
Health-Effects Research at the Crossroads: Molecular-Based Tools for Biologic Dosimetry and Individual Susceptibility
Anthony V. Carrano
Associate Director for Biology and Biotechnology Research
Lawrence Livermore National Laboratory
Livermore, California
These are unique and exciting times for health-effects research. Never before has such a wealth of readily exploitable materials, information, and technologies been available to bioscience researchers. The source of most of these resources has been the international human-genome effort. For the Department of Energy (DOE) Office of Biological and Enivironmental Research (OBER) program to remain competitive, it is essential that it reshape the direction of its health-effects research program. That can be achieved by capitalizing on the science and technology of the genome project. For example, a collection of 70-80% of all human genes is laid out before us now in the form of recombinant-DNA clones. With the unique technologies of molecular biology, bioinformatics, and bioinstrumentation, we can identify the genes responsible for susceptibility or resistance to radiation and chemical exposure and potentially use those genes as individual-specific biologic sentinels. In addition, those gene resources provide the basis for unraveling the mechanisms by which xenobiotic substances might cause ill health. We can also begin to consider large-scale studies to determine the structure and function of proteins, exploiting in the process the unique structural-biology resources that DOE has created. Technologies have arisen that enable miniaturization, massive parallel analysis, and exquisite sensitivity. Animal models are playing an increasing role in improving our understanding of the human gene function and its response to exposure. The current OBER health-effects program has a strong foundation in each of those fields and a proven ability to manage and conduct large-scale, complex projects effectively. The time is now to be bold, to capitalize on the strength of the current scientific program, and to develop and implement a vision for health-effects research in the 21st century.
EXPOSURE AND BIOLOGIC DOSE
Health-effects research began 50 yr ago in the Atomic Energy Commission, the original precursor of DOE, with a primary focus on measuring the health effects of ionizing radiation. Dose measurements were crucial then, and they still are. We know now that there are many steps between exposure and health effect. Exposure can come from the air we breathe, from the food or water we ingest, from our occupations, and from our lifestyles. Our exposures are moderated by factors within our bodies that convert external exposure to biologic doses. The factors that mediate this process can be the proteins that metabolize xenobiotic chemicals, proteins that repair damaged DNA, or other proteins that regulate our cellular processes and make us more or less susceptible to disease. Thus, over the last 50 yr we have learned that the dose measured by instruments is not necessarily the same as the dose that a person experiences. Moreover, 2 persons who receive the same dose might experience very different
biologic responses. We have learned that people are uniquely susceptible or resistant to heath effects. We have spent many years in developing sophisticated instruments that measure physical doses precisely, but their measurements might not reflect the biologic damage incurred by a specific person. That is the challenge to the health-effects program of the future: understanding the basis for individual susceptibility and using the information to minimize the health effects of exposure. The human-genome project will play a key role in identifying genes and providing technologies to address those issues.
We are very good at determining the shape of the dose-response curve at moderate to high doses, but we do poorly at low doses. We have not yet been able to define the part of the dose-response curve that covers the low doses. Why is that? It is not due to our inability to measure physical dose; our instruments are generally exquisitely sensitive. Rather, it is due to our inability to measure biologic doses with sufficient statistical certainty. We also generally observe a scatter in the points of the curve. Individual response—the unique ability of each person to detoxify, repair, or otherwise ameliorate a biologic insult—is probably a major contributor to that scatter. One of the venues for future research would be to understand the differences that make each of us more or less susceptible to effects of an exposure, whether to radiation or to chemicals. DOE can draw on its strengths in genomics and in cellular and structural biology and have a major role in reaching that goal.
Historically, DOE has made important contributions in developing tools for estimating biologic dose. The chromosome-painting technology was developed at a DOE national laboratory and has seen wide acceptance not only in basic research but also in clinical genetics.1 The technology has become very useful for quantifying radiation damage in people and other living systems. It spun directly out of the genome project in that some of the first chromosome-specific painting probes were made from recombinant-DNA libraries established by the DOE-sponsored National Laboratory Gene Library Project in the 1980s.2 Because the technology allows for relatively rapid analysis, many more cells or people can be examined for cytogenetic alterations than was practical with the previous conventional cytogenetic technology. That allows more statistical precision in measurement and facilitates quantification at lower doses. The technique has been applied to many population studies, including those of the atomic-bomb survivors3 and the Chernobyl cleanup workers.4
If one desires to study the pharmacologic response or biologic consequence at the very low end of the dose-response curve, the accelerator mass spectrometry (AMS) methods developed under DOE sponsorship allow us to do it very well for chemical exposure.5 The technology is used to count the number of carbon=14 atoms in the biologic material of interest. Cells in vito, in animals, and in humans can be provided 14C-labeled chemicals at pharmacologically relevant doses to estimate the transport, detoxification, and conversion of the substance under investigation. Studies conducted with biologic AMS have been able to measure effects only one-hundredth or one-thousandth as large as those required by other laboratory methods. For example, it has been possible to measure the transport and fate of benzene in rodent systems at doses less than one-hundredth of the dose of benzene obtained from a single cigarette6 or to measure the adducts formed in DNA from the eating of the equivalent of a well-done hamburger.7 Being able to make measurements at such low doses has illuminated some major differences in pharmacologic response between high and low doses. Such methods as AMS offer a great advantage and might very well become the standard for chemical measurements in vivo as the technology matures.
INDIVIDUAL SUSCEPTIBILITY AND RESISTANCE
A person's genes play a major role in both response to exposure and susceptibility or resistance to disease. Genes encode the information to make proteins, and proteins perform various functions in cells. We need to place a much greater emphasis on understanding the structure of proteins, how they function, and how they might influence susceptibility or resistance to disease. Some of the relevant proteins are the cytochromes, which have a major role in chemical detoxification and modification in the liver and other tissues; flee-radical scavengers that can protect us against radiation damage; and proteins that recognize and repair damaged DNA.
Examples of the role of proteins as susceptibility factors are numerous. A family of cytochrome P450 genes is clustered on human chromosome 19.8 One of these genes, which encodes the protein coumarin hydroxylase, has been isolated and sequenced, and its variation in several population groups has been measured.9
The DNA sequence for this gene varies in the population. One variant sequence is caused by a single base change and results in loss of enzymatic activity; affected persons cannot metabolize coumarin or similar chemicals that use this pathway.
Another example involves DNA-repair proteins. ERCC2 is a gene involved in the repair of DNA damage. Defects in this gene are associated with 3 diseases.10,11 The most severe is xeroderma pigmentosum D (XPD); those affected are extremely sensitive to sunlight and, in fact, develop cancers from such exposure. The second is Cockayne syndrome; the primary phenotype is severe neurologic dysfunction. The third is trichothiodystrophy which exhibits a milder phenotoype and is associated with metabolic deficiencies. The mutations that cause those 3 diseases all occur in ERCC2 and are complex and not well understood. There does not appear to be a single mutation associated with each one. It is known that the product of ERCC2 acts in a complex with other proteins. Therefore, it will be important to understand how proteins interact with each other and how mutations might affect interactions. A critical defect in 1 region of a protein might have a very different effect phenotypically from a critical defect in another region, depending on whether the regions are sites of interaction with other proteins.
In health-effects studies in the future, it will be important to make use of the resources and technologies that have already been developed to examine populations for genetic variation and to relate that variation to susceptibility and resistance to disease. The first step is to identify and sequence the appropriate suite of genes that might confer susceptibility. It is relatively straightforward to identify the 200-300 such genes that we know of today. Others will be added to the list as gene discovery accelerates in the human-genome project. The DNA sequence of the introns, exons, and 5'-and 3'-flanking regions for each gene must be determined. Although it is primarily the coding and regulatory regions that will influence protein production and function, a sufficient amount of intronic sequence is needed to facilitate diagnostics. The second step is to identify populations in which DNA-sequence variation in the genes can be measured. It is important to measure sequence variation not only in disease-susceptible and disease-resistant people, but also in the "normal" population. Subtle gene defects in many people might have more consequence for ill health in a population than would major gene defects in only a few people. Susceptibility genes can be resequenced with efficient, low-cost, state-of-the-art methods. Alternatively, given a sufficient number of regions to be resequenced, mass-hybridization methods could play an important role.12,13 In fact, the "chip based" methods probably will be the methods of choice, once enough genes and potential variants have been identified.
Measuring a sequence variation alone is not likely to be sufficient for determining its role in ill health. Ultimately, we will need information on the consequences of sequence variation for the functions of encoded proteins. That will require a commitment to structural biology to determine the structures of the variant proteins and a commitment to biochemistry to evaluate functions. By making appropriate comparisons in normal, susceptible, resistant, and diseased people, we should be able to make the link between variation and disease. Appropriate animal or other model systems will be useful in validating the findings.
THE IMAGE CONSORTIUM
An important source of genes for studies on susceptibility is the IMAGE (intergrated molecular analysis of genomes and their expansion) Consortium.14 This collaboration of industry, academe, and national laboratories has established the largest public collection of cDNAs and made them available worldwide to the scientific community. In addition to the clone resource, the ends of the cDNAs have been sequenced to provide ESTs (expressed sequence tags), and these are available in a public database, dbEST.15 Over 600,000 cDNAs have been collected, and they are estimated to represent about 80% of all human genes.16 Because these libraries represent material from over 30 human tissues and in many cases are highly redundant for specific genes or gene segments, one can begin to put together entire genes directly on the computer. Given that resource, it is possible to start with sequences known in other species to be involved in DNA repair or chemical detoxification and use them to see whether there are corresponding human genes. This will facilitate the identification and understanding of susceptibility and of other genes in humans. The cDNA collection also constitutes a powerful resource for studies on gene expression in normal and diseased tissue.
MOUSE AND OTHER MODEL SYSTEMS
Despite the tens of million of years of divergence between mouse and human, sequence homology is highly conserved. Comparative sequence analysis is a method for elucidating the functions of human genomic sequences by identifying sequence elements that are evolutionarily conserved between mouse and human. Sequence is expected to be more highly conserved for regions that represent essential biochemical functions. In addition to the coding regions, however, comparative sequence analysis can also identify other functional components of the genome, such as enhancers and other transcriptional regulatory elements. This approach is largely complementary to the sequencing of full-length human cDNAs, inasmuch as it is more likely to identify small, conserved noncoding elements, which are often well upstream of a gene or within introns and whose locations are nearly impossible to predict with conventional computational approaches. For example, the genomic region that contains the DNA-repair gene XRCC1 has been sequenced in both mouse and human.17 The mouse and human genes each contain 17 exons, are 84% identical within the coding regions, and are 86% identical at the amino-acid-sequence level. Intron and exon lengths are highly conserved. In addition to the coding regions, 9 conserved elements were identified between mouse and human, with sequence identities ranging from 65% to 78%. It is interesting to speculate that these conserved noncoding regions are sites of regulation of XRCC1 and hence also play a role in its expression.
DNA sequence homology across species has been identified for genes associated with several human diseases. The human genes for cystic fibrosis, achondroplasia, and myotonic dystrophy share homology with sequences in the mouse, the fruit fly, worms, and yeasts. XPD, Huntington disease, and nonpolyposis colon cancer have corresponding mouse genes. Thus, other species can be useful models for studying the mechanisms of human diseases. For example, we can now begin to pursue a comparative genomics approach to targeted gene biology. Known sequences from other species can be used to query the cDNA database, identified human cDNAs can be obtained from the available IMAGE collection, and these cDNAs can be used as hybridization probes in human and mouse clones to identify the genomic region spanned by these genes. Simultaneously, the full-length cDNAs can be established and used for expression and functional studies. DOE has historically invested in such model studies, and this is a legacy of which it should be proud. With the information emanating at a high rate from the human-genome project, comparative mapping, sequencing, and functional analyses hold enormous promise.
BUILDING THE BRIDGES
The human-genome project has produced and will continue to produce excellent and extensive resources and technologies relevant to health-effects research. These take the form of genomic clones, cDNAs, DNA sequences, bioinformatics tools, automation, and instrumentation. We need to capitalize on those resources in the DOE health-effects research program. A major emphasis might be placed on using the resources to address individual susceptibility and health effects as an overarching theme. That would lead naturally to programs in gene action and function as related to the DOE mission, computational and structural biology for which DOE has extensive resources, and health and environmental studies. Now is the time to capitalize on the genome resources, and DOE can play a major role by applying its successful genomics experience to understand, prevent, and ameliorate health effects of environmental exposures.
ACKNOWLEDGMENT
This work was performed under the auspices of the US Department of Energy by Lawrence Livermore National Laboratory under contract W-7405-ENG-48.
REFERENCES
1. Collins C, Kuo WL, Segraves R, Fuscoe J, Pinkel D, Gray JW. Construction and characterization of plasmid libraries enriched in sequences from single human chromosomes. Genomics 1991;11:997-1006.
2. Van Dilla MA, Deaven LL, Albright KL, Allen NA, Aubuchon MR, Bartholdi MF , Brown NC, Campbell EW, Carrano AV, Clark
LM, Cram LS, Fuscoe JC, Gray JW, Hildebrand CE, Jackson PJ, Jett JH, Longmire JL, Lozes CR, Luedemann ML, Martin JC, McNinch JS, Meincke LJ, Mendelsohn ML, Meyne J, Moyzis RK, Munk AC, Perlman J, Peters DC, Silva AJ, Trask BJ. Human chromosome-specific DNA libraries: construction and availability. Biotechnology 1986;4:537-552.
3. Awa AA, Nakano M, Ohtaki K, Kodama Y, Lucas J, Gray J. Factors that determine the in vivo dose-response relationship for stable chromosome aberrations in A-bomb survivors. J Radiat Res 1992;33 Suppl: 206-214.
4. Inskip PD, Hartshome MF, Tekkel M, Rahu M, Veidebaum T, Auvinen A, Crooks LA, Littlefield LG, McFee AF, Salomaa S, Makinen S, Tucker JD, Sorensen KJ, Bigbee WL, Boice JD Jr. Thyroid nodularity and cancer among Chernobyl cleanup workers from Estonia. Radiat Res 1997;147:225-235.
5. Turteltaub KW, Felton JS, Gledhill BL, Vogel JS, Southon JR, Caffee MW, Finkel RC, Nelson DE, Proctor ID, Davis JC. Accelerator mass spectrometry in biomedical dosimetry: relationship between low-level exposure and covalent binding of heterocyclic amine carcinogens to DNA. Proc Natl Acad Sci USA 1990;87:5288-5292.
6. Creek, MR, Mani C, Vogel JS, Turteltaub KW. Tissue distribution and macromolecular binding of extremely low doses of [14C]-Benzene in B6C3F mice. Carcinogenesis 1997; In press.
7. Turteltaub KW, Vogel JS, Frantz C, Buonarati MH, Felton JS. Low-level biological dosimetry of heterocyclic amine carcinogens isolated from cooked food. Environ Health Perspect 1993;99:183-186.
8. Hoffman SM, Fernandez-Salguero P, Gonzalez FJ, Mohrenweiser HW. Organization and evolution of the cytochrome P450 CYP2A-2B-2F subfamily gene cluster on human chromosome 19. J Mol Evol 1995;41:894-900.
9. Fernandez-Salguero P, Hoffman SM, Cholerton S, Mohrenweiser H, Raunio H, Rautio A, Pelkonen O, Huang JD, Evans WE, Idle JR, et al. A genetic polymorphism in coumarin 7-hydroxylation: sequence of the human CYP2A genes and identification of variant CYP2A6 alleles. Am J Hum Genet 1995;57:651-660.
10. Takayama K, Salazar EP, Lehmann A, Stefanini M, Thompson LH, Weber CA. Defects in the DNA repair and transcription gene ERCC2 in the cancer-prone disorder xeroderma pigmentosum group D. Cancer Res 1995;55:5656-5663.
11. Takayama K, Salazar EP, Broughton BC, Lehmann AR, Sarasin A, Thompson LH, Weber CA. Defects in the DNA repair and transcription gene ERCC2 (XPD) in trichothiodystrophy. Am J Hum Genet 1996;58:263-270.
12. Drmanac R, Drmanac S, Labat I, Crkvenjakov R, Vicentic A, Gemmell A. Sequencing by hybridization: towards an automated sequencing of one million M 13 clones arrayed on membranes. Electrophoresis 1992; 13:566-573.
13. Chee M, Yang R, Hubbell E, Berno A, Huang XC, Stem D, Winkler J, Lockhart DJ, Morris MS, Fodor SP. Accessing genetic information with high-density DNA arrays. Science 1996;274:610-614.
14. Lenaon G, Auffray C, Polymeropoulos M, Soares MB. The I.M.A.G.E. Consortium: an integrated molecular analysis of genomes and their expression. Genomics 1996;33:151-152.
15. Rodriguez-Tome P. Searching the dbEST database. Methods Mol Biol 1997;69:269-283.
16. Frazer KA, Ueda Y, Zhu Y, Gifford VR, Garofalo MR, Mohandas N, Martin CH, Palazzolo MJ, Cheng J, Rubin EM. Computational and biological analysis of 680 kb of DNA sequence from the human 5q31 cytokine gene cluster region. Genome Res 1997;7:495-512.
17. Lamerdin JE, Montgomery MA, Stilwagen SA, Scheidecker LK, Tebbs RS, Brookman KW, Thompson LH, Carrano AV. Genomic sequence comparison of the human and mouse XRCC1 DNA repair gene regions. Genomics 1995;25:547-554.
Discussant
John D. Boice Jr.
International Epidemiology Institute
Rockville, Maryland
Major advances in our understanding of radiation effects1 in the United States occurred in the latter part of the 20th century. Continued studies of atomic-bomb survivors, patients, workers, and the general public have provided new knowledge. Laboratory approaches are being incorporated into epidemiologic investigations to explain more about the biologic mechanism by which radiation causes cancer in humans. Despite great strides in understanding and quantifying radiation effects, there remains a lingering bemusement. Research that will continue into the next millennium might help to dispel perplexities as we build on and extend past accomplishments. The Department of Energy (DOE) and its predecessors and the National Research Council have contributed substantially to our knowledge of radiation effects.
RADIATION EPIDEMIOLOGY
Accomplishments in radiation epidemiology have been compiled and synthesized in the 1994 report of the United Nations Scientific Committee on the Effects of Atomic Radiation2 and in the proceedings of the 1996 annual meeting of the National Council on Radiation Protection and Measurements (NCRP).3 The focus of early studies has been extended and expanded. Leukemia was first studied in pioneering radiologists 4 and is now studied in nuclear-industry workers;5 osteosarcomas in radium-dial painters6 and now in retinoblastoma patients;7 cancers after radiotherapy for ankylosing spondylitis8 and cervical cancer 9 and now after radiotherapy for childhood cancer;10 childhood cancers after prenatal X-rays11 and now adult cancers after in utero irradiation; 12,13 thyroid cancer after thymic irradiation14 and now after Chernobyl releases;15 breast cancer after tuberculosis fluoroscopy16 and now after Hodgkin's disease radiotherapy;17 lung cancer among underground miners18 and now among above-ground residents;19 and cancer among atomic-bomb survivors20—and still among atomic-bomb survivors.21 DOE and the national laboratories have supported key studies over the years, such as those of radium-dial painters, radiation workers, Chernobyl and other Russian workers, and atomic-bomb survivors. DOE has also been in the forefront of biodosimetry research, including classical cytogenetics, FISH technology, and GPA.
Occupational Studies
In 1902, the first cancer death attributed to radiation occurred after occupational exposure of Thomas Edison's assistant, Clarence Dally.22 Except during radiation accidents,23 such heavy exposures are rarely experienced today.
Studies of pioneering radiologists, underground miners, and radium-dial painters contributed to our understanding of radiation effects and to the setting of radiation protection standards.24 Studies of radium-dial painters had been conducted for many years out of the Argonne National Laboratory. Radiation exposures in the workforce today are generally much lower than in the past, and any associated increases in cancer are difficult to detect. Combining and extending studies of occupationally exposed groups, similar to what has been done for underground miners,18 appears to be the approach for the 21st century.
Nuclear-Installation Workers
Studies within the nuclear industry might be able to validate the risk estimates obtained from high-dose and high-dose-rate investigations. For many years, DOE has supported analytic epidemiologic studies of the workforce involved with the Manhattan Project and with later aspects of weapons and energy development. Radiation doses of most workers are recorded, and cumulative exposures of some might reach levels where any adverse effects could be detected. Nearly 100,000 nuclear-industry workers in 3 countries were analyzed, and leukemia, but not
other cancers, was statistically significantly increased.5 Overall, only about 9 of the nearly 4,000 cancer deaths could be attributed to radiation. Even combined worker studies have difficulty in providing risk estimates of useful precision, however, when the range of exposures is narrow. Studies of electric-utility workers at nuclear-power stations might provide additional information on radiation risks because of the relatively high cumulative exposures possible.25,26 More work is required to account for other potential hazards in the workplace, such as asbestos, solvents, and paints.
Plutonium Workers
Little correlation between plutonium intake and cancer mortality has been found in studies of large populations of workers exposed to low levels of plutonium. Studies out of Los Alamos National Laboratory have been informative in this regard.27,28 Apparently, high-dose plutonium and gamma-ray exposures have caused excess lung cancers among over 2,300 workers at the Mayak radiochemical and plutonium production plants in Russia.29-31
Chernobyl-Cleanup Workers
Few epidemiologic studies of the more than 600,000 workers at Chernobyl who cleaned up the environment and entombed the damaged reactor have been published. Biologic-dosimetry studies confirm that the average dose received was about 10cGy,32,33 although doses as high as 35 cGy were permitted because of the need for emergency action. Thyroid screening of 2,400 Estonian cleanup workers with ultrasonography and needle biopsy of nodules revealed no radiation-related increase in thyroid disease.34 No excess incidence of leukemia or other cancers was found among nearly 5,000 workers from Estonia, and only death due to suicide was statistically significantly increased.35 Much larger studies will be required to provide information on the effects of protracted exposures,36 although a recent study of 155,000 Russian cleanup workers also failed to reveal a relationship between leukemia and recorded dose.37 Continuing collaborative research in the Ukraine and elsewhere has been supported by DOE and other agencies.
Radiologic Technologists
Radiologic technologists who likely received much-lower total doses than did radiologists38 have not been reported to be at increased risk for leukemia39 or breast cancer.40 A survey of 145,000 radiologic technologists in the United Stated should provide new information on radiation risks well into the next millennium, accounting for genetic susceptibility41 and other cancer risk factors.42
Airline Crews
Airline crews are exposed to higher levels of cosmic rays than are earth-bound workers, in addition to a variety of physical and chemical carcinogens. Commercial pilots and flights crews might receive annual doses on excess of 5 mSv. Small studies of female flight attendants and male civilian airline pilots have been conducted.43-45 If high-altitude supersonic travel is expanded, there is a potential for increased exposure of workers and of the general population to low-level high-LET radiation.
Atomic-Bomb Survivor Studies
The atomic bombs that ended World War II increased the rate of cancer in surviving populations.21,46 These critically important studies of survivors have been and continue to be supported by DOE and the government of Japan. Risk was somewhat higher among children than adults, among residents of Hiroshima than Nagasaki, and among females than males. The excess relative risk over time has remained constant for adults but is decreasing for survivors exposed as children. The incidences of some cancers—chronic lymphocytic leukemia; non-Hodgkin's
lymphoma; Hodgkin's disease; and cancers of the pancreas, rectum, cervix, testes, and prostate—were not statistically significantly increased.
The dose response for leukemia appears linear-quadratic, whereas a straight line adequately fits the data for all solid cancers combined. About 500 cancers (accounting for 7% of all cancer deaths and 1% of all deaths) were attributed to radiation from the atomic bombs. Recent application of tumor-registry data revealed excess cancers of the liver, thyroid, and skin.46 Little additional information will be obtained on Japanese survivors who were older than about 40 in 1945, inasmuch as most have died. The risk of radiogenic leukemia also appears to have run its course 40 yr after exposure. However, it will be well into the next millennium before lifetime risks among those who were exposed when young are fully assessed.
STUDIES OF PATIENTS GIVEN RADIOTHERAPY
Classic studies of populations medically irradiated for benign diseases (such as ankylosing spondylitis8) and malignant diseases (such as cervical cancer47) began in the 1950s and 1960s. Today, major advances in cancer therapy are allowing patients to live for many years, and the late effects of curative treatments have become important.48 Studies of long-term survivors of Hodgkin's disease,17 childhood cancer,10 breast cancer,49 and other cancers will continue to provide quantitative information on radiation risks that have both clinical and public-health importance. Studies of patients receiving whole-body irradiation for bone marrow transplants will be increasingly important as this modern treatment continues to cure patients. The largest combined study of over 20,000 patients who underwent bone marrow transplantations found substantially increased rates of second cancers attributable to high-dose, whole-body radiotherapy.50
STUDIES OF DIAGNOSTIC RADIATION
Other than radon, medical radiation contributes the most to population exposure. The first report that fractionated, low-dose radiation could result in breast cancer occurred in the 1960s, and modification of risk by dose and age was reported in the 1970s.51 Radiogenic breast-cancer risk decreases with age at exposure, and risk appears minimal for asymptomatic postmenopausal women undergoing screening with X-ray mammography.52 Periodic medical radiation exposures have not been linked to lung cancer and have been linked only weakly to leukemia. Fractionation appears to have little effect on reducing the risk of radiogenic breast cancer in tuberculosis patients repeatedly exposed to fluoroscopy, but it appears to reduce dramatically the risk of radiogenic lung cancer with a dose-rate effectiveness factor closer to 10 than to 2.53
STUDIES OF IODINE-131
It has been known since the 1950s that X-rays in childhood result in high rates of thyroid cancer. Exposure of the thyroid gland in adult life results in little if any cancer risk, whether from external photons54 or internal I-131.55 Thyroid cancer has not been linked to diagnostic uses of I-131, but results of a recent study suggest an association between thyroid nodular disease and I-131 administration.56 The reactor accident at Chernobyl was followed by remarkable excesses of thyroid cancers after childhood exposure, which have been attributed to radionuclides of iodine, including I-131.15,57-59 Other shorter lived isotopes of iodine possibly contributed to thyroid-cancer risk, as they did after the weapons-testing accident in the Marshall Islands in 1954.60,61 The studies of Marshall Islanders have been supported over the years by the Brookhaven National Laboratory. The precise nature of the thyroid-cancer risk among the residents near Chernobyl remains elusive and awaits dose-response evaluations and clarification of the role of screening, the contribution of specific radionuclides, the modifying effect of endemic goiter, and the apparent urban-rural and country differences in risk.57,62 The high excess within 5 yr of exposure seems to have come a bit too early to be consistent with results of prior studies.54 However, if the experience of X-irradiated populations holds, the excess of thyroid cancer should continue far into the 21st century. DOE, in cooperation with the National Cancer Institute and other agencies, is supporting comprehensive studies of thyroid cancer in Belarus and other countries of the former Soviet Union. No increase in leukemia has been attributed to Chernobyl radiation 63,64 or to medical I-131,65 in all likelihood because of the associated low bone-marrow dose.
STUDIES OF RADON AND RADON PROGENY
In 1556, Agricola described the mysterious lung disease afflicting miners of the Black Forest regions of eastern Europe, mentioning that the occupation was so hazardous that some women lost as many as 7 husbands to the dreaded condition.66 Inhaled radon and radon progeny were later indicted as the culprits. Today, underground-miner studies are prominent in assessing the risk posed by low-level general-population exposures to radon,67 although international residential studies might soon provide validation and guidance.68 Studies of underground miners convincingly demonstrate that radon is a potent carcinogen, able to cause lung cancer and to interact with cigarette-smoking in a way that increases risk.18 Estimates based on underground-miner exposures suggest that perhaps 10-14% of all lung cancers in the general population are related to breathing of residential radon. The possibility that radiation causes characteristic mutational features in lung-tumor cells has been investigated in several laboratories. It would be useful to be able to identify environmental carcinogens, including radon, on the basis of the genetic ''fingerprint'' that they leave on cells that become cancerous.69-71 Leukemias and other cancers did not occur in excess among underground miners.72
It has been difficult to detect a radon risk in residential case-control studies largely because the relative risk at 150 Bq m-3 is so low, only about 1.15.73 A recent study in Finland was negative despite a good design and relatively high exposure levels.74 However, even that investigation did not have the statistical power to reject the possibility that miner estimates are appropriate. The value of combining data is seen in a recent meta-analysis of 8 large case-control studies.19 Although the reasons for heterogeneity in risk estimates could not be clearly resolved, the combined estimate of the relative risk at 150 Bq m-3, 1.14, was statistically significant. The authors conclude that, on the basis of current understanding, there is no reason to reject the validity of miner estimates, and future combining of current and continuing studies75 will be required to evaluate fully the risk posed by indoor radon.
Large radon surveys have been conducted in the United Kingdom, where the average level of radon in homes is 20 Bq m-3, and the action level is 10 times the average (200 Bq m-3).76 A small number of homes exceed 2,000 Bq m-3, and 10,000 Bq m-3 was recorded in Cornwall. Although the importance of very low levels will continue to be debated into the 21st century, there should be no debate about the health hazard associated with the very high radon levels that can be experienced in some living quarters.
ENVIRONMENTAL-EXPOSURE STUDIES
Studies of populations exposed to high levels of natural background radiation have largely been negative or noninformative.2,24,77 In general, ecologic studies are the weakest form of epidemiologic research and have provided little insight into radiation risks.2,3,19 Unique studies of populations exposed to high levels of radioactive waste in the southern Urals of Russia, however, are being conducted and should continue into the next century.78 In 1957, a storage tank at the Chelyabinsk nuclear facility exploded (the Kyshtym accident) and released large amounts of radioactive waste into the Techa River. Before the accident, high-level radioactive wastes from the Mayak facility were dumped into the river.79 Leukemia has been reported to be in excess among the 28,000 residents,79 and doses as high as 400 cGy were reported. The studies have the potential to provide new information on the effects of ionizing radiation delivered over long periods. DOE is supporting a number of important initiatives in the southern Urals.
STUDIES OF SUSCEPTIBILITY AND INTERACTION
In the 1920s, osteosarcoma was first identified as a consequence of radium poisoning from excessive ingestion during watch-dial painting among young women.80 Today, radiogenic bone cancer remains a rare but important consequence of high-dose radiation exposure. Research into the 21st century is likely to clarify whether genetic susceptibility can increase the risk of radiogenic cancer, as among children with retinoblastoma.7,81,82 Interaction studies might be increasingly important as we go beyond determining and quantifying radiation risks. 83,84 Do radiotherapy and chemotherapy together increase the risk of leukemia85,86 or bone cancer81? Do
radiotherapy and cigarette-smoking increase the risk of lung cancer?87 Do indoor radon and cigarette-smoking increase the risk of lung cancer as they do in underground miners?18 Do stomach irradiation and surgery potentiate the development of stomach cancer?88 Do radiotherapy and immunosuppression interact to increase the risk of lymphomas or other malignancies?50 Is it possible that low-dose radiation can increase the risk of breast cancer in genetically predisposed persons? The evidence that heterozygosity for ataxia-telangiectasia (AT) can increase the risk of radiogenic breast cancer is not convincing.2 Recently, it has been questioned whether even AT heterozygosity itself is an important risk factor for breast cancer.89,90
CONCLUSION
There will be some clarification of radiation risks in the next century—notably those associated with exposures in childhood, in medicine, and in particular occupations—as well as of the effects of I-131, plutonium, and radon. There will be advances in biologic dosimetry. There will also be new opportunities to study cancer mechanisms. Does radiation interact with environmental carcinogens or underlying genetic predispositions to increase risk? Will the cancer risk after childhood exposure eventually return to normal? Can radiation, whether external or internal, leave molecular signatures that implicate it uniquely as an environmental cause of specific cancers? Can we learn about susceptibility states as we learn about radiation effects? The research focus in the 21st century will build on the foundation that has been laid during the last 100 yr of radiation experience. Continued support from DOE will remain critical, as it has been in the past, to capture the scientific knowledge from studies of atomic-bomb survivors (the childhood exposures), studies in Russia (chronic versus brief exposure effects), and studies of biomarkers of exposure (FISH, GPA, and tooth enamel).
REFERENCES
1. Boice JD Jr, Land CE, Preston DL. Ionizing radiation. In: Schottenfeld D, Fraumeni JF Jr, editors. Cancer epidemiology and prevention. New York: Oxford Press; 1996. p 319-54.
2. United Nations Scientific Committee on the Effects of Atomic Radiation. Sources and effects of ionizing radiation. Publ E.94.IX.11. New York: United Nations; 1994.
3. Boice JD Jr, editor. Implications of new data on radiation cancer risk. Bethesda, MD: National Council on Radiation Protection and Measurements; 1997. p 315.
4. March HC. Leukemia in radiologists. Radiology 1944;43:275-8.
5. Cardis E, Gilbert ES, Carpenter L, and others. Effects of low doses and low dose rates of external ionizing radiation: cancer mortality among nuclear industry workers in three countries. Radiat Res 1995; 142:117-32.
6. Rowland RE, Stehney AF, Lucas HF Jr. Dose-response relationships for female radium dial workers. Radiat Res 1978;76:368-83.
7. Eng C, Li FP, Abramson DH, and others. Mortality from second tumors among long-term survivors of retinoblastoma. J Natl Cancer Inst 1993;85:1121-28.
8. Court Brown WM, Doll R. Leukemia and aplastic anemia in patients irradiated for ankylosing spondylitis. Med Res Council Spec Rep. Series No. 295. HMSO: London; 1957.
9. Boice ID Jr, Engholm G, Kleinerman RA, and others. Radiation dose and second cancer risk in patients treated for cancer of the cervix. Radiat Res 1988;116:3-55.
10. Hawkins MM, Kinnier-Wilson LM, Stovall MA, and others. Epipodophyllotoxins, alkylating agents, and radiation and risk of secondary leukaemia after childhood cancer. Br Med J 1992;304:951-8.
11. Stewart A, Webb J, Hewitt D. A survey of childhood malignancies. Br Med J 1958;1:1495-508.
12. Delongchamp RR, Mabuchi K, Yoshimoto Y, and others. Cancer mortality among atomic bomb survivors exposed in utero or as young children, October 1950-May 1992. Radiat Res 1997;147:385-95.
13. Miller RW, Boice JD Jr. Cancer after intrauterine exposure to the atomic bomb. Radiat Res 1997;147:396-7[Commentary].
14. Hempelmann LH, Hall WJ, Phillips M, and others. Neoplasms in persons treated with x-rays in infancy: fourth survey in 20 years. J Nail Cancer Inst 1975;55:519-30.
15. Williams D, Pinchera A, Karaoglou A, and others, editors. Thyroid cancer in children living near Chernobyl. Report EUR 15248. Luxembourg: CEC; 1993.
16. Boice JD Jr, Preston D, Davis FG, and others. Frequent chest x-ray fluoroscopy and breast cancer incidence among tuberculosis patients in Massachusetts . Radiat Res 1991;125:214-22.
17. Bhatia S, Robison LL, Oberlin O, and others. Breast cancer and other second neoplasms after childhood Hodgkin's disease. N Engl J Med 1996;334:745-51.
18. Lubin JH, Boice JD Jr, Edling C, and others. Lung cancer in radon-exposed miners and estimation of risk from indoor exposure. J Natl Cancer Inst 1995;87:817-27.
19. Lubin JH, Boice JD Jr. Lung cancer risk from residential radon: meta-nalysis of eight epidemiologic studies. J Nail Cancer Inst 1997;89:49-57.
20. Beebe GW, Kato H, Land CE. Studies of the mortality of A-bomb survivors. 6. Mortality and radiation dose, 1950-1974. Radiat Res 1978;75:138-201.
21. Pierce DA, Shimizu Y, Preston DL, and others. Studies of the mortality of A-bomb survivors. Report 12, Part 1. Cancer 1950-1990. Radiat Res 1996; 146:1-27.
22. Brown P. American martyrs to science through the roentgen rays. Springfield, IL: Charles C Thomas; 1993.
23. Saenger EL. Radiation accidents. Ann Emerg Med 1986;15:1061-6.
24. Boice JD Jr, Lubin JH. Occupational and environmental radiation and cancer. Cancer Causes Control 1997;8:312-25.
25. Jablon S, Boice JD Jr. Mortality among workers at a nuclear power plant in the United States. Cancer Causes Control 1993;4:427-30.
26. Muirhead CR, Boice JD Jr, Raddatz CT, and others. Comparison of dose histories for U.S. nuclear power plant workers, based on records held by a major dosimetry service company and on the NRC REIRS database. Health Phys 1996;70:645-50.
27. Voelz GL. Health considerations for workers exposed to plutonium. Occup Med 1991;6:681-694.
28. Voelz GL, Lawrence JNP. A 42-year medical follow-up of Manhattan Project plutonium workers. Health Phys 1991;61:181-190.
29. Koshurnikova NA, Shilnikova N, Okatenko P, and others. The risk of cancer among nuclear workers at the "Mayak" Production Association: preliminary results of an epidemiological study. NCRP Proc 1997;18:113-21.
30. Koshurnikova NA, Bysogolov GD, Bolotnikova MG, and others. Mortality among personnel who worked at the Mayak complex in the first years of its operation. Health Phys 1996;71:90-3.
31. Burkhart W, Kellerer AM, editors. Radiation exposure in the southern Urals. Sci Total Environ 1994;142:1-125.
32. Bigbee WL, Jensen RH, Rahu M, and others. Glycophorin A biodosimetry in Chemobyl cleanup workers from the Baltic countries. Br Med J 1995;312:1078-9.
33. Bigbee WL, Jensen RH, Viedebaum T, and others. Biodosimetry of Chemobyl cleanup workers from Estonia and Latvia using the glycophorin A in vivo somatic cell mutation assay. Radiat Res 1997;147:215-25.
34. Inskip PI, Hartshome MF, Tekkel M, and others. Thyroid nodularity and cancer among Chernobyl cleanup workers from Estonia. Radiat Res 1997;147:225-35.
35. Rahu M, Tekkel M, Veidebaum T, and others. The Estonian study of Chernobyl cleanup workers: II. Incidence of cancer and mortality. Radiat Res 1997;147:641-52.
36. Karaoglou A, Desmet G, Kelly GN, and others, editors. The radiological consequences of the Cherobyl accident. EUR 16544EN. Brussels: European Commission; 1996.
37. Ivanov VK, Tsyb AF, Konogorov AP, and others. Case-control analysis of leukaemia among Chemobyl accident emergency workers residing in the Russian Federation 1986-1993. J Radiol Prot 1997;17:137-57.
38. Wang JX, Inskip PD, Boice JD Jr, and others. Cancer incidence among medical diagnostic x-ray workers in China, 1950 to 1985. Int J Cancer 1990;45:889-95.
39. Jablon S, Miller RW. Army technologists: 29-year follow up for cause of death. Radiology 1978;126:677-9.
40. Boice JD Jr, Mandel JS, Doody MM. Breast cancer among radiologic technologists. JAMA 1995;274:394-401.
41. Struewing JP, Tarone RE, Brody LC, and others. BRCA1 mutations in young women with breast cancer. Lancet 1996;347:1493.
42. Boice JD Jr, Mandel JS, Doody MM, and others. A health survey of radiologic technologists. Cancer 1992;69:586-98.
43. Pukkala E, Auvinen A, Wahlberg G. Incidence of cancer among Finnish airline attendants, 1967-92. Br Med J 1995;311:649-52.
44. Lynge E. Risk of breast cancer is also increased among Danish female airline cabin attendants. Br Med J 1996;312:253.
45. Band PR, Le ND, Fang R, and others. Cohort study of Air Canada pilots: mortality, cancer incidence, and leukemia risk Am J Epidemiol 1996;143:137-43.
46. Thompson DE, Mabuchi K, Ron E, and others. Cancer incidence in atomic bomb survivors. Part II: Solid tumors, 1958-1987. Radiat Res 1994;137:S17-67.
47. Hutchison GB. Leukemia in patients with cancer of the cervix uteri treated with radiation. A report covering the first 5 years of an international study. J Natl Cancer Inst 1968;40:951-82.
48. Curtis RE. Second cancers following radiotherapy for cancer. NCRP Proc 1997;18:79-94.
49. Boice JD Jr, Harvey E, Blettner M, and others. Cancer in the contralateral breast after radiotherapy for breast cancer. N Engl J Med 1992;326:781-5.
50. Curtis RE, Rowlings PA, Deeg HJ, and others. Solid cancers after bone marrow transplantation. N Engl J Med 1997;336:897-904.
51. Boice JD Jr, Monson RR. Breast cancer in women after repeated fluoroscopic examinations of the chest. J Natl Cancer Inst 1977;59:823-32.
52. Howe GR, McLaughlin J. Breast cancer mortality between 1950 and 1987 after exposure to fractionated moderate-dose-rate ionizing radiation in the Canadian fluoroscopy cohort study and a comparison with breast cancer mortality in the atomic bomb survivors study. Radiat Res 1996;145:694-707.
53. Howe GR. Lung cancer mortality between 1950 and 1987 after exposure to fractionated moderate-dose-rate ionizing radiation in the Canadian fluoroscopy cohort study and a comparison with lung cancer mortality in the atomic bomb survivor study. Radiat Res 1995; 142:295-304.
54. Ron E, Lubin JH, Shore RE, and others. Thyroid cancer after exposure to external radiation: a pooled analysis of seven studies. Radiat Res 1995;141:259-77.
55. Hall P, Mattsson A, Boice JD Jr. Thyroid cancer following diagnostic administration of iodine-131. Radiat Res 1996;145:86-92.
56. Hall P, Fürst CJ, Mattsson A, and others. Thyroid nodularity after diagnostic administration of iodine-131. Radiat Res 1996;146:673-80.
57. Nagataki S. Nagasaki symposium on Chernobyl: update and future. Amsterdam: Elsevier; 1994.
58. Kazakov VS, Demidchik EP, Astakhova LN. Thyroid cancer after Chernobyl. Nature 1992;359:21.
59. Stsjazhko VA, Tsyb AF, Tronko ND, and others. Childhood thyroid cancer since accident at Chernobyl. Br Med J 1995;310:801.
60. Conard RA. Late radiation effects in Marshall Islanders exposed to fallout twenty-eight years ago. In: Boice JD Jr, Fraumeni JF Jr, editors. Radiation carcinogenesis: epidemiology and biological significance. New York: Raven Press; 1984. p 57-71.
61. Robbins J, Adams WH. Radiation effects in the Marshall Islands. In: Nagataki S, editor. Radiation and the thyroid. Tokyo: Exerpta Medica; 1989. p 11-24.
62. Boice JD Jr, Linet M. Chemobyl, childhood cancer and chromosome 21. Br Med J 1994;309:139-40; Br Med J 1994;309:1300[Editorial].
63. Ivanov EP, Tolochko G, Lazarev VS, and others. Childhood leukaemia after Chernobyl. Nature 1993;365:702.
64. Parkin DM, Clayton D, Black RJ, and others. Childhood leukaemia in Europe after Chernobyl 5-year follow-up. Br J Cancer 1996;73:1006-12.
65. Hall P, Boice JD Jr, Berg G, and others. Leukaemia incidence after iodine-131 exposure. Lancet 1992;340:1-4.
66. Agricola C. De re metallica. Basel, 1556. New York: Dover Publications; 1950 English reprint (Hoover translation). p 214.
67. Lubin JH, Tomasek L, Edling C, and others. Estimating lung cancer mortality from residential radon using data for low exposures of miners. Radiat Res 1997;147:126-34.
68. Boice JD Jr. Radon, your home or mine? Radiat Res 1997;147:135-7 [Commentary].
69. Vahakangas KH, Samet JM, Metcalf RA, and others. Mutations of p53 and ras genes in radon-associated lung cancer from uranium miners. Lancet 1992;339:576-80.
70. Bennett WP. Does radiation cause molecular signatures? NCRP Proc 1997;18:223-7.
71. De Benedetti VMG, Travis LB, Welsh JA, and others. p53 mutations in lung cancer following radiation therapy for Hodgkin's disease. Cancer Epidemiol Biomark Prey 1996;5:93-8.
72. Darby SC, Whitley E, Howe GR, and others. Radon and cancers other than lung cancer in underground miners: a collaborative analysis of 11 studies. J Natl Cancer Inst 1996;87:378-84.
73. Alavanja MCR, Brownson RC, Benichou J, and others. Attributable risk of lung cancer in lifetime nonsmokers and long-term ex-smokers (Missouri, United States). Cancer Causes Control 1995;6:209-16.
74. Auvinen A, Makelainen I, Hakama M, and others. Indoor radon exposure and risk of lung cancer: a nested case-control study in Finland. J Natl Cancer Inst 1996;88:966-72.
75. Wang Z-Y, Lubin JH, Wang L-D, and others. Radon measurements in underground dwellings from two prefectures in China. Health Phys 1996;70:192-8.
76. Bradley EJ, Lomas PR, Green BMR, and others. Radon in dwellings in England: 1997 review. NRPB-RR293. Chilton: NRPB; 1997.
77. Wang Z, Boice JD Jr, Wei L, and others. Thyroid nodularity and chromosome aberrations among women in areas of high background radiation in China. J Natl Cancer Inst 1990;82:478-85.
78. Kossenko MM, Deyteva MD, Vyushkova OV, and others. Issues in the comparison of risk estimates for the population in the Techa River region and atomic bomb survivors. Radiat Res 1997;148:54-63.
79. Kossenko MM, Degteva MO. Cancer mortality and radiation risk for the Techa River population. Sci Total Environ 1994; 142:73-89.
80. Martland HS. The occurrence of malignancy in radioactive persons: a general review of data gathered in the study of the radium dial painters with special reference to the occurrence of osteogenic sarcoma and the interrelationship of certain blood diseases. Am J Cancer 1931; 15:2435.
81. Tucker MA, D'Angio GJ, Boice JD Jr, and others. 1987 bone sarcomas linked to radiotherapy and chemotherapy in children. N Engl J Med 1987;317:588-93.
82. Wong FL, Boice JD Jr, Abramson DH, and others. Cancer incidence after retinoblastoma: radiation dose and sarcoma risk. JAMA 1997; 278:1262-7.
83. Sankaranarayanan K, Chakraborty R. Cancer predisposition, radiosensitivity and the risk of radiation-induced cancers. I. Background. Radiat Res 1995;143:121-43.
84. Strong LC. Genetic and environmental interactions. Cancer 1977;40:1861-6.
85. Curtis RE, Boice JD Jr, Stovall M, and others. Risk of leukemia after chemotherapy and radiation treatment for breast cancer. N Engl J Med 1992;326:1745-51.
86. Travis LB, Weeks J, Curtis RE, and others. Leukemia following total body irradiation and chemotherapy for non-Hodgkin's lymphoma. J Clin Oncol 1996;14:565-71.
87. van Leeuwen FE, Klokman WJ, Stovall M, and others. Roles of radiotherapy and smoking in lung cancer following Hodgkin's disease. J Natl Cancer Inst 1995;87:1530-7.
88. Griem ML, Kleinerman RA, Boice JD Jr, and others. Cancer following radiotherapy for peptic ulcer. J Natl Cancer Inst 1994;86:842-9.
89. FitzGerald MG, Bean JM, Hedge SR, and others. Heterozygous ATM mutations do not contribute to early onset of breast cancer. Nat Genet 1997;15:307-10.
90. Vorechovsy I, Rasio D, Luo L, and others. The ATM gene and susceptibility to breast cancer: analysis of 38 breast tumors reveals no evidence for mutation. Cancer Res 1996;56:2726-32.
Discussant
Roger McClellan
President, Chemical Industry Institute of Technology
Research Triangle Park, North Carolina
What I want to do in 10 min is try to emphasize a couple of basic points related to one of the themes we have heard about today: the various paradigm shifts that are occurring.
We want to talk a little about risk-based decision-making and about the need to replace fear with scientific information. Much of our decision-making with respect to environmental and occupational factors has been driven by public fear. The scientific community has contributed to the development of that fear, but at the same time we have the paradox that we as scientists have been trying to create the scientific information that can tip the scales and let science and information weigh in the decision-making process.
The radiation arena, for example, illustrates what I'm pointing out here. Radiation is probably the risk factor that we know the most about, but it still ranks at the top of the list of public fears. Why is that? In our zeal to obtain funding to understand radiation, have we contributed to the development of public fear? In doing so, perhaps we have pushed aside concern for other risk factors.
We need to adopt a systems orientation as we look at human health risks, and our range of concern must extend from sources to human health responses. One of the contributions of the radiation community has been such a systems orientation. The understanding of internal dose came from the radiation field.
For many years, our approach to managing risks was pragmatic: we were attempting to get linkages between responses and exposures. Now for perhaps the last 15 yr, in a more-structured risk paradigm, we have attempted to focus on hazard characterization, hazard identification, dose-response relationships, and exposure assessments to characterize risk.
We might have given too much emphasis to identifying hazards without putting them into the perspective of overall risk characterization. But much of what has been developed with regard to that characterization has its roots in the radiation community and in the work that has been supported by the Department of Energy (DOE) and its predecessor organizations.
We have heard about the dilemma of detecting low levels of excess risk and the need to extrapolate. Typically, we are talking in terms of occupational studies, in which we can have confidence in data that show relative risks over 2. But we are squeamish when we get down to relative risks of 1.5. Today, we are starting to probe relative risks of around 1.05 and develop public policy based on them. In fact, our public-policy concerns go down into relative-risk regions where we must deal with extrapolation.
We can now measure not only the compounds in our environment, but what might be called punitive preneoplastic or predisease entities in the human body. How do we interpret those? Scientists have a special responsibility to point out clearly what we know but also to point out where we are extrapolating.
The other extrapolation involves going from the cells in vivo to the complex, integrated human organism. As our ability to probe molecular and cellular events increases, we move into a paradigm shift where we can no longer assume that every change is an adverse effect.
What happens in the complex integrated organism is an interplay of thousands of events. That puts a special responsibility on us as we try to interpret the results of our studies of potentially toxic agents in the cell system or in the laboratory animal system as they might apply to humans, frequently at doses that are much lower than those we are using here.
Dr. Hood has appropriately noted the extent to which we are going to have to continue to try to marry the field of laboratory science with the more population-based science that is needed for risk-based decision-making. A superb retrospective analysis by Samet and colleagues used Philadelphia data from 1974 to 1988. Philadelphia had a population of about 1.5 million. About 50 people a day died. Our challenge is to understand what factors are at play in causing such deaths, and what factors we can influence so that people can have not only a longer life, but also more-productive and more-enjoyable life.
We obviously are concerned that air pollution is one of the factors. Just as the deaths were of varied causes, the air pollutants were varied. But there are trends. If we examine the data closely, we see that some of them are interrelated. A major challenge is to tease out associations between risk factors and mortality patterns.
If we are going to make sense out of all this, we need improvements in the information base across the board, including information on individuals and on what they are dying of. Therein lies the continued tension between our desire collectively to have more information and our desire individually to have privacy. That is related to the ethical issue, and I am pleased to see that in the DOE program and in the National Institutes of Health program ethical issues have been given an important role.
We need to keep in mind that despite our attention to single risk factors, the real concern is not the small individual relative risks, but all the risk factors. A big cofactor in risk for many diseases is cigarette-smoking. We have educated a whole cadre of scientists who started to focus on particular risk factors as graduate students, and they have made a career out of studying those risk factors. They find it easy to talk only about those risk factors and forget the relative association. Radon in homes is a potential calculable risk factor, but it is important to keep in mind that about 80% or so of that risk is also attributable to cigarette-smoking. We need to talk about both, and we need to talk about the others that go along with them.
We have moved into an era in which we are trying to use risk assessments, make better use of risk-based decision-making, and make risk-management decisions. We need to continue to try to identify the research that will improve risk assessments; and we have to continue to seek means to communicate the total risk process and risk characterization to all stakeholders.
Discussant
Kenneth Olden
Director, National Institute of Environmental
Health Sciences and National Toxicology Program
Research Triangle Park, North Carolina
Coauthor
Janet Guthrie
Office of Policy, Planning and Evaluation
National Institute of Environmental Health Sciences
Research Triangle Park, North Carolina
Pity today's environmental-health policy-makers. Not only do they set policy that is guaranteed to make enemies—of those who feel that it is needlessly costly and those who feel that it is needlessly weak—but also they must make decisions on the basis of too little information. And in addition to their having too little information, much of the information that they have is not directly relevant to the real-world situations in which it is applied.
Intelligent environmental-health policy is a necessity. We as a nation have made great strides in studying and treating end-stage disease. Increasingly, though, we are coming to understand the costs of focusing so strongly on end-stage disease, both in terms of economic costs and in terms of the costs of human suffering. It would be much better to prevent diseases in the first place, a possibility that can be realized by better understanding and control of the environmental components of disease. Diseases arise from the complex interaction of our environmental exposures, our individual susceptibility to the effects of exposures, and the age or time at which we experience the exposures. Each of those components must be better understood; armed with a stronger scientific foundation, environmental-health policy that best serves real communities could be developed.
At present there are a number of bottlenecks to developing the ideal world of rational decision-making that would engender confidence among the American people.1 Four of them will be discussed here: The National Institute of Environmental Health Sciences (NIEHS) is committing funds and effort to defining the parameters of individual susceptibility to individual toxicants, to developing innovative toxicity tests that are faster and cheaper that the current models, to exploring how chemical toxicity changes when toxicants are present in mixtures, and to assessing actual environmental exposures as they occur in a sample of the American people. This more complete knowledge base will greatly improve the tools used in designing environmental-health policy.
INDIVIDUAL SUSCEPTIBILITY
There is an urgent need to determine the genetic basis of individual susceptibility to disease, particularly to the environmental components of disease. To that end, NIEHS recently announced plans for a new Environmental Genome Project. It will be a multiyear, multicenter endeavor that will sequence the 200 or more genes that we think predispose people to environmentally associated disease. It will also be a transagency endeavor, requiring collaboration with the National Institute for Human Genome Research, the National Cancer Institute, and others. The first phase of this study is expected to last 5 yr and to cost $60 million. More details on this project are given at the end of this paper.
Clearly, we are not all equal, or homogeneous, with respect to our susceptibility to the effects of environmental exposures. Not everyone who smokes develops lung cancer, and not everyone exposed to carcinogens or toxicants gets sick. It is time to undertake a national effort to identify polymorphisms that account for this interindividual variation, relate the variation to population groups, and incorporate the resulting knowledge into commonsense, science-based knowledge of genes that code for P450 enzymes and DNA-repair enzymes, genes that direct cell-cycle control, genes for the metabolism of nucleic-acid precursors, and genes for signal-transduction systems. (Others in this volume, such as Anthony V. Carrano, have mentioned the importance of several of these genes.)
IMPROVED TOXICITY TESTING
There is an urgent need to develop toxicity tests that are faster and less expensive. The NIEHS National Toxicology Program (NTP) has been advocating this need for several years and has brokered collaborations with the Chemical Industry Institute for Toxicology, the Environmental Protection Agency (EPA), the Food and Drug
Administration, and a number of industry groups. To date, we have identified 2 transgenic-animal models that can identify carcinogens in 6 mo, rather than in the standard 2 yr.2,3 The need for fewer animals and histopathologic slides also reduces the evaluation time of these studies, which currently extends them to 5 yr and more. Studies using these new models can be done at about one-tenth the cost of the 2-yr rodent bioassay.
The models are still being evaluated, but the results are encouraging. One boon to risk assessment is that the new models appear to minimize the strain-specific effects found in conventional rodent models, which so often confound the risk-assessment decisions based on them. On the basis of these successes, we have increased confidence in, and enthusiasm for, our ability to develop models that generate toxicity data faster and less expensively. It is our further hope to improve on even the 6-mo timetable. In the future, models can be developed with recombinant-DNA methods, combinatorial chemistry, knowledge of mechanisms, and structure-based prediction that could provide a high-throughput technology to identify carcinogens and other toxicants in a matter of days.
MIXTURES
Although toxicity studies are conducted on single agents, we are seldom exposed to 1 agent at a time. We live in an environment composed of a complex and changing mixture of natural and synthetic chemicals. Consider the number of chemicals in the air we breathe, in the water we drink, and in the food we consume, and you see how poorly toxicity studies capture the complexity.
The important question is, How do these agents interact? Synergistically, antagonistically, additively? We already know of several examples of synergism of carcinogens, such as that shown by concurrent exposure to cigarette smoke and radon4 and, in the case of liver cancer, to aflatoxin B 1 and hepatitis B virus.5,6
Clearly, then, we have evidence of synergistic interactions, both among chemicals and between chemical and physical agents. That possibility raises concern in light of current awareness that chemicals in our environment can have estrogenlike activity. These environmental estrogens individually have very weak estrogenic activity compared with our bodies' own natural estrogens. What is unclear is their synergistic or multiplicative potential, which will remain bothersome until we complete studies that validate or refute the possibility. At NIEHS, there are numerous efforts to elucidate the potential health effects of environmental estrogens. Furthermore, we plan to release a request for applications that will solicit university-based scientists to propose new studies in complex-mixtures research.
EXPOSURE ASSESSMENT
We do not know what chemicals the American people are exposed to. Exposures are estimated on the basis of EPA toxic-release and production-inventory databases, but the estimates indicate only the potential for exposure, rather than actual exposures. Real-world exposure-assessment studies are needed to address this information gap.
We are all different in our uptake and metabolism of environmental toxicants. We need studies to determine actual biologic exposures; that is, how much of a substance that someone is exposed to is absorbed and distributed systemically. We need to define the biologically effective dose for individual toxicants. We will probably discover that the biologically effective dose is different for different people; this is another aspect of individual susceptibility.
Exposure-assessment studies can and should be done now. NIEHS NTP is developing a partnership with the Centers for Disease Control and Prevention to incorporate more exposure assessment into the NHANES-IV study. Results will help to answer the question, Of the 70,000-80,000 synthetic chemicals in the United States, how many should we be worried about? Is the number 40? 50? 1007 200?
Exposure-assessment data can be analyzed in different ways to determine whether there are important differences in toxicant uptake between population groups. Results can be analyzed by sex, socioeconomic status, or age. Age is an important way to separate and analyze data because children's exposures can vary dramatically from those of adults, both because of their smaller body mass and because of behavioral differences. Analyzing the data
by socioeconomic status could also help us to determine whether the poor, because of where they live and work, have exposure profiles different from those of the rest of the population.
ENVIRONMENTAL GENOME PROJECT
The Environmental Genome Project was officially announced during NIEHS testimony before Congress at appropriation hearings in April 1996. The announcement was received with much enthusiasm and interest and has been the subject of numerous articles in the press.
Since then, NIEHS management has met with Harold Varmus, director of the National Institutes of Health; Francis Collins, director of the National Institute of Human Genome Research; Richard Klausner, director of the National Cancer Institute; and other institute directors. Many have committed funds to the project. In October 1997, we convened a working group and a study group to design the project and develop appropriate safeguards.
Basically, the project will involve sequencing the allelic variants of genes known to control various processes. The technologies developed by the Human Genome Project will enable the Environmental Genome Project to proceed more rapidly than previously has been possible. The multicenter project is expected to involve many of the same centers as did the Human Genome Project. We expect to sequence environmental-susceptibility genes in about 1,000 persons from several population groups. The study group will help to define the numbers needed to ensure a statistically reliable sample.
It is possible that 20-50% of the population will have mutations or polymorphisms of some of the genes that predispose them to disease arising from unique environmental exposures. If that is true, the impact on public health will be enormous. The taxpayers' return on understanding frequently occurring genetic polymorphisms that lead to disease will obviously be much greater than that generated by sequencing and studying rare-disease genes that affect, at most, 1% of the population. Thus, the Environmental Genome Project has the potential to have a tremendous public-health effect.
REFERENCES
1. Olden K, Guthrie J. New frontiers in environmental health research. In: Rom WN, editor. Environmental and occupational Medicine, 3rd ed. Baltimore: Lippincot-Raven, in press.
2. Spalding JW, Momma J, Elwell MR, Tennant RW. Chemically induced skin carcinogenesis in a transgenic mouse line (TG.AC) carrying a v-Ha-ras gene. Carcinogenesis 1993;14:1335-41.
3. Tennant RW, French JE, Spalding JW. Identifying chemical carcinogens and assessing potential risk in short-term bioassays using transgenic mouse models. Environ Health Perspect 1995;103:942-50.
4. Finkelstein MM. Clinical measures, smoking, radon exposure, and risk of lung cancer in uranium miners. Occup Environ Med 1996;53:697-702.
5. Ross RK, Yuan J-M, Yu MC, Wogan GN, Qian G-S, Tu J-T, Groopman JD, Gao Y-T, Henderson BE. Urinary aflatoxin biomarkers and risk of hepatocellular carcinoma. Lancet 1992;339:943-6.
6. Wang J-S, Qian G-S, Zarba A, He X, Zhu Y-R, Zhang B-C, Jacobson L, Gange S J, Munoz A, Kensler TW, Groopman JD. Temporal patterns of aflatoxin-albumin adducts in hepatitis B surface anitgen-positive and antigen-negative residents of Daxin, Qidong County, People's Republic of China. Cancer Epidemiol Biomarkers Prev 1996;5:253-261.
Discussant
E. Morton Bradbury
Life Sciences Division Director, Los Alamos National Laboratory
Los Alamos, New Mexico
and
Professor of Biological Chemistry, School of Medicine
University of California, Davis
The core of structural biology in this country now lies in the unique facilities provided by the Department of Energy (DOE) for the scientific community. The premiere technique is X-ray diffraction, particularly the immense capabilities provided by the synchrotron radiation sources at Brookhaven, Berkeley, and Argonne national laboratories and at Cornell and Stanford universities. Synchrotron radiation sources have 2 major advantages over the conventional X-ray sources available in the laboratories of academic and industrial scientists: The finely collimated synchrotron X-ray beams are at least hundreds of times more intense than conventional X-ray sources, and the wavelengths of synchrotron X-rays are tunable. These capabilities have enormously extended our ability to solve the structures of proteins, nucleic acids, and their multisubunit complexes rapidly.
Several other techniques complement these X-ray capabilities, particularly the neutron beams provided at Argonne, Brookhaven, and Los Alamos national laboratories. X-ray diffraction can locate the heavier atoms (such as carbon, nitrogen, oxygen, and phosphorus) in biologic macromolecules. In contrast, the hydrogen atom is a very weak scatterer of X-rays and can be detected only in the very few crystal structures of the highest resolution. Neutron diffraction complements X-ray diffraction because of its ability to locate hydrogen easily and provide the orientation of water molecules and the direction of hydroxyl groups, data that are important for understanding the catalytic and other functions of proteins and nucleic acids. In elucidating the low-resolution solution structures of large multisubunit protein and protein-nucleic-acid complexes, neutron scatter is the technique of choice because the scattering length of hydrogen has the opposite sign to that of the other atoms found in biologic macromolecules and also to that of deuterium. Those differences can be exploited to determine the spatial relationships of proteins and nucleic acids in complex biologic assemblies. In addition to the above applications, inelastic neutron scatter can be used to study protein dynamics and the role of dynamics in protein functions.
Multidimensional nuclear magnetic resonance (NMR) spectroscopy is used widely in academic and industrial laboratories to solve the solution structures of low-molecular-weight proteins and nucleic acids. This technique is limited to protein molecular weights up to about 30,000 daltons because of the spectral line broadening that accompanies the slower tumbling of larger molecules. The size limit can be extended by the specific labeling of proteins with stable isotopes provided by DOE and National Institutes of Health resources. It can be extended also by increasing the field strengths of the superconducting magnets used with NMR. The Environmental Molecular Sciences Laboratory at Pacific Northwest Laboratory is working with Oxford Magnetics to extend the magnetic-field strength to give a frequency of I GHz for protons, which is probably the limit of the current superconducting metal-alloy wire. This very high field strength, with specific labeling with stable isotopes, will provide users with the most-advanced capabilities for multidimensional NMR.
Other important techniques include the use of the high-field scanning transmission electron microscope at Brookhaven National Laboratory and high-field mass spectrometers at Pacific Northwest Laboratory. The emerging atomic-probe microscopies have the potential to revolutionize structural biology at low and intermediate resolutions. Current interest in these techniques stems from their ability to image hydrated biologic macromolecules adsorbed on fiat surfaces. In addition to the structural information obtained, ligands can be attached to the probe tips and used to examine samples. Under development is a range of atomic-probe tips with different chemical and physical characteristics designed to address specific questions concerning the function of macromolecules, organelles, and cells.
The DOE structural-biology facilities (figure 1) provide resources for both internal and external users in the United States. The responsibilities of the internal users are to extend the capabilities of the facilities and to apply
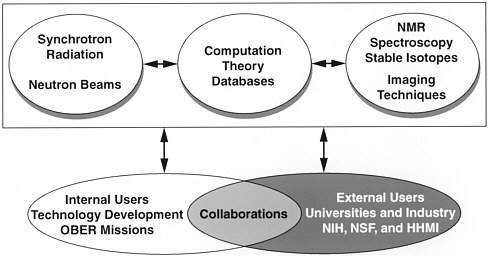
FIGURE 1 DOE structural-biology programs.
them to the research missions of the DOE. In addition, they provide an interface with external users to exploit the facilities efficiently and to collaborate on projects of joint interest.
Structural-biology capabilities similar to those available in this country are enjoyed by scientists in Europe and Japan. However, as discussed in an article in Nature,1 there are serious concerns about the ''neutron drought'' in this country. The situation is exacerbated by the closing down of the High Flux Beam Reactor at Brookhaven National Laboratory; for the future of neutron science in this country, it is hoped that the shutdown does not become permanent. The neutron-spallation source at Los Alamos National Laboratory is being upgraded, but this will only partially alleviate the demand for neutrons by the scientific community. According to the Nature article, there are 3,000 regular neutron users in Europe, compared with 1,000 in this country. Most of the users have backgrounds in condensed-matter physics and chemistry. The smaller contingent of structural biologists uses neutron beams to extend the findings of X-ray crystallography by locating hydrogen atoms, water molecules, and ions and to determine the low-resolution organization of complex multisubunit protein and protein-nucleic-acid complexes.
In comparison with the neutron-beam situation, synchrotron radiation beams are becoming widely available. Synchrotron beams extend our capabilities beyond those of conventional X-ray sources in 3 ways: Regular crystal structures can be solved more rapidly; the structures of biologic macromolecules and their complexes that give small or irregular crystals can be solved; and the structures of complex biologic assemblies, such as viruses and multisubunit complexes, can be solved in a reasonable period. The third is very important because the major trend in biomedical sciences is toward increased complexity. All aspects of DNA processing—transcription, replication, and damage repair involve multiprotein-DNA complexes. The process of transcription in yeast requires no fewer than 50 proteins. In addition, many of those proteins are subjected to reversible chemical modifications, such as phosphorylation. How do we approach such complexity? The starting point for the ultimate understanding of such complex processes includes the structures of the individual components and of their complexes at different stages. Similar structural and temporal complexities are involved in most biologic processes, and there is an urgent need to develop approaches to explain these complexities and the rules governing the assembly of complex biologic systems.
The initiation of the Human Genome Project by the Office of Health and Environmental Research, now the Biological and Environmental Research (BER) program, resulted from recognition that the full understanding of the health effects of DNA damage by radiation and chemicals would require detailed knowledge of the sequence organization and packaging of the human genome. The project has developed into major national and international genome programs, and the sequencing of the human genome is expected to be completed within the next decade. More recently, BER initiated a project directed at sequencing the genomes of microbes relevant to the missions of DOE in the environmental sciences, in environmental remediation, and in the development of responses to biologic threats. Progress in the human-and microbial-genome projects has far exceeded expectations, and sequence data are rapidly being accumulated. Cognizant of these successes, the scientific community is discussing the exploitation of the science embodied in the sequence data. With the increased capabilities in synchrotron radiation facilities, an explosion in the number of structures that will be solved by X-ray diffraction can be predicted with confidence; this has major relevance to the human-and microbial-genome projects and depends on their success.
A major structural problem associated with deciphering the human genome concerns the structure-function relationships of the proteins coded by the very large number of unknown genes in it. Most structures under investigation are of proteins with known functions. These make up a small subset of the proteins coded by the estimated 60,000-100,000 human genes. The problem of understanding the functions of such a large number of unknown proteins is formidable and will be the task of the functional-genomics component of the Human Genome Project and of molecular and cellular biology. The initial phase of functional genomics is to devise strategies to provide information on the possible functions of unknown proteins; for example, an antibody to an unknown protein would allow its subcellular location to be determined. A structural approach to functional genomics would involve the identification of the complete alphabet of structural motifs that are required for specific protein functions, such as DNA binding. From the identification of particular sequence-structure motifs, the possible functions of unknown proteins might be predicted from their gene sequences. In biology, however, there are always exceptions to general rules, and proteins with quite different structures have evolved with identical functions. As proposed by Joel Berendzen and Thomas Terwilliger of Los Alamos National Laboratory; David S. Eisenberg of the University of California, Los Angeles; and Sung-Hou Kim of the University of California, Santa Barbara, the long-term rational approach to functional genomics would be a major national and international effort to determine a representative set of protein structures that would provide the database to model or predict the structures of any protein on the basis of its gene sequence. A structure-determination project of this magnitude could be organized as a protein-structure initiative.
The purpose of such a protein-structure initiative would be to obtain the structures and functions of proteins from their sequences. Such an initiative would require a coordinated effort to obtain the required structural database and informatics. With such a coordinated effort, the "basis set" of protein structures required to predict unknown structures at a coarse level could be obtained in 5-10 yr. A more-comprehensive basis set, which would allow the precise prediction of these structures from their sequences, might take 10-20 yr. The impact of such databases of structure would be enormous for the biotechnology industry and the health industry and for our fundamental understanding of how enzymes and other biomolecules function. Moreover, the comprehensive basis set will provide structural and thereby functional information about genetic polymorphisms and how they contribute to individual susceptibilities to environmental stresses. Existing protein-structure databases should be coordinated with the Human Genome Project in determining priorities for protein-structure determinations. Determination of the structure of an unknown protein would have priority over determination of the structure of a new member of an existing protein family. A protein-structure initiative would require large-scale capabilities for the overexpression, purification, and crystallization of the many unknown proteins identified by the Human Genome Project.
As mentioned previously, a major direction in the biomedical sciences is toward increased complexity. In biologic processes that involve a large number of known proteins assembled into multisubunit protein complexes or protein-nucleic-acid complexes, pooling strategies, such as those proposed by David Torney (Los Alamos National Laboratory), are required to determine the order of assemblies of such complexes. In the case of a protein of unknown function, it is necessary to determine whether it functions alone or in combination with other protein
FIGURE 2 Relationships of DOE facilities and capabilities.
subunits. The identification of a conserved cluster of residues forming a patch on the surface of a protein would be a strong indication that such a patch is involved in interactions with other proteins that would have to be identified. Experimental approaches that can be used for this purpose include the yeast 2-hybrid system and phage-display techniques. Phage-display techniques provide powerful approaches for functional genomics. Phage random-peptide and gene-fragment libraries can be used to identify interacting peptides and proteins. Phage-antibody libraries provide an easy and very rapid approach to the derivation of antibodies against antigens, including those that are highly conserved and difficult to obtain for biologic reasons. This approach is ideally suited to functional genomics and the selection of antibodies against toxins.
In addition to the large number of unknown genes that will be found by the Human Genome Project, unusual noncoding DNA sequences will be identified. For example, the expansions of triplet DNA repeats define a new type of mutation, a DNA structure mutation, in the human genome. The genetic instability due to triplet-repeat expansions is associated with many inherited neurologic diseases. Expanded-triplet repeats are found inside noncoding DNA regions; upstream, downstream, or in the introns of genes; and, less often, in coding-DNA regions. Unusual DNA structures, such as DNA hairpins, can be formed by the DNA strands of repeated trinucleotide sequences upstream or downstream of genes and probably affect the expressions of associated genes. These mutations, therefore, affect not the intrinsic function of the gene product, but the level of its expression. Either the associated gene is totally suppressed or the expression of its protein product is reduced to nonphysiologic levels. When the repeat is a triplet codon and is in the coding region of a gene, it introduces a run of amino acids into the protein, thereby affecting its function. Many unusual noncoding-DNA sequences have been found and will continue to be found by the Human Genome Project, and it will be necessary to determine their structure-function relationships.
Finally, the Human Genome Project will identify DNA sequences involved in essential chromosomal elements, such as telomeres, centromeres, and origins of DNA replication. Because of the perceived links of the
shortening of telomeric DNA repeats with aging and the reactivation of telomerase (the enzyme that maintains the length of telomeric DNA) with rapidly dividing cancer cells, the functions of telomeres are under intense investigation. In addition to the above structural elements, DNA sequences required for the long-range organization of the human genome in chromosomes will be identified by the Human Genome Project. Those sequences are thought to define and isolate DNA loops or chromosomal domains as both structural and genetic units. It is important to note that there is no cross-talk between individual genetic units and the control of individual genes. This phenomenon is determined by the interactions of trans-acting factors with cis-acting DNA-sequence elements and by the topologic state of the DNA loop. It is probable that new sequence elements required for control of gene expression will be identified. Clearly, the structure-function relationships of chromosomal and genetic sequence elements will have to be elucidated for a full understanding of chromosomal organization, structure, and function.
There is little doubt that the future of structural biology in this country lies in the unique facilities provided by DOE. A broad structural-biology initiative would link the capabilities of these facilities with the human-genome and microbial-genome projects through functional genomics and health-effects reasearch as outlined in figure 2.
REFERENCE
1. Delays at Brookhaven reactor worsen US "neutron drought." Nature 1997;388:5033.
Discussant
Lisa Stubbs
Oak Ridge National Laboratory
Oak Ridge, Tennessee
INTRODUCTION: THE MODEL MOUSE
The mouse has a long history as an experimental model in genetic and toxicity studies because it is small, breeds rapidly, and survives well in the laboratory setting. Mice begin to reproduce at the age of about 6 wk, and a female mouse typically produces large litters—10-20 offspring—every 6 wk for the remainder of her reproductive life. A mouse's life span is relatively short, so it is possible to examine large numbers of animals of a particular lineage over the full course of their life cycles. Because both genetics and toxicology are statistical sciences that require data from many individuals for accurate results, mice have been the model of choice for these (and other) fields of study for many years.
Many sophisticated methods of genetic and embryologic manipulation have been developed in the mouse. The most-promising and important of these methods are transgenic techniques, such as gene targeting, which has been exploited very successfully in the last several years. Gene targeting has revolutionized the use of mouse models by permitting us to create mutations in genes that are known or thought to be involved in human disease. Once generated, the mutant mouse can be used to monitor the progress of a disease and to test its response to experimental treatments without subjecting human patients to risk or discomfort. Others have discussed the details of gene targeting and provided examples of how mouse targeted mutations can be used to study human diseases. My focus is on other types and uses of "model mice."
VARIABLE TRAITS, DISEASE SUSCEPTIBILITIES, AND INBRED STRAINS
Gene targeting is best used to study genes that have already been linked to human disease, but targeting techniques are of limited utility in the discovery of new health-related genetic factors. How can we come to identify and understand the functions of the remaining genes? Many disease-related genes have been efficiently tracked in human families, especially genes associated with inherited diseases that are expressed in a simple yes-no fashion, muscular dystrophy, cystic fibrosis, and a small fraction of breast and colon cancers. Those kinds of genes will be most easily and efficiently identified through studies of human patients and human DNA. But many of the genes that are most important to everyday health produce disorders that are more subtle and more variable and whose expression depends heavily on genetic background or environmental influence. These are the genes that are most difficult to study in the human population; but they are relatively easy to study in mice.
One important key to the successful use of mice as genetic models is the existence of stable, well-characterized inbred strains.1 Essentially, inbred strains can be considered as large collections of genetically identical twins. Animals of 2 inbred strains can be quite different from each other, exhibiting different toxin sensitivities, reactions to drugs, disease susceptibilities, and so on. For example, when an experiment is conducted with a group of animals of the C57BL6 strain, genetic variation need not be considered as a factor contributing to the variability of the experiment; the genetic components of a trait will not vary between the individual animals. However, if the same experiment is performed with animals of the 129/Sv strain, consistent differences in response between the 2 C57BL6 groups can be attributed to genetic differences, with a considerable degree of certainty. If all members of a particular inbred strain are fed the same mouse chow, drink the same water, and are housed in similar cages in the same building, the effects of environmental exposure can be minimized. This kind of consistency, which is so crucial to analysis of genetic and toxicity data, cannot be achieved with most animal models, and it certainly is not possible in the human population.
The genetic and physiologic differences between individuals in various inbred strains have been used to track the genetic locations of a number of quantitative-trait loci (QTLs), contributing to phenotypic characteristics as varied as tumor susceptibility, obesity, drug preference, and tendency to addiction.2,3 The QTL alleles contribute
to these and other types of disorders not in an independent yes-no fashion, but in a concerted, generally additive manner. Those factors can be studied more easily with the fixed genetic backgrounds of specific inbred strains. But can these mapped mouse QTL alleles and mutant phenotypes be used with any certainty to help us to identify human genes with similar effects? How similar are mouse and human genes?
EXTRAPOLATING GENETIC INFORMATION FROM HUMAN TO MOUSE AND BACK AGAIN
After years of genetic research, we understand a considerable amount about the structure and genetic organization of mouse chromosomes. As genetic studies have demonstrated, mouse and human chromosomes are similar in gene content and gene organization, with a few important exceptions. To understand how the 2 genomes are related, first imagine that 23 pairs of human chromosomes were shattered into many bits of different sizes—200-300 bits ranging in size from 50 to 500 genes. Next, imagine that those 200-300 bits were reassembled into new structures without the benefit of a map or key to tell us which pieces went with which. Relative to the human genome, mouse chromosomes appear to be assembled in just this way.4 In general, genes have not been added or deleted; there is nearly a 1-to-1 correlation between the genes in mouse chromosomes and the genes in related regions of human chromosomes. In other words, nearly all human genes have a direct mouse counterpart. I say nearly because there are indeed examples of human genes that do not have a direct functional equivalent in mice; there are also mouse genes with no known human gene cognate. 5 Most of the different ones are of a particular type: recently evolved members of gene families that arose from repeated duplications of a single ancestral gene and that were modified by genetic drift and gene conversion. The recently evolved gene-family members might be particularly interesting because in general they represent the acquisition or loss of unique gene function. These "extra" or "missing" genes might therefore be the most-easily traced clues to species-specific differences in development, basic metabolism, disease susceptibilities, and other characteristics.
Within most of the 200-300 lists of chromosomes, however, mouse and human regions contain related genes organized in a very similar manner—in the same order along the chromosome, for example, and at about the same distances from one another.5-7 This genetic similarity provides us quite a lot of predictive power. Because the maps of related mouse and human regions are, in many senses, mirror images of each other, genetic data, DNA sequence data, and other information can be extrapolated from human regions into mouse. Similarly, genetic and physiologic data derived from the analysis of mouse mutations can be used to predict the existence and locations of human genes with specific biologic functions.
For example, consider the structure of human chromosome 19q (as has been mapped by members of the Lawrence Livermore National Laboratory's Human Genome Center8) aligned with the homologous region of proximal mouse chromosome 7. Close examination shows that pairs of related mouse and human genes are found in more or less the same order, and with nearly identical spacing, along the lengths of the 2 related chromosomal regions.7 Because gene content and order have been so well preserved, we can predict with confidence that a new gene mapped to chromosome 19q will have a counterpart in mouse and that it will be in a specific region of chromosome 7. In addition, every new mouse mutation that is assigned to this region of mouse chromosome 7 can be predicted to define a functionally similar transcription unit in human 19q. With a clear understanding of the limitations, we can use the clone resources, mapping data, and sequencing information being collected for human 19q as though they were derived from the related region in the mouse. In many senses—and with caution—the mouse and human genomes can therefore be studied in parallel as 2 highly divergent versions of the same gene set.
SPONTANEOUS AND INDUCED MOUSE MUTATIONS TO PROVIDE A LARGE AND VARIED SELECTION OF HUMAN-DISEASE MODELS
Variety Is the Key: Many Ways to Make a Model Mouse
What kind of mutant resources are available for analysis of acquired and inherited human disease? A remarkable number of known, mapped mouse genetic mutants and variants have been generated and collected over
the last several decades, and well-developed techniques are available for adding to this valuable mutant resource. Transgenic techniques are among the more-recently developed means of deriving new mouse mutants. But most existing mouse models have been generated by more-traditional mutagenesis methods: through exposure of animals to mutagens, such as radiation and chemicals. Many of the existing mouse mutants arose as byproducts of risk-assessment studies, a major focus of efforts funded by the Department of Energy over the past 40 yr. For example, William and Liane Russell and colleagues at Oak Ridge National Laboratory have played a dominant role in establishing the guidelines for radiation and chemical exposures of humans and have done so by monitoring numbers of mutant offspring produced in radiation-and chemical-treated mice. 9,10
A number of the mutant offspring that arose from those mutagen-treated parents expressed disorders that resembled various types of human disease, and the Russells and co-workers wisely saved the mutant animals for future genetic studies. Many other mouse mutants, generated in similar risk-assessment studies in other institutions or arising spontaneously in large breeding colonies, are now the subjects of research carried out by molecular biologists, pathologists, embryologists, and others in institutions around the world.11 These mutants were selected from the animal colonies because they expressed specific types of health-related disorders, and not because of a priori knowledge regarding the nature of the genes involved. Although it can be difficult to link such "randomly generated" mutations to defects in specific genetic loci, the process is becoming less and less difficult as the results of the Human Genome Project begin to unfold and as new information about genome structure, gene location, and gene function begins to flood the public databases. The genes that are found linked to the randomly generated mutations are often novel or are known genes that would not have been expected to be associated with the types of disorders seen in the mutant mice. Therefore, spontaneous, induced, and targeted mutations constitute highly complementary tools for discovering links between genes and hereditary disease. The power of the mouse is its versatility: Targeted mutations can be induced in known genes that are thought to be required for health, and new genes, or more-subtle effects of known genes, can be discovered through analysis of randomly placed mutations that are selected specifically for their phenotypic effects.
Unlike targeted mutations, the genetic defects that arise spontaneously or as a result of mutagen treatments are of many different types—not just "knockouts," but also missense and regulatory mutations that permit the expression of a gene at different levels, in different tissues, or in forms slightly modified from those seen in normal mice. The mutations can alter the function of the protein product in subtle, but nevertheless insidious, ways. Missense mutations, which cause substitution of a single amino acid in an otherwise normal protein, can sometimes produce severe health effects, whereas complete absence of the same protein, as would be caused by a gene knockout, might be relatively benign.11,12 In other cases, where absence of a protein proves lethal, relatively mild missense mutation can provide a means of studying the effects of a gene defect in live-born mice.
CAN GENE'S HEALTH-RELATED FUNCTIONS BE DEDUCED FROM THE PHENOYTPES OF MUTANT MICE?
Are genetic defects expressed by a mouse truly good indicators of the genes that are critical to human disease and susceptibilities? Certainly there are biologic differences between rodents and humans that prevent mice from serving as perfect models. However, a number of related mouse and human gene pairs are known to perform similar or even identical biologic functions. For example, defects in the mouse Kit gene cause pigmentation, hematopoietic, and reproductive defects associated with a classical mouse mutation called dominant white spotting, or W. The relationship between Kit and W was originally discovered when the human gene, KIT, was mapped to the centromeric region of human chromosome 4. This region is known to be related to the W region of mouse chromosome 5; because the gene's properties suggested a link to hematopoietic cell growth, Kit was immediately suspected as the "culprit" gene responsible for the phenotype of W mice. The proof of the genetic connection was established soon after.13,14
In fact, the pigmentation defect seen in W mice is almost identical to that observed in people who inherit what is called the piebald trait, and some of these people, but not all of them, have since been found to carry mutations in the human Kit gene.15 Piebald trait had been difficult to map in human families because, as in mice, a similar type of pigmentation pattern can be caused by mutations in several genes. Establishing the connection between Kit
and W provided the essential link that permitted the genetic cause of one type of piebald trait to be discovered in human patients, a feat that could not be accomplished directly through studies of human families. Since then, several other links between genes and human disease have been established first through analysis of mutations that cause similar symptoms in mice. A recent example is the link between the leptin gene and obesity in ob mutant mice.16 Although such mutations are rare, obese human patients with leptin gene defects recently have been identified,17 providing the only certain link between a single gene and obesity in humans. Genes linked to specific types of human deafness,18 neural-tube defects,19 and other defects have also been discovered through the isolation of genes linked to similar, more easily mapped mouse mutations. As this trend continues, it is likely that more and more human-disease genes will be first discovered in mice.
An Unmet Challenge: Tracing the Causes of Human Birth Defects
One of the remaining unmet challenges in human-health research is in the discovery of the causes of human birth defects. Although some birth defects are inherited, the bulk of human developmental disorders are not strictly, or even primarily, genetic; most are probably due to a combination of genetic susceptibilities and environmental factors. I use the word probably because we can only guess at the causes of most common developmental defects. Most human birth defects arise without warning, without the benefit of diagnosis and prenatal counseling, and without any credible medical explanation.
Because the mouse has long been used as a model for human development, the existing mutant collections are especially rich in animals that have various types of prenatal defects.11,20 Although the abnormalities that are seen in most human infants born with birth defects—such as limb deformities, skeletal malformations, neurologic disorders, and hydrocephalus—are not mainly genetic, the genes that are disrupted in animals with similar disorders can tell us a considerable amount about the potential causes of those human defects. What kinds of proteins are essential to the normal process of limb, skeletal, neural-tube, or nervous-system development? With which molecules might those essential proteins interact, and what kinds of molecules might interrupt their function? The same proteins that are disturbed by genetic defects—or other proteins that are part of the same developmental pathways—are likely also to be inhibited, stimulated, or altered by drags, exposures, or environmental toxicants. Pinning a development function to a specific mutated gene therefore might not directly identify the factors that are key to genetic susceptibilities, but it does provide new tools that can be used to gain molecular access to the most-sensitive prenatal events.20
SUMMARY AND CONCLUSION
The mouse has been studied as a model for many years, but the use of the mouse in analysis of human-health disorders—from outright genetic disease to cancer susceptibility, developmental disorders, quantitative traits, and other problems influenced by genetic background and environmental exposures—is just beginning to reach full potential. As new mutants are created and the human and mouse genomes are further linked through gene mapping, gene sequencing, and basic gene-expression studies, the mouse is certain to become a major tool for linking mapped human genes to biologically relevant, in vivo functions. The combined power of targeted and induced mutagenesis methods, together with well-controlled methods for studying the influence of genetic background and environment on the expression of traits in normal and mutant animals, makes the mouse a unique and valuable resource for the dissection of complex human traits.
REFERENCES
1. Festing MFW. Origins and characteristics of inbred strains of mice. In: Lyon MF, Rastan S, and Brown SDM, editors. Genetic variants and strains of the laboratory mouse, 3rd ed. Oxford University Press, Oxford, U.K.;1996. p. 1536-1576.
2. Plomin R, McClearn GE, Gora-Maslak G, Neiderhiser JM. Use of recombinant inbred strains to detect quantitative trait loci associated with behavior. Behav Genet, 1991;21:99-116.
3. Nagase H, Bryson S, Fee F, Balmain A. Multigenic control of skin turnout development in mice. Ciba Found Symp, 1996; 197:156-68.
4. DeBry RW, Seldin MF. Human/mouse homology relationships. Genomics 1996;33:337-51.
5. Hood L, Koop BF, Rowen L, Wang K. Human and mouse T-cell receptor loci: the importance of comparative large-scale DNA sequence analysis. Cold Spring Harb Symp on Quantit Biol, 1993;58:339-348.
6. Oakey RJ, Watson ML, Seldin MF. Construction of a physical map on mouse and human chromosome 1: comparison of 13 Mb of mouse and 11 Mb of human DNA. Human Mol Genet, 1992;6:613-620.
7. Stubbs L, Carver EA, Shannon ME, Kim J, Geisler J, Generoso EE, Stanford BG, Dunn WC, Mohrenweiser H, Zimmermann W, Watt SE, Ashworth LK. Detailed comparative map of human chromosome 19q and related regions of the mouse genome. Genomics, 1996;35:499-508.
8. Ashworth LK, Batzer MA, Brandriff B, Branscomb E, de Jong P, Garcia E, Games J, Gordon L, Lamerdin JE, Lennon G, Mohrenweiser H, Olsen A, Slezak T, Carrano AV. A metric physical map of human chromosome 19. Nat Genet 1995; 11:422-427.
9. Russell WL. X-ray-induce mutations in mice. Cold Spring Harb Symp Quant Biol 1951; 14:16-22.
10. Russell LB, Russell WL. Frequency and nature of specific-locus mutations induced in female mice by radiation and chemicals: a review. Mutation Res 1992; 296: 107-127.
11. Doolittle DP, Davisson MT, Guidi JN, Green MC. Catalog of mutant genes and polymorphic loci. In: Lyon MF, Rastan S, and Brown SDM, editors. Genetic variants and strains of the laboratory mouse, 3rd ed. Oxford University Press, Oxford, U.K.; 1996. p. 17-854.
12. Rinchik EM, Carpenter DA, Handel MA. Plieotropy in microdeletion syndromes: neurologic and spermatogenic abnormalities in mice homozygous for the p6H deletions are likely due to the dysfunction of a single gene. Proc Natl Acad Sci USA, 1995; 92: 6394-6398.
13. Geissler EN, Ryan MA, Housman DE. The dominant-white spotting (W) locus of the mouse encodes the c-kit proto-oncogene. Cell 1988;55:185-192.
14. Chabot B, Stephenson DA, Chapman VM, Besmer P, Bernstein A. The proto-oncogene c-kit encoding a transmembrane tyrosine kinase receptor maps to the mouse W locus. Nature, 1988;335:88-89.
15. Fleischman RA, Saltman DL, Stastny V, Zneimer S/Deletion of the c-kit protooncogene in the human developmental defect piebald trait. Proc Natl Acad Sci USA, 1991;88:10885-10889.
16. Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature, 1994;372:425-432.
17. Montague CT, Farooqi IS, Whitehead JP, Soos MA, Rau H, Wareham NJ, Sewter CP, Digby JE, Mohammed SN, Hurst JA, Cheetham CH, Earley AR, Barnett AH, Prins JB, O'Rahilly S. Congenital leptin deficiency is associated with severe early-onset obesity in humans. Nature, 1997;387:903-908.
18. Gibson F, Walsh J, Mburu P, Varela A, Brown KA, Antonio M, Beisel KW, Steel KP, Brown SDM. A type VII myosin encoded by the mouse deafness gene shaker-1 . Nature, 1995;374:62-64.
19. Tassabehji M, Read, AP, Newton VE, Harris R, Balling R, Grass P, Strachan T. Waardenberg's syndrome patients have mutations in the human homologue of the Pax-3 paired box gene. Nature, 1992;355:635-636.
20. Culiat CT, Carver EA, Walkowicz M, Rinchik EM, Russell LB, Cacheiro NLA, Generoso W, Stubbs L. Induced mouse chromosomal rearrangements as tools for identifying critical developmental genes and pathways. Reproductive Toxicol 1997;11:345-351.



























