
22
New Technologies for Producing Systemic and Muscosal Immunity by Oral Immunization: Immunoprophylaxis in Meals, Ready-to-Eat
Arthur O. Anderson1
INTRODUCTION
Immunity, inflammation, and nutrition form an interactive system that impacts on the performance of soldiers in stressful or hazardous environments. A deficiency in any component of this system diminishes the effectiveness of the others (Beisel, 1994). Decrements in performance during the first few weeks of deployment frequently are associated with illness caused by endemic infectious disease agents. This risk is enhanced by the threat of possible biological weapon attacks (Mobley, 1995). To minimize infection-induced performance decrements, troops should be immunized against recognized endemic disease and biological warfare threat agents months prior to deployment, preferably during basic training.
Promising new technologies from recombinant DNA and biodegradable polymer research will change the way vaccines are produced and will simplify how immunity is induced and maintained (Michalek et al., 1995; Walker, 1994). Two recent papers demonstrate the feasibility of large-scale agricultural production of recombinant vaccine antigens (Ma et al., 1995) and functional recombinant human antibodies (Haq et al., 1995) in quantities that would satisfy virtually any contingency requirement. Engineered human secretory antibodies could be enterically coated and incorporated into foods for passive protection of soldiers from common diarrheal diseases and enterotoxin reactions that affect nutrition and performance (Hyams et al., 1991, 1993; Sarraf et al., 1997).
The advantages of passive and active immunization2 via the oral route are multiple. Recombinant vaccines may be encapsulated in biodegradable polymers to prolong shelf life and provide controlled-release, targeted delivery or protection from denaturation by stomach acid and intestinal enzymes until the product is absorbed in the gastrointestinal tract (Michalek et al., 1994; Morris et al., 1994). Combining encapsulated vaccines or antibodies with nutritious foods makes them more convenient and acceptable to use and removes the logistical and anxiety factors associated with the need for periodic inoculations. In addition to enhancing soldier performance and autonomy, systems for oral immunization will save time and money. It will no longer be necessary for soldiers to delay deployment so that they may assemble for vaccination. Because the new vaccines can be self-administered, they can be taken without need of medically trained personnel. Oral vaccination also eliminates dangerous medical waste and the risk of contamination, which are concerns when needles are used. Taken together, it is now feasible to provide complete passive and active protective immunity along with good nutrition in Meals, Ready-to-Eat (MREs). These new technologies should become an exciting and active area of applied research with
numerous opportunities for interdisciplinary collaboration and leveraging of the tasks.
CURRENT CONCEPTS AND ISSUES IN IMMUNITY
It may be useful to digress from the primary objective (i.e., that of describing the exciting new technologies) in order to explain relevant concepts and critical issues in immunology. This explanation will help establish the importance of certain new approaches to immunization and reveal how these approaches could benefit soldiers of the twenty-first century.
Concept of Compartmentalization of Immune Responses
Immune responses to vaccines are influenced by the route of immunization (injection or oral), the form of the antigen (live, killed, soluble, peptide subunit, or particulate), and the presence in the vaccine of biologically active elements, such as proteins that mediate specific tissue tropisms (components of the pathogen that enable it to attach to and replicate in specific host cells) or materials included as adjuvants (substances added to enhance antigenicity), vectors, or vehicles (Walker, 1994). For example, differences in immune responses to live versus inactivated viral vaccines may be a function of compartmentalization of immune responses, activities requiring vaccine viability like tropism, or molecular strategies for cell entry and replication (Rubin et al., 1986; Spriggs, 1996). In this review to avoid confusion, general comments about effects of route of administration or vehicles on immune responses will be restricted to responses initiated by simple, unmodified, nonreplicating protein antigens.
There is now substantial evidence supporting the existence of at least two immune systems, a "peripheral" immune system (involving the spleen, lymph nodes, and other nonepithelial tissues) and a "mucosal" immune system (involving the epithelial tissue lining the respiratory and gastrointestinal tracts) (Ogra et al., 1994). These systems operate separately and simultaneously in most species, including humans.
Protective immunity acquired during convalescence usually is referred to as "systemic immunity," but this term is imprecise. Systemic immunity might be a combination of mucosal and peripheral immunity, or it might be dominated by an incomplete form of immunity dictated by a specific pathogen (Mosmann and Sad, 1996; Salgame et al., 1991). For example, if a pathogen stimulates a cytokine cascade that favors antibody production (at the expense of endocytosis and intralysosomal killing), it would continue to prosper within a compartment in which antibodies are ineffective (Finkelman, 1995; Yamamura et al., 1991). Unless a vaccine stimulates the appropriate system or a combination of systems, protective immunity might not be complete.
The concept of anatomic compartmentalization of immunity is supported by observations from several disciplines (Kroemer et al., 1993). The anatomy of antigen uptake and the physiology and biochemistry of lymphocyte recirculation within unique tissue microenvironments may influence significantly the quality of humoral and cellular immune responses.
The way antigens are acquired by individual lymphatic tissues affects the outcome of an immune response. For example, the same antigen may produce qualitatively different immune responses in lymph nodes, spleen, or Peyer's patches (lymphoid tissue aggregates under intestinal mucosa) (Anderson, 1990). Antigens in lymph are filtered, trapped, processed, and presented at the site where lymph passes over fixed antigen-presenting cells in lymph nodes. Such antigen handling by lymph nodes most often results in peripheral immunity, characterized by the appearance of specific immunoglobulin (Ig)G in the blood. Antigens in blood are filtered, trapped, processed, and presented in strategic blood-tissue interfaces in the spleen, which also result in peripheral immunity. However, the spleen microenvironment is somewhat more complicated because it also accommodates circulating antigen-presenting cells and immunoreactive T-and B-cells from other tissues committed to either peripheral or mucosal immunity. In contrast, antigens in the lumens of enteric organs (i.e., the respiratory and gastrointestinal tracts) are nondestructively endocytosed by specialized epithelial cells called M (membraneous)-cells and transported across the cytoplasm in vacuoles onto lymphoid cells in Peyer's patches, where response to antigen presentation triggers commitment to "mucosal immunity," characterized by release of specific IgA into the secretions3 (McGhee et al., 1992).
Lymphocyte traffic patterns, which are regulated by selective expression of adhesion proteins in peripheral or mucosal lymphatic tissues, maintain anatomic segregation of immunologic memory (that enables the immune system to mount a more vigorous and effective response whenever it is restimulated by a specific foreign antigen) by causing antigen-primed cells to return to specific anatomic destinations, where they will encounter conditions that further facilitate expression of peripheral or mucosal immunity (Butcher and Picker, 1996; Ebnet et al., 1996) (Figure 22-1). The multitude of potential conditions includes the prevalence of specific cytokines, adhesion to and costimulation by specific stromal cells, and still, unknown microenvironmental factors intrinsic to those lymphoid compartments that favor commitment of B-cells to specific immunoglobulin isotypes or T-cells to peripheral or mucosal immunity (Anderson, 1990; Rott et al., 1996).
Humoral immunity is mediated by euglobulins called antibodies that are produced locally but act at great distances from where they are made and secreted. Cellular immunity is like hand-to-hand combat. T-cells, natural killer (NK) cells, and "armed" macrophages enter into physical combat with the
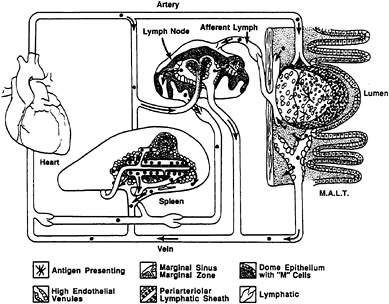
FIGURE 22-1 A simplified drawing of the structures and connections of secondary lymphatic tissues where antigen may most efficiently direct immune responses. Multiple known and unknown factors intrinsic to the microenvironments of lymph node, spleen, and mucosa-associated lymphatic tissue (here, M.A.L.T is represented by Peyer's patch) influence whether a peripheral or mucosal type of response occurs. The drawing also indicates that these tissues are integrated with each other and the rest of the individual through vascular and lymphatic connections and a system of lymphocyte recirculation. Microenvironmental structures in the drawing are identified by the symbols below the drawing.
pathogen and kill it by punching holes into its membranes or exposing it to enzymes. Cellular immunity also is associated with secretion of hormone-like molecules called cytokines and chemokines. These enable the effector cells to perform their duties actively. Many of the same cytokines also have effects on the humoral immune response by affecting B-cell division, differentiation, and maturation. The distinction between systems that regulate humoral immunity and those that regulate cellular immunity should not be confused with anatomic compartmentalization or with commitment to peripheral or mucosal immunity. There is a division of labor between cellular and humoral immunity that is parallel in both the peripheral and mucosal systems. Some of the same cell interactions and cytokines involved in cellular or humoral immunity are also involved in favoring peripheral over mucosal immunity, and vice versa (Abbas et al., 1996; Mosmann and Sad, 1996; Rocken and Shevach, 1996), as will be discussed later.
Compartmentalized Humoral Immunity
All immunoglobulins, whether peripheral or mucosal, function by binding antigen in a pocket formed by the complementarity-determining regions (CDRs) encoded by the immunoglobulin heavy (VH)-and light (VL)-chain variable region genes (Carayannopoulos and Capra, 1993). The pocket formed by the VH-VL CDRs spatially conforms to the surface shape of the antigen that binds to the antibody (Figure 22-2). The method by which the genes for antigen-specific CDRs may be obtained quickly so that recombinant protective antibodies may be produced will be discussed later in this review.
The antibody class conferred by the heavy-chain constant (C) region genes determines how the antibody will function and where it will act. Antibody C-region genes are expressed after rearrangement of the selected heavy-chain gene and attachment to the already rearranged variable, diversity, and joining region genes.4 Thus IgM, IgD, IgG3, IgG1, IgG2a, IgG2b, IgE, and IgA each have heavy chains that control how they participate in immunity, especially with regard to third-party molecular interactions such as Fc receptor binding,5 activation of the complement system, and endosomal transport across mucosal epithelial cells.
Peripheral Immunity and IgG
Antibodies of peripheral immunity protect the parenchymal organs and peripheral anatomic sites that are bathed in tissue fluid and supplied by the
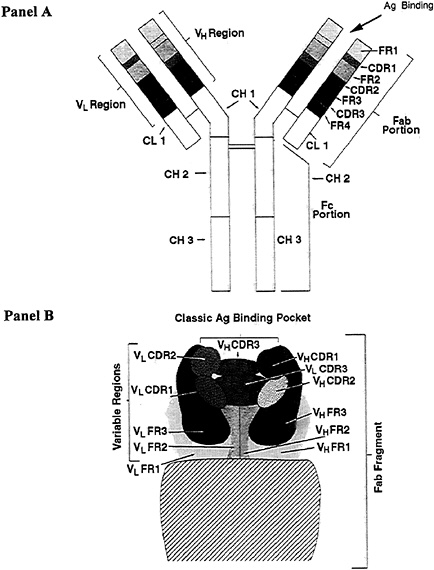
FIGURE 22-2 Diagram of an immunoglobulin molecule. The heavy and light chains of an immunoglobulin molecule are displayed linearly in Panel A, and the antigen-combining site is displayed in native configuration in Panel B. In Panel A, disulfide bonds link the light to the heavy chains, and the heavy chains to each other, to form a dimer. The light-chain constant region 1 is labeled CL 1, and the three heavy-chain constant regions are labeled CH 1–3. Although CH 2 and 3 together form the Fc portion, only the CH 3 domain binds to Fc receptors and controls (defines) the anatomic specialization of the immunoglobulin. The CH 3 region is most important in functions of specific immunoglobulin isotypes. The light-and heavy-chain variable regions are labeled VL and VH, respectively. This linear diagram helps to show the sequential location of the complementarity-determining regions (CDR 1–3) and the framework regions (FR 1–4). Framework regions are relatively conserved portions of the variable region that serve as spacers to position the antigen-binding sites properly when the variable region is folded. Panel B shows how an antigen-binding pocket forms when the VL-VH CDRs are folded into native configuration.
blood microvasculature. These antibodies maximize cellular uptake and internalization of antigens. After pathogens have breached the barriers of the skin and/or mucous membranes, antibodies of the IgM and IgG subclasses work in conjunction with the complement system. This collaboration serves to neutralize, injure, aggregate, and opsonize (to coat the pathogen with antigen-specific antibody and complement so that it can be ingested easily and destroyed by a phagocyte that is bearing receptors for Fc and complement) the pathogens so that they may be engulfed and destroyed by phagocytes.
Except in rare instances where pentameric IgM (complexed with joiner [J] chain) may be secreted across epithelium, most circulating antibodies of the IgM and IgM subclasses work in blood, lymph, and tissue fluids. They do not normally appear in mucosal secretions (Underdown and Mestecky, 1994).
Mucosal Immunity and Secretory IgA
Antibodies of mucosal immunity function outside the body at luminal surfaces of the moist epithelium lining conjunctiva; nasopharynx; oropharynx; gastrointestinal, respiratory, and urogenital tracts; and in the ducts or acini of exocrine glands. The principal antibody involved in mucosal immunity is secretory IgA (Underdown and Mestecky, 1994). This class of antibody requires the cooperation of two cell types for optimal activity in vivo. One cell type, the plasma cell, makes the IgA, while an epithelial cell transports it to the gut lumen where it works (Figure 22-3, Panel A). The plasma cell posttranslationally dimerizes the IgA by joining two molecules with another polypeptide, the J-chain. In addition to holding the two IgA molecules together, the J-chain facilitates binding to a poly-Ig receptor synthesized by and displayed on the abluminal side of epithelial cells. The complex is transported in endosomes to the luminal side of the epithelial cell and secreted. The portion of the poly-Ig receptor retained with secreted IgA is called the secretory component.
Pathogens adapted to infect mucosa express virulence factors that allow them to adhere, colonize, or invade epithelium. Secretory IgA prevents absorption of these viruses, bacteria, and toxins by blocking their adhesion while they are still on the external side of an epithelial barrier. This activity is opposite to that of antibodies associated with peripheral immunity, which aid the host cells to bind pathogens. By preventing cellular attachment of the antigen, IgA enables it to be flushed away in the stream of secreted fluids and mucous washing over the epithelial membranes.
IgA also may facilitate transport of pathogens and toxins out of the body by causing them to be conveyed into bile and other exocrine secretions (Mazanec et al., 1993). At sites where nondegradative endosomal transport delivers a pathogen into the host, such as across M-cells in the dome epithelium of the Peyer's patch (the region of a Peyer's patch where antigen from the gut lumen is transported into the lymphatic tissue) (Neutra and Kraehenbuhl, 1994; Owen
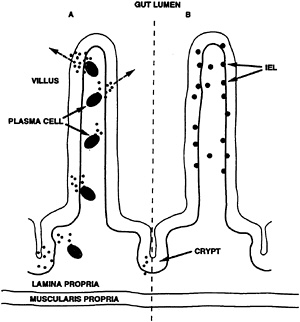
FIGURE 22-3 Diagram of small intestinal mucosa. Panel A shows the synthesis of secretory IgA by plasma cells and transport to the intestinal lumen by epithelial cells (dots and interrupted arrows). Panel B shows an intestinal villus. Large black dots represent sites where intraepithelial lymphocytes (IEL) involved in cellular prophylaxis, thymic independent T-cell maturation, or tolerance induction usually are found.
and Jones, 1974), antigen-specific IgA recently has been shown to neutralize viral pathogens while they are in the endosome (Mazanec et al., 1992).
IgA is the predominant antibody manufactured by the body. This escaped appreciation for many years because blood contains a relatively low concentration of IgA compared with other immunoglobulins. However, 75 percent of the antibody-producing cells in the body are making IgA, and most of this IgA is released on a continuous basis into gastrointestinal fluid, saliva, tears, urine, and other secretions. In humans, up to 40 mg/kg body weight of IgA is manufactured and secreted daily, which is many orders of magnitude greater than that of all other immunoglobulin isotypes (Brandtzaeg, 1994).
Compartmentalized Cell-Mediated Immunity
T-lymphocytes involved in peripheral and mucosal cell-mediated immunity compromise numerous functional subclasses of increasing diversity (Punt and Singer, 1996). Both T-helper cells (CD4) and cytotoxic T-suppressor cells (CD8) may assume immunoregulatory roles during immune responses, or they may differentiate into effector cells that exhibit segregated traffic patterns and functions (Anderson and Shaw, 1996; Ebnet et al., 1996; Salgame et al., 1991).
The profile of cytokines secreted by T-cells dictates immunoreactive cell commitment to either peripheral or mucosal immune functions. Gamma interferon from T-helper 1 (Th1) cells enhances peripheral immunity and suppresses mucosal immunity. IL-4 from T-helper 2 (Th2) cells enhances mucosal immunity and suppresses peripheral immunity. Other cytokines can further influence this divergence or lead to partial or complete compromises.
Peripheral T-Cells. T-cells committed to peripheral immunity circulate in the blood and help B-or T-cell precursors differentiate into antibody-secreting cells or cytotoxic effector cells respectively. These T-cells also release a number of cytokines that activate and arm mononuclear phagocytes and natural killer cells to destroy intracellular parasites.
Mucosal T-Cells. Mucosally committed T-cells enter Peyer's patches, the lamina propria (the layer of connective tissue underlying the mucosal epithelium), and the intraepithelial compartment (Figure 22-3, Panel B). There is considerable phenotypic diversity among these mucosal T-cells. The subclass of mucosa-homing T-cells known as intraepithelial lymphocytes is believed to play a role in protecting mucosal surfaces from being injured by infectious pathogens or parasites (London et al., 1987). However, some of the diverse cells that populate the intraepithelial compartment include thymic-independent T-cells, which enter the epithelium to complete maturation (Punt and Singer, 1996). Many of these cells display phenotypic markers that reflect intermediate stages of T-cell differentiation.
CD8 T-Cell Heterogeneity. Heterogeneity of T-cells of the suppressor-cytotoxic phenotype (CD8) has been described, especially for those cells located within the mucosal epithelium. The CD8 membrane marker is expressed as a heterodimeric complex consisting of an alpha and a beta chain (i.e., CD8ab). Within the intraepithelial compartment, CD8+ cells6 are commonly found expressing two alpha chains (CD8aa). T-cells bearing CD8ab and CD8aa may exhibit separate traffic patterns, as suggested by the observation that they are found in peripheral and mucosal compartments in different ratios. They also may participate in regulating the balance between peripheral and mucosal immunity to specific antigens because of their nominal "suppressor" activity (Salgame et al., 1991). There is some indication from basic studies that these cells exhibit organ-selective commitments.
CD8 Homing and Cross-Regulation. When mice with severe combined immunodeficiency (SCID) (which lack mature T-or B-cells) are reconstituted with lymphocytes derived from CB17 mice (normal parent strain of SCID), the fate of cells derived from a particular tissue may be traced in vivo (Hilbert et al., 1994). If residence in that tissue imprints functional commitment or organ selectivity, then that will be reflected by the phenotype and homing characteristics of the cells after transfusion. Normal lymphoid cells derived from CB17 lymph nodes or Peyer's patches were transfused into syngeneic SCID mice. Donor CD8ab T-cells, extracted from CB17 lymph nodes, lodged in lymph node para-cortex and in the intestinal lamina propria of SCID recipients. The CD8ab lymph node cells did not enter the intraepithelial compartment but remained near plasma cells in the lamina propria. The preponderant CD8+ phenotype in CB17 Peyer's patch and intraepithelial lymphocytes (IEL) is CD8aa. Infusion of Peyer's patch cells resulted in excellent reconstitution of SCID IEL, especially of CD8aa T-cells. Thus, the homing preference of these putative suppressor cell types is consistent with the possibility of their playing a role in antigen-specific cross-regulation of peripheral versus mucosal immunity as discussed later.
Functions of CD8+ Intraepithelial Lymphocytes. The exact functions of IEL subpopulations are not yet known, but animal experiments and ex vivo cellular cytotoxicity assays suggest that some IEL exhibit cellular cytotoxicity directed against viral antigens on infected mucosal epithelial cells (Cuff et al., 1993; London et al., 1987). IEL also are thought to be involved in initiating tolerance (Elson et al., 1995; Gelfanov et al., 1996; Sim, 1995), but it is not clear whether all IEL, or a unique subset of IEL, induce tolerance to food antigens. Presentation of antigen in a manner similar to that of nonclassical major histocompatibility (MHC) antigens7 displayed on basolateral membranes of intestinal epithelial cells in the absence of costimulatory signals8 (Robey and Allison, 1995) may be involved in triggering tolerogenic T-cells (T-cells that induce immunological tolerance; the concept of tolerance will be discussed later) (Matzinger, 1994). There are many basic questions to be answered in the rapidly enlarging field devoted to IEL function.
Immune Assessment by T-Cell Phenotypes. Monoclonal antibodies are now available that enable phenotypic distinction of the class and potential function of T-cells, including their commitment to being peripheral or mucosal cells. The relative phenotypes displayed on a class of T-cells differ depending on whether they are immature, resting, activated, naive, or memory cells.9 Other phenotypic markers make it possible to determine the percentages of T-cells committed to lodge in secondary lymphatic tissues, skin, inflammatory sites, parenchymal organs, mucosal lamina propria, or intraepithelial compartments of mucosa compared with the percentage of T-cells that will continue to circulate in the blood (Altman et al., 1996; Rott et al., 1996).
Compartmentalized Immunity and Lymphocyte Traffic
Immunity is dependent upon continuous movement of cells through blood, tissue and lymph (Anderson and Shaw, 1996). Lymphoid cells traffic to the secondary lymphoid organs of the spleen, lymph nodes, and Peyer's patches in order to encounter antigens acquired from the environment via blood, lymph, or across mucous membranes. Where, and by which cells, antigens are presented to the trafficking cells has a significant influence on the outcome of the immune response with respect to antibody isotype commitment and future homing preference of memory and effector lymphoid cells (Anderson and Shaw, 1996; Butcher and Picker, 1996; Husband and Gowans, 1978; Rott et al., 1996).
Random and segregated traffic patterns are essential for efficient operation of the two separate but overlapping immune systems operating in mammalian species. Without knowing about lymphocyte recirculation and homing, it is difficult to understand how peripheral and mucosal immune responses may be segregated yet interact for cross-regulation. An enormous number of lymphocytes are involved in this process (Ebnet et al., 1996).
The feat of coordinating the anatomically dispersed immune system comprised of mobile, circulating, individual, and extremely diverse cells depends upon movement and a system of membrane recognition and activation signals. A mixture of integrins, selections,10 and receptors for chemokines (chemotactic cytokines) expressed by lymphocytes and endothelial cells are involved in precipitating selective emigration of lymphoid subsets from the blood in tissues where specific counterreceptors (receptors that are also ligands) are displayed on
luminal surfaces of endothelial cells. These recognition events may happen in skin, mucosa, or specific secondary lymphatic tissues, such as Peyer's patch or peripheral lymph node. Receptor-ligand interactions allow these cells to find their way around the body, as well as to determine where to adhere to endothelium, when to migrate, and how to find their site of action within tissue (Anderson and Shaw, 1996; Gretz et al., 1996; Rott et al., 1996).
A consensus hypothesis (Figure 22-4) has been offered to explain the overall process of emigration and to propose ways to explain organ-selective migration as a ''combinatorial mechanism"11 (Butcher, 1991). Circulating lymphocytes use adhesive selections to roll on endothelial-cell luminal surfaces; they become loosely tethered using Selectin-glycam/CD34 interactions.12 Upon activation either by receptors expressed on the endothelial surface or by chemokines, the binding characteristics of these receptors are changed very rapidly (within milliseconds) from low affinity to high affinity (Anderson and Shaw, 1993; Ebnet et al., 1996). Different specific receptor-counterreceptor interactions are responsible for each stage and mediate recognition, binding, and emigration of cells from the blood.
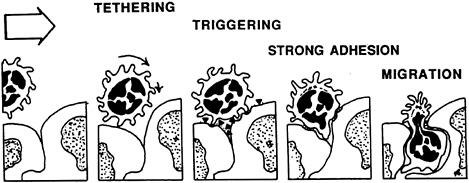
FIGURE 22-4 Diagram of the adhesion cascade. The rolling cell binds, then begins to migrate between endothelial cells and enters the tissue. SOURCE: Adapted from Anderson and Shaw (1993).
Recirculation Is the Integrating Principle. Recirculation of a precursor pool of uncommitted lymphocytes, from the blood into lymph nodes or mucosal lymphatic tissues and then back to the blood again, forms the basis of immuno-surveillance and integration of immune functions across the segregated systems. The magnitude of cell traffic reflected by the number of cells returned to the blood in efferent lymph is enormous. Enough lymphocytes recirculate from lymph to blood to replace the total blood lymphocyte pool from 10 to 48 times every 24 hours (Anderson and Shaw, 1996; Ebnet et al., 1996).
Compartmentalized Traffic Is Acquired. While naive lymphocytes appear to access peripheral lymphoid tissues randomly during recirculation, memory lymphocytes selectively return to the tissues where they initially were stimulated by antigen. Precursor or naive lymphocytes (those that have not seen antigen in a lymphatic tissue or participated in an immune response) enter all tissues, especially secondary lymphatic tissues, such as Peyer's patch, peripheral lymph node, or spleen, without any organ-selective bias. In the short period after activation by antigen, lymphocytes behave like inflammatory cells and avoid returning to secondary lymphoid tissues, preferring to lodge in skin, gut, or inflammatory sites as "acute memory" cells. After maturation occurs, some of these memory cells follow an organ-selective traffic route determined by the tissue in which that particular lymphocyte encountered the signals to divide, differentiate, and mature.
When activated by antigen (displayed by an antigen-presenting cell) in any secondary lymphatic tissue, lymphocytes proliferate and undergo many of the changes associated with differentiation. For a few days after their activation, they radically change their cell-surface expression of molecules involved in adhesion. When these cells emerge from lymphatic tissue as memory cells, their pattern of migration is markedly different. As new memory cells, lymphocytes preferentially home to specific nonlymphoid tissues, such as skin or gut (rather than the traffic areas of secondary lymphoid tissues). This behavior fits reasonably well with the increases seen in expression of "inflammatory" adhesion molecules, concomitant with decreases in L-selectin associated with recirculation. Memory CD4 (T-helper) cells express more VLA-4 and LFA-1 and have increased capacity to bind to their ligands VCAM-1 and ICAM-1, the expression of which is increased at sites of inflammation.
Conditions Influencing T-Cell Compartmentalization
Secondary lymphoid tissues that drain peripheral or mucosal tissues differ from each other and therefore differentially influence maturation of T-cells (Cahill et al., 1977). If lymphocytes are activated in lymph nodes that drain skin, they become specialized to migrate preferentially into skin and serve the immunologic needs of skin (Mackay, 1991; Mackay et al., 1992). Conversely, if lym-
phocytes are activated in lymphatic tissues sampling antigens from the gastrointestinal (GI) tract, they become specialized to migrate preferentially into the GI tract and serve its immunologic needs (Abernethy et al., 1991; Kimpton et al., 1989).
Adhesion Molecules. Expression of specific adhesion proteins and selective secretion of cytokines in peripheral and mucosal lymphoid tissue help to create unique microenvironments (Ebnet et al., 1996). Somehow an influence of this microenvironment is imprinted on the lymphocytes differentiating therein.
Differentiation in mucosal lymph nodes results in the induction of integrins that may predispose T-cells to interact with vascular addressins 13 expressed on GI tract venular endothelium and therefore migrate into the GI tract; included are both the integrins aeß7 for CD8+ IEL cells, which bind to E-cadherin14 on mucosal epithelial cells (Cepek et al., 1994; Karecla et al., 1995), and a4ß7 for CD4+ lymphocytes, which bind to mucosal addressin cell adhesion molecule (MADCAM)-1 on mucosal venular endothelium and splenic marginal sinus (Berlin et al., 1993; Hamann et al., 1994; Kraal et al., 1995; Lyons and Parish, 1995). Conversely, T-cells maturing in skin-draining lymph nodes preferentially retain expression of L-selectin (and the sialylated CLA [cutaneous lymphocyte-associated] antigen, recognized by HECA [human endothelial cell-associated] 452 monoclonal antibody, which binds to endothelial E-selectin). Both of these integrins appear to facilitate subsequent entry of lymphocytes into the skin or lymph node, where L-selectin binds to carbohydrates of the endothelial sialomucins15 GlyCAM and CD34.
The leukocyte integrin a4 ß1 (VLA-4) is activated by a signal emanating from the endothelium and mediates strong binding to VCAM-1 expressed on HEV surfaces. This is thought to facilitate leukocyte emigration between endothelial cells. These differences in the phenotype of emerging T-cells must reflect local differences in the microenvironment. Such differences have not been defined clearly; however, transforming growth factor (TGF)-ß is believed to be better expressed in mucosal sites and contributes to the distinctive phenotypes of resident cells. To illustrate, TGF-ß1 upregulates and maintains expression of
aeß7 integrin in T-cells destined to enter the intraepithelial compartment, where it anchors T-cells to E-cadherin on epithelial cells (Cepek et al., 1994).
Conditions Influencing B-Cell Compartmentalization
Primary B-cells expand rapidly in primary mucosal lymphoid follicles of very young mammals and undergo diversifying mutations in the genes encoding the antigen-binding sites during V-D-J rearrangement (Weinstein et al., 1994a). They are then subject to positive selection to generate an antibody repertoire biased toward antigens prevalent in the environment; progeny that do not bind antigen in this milieu undergo apoptosis (programmed cell death). Survivors leave the follicles in efferent lymph and enter the blood where they circulate and recirculate as small precursor IgM+/IgD+ B-cells. They migrate continuously into all secondary lymphoid tissue until triggered to divide by the appropriate antigen and costimulatory signals.
The subsequent differentiation of circulating B-cells depends on whether they are first activated in peripheral lymphoid tissue (lymph nodes and spleen) or in mucosal sites (such as Peyer's patches), where they are influenced by prevalent cytokines and unique stromal and accessory cell16 populations in the respective tissues (Weinstein and Cebra, 1991; Weinstein et al., 1991).
Some precursor B-cells will be first activated in spleen or lymph node. In that process, they are programmed toward one of the IgG isotypes (rather than IgA). Daughter cells from this clonal expansion leave the lymph node in the efferent lymph, return to the blood, and seed the spleen, bone marrow, sites of inflammation, and other lymph nodes as plasma cells and "memory" B-lymphoblasts (Cebra et al., 1977; Kantele et al., 1997; Quiding-Jarbrink et al., 1995).
Other precursor B-cells will be activated first in Peyer's patch. Those cells become committed to rearrange their immunoglobulin heavy-chain genes to express IgA (rather than IgG isotypes noted above); despite having made this commitment, the switch to IgA often is delayed for several days. These cells leave in the efferent lymph, pass through the mesenteric lymph node (where they may be subject to immunoregulation17 by T-cells they encounter there), and return to the blood. These cells selectively migrate to the spleen, which serves as an auxiliary site for clonal expansion prior to dissemination of the daughter cells
via the blood into mucosal lamina propria. This process takes 5 to 7 days, at least 4 trips in the blood, and 2 to 3 cell generations (Cebra et al., 1977).
Immune Assessment by B-Cell Phenotyping. Between 2 and 4 days after antigen exposure, the orderly appearance of significant quantities of recirculating (naive precursor B-cells) and nonrecirculating (memory or preplasma cells) cells in the blood varies in frequency and may provide a valid window for monitoring the status of immune responses before serum antibody can be measured (Anderson and Shaw, 1996). Analysis using a flow cytometer may be performed on cells isolated from circulating blood. Two-color FACScan (fluorescence activated cell sorter) analysis will pair red fluorochrome-labeled antigen18 (which binds to surface immunoglobulins on specific B-cells) with green fluorochrome-labeled antibodies, which can identify whether the circulating cells are committed to peripheral or mucosal immunity or some other phenotypic marker (Quiding-Jarbrink et al., 1995).
It is relatively easy to label recombinant or highly purified antigen for use in identifying and counting antigen-specific B-cells. Numerous monoclonal antibodies are available to assist in identifying the functional commitment of cells, including whether the surface immunoglobulins include IgD (indicative of a precursor or primary B-cell), or an IgG or IgA isotype (indicative of a secondary or memory B-cell committed to peripheral or mucosal immunity, respectively). With more experience using flow cytometry, the status of an immune response will be able to be estimated as it evolves. Techniques have even been developed for detection of cytoplasmic proteins by FACScan, including cytokines secreted by T-cells that influence B-cell responses (Assenmacher et al., 1994; Openshaw et al., 1995). This is especially important for estimating the potential for mucosal IgA responses because appearance of surface IgA-positive cells in the blood precedes lodging and differentiation into antibody secretion by several days.
ISSUES FOR CONVENTIONAL MODES OF VACCINATION
Complicated Diseases Require Peripheral and Mucosal Protective Immunity
Protection from diseases that threaten military personnel requires complex forms of immunity (Mobley, 1995). The nature of the vaccine antigen and route of its administration may confer anatomic compartmentalization to the immune response, restricting it to peripheral or mucosal immunity. Some infectious disease organisms spread parenterally to involve parenchymal organs but do not infect the epithelial cells lining respiratory, gastrointestinal, or urogenital tracts. Protection against these pathogens depends on circulating IgM and IgG antibodies19 and/or peripheral cell-mediated immunity for resolution. Other disease agents principally infect mucosal epithelium, causing serious nutritional and physiologic alterations, but in these diseases, circulating antibodies of the IgG class are not protective. Instead, secretory IgA and/or intraepithelial cytotoxic lymphocytes are needed for protection (Ogra et al., 1994).
In most biological warfare scenarios, every mucosal surface of the body could be a portal of entry for aerosol-disseminated or ingested biological agents. Once an agent enters the body through the mucosa of the eyes, nose, throat, lungs, and gastrointestinal tract, it is able to disseminate and initiate systemic parenchymal, as well as mucosal pathology. Parenteral immunization, the most common route of vaccination, usually elicits a peripheral immune response, with protective IgM-IgG antibodies and peripheral cell-mediated immunity. Parenteral immunization usually fails to stimulate mucosal lymphatic tissues to generate protective IgA antibodies or antigen-specific IEL. Many hazardous agents infect across the mucosa but spread through the systemic circulation. Protection against these agents requires vaccines that induce both peripheral and mucosal immune responses.
Cross-Regulation Is an Issue with Conventional Modes of Vaccination
Early studies suggested that the route of vaccination was crucial for determining whether protective peripheral or mucosal immunity would develop. Furthermore, immunization methods and routes for inducing mucosal immunity
often delayed or prevented induction of peripheral immunity and vice versa (Chase, 1946; MacDonald, 1982; Mattingly and Waksman, 1978). This phenomenon, which may be termed cross-regulation (Figure 22-5), was first seen by Pierce and Koster (Koster and Pierce, 1983; Pierce, 1978) and Hamilton et al. (1979) when they were testing a nontoxic cholera vaccine candidate. They found that when the initial priming occurred by vaccinating via a parenteral route and the second inoculation of antigen was given by an enteric route, the ability to elicit immunity in the mucosa was reduced. But parenteral priming followed by a second parenteral dose resulted in a strong booster response.
It is important to emphasize that the vaccine alluded to above was most likely denatured cholera toxin. In contrast, recent adjuvant studies with nondenatured cholera toxin proteins (that retain the capacity to bind GM1 ganglioside, the mechanism by which cholera toxin binds to intestinal epithelial cells) show that the proteins have great promise as vaccine vehicles (Dertzbaugh and Elson, 1993a; Dertzbaugh and Van Meter, 1996; Elson and Dertzbaugh, 1994; Elson et al., 1995; Walker, 1994). Cholera toxin and its B-subunit have the special ability to overcome cross-regulation of immunity and are components of several of the new vaccine technologies that will be discussed later in this report.
When Brown et al. (1981) empirically tested the effect of route of immunization of a formalin-inactivated Rift Valley fever virus vaccine on protection from parenteral or mucosal challenge, they found that the nonreplicating vac-
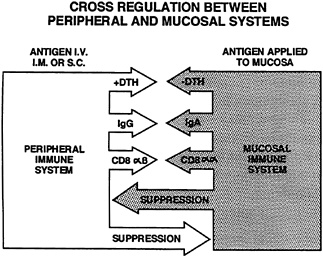
FIGURE 22-5 The dichotomy of immune systems involved in responding to vaccine antigens. Each immune system has its own stereotypical way of responding to antigen that is delivered by parenteral (left) or oral (right) routes of immunization. The predominant antibody type (IgG or IgA) and effector T-cell type (CD8ab vs. CD8aa) are shown. Each system has the ability to suppress the other by numerous mechanisms. Delayed hypersensitivity (DTH), regulated by preponderant Th1-Th2 cytokines, is not displayed in healthy mucosal tissues.
cine elicited an immune response that protected mice from challenge by parenteral route but not from challenge by aerosol. This asymmetric immunity also was found for the C84 Venezuelan equine encephalitis virus vaccine (Jahrling and Stephenson, 1984). These experiments were repeated using a matrix design that tested both routes of immunization against homologous and heterologous challenge routes (Anderson et al., 1987a, b; Hart et al., 1995; Unpublished data, M. L. M. Pitt and A. O. Anderson, U.S. Army Medical Research Institute of Infectious Disease, Fort Detrick, Md., 1987). Upon heterologous challenge, viral pathogenesis (target organs affected and time course of disease evolution) was altered. Protection from encephalitis was deficient when subcutaneously immunized mice were challenged by aerosol. Moreover, when mucosally immunized mice were challenged by subcutaneous inoculation, the onset of viral infection of the liver extended over an 18-d time course, where, ordinarily, the liver is affected only during the first 3 to 6 days. The levels of specific IgA or IgG in serum and secretions corresponded to whether the challenged animals experienced protection and/or altered pathogenesis (Anderson et al., 1987b).
Oral Tolerance and Mucosal Immunity
There is no easy way to explain this phenomenon. The mucosal immune system is known to be the site of priming for two paradoxically opposite purposes, that is, tolerance (immunological nonresponsiveness or suppression to an antigen with proven immunogenicity) and mucosal immunity. The usual response of the GI tract to antigens is tolerance rather than immunity (Chen et al., 1995). The mechanisms of oral tolerance remain unclear. Tolerance could be primed in Peyer's patches or in the intraepithelial compartment, or IELs may be good candidates for communicating a tolerogenic signal. Epithelial cells can present antigens, but without important costimulatory molecules, the cells that see this antigen may die or be rendered nonresponsive (Matzinger, 1994). It is thought that tolerance can occur through active suppression (cells or cytokines that induce nonresponsiveness), anergy (live nonresponsive cells), or apoptosis of antigen-reactive cells.
Tolerance Versus Cross-Regulation. The healthy GI tract is bathed in an enormously diverse collection of environmental antigens, yet only certain antigens stimulate active mucosal, peripheral, or combined mucosal and peripheral immune responses. Somehow the tissue is able to distinguish pathogens (dangerous) from normal flora (safe) and food antigens (safe) (Matzinger, 1994). Inducible chemokines may be the mucosal "danger" signal (Eckmann et al., 1993; Oppenheim et al., 1991). Pathogens that bind and/or invade mucosal epithelium cause epithelial cells to release cytokines and chemokines that attract inflammatory and/or immune cells, or cause epithelial cells to express proteins
(classical and nonclassical MHC20) that target antigens for induction of immunity (Agace et al., 1993; Eckmann et al., 1993; McCormick et al., 1993). Mucosal antigens that lack costimulatory activity are programmed for tolerance (Bendelac, 1995; Kagnoff, 1996; Shroff et al., 1995). When tolerance results from mucosal immunization, it does not appear to diminish stimulation of B-cells committed to IgA secretion (Shroff et al., 1995); however, in contrast to cross-regulation, mucosal tolerance (Mowat, 1994) is most likely responsible for preventing systemic responses to common food antigens, but it has different conditions of triggering than that for cross-regulation.
Tolerance Versus Suppression. What some regard as mucosal or peripheral tolerance could merely represent a temporary induction of antigen-specific suppression. The suppression of systemic delayed cutaneous hypersensitivity and the reduction of IgG expression induced by mucosal priming lasts less than 3 months (Anderson et al., 1991; Koster and Pierce, 1983). In contrast, true tolerance persists until some stimulus breaks its effect. Peripheral priming with antigens in peripheral lymph nodes results in reduced mucosal immune responses comparable to that mentioned above (Anderson et al., 1987b; Anderson et al., 1991). This "yin-yang" effect on peripheral versus mucosal immunity suggests that cross-regulation might not be tolerance, but tolerance may be influenced indirectly by T-helper cells secreting cytokines that exert positive or negative feedback control (Figure 22-6). This suppression may require cell contact, or it may exert its effect through local cytokine commitment that prevents T-cell help of the required variety. For example, what is needed may be IL-4, but what is available is interferon gamma (IFNγ), and this difference is interpreted as suppression.
T-Helper Subclasses and Cross-Regulation. Because cross-regulation may influence immunity during the short period of time after oral or parenteral immunization, it is of value to discuss the possible role of T-helper cell subclasses in cross-regulation (Street and Mosmann, 1991; Swain et al., 1991; Trinchieri, 1993). T-helper cells segregate into two subsets depending on whether the cells secret IFNγ and interleukin-2 (IL-2) (T-helper 1, Th1), or IL-4, IL-5, IL-6, and IL-10 (T-helper 2, Th2) (Mosmann and Sad, 1996). This distinction is important because the cytokines determine the kind of help provided by the respective T-helper cell types. This dualistic system, which enables the achievement of harmony between opposing cytokine influences, is illustrated graphically in Figure 22-6.
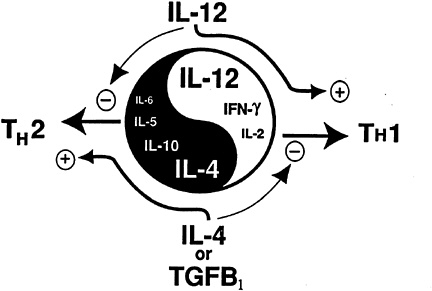
FIGURE 22-6 This diagram, resembling the oriental symbol for yin and yang, represents microenvironmental factors that stimulate T-cells to express either T-helper 1 (Th1) or T-helper 2 (Th2) cytokines, which are associated with cellular and humoral immunity, respectively. Other vectorial combinations of cytokines across this dualistic system may contribute to cross-regulation of immune responses associated with parenteral or mucosal immunization. The cytokines IL-12 and TGF-β1 are predominant influences in peripheral and mucosal lymphatic tissues. Thus, vectorial expression of these cytokines affect T-and B-cells in such a way that proliferating B-cells become committed to secrete peripheral IgG or mucosal IgA, respectively.
The central object in Figure 22-6, which resembles the symbol for yin and yang, represents the symmetric opposing effects of cytokines on lymphatic tissue microenvironments. In "peripheral" lymphoid environments, such as in lymph nodes draining the skin, IL-12 (Kang et al., 1996) secreted by Langerhans cells favors differentiation of T-cells that secrete IFNγ and IL-2. In addition to favoring cellular immunity, these Th1 cytokines favor production of IgG isotopes by B-cells. The mucosal environment in the Peyer's patch is affected by high local concentrations of TGF-β1. This cytokine favors development of T-cells that secrete IL-4 and other Th2 cytokines. IL-4 negatively affects peripheral cellular immunity at the same time that it favors development of B-cells committed to IgA secretion. Furthermore, TGF-β1 has been shown to directly affect immunoglobulin gene class switching to favor selection of α-heavy-chain genes. Thus, environmental influences, of which cytokines are a prominent few, affect the direction of the immune response toward cellular (Th1) or humoral immunity when the vector is oriented along the extreme opposites. However, intermediate vectorial orientations productive of mixed secretion of cytokines can produce mucosal or peripheral immunity when IL-4 and IFNγ moderately predominate among other microenvironmental cues. Thus, the relative concen-
tration of T-helper types in lymph nodes or Peyer's patches influences selective expression of IgG or IgA immunoglobulin isotypes, respectively (Stavnezer, 1995). Anatomic compartmentalization of T-helper subclasses is of value in considering possible mechanisms of cross-regulation.
When Th1 responses are predominant (as they are in skin-draining lymph nodes influenced by Langerhans cells that secrete IL-12 [Kang et al., 1996]), T-helper cells secrete IL-2 and IFNγ , resulting in selective expression of IgG immunoglobulins and activation of cytotoxic T-cells and armed mononuclear phagocytes (Ariizumi et al., 1995; Kange et al., 1996; Weinstein et al., 1991). Where Th2 responses are predominant (as they are in mucosal sites), T-helper cells secrete IL-4, IL-5, IL-6, and IL-10, resulting in selective expression of different immunoglobulin isotypes, including IgA (Hiroi et al., 1995; Lebman and Coffman, 1994; Xu-Amano et al., 1993), and downregulation of peripheral cell-mediated immunity at the same time that there is increased development of cytotoxic T-cells that selectively home to mucosal intraepithelial sites (Ke and Kapp, 1996). The T-helper cell types provide positive and negative feedback control on commitment of B-cells to express specific antibody isotypes. For example, IFNγ suppresses commitment to IgA, and IL-4 suppresses commitment to IgG. Whether this is a direct effect on the B-cell or an indirect effect mediated by T-cells is a subject of great interest (Abbas et al., 1996; Finkelman, 1995; Lebman and Coffman, 1994).
Anatomic Compartmentalization of T-Helper Subclass Commitment. In mucosal sites, abundance of the cytokine TGF-β1 programs Th0 cells to develop into Th2 cells (Lebman and Coffman, 1994; Young et al., 1994). The cytokines secreted by Th2 cells contribute to expansion and differentiation of B-cells committed to IgA expression. TGF-β1 also contributes to selective expression of IgA antibodies by favoring immunoglobulin heavy-chain gene switching to IgA, and by suppressing expression of other isotypes (Lebman and Coffman, 1994; Stavnezer, 1995). TGF-β1 is not widely distributed in peripheral lymph nodes where there is selective expression of Th1 cellular responses and IgG antibodies. Recent publications suggest that Langerhans cells from the periphery may have a role in Th1 commitment by secretion of IL-12 and IL-1β-converting enzyme (Ariizumi et al., 1995; Kang et al., 1996). Other relationships of Th1-Th2 responses may be dependent upon which antigen-presenting cell type migrates into a site and conditions the microenvironment (Morikawa et al., 1995).
When this system is analyzed in vitro at the single-cell level, specific T-helper cell types or cytokine profiles are not sufficient to cause uncommitted IgM+, IgD+, or IgM-only B-cells to switch to IgA (Kotloff and Cebra, 1988; Schrader et al., 1990). However, when antigen-presenting cell types or stromal cells are added, switching occurs. Thus the cross-regulation is related to a sum of multiple ''microenvironmental" factors unique to the tissue where the im-
mune response is initiated (Weinstein and Cebra, 1991; Weinstein et al., 1991). Resolution of this on a molecular level awaits further research observations.
Cross-Regulation Is Incompatible with Complete Protective Immunity
Cross-regulation is not a desirable feature of conventional modes of vaccination. Effective ways to overcome the cross-regulation that prevents simultaneous induction of mucosal and peripheral immunity have been sought urgently. Not all vaccine antigens exhibit this behavior, especially those that have so-called adjuvant effects. It has been shown that modifying the vaccines with immunological adjuvants was a partial solution, but it was required that the adjuvants be used at optimal doses, which were different for each vaccine tested (Anderson, 1985; Anderson and Reynolds, 1979; Anderson and Rubin, 1985; Anderson et al., 1987b). Adjuvants were not effective with all antigens, and it was necessary to perform empirical studies to select the optimal combinations. Certain adjuvants (such as the lipoidal amine avridine) also potentiated the undesirable cross-regulation observed with native antigen (Anderson et al., 1985, 1987b). For example, if an antigen given orally elicited a good IgA response in secretions but failed to elicit or slightly depress specific IgG in serum, use of an adjuvant with the antigen would increase the IgA response and further decrease or block the IgG response in serum.
SOLUTIONS TO CROSS-REGULATION FROM NEW TECHNOLOGIES
Although the approach of this group was to define cross-regulation, other scientists working with biodegradable microspheres and enterotoxin-derived vaccine delivery systems were finding that they could induce simultaneous peripheral and mucosal immunity. Antigens administered orally in biodegradable microspheres composed of poly(lactide/glycolide) copolymer, or antigens associated with carrier proteins derived from cholera toxin (CT) or E. coli heat-labile enterotoxin (LT), induced simultaneous peripheral and mucosal immunity (Eldridge et al., 1990; Elson and Ealding, 1984a, b). The same or similar antigens given without these particular vehicles yielded cross-regulation. In addition, antigens conjugated with the B-subunits of CT or LT triggered acceptable peripheral and mucosal immune responses that were unattainable when antigen was given alone (Dertzbaugh and Elson, 1991; Elson and Dertzbaugh, 1994; Holmgren et al., 1993). Taken together, this research provided evidence that cross-regulation could be overcome using nontoxic and biodegradable vaccine vehicles.
Solutions from Microencapsulation Technology
The first technology to be highlighted is microencapsulation of vaccines with biodegradable polymers for immunization of the peripheral and mucosal immune systems. There are a number of recent reviews (in new journals committed to the field) that cover all the new microencapsulation systems, polymer chemistry (Gombotz and Pettit, 1995; Langer, 1990), and biologic applications (Michalek et al., 1994; Morris et al., 1994; Walker, 1994). After reports of success in potentiating immune responses with adjuvants composed of particulate antigens, numerous particulates were tested for vaccine-enhancing activities. The chemical composition of particles that were tested included polystyrene, latex, poly(methylmethacrylate), polyacrylamide, poly(buty1-2-cyanoacrylate), alginate, ethyline-vinyl acetate copolymer, and poly(lactide glycolide) copolymer (Michalek et al., 1994). These polymers exhibited a broad range of utility, toxicity, and biodegradability. This discussion will be restricted to observations about the use of polymers comprising lactic and glycolic acids (natural products of energy metabolism) as vehicles because they have been tested for efficacy in initiating peripheral and mucosal immune responses. Poly(lactide/glycolide) copolymer originally was developed for biodegradable suture material (Morris et al., 1994). It is licensed by the U.S. Food and Drug Administration for that use and has been used with no serious adverse sequelae for many years in human subjects. During the past 10 years, there has been exponential development of microencapsulation technology for the delivery of drugs and hormones and for other uses, such as the scratch-and-sniff advertisements found in magazines.
Biodegradable Microspheres Have Adjuvant Effects
The technology was first successfully applied to parenteral and oral immunization with vaccines by John Eldridge in the early 1980s. In addition to demonstrating that encapsulation protected the antigen from deterioration or degradation in the GI tract, Eldridge revealed that microencapsulation provided an "adjuvant" effect. The same quantity of antigen in microcapsules gave more vigorous immune responses than antigen given alone (Eldridge et al., 1990; Michalek et al., 1994; Walker, 1994).
Many of Eldridge's vaccine formulations were inoculated parenterally to determine antigen-release kinetics. The rate of particle degradation (a function of molecular weight, surface area, and ratio of lactide to glycolide in polymer composition) could be utilized effectively to control the rate of antigen release. It was discovered that microcapsule formulations could be constructed that released antigens in pulses, which mimicked a multiple-inoculation schedule although the product was administered only once. This line of inquiry has prospered, and new, improved formulations have been developed that facilitate encapsulation of aqueous-phase antigens and various water-in-oil or water-in-oil-in-water emulsions (Yan et al., 1994).
Microencapsulated Vaccines Overcome Cross-Regulation
When vaccine antigen in poly(lactide/glycolide) copolymer was administered orally in various size ranges between 1 and greater than 10 µm, particles of over 10-µm diameter were not absorbed in the gut. The larger absorbable particles (diameters > 5 to < 10 µm) arrested in Peyer's patches, whereas the smaller particles (> 1 to < 5 µm) were disseminated to mesenteric lymph node and spleen (Eldridge et al., 1990). This bimodal anatomic distribution of particles was accompanied by simultaneous elicitation of IgG and IgA immune responses. Hydrophobicity, contributed by the chemical composition of the polymer, enhanced uptake of the particles in Peyer's patches, and hydrophilicity favored dissemination. Thus, the use of particles in a range of sizes appeared to be the critical determining factor that enabled simultaneous induction of peripheral and mucosal immune responses and the avoidance of cross-regulation.
Microencapsulation with biodegradable polymers is not a technology foreign to food and nutrition research (Dunne, 1994). Preparation of this manuscript brought to light that a previous meeting of the Committee on Military Nutrition Research (IOM, 1994) had included a presentation on the use of these materials for food additives. The present use of microcapsules as vaccine vehicles could fit into the area of "medical food additives." Encapsulation of antibodies for passive prophylaxis requires less technology than that for vaccines. The sole objective is to bring the antibodies past the denaturing effects of gastric acid, and for this the antibodies need only be incorporated in standard "enteric" coatings. How these recombinant human antibodies are made is another exciting technology that will be covered along with antigen-antibody expression in transgenic plants.
Solutions from Enterotoxin-Derived Vaccine Carriers
Cholera toxin (CT) has important pharmacological effects. The toxin is composed of two subunits: a toxigenic A-subunit and the B-subunit (CTB), a pentameric ring structure that mediates binding of the toxin to the surface of eukaryotic cells. This has been known since it was determined that the watery diarrhea of cholera was not caused by intestinal epithelial cell injury (Gangarosa et al., 1960). The prodigious fluid output was produced by a pharmacological effect of CT on intestinal epithelial cells. After CT binds monosialyl GM1 ganglioside on epithelial cells via the pentameric B-subunit, the toxic A-subunit enters the cell and ADP-ribosylates the adenylate cyclase regulatory protein Gs. The result is runaway adenylate cyclase activity, which causes the absorptive epithelial cells to secrete massive amounts of fluid (Holmgren, 1981; Spangler, 1992).
Adjuvant Activity of Cholera Toxin
CT is a potent immunogen regardless of route of administration (Fuhrman and Cebra, 1981; Pierce and Gowans, 1975). It is one of the few antigens that will yield a potent IgA immune response when administered orally to experimental animals. CT never induces tolerance or cross-regulation in vivo. Furthermore, it can prevent a tolerogenic antigen from inducing tolerance when the two are coadministered. Cholera toxin may lose its ability to increase immunogenicity of nominal antigens if its B-subunit (CTB) is denatured or blocked, adversely affecting the GM1 ganglioside-binding capacity that is essential for the adjuvant effect (Dertzbaugh and Elson, 1993a; Sun et al., 1994).
Chemical conjugation (cross-linking) of antigens to CT or CTB can cause relatively poor mucosal immunogens to stimulate peripheral and mucosal immune responses strongly (Dertzbaugh and Elson, 1991, 1993b; Elson and Dertzbaugh, 1994; Elson and Ealding, 1984 a, b; Holmgren et al., 1993). The adjuvant activity of CT can be obtained if CT is mixed with antigen (Lehner et al., 1992; Ruedl et al., 1996; Stok et al., 1994). On the other hand, nontoxic CTB must be linked to an antigen to produce an adjuvant effect. Sometimes the procedure used to link antigens to CTB chemically damages the tertiary structure of the pentameric B-subunit ring and reduces adjuvanticity. An objective of Dertzbaugh and this laboratory has been to develop a bacterial expression system that will fuse an antigenic construct genetically to CTB in such a way that it can be expressed without alteration of the native configuration of the antigen or the B-subunit. A similar approach already has been reported to have had some success (Hajishengallis et al., 1995; Wu and Russell, 1994). Dertzbaugh and this group have been focusing on the benefits of using the nontoxic B-subunit (with or without molecular spacers derived from the nontoxic A2-peptide). Using constructs that lack the A1 ADP-ribosylates will avoid the necessity of dealing with issues related to toxicity.
Immunohistochemistry (Unpublished data, A. O. Anderson and M. T. Dertzbaugh, U.S. Army Medical Research Institute of Infectious Diseases, Fort Detrick, Md., 1992) reveals that some of the increased antibody production found when CT is used as an adjuvant may result from facilitated secretion by a few plasma cells rather than an increase in the number of cells secreting antibody. It has been observed that CT induces B-cells to secrete antibody when they are still in intermediate stages of migration and differentiation. Normally, IgM-or IgG-secreting cells are seen infrequently in gut lamina propria. During the first 24 hours after oral CT treatment, IgM+, IgG+, and IgA+ cells show marked accumulation of antibodies in surrounding lamina propria, indicative of increased secretion. These effects of CT holotoxin (the complete toxin molecule, possessing all subunits) may be pharmacological rather than immunological if CT causes plasma cells to hypersecrete by a mechanism similar to that which causes diarrhea through epithelial hypersecretion.
Cholera Toxin and E. Coli Heat-Labile Enterotoxin
CT and heat-labile enterotoxins (LTs) of E. coli are very homologous in amino acid sequence (CT and LT have 80% amino acid homology [Dallas and Falkow, 1980]) and tertiary structure (Burnette, 1994; Sixma et al., 1991; Zhang et al., 1995). LT is believed to have less potent toxicity than CT, but both cause diarrhea at relatively low doses in human subjects. The LT-1 enterotoxin has been used in similar adjuvant studies and found to have effects virtually identical to those of CT (Clements et al., 1988).
One critical difference between CT and LT is that the A-subunit of CT is cleaved posttranslationally into an enzymatic A1- and a nontoxic A2-peptide, whereas the A1 and A2 of LT must be cleaved by proteases encountered in the environment for toxicity to result. A report by Lycke et al. (1992) indicated that mutations that destroy the ADP-ribosylating activities of CT or LT also destroy the adjuvanticity. However, Dickinson and Clements (1995) were able to dissociate adjuvanticity from toxicity by creating a mutant that is resistant to proteolytic activation. The R192G mutant LT has an arginine substituted for a glycine at the 192nd amino acid from the N terminus, rendering the site resistant to proteolysis. This mutant LT (mLT) retains its ability to function as an adjuvant when given orally. Furthermore, its adjuvant activity extends to the potentiation of antigen-specific IgG serum and IgA mucosal antibody responses in mice (Dickinson and Clements, 1995). It also prevented induction of tolerance (cross-regulation) to nominal antigens. This promising product is moving rapidly through preclinical and clinical safety and efficacy trials (Personal communication, CDR D. Scott, Naval Medical Research Institute, Bethesda, Md., 1996, and J. Clements, Tulane University, New Orleans, La., 1996).
Effects of CT-LT on Mucosal T-Cell Compartments
The mechanisms that enable CT or LT to overcome cross-regulation, prevent tolerance, and potentiate peripheral and mucosal immune responses are not known. In vitro and in vivo studies of the cells, cytokines, and tissues affected by CT or LT have not revealed many similarities to effects of traditional adjuvants, such as Freund's complete adjuvant21 (Anderson, 1985; Anderson et al., 1987b; Freund, 1956). Perhaps CT and LT merely remove the effects of CD8+ cells and improve the natural immune processes described earlier in this chapter, such as antigen presentation, cytokine secretion, and lymphocyte traffic.
Effect of Cholera Toxin or Cholera Toxin B-Subunit on Lymphoid Tissue Compartments
When T-and B-cell compartments in lymphatic tissues from mice that had received CT or CTB orally were examined by immunohistochemistry, there were no glaring changes in T-and B-cell populations in lymph nodes, spleen, or Peyer's patches. However, CT and CTB very quickly depleted the CD8+ cells in the intraepithelial compartment, cells that are normally very prevalent. The rapid 80 percent reduction in CD8+ IEL between 12 and 72 hours was impressive. Equally impressive was the finding that between 10 and 14 days later, the intraepithelial compartment was repopulated. Depletion of IEL primarily affected CD8+ cells, but other phenotypes also must have left the epithelium. The CD4+ cells in Peyer's patches and lamina propria stayed the same or increased in number during the drop in CD-8+ cells. The depletion of IEL was triggered by doses of CT or CTB that have been shown to be mucosal adjuvants in other studies (Elson and Ealding, 1984a, b). These anatomic findings were consistent with a hypothesis that CT-LT achieves its adjuvant effect by altering the T-cell regulatory environment in mucosal lymphoid compartments to support mucosal immunity, prevent tolerance induction, and ameliorate cross-regulation (Elson et al., 1995).
Solutions to Quantity and Cost Issues for CT-LT Vaccine Carriers
The idea of developing new vaccines that incorporate recombinant antigens and/or biologically active carriers raises a few questions. One question concerns yield. Bacteria grow rapidly, but it takes a lot of bacteria a long time to produce only a handful of product, and the cost of operating large fermenters is not trivial. Contamination of product with bacterial endotoxin also may affect the yield. The product must be purified free of endotoxin, and that could result in the loss of some of the product.
Another of the factors that might limit interest in developing recombinant vaccines and therapeutic antibodies is cost. If often has been stated that, with luck, vaccines take 10 years or more from discovery to FDA licensure. Many people start to develop vaccines, but few vaccines become licensed because anywhere along the way the product may fail. If it fails during the preclinical phase, money will be saved. Many other vaccines fail during Phase I safety trials in human volunteers, if adverse reactions are unacceptable. The worst case would be failure during field testing when a vaccine that was safe and efficacious in experimental animals failed to corroborate its efficacy in humans.
Pharmaceutical industries face these realities continually, but the public (the customers) cannot understand why every start does not necessarily translate into a new beneficial product for them. All of this costs time, personal commitment, and lots of money. This becomes an especially frustrating concept if one considers that the vaccines needed most, such as malaria vaccines, are against disease
threats that are endemic in countries whose financial resources preclude any chance of buying the product.
The next section will show how both of these issues may be resolved by producing antigens or antibodies in plants.
SOLUTIONS FROM PRODUCTION IN TRANSGENIC PLANTS
Rapid progress in plant biotechnology will result in contributions to vaccine production from agriculture. Recent results suggest that genetically engineered plants may be used to produce vaccines against human diseases (Arntzen et al., 1994; Haq et al., 1995; Mason et al., 1992). A concern is that plant-produced vaccines will require purification free of alkaloids and other toxic plant materials. Two approaches may be used to resolve this. One is to use plant vectors that incorporate the protein into a tissue that lacks toxic alkyloids, for example, bananas or soybeans (Arntzen et al., 1994; McCabe et al., 1988). Another approach would be to use the same technology used to incorporate the vaccine construct into plants to remove the genes for the toxic substances genetically (Moffat, 1995; Morris et al., 1994; Personal communication, M. B. Hein, Scripps Research Institute, La Jolla, Calif., 1995).
If the potential concerns about plant alkaloids can be resolved quickly, production of vaccines in plants will be a significant improvement over current methods. Such vaccines might be cheaper than those now available because plants are easier to grow in large quantities than are cultured animal, insect, bacterial, or yeast cells now used to make most vaccines (Moffat, 1995; Morris et al., 1994).
The details of inserting antigenic constructs into plants as expression vectors have been worked out, as well as systems for the use of baculoviruses, bacteria, or yeast to produce products. The discovery that plant tumors (called galls) were produced by a bacterium called Agrobacterium tumefasciens is responsible for this rapid progress in developing plants as expression vectors (Horsch et al., 1988; Jefferson et al., 1987). In addition, new technologies for inserting genes into plants have been developed to overcome limited host-range specificity of Agrobacterium vectors (McCabe et al., 1988; Owens and Cress, 1985; Pedersen et al., 1983).
The plant biotechnology field has been working out the requirements for inserting and expressing genes in plants since the late 1970s. The recent break-through has been spurred by demonstration of efficient production of heterologous proteins in plants (Gasser and Fraley, 1989; Hiatt et al., 1989). This may produce a potential added benefit for worried tobacco farmers because the easiest plant vector to transform is the tobacco plant. As people progressively stop smoking, the new technology might provide important replacement crops for tobacco growers if the toxic alkaloids in tobacco leaf can be cloned out easily.
New biotechnology companies are well positioned to profit from the anticipated economic benefits because all that would be needed to scale-up produc-
tion of plant-made protein to the amounts needed for a commercial vaccine is to add acreage. Cheaper vaccines that are available in large quantities make vaccination of an entire expeditionary force feasible in contingency situations. It is anticipated that use of transgenic plants as expression vectors might provide all the protein needed during one growing season, while conventional vaccine production techniques would require much more time. These vaccines potentially will be safer because production in plants virtually eliminates the problem of contamination with animal viruses.
Another line of research is the development of "edible vaccines" (Arntzen et al., 1994). These vaccines are designed with the expectation that no purification step is required, and the vaccine components are expressed in final form in the interstices of the plant. The use of a CTB or LT vaccine carrier would target the antigen construct to be taken up and processed in mucosal lymphatic tissues, where the biological effects of the carrier should program the response for peripheral and mucosal immunity.
ANTIBODY NEEDED FOR PASSIVE PROTECTION
Passive transfer of immunity by serum injection has become a routine medical practice after its original description over 100 years ago (Cohn et al., 1946; von Behring and Kitasato, 1890). Passive immunoprophylaxis by parenteral infusion (for "peripheral" protective immunity) using IgG antibodies also has become very common (Cryz, 1991). In a military scenario, the benefit of passive protection by antibodies raised by conventional means would appear to be limited by cost and availability of sufficient quantities. Except under very unusual circumstances, it would be impractical to produce industrial quantities of prophylactic antibodies in humans prospectively (DasGupta, 1993; Franz et al., 1993; Metzger and Lewis, 1979). Antibodies prepared in other species (such as horse, cow, or pig) may be given without risk for passive oral protection (Losonsky et al., 1985), but the risk of serum sickness (multiple organ damage caused by host antibodies forming complexes with the "foreign" animal proteins) (or other adverse reactions) would preclude any sequential parenteral use (Renegar and Small, 1994; Tsunemitsu et al., 1989).
Solutions for Quantities of Antibody Needed
Once again, use of transgenic plants to express recombinant human antibodies should provide a solution (Hiatt et al., 1989; Ma et al., 1995). This final section will describe how high-affinity antigen-combining sites of immunoglobulins can be obtained rapidly using recombinant DNA technology, genetically fused with human light-and heavy-chain constant regions, and expressed as functional antibodies in plants.
Preselection of "High Affinity-Ig" B-Cells for Complementarity-Determining Region Isolation and Expression in Transgenic Plants
After in vivo antigenic stimulation in mammals, B-cells undergo molecular and cellular changes within germinal centers of lymphoid follicles. An important change that B-cells undergo is "affinity maturation," whereby B-cells expressing specific antibodies for a given antigen undergo progressive changes that result in selection of cells capable of making antibodies with higher binding affinity (Eisen and Siskind, 1964; Gearhart, 1993).
Affinity maturation occurs through a process of somatic hypermutation, whereby single base pairs are substituted in the DNA sequence of immunoglobulin variable regions, specifically in the CDRs. The specific base substitutions occur at "hot" spots in the CDR gene sequences, which could serve as markers for immunoglobulins with increased antigen affinity (Betz et al., 1993). The DNA of such cells may be rescued selectively for development of high-affinity antibody.
After it is isolated from B-cells in tissue sections, DNA from immunoglobulin V-region genes can be cloned into filamentous phage display vectors (Winter et al., 1994). This makes it possible to study the effect of individual nucleotide changes on the affinity of antibodies from B-cells isolated at specific sites within the reactive lymphoid tissue (Bakker et al., 1995). Genomic DNA from B-cells isolated from rabbit appendix germinal centers has been cloned and sequenced, and single-point mutations in germline V-gene sequences that occurred as part of affinity maturation could be followed (Weinstein et al., 1994a, b). This procedure currently is being used to isolate antigen-specific CDRs after deliberate immunization with selected antigens including the F-1 antigen of Yersinia pestis.
Polymerase chain reaction (PCR) amplification and cloning of immunoglobulin VH-regions permit rapid isolation of CDRs encoding antigen-binding sites (Jacob et al., 1991, 1993; Marks et al., 1991; Weinstein et al., 1994a, b). Germinal centers are oligoclonal populations of dividing B-cells found in characteristic anatomic locations in lymphoid tissues. Sampling cells from the different regions of a germinal center (identified as specific for nominal antigen by labeled antigen-binding to surface antibody on B-cells in frozen tissue sections) will reduce the number of CDR clones that must be screened in order to select the ones encoding high-affinity antigen binding (Unpublished data, A. O. Anderson and P. D. Weinstein, U.S. Army Medical Research Institute of Infectious Diseases, Fort Detrick, Md., 1995). This is an improvement over random selection from large V-gene libraries (Marks et al., 1991). In addition, there are methods of converting low-affinity binding sites to high-affinity by directed mutagenesis (Betz et al., 1993; Griffiths et al., 1994; Wong et al., 1995).
Making Recombinant Human Antibodies in Transgenic Plants
Expression of the selected CDRs on conserved framework structures of human immunoglobulins in transgenic plants should allow production of large amounts of protective antibodies (Ma et al., 1995). The example that proved that functional antibodies could be made in plants solved many potential problems. The objective was to produce secretory IgA that was specific for a streptococcal antigen involved in dental caries.
It was a good test of feasibility because there are critical genetic and immunoglobulin structural requirements for IgA molecules to function in vivo. It is not sufficient to be successful in cloning a functional VH-VL CDR complex. One also must account for the fact that mammalian immunoglobulin genes are divided among different chromosomes, and the molecule is assembled posttranslationally. The assembled IgA heavy-chain structure must not interfere with dimerization because failure to dimerize interferes with secretion by epithelial cells, antibody function, or both. The dimer must link up with the J-chain so the complex can bind to the extracellular polymeric immunoglobulin receptor, which is involved in translocating the antibody across epithelium. What Ma and his colleagues (1995) accomplished truly was amazing. They focused on conferring the ability on transgenic plants to assemble secretory IgA molecules as though they already had been secreted by the epithelial cell. They cleverly used the Mendelian genetics of their transgenic plants to divide up the task of inserting a lot of mammalian genetic material. The group made four separate transgenic plant lines, each of which received a gene for only a part of the secretory IgA. After they were certain that no genes were altered by the process, they crossed the plants the way Greggor Mendel crossed his peas. The sequence of matings is shown in Figure 22-7. Plants have a useful idiosyncrasy. The progeny efficiently produced the anticipated antibody fully assembled in functional form and in ample quantities. Where these antibodies accumulated in the transgenic plants suggested that purification of the product free of plant material should be relatively easy.
Research directed toward application of this technology will be tremendously valuable because present methods of antibody production require time-consuming immunization procedures in large animals and result in a product of limited sequential use in humans. Furthermore, the use of plants expressing a transgene that produces human antibodies would permit the quantity of antibody attainable to be limited only by the size of the crop or number of animals bred.
The sequence of development would depend upon the ease by which one rapidly could select high-affinity CDRs that bind and neutralize a pathogen, construct recombinant human antibodies of IgA or IgG isotypes using the selected CDR, and determine the affinity of the antibody constructs selected for expression in transgenic plant crops (Bakker et al., 1995; Winter et al., 1994). Depending upon the ease of isolation and purification of the product, such protective antibodies could be quickly prepared against disease threats that are en-
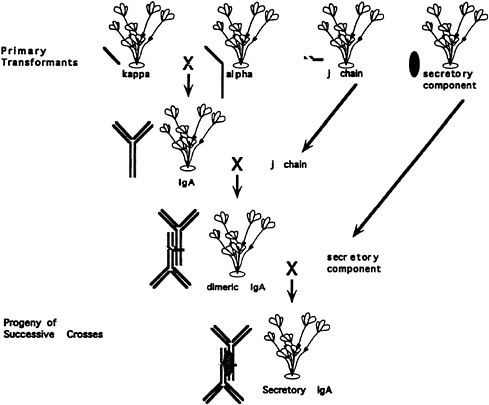
FIGURE 22-7 This schematic diagram illustrates the process for generating transgenic plants expressing assembled immunoglobulins. Individual plants were first transformed with one of four genes encoding kappa-light chain, alpha-heavy chain, J-chain, or secretory component. Plants expressing the kappa or alpha chain were first crossed to generate progeny containing assembled IgA. Successive crosses of IgA producing plants with J-chain-containing plants yielded progeny with assembled dimeric IgA. These plants were then crossed with plants expressing a truncated form of the polyimmunoglobulin receptor (secretory component) to generate progeny containing secretory IgA. SOURCE: M. Hein (Scripps Research Institute, La Jolla, Calif., 1995), used with permission.
demic worldwide. The shortened time interval and capacity for rapid scale-up should make these products inexpensive to produce in large quantities. Incorporating enterically coated recombinant antibodies in MREs could be used to bring about a significant reduction in morbidity or disease-related decrements in performance in soldiers deployed anywhere in the world.
AUTHOR'S CONCLUSIONS AND RECOMMENDATIONS
The study of mucosal immunology has contributed to the understanding of concepts and issues of host resistance that is needed for protection of the gastro-
intestinal tract and other mucosa that provide for optimal nutrition. An added benefit of support for this discipline has been a better understanding of how vaccines can be used more effectively.
The new technologies of vaccine microencapsulation, use of the nontoxic B-subunit of cholera or E. coli enterotoxin carriers for recombinant vaccine constructs, and incorporation of recombinant vaccine constructs into edible plants have become feasible, and this technology can be studied for acceptability for delivery of vaccines to military personnel.
The above technologies overcome cross-regulation and induce immune responses with circulating IgG, as well as secretory IgA. This should provide complete active prophylaxis without need for injections or time-consuming vaccine schedules.
Expression of functional recombinant human antibodies in plants has been demonstrated. This makes feasible the production of industrial quantities of protective recombinant antibody for use in passive immunoprophylaxis. Such antibody, when protected from stomach acid by enteric coatings or encapsulation, could be incorporated into meals for protection of troops from endemic causes of dysentery.
The following recommendations can be made:
-
Institute a rational policy for immunizing military recruits against threat agents and selected geographic pathogens.
-
Establish a Science and Technology Objective for adaptation of militarily relevant vaccines for oral administration through application of the above technology.
-
To achieve this goal, encourage cooperative collaboration between the U.S. Army Research Institute of Environmental Medicine; U.S. Army Natick Research, Development and Engineering Center; and U.S. Army Medical Research Institute of Infectious Diseases.
-
Increase funding support for technology development that may not be linked to specific threat agents.
REFERENCES
Abbas, A.K., K.M. Murphy, and A. Sher 1996 Functional diversity of helper T lymphocytes. Nature 383:787–793.
Abernethy, N.J., J.B. Hay, W.G. Kimpton, E. Washington, and R.N.P. Cahill 1991 Lymphocyte subset-specific and tissue-specific lymphocyte-endothelial cell recognition mechanisms independently direct the recirculation of lymphocytes from blood to lymph in sheep. Immunology 72:239–245.
Agace, W., S. Hedges, U. Andersson, J. Andersson, M. Ceska, and C. Svanborg 1993 Selective cytokine production by epithelial cells following exposure to Escherichia coli. Infect. Immun. 61:602–609.
Altman, J.D., P.A.H. Moss, P.J.R. Goulder, D.H. Barouch, M.G. McHeyzer-Williams, J.I. Bell, A.J. McMichael, and M.M. Davis 1996 Phenotypic analysis of antigen-specific T lymphocytes. Science 274:94–96.
Anderson, A.O. 1985 Multiple effects of immunological adjuvants on lymphatic microenvironments. Int. J. Immunother. 1:185–195.
1990 Structure and organization of the lymphatic system. Pp. 14–45 in Immunophysiology. The Role of Cells and Cytokines in Immunity and Inflammation, J.J. Oppenheim and E. Shevach, eds. New York: Oxford University Press.
Anderson, A.O., and J.A. Reynolds 1979 Adjuvant effects of the lipid amine CP-20, 961 (on lymphoid cell traffic and antiviral immunity). J. Reticuloendothel. Soc. 350:33–41.
Anderson, A.O., and D.H. Rubin 1985 Effect of avridine on enteric antigen uptake and mucosal immunity to reovirus. Adv. Exp. Med. Biol. 186:579–590.
Anderson, A.O., and S. Shaw 1993 T-cell adhesion to endothelium: The FRC conduit system and other anatomic and molecular features which facilitate the adhesion cascade in lymph node. Sem. Immunol. 5:271–282.
1996 Lymphocyte trafficking. Pp. 39–49 in Clinical Immunology, Principles and Practice, R.R. Rich, T.A. Fleisher, B.D. Schwartz, W.T. Shearer, and W. Strober, eds. St. Louis, Mo.: Mosby-Year Book, Inc.
Anderson, A.O., T.T. McDonald, and D.H. Rubin 1985 Effect of orally administered avridine on enteric antigen uptake and mucosal immunity. Int. J. Immunother. 1:107–115.
Anderson, A.O., L.F. Snyder, M.L.M. Pitt, and O.L. Wood 1987a Mucosal priming alters pathogenesis of Rift Valley fever. Adv. Exp. Med. Biol. 237:727–733.
Anderson, A.O., O.L. Wood, A.D. King, and E.H. Stephenson 1987b Studies on anti-viral mucosal immunity using the lipoidal amine adjuvant avridine. Adv. Exp. Med. Biol. 216B:1781–1790.
Anderson, G.W., Jr., J.O. Lee, A.O. Anderson, N. Powell, J.A. Mangiafico, and G. Meadors 1991 Efficacy of a Rift Valley fever virus vaccine against an aerosol infection in rats. Vaccine 9:710–714.
Ariizumi, K., T. Kitajima, O.R. Bergstresser, and A. Takashima 1995 Interleukin-1ß converting enzyme in murine Langerhans cells and epidermal-derived dendritic cell lines. Eur. J. Immunol. 25:2137–2141.
Arntzen, C.J., H.S Mason, J. Shi, T.A. Haq, M.K. Estes, and J.D. Clements 1994 Production of candidate oral vaccines in edible tissues of transgenic plants. Pp. 339–344 in Vaccines '94, F. Brown, R.M. Chanock, H.S. Ginsberg, and R.A. Lerner, eds. Cold Spring Harbor, New York: Cold Spring Harbor Laboratory.
Assenmacher, M., J. Schmitz, and A. Radbruch 1994 Flow cytometric determination of cytokines in activated murine Th lymphocytes. Expression of Il-10 in IFN-gamma and in IL-4 expressing cells. Eur. J. Immunol. 24:1097–1101.
Bakker, R., E. Lasonder, and N.A. Bos 1995 Measurement of affinity in serum samples of antigen-free, germ-free and conventional mice after hyperimmunization with 2,4-dinitrophenyl keyhole limpet hemocyanin, using surface plasmon resonance. Eur. J. Immunol. 25:1680–1686.
Beisel, W.R. 1994 Endocrine and immune system responses to stress. Pp. 177–207 in Food Components to Enhance Performance, An Evaluation of Potential Performance-Enhancing Food Components for Operational Rations, B.M. Marriott, ed. A report of the Committee on Military Nutrition Research, Food and Nutrition Board, Institute of Medicine. Washington, D.C.: National Academy Press.
Bendelac, A. 1995 CD1: Presenting unusual antigens to unusual T lymphocytes. Science 269:185–192.
Berlin, C., E.L. Berg, M.J. Briskin, D.P. Andrew, P.J. Kilshaw, B. Holzmann, I.L. Weissman, A. Hamann, and E.C. Butcher 1993 a4ß7 integrin mediates lymphocyte binding to the mucosal vascular addressin MAdCAM-1. Cell 74:185–195.
Betz, A.G., M.S. Neuberger, and C. Milstein 1993 Discriminating intrinsic and antigen-selected mutational hotspots in immunoglobulin V genes . Immunol. Today 14:405–411.
Brandtzaeg, P. 1994 Distribution and characteristics of mucosal immunoglobulin-producing cells. Pp. 251–262 in Handbook of Mucosal Immunology, P.L. Ogra, J. Mestecky, M.E. Lamm, W. Strober, J.R. McGhee, and J. Bienenstock, eds. Boston: Academic Press, Inc.
Brown, J.L., J.W. Dominik, and R.L. Morrissey 1981 Respiratory infectivity of a recently isolated Egyptian strain of Rift Valley fever virus. Infect. Immun. 33:848–853.
Burnette, W. Neal 1994 AB5 ADP-ribosylating toxins: Comparative anatomy and physiology. Structure 2:151–158.
Butcher, E.C. 1991 Leukocyte endothelial cell recognition: Three or more steps to specificity and diversity. Cell 67:1033–1036.
Butcher, E.C., and L.J. Picker 1996 Lymphocyte homing and homeostasis. Science 272:60–66.
Cahill, R.N.P., D.C. Poskitt, H. Frost, and Z. Trnka 1977 Two distinct pools of recirculating T lymphocytes: Migratory characteristics of nodal and intestinal T lymphocytes . J. Exp. Med. 145:420–428.
Carayannopoulos, L., and J.D. Capra 1993 Immunoglobulins. Structure and function. Pp. 293–314 in Fundamental Immunology, 3d ed., W.E. Paul, ed. New York: Raven Press, Ltd.
Cebra, J.J., P.J. Gearhart, R. Kamat, S.M. Robertson, and J. Tseng 1977 Origin and differentiation of lymphocytes involved in the secretory IgA responses. Cold Spring Harbor Symp. Quant. Biol. 41:201–215.
Cepek, K.L., S.K. Shaw, C.M. Parker, G.J. Russell, J.S. Morrow, D.L. Rimm, and M.B. Brenner 1994 Adhesion between epithelial cells and T lymphocytes mediated by E-cadherin and the aEß7 integrin. Nature 372:190–193.
Chase, M.W. 1946 Inhibition of experimental drug allergy by prior feeding of the sensitivity agent. Proc. Soc. Exp. Biol. Med. 61:257–259.
Chen, Y., J. Inobe, R. Marks, P. Gonnella, V.K. Kuchroo, and H.L. Weiner 1995 Peripheral deletion of antigen-reactive T-cells in oral tolerance. Nature 376:177–189.
Clements, J.D., N.M. Hartzog, and F.L. Lyon 1988 Adjuvant activity of Escherichia coli heat-labile enterotoxin and effect on the induction of oral tolerance in mice to unrelated protein antigens. Vaccine 6:269–277.
Cohn, E.J., L.E. Strong, and L.W. Hughes, Jr. 1946 Preparation and properties of serum and plasma proteins. 4. A system for the separation of fractions of proteins and lipoprotein components of biological tissues and fluids. J. Am. Chem. Soc. 8:459.
Cryz, S.J., Jr., ed. 1991 Vaccines and Immunotherapy. New York: Pergamon Press.
Cuff, C.E., C.K. Cebra, D.H. Rubin, and J.J. Cebra 1993 Developmental relationship between cytotoxic a/ß T-cell receptor-positive intraepithelial lymphocytes and Peyer's patch lymphocytes. Eur. J. Immunol. 23:1333–1339.
Dallas, W.S., and S. Falkow 1980 Amino acid sequence homology between cholera toxin and Escherichia coli heat-labile toxin. Nature (London) 288:499–501.
DasGupta, B.R., ed. 1993 Botulinum and Tetanus Neurotoxins, Neurotransmission and Biomedical Aspects. New York: Plenum Press.
Dertzbaugh, M.T., and C.O. Elson 1991 Cholera toxin as a mucosal adjuvant. Pp. 119–131 in Topics in Vaccine Adjuvant Research, D.R. Spriggs and W.C. Koff, eds. Boca Raton, Fla.: CRC Press.
1993a Reduction in oral immunogenicity of cholera toxin B subunit by N-terminal peptide addition. Infect. Immun. 61(2):384–390.
1993b Comparative effectiveness of the cholera toxin B subunit and alkaline phosphatase as carriers for oral vaccines. Infect. Immun. 61(1):48–55.
Dertzbaugh, M.T., and C.J. Van Meter 1996 The mucosal immunogenicity of cholera toxin B-subunit is dependent on its ability to bind to ganglioside. Unpublished manuscript. Fort Detrick, Md.: U.S. Army Medical Research Institute of Infectious Diseases.
Dickinson, B.L., and J.D. Clements 1995 Dissociation of Escherichia coli heat-labile enterotoxin adjuvanticity from ADP-ribosyltransferase activity. Infect. Immun. 63(5):1617–1623.
Dune, C.P. 1994 Biochemical strategies for ration design: Concerns of bioavailability. Pp. 93–109 in Food Components to Enhance Performance, An Evaluation of Potential Performance-Enhancing Food Components for Operational Rations, B.M. Marriott, ed. A report of the Committee on Military Nutrition Research, Food and Nutrition Board, Institute of Medicine . Washington, D.C.: National Academy Press.
Ebnet, K., E.P. Kaldjian, A.O. Anderson, and S. Shaw 1996 Orchestrated information transfer underlying leukocyte endothelial interactions. Ann. Rev. Immunol. 14:155–177
Eckmann, L., M.F. Kagnoff, and J. Fierer 1993 Epithelial cells secrete the chemokine interleukin-8 in response to bacterial entry. Infect. Immun. 61:4569–4574.
Eisen, H.N., and G.W. Siskind 1964 Variations in affinities of antibodies during the immune response. Biochemistry 3:996–1008.
Eldridge, J.H., C.J. Hammond, J.A. Meulbroek, J.K. Staas, R.M. Gilley, and T.R. Tice 1990 Controlled vaccine release in the gut-associated lymphoid tissues. I. Orally administered biodegradable microspheres target the Peyer's patches. J. Controlled Release 11:205–214
Elson, C.O., and M.T. Dertzbaugh 1994 Mucosal adjuvants. Pp. 391–401 in Handbook of Mucosal Immunology, P.L. Ogra, J. Mestecky, M.E. Lamm, W. Strober, J.R. McGhee, and J. Bienenstock, eds. Boston: Academic Press, Inc.
Elson, C.O., and W. Ealding 1984a Cholera toxin feeding did not induce oral tolerance in mice and abrogated oral tolerance to an unrelated protein antigen. J. Immunol. 133:2892–2897.
1984b Generalized systemic and mucosal immunity in mice after mucosal stimulation with cholera toxin. J. Immunol. 132:2736–2741.
Elson, C.O., S.P. Holland, M.T. Dertzbaugh, C.F. Cuff, and A.O. Anderson 1995 Morphologic and functional alterations of mucosal T-cells by cholera toxin and its B subunit. J. Immunol. 154:1032–1040.
Finkelman, F.D. 1995 Relationships among antigen presentation, cytokines, immune deviation, and autoimmune disease. J. Exp. Med. 182:279–282.
Franz, D.R., L.M. Pitt, M.A. Clayton, J.A. Hanes, and K.J. Rose 1993 Efficacy of prophylactic and therapeutic administration of antitoxin for inhalation botulism. Pp. 473–476 in Botulinum and Tetanus Neurotoxins, B.R. DasGupta, ed. New York: Plenum Press.
Freund, J. 1956 The mode of action of immunological adjuvants. Adv. Tuberc. Res. 7:130–148.
Fuhrman, J.A., and J.J. Cebra 1981 Special features of the priming process for a secretory IgA response. J. Exp. Med. 153:534–544.
Gangarosa, E.J., W.R. Beisel, C. Benyajati, H. Sprinz, and P. Piyaratn 1960 The nature of the gastrointestinal lesion in Asiatic cholera and its relation to pathogenesis: A biopsy study. Am. J. Trop. Med. Hyg. 9:125–135.
Gasser, C.S., and R.T. Fraley 1989 Genetically engineering plants for crop improvement. Science 244:1293–1299.
Gearhart, P.J. 1993 Somatic mutation and affinity maturation. Pp. 865–885 in Fundamental Immunology, 3rd ed., W.E. Paul, ed. New York: Raven Press, Ltd.
Gelfanov, V., V. Gelfanova, Y-G. Lai, and N-S. Liao 1996 Activated ab-CD8+, but not TCR-ab+murine intestinal intraepithelial lymphocytes can mediate perforin-based cytotoxicity, whereas both subsets are active in FAS-based cytotoxicity. J. Immunol. 156:35–41.
Gombotz, W.R., and D.K. Pettit 1995 Biodegradable polymers for protein and peptide drug delivery. Bioconjugate Chem. 6:332–351.
Gretz, J.E., E.P. Kaldjian, A.O. Anderson, and S. Shaw 1996 Sophisticated strategies for information encounter in the lymph node: The reticular network as a conduit of soluble information and a highway for cell traffic. J. Immunol. 157:495–499.
Griffiths, A.D., S.C. Williams, O. Hartley, I.M. Tomlinson, P. Waterhouse, W.L. Crosby, R.E. Kontermann, P.T. Jones, N.M. Low, T.J. Allison, T.D. Prospero, H.R. Hoogenboom, A. Nissim, J.P.L. Cox, J.L. Harrison, M. Zaccolo, E. Gherardi, and G. Winter 1994 Isolation of high affinity human antibodies directly from large synthetic repertoires. EMBO J. 13:3245–3260.
Hajishengallis, G., S.K. Hollingshead, T. Koga, and M.W. Russell 1995 Mucosal immunization with a bacterial protein antigen genetically coupled to cholera toxin A2/B subunits. J. Immunol. 154:4322–4332.
Hamann, A., D.P. Andrew, D. Jablonski-Westrich, B. Holzmann, and E.C. Butcher 1994 Role of a4-integrins in lymphocyte homing to mucosal tissues in vivo. J. Immunol. 152:3282–3292.
Hamilton, S.R., J.H. Yardley, and G.D. Brown 1979 Suppression of local intestinal immunoglobulin A immune response to cholera toxin by subcutaneous administration of cholera toxoids. Infect. Immun. 24:422–426.
Haq, T.A., H.S. Mason, J.D. Clements, and C.J. Arntzen 1995 Oral immunization with a recombinant bacterial antigen produced in transgenic plants. Science 268:714–716.
Hart, M.K., W. Pratt, F. Panelo, K. Stephan, L. Aikens, R. Tammariello, and M. Dertzbaugh 1995 Dissociation between serum neutralizing antibodies and protection from airborne VEE virus in immunized mice. Clin Immunol. Immunopathol. 76:S97
Hiatt, A., R. Cafferkey, and K. Bowdish 1989 Production of antibodies in transgenic plants. Nature 342:76–78.
Hilbert, D.M., K.L. Holmes, A.O. Anderson, and S. Rudikoff 1994 Long-term lymphoid reconstitution of SCID mice suggests self renewing B-and T-cell populations in peripheral and mucosal tissues. Transplantation 58:466–475.
Hiroi, T., K. Fujihashi, J.R. McGhee, and H. Kiyono 1995 Polarized Th2 cytokine expression by both mucosal gs and ab T-cells. Eur. J. Immunol. 25:2743–2751.
Holmgren, J. 1981 Actions of cholera toxin and the prevention and treatment of cholera. Nature 292:413–417.
Holmgren, J., N. Lycke, and C. Czerkinsky 1993 Cholera toxin and cholera B subunit as oral-mucosal adjuvant and antigen vector systems. Vaccine 11(12):1179–1184.
Horsch, R.B., J. Fry, N. Hoffmann, J. Neidermeyer, S.G. Rogers, and R.T. Fraley 1988 Leaf disc transformation. In Plant Molecular Biology Manual A5:1–9, S.B. Gelvin, R.A. Schilperoort, and D.P.S. Verma, eds. Dordrecht, Belgium: Kluwer Academic Publishers.
Husband, A.J., and J.L. Gowans 1978 The origin and antigen-dependent distribution of IgA-containing cells in the intestine. J. Exp. Med. 148:1146–1160.
Hyams, K.C., A.L. Bourgeois, B.R. Merrell, P. Rozmajzl, J. Escamilla, S.A. Thronton, G.M. Wasserman, A. Burke, P. Echeverria, K.Y. Green, A.Z. Kapikian, and J.N. Woody 1991 Diarrheal disease during Operation Desert Shield. N. Eng. J. Med. 325:1423–1428.
Hyams, K.C., J.D. Malone, A.Z. Kapikian, M.K. Estes, X. Jiang, A.L. Bourgeois, S. Paparello, R.E. Hawkins, and K.Y. Green 1993 Norwalk virus infection among Desert Storm troops. J. Infect. Dis. 167:986–987.
IOM (Institute of Medicine) 1994 Food Components to Enhance Performance, An Evaluation of Potential Performance-Enhancing Food Components for Operational Rations, B.M. Marriott, ed. A report of the Committee on Military Nutrition Research, Food and Nutrition Board. Washington, D.C.: National Academy Press.
Jacob, J., G. Kelsoe, K. Rajewsky, and U. Weiss 1991 Intraclonal generation of antibody mutants in germinal centers. Nature 354:389–392.
Jacob, J., J. Przylepa, C. Miller, and G. Kelsoe 1993 In situ studies of the primary immune response to (4-hydroxy-3-nitrophenyl)acetyl. III. The kinetics of V region mutation and selection in germinal center B-cells. J. Exp. Med. 178:1293–1307.
Jahrling, P.B., and E.H. Stephenson 1984 Protective efficacies of live attenuated and formaldehyde-inactivated Venezuelan equine encephalitis virus vaccines against aerosol challenge in hamsters. J. Clin. Microbiol. 19:429–431.
Jefferson, R.A., T.A. Kavanagh, and M.W. Bevan 1987 GUS fusions: ß-glucuronidase as a sensitive and versatile gene fusion marker in higher plants. EMBO J. 6(13):3901–3907.
Kagnoff, M.F. 1996 Mucosal immunology: New frontiers. Immunol. Today 17:57–59.
Kang, K., M. Kubin, K.D. Cooper, S.R. Lessin, G. Trinchieri, and A.H. Rook 1996 IL-12 synthesis by human Langerhans cells. J. Immunol. 156:1402–1407.
Kantele, A., J.M. Kantele, E. Savilahti, M. Westerholm, H. Arvilommi, A. Lazarovits, E.C. Butcher, and P.H. Makela 1997 Oral, but not parenteral, typhoid vaccination induces circulating antibody-secreting cells that all bear homing receptors directing them to the gut. J. Immunol. 158:574–579.
Karecla, P.I., S.J. Bowden, S.J. Green, and P.J. Kilshaw 1995 Recognition of E-cadherin on epithelial cells by the mucosal T-cell integrin aM290ß7 (aEß7). Eur. J. Immunol. 25:852–856.
Ke, Y., and J.A. Kapp 1996 Oral antigen inhibits priming of CD8+ CTL, CD4+ T-cells, and antibody responses while activating CD8+ suppressor T-cells. J. Immunol. 156:916–921.
Kimpton, W.G., E.A. Washington, and R.N.P. Cahill 1989 Recirculation of lymphocyte subsets (CD5+, CD4+, CD8+, SBU-T19+ and B-cells) through gut and peripheral lymph nodes. Immunology 66:69–75.
Koster, F.T., and N.F. Pierce 1983 Parenteral immunization causes antigen-specific cell-mediated suppression of an intestinal IgA response. J. Immunol. 131:115–119.
Kotloff, D.B., and J.J. Cebra 1988 Effect of TH-lines and clones on the growth and differentiation of B-cell clones in microculture. Mol. Immunol. 25:147–155.
Kraal, G., K. Schornagel, P.R. Streeter, B. Holzmann, and E.C. Butcher 1995 Expression of the mucosal vascular addressin, MAdCAM-1, on sinus-lining cells in the spleen. Am. J. Pathol. 147:763–771.
Kroemer, G., E. Cuende, and C-A. Martinez 1993 Compartmentalization of the peripheral immune system. Adv. Immunol. 53:157–216.
Langer, R. 1990 New methods of drug delivery. Science 249:1527–1533.
Lebman, D.A., and R.L. Coffman 1994 Cytokines in the mucosal immune system. Pp. 243–250 in Handbook of Mucosal Immunology, P.L. Ogra, J. Mestecky, M.E. Lamm, W. Strober, J.R. McGhee, and J. Bienenstock, eds. Boston: Academic Press, Inc.
Lehner, T., L.A. Bergmeier, C. Panagiotidi, L. Tao, R. Brookes, L.S. Klavinskis, P. Walker, R.G. Ward, L. Hussain, A.J.H. Gearing, and S.E. Adams 1992 Induction of mucosal and systemic immunity to a recombinant simian immunodeficiency viral protein. Science 258:1365–1369.
London, S.D., D.H. Rubin, and J.J. Cebra 1987 Gut mucosal immunization with reovirus serotype l/L stimulates virus-specific cytotoxic T-cell precursors as well as IgA memory cells in Peyer's patches. J. Exp. Med. 165:830–847.
Losonsky, G.A., J.P. Johnson, J.A. Winkelstein, and R.H. Yolken 1985 Oral administration of human serum immunoglobulin in immunodeficient patients with viral gastroenterities. J. Clin. Invest. 76:2362–2367.
Lycke, N., T. Tsuji, and J. Holmgren 1992 The adjuvant effect of Vibrio cholerae and Escherichia coli heat-labile enterotoxins is linked to their ADP-ribosyltransferase activity. Eur. J. Immunol. 22:2277–2281.
Lyons, A.B., and C.R. Parish 1995 Are murine marginal-zone macrophages the splenic white pulp analog of high endothelial venules? Eur. J. Immunol. 25:3165–3172.
Ma, J. K.-C., A. Hiatt, M. Hein, N.D. Vine, F. Wang, P. Stabila, C. van Dolleweerd, K. Mostov, and T. Lehner 1995 Generation and assembly of secretory antibodies in plants. Science 268:716–719.
MacDonald, T.T. 1982 Enhancement and suppression of Peyer's patch immune response by systemic priming. Clin. Exp. Immunol. 49:441–448.
Mackay, C.R. 1991 Skin-seeking memory T-cells. Nature 349:737–738.
Mackay, C.R., W.L. Marston, L. Dudler, O. Spertini, T.F. Tedder, and W.R. Hein 1992 Tissue-specific migration pathways by phenotypically distinct subpopulations of memory T-cells. Eur. J. Immunol. 22:887–895.
Marks, J.D., H.R. Hoogenboom, T.P. Bonnert, J. McCafferty, A.D. Griffiths, and G. Winter 1991 By-passing immunization. Human antibodies from V-gene libraries displayed on phage. J. Mol. Biol. 222:16007–16010.
Mason, H.S., D.M. Lam, and C.J. Arntzen 1992 Expression of hepatitis B surface antigen in transgenic plants. Proc. Natl. Acad. Sci. USA 89:11745–11749.
Mattingly, J.A., and B.H. Waksman 1978 Immunologic suppression after oral administration of antigen. I. Specific suppressor cells formed in rat Peyer's patches after oral administration of sheep erythrocytes and their systemic migration. J. Immunol. 121:1878–1883.
Matzinger, P. 1994 Tolerance, danger, and the extended family. Annu. Rev. Immunol. 12:991–1045.
Mazanec, M.B., C.S. Kaetzel, M.E. Lamm, D. Fletcher, and J.G. Nedrud 1992 Intracellular neutralization of virus by immunoglobulin A antibodies. Proc. Natl. Acad. Sci. USA 89:6901–6905.
Mazanec, M.B., J.G. Nedrud, C.S. Kaetzel, and M.E. Lamm 1993 A three-tiered view of the role of IgA in mucosal defense. Immunol. Today 14:430–435.
McCabe, D.E., W.F. Swain, B.J. Martinell, and P. Christou 1988 Stable transformation of soybean (Glycine max) by particle acceleration. Bio/Tech. 6:923–926.
McCormick, B.A., S.P. Colgan, C. Delp-Archer, S.I. Miller, and J.L. Madara 1993 Salmonella typhimurium attachment to human intestinal epithelial monolayers: Transcellular signaling to subepithelial neutrophils . J. Cell Biol. 123:895–907.
McGhee, J.R., J. Mestecky, M.T. Dertzbaugh, J.H. Eldridge, M. Hirasawa, and H. Kiyono 1992 The mucosal immune system: From fundamental concepts to vaccine development. Vaccine 10:75–88.
Metzger, J.F., and G.E. Lewis, Jr. 1979 Human-derived immune globulins for the treatment of botulism. Rev. Infect. Dis. 1:689–692.
Michalek, S.M., J.H. Eldridge, R. Curtiss III, and K.L. Rosenthal 1994 Antigen delivery systems: New approaches to mucosal immunization. Pp. 373–380 in Handbook of Mucosal Immunology, P.L. Ogra, J. Mestecky, M.E. Lamm, W. Strober, J.R. McGhee, and J. Bienenstock, eds. Boston: Academic Press, Inc.
Michalek, S.M., N.K. Childers, and M.T. Dertzbaugh 1995 Vaccination strategies for mucosal pathogens. Pp. 269–301 in Virulence Mechanisms of Bacterial Pathogens, 2nd ed., J.A. Roth, Ed. Washington, D.C.: American Society for Microbiology.
Mobley, J.A. 1995 Biological warfare in the twentieth century: Lessons from the past, challenges for the future. Mil. Med. 160:547–553.
Moffat, A.S. 1995 Exploring transgenic plants as a new vaccine source. Science 268:658–660.
Morikawa, Y., K. Tohya, H. Ishida, N. Matsuura, and K. Kakudo 1995 Different migration patterns of antigen-presenting cells correlate with Th1/Th2-type responses in mice. Immunology 85:575–581.
Morris, W., M.C. Steinhoff, and P.K. Russell 1994 Potential of polymer microencapsulation technology for vaccine innovation. Vaccine 12:5–11.
Mosmann, T.R., and S. Sad 1996 The expanding universe of T-cell subsets: Th1, Th2 and more. Immunol. Today 17:138–146.
Mowat, A.M. 1994 Oral tolerance and regulation of immunity to dietary antigens. Pp. 185–201 in Handbook of Mucosal Immunology, P.L. Ogra, J. Mestecky, M.E. Lamm, W. Strober, J.R. McGhee, and J. Bienenstock, eds. Boston: Academic Press, Inc.
Neutra, M.R., and J-P Kraehenbuhl 1994 Cellular and molecular basis for antigen transport in the intestinal epithelium. Pp. 27–39 in Handbook of Mucosal Immunology, P.L. Ogra, J. Mestecky, M.E. Lamm, W. Strober, J.R. McGhee, and J. Bienenstock, eds. Boston: Academic Press, Inc.
Ogra, P.L., J. Mestecky, M.E. Lamm, W. Strober, J.R. McGhee, and J. Bienenstock, eds. 1994 Handbook of Mucosal Immunology. Boston: Academic Press, Inc.
Openshaw, P.J.M., E.E. Murphy, N.A. Hosken, V. Manio, K. Davis, and A. O'Garra 1995 Heterogeneity of intracellular cytokine synthesis at the single cell level in polarised T helper 1 and T helper 2 populations. J. Exp. Med. 182:1357–1367.
Oppenheim, J.J., C.O.C. Zachariae, N. Mukaida, and K. Matsushima 1991 Properties of the novel proinflammatory supergene ''intercrine" cytokine family. Annu. Rev. Immunol. 9:617–648.
Owen, R.L., and A.L. Jones 1974 Epithelial cell specialization within human Peyer's patches: An ultrastructural study of intestinal lymphoid follicles. Gastroenterology 66:189–203.
Owens, L., and D.E. Cress 1985 Genotypic variability of soybean response to Agrobacterium strains harboring the Ti or Ri plasmids. Plant Physiol. 77:87–94.
Pedersen, H.C., J. Christiansen, and R. Wyndaele 1983 Induction and in vitro culture of soybean crown gall tumors. Plant Cell Rep. 2:201–204.
Pierce, N.F. 1978 The role of antigen form and function in the primary and secondary intestinal immune responses to cholera toxin and toxoid in rats. J. Exp. Med. 148:195–206.
Pierce, N.S., and J.L. Gowans 1975 Cellular kinetics of the intestinal immune response to cholera toxoid in rats. J. Exp. Med. 142:1550–1563.
Punt, J.A., and S. Singer 1996 T-cell development. Pp. 157–175 in Clinical Immunology Principles and Practice, R.R. Rich, T.A. Fleisher, B.D. Schwartz, W.T. Shearer, and W. Strober, eds. Boston: Mosby.
Quiding-Jarbrink, M., M. Lakew, I. Nordstrom, J. Bachereau, E. Butcher, J. Holmgren, and C. Czerkinsky 1995 Human circulating specific antibody-forming cells after systemic and mucosal immunizations: Differential homing commitments and cell surface differentiation markers. Eur. J. Immunol. 25:322–327.
Renegar, K.B., and P.A. Small, Jr. 1994 Passive immunization: Systemic and mucosal. Pp. 347–356 in Handbook of Mucosal Immunology, P.L. Ogra, J. Mestecky, M.E. Lamm, W. Strober, J.R. McGheee, and J. Bienenstock, eds. Boston: Academic Press, Inc.
Robey, E., and J.P. Allison 1995 T-cell activation: Integration of signals from the antigen receptor and costimulatory molecules. Immunol. Today 16:306–310.
Rocken, M., and E.M. Shevach 1996 Immune deviation—The third dimension of nondeletional T-cell tolerance. Immunol. Rev. 149:175–194.
Rott, L.S., M.J. Briskin, D.P. Andrew, E.L. Berg, and E.C. Butcher 1996 A fundamental subdivision of circulating lymphocytes defined by adhesion to mucosal addressin cell adhesion molecule-1 . J. Immunol. 156:3727–3736.
Rubin, D.H., M.A. Eaton, and A.O. Anderson 1986 Reovirus infection in adult mice: The virus hemagglutinin determines the site of intestinal disease. Microb. Pathog. 1:79–87.
Ruedl, C., C. Rieser, N. Kofler, G. Wick, and H. Wolf 1996 Humoral and cellular immune responses in the murine respiratory tract following oral immunization with cholera toxin or Escherichia coli heat-labile enterotoxin. Vaccine 14:792–798.
Salgame, P., J.S. Abrams, C. Clayberger, H. Goldstein, J. Convit, R.L. Modlin, and B.R. Bloom 1991 Differing lymphokine profiles of functional subsets of human CD4 and CD8 T cell clones. Science 254:279–282.
Sarraf, P., R.C. Frederich, E.M. Turner, G. Ma, N.T. Jaskowiak, D.J. Rivet III, J.S. Flier, B.B. Lowell, D.L. Fraker, and H.R. Alexander 1997 Multiple cytokines and acute inflammation raise mouse leptin levels: Potential role in inflammatory anorexia. J. Exp. Med. 185:171–175.
Schrader, C.E., A. George, R.L. Kerlin, and J.J. Cebra 1990 Dendritic cells support production of IgA and other non-IgM isotypes in clonal microculture. Int. Immunol. 2(6):563–570.
Shroff, K.E., K. Meslin, and J.J. Cebra 1995 Commensal enteric bacteria engender a self-limiting humoral mucosal immune response while permanently colonizing the gut. Infect. Immun. 63:3904–3913.
Sim, G-K. 1995 Intraepithelial lymphocytes and the immune system. Adv. Immunol. 58:297–343.
Sixma, T.K., S.E. Pronk, K.H. Kalk, E.S. Wartna, B.A.M. vanZanten, B. Witholt, and W.G. Hol 1991 Crystal structure of a cholera toxin-related heat-labile enterotoxin from E. coli. Nature (London) 351:371–377.
Spangler, B.D. 1992 Structure and function of cholera toxin and the related Escherichia coli heat-labile enterotoxin. Microbiol. Rev. 56:622–647.
Spriggs, M.K. 1996 One step ahead of the game: Viral immunomodulatory molecules. Annu. Rev. Immunol. 14:101–130.
Stavnezer, J. 1995 Regulation of antibody production and class switching by TGF-beta. J. Immunol. 155:1647–1651.
Stok, W., P.J. van der Heijden, and A.T.J. Bianchi 1994 Conversion of orally induced suppression of the mucosal immune response to ovalbumin into stimulation by conjugating ovalbumin to cholera toxin or its B subunit. Vaccine 12:521–526.
Street, N., and T.R. Mosmann 1991 Functional diversity of T lymphocytes due to secretion of different cytokine patterns. FASEB J. 5:171–177.
Sun, J-B., J. Holmgren, and C. Czerkinsky 1994 Cholera toxin B subunit: An efficient transmucosal carrier-delivery system for induction of peripheral immunological tolerance. Proc. Natl. Acad. Sci. USA 91:10795–10799.
Swain, S.L., G. Huston, S. Tonkonogy, and A. Weinberg 1991 Transforming growth factor-ß and IL-4 cause helper T-cell precursors to develop into distinct effector helper cells that differ in lymphokine secretion pattern and cell surface phenotype. J. Immunol. 147:2991–3000.
Trinchieri, G. 1993 Interleukin-12 and its role in the generation of TH1 cells. Immunol. Today 14:335–338.
Tsunemitsu, H., M. Shimizu, T. Hirai, H. Yonemichi, T. Kudo, K. Mori, and S. Onoe 1989 Protection against bovine rotaviruses in newborn calves by continuous feeding of immune colostrum. Nippon Juigaku Zasshi 51:300–308.
Underdown, B.J., and J. Mestecky 1994 Mucosal immunoglobulins. Pp. 79–97 in Handbook of Mucosal Immunology, P.L. Ogra, J. Mesteck, M.E. Lamm, W. Strober, J.R. McGhee, and J. Bienenstock, eds. Boston: Academic Press, Inc.
von Behring, E., and S. Kitasato 1890 On the acquisition of immunity against diphtheria and tetanus in animals. Deutsch. Med. Wochenschr. 16:1113–1114.
Walker, R.I. 1994 New strategies for using mucosal vaccination to achieve more effective immunization. Vaccine 12(5):387–400.
Weinstein, P.D., and J.J. Cebra 1991 The preference for switching to IgA expression by Peyer's patch germinal center B-cells is likely due to the intrinsic influence of their microenvironment. J. Immunol. 147:4126–4135.
Weinstein, P.D., P.A. Schweitzer, J.A. Cebra-Thomas, and J.J. Cebra 1991 Molecular genetic features reflecting the preference for isotype switching to IgA expression by Peyer's patch germinal center B-cells. Int. Immunol. 3:1253–1263.
Weinstein, P.D., A.O. Anderson, and R.G. Mage 1994a Rabbit IgH sequences in appendix germinal centers: VH diversification by gene conversion-like and hypermutation mechanisms. Immunity 1:647–659.
Weinstein, P.D., R.G. Mage, and A.O. Anderson 1994b The appendix functions as a mammalian bursal equivalent in the developing rabbit. Adv. Exp. Med. Biol. 355:249–254.
Winter, G., A.D. Griffiths, R.E. Hawkins, and H.R. Hoogenboom 1994 Making antibodies by phage display technology. Annu. Rev. Immunol. 12:433–455.
Wong, Y.W., P.H. Kussie, B. Parhami-Seren, and M.N. Margolies 1995 Modulation of antibody affinity by an engineered amino acid substitution. J. Immunol. 154:3351–3358.
Wu, H-Y., and M.W. Russell 1994 Comparison of systemic and mucosal priming for mucosal immune responses to a bacterial protein antigen given with or coupled to cholera toxin (CT) B subunit, and effects of pre-existing anti-CT immunity. Vaccine 12(3):215–222.
Xu-Amano, J., H. Kiyono, R.J. Jackson, H.F. Staats, K. Fujihashi, P.D. Burrows, C.O. Elson, S. Pillai, and J.R. McGhee 1993 Helper T-cell subsets for immunoglobulin A responses: Oral immunization with tetanus toxoid and cholera toxin as adjuvant selectively induces Th2 cells in mucosa-associated tissues. J. Exp. Med. 178:1309–1320.
Yamamura, M., K. Uyemura, R.J. Deans, K. Weinberg, T.H. Rea, B.R. Bloom, and R.L. Modlin 1991 Defining protective response to pathogens: Cytokine profiles in leprosy lesions. Science 254:277–279.
Yan, C., J.H. Resau, J. Hewetson, M. West, W.L. Rill, and M. Kende 1994 Characterization and morphological analysis of protein-loaded poly(lactide-coglycolide) microparticles prepared by water-in-oil-in-water emulsion technique. J. Controlled Release 32:231–241.
Young, H.A., P. Ghosh, J. Ye, J. Lederer, A. Lichtman, J.R. Gerard, L. Penix, C.B. Wilson, A.J. Melvin, M.E. McGurn, D.B. Lewis, and D.D. Taub 1994 Differentiation of the T-helper phenotypes by analysis of the methylation state of the IFN-ß gene. J. Immunol. 153:3603–3610.
Zhang, R.G., M.L. Westbrook, S. Nance, B.D. Spangler, G.G. Shipley, and E.M. Westbrook 1995 The three-dimensional crystal structure of cholera toxin. J. Mol. Biol. 251:563–573.
DISCUSSION
ROBERT NESHEIM: Are there any questions? I have a question. How stable are these antibodies in the plant materials?
ARTHUR ANDERSON: The antibodies are very stable when expressed in plant materials. The Streptococcus mutans-specific IgA that Ma et al. (1995) expressed in transgenic tobacco plants had been secreted into a plant compartment that made it very easy to extract. All they had to do was freeze, grind up the leaves, centrifuge at 15,000g to remove fiber and large plant molecules, and the antibodies were precipitated out of soluble proteins with 40 and 60 percent (NH4)2SO4.
ROBERT NESHEIM: The antibody genes can just be put into foods? What happens with heat processing?
ARTHUR ANDERSON: That could be a problem with protein antigens or antibodies expressed in plants. In the experiment of Haq et al. (1995), the mice first
were given crude tobacco leaf extract. In later experiments, they ate raw potato tubers. If the tubers were cooked, as we like them, the antigen would be denatured and may not induce immunity. This is also a problem with cooking antibodies.
Everything I said about use of cholera toxin B-subunit as a carrier for oral vaccines absolutely depends on it retaining its conformational integrity. If the B-subunit pentameric ring is denatured and it cannot bind host cell membrane GM1 gangliosides, then everything I said is incorrect. However, Arntzen has been working on developing foods like transgenic bananas that could be eaten raw. Banana also could be stabilized and dried using sugars and lyophilization. This would enable it to be microencapsulated in an enteric coating to get it past the denaturing effect of stomach acid.
These are issues that people who prepare Meals, Ready-to-Eat (MREs) are most ready to test. I have not done all of the components of the vision I presented. However, the literature says that the field is ready to apply this new technology for protection of soldiers.
Obviously, I would first like to protect a soldier against the common mucosal pathogens that cause intestinal pathology. I would use recombinant antibodies from transgenic plants incorporated into every MRE to provide passive protection. At the same time, I would periodically give the soldiers "special MREs" or "vaccine candy bars" with the cholera B-subunit linked vaccines to enable development of acquired active immunity.
There is no reason to suspect that oral passive antibody would block development of immunity with encapsulated or carrier-linked antigens.
GABRIEL VIRELLA: If the ganglioside is not expressed, how does the recombinant cholera toxin B-subunit get to the right cells?
ARTHUR ANDERSON: Cholera toxin B-subunit (CTB) binds to GM1 ganglioside on all eukaryotic cells. The sialomucins on absorptive epithelial cells interfere slightly with binding, which causes the carrier to bind to M-cells over mucosal lymphatic tissues or to IEL [intraepithelial lymphocytes] cells, some of which have higher GM1 ganglioside in their membranes. The mucosal M-cells are exactly where we want the antigen to bind because this is where antigens must be taken up for a mucosal immune response to result.
The CTB binds more in the proximal small bowel than in the distal bowel because binding depletes the material before it reaches the distal bowel. However, proximal bowel binding has produced the desired results. In sheep, the distal Peyer's patches in the ileum have a different function from those in the proximal bowel. We do not know if having antigens bind to distal bowel lymphatic tissues might change the ratio of antigen priming to tolerance induction, for example.
On the other hand, there is a great deal of interest in using enterotoxin-derived carriers in stimulating immunity via intranasal route, which accesses the tonsils and adenoids as antigen-reactive mucosal lymphatic tissues.
DOUGLAS WILMORE: There is another route by which antigens can enter the gut, and that is by paracellular channels—gaps that form when the gut is under-perfused and as a result of the effects of acidosis on mucosal integrity. People are now debating how the influence of endotoxin or other products affect antigens coming up those pathways. Do you see other mechanisms to protect the gut in that way?
ARTHUR ANDERSON: There has been research about particulate antigens, such as bacteria and yeast, gaining access to the blood circulation by mechanisms you referred to. Griffiss has shown that levels of IgA in the circulating blood appear to dampen the effects of bacteremia on complement activation. There is a blood threshold maintained by IgA levels, which, when exceeded, allows endotoxin to unleash its effects on cytokine cascades, complement activation, and bradykinin activation. Endotoxin and yeast and bacteria normally enter your blood during eating and eliminating and brushing one's teeth. Most people do not show any physiological reactions to this as long as their IgA levels are normal. On the other hand, IgA-deficient people are very susceptible to endotoxin-mediated shock.
Endotoxin-containing immunogens actually have been shown to down-modulate mucosal immunity. This has been confirmed by using C3H/HeN and C3H/HeJ mouse substrains. The C3H/HeJ mice lack the gene for cytokine response to endotoxin. These mice are resistant to induction of endotoxin shock. They also mount very potent IgA responses. The endotoxin-susceptible C3H/HeN mouse shows dose-related decrements in IgA production related to endotoxin added to the GI tract. There are a series of publications on this coming from Gerry McGhee's laboratory in Birmingham, Alabama.
JOHANNA DWYER: I remember that, a long time ago, one of the formula companies wanted to put antibodies in infant formulas, and I think it foundered. One of the concerns was that some infants might get leaky guts or something, which prevents complete benefit of the antibodies, and they still get sick. Does what you propose have such a danger? Can it be a two-edged sword?
ARTHUR ANDERSON: That is a good question. What I have presented about conventional modes of immunization already creates a two-edged sword. In my review, I hope I have narrowed down the alternatives to reduce this risk.
What you referred to is a phenomenon which only occurs in infants. It is called the phenomenon of gut closure. New babies do not have all the functional proteases for a while after they are born. The phenomenon relates to the fact that relatively intact proteins can be directly absorbed across the mucosa. After the mucosa matures to a point where most proteases are released, only proteins with mechanisms to resist protease degradation can get across. This, I believe, is what gut closure means. If non-IgA antibodies to pathogens are put in milk, some of those antibodies could actually enable concentration and absorption of viral pathogens in newborn babies. This would result in a paradoxical adverse effect.
Normal cow's milk already has secretory IgA against many pathogens, so adding antibodies would not really help. If the antibodies were not dimeric IgA with J-chain and secretory component, they also would not help. The added IgA was too expensive because of its method of preparation; and once you start, you have to keep giving it. I would not have done it with the technology available then, either.
I also want to comment on the double-edged sword of using enterotoxin carriers (CT/LT). Both CTB and LT/mLT have been tested in phase I trials in humans. These vaccines have been tolerated well, and dose finding studies have produced benign toxicities. I have enjoyed being a voyeur to the vaccine trials of products developed by Jan Holmgren, John Mekalanos, and John Clements and tested in the Clinical Studies unit of USAMRIID by Dave Taylor and Dan Scott, respectively. In normal unvaccinated subjects, cholera holotoxin or Vibrio cholerae infection is going to cause cramping and diarrhea. When CTB, with or without killed Vibrios, or LTB is given to unvaccinated volunteers, no signs or symptoms result. I have not referred to all the individual studies which show that oral use of CT/LT or CTB with antigen result in both IgA and IgG immune responses. I don't see a double-edged sword there, unless B-subunit loses GM1-binding activity.
DAVID SCHNAKENBERG: How often do you have to present antibodies by the oral route? You said IgA could be put in MREs. To get the effect, do you have to administer it daily?
ARTHUR ANDERSON: There is pretty wide variability in gut transit time. In the mouse, it is about 6 hours. In human beings, it can be anything from hours to days. If you use enterically coated antibodies in three meals a day, there should be sufficient antibody present all the time. IgA binds to intestinal mucin. The mucin moves more slowly than does the fecal stream, which should allow a buildup of IgA.
DAVID SCHNAKENBERG: What is the lag time from dosage to effective protection? If you are using this only to a point of deployment, is this something you start 2 weeks ahead of time?
ARTHUR ANDERSON: If you are referring to passive antibody protection, it would be adequate to give soldiers the antibodies in their meals en route. By the time they arrive, their intestines would be protected. If you are referring to active protection with vaccines, it is preferrable that the initial priming doses be given as long in advance as possible. I would say it is best to immunize troops during basic training. It is not essential that antibody levels be maintained. Desert Shield/Desert Storm taught us to trust immunological memory. There were people who got only one dose of vaccine and people who got a full series of shots before going overseas; both had sufficient antibody titers to be protective when given a booster 24 to 36 months later. Circulating memory cells will ensure that soldiers ready to deploy will have a return in protective antibody titer within 3 days of the booster. That is what I believe we need to do.
HARRIS LIEBERMAN: There is a special category of foods, I believe, of medical foods. It sounds like the kinds of things you are talking about might go into that category, a special sort of deployment ration that is essentially a medical food. I don't think you would want to put antigens in all the MREs, but you would want to have a special category.
ARTHUR ANDERSON: I agree. It is not intended that chronic enteric exposure to vaccine antigens be given. That is one of the ways tolerance to food antigens develops. However, your idea of preparing an easily identifyable MRE as a medical food, with instructions on the cover about frequency of use for optimal immunity, would be useful in assuring the proper use by deployed troops. How this would be utilized effectively is something your institute is better suited for determining.


















































