
The Chemistry of Signal Transduction
JON CLARDY
Extracellular molecules can influence intracellular processes, and whether we refer to this as signal transduction or chemical ecology depends on context. For example, the conjugation of two cells of the bacterial species Streptomyces faecalis induced by an "aggregation substance" on the surface of one and a "binding substance" on the surface of the other is a textbook example of microbial chemical ecology (1). The activation of a resting helper T-cell through the stimulation of a receptor on its surface by an antigen on the surface of another cell has become one of the best studied examples of signal transduction (2, 3). In both cases a recognition between complementary elements on the surfaces of two different cells generates the potential for altered cellular function. Many other examples attest to the fundamental similarity between chemical ecology and signal transduction. (i) When an external molecule stimulates the IgE receptor on the surface of a mast cell, an intracellular signal leads to the release of histamine in a process referred to as degranulation (2). (ii) When a male gamete cell of the water mold Allomyces detects the sesquiterpene diol sirenin, it responds by swimming along the sirenin concentration gradient to find the female source (1). Mast-cell degranulation is usually studied as an example of signal transduction and Allomyces navigation as an example of chemical ecology, but in both
Jon Clardy is Horace White Professor of Chemistry at Cornell University, Ithaca, New York.
cases an external molecule dramatically affects cellular function. Our goal in studying any of these processes is the same: a molecule-by-molecule accounting of information transfer in biological systems.
While the prospect of analyzing every possible biological signal is daunting, the recognition that nature tends to use the same basic mechanism in a variety of guises makes the task less formidable. The repeated use of a common pathway is nicely illustrated by the immunosuppressive agents FK506, rapamycin, and cyclosporin. Their ability to disrupt signaling in T-cells leads to immunosuppressive activity, but the same molecules that disrupt signals in T-cells also prevent the degranulation of mast cells and inhibit the proliferation of yeast (2, 4). We could equally well call them antifungal, insecticidal, antiinflammatory, antiallergic, or antiretroviral as well as immunosuppressive agents (2, 4).
Another connection between cellular signal transduction and chemical ecology is the essential role played by natural products—secondary metabolites with no known role in the internal economy of the producing organism (5). Cyclosporin A (CsA), FK506, and rapamycin are all microbial natural products that are probably synthesized to chemically defend their producing organism (5). Studying these natural products as microbial chemical warfare agents would unarguably qualify as chemical ecology. However, the similarity of signaling pathways allows us to use these same natural products as probes of cellular signaling or as important chemotherapeutic agents in human disease.
The rest of this paper will focus on the factors affecting one step in one signaling pathway in resting helper T cells. The inquiry may appear overly specialized, but the strategy nature employs, using a small natural product to link two much larger proteins, has only recently been appreciated. Now that we recognize the strategy, we can expect to see it again.
SIGNAL TRANSDUCTION IN T CELLS AND THE ROLE OF NATURAL PRODUCTS
The signal to activate a resting helper T-cell can be divided into three parts: the extracellular recognition of an antigen by the membrane-spanning T-cell receptor (TCR), the cytoplasmic signal transduction cascade that transmits the recognition information to the nucleus, and the activation of genes in the nucleus (3, 6). The TCR recognizes the foreign antigen, a processed peptide held in the cleft of the major histocompatibility complex (MHC) protein on the surface of the antigen-presenting cell (7, 8). Some additional interactions be-
tween T-cell surface proteins such as CD4 or CD8 with the MHC protein are needed to start the signal on its way to the nucleus. The progression of the signal from the interior portion of the TCR to the nucleus is called the cytoplasmic signal transduction cascade—a series of steps that are imperfectly understood (6). Ultimately, the signal results in the expression of a gene and the production of a gene product. The interleukin 2 (IL-2) gene is activated when the nuclear factor of activated T-cells (NF-AT) binds to the IL-2 promoter region (9, 10).
Some parts of the signal process can be studied more easily than others. Much has been learned about the membrane-associated events involving MHC and TCR as well as events in the nucleus involving NF-AT and IL-2, but the cytoplasmic series of steps is less well defined. The study of cytoplasmic signal transduction, as well as a number of other biological processes, has progressed only to the extent that highly specific cellpermeant agents are available to inhibit or otherwise modify normal biological processes (11, 12). By far the most fruitful source of such agents is the realm of natural products. Natural products represent a library of tremendous chemical diversity and proven biological utility for—at the risk of tautology—only those natural products that convey some survival benefit are likely to have survived (5). Our understanding of the inhibition of the cytoplasmic signal transduction cascade of T-cells originated in the discovery of three natural products: CsA, FK506, and rapamycin (Figure 1).
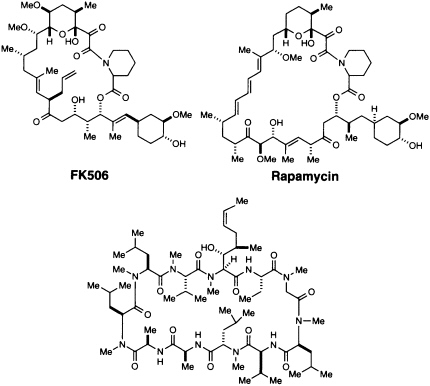
The discovery and utilization of CsA by Sandoz initiated a series of important advances in understanding and controlling immunosuppression (4, 13). Since 1983 this hydrophobic peptide has been widely used in clinical transplantation, and its introduction led to a remarkable increase in survival rates of transplanted livers and hearts (4). CsA (Figure 1) is produced by a variety of fungi imperfecti, notably Tolypocladium inflatum (formerly Trichoderma polysporum) isolated in Norway (13). FK506 (Figure 1) was discovered in a directed screening program at the Fujisawa Pharmaceutical Company in 1987 (14). It is produced by Streptomyces tsukubaensis, a species discovered in a soil sample from Tsukuba, Japan (15). While its biological effects are essentially identical to those of CsA, the two structures are chemically quite different (Figure 1). Rapamycin (Figure 1) was first described in 1975 as an antifungal agent (15-17). It was isolated from Streptomyces hygroscopicus from Easter Island, and the name rapamycin comes from Rapa Nui, the native name for Easter Island. All three agents have a variety of biological effects, but early studies showed that they interfere with a cytosolic signaling step and thus could be used to probe the cytoplasmic signal transduction pathway.
Cyclosporin A [CsA]
FIGURE 1 Structures of CsA, FK506, and rapamycin.
IMMUNOPHILINS AND THEIR COMPLEXES
The inhibitory natural products were used to identify their cellular targets, and in each case a highly specific binding protein was identified.
-
A cytosolic binding protein for CsA was first isolated in 1984 and named cyclophilin, later cyclophilin A (CyPA), in reference to its high-affinity for CsA (18). CyPA is a basic, abundant protein with a mass of 18 kDa, and it is found in a variety of tissues. The first clue to its function came in 1989 when two independent groups isolated the enzyme that catalyzes peptidyl proline isomerization/peptidylprolyl cis-trans isomerase (EC 5.2.1.8; PPIase), in protein chains and (re)discovered CyPA (19, 20). CyPA is a potent PPIase, and its enzymatic activity is strongly inhibited by CsA.
-
The cellular target of FK506 was identified by two independent
-
groups in 1989 and named FKBP, later FKBP12, for the FK506-binding protein with a mass of 12 kDa (21, 22). FKBP12 is also a potent PPIase, and FK506 strongly inhibits its activity (Ki = 0.4 nM).
-
Rapamycin also appears to have a major cytoplasmic target, FKBP12 (21-23). Rapamycin binds to FKBP12 and inhibits its PPIase activity slightly better than does FK506 (Ki = 0.2 nM). FK506 and rapamycin compete for the same binding site in FKBP12.
Immunophilin is the generic term for a binding protein for an immunosuppressive agent, and all currently known immunophilins belong to the cyclophilin or FKBP families. The two families of immunophilins—like the small molecules they bind—don't have any readily apparent relationship. FKBP12 and CyPA have no sequence similarity, CsA does not inhibit FKBP12, and FK506 does not inhibit CyPA.
Since both known targets of the immunosuppressive drugs are PPIases and all known PPIases are immunophilins (24), the hypothesis that PPIase activity is involved in signal transduction and that inhibition of PPIase activity is the convergent step in immunosuppression seemed plausible. However, other studies show that the PPIase hypothesis is not tenable (6). The main lines of contradictory evidence are that (i) relatively low levels of drug are required for immunosuppression—drug levels at which only a fraction of the PPIase activity was eliminated; (ii) synthetic analogs of both FK506 and CsA are potent PPIase inhibitors but have no immunosuppressive activity; (iii) while both FK506 and rapamycin bind the same protein, FKBP12, they affect different stages of the T-cell cycle—only FK506 inhibits the activation step leading to IL-2 production, and (iv) the yeast Saccharomyces cerevisiae, normally sensitive to FK506 and CsA, remains viable and insensitive to either drug when FKBP or CyPA is knocked out chemically or genetically (25, 26).
In the currently accepted model, the complex of the immunosuppressive agent with its cognate protein inhibits cytoplasmic signal transduction (6). FK506, CsA, and rapamycin form complexes with immunophilins, and these complexes possess immunosuppressive activity through their ability to interact with another target. The FKBP12-FK506 and the CyPA-CsA complexes have the same target, the abundant serine-threonine phosphatase calcineurin, and thus have essentially identical biological effects (27, 28). Calcineurin is inhibited by FKBP12-FK506 and CyPA-CsA at nanomolar concentrations, but the individual components show no inhibitory activity. Thus the natural product adds a new function to its binding protein, and how this acquired function works in atomic detail has attracted several groups of investigators. FKBP12-rapamycin interacts with a protein called FRAP, which is not as well characterized as calcineurin (29).
STRUCTURAL STUDIES ON IMMUNOPHILIN-IMMUNOSUPPRESSANT COMPLEXES
Both NMR and x-ray studies have been done on free FKBP12, CyPA, and a variety of complexes (30). These studies have been reviewed recently, and the remainder of this paper will focus on relatively recent work in the FKBP area (30). Unfortunately, there are no structural studies on the FKBP12-FK506-calcineurin or CyPA-CsA-calcineurin complexes, so our understanding of the interactions is indirect and incomplete. Nevertheless, the outline, if not the complete details, of an answer is apparent.
Both NMR and x-ray structural studies on FKBP12 and its complexes show that the protein folds as a five-stranded ß-sheet wrapped around a short a-helix with an overall shape resembling an ice cream cone (Figure 2 Left) (31-35). The ligands bind in a hydrophobic cavity between the helix and sheet in a pocket flanked by three loop regions. The protein core is composed exclusively of hydrophobic residues, many of which are highly conserved among FKBPs from different organisms. Although the protein is relatively small, it contains many structural motifs: ß-sheet, a-helix, 310-helix, and an assortment of turns. The region where FK506 and FKBP12 form a composite binding surface for calcineurin is of greatest interest. The composite binding surface is the surface that interacts with calcineurin, and it must contain elements of both FK506 and FKBP12, since changes in either one can lead to complexes that do not bind to calcineurin. The loop regions near the binding pocket are of special interest: the 40s loop formed by a bulge in the fifth ß-strand and the 80s loop between the second and third ß-strands (36).
The high-resolution x-ray structure of the FKBP12-FK506 complex shows half of FK506's solvent-accessible surface area buried in FKBP12; the other half of the ligand is exposed. Thus, FK506 has two domains: the binding domain that interacts with FKBP12 and the effector domain that can interact with a second protein. Formation of the complex appears to have structural consequences for FKBP12. Solution NMR studies of free FKBP12 have been reported, and these studies contain valuable insights into the protein's dynamic behavior (refs. 31, 32, 37-40; S. W. Michnick, M. K. Rosen, M. Karplus and S. L. Schreiber, unpublished data). Free FKBP12 has restricted motion for the hydrophobic core, whereas several residues in the 40s and 80s loops display higher mobility. When FK506 binds to FKBP12, both the 40s and 80s loops have well-defined structures, and a picture has emerged in which the 40s and 80s loops are relatively flexible in free FKBP12 but take on a structure when FK506 is bound (36). Several studies have shown that the structure of FKBP12 shows few if any changes that depend on the bound ligand (41). Thus, the x-ray structure of the FKBP12-rapamycin complex shows no significant
protein conformational changes—a conclusion reinforced by a subsequent studies (42, 43).
A detailed analysis of FKBP13, another member of the FKBP family, has led to important insights into the composite binding surface (44). FKBP13 was described by Schreiber's group in 1991 and used as the basis for an intriguing set of mutational studies (45-47). FKBP12 and FKBP13 are remarkably similar (43% amino acid identity), and the 92-amino acid C-terminal sequence of FKBP13 has 46 identical and 20 related residues when compared with FKBP12 (45). The two proteins show exact identity for all amino acids lining the FK506-binding pocket. However, embedded in this overall similarity are differences that result in a composite binding surface for FKBP13-FK506 that interacts only weakly with calcineurin (Ki= 1500 nM vs. 7.9 nM for FKBP12-FK506) (45). A series of chimeric FKBPs identified the 80s loop as the most important protein region for interaction with calcineurin. A chimeric protein with the 80s loop of FKBP13 replacing the corresponding residues of FKBP12 weakly inhibits calcineurin (Ki = 580 nM) (47). The complementary chimeric protein, in which the 80s loop of FKBP12 replaces the corresponding residues of FKBP13, is an effective calcineurin inhibitor (Ki = 27 nM) (47). Additional mutational studies patterned on the FKBP13 sequence point to the tip of the 80s loop as the key structural feature (46). The change of only two residues, proline rather than glycine at position 89 and lysine rather than isoleucine at position 90, in FKBP13 confers calcineurin-binding activity (Ki = 13 nM) (46).
Mutational studies on the 40s loop gave different, but no less interesting, results. The complete substitution of the 40s loop of FKBP12 by that of FKBP13 does not abolish activity; the resulting chimeric FKBP inhibits calcineurin (Ki = 19 nM) (47). In this chimeric protein, residue 42 is glutamine. However, the single amino acid change Arg-42 to glutamine in FKBP12 diminishes calcineurin inhibition by 2 orders of magnitude. A recently completed x-ray analysis of the FKBP13-FK506 complex adds a detailed structural understanding to these mutational results (44).
Figure 2 Right shows the overall structure of the FKBP13-FK506 complex, and a more detailed view comparing FKBP12-FK506 with FKBP13-FK506 is given in Figure 3. In FKBP12, Ile-90 contributes to a hydrophobic groove created by Phe-36 and the hemiketal ring of FK506 (Figure 3). In the structure of the FKBP13-FK506 complex, a lysine projects out from the 80s loop, covers this groove, and disrupts the hydrophobic pocket.
The structural analysis of the 40s loop is more complex. There are only minor differences at residues 38 and 43-45, but from residue 39 to residue 42 the FKBP13 backbone is displaced by up to 2 Å from that of FKBP12. How can the extensive modifications of the chimeric FKBP result in a
calcineurin-inhibiting complex, while the single mutation at Arg-42 results in a noninhibitory complex? The available data simply don't define a unique model (12). In one model Arg-42 makes a crucial contact with calcineurin. When this contact is lost, binding is diminished. A second model assumes that Arg-42 does not make a contact with calcineurin; it forms part of an Arg-Asp-Tyr triad that keeps the 40s loop from interfering with calcineurin binding. The single Arg-42 mutation allows the 40s loop to interfere with binding whereas the wholesale change of residues provides a different organizational motif. The different organizational motif could include bound waters, conformational restriction from Pro-41, and an interaction between Leu-40 and His-25.
While not all the details of the composite binding surface of the FKBP12-FK506 complex are known, the general way in which FK506 adapts FKBP12 to form such a surface is becoming clear.
FK506 AND PEPTIDOMIMICRY
The discovery of FKBP12 as an abundant cytosolic protein found in a wide variety of cells suggests that it has an important role. What is that role and what is the natural ligand for FKBP12? We are exploring an approach to this teleological question based on the assumption that FK506 mimics an endogenous, probably peptidyl, substance (48).
The possibility that FK506 and rapamycin (Figure 1) mimic peptide ligands for or substrates of FKBP12 is suggested by the structural similarity between the pyranose ring, a-keto amide, and the homoprolyl fragments of FK506 on one hand and the optimal Leu-Pro and Val-Pro substrates for PPIase activity on the other (12). The peptide analogy is amplified by a pair of hydrogen bonds between FKBP12 and FK506 (Figure 4A) revealed in the high-resolution x-ray analysis (33, 41). This peptidomimetic analysis of FK506 leaves a stereochemical puzzle, the stereochemistry of the substituent at C-26, which has the nonnatural configuration compared with the natural peptide (note R in Figure 4A and R' in Figure 4B).
We recently completed a high-resolution x-ray diffraction analysis of an FK506-peptide hybrid bound to FKBP12 (48). The hybrid ligands were developed around the a-keto homoprolyl moiety found in FK506-an element that appears crucial for tight binding. The adjoining amino acids were optimized to give maximum binding, and finally the variable-length tethers were added (Figure 4C). A 210-nm binder was selected for careful structural analysis. The resulting structure shows that many features first identified in FKBP12-FK506 persist in this hybrid structure (48). The homoprolyl ring is the most deeply buried and the tether is on the outside of the protein. The structure also has two hydrogen bonds between the
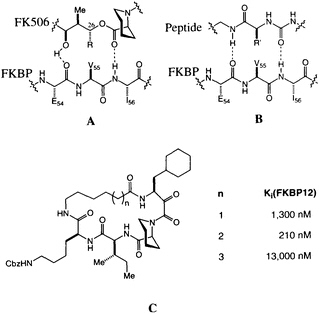
FIGURE 4 (A) Schematic of the original peptidomimetic analysis of FK506. Note the hydrogen bonds to Ile-56 and Glu-54. (B) Schematic of a putative peptidyl ligand binding to FKBP12, based on A. Note the difference in stereochemistry for R and R'. (C) Structure of the cyclic peptide-FK506 hybrids and their inhibitory constants for FKBP12 PPIase inhibition.
homoprolyl C==O and Ile-56 and the main-chain C==O of Glu-54 and the lysine NH of the hybrid ligand. The dipeptide fragment of the hybrid ligand binds to FKBP12 by forming a short, two-stranded antiparallel sheet. If the structures of FKBP12-FK506 and FKBP12-hybrid are superimposed, a previously unappreciated feature becomes clear. The trisubstituted double bond of FK506, which was earlier considered to be the ''side-chain" substituent (R in Figure 4A), has an orientation and shape similar to the isoleucine amide of the hybrid. The atoms of the macrocyclic ring of FK506, which were earlier considered to be "main chain," closely follow the path of the isoleucine side chain of the hybrid. The structure suggests that the trisubstituted double bond of FK506 is an amide surrogate and its ring atoms mimic an amino acid side chain (Figure 5). Reversing the roles of these two groups in FK506 resolves the stereochemical puzzle, since the stereochemistry at C-26 now corresponds to the natural stereochemistry of an amino acid.
The structure affords other insights into the possible binding of
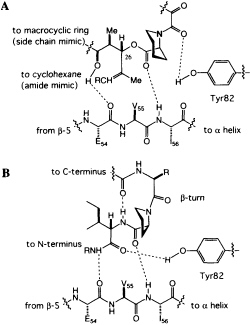
FIGURE 5 (A) Schematic of the revised peptidomimetic analysis of FK506. (B) Model of a peptide bound to FKBP12.
peptides to FKBP12. The phenolic hydrogen of Tyr-82 forms a hydrogen bond with the amide carbonyl of the dicarbonyl unit in FK506 (Figure 5A). In the hybrid structure, the phenolic hydrogen forms a hydrogen bond with the C==O of the isoleucine fragment (Figure 5B), an arrangement that would not have been possible for FK506. An intramolecular hydrogen bond from the linker carbonyl and the isoleucine NH suggests that peptides bind to FKBP12 with a ß-turn (Fig. 5B), a conclusion independently reached by a theoretical analysis of the PPIase mechanism (49).
BROADER PERSPECTIVE
The desire to understand the molecular basis of information transfer in biological systems unifies many seemingly disparate disciplines, and strategies discovered in one discipline are likely to be relevant to all. The use of the natural products CsA, FK506 and rapamycin has greatly enhanced understanding of cytoplasmic signal transduction in T-cells, but more importantly they have sketched an initial picture of how a small molecule can simultaneously interact with two large molecules. Chemical
ecology has benefited from this conceptual framework, as demonstrated by the recent suggestion that pheromones mediate the interaction of a pheromone-binding protein with another protein target (50). We can expect additional examples.
SUMMARY
Several disciplines, including chemical ecology, seek to understand the molecular basis of information transfer in biological systems, and general molecular strategies are beginning to emerge. Often these strategies are discovered by a careful analysis of natural products and their biological effects. Cyclosporin A, FK506, and rapamycin are produced by soil microorganisms and are being used or considered as clinical immunosuppressive agents. They interrupt the cytoplasmic portion of T-cell signaling by forming a complex with a binding protein—FKBP12 in the case of FK506 and rapamycin and cyclophilin A (CyPA) in the case of cyclosporin A (CsA). This complex in turn inhibits a protein target, and the best understood target is calcineurin, which is inhibited by FK506-FKBP12 and CyPA-CsA. Mutational and structural studies help define how FK506-FKBP12 interacts with calcineurin, and the results of these studies are summarized. The existence of strong FK506-FKBP12 binding suggests that FK506 is mimicking some natural ligand for FKBP12. Synthetic and structural studies to probe this mimicry are also described.
I am grateful to Stuart L. Schreiber for generously sharing unpublished information from his laboratory. L. Wayne Schultz helped prepare the illustrations. I am grateful to the National Institutes of Health (Grant R01-CA59021) for financial support.
REFERENCES
1. Agosta, W.C. (1992) Chemical Communication: The Language of Pheromones (Freeman, New York), pp. 14-18, 29-31.
2. Rosen, M. K. & Schreiber, S. L. (1992) Angew. Chem. Int. Ed. Engl. 31, 384-400.
3. Sigal, N. H. & Dumont, F. J. (1992) Annu. Rev. Immunol. 10, 519-560.
4. Fliri, H., Bauman, G., Enz, A., Kallen, J., Luyten, M., Mikol, V., Movva, R., Quesniauz, V., Schreier, M., Walkinshaw, M., Wenger, R., Zenke, G. & Zurini, M. (1993) Ann. N.Y. Acad. Sci. 696, 39-47.
5. Williams, D. H., Stone, M. J., Hauck, P. R. & Rahman, S. K. (1989) J. Nat. Prod. 52, 1189-1208.
6. Schreiber, S. L. (1991) Science 251, 283-287.
7. Stern, L. J., Brown, J. H., Jardetzky, T. S., Gorga, J. C., Urban, R. G., Strominger, J. L. & Wiley, D. C. (1994) Nature (London) 368 , 215-221.
8. Chicz, R. M. & Urban, R. G. (1994) Immunol. Today 15, 155-159.
9. Emmel, E. A., Verweij, C. L., Durand, D. B., Higgins, K. M., Lacy, E. & Crabtree, G. R. (1989) Science 246, 1617-1620.
10. Flanagan, W.F., Corthesy, B., Bram, R.J. & Crabtree, G.R. (1991) Nature (London) 352, 803-807.
11. Schreiber, S. L., Albers, M. W. & Brown, E. J. (1993) Acc. Chem. Res. 26, 412-420.
12. Rosen, M. K. (1993) Ph.D. dissertation (Harvard Univ., Cambridge, MA).
13. Rüegger, A., Kuhn, M., Lichti, H., Loosli, H.-R., Huguenin, R., Quiquerez, C. & von Wartburg, A. (1976) Helv. Chim. Acta 59, 1075-1092.
14. Tanaka, H., Kuroda, A., Marusawa, H., Hatanaka, H., Kino, T., Hoto, T. & Hashimoto, M. (1987) J. Am. Chem. Soc. 109, 5031-5033.
15. Vezina, C., Kudelski, A. & Sehgal, S. N. (1975) J. Antibiot. 28, 721-726.
16. Sehgal, S. N., Baker, H. & Vezina, C. (1975) J. Antibiot. 28, 727-732.
17. Swindells, D.C.N., White, P.S. & Findlay, J.A. (1978) Can. J. Chem. 56, 2491-2492.
18. Handschumacher, R. E., Harding, M.W., Rice, J., Drugge, R.J. & Speicher, D. W. (1984) Science 226, 544-547.
19. Fischer, G., Wittman, L. B., Lang, K., Kiefhaber, T. & Schmid, F. X. (1989) Nature (London) 337, 476.
20. Takahashi, N. T., Hayano, T. & Suzuki, M. (1989) Nature (London) 337, 473-475.
21. Harding, M.W., Galat, A., Uehling, D.E. & Schreiber, S. L. (1989) Nature (London) 341, 758-760.
22. Siekierka, J. J., Hung, S. H. Y., Poe, M., Lin, C. S. & Sigal, N. H. (1989) Nature (London) 341, 755-757.
23. Fretz, H., Albers, M. W., Galat, A., Standaert, R. F. & Schreiber, S. L. (1991) J. Am. Chem. Soc. 113, 1409-1411.
24. Rahfeld, J.-U., Schierhorn, A., Mann, K. & Fischer, G. (1994) FEBS Lett. 343, 65-69.
25. Wiederrecht, G., Brizuela, L., Elliston, D., Sigal, N. & Siekierka, J. (1991) Proc. Natl. Acad. Sci. USA 88, 1029-1033.
26. Tropschug, M., Barthelmess, I. & Neupert, W. (1989) Nature (London) 342, 953-955.
27. Liu, J., Farmer, J. D., Lane, W. S., Friedman, J., Weissman, I. & Schreiber, S. L. (1991) Cell 66, 807-815.
28. Liu, J., Albers, M., Wandless, T. J., Luan, S., Alberg, D. A., Belshaw, P. J., Cohen, P., MacKintosh, C., Klee, C. B. & Schreiber, S. L. (1992) Biochemistry 31, 3896-3901.
29. Brown, E. J., Albers, M. W., Shin, T. B., Ichickawa, K., Keith, C. T., Lane, W. S. & Schreiber, S. L. (1994) Nature (London) 369, 756-758.
30. Clardy, J. (1994) Perspect. Drug Discovery Des. 2, in press.
31. Moore, J. A., Peattie, D. A., Fitzgibbon, M.J. & Thomson, J. A. (1991) Nature (London) 351, 248-250.
32. Michnick, S. W., Rosen, M. K., Wandless, T. J., Karplus, M. & Schreiber, S. L. (1991) Science 251, 836-839.
33. VanDuyne, G. D., Standaert, R. F., Karplus, P. A., Schreiber, S. L. & Clardy, J. (1991) Science 251, 839-842.
34. VanDuyne, G. D., Standaert, R. F., Schreiber, S. L. & Clardy, J. (1991) J. Am. Chem. Soc. 113, 7433-7434.
35. Weber, C., Wider, G., Freyberg, B. v., Traber, R., Braun, W., Widmer, H. & Wüthrich, K. (1991) Biochemistry 30, 6563-6574.
36. Clardy, J. (1993) Ann. N.Y. Acad. Sci. 685, 37-46.
37. Lepre, C. A., Thomson, J. A. & Moore, J. M. (1992) FEBS Lett. 302, 89-96.
38. Meadows, R. P., Nettesheim, D. G., Xu, R. T., Olejniczak, E. T., Petros, A. M.,
Holzman, T.F., Severin, J., Gubbins, E., Smith, H. & Fesik, S.W. (1993) Biochemistry 32, 754-765.
39. Petros, A. M., Gampe, R. T., Jr., Gemmecker, G., Neri, P., Holzman, T. F., Edalji, R., Hochlowski, J., Jackson, M., McAlpine, J., Luly, J. R., Pilot-Matiaas, T., Pratt, S. & Fesik, S. W. (1991) J. Med. Chem. 34, 2925-2928.
40. Petros, A. M., Luly, J. R., Liang, H. & Fesik, S. W. (1993) J. Am. Chem. Soc. 115, 9920-9924.
41. VanDuyne, G. D., Standaert, R. F., Karplus, P. A., Schreiber, S. L. & Clardy, J. (1993) J. Mol. Biol. 229, 105-124.
42. Becker, J. W., Rotonda, J., McKeever, B. M., Chan, H. K., Marcy, A. I., Wiederrecht, G., Hermes, J. D. & Springer, J. P. (1993) J. Biol. Chem. 268, 11335-11339.
43. Holt, D. A., Luengo, J. I., Yamashita, D. S., Oh, H.-J., Konialian, A. L. , Yen, H.-K., Rozamus, L. W., Brandt, M., Bossard, M. J., Levy, M. A., Eggleston, D. S., Liang, J., Schultz, L. W., Stout, T. J. & Clardy, J. (1993) J. Am. Chem. Soc. 115, 9925-9938.
44. Schultz, L. W., Martin, P. K., Liang, J., Schreiber, S. L. & Clardy, J. (1994) J. Am. Chem. Soc. 116, 3129-3130.
45. Jin, Y.-J., Albers, M. W., Lane, W. S., Bierer, B. E., Schreiber, S. L. & Burakoff, S. J. (1991) Proc. Natl. Acad. Sci. USA 88, 6677-6681.
46. Rosen, M. K., Yang, D., Martin, P. K. & Schreiber, S. L. (1993) J. Am. Chem. Soc. 115, 821-822.
47. Yang, D., Rosen, M. K. & Schreiber, S. L. (1993) J. Am. Chem. Soc. 115, 819-820.
48. Ikeda, Y., Schultz, L. W., Clardy, J. & Schreiber, S. L. (1994) J. Am. Chem. Soc. 116, 4143-4144.
49. Fischer, S., Michnick, S. W. & Karplus, M. (1993) Biochemistry 32, 13830-13837.
50. Du, G., Ng, C.-F. & Prestwich, G. D. (1994) Biochemistry 33, 4812-4819.














