
15
The History of a Genetic System
Aleksandar Popadic/and Wyatt W. Anderson
Nineteen forty-four was a special year in the history of evolutionary biology. George Gaylord Simpson published Tempo and Mode in Evolution (1944), bringing paleontology and the concepts of macroevolution into the modern synthesis of evolutionary theory. Nineteen forty-four also saw the publication of Contributions to the Genetics, Taxonomy, and Ecology of Drosophila pseudoobscura and Its Relatives by Theodosius Dobzhansky and Carl Epling (Dobzhansky and Epling, 1944). This monograph presented a detailed analysis of chromosomal polymorphism for inversions in D. pseudoobscura and Drosophila persimilis, summarizing earlier studies and presenting extensive new data. These inversions, often referred to as gene arrangements, constitute a genetic system that has played a prominent role in studies of population genetics and evolutionary biology during the past 50 years. In one of the three papers making up the 1944 monograph, Epling reasoned on biogeographical grounds that the distribution pattern of these gene arrangements was ancient, dating from perhaps the Miocene. It follows from Epling's hypothesis that the gene arrangements themselves are ancient. Simpson was one of the principals in a lively correspondence that ensued between Epling and other evolutionary biologists about the age of this genetic system. Simpson, Mayr, and Stebbins published their views as a Symposium on the Age of the Distribution Pattern of the Gene Arrangements in Drosophila pseudoobscura in 1945 (Mayr et al. , 1945).
Aleksandar Popadic/ is a Ph.D. candidate in genetics and Wyatt W. Anderson is professor of genetics at the University of Georgia, Athens.
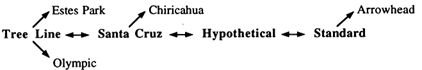
Figure 1 Cytogenetic phylogeny of the D. pseudoobscura gene arrangements examined in the present study.
Stebbins supported Epling's hypothesis and argued that the inversion system was quite old, while Mayr disagreed with Epling and favored a more recent origin of the distribution pattern. Simpson concluded that the age of the system could not be determined from the available evidence. It thus seems appropriate in this colloquium honoring the 50th anniversary of Simpson's seminal work to address a question about the history of the D. pseudoobscura inversions, not quite the question of age that Simpson considered, but rather the related question of which gene arrangement was the ancestral one.
In D. pseudoobscura, the third chromosome is polymorphic for more than 40 gene arrangements resulting from overlapping paracentric inversions (Olvera et al., 1979), which can be ordered in a phylogeny based on the breakpoints of inversions under the parsimonious assumption that each inversion arose only once (Figure 1). With the single exception of Hypothetical, all of the gene arrangements necessary to reconstruct the complete phylogeny have been observed in nature. Four of these arrangements—Standard (ST), Hypothetical (HY), Santa Cruz (SC), and Tree Line (TL)—are central to the phylogeny, with all the others being their one- or two-step derivatives. The tree in Figure 1 is unrooted, and the question of which of these four gene arrangements is ancestral has remained unanswered for more than 50 years, inasmuch as cytogenetic, biogeographic, and electrophoretic data have not consistently supported a single hypothesis.
ST was the first arrangement proposed as ancestral (Sturtevant and Dobzhansky, 1936), because it is the only one shared by D. pseudoobscura and its sibling species, Drosophila persimilis. HY was suggested (Dobzhansky and Sturtevant, 1938) because its inverted region resembles the banding pattern of the homologous chromosome in Drosophila miranda , a related species more distant from D. pseudoobscura than is D. persimilis. Historically, SC has received less attention than the other arrangements, although it too has been suggested (Dobzhansky and Epling, 1944, Brown, 1989). More recently, TL has been considered a favorite candidate for the ancestral gene arrangement on the basis of its distribution pattern (Wallace, 1966; Olvera et al., 1979; Wallace, 1988) and comparison of alleles at protein loci in D. pseudoobscura, D. persimilis,
and D. miranda (Olvera et al., 1979) and because it pairs more fully with the miranda homolog of the pseudoobscura third chromosome in interspecies hybrids than does either SC or ST (Wallace, 1966; Morrow, 1970; Wallace, 1988). The only informative molecular data on this topic come from a recent analysis of restriction site polymorphism (RSP) within these arrangements (Aquadro et al., 1991) that produced two important results. First, the phylogeny based on RSP data corroborates the cytogenetic phylogeny. Second, it was estimated that the TL branch diverged from the SC-ST group about 1.7 million years ago. The greater depth of the TL branch and its early splitting have been used to support the ancestral status of the TL arrangement, which represents the consensus view today (Aquadro et al., 1991; Powell, 1992).
We decided to approach the question of the ancestral arrangement in the following way. First, an independent assessment of the phylogenetic relationships among the central gene arrangements was made on the basis of nucleotide sequences flanking the amylase 1 (Amy1) gene, which is located within the inversions. Second, a framework of competing hypotheses regarding the ancestral type was devised on the basis of the known relationships among central arrangements, and each of these hypotheses was compared with the empirically derived phylogenies. Third, several additional chromosomes were added to our earlier RSP data set (Aquadro et al., 1991) and analyzed by several different methods to determine whether this more extensive phylogenetic analysis still supported an ancestral status for TL.
Materials And Methods
Lines of D. pseudoobscura stocks homozygous for the third chromosome were constructed using balancer stocks (Pavlovsky and Dobzhansky, 1966). Salivary glands were dissected from third-instar larvae, 30 per line, and gene arrangements were diagnosed from squash preparations of the polytene chromosomes. The six strains used for determining DNA sequences flanking Amy1 are as follows: ST, Ayala reference strain, from northern California; SC, strain BAJA 859#3, from Baja California, Mexico; Chiricahua (CH), strain AH 87#2, from northern California; TL, strain AH 73#2, from northern California; Estes Park (EP), strain BC p430#4, from British Columbia, Canada; Olympic (OL), strain s14AR-D; from British Columbia, Canada. Three SC strains from Michoacan, Mexico, were added to the original RSP data set: strain MEX z67w, strain MEX z53y, and strain MEX z13w. Restriction mapping of these strains was carried out as described previously (Aquadro et al., 1991). In addition to the inversions already identified, the RSP data set includes Arrowhead (AR).
Isolation of total genomic DNA was accomplished by extraction from freshly ground flies and purification by CsCl density gradient centrifugation (Bingham et al., 1981). Genomic DNA was digested with HindIII and EcoRI restriction enzymes and then loaded onto a 5–30% sucrose step gradient (Ausubel et al., 1987). The fraction containing 5- to 6-kb fragments was cloned into the vector pBluescript SK—(Stratagene) and transformed by high-voltage electroporation (Maniatis et al., 1982, Dower et al., 1988). D. pseudoobscura clones containing Amy homologous sequences were isolated from a genomic library by colony hybridization (Maniatis et al., 1982) using the plasmid pFA4 (Brown et al., 1990) containing the D. pseudoobscura Amy1 coding sequence as probe. All sequences were determined using an automatic sequencer (Applied Biosystems 373A) following the manufacturer's protocol. In all cases both strands were sequenced. Some regions were sequenced again manually (Sanger et al., 1977) using the Sequenase DNA sequencing kit (United States Biochemical). Sequenced regions include 667 nucleotides (from bases 701 to 35) upstream of the start codon and 391 nucleotides (from bases 62 to 452) downstream of the stop codon. These sequences have been deposited in GenBank (accession numbers U09746–U09757). Sequences were aligned with each other using the GENALIGN program (IntelliGenetics) and checked again by eye. Sequence divergence estimates were calculated as direct counts of nucleotide sequence differences, since no correction is needed for differences as small as those in our study (Nei, 1987).
The phylogenetic analysis was carried out using the neighbor-joining (NJ) and maximum likelihood (ML) methods in the PHYLIP package (Felsenstein, 1993), the NJ method in the MEGA package (Kumar et al., 1993), and the maximum parsimony (MP) method in PAUP (Swofford, 1991). For the RSP data, we excluded strains with a redundant restriction pattern, which reduced the number of strains that were phylogenetically analyzed from 33 to 21. To bootstrap the NJ tree derived from RSP data, we followed the advice kindly given to us by Walter Fitch. First, "1" and "0" in the data set were replaced with "A" and "T," respectively. Second, the SEQBOOT program (PHYLIP) was used to produce 100 bootstrapped data sets. Third, the DNADIST, NEIGHBOR, and CONSENSE programs (PHYLIP) were used in succession to produce the bootstrap values.
Results And Discussion
A Test of Hypotheses Regarding the Ancestral Gene Arrangement. Amylase in D. pseudoobscura is a family of three genes, located within the inverted region in most gene arrangements (Aquadro et al., 1991). The Amy1 gene
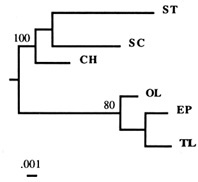
Figure 2 Dendrogram based on the combined flanking sequences of the Amy1 gene, obtained by the NJ algorithm. A separate analysis of the two (5' and 3') flanking regions generated the same basic topology. Numbers refer to the percentage of times a node was supported in 200 bootstrap replications.
is the only copy present in all arrangements, and it has been suggested that the Amy2 and Amy3 copies arose by duplication of Amy1 (Brown et al., 1990). In this study, we used only 5' and 3' flanking regions specific to the Amy1 gene.
A phylogenetic analysis of each flanking region generated the same branching topology, so we combined the two regions (Figure 2). In the deduced phylogeny, the TL arrangement splits off early from the SC–ST group, a finding concordant with the results of the previous RSP data analysis (Aquadro et al., 1991). An intuitive interpretation of this phylogeny is that the TL arrangement is ancestral to both SC and ST arrangements. and the SC–ST group calculated from our sequence data agrees well with that based on RSP data (2.9%). These findings seem to support the ancestral status of TL. What they do not provide is an exclusion of other gene arrangements as potential ancestors. Thus, we are still left with the following questions: Are our results consistent with another arrangement than TL being ancestral? And what about the HY arrangement, which has never been found in nature? Since no data are available for it, the phylogenies in Figure 2 cannot be used to rule out the possibility that HY was in fact ancestral.
To settle these questions, we developed a framework of competing hypotheses based on the assumption that each inversion type is monophyletic in origin, which accords with the RSP data (Aquadro et al., 1991), and the assumption that the relationships among the four central arrangements (Figure 1) are derived parsimoniously, with each inversion arising by two breakpoints from its parental arrangement. Under these assumptions, we successively chose each of the four possible arrangements to represent the ancestral type and asked in each case what the branching pattern of the resulting phylogenetic tree would be.
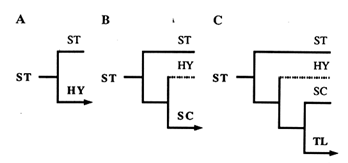
Figure 3 An illustration of the phylogeny that would result from three successive inversion events, beginning with the ST gene arrangement. In A, an inversion of the ST gene arrangement gives HY; in B, an inversion of HY gives SC; and in C, an inversion of SC gives TL.
For the reader not familiar with the construction of phylogenies, this reasoning is shown in Figure 3. By applying this approach to each of the four central arrangements, we generated the six phylogenies displayed in Figure 4. Since the ST and TL arrangements are at the ends of the phylogeny, each produces only one tree (Figure 4A and B). Because they are located in the middle of the phylogeny, the HY and SC arrangements have two possibilities for the first node, thus producing two possible trees each (Figure 4 C1, C2, D1, and D2).
Of the six possible trees, four are incompatible with the empirically obtained tree in Figure 2, which rules out the scenarios based on the ancestral status of ST and HY (Figure 4A, C1, and C2), as well as the one assuming both that SC is ancestral and that the first node leads to HY (Figure 4D2). Although the HY arrangement has never been found in nature, we are still able to exclude it as a potential ancestor. Each of the two remaining trees agrees with the empirical tree. One of them (Figure 4B) follows from the assumption that TL is ancestral, while the other (Figure 4D1) represents the case where SC is the ancestor and the first node leads to TL. On the basis of this analysis, then, either the TL or the SC arrangement could be ancestral.
Could SC be the Ancestral Arrangement? In the previous section, we used Amy flanking regions to generate the empirical phylogeny. These regions appear to be functionally unconstrained, with sequence divergence levels comparable to that of the RSP data. This makes them useful for phylogenetic analysis. At the present moment, however, the lack of information from the outgroup species, D. miranda, limits our ability to distinguish between the two candidates (TL and SC) on the basis of sequence data. This leaves the RSP data as the only source of further
insight into the ancestral arrangement. Therefore, we decided to reanalyze the data set from the original RSP study with the following additions. First, we added three more SC strains to assure that SC was adequately represented, since the original data set contained only two SC strains. Second, the data were analyzed with the NJ, MP, and ML methods. All three methods have been shown (Jin and Nei, 1991) to be superior in finding the correct tree topology over the unweighted pair-group method of averages (UPGMA) algorithm used previously. It
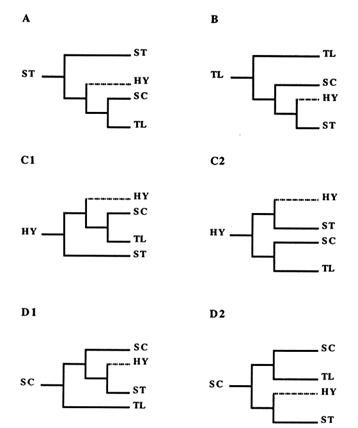
Figure 4 The six possible scenarios showing relationships among ST, HY, SC, and TL gene arrangements. In A, ST is ancestral, whereas in B, TL occupies that position. In C1 and C2, HY is the ancestor and the first bifurcation leads to ST and SC. In D1 and D2, SC is the ancestral type with the first bifurcation leading to TL and HY. Branches leading to HY are indicated by dotted lines, since this gene arrangement has not been found in nature.
is especially instructive to see if the phylogenetic analysis of the enlarged RSP data set also supports an ancestral status for TL.
The NJ tree (Figure 5A) shows one SC strain and all of CH strains as being closest to the outgroup (D. miranda), with very short branch lengths. The rest of the SC strains join either the TL or the ST arrangements. In contrast, the ST and TL arrangements form separate clades (along with their derivatives). Except for the short branches that are replaced with a multifurcation at the ancestral node, the same tree topology was generated with the ML method (Figure 5B). The parsimony analysis was performed both on the complete data set (results not shown) and on selected subsets of the data, two of which are shown (Figure 6A, B). Again, all of the MP trees show no difference from the previously generated NJ and ML trees. Therefore, the same basic topology was generated with the three different methods, although each method is based on a different set of assumptions. The major characteristic of this common topology is that while the TL and ST arrangements (and their derivatives) form separate clades, the SC arrangements are spread throughout the phylogeny. One SC arrangement is closest to the outgroup
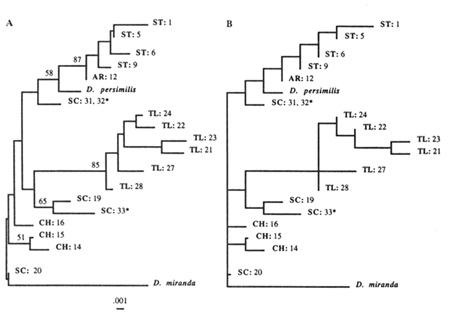
Figure 5 Phylogenetic trees inferred from the RSP data. Taxa are labeled as in the previous study (Aquadro et al., 1990), with three new SC strains indicated by an asterisk. (A) NJ tree. Numbers refer to the bootstrap values as percentages for 100 replicates. (B) Maximum likelihood tree.
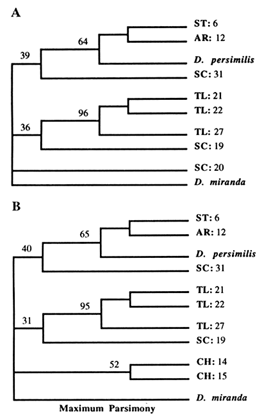
Figure 6 MP trees. Numbers refer to parsimony bootstrap values in percentages (out of 100 replicates). Branch lengths are not proportional to the number of nucleotide changes. (A) This tree is a strict consensus of 10 trees of length 25. The consistency index was 0.80. (B) Strict consensus of 10 trees of length 27, with a consistency index of 0.78.
(according to the NJ and ML methods), while the rest join either the TL or ST clades. Also, the CH arrangements split off from the ancestral node, without joining any of the SC strains. These results are somewhat different than those obtained with the UPGMA method in the previous study (Aquadro et al., 1991). This discrepancy is due partially to the addition of three more SC strains, after which the scattering of SC arrangements throughout the phylogeny becomes obvious, and partially to the varying rate of evolution among different arrangements, combined with the low level of observed divergence (at most 4%). Recently, it has been shown that the UPGMA method performs inadequately under either circumstance, especially for RSP data (Jin and Nei, 1991).
We decided to investigate this matter further by modifying our framework of competing hypotheses so it would take into account all arrangements represented in the RSP data. Two main changes were inclusion of both the outgroup (D. miranda) as well as derivatives of the
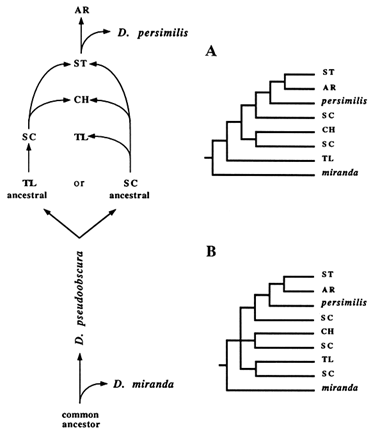
Figure 7 The two possible scenarios showing the relationships among all gene arrangements represented in the RSP data set. In A, TL is assumed to be ancestral; in B, SC is assumed to be ancestral.
SC and ST arrangements. Also, for simplicity, the HY inversion was not included. By following the same sort of reasoning depicted in Figure 4, we derived two trees that assume that either TL or SC was the ancestral gene arrangement (Figure 7A, B). The main difference between them is the branching pattern at the ancestral node. If TL is the ancestral arrangement, then TL and D. miranda should share a common ancestor, whereas SC and D. miranda should not. Phylogenetically, this scenario would require TL to branch off immediately after the outgroup. In contrast, the ancestry of SC predicts an unresolved branching pattern, represented with a trifurcation at the ancestral node. This same lack of resolution can be observed in all empirical trees, combined with the dispersal of SC throughout the phylogeny.
Therefore, comparison of the two competing hypotheses with the empirical results is more supportive of an ancestral status for SC. George Gaylord Simpson would probably have been pleased to know that 50 years after he considered the history of gene arrangements in D. pseudoobscura, a molecular evolutionary approach has brought us close to an understanding of the origin of this genetic system.
Summary
Although the chromosomal polymorphism for inversions in Drosophila pseudoobscura is one of the best studied systems in population genetics, the identity of the ancestral gene arrangement has remained unresolved for more than 50 years. There are more than 40 gene arrangements, and 4 of them (Standard, Hypothetical, Santa Cruz, and Tree Line) have been considered as candidates for the ancestral type. We propose a framework of competing hypotheses to distinguish among the alternatives. Two conclusions come from contrasting each hypothesis with the results from DNA sequencing and restriction mapping. First, not only Standard but also Hypothetical can be excluded as the ancestral gene arrangement. Second, although either Tree Line or Santa Cruz could be the ancestral type, the available data provide greater support for Santa Cruz.
We thank Danijela Popadic for technical assistance and J. C. Avise, F. Ayala, M. Arnold, M. Ball, Y. Fu, J. Hamrick, E. McCarthy, J. McDonald, R. Meagher, and B. Wallace for comments on the manuscript; B. Bowen provided an especially helpful review. We also thank M. T. Clegg, W. M. Fitch, and E. Mayr for helpful suggestions about our analysis. This work was supported in part by a grant from the National Science Foundation (BSR-8516188 to W.W.A.).
References
Aquadro, C. F., Weaver, A. L., Schaeffer, S. W. & Anderson, W. W. (1991) Molecular evolution of inversions in Drosophila pseudoobscura : the amylase gene region. Proc. Natl. Acad. Sci. USA 88, 305–309.
Ausubel, F. M., Brent R., Kingston, R. E., Moore, D. D., Seidman, J. G., Smith, J. A. & Struhl, K. (1987) Current Protocols in Molecular Biology (Wiley/Green, New York).
Bingham, P. M., Levis, R. & Rubin, G. M. (1981) Cloning of DNA sequences from the white locus of D. melanogaster by a novel and general method. Cell 25, 693–704.
Brown, C. J. (1989) Ph.D. thesis (Univ. of Georgia, Athens).
Brown, C. J., Aquadro, C. F. & Anderson, W. W. (1990) DNA sequence evolution of the amylase multigene family in Drosophila pseudoobscura. Genetics 126, 131–138.
Dobzhansky, T. & Sturtevant, A. H. (1938) Inversions in the chromosomes of Drosophila pseudoobscura. Genetics 23, 28–64.
Dobzhansky, T. & Epling, C. (1944) Contributions to the genetics, taxonomy, and ecology of Drosophila pseudoobscura and its relatives. Carnegie Inst. Washington Publ. 554.
Dower, W. J., Miller, J. F. & Ragsdale, C. W. (1988) High efficiency transformation of E. coli cells by high voltage electroporation. Nucleic Acids Res. 16, 6127.
Felsenstein, J. (1993) PHYLIP, Phylogeny Inference Package (Univ. of Washington, Seattle), Version 3.5c.
Jin, L. & Nei, M. (1991) Relative efficiencies of the maximum-parsimony and distance-matrix methods of phylogeny construction for restriction data. Mol. Biol. Evol. 8, 356–365.
Kumar, S., Tamura, K. & Nei, M. (1993) MEGA, Molecular Evolutionary Genetics Analysis (Pennsylvania State Univ., University Park, PA), Version 1.
Maniatis, T., Fritsch, E. F. & Sambrook, J. (1982) Molecular Cloning: A Laboratory Manual (Cold Spring Harbor Lab. Press, Plainview, NY).
Mayr, E., Stebbins, G. L. & Simpson, G. G. (1945) in Symposium on age of the distribution pattern of the gene arrangements in Drosophila pseudoobscura. Lloydia 8, 69–108.
Morrow, D. (1970) M.S. thesis (Cornell Univ., Ithaca, NY).
Nei, M. (1987) Molecular Evolutionary Genetics (Columbia Univ. Press, New York).
Olvera, O., Powell, J. R., de la Rosa, M. E., Salceda, V. M., Gaso, M. I., Guzman, J., Anderson, W. W. & Levine, L. (1979) Population genetics of Mexican Drosophila VI. Cytogenetic aspects of the inversion polymorphism in Drosophila pseudoobscura. Evolution 33, 381–395.
Pavlovsky, O. & Dobzhansky, T. (1966) Genetics of natural populations. XXXVII. The coadapted system of chromosomal variants in a population of Drosophila pseudoobscura. Genetics 53, 843–854.
Powell, J. (1992) Inversion polymorphism in Drosophila pseudoobscura and Drosophila persimilis. In Drosophila Inversion Polymorphism, eds. Krimbas, C. & Powell, J. R. (CRC, London), pp. 73–125.
Sanger, F., Nicklen, S. & Coulson, A. R. (1977) DNA sequencing with chain terminating inhibitors. Proc. Natl. Acad. Sci. USA 74, 5463–5467.
Simpson, G. G. (1944) Tempo and Mode in Evolution (Columbia Univ. Press , New York).
Sturtevant, A. H. & Dobzhansky, T. (1936) Inversions in the third chromosome of wild races of Drosophila pseudoobscura, and their use in the study of the history of the species. Proc. Natl. Acad. Sci. USA 22, 448–450.
Swofford, D. L. (1991) PAUP, Phylogenetic Analysis Using Parsimony Illinois Nat. Hist. Surv., Champaign, IL), Version 3.0s.
Wallace, B. (1966) Chromosomes, Giant Molecules, and Evolution (Norton, New York).
Wallace, B. (1988) In defense of verbal arguments. Perspect. Biol. Med. 31, 201–211.












