
The Chemistry of Gamete Attraction: Chemical Structures, Biosynthesis, and (A)biotic Degradation of Algal Pheromones
WILHELM BOLAND
In 1971, Müller, Jaenicke, and colleagues (1) isolated the first pheromone of a marine brown alga. The compound was collected from laboratory cultures of fertile female gametophytes of the cosmopolitan brown alga Ectocarpus siliculosus. Soon after release, the originally motile female microgametes begin to settle on a surface and start to secrete a chemical signal. The biological function of this pheromone is the improvement of mating efficiency by attraction of the flagellated, motile males. The chemical structure of the signal compound was established as 6-(1Z)-(butenyl)cyclohepta-1,4-diene (ectocarpene; Figure 1 and Table 1). In the following years, this compound proved to be the progenitor of a whole series of C11 hydrocarbons, which are involved as signals in the sexual reproduction of brown algae. Up to now, 11 such pheromones (Table 1) but more than 50 stereoisomers of the parent compounds (cf. Figure 1) have been identified within the pheromone bouquets of >100 different species of brown algae (2-6). In the highly evolved orders Laminariales, Desmarestiales, and Sporochnales, the sexual pheromones first induce spermatozoid release from antheridia prior to attraction and fertilization (7, 8). Other species, in particular those from the genus Dictyopteris, produce large amounts of the same C11 hydrocarbons in their thalli. In the case of the Mediterranean phaeophyte Dictyopteris membranaceae (9), the compounds are released into the environment (10), and
Wilhelm Boland is chair of the Department of Bioorganic Chemistry at the Institut für Organische Chemie in Bonn, Germany.
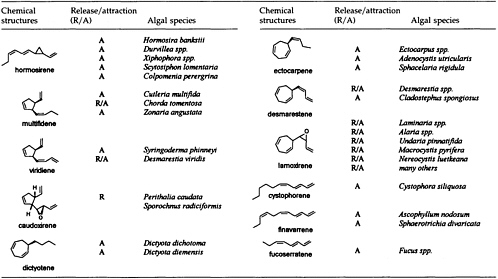
here they may interfere with the communication systems of other brown algae (11) or act as feeding deterrents against herbivores (12). Thus, a single C11 hydrocarbon (Table 1) may have at least three well-defined biological functions: (i) synchronization of the mating of male and female cells by the controlled release of male spermatozoids, (ii) enhancement of the mating efficiency by attraction, and (iii) chemical defense of the plant due to the presence of high amounts of pheromones within and release from the thalli into the environment.
Interestingly, the occurrence of the C11 hydrocarbons is not limited to the order of the marine brown algae. The same compounds have been found in cultures of diatoms (13) or among the volatiles released during blooms of microalgae in freshwater lakes (14). Furthermore, there is a steadily increasing number of reports on the occurrence of C11 hydrocarbons in roots, leaves, blossoms, or fruits of higher plants (15-17), although no attempts have been made to attribute a specific biological function to the C11 hydrocarbons in these higher plants.
This review tries to summarize our present knowledge of the chemistry of the pheromones of marine brown algae. The biochemical aspects of signal perception and transduction as well as the species-specific recognition of male and female gametes will be not discussed. The major topics will be the demonstration of the structural diversity in this group of C11 hydrocarbons, the different modes of their biosynthesis in lower and higher plants, and, finally, their (a)biotic degradation in an aqueous environment.
BROWN ALGAE AND THEIR PHEROMONES
Since the isolation and characterization of ectocarpene as the first volatile pheromone of a brown alga, 10 additional bioactive compounds have been isolated from female gametes or eggs of brown algae (Table 1). The isolation technique exploits the volatility of the compounds by collecting them in a closed system with a continuously circulating stream of air, which is passed over a very small carbon trap (1.5-5 mg) (18). Solvent desorbtion of the carbon with CH2Cl2 or CS2 (˜30 µl) provides solutions of the volatiles suitable for analysis by combined gas chromatography/mass spectrometry. A complex pattern of volatiles, obtained from fertile female gynogametes of the Mediterranean phaeophyte Cutleria multifida, is shown in Figure 1 (19). The identification of the individual compounds follows from mass spectrometry and from comparison with synthetic reference compounds (2). The biological activity of the identified compounds is assayed by exposing microdroplets of a water immiscible, high density solvent with known concentrations of the compounds to male gametes in sea water (20). After 4 min in the dark, the
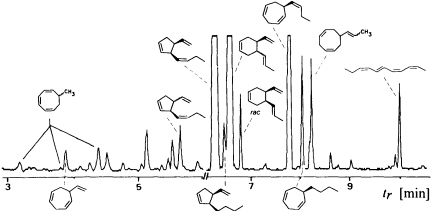
FIGURE 1 Gas chromatographic separation of the collected volatiles from fertile gynogametophytes of C. multifida. Conditions: fused silica column SE 30 (10 m x 0.32 mm); 40°C isotherm for 2 min, then at 10°C/min to 250°C; injection port: 250°C; detector: Finnigan ion trap, ITD 800; transfer line at 270°; electron impact (70 eV); scan range, 35-250 Da/sec. Due to the very low concentration of the low boiling point compounds, the total ion current in the range 3-6 min is enhanced by a factor of 6. Structures of the relevant hydrocarbons are given. Three configurational isomers appear for 7-methylcycloocta-1,3,5-triene owing to the high temperatures of the gas chromatographic separation.
population of the male gametes above the droplets of pure solvent and those containing the bioactive substances is documented by flash photography. For highly active pheromones, massive accumulation of males is observed in the range between 1 and 1000 pmol; the calculated values are valid for the saturated solvent/water interphase (21). Of the large number of the structurally very diverse compounds of Figure 1, the male gametes of C. multifida respond only to multifidene at a threshold level of 6.5 pmol and to ectocarpene at 900 pmol. The other compounds are virtually inactive. This also holds for the configurational isomers of multifidene, such as (E)-butenyl and 3,4-trans-disubstituted analogues, which on average exhibit only 1% the activity of the pheromone (21). Interestingly, even the activity difference between the natural (+)-(3S,4S)- and the synthetic (—)-(3R,4R)-multifidene is of the same order (22). Following this general sequence of isolation, characterization, synthesis, and bioassay, the pheromones of >100 species of brown algae have been determined (cf. Table 1) (3-6).
The ability of a compound to induce mass release of male gametes is approached by exposing mature male gametophytes of a species to particles of porous silica previously loaded with the test substances.
Quantitative data are available by placing droplets of an inert solvent with known concentrations of the test substance into close vicinity of the fertile gynogametophyte. For example, the mass release of male gametes of Laminaria digitata occurs within 8-12 sec at a threshold of ˜50 pmol of lamoxirene (8, 23, 24), and male gametes of Perithalia caudata are released within ˜10 sec, triggered by caudoxirene at concentrations down to 30 pmol (25).
Considering the large number of plant species and the limited number of only 11 different pheromones, it becomes obvious that these signals cannot be specific at the level of the species or even the genus. Moreover, ectocarpene, hormosirene, and dictyotene are typically present in most of the pheromone blends, and, hence, their presence may reflect nothing but a phylogenetic reminiscence of a biosynthetic pathway that converts a whole array of appropriate precursor molecules (see below) into olefinic hydrocarbons. The quantitative determination of the released volatiles from signaling females shows that females of E. siliculosus (26) release within 1 h ˜0.6 fmol of ectocarpene per individual; ˜75 fmol per h per egg of hormosirene was reported (27) as the initial rate of the secretory capacity of individual females of Hormosira banksii.
Much larger amounts of volatiles are present in thalli of several members of the genus Dictyopteris, but their production is obviously not linked to sexual events (ref. 28 and references cited therein). Interestingly, Dictyopteris divaricata from Japan (29) and Dictyopteris zonarioides from California (30, 31) produce sesquiterpenoids instead of C11 hydrocarbons. In the case of the Mediterranean D. membranaceae, the compounds are continuously released to the environment. The thalli of this alga (1 kg wet weight) release 10-50 mg of C11 hydrocarbons into seawater within 24 h (10). This exceeds by far the required threshold concentrations for chemotaxis, and one may suspect that the compounds can act as ''mating disruptants" in the control of the habitat. This idea, however, awaits experimental confirmation. According to recent analyses, the occurrence of large amounts of C11 hydrocarbons in thalli of brown algae is not limited to the genus Dictyopteris. Two Fucales from the Red Sea—namely, Sargassum asporofolium and Sargassum latifolium (S. Fatallah and W.B., unpublished data)—contain, besides sesquiterpenes, ectocarpene and dictyotene as the major volatiles.
Recent advances in gas chromatographic separations of enantiomers allow precise determination of the enantiomeric purity of the algal pheromones. The cis-disubstituted cyclopentenes, such as multifidene, viridiene, and caudoxirene, are of high optical purity [=95% enantiomeric excess (e.e.)] whenever they have been found (32, 33). The situation is different with the cyclopropanes and the cycloheptadienes, as shown in Table 2 and Figure 1. Hormosirene from female gametes or thalli of
TABLE 2 Enantiomer composition of hormosirene from secretions of female gametes or thalli of brown algae
|
Genus and species |
Origin |
Major enantiomer |
ee % |
|
Dictyopteris acrostichoides |
Sorrento, Australia |
(-)-(1R,2R) |
74.2 |
|
D. membranaceae |
Villefranche, French Mediterranean |
(-)-(1R,2R) |
71.2 |
|
D. prolifera* |
Hikoshima, Japan |
(-)-(1R,2R) |
90.0 |
|
D. undulata* |
Hikoshima, Japan |
(-)-(1R,2R) |
92.0 |
|
Analipus japonicus* |
Muroran, Japan |
(+)-(1S,2S) |
66.0 |
|
A. japonicus* |
Akkeshi, Japan |
(+)-(1S,2S) |
90.0 |
|
Durvillaea potatorum |
Sorrento, Australia |
(-)-(1R,2R) |
51.7 |
|
Haplospora globosa |
Halifax, Nova Scotia |
(+)-(1S,2S) |
83.3 |
|
Hormosira banksii |
Flinders, Australia |
(-)-(1R,2R) |
82.8 |
|
Xiphophora gladiata |
Hobart, Tasmania |
(-)-(1R,2R) |
72.3 |
|
X. chondrophylla |
Flinders, Australia |
(-)-(1R,2R) |
82.0 |
|
References are given in the text. Examples marked with an asterisk are taken from ref. 31. |
|||
brown algae of different geographic origin proved to be secreted always as a well-defined mixture of enantiomers (34). The enantiomeric composition of hormosirene from gametes of Analipus japonicus varies depending even on the locality within Japan (31). In contrast, the e.e. of hormosirene, present as a by-product (4-6%) in the pheromone bouquet of E. siliculosus, collected at various localities from all over the world, seems to remain constant (90% ± 5% e.e.; W.B. and D. G. Muller, unpublished data). If less common configurations of the pheromones are concerned, as, for example, in C11 hydrocarbons with (E)-butenyl or (E,E)-hexadienyl substituents, the optical purity is often very low. 6-(1E)-(Butenyl)cyclohepta-1,4-diene from the Australian Dictyopteris acrostichoides exhibits an e.e. of only 26% (10); the cis-disubstituted cyclohexene from the blend of C. multifida (Figure 1) is virtually racemic (19). It is tempting to assume that for marine brown algae the production of characteristic enantiomeric mixtures represents a simple means for individualization of the signal blends, although up to now there is no experimental confirmation for this hypothesis.
BIOSYNTHESIS OF C11HYDROCARBONS IN HIGHER AND LOWER PLANTS (PHAEOPHYCEAE)
Given the absence of methyl branches and according to the suggestive positions of the double bonds within the two acyclic C11 hydrocarbons undeca-(1,3E,5Z)-triene and undeca-(1,3E,5Z,8Z)-tetraene, their origin from fatty acids is highly probable. In the case of higher plants, the
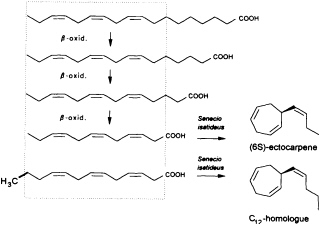
FIGURE 2 Biosynthesis of ectocarpene in the higher plant S. isatideus (Asteraceae). ω3 fatty acids and their degradation products (β-oxidation) comprise the structural elements for the biosynthesis of ectocarpene in the higher plant S. isatideus. The structurally related trideca-3,6,9-trienoic acid is metabolized by analogy into the C12 homoectocarpene.
precursors might be expected to come from the pool of the ω3 and ω6 fatty acids with a total of 18-12 carbon atoms (Figure 2). In marine brown algae the family of unsaturated C20 fatty acids provides another, potentially abundant source of suitable precursors (35).
Interestingly, the first biosynthetic experiments with [3H]linolenic acid and the terrestrial plant Senecio isatideus (Asteraceae) as a model system for the biosynthesis of algal pheromones were unsuccessful. If, however, labeled dodeca-3,6,9-trienoic acid is administered, a rapid transformation into ectocarpene takes place (36). Nevertheless, the C12 acid ultimately derives from linolenic acid via three β-oxidations, since labeled tetradeca-5,8,11-trienoic acid, which requires only one β-oxidation, is converted into labeled ectocarpene albeit with very low efficiency (37).
Mechanistic insight into this process was obtained by administration of labeled trideca- or undeca-3,6,9-trienoic acid instead of the natural C12 precursor (Figure 2). In this case, the artificial 2Hn, metabolites can be analyzed by mass spectrometry without interference from the plants' own 1H metabolites, since a homo- or norectocarpene is formed. The sequence of the oxidative decarboxylation/cyclization reaction proceeds without loss of 2H atoms from the double bonds but with loss of a single 2H atom from certain methylene groups of the precursor acids (Figure 3). If C(1) and a 2H atom from C(5) of the labeled precursor is lost, finavarrene is the product of the reaction channel. If the methylene group
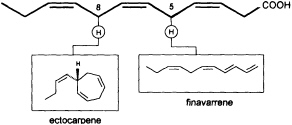
FIGURE 3 C11 hydrocarbons from dodeca-3,6,9-trienoic acid. Loss of C(1) and a single hydrogen from C(5) yield the acyclic hydrocarbon finavarrene. Decarboxylation and loss of a single hydrogen from C(8) results in (6S)-ectocarpene. No other hydrogen atoms are lost during the biosynthetic sequence.
at C(8) is involved, ectocarpene is formed with simultaneous loss of C(1), the latter probably as CO2 (36). Administration of (8R)-or (8S)-[2H]trideca-3,6,9-trienoic acids (38) yields (6S)-homoectocarpene with exclusive loss of the C(8) HR. No intramolecular isotope effect is observed. The removal of the hydrogen atom from C(5) en route to finavarrene proceeds with the same side specificity. Considering the absolute configuration of ectocarpene as (6S) and taking into account the established preference of the enzyme(s) for the removal of the C(8) HR, the course of the reaction from dodecatrienoic acid to the C11 hydrocarbon ectocarpene can be rationalized as depicted in Figure 4. The acid is assumed to fit into the active site of the enzyme in a U-shaped manner, exposing the C(8) HR to a reactive functional group X. The sequence could be initiated by removal of a single electron from the carboxyl group. Subsequent decarboxylation would generate an allyl radical, which can interact with the C(6)=C(7) double bond generating a cyclopropyl intermediate. Final
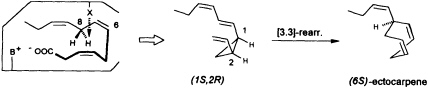
FIGURE 4 Ectocarpene as the product of a [3.3]-sigmatropic rearrangement. The fatty acid accommodates to the active center of the enzyme in a U-shaped fashion. Decarboxylation in conjunction with loss of the C(8) HR hydrogen atom yields, after cyclization between C(4) and C(6) of the precursor, the thermolabile (1S,2R)-cyclopropane. A subsequent spontaneous [3.3]-sigmatropic rearrangement (Cope rearrangement) proceeds via the cis-endo transition state and yields (6S)-ectocarpene.
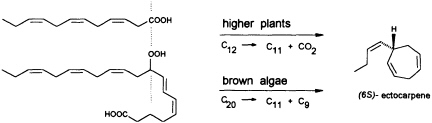
FIGURE 5 Biosynthesis of C11 hydrocarbons in higher and lower plants. The similar pattern of functionalization of dodeca-3,6,9-trienoic acid and 9-HPEPE is shown.
removal of the C(8) HR as a radical by the active center of the enzyme terminates the sequence, and the disubstituted cyclopropane (1S,2R) is released as the first product. The compound is thermolabile and rearranges via a cis-endo transition state to (6S)-ectocarpene.
In contrast to the terrestrial plant S. isatideus, female gametes of the marine brown algae (model system: female gametes of E. siliculosus) do not utilize dodeca-3,6,9-trienoic acid for production of the C11 hydrocarbons. Instead, the marine plants exploit the pool of unsaturated C20 acids (20:4 → 20:6) for the production of their pheromones. [2H8]Arachidonic acid is very effectively transformed into 6-[2H4]butylcyclohepta-1,4-diene (dictyotene) by a suspension of female gametes of E. siliculosus. A synthetic sample of [2H6]nonadeca-8-11,14,17-tetraenoic acid, which can be thought of as a 20-noranalogue of icosa-5,8,11,14,17-pentaenoic acid, gives the corresponding norectocarpene, together with a labeled norfinavarrene, in high yield (39, 40). Since the icosanoids are not cleaved to unsaturated C12 precursors, their primary functionalization is assumed to be achieved by a 9-lipoxygenase yielding 9-hydroperoxyicosa(5Z,7E,11Z,14Z,17Z)-pentaenoic acid (9-HPEPE), which mimics the functionalization pattern of dodeca-3,6,9-trienoic acid (Figure 5). Assuming a homolytic cleavage of the hydroperoxide (41) as shown in Figure 6, once again, the disubstituted cyclopropane will be released from the active center as a thermolabile intermediate. Although this mechanistic hypothesis has not yet been experimentally confirmed, the concept nevertheless provides a valuable platform for the systematic derivation of all the known C11 hydrocarbons from the fatty acid precursors. It is also conceivable that the oxidative decarboxylation/cyclization of dodeca-3,6,9-trienoic acid in higher plants could proceed via a peroxy acid.
The unusual stereochemistry of the acyclic undeca-(2Z,4Z,6E,8Z)tetraene (giffordene; cf. Figure 1), the major volatile from the brown alga Giffordia mitchellae, follows from the same concept (40, 42). If the 9-HPEPE
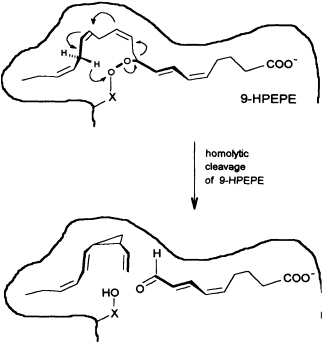
FIGURE 6 Speculative mechanism of C11 hydrocarbon biosynthesis from fatty acid hydroperoxides in algae. Homolytic cleavage of the hydroperoxide is assumed to give an allyl radical, which cyclizes to the thermolabile (1S,2R)-cyclopropane. The sequence is terminated by transfer of a hydrogen radical from C(16) to the -X-O function. The cyclopropane rearranges to (6S)-ectocarpene as shown in Figure 4.
precursor is forced by the enzyme into an appropriate cisoid conformation, homolytic cleavage of the peroxide generates the thermolabile undeca-(1,3Z,5Z,8Z)-tetraene. The latter suffers a spontaneous [1.7]-hydrogen shift, and it is the helical transition state structure of this antarafacial hydrogen shift that accounts for the observed stereochemistry of the product (Figure 7a). The concept is strongly supported by the simultaneous occurrence of small traces of the more stable (1,3Z,5Z)-undecatriene in the same bouquet. A low temperature synthesis (−30°C) of the postulated undeca-(1,3Z,5Z,8Z)-tetraene has been developed, and from the kinetic data a half-life of ≈2.5 h is calculated for this compound under the conditions of the natural environment (18°C) (G. Pohnert and W.B., unpublished data). The activation energy of this [1.7]-hydrogen shift (Ea = 67.4 kJ·mol−1; ∆S298 = −91.9 J·mol−1·K−1) is considerably
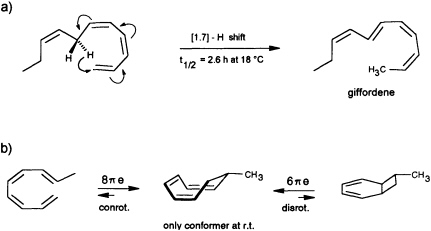
FIGURE 7 Pericyclic reactions in the biosynthesis of giffordene and 7-methylcyclooctatriene. (a) The [1.7]-hydrogen shift of the thermolabile undeca-(1,3Z,5Z,8Z)-tetraene generates undeca-(2Z,4Z,6E,8Z)-tetraene (giffordene), the major product of the brown alga G. mitchellae. (b) The thermolabile nona-(1,3Z,5Z,8E)-tetraene cyclizes at ambient temperature rapidly to 7-methylcycloocta-1,3,5-triene. At ambient temperature, the bicyclic isomer does not contribute to the equilibrium (requires =80°C).
lower than that of the well studied [1.7]-hydrogen shift from previtamin D3 to vitamin D3 (43). Both of these pericyclic reactions do not appear to be catalyzed by enzymes.
Yet another pericyclic reaction may account for the biosynthesis of 7-methylcycloocta-1,3,5-triene, present at a trace level in the hydrocarbon blend from C. multifida (Figure 1). If a nona-(1,3Z,5Z,7 E)-tetraene were produced by homolytic cleavage of a suitable fatty acid hydroperoxide, this acyclic olefin should readily undergo an 8πe electrocyclic ring closure (Figure 7b). The same reaction has been postulated previously as the key step within the biosynthetic sequence en route to the endriandric acids from the Australian tree Endriandra introrsa (45). Again, a low temperature synthesis (−30°C) of the nona-(1,3Z,5Z,7E)-tetraene allowed the determination of the kinetic data. The half-life of the acyclic precursor is limited to a few minutes at ambient temperature, and the activation energy is significantly lower (Ea = 59.4 kJ·mol−1; ∆S273 = −89.7 J·mol−1·K−1 ) than those reported for the electrocyclization of a series of isomeric decatetraenes (44). The data follow a predicted trend based on theoretical calculations (46). In contrast to the three signals that were obtained by gas chromatographic analysis at higher temperature
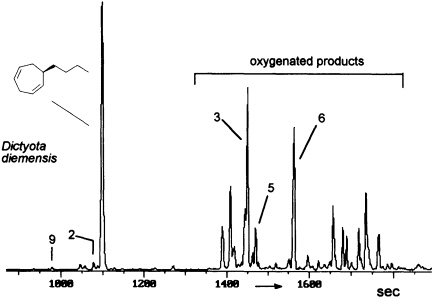
FIGURE 8 Gas chromatographic separation of the volatiles of D. diemensis egg extracts (47). Conditions: fused silica column OV 1 (10 m x 0.32 mm); 50°C isotherm for 2 min, then at 10°C/min to 250°C; injection port: 250°C; detector: Finnigan ion trap, ITD 800; transfer line at 270°C; electron impact (70 eV); scan range, 35-250 Da/sec. For identity of numbered compounds refer to Figure 9.
(Figure 1), 1H NMR studies show that at ambient temperature, the monocyclic 7-methylcyclooctatriene is present as the only isomer (no evidence for an equilibrium with a bicyclic cyclohexadiene).
(A)BIOTIC DEGRADATION OF ALGAL PHEROMONES
The degree and extent of the abiotic degradation of the pheromones becomes immediately obvious when samples from algae releasing cyclohepta-1,4-dienes (such as dictyotene or ectocarpene) are collected. For example, volatiles collected from cultures of fertile gynogametophytes of Dictyota diemensis exhibit, besides dictyotene, a complex pattern of oxygenated compounds, as shown in Figure 8 (47). Since the concentrations of the oxygenated products are even lower than that of the genuine pheromone, the enrichments from natural sources can not be used for an exhaustive structure elucidation.
However, the pattern of Figure 8 can be perfectly mimicked by a chemical model system with dictyotene as the substrate and iodosylben-
zene (Ph-I==O)/manganese tetraphenylporphyrin (TPPMn) as the oxidant (48). An aqueous system, containing huminic acids and traces of Cu(I) (49), gives similar results (50). Since these abiotic degradations can be carried out on a gram scale, the by-products shown in Figure 8 become available as pure compounds after extensive chromatographic separations. Up to now, more than 20 individual compounds have been characterized (50). Their structures fit into a general scheme of an oxidative sequence starting with a pentadienyl radical as depicted in Figure 9. Once generated, the radical reacts at all possible positions with oxygen and yields the three isomeric alcohols as the primary products. Further oxidation of the alcohols provides the dihydrotropones, which were first isolated from the two brown algae Dictyopteris australis and Dictyopteris plagiogramma (28, 51). Elimination of water generates the alkyltropilidenes. If singlet oxygen is involved, the corresponding hydroperoxides will be formed. Their subsequent decomposition may be responsible for the formation of the ketoepoxide and for the fragmentation of the carbon framework. The isomeric butylbenzaldehydes and the substituted furane fit into the same reaction channel. The butylbenzenes result from decarbonylation of the alkylbenzaldehydes. Butylbenzene and the alkyltropilidenes are remarkably attractive for male gametes of E. siliculosus; the alcohols and ketones are not. Owing to their ability to act as Michael acceptors, the dihydrotropones and tropones, which represent the major degradation products of the cycloheptadienes, may be important as chemical defenses (e.g., feeding deterrents) of those brown algae that are capable of synthesizing them. Similar degradative routes can be expected for all the other C11 hydrocarbons, and it seems likely that chemical model systems like Ph-I==O/TPPMn can provide all the required reference substances.
Exploratory experiments with dictyotene and suspensions of male gametes of E. siliculosus showed a significantly enhanced production of the 6-butylcyclohepta-2,4-dienol and its isomers. This indicates that a biological degradative pathway does exist and that this pathway follows the same oxidative sequence as the abiotic route. However, final conclusions about the biotic contribution to the pheromone transformation cannot be drawn before careful analysis of the degree of enantioselectivity of the biotic reaction.
SUMMARY
Female gametes of marine brown algae release and/or attract their conspecific males by chemical signals. The majority of these compounds are unsaturated, nonfunctionalized acyclic, and/or alicyclic C11 hydrocarbons. Threshold concentrations for release and attraction are generally
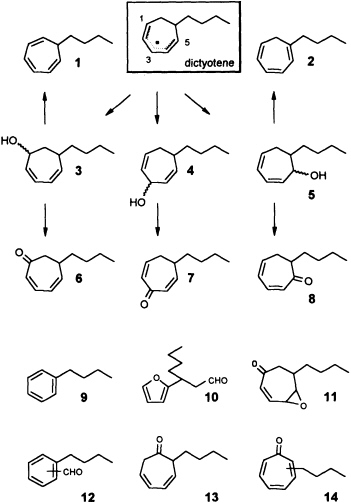
FIGURE 9 Oxidative degradation of dictyotene by TPPMn/Ph-I-O. Compounds 1-14 are isolated products of an oxidative degradation of dictyotene by a radical pathway induced by the system TPPMn/Ph-I-O. Most of these compounds are present among the oxygenated derivatives of dictyotene from natural sources (cf. Figure 8).
observed in the range of 1-1000 pmol. The blends may contain various configurational isomers of the genuine pheromones as well as mixtures of enantiomers. Higher plants produce the C11 hydrocarbons from dodeca3,6,9-trienoic acid; brown algae exploit the family of icosanoids for biosynthesis of the same compounds. The biosynthetic routes comprise several spontaneously occurring pericyclic reactions such as [3.3]-sigma-tropic rearrangements, [1.7]-hydrogen shifts, and electrocyclic ring closures. All pheromones are (a)biotically degraded by ubiquitous oxidative pathways involving singlet oxygen or hydroxyl radicals, which may be produced through the agency of heavy metals, huminic acids, or light.
REFERENCES
1. Müller, D.G., Jaenicke, L., Donike, M. & Akintobi, T. (1971) Science 171, 815-817.
2. Jaenicke, L. & Boland, W. (1982) Angew. Chem. Int. Ed. Engl. 94 , 643-653.
3. Maier, I. & Müller, D. G. (1986) Biol. Bull. 170, 145-175.
4. Boland, W. (1987) Biol. Unserer Zeit 17, 176-185.
5. Jaenicke, L. (1988) Bot. Acta 101, 149-159.
6. Maier, I. (1993) Plant Cell Environ. 16, 891-907.
7. Maier, I. & Müller, D. G. (1982) Protoplasma 113, 137-143.
8. Maier, I. (1987) in Algal Development (Molecular and Cellular Aspects), eds. Wiessner, W., Robinson, D. G. & Starr, R. C. (Springer, Berlin), pp. 66-74.
9. Boland, W. & Müller, D. G. (1987) Tetrahedron Lett. 28, 307-310.
10. Wirth, D., Fischer-Lui, I., Boland, W., Icheln, D., Runge, T., König, W. A., Phillips, J. & Clayton, M. (1992) Helv. Chim. Acta 75, 734-744.
11. Boland, W., Marner, F.-J., Jaenicke, L., Müller, D. G. & Fölster, E. (1983) Eur. J. Biochem. 134, 97-103.
12. Hay, M. E., Duffy, J. E., Fenical, W. & Gustafson, K. (1988) Mar. Ecol. Prog. Ser. 48, 185-192.
13. Derenbach, J. B. & Pesandeo, D. (1986) Mar. Chem. 19, 337-432.
14. Jüttner, F. & Wurster, K. (1984) Limnol. Oceanogr. 29, 1322-1324.
15. Bohlmann, F., Zdero, C., Berger, A., Suwita, A., Mahanta, P. & Jeffrey, C. (1979) Phytochemistry 18, 79-93.
16. Boland, W., Jaenicke, L. & Brauner, A. (1982) Z. Naturforsch. 37C, 5-9.
17. Kollmannsberger, H. & Berger, R. G. (1992) Chem. Mikrobiol. Technol. Lebensm. 14, 81-86.
18. Grob, K. & Zürcher, F. (1976) J. Chromatogr. 117, 285-294.
19. Keitel, J., Fischer-Lui, I., Boland, W. & Müller, D. G. (1990) Helv. Chim. Acta 73, 2101-2112.
20. Müller, D. G. (1976) Z. Pflanzenphysiol. 80, 120-130.
21. Boland, W., Jakoby, K., Jaenicke, L. & Müller, D. G. (1980) Z. Naturforsch. 36C, 262-271.
22. Boland, W., Jaenicke, L. & Müller, D.G. (1981) Justus Liebigs Ann. Chem. 2266-2271.
23. Müller, D. G., Gassmann, G. & Lüning, K. (1979) Nature (London) 279, 430-431.
24. Marner, F.-J., Müller, B. & Jaenicke, L. (1984) Z. Naturforsch. 39C, 689-691.
25. Müller, D. G., Boland, W., Becker, U. & Wahl, T. (1988) Biol. Chem. Hoppe-Seyler 369, 655-659.
26. Müller, D. G. & Schmid, C. E. (1988) Biol. Chem. Hoppe-Seyler 369, 647-653.
27. Maier, I. & Clayton, M. N. (1993) Bot. Acta 106, 344-349.
28. Moore, R. E. (1977) Acc. Chem. Res. 10, 40-47.
29. Kajiwara, T., Hatanaka, A., Kodama, K., Ochi, S. & Fujimura, T. (1991) Phytochemistry 30, 1805-1807.
30. Fenical, W. & Sims, J. J. (1972) Phytochemistry 11, 1161-1163.
31. Kajiwara, T., Kodama, K. & Hatanaka, A. (1993) in Volatile Attractants from Plants, ACS Symposium Series 525 (Am. Chem. Soc., Washington, DC), pp. 103-120.
32. Boland, W., König, W. A. & Müller, D. G. (1989) Helv. Chim. Acta 72, 1288-1292.
33. Wirth, D., Boland, W. & Müller, D. G. (1992) Helv. Chim. Acta 75, 751-758.
34. Boland, W., Flegel, U., Jordt, G. & Müller, D. G. (1987) Naturwissenschaften 74, 448-449.
35. Schmid, C. E., Eichenberger, W. & Müller, D. G. (1991) Biol. Chem. Hoppe-Seyler 372, 540-544.
36. Boland, W. & Mertes, K. (1985) Eur. J. Biochem. 147, 83-91.
37. Stratmann, K. (1992) Ph.D thesis (University of Karlsruhe, Karlsruhe, F.R.G.).
38. Neuman, C. & Boland, W. (1990) Eur. J. Biochem. 191, 453-459.
39. Stratmann, K., Boland, W. & Müller, D. G. (1992) Angew. Chem. Int. Ed. Engl. 31, 1246-1248.
40. Stratmann, K., Boland, W. & Müller, D. G. (1993) Tetrahedron 49, 3755-3766.
41. Gardner, H. W. (1991) Biochem. Biophys. Acta 1084, 221-239.
42. Boland, W., Jaenicke, L. & Müller, D. G. (1987) Experientia 43 , 466-467.
43. Hanewald, K. H., Rappoldt, M. P. & Roborgh, J. R. (1961) Rec. Trav. Chim. PaysBas 80, 1003-1014.
44. Huisgen, R., Dahmen, A. & Huber, H. (1967) J. Am. Chem. Soc. 89, 7130-7131.
45. Bandaranayake, W. M., Banfield, J. E. & Black, D. S. C. (1980) J. Chem. Soc. Chem. Commun. 902-903.
46. Thomas, B. E., Evanseck, J. D. & Houk, K. N. (1993) J. Am. Chem. Soc. 115, 4165-4169.
47. Phillips, J. A., Clayton, M. N., Maier, I., Boland, W. & Müller, D. G. (1990) Phycologia 29, 367-379.
48. McMurry, T. J. & Groves, J. T. (1986) in Cytochrome P-450, Structure, Mechanism, and Biochemistry, ed. Ortiz de Montellano, P.R. (Plenum, New York), pp. 1-28.
49. Mopper, K. & Zhou, X. (1990) Science 250, 661-662.
50. Erbes, P. (1992) Ph.D. thesis (University of Karlsruhe, Karlsruhe, F.R.G.).
51. Moore, R. E. & Yost, G. (1973) J. Chem. Soc. Chem. Commun. 937-938.
















