
This paper was presented at a colloquium entitled “Genetics and the Origin of Species,” organized by Francisco J.Ayala (Co-chair) Walter M.Fitch (Co-chair), held January 30–February 1, 1997, at the National Academy of Sciences Beckman Center in Irvine, CA.
Neutral behavior of shared polymorphism
ANDREW G.CLARK
Institute of Molecular Evolutionary Genetics, Department of Biology, 208 Mueller Laboratory, Pennsylvania State University, University Park, PA 16802
ABSTRACT Several cases have been described in the literature where genetic polymorphism appears to be shared between a pair of species. Here we examine the distribution of times to random loss of shared polymorphism in the context of the neutral Wright-Fisher model. Order statistics are used to obtain the distribution of times to loss of a shared polymorphism based on Kimura’s solution to the diffusion approximation of the Wright-Fisher model. In a single species, the expected absorption time for a neutral allele having an initial allele frequency of 1/2 is 2.77 N generations. If two species initially share a polymorphism, that shared polymorphism is lost as soon as either of two species undergoes fixation. The loss of a shared polymorphism thus occurs sooner than loss of polymorphism in a single species and has an expected time of 1.7 N generations. Molecular sequences of genes with shared polymorphism may be characterized by the count of the number of sites that segregate in both species for the same nucleotides (or amino acids). The distribution of the expected numbers of these shared polymorphic sites also is obtained. Shared polymorphism appears to be more likely at genetic loci that have an unusually large number of segregating alleles, and the neutral coalescent proves to be very useful in determining the probability of shared allelic lineages expected by chance. These results are related to examples of shared polymorphism in the literature.
Shared polymorphism may be formally defined as follows: suppose species A has two alleles at a locus, A1 and A2, and species B also has two alleles at the homologous locus, labeled B1 and B2. Shared polymorphism, occurs if alleles A1 and B1 cluster together and are significantly divergent from alleles A2 and B2, which also cluster together. The biological conclusions to be drawn from shared polymorphism depend on the chance that neutral alleles can exhibit this property. Formally, shared polymorphism may arise either when there was a polymorphism in the population ancestral to the two species examined today, and that polymorphism has been maintained through the two distinct species’ lineages, or by more recent parallel generation of similar alleles. If the identity of alleles is well described, as is the case for DNA sequences, it may be extremely unlikely that multiple parallel mutations had occurred. In such cases, polymorphism maintained in both lineages since the time of the common ancestor is the most plausible explanation. Cases of shared polymorphism generally involve genes whose function suggests a mechanism whereby strong natural selection acts to maintain a highly diverse set of alleles. If a gene of unknown function exhibits interspecific shared polymorphism, we would like to know whether it is appropriate to argue that the gene is likely to be undergoing a similar pattern of diversity-enhancing selection.
One of the more striking patterns of naturally occurring genetic variation documented by Dobzhansky (1) is the polymorphism of third-chromosome inversions in Drosophila pseudoobscura. The broad geographic distribution and temporal stability of this polymorphism led Dobzhansky and others to conclude that the polymorphism is stably maintained by natural selection. Laboratory cage experiments suggested that the inversions differed significantly in fitness (2). Analysis of restriction site variation on the third-chromosome inversions revealed that the molecular phylogeny is concordant with the previously inferred inversion phylogeny (based on overlaps in the inversions), and that the persimilis allele clusters within the range of pseudoobscura alleles (3). The inversion polymorphism was estimated to be about 2 million years old, which is about the age of the pseudoobscura-miranda split (3). Given the widespread nature of the pseudoobscura-inversion polymorphisms, and their evidently ancient origin, it becomes an intriguing question why D. persimilis, which had a common ancestor with D. pseudoobscura only 2 million years ago, does not share the polymorphism for at least some of the pseudoobscura third-chromosome inversions. In fact the two species have only the standard arrangement in common. It appears that the answer lies in the demographics of the process of speciation itself, and an important conclusion of this analysis is that searches for shared polymorphism in species young enough to expect some sharing even of strictly neutral genes may shed considerable light on these demographic processes.
One of the most striking examples of shared polymorphism (or “trans-species” polymorphism) can he found among alleles of the class I and class II major histocompatibility complex (MHC) genes. Allele sharing was first noticed in constructing gene trees of MHC class I alleles and finding that the human and chimp alleles are often more closely related than they are to other alleles (4, 5). Statistical significance of the shared polymorphism was verified by showing that neighbor-joining trees yield clusters of alleles that are significantly divergent from other clusters of alleles by bootstrap tests, and each cluster bears alleles from both species (6). Such shared polymorphism can be found in gorilla as well (7). Even more striking is the degree of allele sharing in the DRB genes, which exhibit not only allele sharing, but remnants of haplotype structure seems to be shared (8, 9). In all these cases, the exceptionally long-lived polymorphism is thought to have been maintained by natural selection favoring diversity, particularly in the peptide binding region of the MHC molecules. The argument has been made that such polymorphisms are not consistent with the neutral theory, given the time back to the common ancestor of humans and other primates (10).
Shared polymorphism between humans and chimpanzees is striking because it implies that the polymorphisms have been maintained in both species since the time of common ancestry, or about 4 million years. Assuming a generation time of 20
© 1997 by The National Academy of Sciences 0027–8424/97/947730–5$2.00/0 PNAS is available online at http://www.pnas.org.
years and a long-term effective population size of 10,000, this represents 20 N generations. The significance of shared polymorphism between humans and our closest relatives is suspected when one compares this figure of 20 N generations to the expected fixation time for a neutral polymorphism (4 N generations). But it is necessary to determine the distribution of time to loss of shared polymorphism, as there may be a long tail with substantial probability density for much greater durations of sharing.
Shared polymorphism in the plant kingdom is even more striking, especially in self-incompatibility genes that have generated appallingly diverse alleles. In the Solanaceae, shared polymorphism of S-alleles may be as old as 70 million years and is accompanied by within-species divergence of allelic lineages of over 50% at the amino acid level (11). Population genetic models of self-incompatibility show that the coalescence time of alleles varies inversely with the rate of origination of novel functional alleles, and that for reasonable estimates of the rate of origination of new alleles, such extremely old polymorphisms are not unlikely (12). Interdigitation of alleles from different species continues as more species are added to the list of sequenced S-alleles (13).
Closely related species might be expected to show higher levels of shared polymorphism, and in some cases this is borne out. In the species clade of Drosophila melanogaster, simulans, sechellia, and mauritiana, only simulans and mauritiana appear to exhibit substantial levels of shared polymorphism as revealed from gene trees of yp2, per, and zeste (14, 15). Sequence divergence does not suggest that mauritiana is younger than sechellia, yet the level of shared polymorphism is much greater between mauritiana and simulans than it is between sechellia and simulans. This observation suggests that the historical population size of sechellia has been much smaller than that of mauritiana, either through a small founding population or a long-term small population size.
Expected Persistence of Neutral Shared Polymorphism
When a highly diverse species splits into two species, then depending on the largely unknown demographic aspects of the splitting process, the two new species may bear a sizable proportion of the polymorphism present in the original species. Assuming the reproductive barrier is complete, there follows a period of time during which this shared polymorphism is lost. Relatively little theoretical attention has been paid to the dynamics of this loss, but it appears that some interesting issues arise in this analysis. Considering first a classical approach, imagine two alleles, A and a, segregating in two independent populations, each having N diploid individuals and an initial allele frequency p for the A allele. The distribution of fixation time for a neutral allele was obtained by Kimura (16), and it is an ungainly expression involving the hypergeometric function:
f(1,t)=p+∑i(2i+1)pq(−1)i
F(1−i, i+2, 2, p)e−[i(i+1)/4N]t. [1]
If the same Wright-Fisher model is applied to two independent populations, the shared polymorphism is lost as soon as absorption occurs in either species. If f(p,t) is the probability density function of absorption time in one population [and F(p,t) is the corresponding cumulative distribution function], then g(p,t)=n[(1−F(t)]n−1f(t) is the density for the first absorption in a collection of n identically behaved populations. For our case, n=2 and we get:
g(p,t)=2[1−F(p,t)]f(p,t) [2]
for the probability density of time to loss of shared polymorphism. Fig. 1 shows the probability density for time to loss of shared polymorphism in this context: The important point is that, while it is true that the mean time to loss of shared polymorphism is rather short (1.7 N generations), the density has a long tail to the right, so that with finite probability shared neutral polymorphism, can last much longer. In particular, 5% of the time-shared, polymorphism is retained until 3.8 N generations, and 1% of the time it is maintained until 5.3 N generations.
Loss of Shared Polymorphic Sites Over Time
The above discussion relates to the case in which there are two clearly distinct allelic lineages. Often molecular data will not be quite so clear, because each allelic lineage will have suffered many mutations since the time of common ancestry. Molecular data allow one to ask how many of the segregating sites in the two species are shared. The two species will undergo a period of random genetic drift in which shared polymorphic sites go to fixation in one or the other species. If the sites undergo random fixation slowly enough that intragenic recombination is likely between rounds of fixation, then it may be acceptable to consider the case of adjacent sites being lost independent of flanking sites. This results in a process of decay in the number of shared polymorphic sites that follows an approximately geometric distribution, and a simulation of the process is shown in Fig. 2.
Number of Shared Polymorphic Sites Expected at Steady State
The process of loss of shared polymorphism does not continue until there are zero sites, because by chance one expects to have some shared sites, particularly if both species are highly polymorphic. Consider first the chance of observing shared polymorphic sites if those sites are randomly and independently scattered along the sequence. Suppose one samples two sequences of length S from, each of two species, and there are s1 sites that differ between the pair of alleles in species 1 and s2 sites that differ in the pair of alleles in species 2. If the polymorphic sites are uniformly and independently distributed, then the probability that k sites are polymorphic in both species (i.e., that there are k shared polymorphic sites) is given by the hypergeometric density:
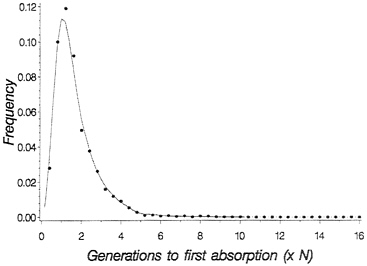
FIG. 1. Probability density for time to loss of shared polymorphism based on numerical integration of Kimura’s (16) density for absorption time. Dots are simulated values for 1,000 replicate populations with 2 N=50, with initial allele frequency p=0.5.
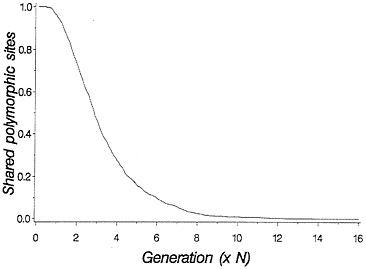
FIG. 2. Loss of shared polymorphic sites in two species initially segregating 1,000 shared polymorphic sites. One thousand replicate populations with 2N=50 were simulated forward in time. All shared sites were assumed to be unlinked.
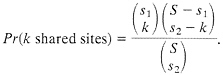
[3]
One-third of the time a site that is segregating in two species will be segregating for the same pair of nucleotides (assume they share one nucleotide through common ancestry). When one examines multiple alleles, the expected number of sites that have shared polymorphism is a more complex expression, and it is easiest to obtain this null distribution by resampling the data by computer. Fig. 3 illustrates this for some Drosophila and human-chimp MHC data. In both cases, the observed number of shared polymorphic sites exceeds all values in the null distribution obtained by computer resampling, just as had been observed in the case of S-alleles by Ioerger et al. (11).
The Neutral Coalescent and Shared Polymorphism
Fig. 4 illustrates one way to conceptualize the problem of shared polymorphism in the context of the coalescent. If two species each coalesce to a common ancestral allele more recently than the time at which they share a common ancestor, then there is not shared polymorphism (Fig. 4A). On the other hand, if they do not have this recent coalescence event, then they share polymorphism (Fig. 4B).
The chance that two alleles had two distinct ancestors the previous generation is ![]() for a diploid population. The chance that three alleles had three distinct ancestors is
for a diploid population. The chance that three alleles had three distinct ancestors is ![]()
![]() , and the chance that n alleles had n ancestors is
, and the chance that n alleles had n ancestors is
(17). This gives rise to the expression that the probability that the first coalescence occurred t+1 generations in the past is
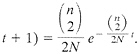
[4]
Now consider a sample drawn from two distinct populations each with n alleles. Initially let there be n shared allelic lineages in the samples. The probability that they both have n ancestral alleles the previous generation is ![]() , because the
, because the
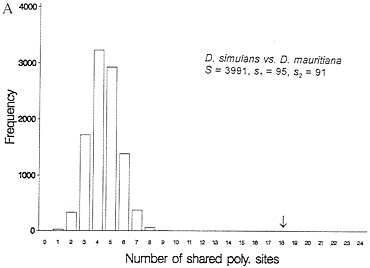
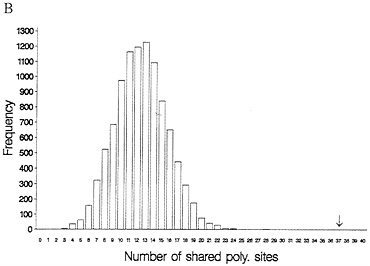
FIG. 3. Observed and expected numbers of shared polymorphic sites under the assumption of a random distribution of polymorphic sites in each species. (A) Data from Drosophila simulans and D. mauritiana for the yp2, zeste, and per genes (15). (B) Number of shared polymorphic sites in a sample of 17 human and seven chimpanzee MHC class I A alleles.
process of coalescence and sampling are independent in the two species. When there is a coalescence in either species, then n-1 shared lineages are left. At this point the process starts anew with n-1 shared lineages, until one or the other population has another coalescence. Eventually there will be two shared lineages, and the next coalescence results in loss of the shared polymorphism. From this it is not difficult to show:
[5]
This equation gives the recursion for the time to loss of shared polymorphism. Fig. 5 plots a solid line for this time to loss of shared polymorphism and also plots simulated points, showing an excellent agreement. With this theory, we can now make statements about the probability of observing a particular level of shared polymorphism given data on two species, provided we know their time of divergence.
Discussion
The problem of shared polymorphism and the distribution of numbers of shared polymorphic sites is closely related to the problem of persistence time of polymorphism in a single species. Related problems have been studied in the past, and
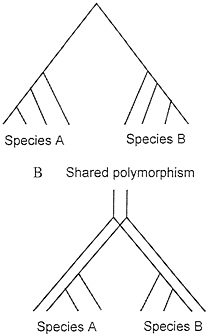
FIG. 4. (A) Coalescence of all variation to a single common ancestral allele at a more recent time than the common ancestor of the two species. (B) Shared polymorphism arising from lack of complete coalescence of allelic lineages in the timespan since speciation.
some insights can be drawn from those. The reverse of this problem was studied by Nei and Li (18), who determined the probability of identical monomorphism in a pair of species that descended from a common ancestor. They found, given reasonable estimates of population size and mutation rates, that monomorphism for the same allele in humans and chimpanzees is not unlikely under neutrality. Griffiths and Li (19) determined properties of fixation in pairs of lineages under more general initial conditions, that is, rather than conditioning the distribution of absorption time on initial allele frequency, as Kimura (16) did, they considered the starting condition to be the steady-state infinite alleles frequency spectrum. They went on to ask about the number of common alleles shared between two populations that descended from a common ancestor. Computer simulations were done forward in time, scoring the allele distribution at times separated by 2t
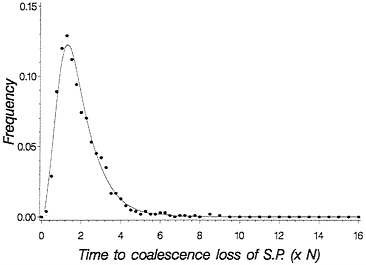
FIG. 5. Probability density for time to loss of shared polymorphism based on time of first coalescence for a pair of species. The solid line is obtained from Eq. 5, and the dots are from simulating the process of drift forward in time in two finite populations each of size 2 N= 50. One thousand replicate population pairs were followed.
generations, a process formally equivalent to simulating two populations each for t generations from a common starting point. Although they did not record the frequency of shared polymorphism, their results were consistent with the current results in demonstrating that the persistence time of alleles can have a long tail. For example, when θ=0.1, even after 40 N generations, the probability that two species share an allele is 15% (19).
Perhaps even more important will be an analysis of the distribution of shared polymorphism under different models of selection. With symmetric overdominant selection, the gene tree has the same branching topology as a neutral gene, but with potentially much deeper times of coalescence (20). Computer simulation of allelic genealogies under over dominant selection showed that with mutation rates, effective sizes, and selection coefficients that seem plausible for MHC loci, the coalescence times may be on the order of tens of millions of years (10). Under conditions where within-species coalescence times are so long, it is reasonable to expect the duration of shared polymorphism between species to be greatly lengthened also.
S-alleles and MHC clearly show shared polymorphism due to strong selection to maintain the variation. The basis for the selection in both cases is that the protein products of these genes accrue a fitness advantage to the bearer of those alleles if they are heterozygous or otherwise more diverse. This implies that the selection acts on coding sequences and should impact replacement sites more than silent sites. If one constructs a 2×2 table of shared vs. nonshared polymorphisms at silent vs. replacement sites, a significant χ2 may be consistent with this mode of selection. In both cases, this test is significant (21).
In the Drosophila melanogaster group species, in appears that simulans and mauritiana are recently enough diverged to maintain many shared polymorphisms by common ancestry of neutral variation. Such recent common ancestry allows an exciting opportunity to examine many genes and characterize the distribution of shared polymorphism across those genes. Genes having stronger purifying selection will lose the shared polymorphism faster than neutral genes, and genes having any sort of selection that maintains diversity will have an excess of shared polymorphism.
The future of human genetics will see an explosion in interest in inferences about ancestral history than can be drawn from extant genetic variation. Coupling these studies to analysis of chimpanzee and other primate polymorphism is likely to be extremely informative. In the first place, shared polymorphism is an excellent filter for searching for genes under strong selection to maintain polymorphism. The divergence time of humans and chimpanzee is estimated to be about 20 N generations ago—long enough that very few neutral shared polymorphisms will be left. Polymorphisms that are shared will be the targets of study to find what functional aspect of those genes results in such longevity of within-species polymorphism. The consideration of polymorphism in populations of ancestral humans is essential to the use of extant human genetic variation to test hypotheses about human origins. Shared polymorphism may provide an unusual opportunity to test ideas about the demographic changes that occurred in the early history of emerging species.
This work was supported by National Science Foundation Grants DEB 9419631 and DEB 9527592.
1. Dobzhansky, Th. (1937) Genetics and the Origin of Species (Columbia Univ. Press, New York).
2. Anderson, W.W., Oshima, C., Watanabe, T., Dobzhansky, Th. & Pavlovsky, O. (1968) Genetics 58, 423–434.
3. Aquadro, C.F., Weaver, A.L., Schaeffer, S.W. & Anderson, W.W. (1991) Proc. Natl. Acad. Sci. USA 88, 305–309.
4. Lawlor, D.A., Ward, F.E., Ennis, P.D., Jackson, A.P. & Parham, P. (1988) Nature (London) 335, 268–271.
5. Mayer, W.E., Jonker, M., Klein, D., Ivanyi, P., van Seventer, G. & Klein, J. (1988) EMBO J. 7, 2765–2774.
6. Nei, M. & Rhzetsky, A. (1991) in Evolution of MHC Genes, eds. Klein, J. & Klein, D. (Springer, Heidelberg), pp. 13–27.
7. Vendetti, C.P., Lawlor, D.A., Sharma, P. & Chorney, M.J. (1996) Hum. Immunol. 49, 71–84.
8. Goagora, R., Fisueroa, F. & Klein, J. (1996) Hum. Immunol. 51, 23–31.
9. Satta, Y., Mayer, W.E. & Klein, J. (1996) Hum. Immunol. 51, 1–12.
10. Takahata, N. & Nei, M. (1990) Genetics 124, 967–978.
11. Ioergen, T.R., Clark, A.G. & Kao, T.-h. (1990) Proc. Natl. Acad. Sci. USA 87, 9732–9735.
12. Clark, A.G. & Kao, T.-h. (1994) in Genetic Control of Self-Incompatibility and Reproductive Development in Flower Plants, eds. Williams, E.G., Clarke, A.E. & Knox, R.B. (Kluwer, Dordrecht), pp. 220–242.
13. Richman, A.D., Uyenoyama, M.K. & Kohn, J.R. (1996) Science 273, 1212–1216.
14. Hey, J. & Kliman, R.M. (1993) Mol. Biol. Evol. 10, 804–822.
15. Kliman, R.M. & Hey, J. (1993) Genetics 133, 375–387.
16. Kimura, M. (1955) Proc. Natl. Acad. Sci. USA 41, 144–150.
17. Tajima, F. (1983) Genetics 105, 437–460.
18. Nei, M. & Li, W.-H. (1975) Genet. Res. 26, 31–43.
19. Griffiths, R.C. & Li, W.-H. (1983) Theor. Pop. Biol. 23, 19–33.
20. Takahata, N. (1990) Proc. Natl. Acad. Sci. USA 87, 2419–2423.
21. Clark, A.G. & Kao, T.-h. (1991) Proc. Natl. Acad. Sci. USA 88, 9823–9827.





