
18
Neuroendocrine Consequences of Systemic Inflammation
Seymour Reichlin1
Introduction
Bacterial and viral infections, bacterial toxins, and severe tissue injury induce a relatively stereotypical pathophysiologic response manifested by fever, catabolism, and sickness behavior. If mild, sickness behavior is manifested by anorexia, drowsiness, and impaired cognition; if severe, delirium, stupor, and coma can supervene. All organ systems are altered by acute and chronic inflammatory states. The dramatic changes in liver function, termed the acute-phase response include suppression of synthesis of albumin, transthyretin, transferrin, and ceruloplasmin, and increase in synthesis of several proteins including fibrinogen, β-2 microglobulin, serum amyloid antecedent (SAA), and C-reactive protein (Dinarello and Wolff, 1993). Chronic inflammation, such as that seen in rheumatoid arthritis, induces a less severe and persistent version of
the acute-phase response. When the acute-phase response is mild, functional capacity is suppressed; when severe, prostration and death can occur.
These changes are mediated by a flood of polypeptide molecules, the inflammatory cytokines, which are released into the circulation by lymphocytes, monocytes, macrophages, and endothelial cells and are produced locally in tissues by resident macrophages and several other types of parenchymal cells. Among the most important of the cytokines released during inflammatory stress are interleukin (IL)-1, IL-2, IL-6, tumor necrosis factor-alpha (TNF-α), interferon-gamma (INF-γ), and several cytokines with intrinsic anti-inflammatory activity, including IL-1 receptor antagonist (IL-1ra), transforming growth factor-β, and soluble TNF receptor (Dinarello, 1994; Dinarello and Wolff, 1993). Cytokines are highly synergistic, and highly potent (Figure 18-1).
Are cytokine responses helpful to survival? That is certainly the case in experimental sepsis in animals where the cytokine effects can be neutralized early in infection. In humans, preliminary studies of extremely sick septic patients showed improved survival after blockade of IL-1 (with IL-1ra) (Fisher et al., 1994b), but in a larger study with patients of varying severity of sepsis, there was no benefit overall (Fisher et al., 1994a). However, subgroup analysis of the larger study suggests that IL-1ra may have been of benefit in a subgroup of the most severely sick individuals (Fisher et al., 1994a). The possible usefulness of the TNF component of response has been reviewed (van der Poll and Lowry, 1995).
Many neuroendocrine functions are profoundly altered during states of inflammatory disease (Reichlin, 1993, 1994, 1995; Sternberg, 1992; Wilder, 1995) (Table 18-1). Some have positive homeostatic value, while others may contribute to the deleterious impact of inflammation. This chapter focuses on changes in the pituitary and in pituitary target hormones; abnormalities in the pancreas, bone, and brain (all of which occur in sepsis or after exposure to inflammatory cytokines) are not considered further. Many of the endocrine responses observed in inflammation are due in part to the associated decrease in nutrient consumption. These are pointed out in sections below.
Hypothalamic-Pituitary-Adrenal Activation
The classical pituitary-adrenal response to stress can be induced by infection or by the injection of bacterial toxin (Kimball et al., 1968; Michie et al., 1990) (Figures 18-1, 18-2). Lipopolysaccharides (LPS) (and other toxins) act on pituitary-adrenal function by stimulating cytokine release. IL-1, IL-2, IL-6, and TNF-α are all capable of activating corticotropin-releasing hormone (CRH) secretion (Kakuscska et al., 1993; Sapolsky et al., 1987). Systemic LPS is also capable of activating central IL-1 neuronal pathways (Breder et al., 1988; Lechan et al., 1990). Toxin-induced cytokine release stimulates secretion of the hypothalamic neuropeptides, CRH, and vasopressin (VP), which synergize at
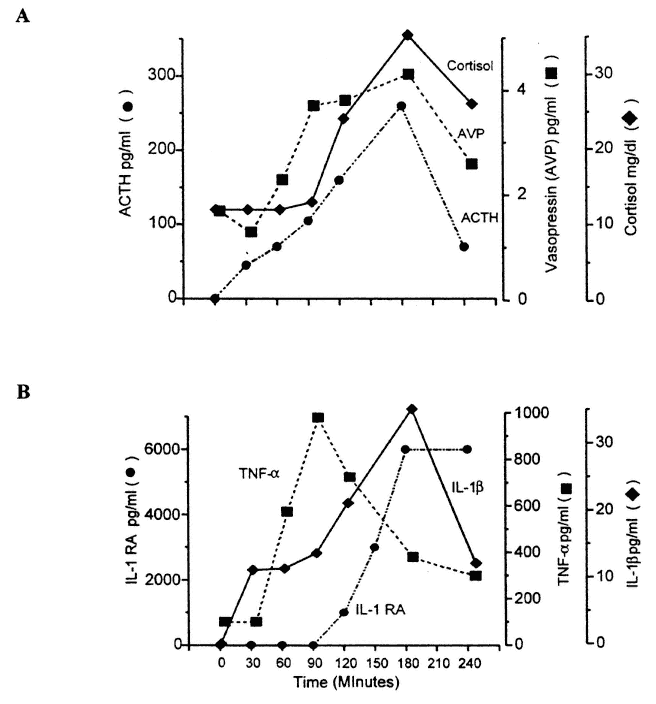
FIGURE 18-1
Composite diagram to illustrate the response of acute-phase cytokines, pituitary-adrenal, and neurohypophysial hormones to the injection of Escherichia coli endotoxin. SOURCE: Figure reprinted from Reichlin (1998) with permission. (A) Changes in adrenocorticotrophic hormone (ACTH) cortisol, and arginine vasopressin (AVP) following injection of E. coli endotoxin. Both ACTH and AVP are activated within 30 minutes, followed approximately 30 minutes later by increased cortisol secretion. As discussed in the text, endotoxin acts by mobilizing peripheral release of inflammatory cytokines, tumor necrosis factor (TNF), and interleukin (IL)-1, which act directly and indirectly on the hypothalamus. Figure was redrawn from the published data of Michie et al. (1990). (B) Time course of appearance in the blood of IL-1β, TNF-α, and IL-receptor antagonist (ra) following intramuscular injection of E. coli endotoxin. Redrawn from the published data of Dinarello and Wolff (1993) and Granowitz et al. (1993). Note that TNF-α appears first and then rapidly falls, that IL-1Β next appears, which is then followed approximately 90 minutes later by IL-1ra. The concentration of IL-1ra is approximately 100 times greater than that of IL-1, giving a molar ratio sufficient to ensure that IL-1 effects are fully neutralized.
TABLE 18-1 Neuroendocrine and Endocrine Consequences of Inflammatory Disease
|
Pituitary-adrenal activation |
|
Syndrome of inappropriate ADH secretion (SIADH) |
|
Sick euthyroid syndrome |
|
Sick gonad syndrome |
|
Growth hormone increase/decrease |
|
Impaired insulin secretion, insulin resistance |
|
Sick bone syndrome |
|
Sick brain syndrome |
the pituitary level to increase the secretion of adrenocorticotrophic hormone (ACTH). Toxin can also act less directly through stimulation of vagal afferents in the liver bed and elsewhere in the abdomen (Watkins et al., 1995b). The vagal afferent system, which acts through chemosensory vagal paraganglia is especially vulnerable to toxic products arising in the abdomen viscera. Central activation of CRH neurons leads to stimulation of the peripheral autonomic nervous system (Irwin et al., 1990) with enhanced release of epinephrine from the adrenal medulla and of norepinephrine from sympathetic nerve endings. Epinephrine synergizes with CRH and VP in stimulating ACTH release.
The consequent release of glucocorticoids in turn modulates the intensity of the acute response, virtually all of whose components are inhibited by glucocorticoids (Munck et al., 1984). These include inhibition of cytokine secretion by immunocompetent cells and the consequent loss of lymphocyte reactions that are secondary to cytokine actions. Among the factors that are inhibited are INF-γ, granulocyte-monocyte stimulating factor, IL-1, IL-2, IL-3, IL-6, and TNF-α. The secretion of many inflammatory mediators by activated lymphocytes and macrophages is also inhibited. These include bradykinin, serotonin, and histamine and the tissue-destructive enzymes collagenase, elastase, plasminogen activator, and prostaglandins. Natural killer (NK) cell activity is also suppressed by glucocorticoids (Callawaert et al., 1991).
Although these anti-inflammatory effects of glucocorticoids are the basis of its widespread clinical use to treat immune disorders, they were for many years looked on as pharmacological side effects and not relevant to physiological function. However, in one view of Munck and colleagues (1984), the pituitary-adrenal response to stress evolved as a mechanism to suppress and modulate an overexuberant inflammatory response to toxins, antigens, and invading organisms. Intense mobilization of cytokines and inflammatory mediators may be fatal.
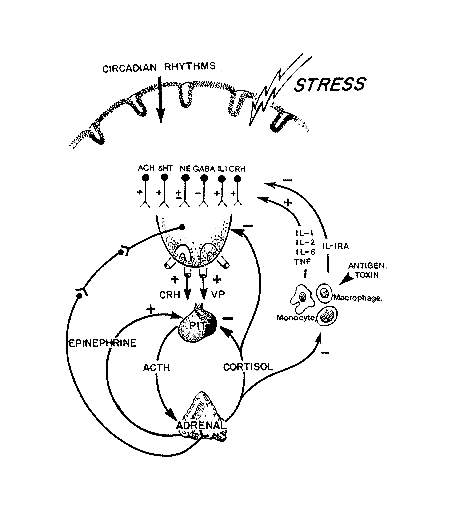
FIGURE 18-2
Schematic outline of the neuroendocrine factors that regulate the secretion of the adrenal cortex.
SOURCE: Figure reprinted from Reichlin (1998) with permission. Adrenocorticotrophic hormone (ACTH) release from the pituitary is stimulated by corticotropin-releasing hormone (CRH) and vasopressin (VP) acting synergistically. Circulating epinephrine (the response of the activated sympathetic nervous system) is also synergistic for ACTH release. The secretion of ACTH is inhibited at the pituitary level by circulating glucocorticoids, and at the hypothalamic level, CRH and VP are also under negative feedback control by glucocorticoids. VP and CRH neurons are in turn subject to a wide range of influences—circulating cytokines, prostaglandins, and many neurotransmitters. Some such as acetylcholine (ACH) are stimulatory, and others such as γ-amino butyric acid (GABA) are inhibitory. Interacting with the hypothalamic-pituitary-adrenal axis is the peripheral immune system (as well as endothelia and other structures), which releases cytokines into the blood which then can activate the adrenal. Each of the major inflammatory cytokines, interleukin (IL)-1 (α or β), IL-2, IL-6, and tumor necrosis factor (TNF)-α are capable of activating ACTH release. The peripheral immune system provides a chemosensory system by which the presence of foreign molecules can be communicated to the brain and can induce an appropriate response, a model of "bidirectional communication between the brain and the immune system" Blalock (1989).
An excellent example of the validity of the Munck hypothesis has come from the study of immune responses in the Lewis strain of rats, which have a genetic defect in hypothalamic CRH synthesis (Chrousos, 1995; Sternberg, 1992; Wilder, 1995). Rats of the Lewis strain develop acute arthritis when injected with streptococcal cell wall suspensions, whereas rats of the Fischer strain do not. Lewis rats are susceptible to many other forms of induced autoimmune disease. Lewis rats differ from Fischer rats in that Lewis rats do not show increased adrenal glucocorticoid release when challenged with antigen and do not increase their hypothalamic content of mRNA coding for CRH as do Fischer rats. Measures that inhibit pituitary-adrenal function in the Fischer rat convert their immune responses to those resembling the Lewis (susceptible) strain. Clinical studies have tested this hypothesis in patients with established rheumatoid arthritis (Chikanza et al., 1992), chronic fatigue syndrome (Demitrack, 1994; Goldenberg, 1993; Manu et al., 1992), and fibromyalgia (Crofford et al., 1994; Goldenberg, 1993; Griep et al., 1993). Studies in rheumatoid arthritis (RA) patients are the most convincing; patients with RA undergoing major joint surgery have a lower or absent glucocorticoid response as compared with patients with osteoarthritis undergoing the same procedure. Moreover, patients with RA have a higher IL-1 response to the surgical procedure, which suggests that glucocorticoids are not damping their stress response. Results of studies of patients with chronic fatigue syndrome or fibromyalgia are ambiguous, though they do suggest some impaired adrenal function in some patients. Whether or not there is a pituitary-adrenocortical deficit in patients with these disorders, current studies do not distinguish whether they arise as secondary or primary abnormalities.
Although it is well established that patients with adrenal deficiency are more likely to die during sepsis than those with normal adrenal function, most studies show that treatment of sepsis with glucocorticoids in patients with normal adrenal function does not improve survival, and in fact, it may increase mortality (Sheagren, 1991). These observations suggest that otherwise healthy individuals respond to sepsis with an appropriate pituitary-adrenal response.
Syndrome of Inappropriate Antidiuretic Hormone Secretion
Characteristic of the response to sepsis is an increase in secretion of VP (antidiuretic hormone, ADH). Lobar pneumonia has been shown to be accompanied by elevated VP levels and by impaired water excretion (Dreyfuss et al., 1988), changes that are secondary to activation of central vasopressinergic pathways. These insights are not new. It has been known since the introduction of serum chloride measurements into clinical chemistry that serum chloride levels in patients with bacterial pneumonia are reduced (secondary to hemodilution) and that the crisis of recovery in lobar pneumonia (in the preantibiotic era) was heralded by a sudden increase in serum chloride
concentration (''chloride shift''). A possible homeostatic value of VP is its ability to inhibit the pyrogenic effects of bacterial toxin (Naylor et al., 1987). There is a downside to the VP response because it stimulates the kidney to delay water excretion, which in septic and elderly individuals enhances the vulnerability to overhydration.
Pituitary-Thyroid Regulation in Inflammatory Disease
The sick euthyroid syndrome is the most common form of thyroid abnormality encountered in populations of acute and chronically ill individuals. It is characterized by low circulating levels of triiodothyronine (T3), low or low normal blood levels of thyroxine (T4), and depressed thyroid hormone binding proteins (Wartofsky and Burman, 1982). Concentrations of thyroid hormones in tissues obtained at autopsy from patients dying with the sick euthyroid syndrome are lower than those of individuals who died suddenly (Arem et al., 1993). The most striking neuroendocrine feature of this disorder (and the one most useful in clinical differentiation from primary thyroid abnormalities) is that the low circulating thyroid hormones do not induce elevation of plasma thyroid-stimulating hormone (TSH) levels as would normally be the case through the operation of the hypothalamic-pituitary thyroid negative feedback system.
The syndrome can be reproduced to a degree in rats by injections of bacterial toxin, IL-1, or TNF-α (Pang et al., 1989) and in humans by injections of TNF-α (van der Poll and Lowry, 1995; van der Poll et al., 1990). In a detailed analysis of the cytokine etiology of the sick euthyroid state in the mouse, none of the cytokines tested individually (TNF-α, IL-1α, IL-6, and INF-γ) were as potent as bacterial toxin in inducing the disorder. Nor were these cytokines as potent when tested together (Boelen et al., 1995). In sick euthyroid humans, depression of the T3 level was proportional to circulating levels of TNF (Mooradian et al., 1990) and was a function of serum IL-6 concentration only in very severely ill individuals (Boelen et al., 1993). Blockade of IL-1 effects with one IL-1 receptor antagonist did not prevent the effects of endotoxium or pituitary-thryoid function (van der Poll et al., 1995). This observation suggests that a number of other cytokines are involved in the diagnosis.
Sick euthyroidism comes about through cytokine-induced alterations at every level of the pituitary-thyroid axis: hypothalamic, pituitary, thyroid gland itself, and peripheral thyroid metabolism (Reichlin, 1993, 1994). In the hypothalamus, thyrotropin-releasing hormone (TRH) synthesis is inhibited (Kakuscska et al., 1994), and somatostatin synthesis enhanced (Scarborough et al., 1989). Within the pituitary, IL-1 is induced in a subpopulation of thyrotrope cells (Koenig et al., 1990) and exerts autocrine and paracrine inhibition of responsiveness to TRH (Sumita et al., 1994). In the thyroid, a number of metabolic abnormalities are induced—impaired peroxidation and impaired
cAMP responsiveness, which leads to reduced TRH responsiveness—while in the periphery, reduced conversion of T4 to T3 and reduced hepatic synthesis of transthyretin and albumin lead to the characteristic low T3 and, in severe cases, low T4 levels (Reichlin, 1993). Starvation contributes to the abnormal T4 to T3 conversion; in uninfected individuals, fasting by itself is sufficient to decrease T4 to T3 conversion with restoration by carbohydrate supplementation (Vagenakis et al., 1975). In septic patients with low serum T3 values, hyperalimentation restored T3 values to normal (Richmand et al., 1980).
In contrast to other chronic inflammatory diseases, patients with AIDS manifest elevation of thyroxine-binding globulin as the disease progresses, and the low T3 syndrome does not appear until patients approach a terminal state when malnutrition and cachexia dominate the clinical picture (Lambert, 1994).
Still unsettled is whether these decreases in thyroid function are homeostatically valuable. In experimental streptococcal pneumonia in the rat, low thyroid levels were associated with reduced mortality, whereas high levels increased vulnerability (Reichlin and Glaser, 1958). In another model of sepsis, pulmonary surfactant levels were restored to normal by T3 supplementation (Dulchavsky et al., 1995). Studies in the human are limited. Thyroid hormone treatment had no benefit (nor did harm) to a relatively small number of septic individuals with the sick euthyroid syndrome (Brent and Hershman, 1986), but in volunteers whose T3 levels were reduced by a period of fasting, T3 supplementation increased the magnitude of negative nitrogen balance (Gardner et al., 1979). These studies are admittedly difficult to carry out, and the results are less than convincing. At this time, there is no evidence that thyroid supplementation is beneficial in patients with sick euthyroid disease, but the question has not been resolved definitively.
Pituitary-Gonadal Regulation
Burns and severe inflammatory illness induce a dramatic fall in sex steroids (Dong et al., 1992). TNF-α also inhibits gonadal secretion (van der Poll et al., 1993). As in the case of pituitary-thyroid dysfunction in sepsis, cytokine suppression of pituitary-gonad function is exerted at many levels. IL-1 reaching the gonads through the circulation, or released from Leydig cells within the testis as an autocrine-paracrine secretion, inhibits steroidogenesis and other cell functions (Syed et al., 1993). In the ovary, intrinsic cytokines, IL-6, TNF-α, and IL-1 regulate steroidogenesis, maturation, atresia, and apoptosis of ovarian cells. Indeed, cytokine-induced apoptosis of granulosa cells is the triggering event that culminates in follicular atresia. At the level of the hypothalamus, IL-1 inhibits pulsatile secretion of gonadotropin-releasing hormone (GnRH), which leads to low gonadotropin secretion and low levels of sex steroids (Rivest and Rivier, 1995; Shalts et al., 1991). These effects are probably mediated at the hypothalamic level by the induction of CRH and/or VP (Rivest and Rivier, 1995; Shalts et al., 1991).
Currently, no useful data exist to indicate that low gonadotropin-sex steroid secretion is homeostatically valuable or harmful in the human or in other species. One could argue that testosterone deficiency might be harmful in that it would increase wasting of muscle mass, but suitable studies of anabolic steroid treatment in sepsis have not been published.
Starvation, in the absence of sepsis is also capable of inhibiting gonadal function in both men (Samuels and Kramer, 1996) and women (Olson et al., 1995). Short-term starvation reduces the magnitude of pulsatile gonadotropin release in both sexes but the degree of gonadotropin suppression is greater in men than in women.
Growth Hormone in Sepsis
In humans, a single injection of LPS induces a sharp increase in growth hormone (GH) (Kimball et al., 1968; Wagner et al., 1975), while in rats, cytokines in low dosage stimulate (Payne et al., 1992) and in large doses, suppress GH (Payne et al., 1992; Peisen et al., 1995; Wada et al., 1995). Severe sepsis is associated with elevated GH levels (Bentham et al., 1993) or normal levels (Voerman et al., 1992a, 1993), and in one particular chronic inflammatory illness, RA, daily GH secretion was reduced (Rall et al., 1996).
However, GH levels alone may not reflect actual GH effects as expressed at the tissue level. GH effects on protein metabolism are mediated through the secretion of somatomedin C (insulin-like growth factor I), a liver-derived protein whose secretion is dependent both on GH stimulation and on the availability of metabolic substrate. For example, in states of nutritional deficiency (protein-calorie malnutrition, kwashiorkor, anorexia nervosa), blood levels of GH are markedly elevated, but somatomedin C concentrations are low, which suggests that tissue "effective" levels of GH are low.
In humans with sepsis, somatomedin C levels are low and can be restored to normal by hyperalimentation (Bentham et al., 1993) or by GH treatment (Voerman et al., 1992b). In what may be an analogous situation—the catabolic state associated with large burns—blood GH levels are elevated, somatomedin values are low, but the administration of additional amounts of GH appear to improve survival and graft healing and to shorten the duration of hospitalization (Knox et al., 1995; Nguyen et al., 1996). One conclusion therefore is that the elevated GH seen in sepsis is homeostatically valuable but that the normal response is suboptimal. Hepatic somatomedin-C response to GH is probably reduced in burns and sepsis; for this reason, somatomedin C (which would bypass the liver) may possibly be a more useful agent than GH in this setting. In a murine model of sepsis, both GH and somatomedin C enhanced host defense (Inoue et al., 1995).
Author's Summary and Conclusions
Activation of inflammatory cytokines by toxins or products of cell injury (as in burns and physical trauma) leads to a variety of metabolic and endocrine changes, mediated in part by the direct action of cytokines on tissue function and by changes in pituitary-endocrine end organ function. Some of the endocrine responses, such as pituitary-adrenal activation and GH hypersecretion, are homeostatically valuable and promote survival or healing. Other responses may be harmful. Increased VP secretion may modulate fever but create water intolerance and susceptibility to hypo-osmolar states. Loss of gonadal activity has possible catabolic consequences. Reduced supply of the active form of thyroid hormone to tissues may conserve metabolic demand but could interfere with some normal thyroid hormone-dependent functions. Cytokine-driven responses of the neuroendocrine system resemble those seen in starvation: reduced thyroid function, reduced levels of GH-dependent peptides, and suppression of gonadal function.
Areas for possible additional study in soldiers under stress include the use of gonadal steroids or anabolic steroids and the use of GH and/or somatomedin C. Newly developed devices the size of a fountain pen make injections feasible under field conditions. Glucocorticoid treatment will be unlikely to help manage stress in individuals whose underlying adrenal reserve is normal. Studies designed specifically to evaluate thyroid status under field conditions of stress and malnutrition would be of value, but on the basis of the limited information on treatment of sick euthyroid patients with thyroid hormone, it is unlikely that this form of therapy will be helpful. Cytokine-mediated changes in neuroendocrine activity interacting with poor nutrition could well impair immunological function and overall resistance to stress.
References
Arem, R., G.J. Wiener, S.G. Kaplan, H.S. Kim, S. Reichlin, and M.M. Kaplan. 1993. Reduced tissue thyroid hormone levels in fatal illness. Metab. Clin. Exp. 42:1102-1108.
Bentham, J., J. Rodriguez-Arnao, and R.J. Ross. 1993. Acquired growth hormone resistance in patients with hypercatabolism. Horm. Res. 40:87-91.
Blalock, J.E. 1989. A molecular basis for bidirectional communication between the immune and neuroendocrine systems. Physiol. Rev. 69:1-32.
Boelen, A., M.C. Platvoet-Ter Schiphorst, O. Bakker, and W.M. Wiersinga. 1995. The role of cytokines in the lipopolysaccharide-induced sick euthyroid syndrome in mice. J. Endocrinol. 146:475-483.
Boelen, A., M.C. Platvoet-Ter Schiphorst, and W.M. Wiersinga. 1993. Association between serum interleukin-6 and serum 3,5,3'-triiodothyronine in nonthyroidal illness. J. Clin. Endocrinol. Metab. 77:1695-1699.
Breder, C.D., C.D. Dinarello, and C.B. Saper. 1988. Interleukin-1 immunoreactive innervation of the human hypothalamus. Science 240:321-324.
Brent, G.A., and J.M. Hershman. 1986. Thyroxine therapy in patients with severe nonthyroidal illnesses and low serum thyroxine concentration. J. Clin. Endocrinol. Metab. 63:1-8.
Callewaert, D.M., V.K. Moudgil, G. Radcliff, and R. Waite. 1991. Hormone specific regulation of natural killer cells by cortisol: Direct inactivation of the cytotoxic function of cloned human NK cells without an effect on cellular proliferation. FEBS Lett. 285:108 015-110.
Chikanza, I.C., P. Petrou, G. Kingsley, G. Chrousos, and G.S. Panayi. 1992. Defective hypothalamic response to immune and inflammatory stimuli in patients with rheumatoid arthritis. Arthritis Rheum. 35:1281-1288.
Chrousos, G.P. 1995. The hypothalamic-pituitary-adrenal axis and immune-mediated inflammation. N. Engl. J. Med. 332:1351-1362.
Crofford, L.J., S.R. Pillemer, K.T. Kalogeras, J.M. Cash, D. Michelson, M.A. Kling, E.M. Sternberg, P.W. Gold, G.P. Chrousos, and R.L. Wilder. 1994. Hypothalamic-pituitary-adrenal axis perturbations in patients with fibromyalgia. Arthritis Rheum. 37:1583-1592.
Demitrack, M.A. 1994. Chronic fatigue syndrome a disease of the hypothalamic-pituitary-adrenal axis? Ann. Med. 26:1-5.
Dinarello, C.A. 1994. The biological properties of interleukin-1. Euro. Cytokine Netw. 5:517-531.
Dinarello, C.A., and S.M. Wolff. 1993. The role of Interleukin-1 in disease. New Engl. J. Med. 328:106-113.
Dong, Q., F. Hawker, D. McWilliam, M. Bangah, H. Bureger, and D.J. Handelsman. 1992. Circulating immunoreactive inhibin and testosterone levels in men with critical illness. Clin. Endocrinol. 36:399-404.
Dreyfuss, D., F. Leviel, M. Paillard, J. Rahmani, and I. Coste. 1988. Acute infectious pneumonia is accompanied by a latent vasopressin-dependent impairment of renal water excretion. Am. Rev. Resp. Dis. 138:583-585.
Dulchavsky, S.A., S.M. Ksenzenko, A.A. Saba, and L.N. Diebel. 1995. Triiodothyronine (T3) supplementation maintains surfactant biochemical integrity during sepsis. J. Trauma 39:53-57.
Fisher, Jr., C.J., J.F. Dhainaut, S.M. Opal, J.P. Pribble, R.A. Balk, G.J. Slotman, T.J. Iberti, E.C. Rackow, M.J. Shapiro, R.L. Greenman et al. 1994a. Recombinant human interleukin 1 receptor antagonist in the treatment of patients with sepsis syndrome. Results from a randomized, double-blind, placebo-controlled trial. Phase III rhIL-1ra Sepsis Syndrome Study group. J. Am. Med. Assoc. 271:1836-1843.
Fisher, Jr., C.J., G.J. Slotman, S.M. Opal, J.P. Pribble, R.C. Bone, G. Emmanuel, D. Ng, D.C. Bloedow, and M.A. Catalano. 1994b. Initial evaluation of human recombinant interleukin-1 receptor antagonist in the treatment of sepsis syndrome: A randomized, open-label, placebo-controlled multicenter trial. The IL-1RA Sepsis Syndrome Study Group. Crit. Care Med. 22:12-21.
Gardner, D.F., M.M. Kaplan, C.A. Stanley, and R.D. Utiger. 1979. Effect of triiodothyronine replacement on the metabolic and pituitary responses to starvation. N. Engl. J. Med. 300:579-584.
Goldenberg, D.L. 1993. Fibromyalgia, chronic fatigue syndrome, and myofascial pain syndrome. Curr. Opin. Rheumatol. 5:199-208.
Granowitz, E.V., R. Porat, J.W. Mier, S.F. Orencole, M.V. Callahan, J.G. Cannon, E.A. Lynch, K. Ye, D.D. Poutsiaka, E. Vannier et al. 1993. Hematologic and immunomodulatory effects of an interleukin-1 receptor antagonist coinfusion during low-dose endotoxemia in healthy adults. Blood 82:2985-2990.
Griep, E.N., J.W. Boersma, and E.R. de Kloet. 1993. Altered reactivity of the hypothalamic-pituitary-adrenal axis in the primary fibromyalgia syndrome. J. Rheumatol. 20:469-474.
Inoue, T., H. Saito, R. Fukushima, T. Inaba, M.T. Lin, K. Fukatsu, and T. Muto. 1995. Growth hormone and insulin-like growth factor I enhance host defense in a murine sepsis model. Arch. Surg. 130:1115-1122.
Irwin, M.T., R.L. Hauger, L. Jones, M. Provencio, and K.T. Britton. 1990. Sympathetic nervous system mediates central corticotropin-releasing factor induced suppression of natural killer cytotoxicity. J. Pharmacol. Exp. Ther. 255:101-107.
Kakuscska, I., L.I. Romero, B.D. Clark, J.M. Rondeel, Y. Qi, S. Alex, C.H. Emerson, and R.M. Lechan. 1994. Suppression of thyrotropin-releasing hormone gene expression by interleukin-1β in the rat: Implications for nonthyroidal illness. Neuroendocrinology 59:129-137.
Kakuscska, I., Y. Qi, B.D. Clark, and R.M. Lechan. 1993. Endotoxin-induced corticotropin-releasing hormone gene expression in the hypothalamic paraventricular nucleus is mediated centrally by interleukin-1 . Endocrinology 133:815-821.
Kimball, H.R., M.B. Lipsett, W.D. Odell, and S.M. Wolff. 1968. Comparison of the effects of the pyrogens, etiocholanolone and bacterial endotoxin on plasma cortisol and growth hormone in man. J. Clin. Endocrinol. Metab. 28:337-342
Knox, J., R. Demling, D. Wilmore, P. Sarraf, and A. Santos. 1995. Increased survival after major thermal injury: The effect of growth hormone therapy in adults. J. Trauma 39:526-530.
Koenig, J.I., K. Snow, B.D. Clark, R. Toni, J.G. Cannon, A.R. Shaw, C.A. Dinarello, S. Reichlin, S.L. Lee, and R.M. Lechan. 1990. Intrinsic pituitary interleukin-1β is induced by bacterial lipopolysaccharide. Endocrinology 127:3053-3058.
Lambert, M. 1994. Thyroid dysfunction in HIV infection. Baillieres Clin. Endocrinol. Metab. 8:825-835.
Lechan, R.M., R. Toni, B.D. Clark, J.G. Cannon, A.R. Shaw, C.A. Dinarello, and S. Reichlin. 1990. Immunoreactive interleukin-1β localization in the rat forebrain. Brain Res. 514:135-140.
Manu, P., T.J. Lane, and D.A. Matthews. 1992. The pathophysiology of chronic fatigue syndrome: Confirmations, contradictions, and conjectures. Int. J. Psychol. Med. 22:397-408.
Michie, H.R., J.A. Majzoub, S.T. O'Dwyer, A. Revhaug, and D.W. Wilmore. 1990. Both cyclooxygenase-dependent and cyclooxygenase-independent pathways mediate the neuroendocrine response in humans. Surgery 108:254-259.
Mooradian, A.D., R.L. Reed, D. Osterweil, R. Schiffman, and P. Scuderi. 1990. Decreased serum triiodothyronine is associated with increased concentrations of tumor necrosis factor. J. Clin. Endocrinol. Metab. 71:1239-1242.
Munck, A., P.M. Guyre, and N.J. Holbrook. 1984. Physiological functions of glucocorticoids in stress and their relation to pharmacological actions. Endocrinol. Rev. 5:25-44.
Naylor, A.M., K.E. Cooper, and W.L. Veale. 1987. Vasopressin and fever: Evidence supporting the existence of an endogenous antipyretic system in the brain. Can. J. Physiol. Pharm. 65:1333-1338.
Nguyen, T.T., D.A. Gilpin, N.A. Meyer, and D.N. Herndon. 1996. Current treatment of severely burned patients. Ann. Surg. 223:14-25.
Olson, B.R., T. Cartledge, N. Sebring, R. Defensor, and L. Nieman. 1995. Short-term fasting effects luteinizing hormone secretory dynamics but not reproductive function in normal-weight sedentary women. J. Clin. Endocrinol. Metab. 80:1187-1193.
Pang, X.P., J.M. Hershman, C.J. Mirell, and A.E. Pekary. 1989. Impairment of hypothalamic-pituitary-thyroid function in rats treated with human recombinant tumor necrosis factor-α (cachectin). Endocrinology 125:76-84.
Payne, L.C., F. Obal, Jr., M.R. Opp, and J.M. Krueger. 1992. Stimulation and inhibition of growth hormone secretion by interleukin-1β: The involvement of growth hormone-releasing hormone. Neuroendocrinology 56:118-123.
Peisen, J.N., K.J. McDonnell, S.E. Mulroney, and M.D. Lumpkin. 1995. Endotoxin-induced suppression of the somatotropic axis is mediated by interleukin-1β and corticotropin-releasing factor in the juvenile rat. Endocrinology 136:3378-3390.
Rall, L.C., N.T. Lundgren, S. Reichlin, J.D. Veldhuis, and R. Roubenoff. 1996. Growth hormone (GH) kinetics in aging and chronic inflammation. FASEB J. 10:A754.
Reichlin, S. 1993. Neuroendocrine-immune interactions. N. Engl. J. Med. 329:1246-1253.
Reichlin, S. 1994. Neuroendocrine consequences of systemic inflammation. Pp. 83-96 in Advances in Endocrinology and Metabolism, E.L. Mazzaferri, R.S. Bar, and R.A. Kreisberg, eds. St. Louis: Mosby.
Reichlin, S. 1995. Endocrine-Immune Interaction. Pp. 2964-3012 in Endocrinology, L.J. DeGroot, ed. Philadelphia: W.B. Saunders Company.
Reichlin, S. 1998. Neuroendocrinology. In William's Textbook of Endocrinology, 9th ed. [in press], J.D. Wilson, D. Foster, P.R. Larsen, and H. Kronenberg, eds. Philadelphia: W.B. Saunders Company.
Reichlin, S., and R.J. Glaser. 1958. Thyroid function in experimental streptococcal pneumonia in the rat. J. Exp. Med. 107:219-236.
Richmand, D.A., M.E. Molitch, and T.F. O'Donnell. 1980. Altered thyroid hormone levels in bacterial sepsis: The role of nutritional adequacy. Metab. Clin. Exp. 29:936-942.
Rivest, S., and C. Rivier. 1995. The role of corticotropin-releasing factor and interleukin-1 in the regulation of neurons controlling reproductive functions. Endocr. Rev. 16:177-199.
Romero, L.I., J.B. Tatro, J.A. Field, and S. Reichlin. 1996. Roles of IL-1 and TNF-α in endotoxin-induced activation of nitric oxide synthase in cultured rat brain cells. Am. J. Physiol. 270:R326-R332.
Samuels, M.H., and P. Kramer. 1996. Differential effects of short-term fasting on pulsatile thyrotropin, gonadotropin, and α-subunit secretion in healthy men-a clinical research center study. J. Clin. Endocrinol. Metab. 81:32-36.
Sapolsky, R., C. Rivier, G. Yamamoto, P. Plotsky, and W. Vale. 1987. Interleukin-1 stimulates the secretion of hypothalamic corticotropin-releasing factor. Science 238:522-524.
Scarborough, D.E., S.L. Lee, C.A. Dinarello, and S. Reichlin. 1989. Interleukin-1 stimulates somatostatin biosynthesis in primary cultures of fetal rat brain. Endocrinology 126:3053-3058.
Shalts, E., Y-J. Feng, and M. Ferrin. 1991. Vasopressin mediates the interleukin-1a-induced reduced decrease in luteinizing hormone secretion in the ovariectomized rhesus monkey. Endocrinology 131:153-158.
Sheagren, J.N. 1991. Corticosteroids for the treatment of septic shock. Infect. Dis. Clin. North Am. 5:875-882.
Sternberg, E.M. 1992. The stress response and the regulation of inflammatory disease. Ann. Int. Med. 117:854-866.
Syed, V., N. Gérard, and A. Kaipia. 1993. Identification of an interleukin-6 like factor in rat seminiferous tubule. Endocrinology 132:293-299.
Sumita, S., Y. Ujike, A. Namiki, H. Watanabe, M. Kawamata, A. Watanabe, and O. Satoh 1994. Suppression of the thyrotropin response to thyrotropin-releasing hormone and its association with severity of critical illness. Crit. Care Med. 22:1603-1609.
Vagenakis, A.G., A. Burger, G.I. Portnoy, M. Rudolph, J.R. O'Brian, F. Azzizi, R.A. Arky, P. Nicod, S.H. Ingbar, and L.E. Braverman. 1975. Diversion of peripheral thyroxine metabolism from activating to inactivating pathways during complete fasting. J. Clin. Endocrinol. Metab. 41:191-194.
van der Poll, T., and S.F. Lowry. 1995. Tumor necrosis factor in sepsis: Mediator of multiple organ failure or essential part of host defense? Shock 3:1-12.
van der Poll, T., J.A. Romijn, E. Endert, and H.P. Sauerwein. 1993. Effects of tumor necrosis factor on the hypothalamic-pituitary-testicular axis in healthy men. Metabolism 42:303-307.
van der Poll, T., J.A. Romijn, W.M. Wiersinga, and H.P. Sauerwein. 1990. Tumor necrosis factor: A putative mediator of the sick euthyroid syndrome in man. J. Clin. Endocrinol. Metab. 71:1567-1572.
van der Poll, T., K.J. Van Zee, E. Endert, S.M. Coyle, D.M. Stiles, J.P. Pribble, M.A. Catalano, L.L. Moldawer, and S.F. Lowry. 1995. Interleukin-1 receptor blockade does not affect endotoxin-induced changes in plasma thyroid hormone and thyrotropin concentrations in man. J. Clin. Endocrinol. Metab. 80:1341-1346.
Voerman, H.J., A.B. Groenveld, H. de Boer, R.J. Strack van Schijndel, J.P. Nauta, E.A. van der Veen, and L.G. Thijs. 1993. Time course and variability of the endocrine and metabolic response to severe sepsis. Surgery 114:951-959.
Voerman, H.J., R.J. Strack van Scijndel, A.P. Groenevelkd, H. de Boer, J.P. Nauta, and L.G. Thijs. 1992a. Pulsatile hormone secretion during severe sepsis: Accuracy of different blood sampling regimens. Metabolism 41:934-940.
Voerman, H.J., R.J. van Schijndel, A.B. Groeneveld, H. de Boer, J.P. Nauta, E.A. van der Veen, and L.G. Thijs. 1992b. Effects of recombinant human growth hormone in patients with severe sepsis. Ann. Surg. 216:648-655
Wada, T., M. Sato, M. Niimi, M. Tamaki, T. Ishida, and J. Takahara. 1995. Inhibitory effects of interleukin-1 on growth hormone secretion in conscious male rats. Endocrinology 136:3936-3941.
Wagner, H., E. Zierden, and W.H. Hauss. 1975. Effects of synthetic somatostatin on endotoxin-induced changes of growth hormone, cortisol and insulin in plasma, blood sugar and blood leukocytes in man. Klin. Wochenschr. 53:539-541.
Wartofsky, L. and K.D. Burman. 1982. Alterations in thyroid function in patients with systemic illness: the "euthyroid sick syndrome." Endocr. Rev. 3(2):164-217.
Watkins, L.R., S.F. Maier, and L.E. Goehler. 1995b. Cytokine-to-brain communication: A review and analysis of alternative mechanisms. Life Sciences 57:1011-1026.
Wilder, R.L. 1995. Neuroendocrine-immune system interactions and autoimmunity. Ann. Rev. Immunol. 13:307-338.
Discussion
LEONARD KAPCALA: In the IL-1 receptor antagonist studies in which no benefits were demonstrated, I wonder whether their treatment may be too late.
SEYMOUR REICHLIN: That is a very good point. First of all, in several species of experimental animals, IL-1 receptor antagonist has really very excellent effects in preventing death, so that if you inject the material prior to challenge, it is effective.
Secondly, in the preliminary studies with a small number of patients treated with IL-1 receptor antagonist, very, very sick patients were used, and these patients did have a beneficial effect. Then when the study was expanded, individuals were included who were less septic than the original series, and in that group the interleukin receptor antagonist had no effect. In that study, those
in the subgroup who were very, very sick had a beneficial effect. What that means to me is that under most circumstances, you make about as much IL-1 as you need, and that the extreme septic people make too much, but you would have to know that at the time you make the diagnosis, and also if the acute illness had gone on for 2 days it may be too late to do anything.
I think that is the practical point.
WILLIAM BEISEL: I was certainly glad to hear your presentation, to hear the interpretations of modern-day cytokinology by a classic endocrinologist. It was a delightful presentation.
SEYMOUR REICHLIN: Thank you. I have to tell you that Dr. Beisel was an endocrinologist in his former life.
WILLIAM BEISEL: I want to point out the committee interest in many of these cytokines—or my personal interest in what I call cytokine-induced malnutrition. This is the acute problem of loss of nitrogen and loss of vitamins and loss of minerals such as zinc, sequestration of iron and zinc. All of those metabolic effects have acute effects on the soldier in a short period of time, and so, in addition to all their beneficial effects and their deleterious effects that can lead to death, I think for the mild infections that our committee is concerned mostly about, how many nutrients are lost and what this has done. . .
SEYMOUR REICHLIN: I would underline what Dr. Beisel said, that in addition to a metabolic disturbance caused by neuroendocrine effects, several of the cytokines, particularly TNF-α have profound direct metabolic effects at the tissue level.
They produce profound catabolic disturbance, which may actually be more important than the effects that are mediated through the classical endocrine system. I would definitely emphasize the cachectin type of responses.
NED BERNTON: I have a question and a comment. Dr. Beisel many years ago worked in volunteers with the spread of infections. Since blood draws in major studies have always been done in the morning, it is very hard to get a feel for what is going on in the circadian rhythm of cortisol secretion, and if indeed that reflects a systemic inflammatory response, and we did some preliminary studies with salivary cortisol that suggested that in some of the subjects, the circadian rhythm was lost, but measuring salivary cortisol levels may be a very interesting way to gain data tracking the circadian rhythm, which would make a lot more sense. And my other question is as IGF [insulin-like growth factor]-I is decreased by malnutrition and infection, would IGF (as a marker of GH
secretion that is not subject to the pulsatile variation of GH) possibly give a useful indication of what was purely nutritional and what was due to hypothalamic-pituitary functional changes?
SEYMOUR REICHLIN: Dr. Bernton's point here I think is extremely valuable. Because of the problems of sampling, you don't really get a clear picture of the changes in circadian rhythm. As a matter of fact, I have been unable to find any studies of melatonin rhythms in the conditions of severe stress such as combat training.
The second question you raised, I think is also important. Somatomedin C levels should be measured. Treatment with this hormone may be beneficial. Is it reasonable to consider genetic hormone treatment under these conditions?
The Genentech Company now sells a fountain pen-size syringe for injecting growth hormone in kids with dwarfism that you can just put in your pocket and give yourself a shot once a day.
DOUGLAS WILMORE: Sy, I wonder if you and Jeff [Rossio] could help the committee and talk about both cytokine and endocrine responses in patients fed different types of nutrients or people who are underfed? A classic beginning would be taking fish oil and not seeing IL-1 elaboration from monocytes, but I wonder if you could expand on that because that is really what we are charged with, it seems to me, to think about how we can modulate nutrient intake and maybe in fact modulate these effector messages.
SEYMOUR REICHLIN: I want to make one comment and turn it over to Jeff, because this is a field I really don't know enough about, but in the study of sick euchyroidism that Mark Molitch (Richmand, O'Donnell, and Molitch), they repleted the patients with hyperalimentation.
They found that carbohydrate replacement in sick euthyroid patients reversed the low T3 levels. It would be, from my point of view, extremely important to look at the other side of the coin, that is, the effect of nutritional status on cytokine production. I don't think there is anything on that, is there?
JEFFREY ROSSIO: I am not aware of anything. You did a study with Charles Dinarello, didn't you, where you gave fish oil supplements and showed that there was a lower IL-1 response?
SEYMOUR REICHLIN: I wasn't involved in that study, but Dinarello did it.
JEFFREY ROSSIO: But wasn't that with your material?
SEYMOUR REICHLIN: It was in collaboration with Sheldon Wolff. That is a very relevant experiment. I don't think that has been done.
JONATHAN KUSHNER: I had a question along the same line until you pointed out the similarities between starvation and inflammation on pituitary secretion. Do you think that in pure starvation any of those effects are cytokine mediated, or do you know?
SEYMOUR REICHLIN: Many of the changes we are talking about can be induced by starvation. Some are hypogonadism, sick euthyroid, increased cortisol, decreased IGF-I. For sure, those three, at least are identical, and there is a starvation component even in those who are infected.
LEONARD KAPCALA: Relevant to that, there are a lot of data showing that insulin can limit the hypogonadism but the insulin only acts centrally. If you administer it peripherally, you don't get that effect, suggesting that insulin acting locally is required for the induction of IL-1 in the brain.
SEYMOUR REICHLIN: Dr. Kapcala is making a point I would like to emphasize. We have done a lot of work—which I didn't show—comparing interleukin with toxins. And LPS, which is a paradigm toxin, is much more potent on most of these reactions than is interleukin or any single interleukin that is available or in combination. Also, effects that we observed with LPS can be blocked only partially with TNF-α, soluble receptor, and IL-1 receptor antagonist (Romero et al., 1996).
The effects of bacterial toxin are mediated by many factors. There are probably some mediators that we still don't even recognize, there are receptors for LPS itself, and there is a direct effect of LPS as a cytokine.
| This page in the original is blank. |


















