
14
Explaining Low Levels of DNA Sequence Variation in Regions of the Drosophila Genome with Low Recombination Rates
Richard R. Hudson
Recent developments in biotechnology have led to a huge burst in available DNA sequence information. DNA sequences from a large number of loci from a large number of taxa are currently available and more sequences are becoming available. With this information, much has been learned about the rates of molecular evolution in different taxa, at a variety of loci, and at different kinds of sites in the genome. Although the territory is large and much remains to be explored, some descriptive generalizations are now possible. In contrast, the population genetic processes underlying this molecular evolution remain almost entirely obscure. In other words, quite a bit is known about the tempo of molecular evolution, but very little is known about the mode. Understanding the population genetic forces that are most important is made exasperatingly difficult by the fact that very small selective effects, much too small to be directly measured, can be the determining factor in evolution. As a consequence, some effort has been devoted to making inferences about the evolutionary process by indirect means such as analyzing the patterns of molecular divergence and polymorphism in a variety of taxa and loci. Excellent recent reviews of empirical and theoretical aspects of these issues are available (Kimura, 1983; Gillespie, 1991). I will describe here some recently collected data and some efforts to make inferences from that data about underlying
Richard R. Hudson is professor of ecology and evolutionary biology at the University of California, Irvine.
population genetic processes. Several hypotheses and population genetic models that may account for the data will be discussed. Most of the discussion will concern a "background selection" model that may account for some important aspects of the data.
The Data
The data that will be considered here are primarily DNA polymorphism data from a number of loci primarily in Drosophila melanogaster and Drosophila simulans. The salient feature of these data, the focus of this paper, is the following: Regions of the Drosophila genome with low rates of recombination per base pair exhibit low levels of polymorphism within populations (Aguadé et al., 1989; Stephan and Langley, 1989; Berry et al., 1991; Begun and Aquadro, 1992; Martín-Campos et al., 1992; Stephan and Mitchell, 1992; Langley et al., 1993). A summary of these data that display the remarkable correlation between recombination rates and levels of polymorphism is given by Begun and Aquadro (1992) and Aquadro et al., (1994). In the following paragraphs, three hypotheses to account for these data will be described. They are (i) a strictly neutral hypothesis, (ii) a hitchhiking with selective sweeps of advantageous mutations hypothesis, and (iii) a background selection of deleterious mutations hypothesis.
A Strictly Neutral Hypothesis
A very simple though interesting hypothesis to explain the correlation of recombination rates with polymorphism levels is a completely neutral one: Regions of low recombination might have low levels of polymorphism because they have low neutral mutation rates. These low neutral mutation rates in regions of low recombination might result from high average levels of constraint in those parts of the genome or because the spontaneous mutation rates are low there. Either of these possibilities would be interesting and surprising, since there is no a priori reason to suspect that mutation rates are lower or constraints are higher in regions of low recombination. Fortunately, there is a simple and powerful way to test this strict neutral interpretation: Examine the levels of divergence between species in these regions of high and low recombination. Under our neutral interpretation, regions of low recombination ought to have low levels of divergence compared to the levels of divergence in regions of high recombination, because the rate divergence under the neutral model is equal to the neutral mutation rate. The data in this regard are quite clear. Regions of low recombination are not diverging more slowly between species than are regions of high recombination (Berry et al.,
1991; Begun and Aquadro, 1992; Martín-Campos et al., 1992; Langley et al., 1993). We therefore reject our strict neutral hypothesis, concluding that regions of low recombination do not have low levels of variation because they have low neutral mutation rates.
Hitchhiking With Favorable Mutations Hypothesis
Another hypothesis that has been proposed to explain the pattern of sequence variation is the hitchhiking or "selective sweeps" model (Maynard Smith and Haigh, 1974; Kaplan et al., 1989; Stephan et al. , 1992; Wiehe and Stephan, 1993). Under this model, the low levels of polymorphism in regions of low recombination are due to the hitchhiking effect of selectively advantageous mutants that sweep through the population and, in the process, eliminate variation at tightly linked sites. In regions of low recombination, large chunks of DNA are swept to fixation by such selection events, whereas in regions of high recombination, only small chunks are swept to fixation. If such selection events are steadily occurring in both high and low recombination regions, the result will be a lower steady-state level of variation in regions of low recombination. If this selective sweeps interpretation is correct, it should be possible to estimate some of the parameters of the population genetic process involved. Indeed, Wiehe and Stephan (1993) have developed a method to estimate an important rate parameter from the patterns of reduced variation. (They assume that most of the variation that is seen is neutral but that the levels of variation are, in some cases, strongly affected by occasional selective sweeps.) The development of this estimation method is quite significant, demonstrating an additional way in which inferences about the mode of molecular evolution can be made from patterns of polymorphism and divergence. Further work is clearly warranted to investigate other properties of this model and to assess the robustness of the rate estimates. Although there remains much to be investigated, this model can account for some important features of the data.
Background Selection Model
Recently, a quite different hypothesis, referred to as the background selection model, has been proposed to account, at least in part, for the low levels of variation in regions of low recombination (Charlesworth et al., 1993). In this model, as with the selective sweeps model, one focuses on the level of neutral variation that will be maintained at a locus embedded in the midst of a large number of other loci at which mutations can occur that are not selectively neutral. Figure 1A illustrates
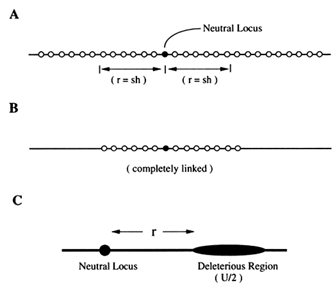
Figure 1 Three models considered in the analysis of the background selection. In each case, a neutral locus (indicated by a solid circle) is linked to other loci at which deleterious mutations can occur. In models A and B, the loci at which deleterious mutations can occur are indicated by the open circles. In model A, recombination can occur between any of the indicated loci. The rate of recombination (r) between the neutral locus and the site indicated by the arrows is assumed to equal sh. (sh is defined to be the decrement in fitness produced by a single heterozygous deleterious mutation; that is, the relative fitness of an individual heterozygous for a single deleterious mutation is assumed to be 1—sh.) In model B, all loci are completely linked. In model C, the rate of recombination between the neutral locus and the deleterious region is assumed to equal r. The total deleterious mutation rate in the deleterious region is assumed to equal U/2.
this model. (The parameters r and sh indicated on the figure will be defined later.) The locus at which neutral variation is being monitored will be referred to as the neutral locus. The central question, at least initially, will be, To what extent is the mean level of variation at the neutral locus affected by the natural selection that occurs at the linked loci. I will quantify this effect of selection at linked loci by the ratio R = π/π0, where π is the expected nucleotide diversity at the neutral locus under the model with selection at linked loci and π0 is the expected nucleotide diversity at the neutral locus in the absence of selection at linked loci. [Nucleotide diversity is a commonly employed measure of the amount of DNA polymorphism in a sample (Chapter 10 of Nei,
1987)]. Under the selective sweep model, the mutations occurring at the linked loci are favorable mutations that more or less regularly sweep through the population. In contrast, under the background selection model, the mutations that affect fitness are all assumed to be deleterious.
In a sufficiently large population, recurrent deleterious mutation and natural selection lead to an equilibrium, with mutation continually adding deleterious alleles and natural selection removing them. For a particular region of the genome, the population at this equilibrium can be characterized by a vector, f = (f0, f1, f2, …), in which fi is the frequency of chromosomes in the population that carry i deleterious mutations in the region being considered. The equilibrium value of f will depend on the mutation rate and the fitness of genotypes with different numbers of mutations. For the case where all loci are completely linked, as indicated in Figure 1B, the ratio R is equal to f0, the frequency of chromosomes that carry no deleterious mutations (Charlesworth et al., 1993). For a wide range of mutation rates and selection coefficients, the effect of background selection is simply to reduce the effective population size from N to f0N. For at least one model, an explicit expression for f0 has been obtained. Namely, if the deleterious effect of a single mutation in heterozygous state is sh and, further, if an individual heterozygous at i loci has relative fitness (1 - sh) i, then the equilibrium value of f0 is exp(-U/2sh), where U/2 is the total (haploid) deleterious mutation rate at the completely linked loci (Kimura and Maruyama, 1966; Crow, 1970). Thus for this model and without recombination, the effect of background selection on levels of variation can be calculated.
With recombination, as in the model of Figure 1A, it is difficult to obtain analytical results, and a simpler model, shown in Figure 1C, is used to gain insight initially. This simpler model with a neutral locus linked to a deleterious region with recombination rate, r, between them has been analyzed using a coalescent approach (Hudson and Kaplan in press). Except in special cases, simple analytic expressions are not obtained, but numerical results are easily obtained. The effect of recombination is shown in Figure 2, for two cases. The important properties are depicted in this figure: (i) if r « sh, the model behaves as if the deleterious region and the neutral locus were completely linked, that is R = f0 = exp(-U/2sh), and (ii) if r » sh, then it is as if the deleterious region were not there—that is, R = 1. In addition, the transition from one situation to the other occurs fairly quickly, as r is increased. This suggests an approximation. Perhaps the effect of background selection in the difficult model of Figure 1A is approximately the same as for the model shown in Figure 1B, in which all loci more than sh recombination units away have been removed, and all the other loci
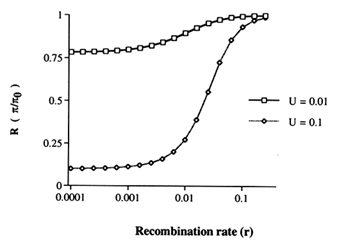
Figure 2 R, the ratio of nucleotide diversity with background selection to nucleotide diversity without background selection, is plotted as a function of r, the recombination rate between the neutral locus and the deleterious region. The model assumed is that depicted in Figure 1C. For these calculations, N = 3200 and sh = 0.02.
are assumed completely linked to the neutral locus. If this conjecture is correct, with the arrangement of loci depicted in Figure 1A, and if the multiplicative interaction model is assumed, the value of R would be
where Ue/2 is the rate of deleterious mutation in the region within sh recombination units on either side of the neutral locus.
Now, some implications of this tentative approximation for patterns of variation seen in Drosophila can be explored. Since recombination usually occurs only in females in Drosophila, the effective recombination rate over many generations for autosomal genes is one-half the rate measured in females. Thus, in Drosophila, Ue would be the total deleterious mutation rate for loci within 2sh × 100 centimorgans of the neutral locus. If we denote the deleterious mutation rate of the entire diploid genome by UT and the fraction of genome within z centimorgans of a locus by F(z) and if the deleterious mutation rate is more or less constant per base pair across the genome, then Ue can be written as F(200sh)UT, and substituting into equation 1,
UT in Drosophila has been estimated as approximately 1.0 (Mukai et al., 1972; Houle et al., 1992) and sh has been estimated to be 0.02 (Crow and
Simmons, 1983). Hence, R can be written as exp[-F(4)/0.04]. To predict R for any locus, one need only determine F(4), the fraction of the genome within 4 centimorgans of the locus. From published genetic and cytological maps, F(z) can be estimated for many loci. For example, consider the cta locus (located at cytological position 40F) near the base of the second chromosome. A region 4 centimorgans in each direction from this locus extends from cytological position 35C to about 44E, which is about 9% of the genome. That is, F(4) for the cta locus is about 0.09. Therefore, we expect R for cta to be about exp(-0.09/0.04) = 0.11. For adh located at position 35B, the region within 4 centimorgans of adh extends from about 33A to 37B, or about 4% of the genome. Therefore, we expect R for adh to be exp(-0.04/0.04) = 0.37.
Note that in a region with uniform recombination rates, F(200sh) will be proportional to sh, the constant of proportionality being the fraction of the genome per 400 centimorgans in the region. Consider a neutral locus in a large region with uniform recombination rate. Let rbp denote the recombination rate per base pair in females in the neighborhood of the neutral locus. The number of base pairs within 400sh centimorgans of neutral locus is 4sh/rbp. By using the fact that the haploid genome is about 3 × 108 base pairs in size, F(200 sh) = 4sh/(rbp × 3 × 108). By substituting this into equation 2 and assuming that UT = 1.0, one obtains
Note that the parameter sh has canceled out. It should be emphasized that this simplified expression is only applicable if the recombination rate per base pair is roughly constant throughout the region 200sh centimorgans on each side of the neutral locus. This will clearly not be true for loci near the tips and centromeres of chromosomes. If the recombination rate per base pair is 10-8 [a typical rate of recombination for regions away from tips and bases of chromosomes (Chovnick et al., 1977)], then R = exp(-0.67) = 0.5. Thus, even in regions of normal recombination, it appears that background selection could have a substantial effect on standing levels of neutral variation.
Further support for the idea that deleterious mutations are reducing variation in regions of normal recombination is obtained from the observation of transposable element insertion polymorphism from the adh region of Drosophila melanogaster. In a restriction site polymorphism survey of a 15-kb region including the adh locus (Aquadro et al., 1986), it was found that 20% of their chromosomes carried one or more transposable-element insertions. Particular insertions were almost all rare and several arguments suggest that these insertions are destined to be
lost due to some form of natural selection (Golding, 1987). If these transposable-element insertions are indeed deleterious, then the background selection operating on these transposable elements should by itself reduce the level of variation in the adh region by 20%. Other transposable elements at somewhat larger distances from adh should also have an effect unless the selection against them is very weak. Other forms of deleterious mutations are also presumably occurring in this region and would make R even smaller.
To analyze X chromosome-linked loci, slightly modified equations are required for f0 (for the no-recombination case, see Charlesworth et al., 1993). Using these modified equations and methods analogous to those described above, one can calculate R values for loci at the tip and base of the X chromosome for which data are available. The predicted R, obtained in this way, for the yellow-achaete-scute region is 0.7. This value is considerably above the very low levels of variation actually observed in this region. Similarly, the low levels of polymorphism observed on the fourth chromosome of Drosophila melanogaster are not predicted by the background selection model. These are the same conclusions reached by Charlesworth et al., (1993) using slightly different methods.
In summary the background selection model predicts substantial reductions in polymorphism in some regions of the genome. However, the extreme reductions observed in the tip and base of the X chromosome and the fourth chromosome are not predicted with the versions of the model currently analyzed and the best available estimates of the parameters.
Discussion and Conclusion
The hitchhiking model appears to be able to account for the major features of the data. Current models of background selection do not appear to be able to account for the very large reductions in polymorphism levels observed in some regions of the Drosophila genome. However, it appears quite probable that background selection does have a substantial effect on some loci and analyses of observed patterns of variation need to take it into consideration. Other aspects of the data such as the frequency spectrum of the observed polymorphisms and the patterns of geographic variation are beginning to be analyzed and may shed additional light on the underlying process (Stephan and Mitchell, 1992; Begun and Aquadro, 1993).
These analyses of molecular divergence and polymorphism illustrate some of the difficulties and the promise of indirect analysis of molecular data. To make progress toward understanding the popu-
lation genetic processes that produce the patterns of molecular polymorphism and the patterns of divergence observed requires a continual interplay between collection and analysis of informative data sets and the consideration of appropriate models. There is clearly a long way to go on the road to understanding the mode of molecular evolution.
Summary
Different regions of the Drosophila genome have very different rates of recombination. For example, near centromeres and near the tips of chromosomes, the rates of recombination are much lower than in other regions. Several surveys of polymorphisms in Drosophila have now documented that levels of DNA polymorphism are positively correlated with rates of recombination; i.e., regions with low rates of recombination tend to have low levels of DNA polymorphism within populations of Drosophila. Three hypotheses are reviewed that might account for these observations. The first hypothesis is that regions of low recombination have low neutral mutation rates. Under this hypothesis between-species divergences should also be low in regions of low recombination. In fact, regions of low recombination have diverged at the same rate as other regions of the genome. On this basis, this strictly neutral hypothesis is rejected. The second hypothesis is that the process of fixation of favorable mutations leads to the observed correlation between polymorphism and recombination. This occurs via genetic hitchhiking, in which linked regions of the genome are swept along with the selectively favored mutant as it increases in frequency and eventually fixes in the population. This hitchhiking model with fixation of favorable mutations is compatible with major features of the data. By assuming this model is correct, one can estimate the rate of fixation of favorable mutations. The third hypothesis is that selection against continually arising deleterious mutations results in reduced levels of polymorphism at linked loci. Analysis of this background selection model shows that it can produce some reduction in levels of polymorphism but cannot explain some extreme cases that have been observed. Thus, it appears that hitchhiking of favorable mutations and background selection against deleterious mutations must be considered together to correctly account for the patterns of polymorphism that are observed in Drosophila.
References
Aguadé, M., Miyashita, N. & Langley, C. H. (1989) Reduced variation in the yellow-achaete-scute region in natural populations of Drosophila melanogaster. Genetics 122, 607–615.
Aquadro, C. F., Begun, D. J. & Kindahl, E. C. (1994) Selection, recombination, and DNA polymorphism in Drosophila. In Alternatives to the Neutral Model, ed. Golding, G. B. (Chapman & Hall, New York), pp. 46–56.
Aquadro, C. F., Deese, M. M., Bland, C. H., Langley, C. H. & Laurie-Ahlberg, C. C. (1986) Molecular population genetics of the alcohol dehydrogenase gene region of Drosophila melanogaster. Genetics 114, 1165–1190.
Begun, D. J. & Aquadro, C. F. (1992) Levels of naturally occurring DNA polymorphism correlate with recombination rates in D. melanogaster. Nature (London) 356, 519–520.
Begun, D. J. & Aquadro, C. F. (1993) African and North American populations of Drosophila melanogaster are very different at the DNA level. Nature (London) 365, 548–550.
Berry, A. J., Ajioka, J. W. & Kreitman, M. (1991) Lack of polymorphism on the Drosophila fourth chromosome resulting from selection. Genetics 129, 1111–1117.
Charlesworth, B., Morgan, M. T. & Charlesworth, D. (1993) The effect of deleterious mutations on neutral molecular variation. Genetics 134, 1289–1303.
Chovnick, A., Gelbart, W. & McCarron, M. (1977) Organization of the Rosy locus in Drosophila melanogaster. Cell 11, 1–10.
Crow, J. F. (1970) in Mathematical Topics in Population Genetics, ed. Kojima, K.-I. (Springer, Berlin), pp. 128–177.
Crow, J. F. & Simmons, M. J. (1983) in The Genetics and Biology of Drosophila, eds. Ashburner, M., Carson, H. L. & Thompson, J. N. (Academic, London), pp. 1–35.
Gillespie, J. H. (1991) The Causes of Molecular Evolution (Oxford Univ. Press, New York).
Golding, G. B. (1987) The detection of deleterious selection using ancestors inferred from a phylogenetic history. Genet. Res. 49, 71–82.
Houle, D., Hoffmaster, D. K., Assimacopoulos, S. & Charlesworth, B. (1992) The genomic mutation rate for fitness in Drosophila. Nature (London) 359, 58–60.
Hudson, R. R. & Kaplan, N. L. (1994) Gene trees with background selection. In Alternatives to the Neutral Model, ed. Golding, G. B. (Chapman & Hall, New York), pp. 140–153.
Kaplan, N. L., Hudson, R. R. & Langley, C. H. (1989) The ''hitchhiking effect" revisited. Genetics 123, 887–899.
Kimura, M. (1983) The Neutral Theory of Molecular Evolution (Cambridge Univ. Press, Cambridge, U.K.).
Kimura, M. & Maruyama, T. (1966) The mutational load with epistatic gene interactions in fitness. Genetics 54, 1337–1351.
Langley, C. H., MacDonald, J., Miyashita, N. & Aguadé, M. (1993) Lack of correlation between interspecific divergence and intraspecific polymorphism at the suppressor of forked region in Drosophila melanogaster and Drosophila simulans. Proc. Natl. Acad. Sci. USA 90, 1800–1803.
Martín-Campos, J. M., Comeron, J. P., Miyashita, N. & Aguadé, M. (1992) Intraspecific and interspecific variation at the y-ac-sc region of Drosophila simulans and Drosophila melanogaster. Genetics 130, 805–816.
Maynard Smith, J. & Haigh, J. (1974) The hitchhiking effect of a favorable gene. Genet. Res. 23, 23–35.
Mukai, T., Chigusa, S. I., Mettler, L. E. & Crow, J. F. (1972) Mutation rate and dominance of genes affecting viability in Drosophila melanogaster. Genetics 72, 335–355.
Nei, M. (1987) Molecular Evolutionary Genetics (Columbia Univ. Press, New York).
Stephan, W. & Langley, C. H. (1989) Molecular genetic variation in the centromeric region of the X chromosome in three Drosophila ananassae populations. I. Contrasts between the vermillion and forked loci. Genetics 121, 89–99.
Stephan, W. & Mitchell, S. J. (1992) Reduced levels of DNA polymorphism and fixed between-population differences in the centromeric region of Drosophila ananassae. Genetics 132, 1039–1045.
Stephan, W., Wiehe, T. H. E. & Lenz, M. W. (1992) The effect of strongly selected substitutions on neutral polymorphism—analytical results based on diffusion theory. Theor. Popul. Biol. 41, 237–254.
Wiehe, T. H. E. & Stephan, W. (1993) Analysis of a genetic hitchhiking model, and its application to DNA polymorphism data from Drosophila melanogaster. Mol. Biol. Evol. 10, 842-854.
| This page in the original is blank. |












