, and P.F.Wang 1974 Increase in tissue concentration of acetylcholine in guinea pigs in vivo induced by administration of choline. Life Sci. 14:921–927.
Haubrich, D.R., P.F.Wang, D.E.Clody, and P.W.Wedeking 1975a Increase in rat brain acetylcholine induced by choline or deanol. Life Sci. 17:975–980.
Haubrich, D.R., P.F.Wang, and P.W.Wedeking. 1975b Distribution and metabolism of intravenously administered choline[methyl-3-H] and synthesis in vivo of acetylcholine in various tissues of guinea pigs. J. Pharmacol. Exp. Ther. 193:246–255.
Hershey, J.M. and S.Soskin. 1931 Substitution of “lecithin” for raw pancreas in a diet of depancreatized dog. Am. J. Physiol. 93:657–658.
Heymans, F., S.C.Da, N.Marrec, J.J.Godfroid, and M.Castagna 1987 Alkyl analogs of diacylglycerol as activators of protein kinase C. FEBS Lett. 218:35–40.
Hoffbauer, F.W., and F.G.Zaki 1965 Choline deficiency in the baboon and rat compared. Arch. Pathol. 79:364–369.
Horne, D.W., R.J.Cook, and C.Wagner. 1989 Effect of dietary methyl group deficiency on folate metabolism in rats. J. Nutr. 119:618–621.
Kaminski, D.L., E.J.Mueller, and M.Jellinek 1980 Effect of small intestinal bypass on hepatic lipid accumulation in rats. Am. J. Physiol. 239:G358–G362.
Kapp, J., M.J.Mahaley, and G.L.Odom 1970 Experimental evaluation of potential spasmolytic drugs. J. Neurosurg. 32:468–472.
Ketola, H.G. 1976 Choline metabolism and nutritional requirement of lake trout (Salvelinus namaycush). J. Anim. Sci. 43:474–477.
Ketola, H.G., and M.C.Nesheim 1974 The influence of dietary protein and methionine levels on the requirement for choline by chickens. J. Nutr. 104:1484–1486.
Ketola, H.G., and R.J.Young 1973 The need for dietary choline by young Japanese quail. Poult. Sci. 52:2362–2363.
Khan, W.A., G.C.Blobe, and Y.A.Hannun. 1992 Activation of protein kinase C by oleic acid. J. Biol. Chem. 267:3605–3612.
Loffelholz, K. 1981 Release of acetylcholine in the isolated heart. A Review. Am. J. Physiol. 240:H431–H440.
Lombardi, B. 1971 Effects of choline deficiency on rat hepatocytes. Fed. Proc. 30:139–142.
Lombardi, B., P.Pani, and F.F.Schlunk 1968 Choline-deficiency fatty liver: Impaired release of hepatic triglycerides. J. Lipid Res. 9:437–46.
Malthe, S.D., and F.Fonnum 1972 Multiple forms of choline acetyltransferase in several species demonstrated by isoelectric focusing. Biochem. J. 127:229–236.
McIntire, J.M., B.S.Schweigert, and C.A.Elvehjem 1944 The choline and pyridoxine content of meats. J. Nutr. 28:219–223.
Meck, W.H., R.A.Smith, and C.L.Williams 1988 Pre- and postnatal choline supplementation produces long-term facilitation of spatial memory. Dev. Psychobiol. 21:339–353.
Meck, W.H., R.A.Smith, and C.L.Williams 1989 Organizational changes in cholinergic activity and enhanced visuospatial memory as a function of choline administered prenatally or postnatally or both. Behav. Neurosci. 103:1234–1241.
Meldrum, E., P.J.Parker, and A.Carozzi 1991 The Ptd-Ins-PLC superfamily and signal transduction. Biochim. Biophys. Acta 1092:49–71.
Merrill, A.H. 1992 Ceramide: A new lipid “second messenger”? Nutr. Rev. 50:78–80.
Merrill, A.H. and V.L.Stevens 1989 Modulation of protein kinase C and diverse cell functions by sphingosine- a pharmacologically interesting compound linking sphingolipids and signal transduction. Biochim. Biophys. Acta 1010:131–139.
Mervis, R., L.Horrocks, P.Demediuk, L.Wallace, D.R.Meyer, S.Beall, K.Caris, and E.Naber 1985 Neurobehavioral effects of chronic choline-containing diets on the adult and aging C57BL/6NNIA mouse brain. Ann. N.Y. Acad. Sci. 444:469–70.
Miller, L.G., D.J.Greenblatt, R.B.Roy, F.Lopez, and L.Wecker 1989 Dietary choline intake modulates benzodiazepine receptor binding and gamma-aminobutyric acid A receptor function in mouse brain. J. Pharmacol. Exp. Ther. 248:1–6.
Mudd, S.H. and J.R.Poole 1975 Labile methyl balances for normal humans on various dietary regimens. Metab. Clin. Exp. 24:721–735.
Newberne, P.M. and A.E.Rogers 1986 Labile methyl groups and the promotion of cancer. Annu. Rev. Nutr. 6:407–432. Nishizuka, Y.
1986 Studies and perspectives of protein kinase C. Science 233:305–312.
Nishizuka, Y. 1992 Intracellular signaling by hydrolysis of phospholipids and activation of protein kinase C. Science 258:607–614.
Poirier, L.A., P.H.Grantham, and A.E.Rogers 1977 The effects of a marginally lipotrope-deficient diet on the hepatic levels of S-adnosylmethionine and on the urinary metabolites of 2-acetylaminofluorene in rats. Can. Res. 37:744–748.
Pomfret, E.A., K.A.daCosta, L.L.Schurman, and S.H.Zeisel 1989 Measurement of choline and choline metabolite concentrations using high-pressure liquid chromatography and gas chromatography-mass spectrometry. Anal. Biochem. 180:85–90.
Pomfret, E.A., K.A.daCosta, and S.H.Zeisel. 1990 Effects of choline deficiency and methotrexate treatment upon rat liver. J. Nutr. Biochem. 1:533–541.
Rama Sastry, B.V., and G.I.Henderson 1972 Kinetic mechanisms of human placental choline acetyltransferase. Biochem. Pharmacol. 21:787–802.
Ridgway, N.D., and D.E.Vance 1987 Purification of phosphatidylethanolamine N-methyltransferase from rat liver. J. Biol. Chem. 262:17231–17239.
Sahley, C.L., S.R.Barry, and A.Gelperin 1986 Dietary choline augments associative memory function in Limax maximus. Neurobiol. 17:113–120.
Sandage, B.W., R.N.Sabounjian, R.White, and R.J.Wurtman 1992 Choline citrate may enhance athletic performance. Physiologist 35:236a.
Schmidt, D.E., and L.Wecker 1981 CNS effects of choline administration: Evidence for temporal dependence. Neuropharmacology 20:535–539.
Selhub, J., E.Seyoum, E.A.Pomfret, and S.H.Zeisel 1991 Effects of choline deficiency and methotrexate treatment upon liver folate content and distribution. Cancer Res. 51:16–21.
Sheard, N.F., J.A.Tayek, B.R.Bistrian, G.L.Blackburn, and S.H.Zeisel 1986 Plasma choline concentration in humans fed parenterally. Am. J. Clin. Nutr. 43:219–2–24.
Shivapurkar, N., and L.A.Poirier 1983 Tissue levels of S-adenosylmethionine and S-adenosylhomocysteine in rats fed methyl-deficient, amino acid-defined diets for one to five weeks. Carcinogenesis 4:1051–1057.
Singh, G.S. 1973 Letter: Action of choline on the rat blood pressure. Indian J. Physiol. Pharmacol. 17:125.
Sitaram, N., H.Weingartner, E.D.Caine, and J.C.Gillin 1978a Choline: Selective enhancement of serial learning and encoding of low imagery words in man. Life Sci. 22:1555–1560.
Sitaram, N., H.Weingartner, and J.C.Gillin 1978b Human serial learning: Enhancement with arecholine and choline impairment with scopolamine. Science 201:274–276.
Stabel, S., and P.J.Parker 1991 Protein kinase C. Pharmac. Ther. 51:71–95.
Svardal, A.M., P.M.Ueland, R.K.Berge, A.Aarsland, N.Aarsaether, E.P.Lonning, and H.Refsum 1988 Effect of methotrexate on homocysteine and other compounds in tissues of rats fed a normal or a defined, choline-deficient diet. Cancer Chemother. Pharmacol. 21:313–318.
Tani, H., S.Suzuki, M.Kobayashi, and Y.Kotake 1967 The physiological role of choline in guinea pigs. J. Nutr. 92:317–324.
Trommer, B.A., D.E.Schmidt, and L.Wecker 1982 Exogenous choline enhances the synthesis of acetylcholine only under conditions of increased cholinergic neuronal activity. J. Neurochem. 39:1704–1709.
Wakelam, M.J.O., S.J.Cook, S.Currie, S.Plamer, and R.Plevin 1991 Regulation of the hydrolysis of phosphatidylcholine in Swiss 3T3 cells. Biochem. Soc. Trans. 19:321–324.
Wallace, L.J., M.G.Kolta, M.C.Gerald, and R.F.Mervis 1985 Dietary choline affects response to acetylcholine by isolated urinary bladder. Life Sci. 36:1377–1380.
Wecker, L. 1986 Neurochemical effects of choline supplementation. Can. J. Physiol. Pharmacol. 64:329–333.
Wecker, L. 1988 Influence of dietary choline availability and neuronal demand on acetylcholine synthesis by rat brain. J. Neurochem. 51:497–504.
Wecker, L. 1990 Dietary choline: A limiting factor for the synthesis of acetylcholine by the brain. Adv. Neurol. 51:139–145.
Wecker, L., and W.D.Dettbarn 1979 Relationship between choline availability and acetylcholine synthesis in discrete regions of rat brain. J. Neurochem. 32:961–967.
Wecker, L., G.Cawley, and S.Rothermel 1989 Acute choline supplementation in vivo enhances acetylcholine synthesis in vitro when neurotransmitter release is increased by potassium. J. Neurochem. 52:568–575.
Weihrauch, J.L. and Y.S.Son 1983 The phospholipid content of foods. J. Amer. Oil Chem. Soc. 60:1971–1978.
Weinstein, I.B. 1990 The role of protein kinase C in growth control and the concept of carcinogenesis as a progressive disorder in signal transduction. Adv. Second Messenger Phosphoprotein Res. 24:307–316.
White, H.L. and C.J.Cavallito 1970 Choline acetyltransferase. Enzyme mechanism and mode of inhibition by a styrylpyridine analogue. Biochim. Biophys. Acta 206:343–358.
White, H.L. and J.C.Wu 1973 Kinetics of choline acetyltransferases (EC 2.3.1.6) from human and other mammalian central and peripheral nervous tissues. J. Neurochem. 20:297–307.
Wong, E.R., and W.Thompson 1972 Choline oxidation and labile methyl groups in normal and choline-deficient rat liver. Biochim. Biophys. Acta 260:259–271.
Wurtman, R.J., F.Hefti, and E.Melamed 1980 Precursor control of neurotransmitter synthesis. Pharmacol. Rev. 32:315–335.
Yao, Z.M., and D.E.Vance 1988 The active synthesis of phosphatidylcholine is required for very low density lipoprotein secretion from rat hepatocytes. J. Biol. Chem. 263:2998–3004.
1989 Head group specificity in the requirement of phosphatidylcholine biosynthesis for very low density lipoprotein secretion from cultured hepatocytes. J. Biol. Chem. 264:11373–11380.
Zeisel, S.H. 1981 Dietary choline: Biochemistry, physiology, and pharmacology. Ann. Rev. Nutr. 1:95–121.
1988 “Vitamin-like” molecules: Choline. Pp. 440–452 in Modern Nutrition in Health and Disease, 7th ed. Philadelphia: Lea & Febiger.
1990 Choline deficiency. J. Nutr. Biochem. 1:332–349.
Zeisel, S.H. and J.K.Blusztajn 1994 Choline and human nutrition Ann. Rev. Nutr. 14:269–296.
Zeisel, S.H., J.H.Growdon, R.J.Wurtman, S.G.Magil, and M.Logue 1980 Normal plasma choline responses to ingestion of lecithin. Neurology 30:1226–1229.
Zeisel, S.H., D.Char, and N.F.Sheard 1986 Choline, phosphatidylcholine and sphingomyelin in human and bovine milk and infant formulas. J. Nutr. 116:50–58.
Zeisel, S.H., T.Zola, K.A.daCosta, and E.A.Pomfret 1989 Effect of choline deficiency on S-adenosylmethionine and methionine concentrations in rat liver. Biochem. J. 259:725–729.
Zeisel, S.H., K.A.daCosta, P.D.Franklin, E.A.Alexander, J.T.Lament, N.F.Sheard, and A.Beiser 1991 Choline, an essential nutrient for humans. FASEB J. 5:2093–2098.
DISCUSSION
RICHARD WURTMAN: I want to congratulate you on a fine review. In the athlete study, I think that the actual choline base is only 1 gram. If it was 3 grams, I think I would know. But my question is this. If we all need 6 to 12 grams of phosphatidylcholine—and that is made up of one-eighth choline—that means we are taking in about three-quarters of a gram to a gram and a half of choline per day; suppose we decided to raise choline intake to 3 grams a day, which would not be crazy if you considered my grandfathers, both of whom, I am told, ate two eggs for breakfast each day. I think the consequence of reducing dietary cholesterol is concurrently to reduce dietary choline. My question is, what is the down side of giving people 3 grams of choline per day as opposed to the gram or gram and a half that they are getting now?
STEVEN ZEISEL: I think that our exposure to choline is greater than the amount that we eat. I agree that human deficiency studies suggest that about 500 milligrams to a gram of choline must be what normal humans eat, but we are exposed to more because reabsorbed bile phosphatidylcholine is about equal to dietary phosphatidylcholine on any single day. It is just recirculated over and over again, so that an individual is exposed to twice as much. Ingesting 3 g choline (3 times normal dietary intake) should not be harmful if you do not give this as free choline. The problem with free choline is that the absorption system from the gut is saturated at about 4 mm of choline in humans. Above that concentration, gut bacteria convert it to trimethylamine which makes you smell like a rotten fish. Choline and glycerophosphorcholine should not cause these problems because they are not broken down to free choline within the gut, and bacteria cannot utilize them to make trimethylamine.
I do not know of any studies that suggest any significant toxicity from taking 3 g of choline for a long period of time. There were a number of studies done in patients with Alzheimer’s disease and controls in which they gave phosphatidylcholine at does delivering about 3 g choline per day for years and the investigators did not report low blood pressure, tachycardia, or slowed heart rate. They did not report any other major problems except for occasional depression, which is probably normal in the population being studied.
EDWARD HORTON: Steve, thank you for a very stimulating, provocative lecture. I am fascinated by the data that choline deficiency or low choline levels may be associated with easy fatigability of muscle. There is preliminary evidence, and not very strong evidence, that activation of protein kinase C may be involved in the translocation and activation of the glucose transport system of the skeletal muscle, and so it would be very interesting to look to see whether there is a defect in the glucose transport system in choline-deficient animals.
STEVEN ZEISEL: I would be glad to study this.
ALLISON YATES: Health food stores have been selling lecithin over the years, and there has not been any definitive analyses of its effects. I am not aware of any studies showing changes in people who are dosing themselves with lecithin.
STEVEN ZEISEL: Lecithin is not phosphatidylcholine. What is sold in health food stores is only about 35 percent phosphotidylcholine, and people are not taking in very significant doses when they ingest a gram of that; they are taking in 200 milligrams of phosphatidylcholine, which is probably not changing their dietary intake a whole lot. A lot of lecithin is added to manufactured foods; chocolate bars have it. That lecithin is not pure phosphatidylcholine, but rather it too is approximately 35 percent phosphatyidyl choline.
RONALD JANDACEK: The studies in prenatal rats fascinated me. The closest analogy that I can think of in human studies is a study in which the investigators studied extensively the children of mothers who were well nourished except for brief periods of time during pregnancy. Those studies probably covered all the areas you are talking about. Although it has been a long time since I read that book, I remember that the investigators saw very few behavioral effects.
STEVEN ZEISEL: Again, when you starve, you break down cells and you release phosphatidylcholine choline, so it is not the same thing as being choline deficient. I think a better model is the following: human milk contains a huge amount of choline, especially during the first few days of life. There has been a recent study of premature infants in England in which they were bottle fed human milk, which has a high choline content, or infant formula, which has about a fourfold lower choline content. They reported that IQ was significantly worse in the premature infants fed the infant formula than in those fed breast milk. I think the defferntial was 8 I.Q. points.
ROBERT NESHEIM: Does choline taste terrible? What is the problem of adding it to a food product?
STEVEN ZEISEL: Choline is added to infant formula. Most people would add it as phosphatidylcholine which does not have the bitter taste of choline.
RICHARD WURTMAN: A firm in Massachusetts sells citrus drinks, and it is now making a choline supplement in a citrus drink for use in this type of testing. I have tasted it and I cannot taste the choline. I think a good food chemist can disguise anything.
JOHN VANDERVEEN: Steve Zeisel, maybe you can help us out on a regulatory problem that has been a thorn in the side of the compliance people for years. That is, they have been insisting that the source of choline be labeled on boxes or whatever as soy lecithin. Then the industry argues that all the lecithin that is available really comes from soy and not from any other source and therefore it should only be lecithin. Is there any reason to debate the distinction?
STEVEN ZEISEL: The reason is that phosphatidylcholine does not only contain choline, it contains two fatty acids. In soy lecithin they are unsaturated fatty acids. If part of efficacy has to do with the fatty acid constituents, you would want to know the difference. One thing I did mention, right from the story of total parenteral nutrition, is that there is a study from Emory University that came out a couple of months ago in which they showed that patients treated with normal intravenous solutions with lipid emulsions developed fatty liver and liver dysfunction. The investigators found that giving phosphatidylcholine as an oral supplement completely reversed the fatty liver, and in the people given placebo, there was no reversion of fatty livers. I would suggest that total parenteral nutrition is not delivering enough choline and that these people are deficient.
DAVID SCHNAKENBERG: What mechanism are you proposing for the effect of 3 hours of exercise on plasma choline levels?
STEVEN ZEISEL: I think either muscle, being 45 percent of lean body weight, is using a great deal as a methyl donor or to make acetylcholine. It could be as simple an explanation as a shift in location of the choline pool because of water shifts.
RICHARD WURTMAN: I think you are right. It is a very good research question with several possibilities. I think some experimentation could come
up with an answer just in terms of what we are doing. There has been no formal approach to this. I would like to establish if we could go to that.
DAVID SCHNAKENBERG: How big a drop is this?
RICHARD WURTMAN: We did this in a total of 45 runners in the Boston Marathon in 2 successive years. The average decrease in plasma choline level was about 45 percent, but for some of them, the level was down by about 60 percent. Everybody showed a drop. I think only 1 person out of the 45 did not show a drop, but in some the drop was small, 20 percent.
The other thing we did was to run people 5, 10, 15, and 20 miles. There was not a linear drop in plasma choline levels with distance. You do not really see much decrease at all until about 15 miles, and then between 15 and 20 miles there was a decrease. Again, I think it would be useful to determine the mechanism, because that may tell a lot about muscle physiology and body chemistry in terms of exercise. It would not be difficult, I think, to set up studies to find out.
JOHN VANDERVEEN: I am just curious. Has anyone made any estimates about the total amount of choline in the body?
STEVEN ZEISEL: Yes I have. I do not remember the exact number but as I said, I think it is about 4 percent of dry body weight.


























