
2
Software Development for Computational Chemistry: Does Anything Remain to Be Done?
Peter R. Taylor
San Diego Supercomputer Center and University of California, San Diego
We consider the state of the art of computational chemistry and then discuss to what extent this state of the art meets the requirements of the chemistry community. Our overview is necessarily broad and somewhat superficial, but it supports the view that while computational chemistry is a mature and very successful field, considerable effort is still needed at the level of fundamental research into methods, algorithms, and implementation, and in training students in these areas.
For the purposes of this paper, we consider computational chemistry to comprise the study of the structure, properties, and dynamics of chemical systems. We recognize that this somewhat narrow definition does not properly incorporate areas such as process modeling that are also of importance to the chemical industry, but the discussions at the workshop strongly suggest that the successes and, not “failures," but let us say "unmet expectations," in these other areas are not different in cause or in possible remedies from those in our narrower definition of computational chemistry. With this definition, then, we can argue with some justification that computational chemistry is one of the great scientific success stories of the past decades. Twenty-five years ago quantum-chemical calculations were performed by quantum chemistry specialists, and the results of such calculations were rarely and with difficult acceptance published in general chemistry journals such as the Journal of the American Chemical Society. At that time calculations on molecules of more than a dozen atoms were a rarity, and the accuracy of the predictions was often overstated and seldom convincing to experimentalists. We need not labor the point here: suffice it to say that the situation today is exactly the reverse. Indeed, the field of computational chemistry has just been recognized by the award of the 1998 Nobel Prize in chemistry to Pople and Kohn. Might it not, therefore, be time to declare success in this endeavor, and that the field is essentially complete? In this essay, we argue not only that this would be a grievous mistake, but also that considerable additional effort is required in a number of areas for computational chemistry to reach its full potential. Our field is mature, not complete; ripe with opportunities, not sterile.
The major activities in computational chemistry can be classified as molecular electronic structure (quantum chemistry), reaction dynamics, and molecular dynamics. These are used to calculate, respectively, the properties of individual molecules or groups of molecules, kinetic information and reaction
pathways for chemical reactions, and the properties and dynamics of large molecules or large assemblies of molecules. All of these activities have been extraordinarily successful over the past decades and have firmly established computational chemistry as a third methodology alongside experiment and theory. Computational chemists have also been among the foremost users of computer hardware, with substantial requirements for computer time, memory, and disk space, and with a history of exploiting both early access to new architectures and novel solutions to reduce the cost of performing their research. They have successfully exploited new vector and parallel computer architectures as they have become available, and at the same time have developed new algorithms to efficiently use first minicomputers and later RISC workstations, and most recently clusters of commodity PCs. The advent of ASCI-class computing resources presents a new opportunity—a vast increase in computational capability to be exploited. However, it is not obvious that the community is ready to use such massively parallel machines, not the least because even our scalable algorithms have generally been tested in situations only up to a few hundred processors. To use the full power of an ASCI-class machine will require successfully harnessing at least an order of magnitude more processors. Will our current algorithms, and at an even higher level, the computational chemistry methods they implement, be suitable for such architectures? It is these questions we concentrate on here, not the detail issues of whether message-passing implementations are more or less appropriate than shared-memory implementations of our methods. We demonstrate, for example, that much of our current methodology is incapable of extending the accuracy of our description of molecular systems beyond what we can currently achieve, and that new methods must be sought.
We take quantum chemistry, as defined above, as an example. The information for nonempirical dynamics calculations comes from quantum-chemical calculations, so in this sense quantum chemistry is fundamental to nonempirical computational chemistry. Typical quantum chemistry calculations, in 1999, treat a single molecule or perhaps a group of molecules, in vacuo, at a temperature of 0 K. The accuracy achievable varies with the size of the molecule (see Figure 2.1), but the highest-accuracy work is comparable to experimental accuracy for many properties. A simple way to represent the relationship between molecular size and accuracy of results is a graph generally referred to as a Pople diagram.
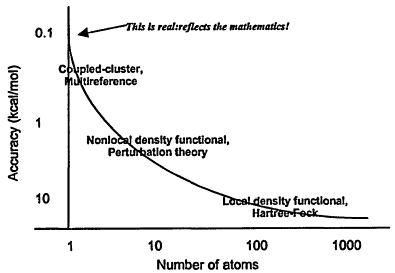
Figure 2.1
Size/accuracy relationship in ab initio calculations.
Figure 2.1 shows the accuracy achievable for different sizes of molecule (or assembly of molecules) and the most common quantum-chemical methods used to achieve that accuracy. One important aspect of Figure 2.1 is the intersection of the curve with the ordinate axis at around 0.1 kcal/mol. This is not a resolution artifact or a mistake in the figure—it reflects a fundamental limitation in our ability to describe the quantum-mechanical motion of the electrons in a molecule, specifically, the cusp behavior as two electrons approach one another. The methods listed in the figure are all based on expansions in one-electron functions, and such expansions are inherently incapable of describing the cusp in a finite number of terms. Increasing the number of terms cannot proceed indefinitely, not only because the calculations become impossibly large, but also because the resulting expansion function sets become linearly dependent. Here is one example of a fundamental problem that must be addressed in order to increase our computational chemistry capabilities—we must develop new methods that better approximate the molecular wave function.
An immediate question might be, Is such increased accuracy really necessary? A flippant response would be that everyone would like more accuracy: like money, or network bandwidth, it is something one cannot have too much of. A more realistic answer would be that an organic chemist planning a synthesis might find herself in a situation where a difference in barrier heights of a few tenths of a kcal/ mol determines whether the ratio of desired product to useless alternative is 9:1 or 1:9. Barrier heights known to 1 kcal/mol or so would be quite useless in this situation. And, as noted above, the accuracy of computed quantum-chemical energies determines the reliability of nonempirical dynamics calculations, so if more accurate dynamics calculations are desired, the accuracy of the quantum-chemical calculations must be improved.
The other axis in Figure 2.1 is molecular size. Again, the crucial question is how to increase the size of the system we can treat. This is not merely in order to treat larger molecules per se, although this is one consideration. Another important issue is the realistic treatment of environment. Most chemistry takes place in the condensed phase or in a gas phase of some density, not in a vacuum. Although some progress has been made in describing solvent effects by embedding the system in a dielectric medium, there is good evidence that detailed solute-solvent interactions are so important in many situations that some solvent molecules must be treated explicitly. We must therefore increase the size of our systems to include perhaps tens of solvent molecules, and thus must develop methods to handle (much) larger systems.
We reiterate that we are not suggesting here that access to much more powerful computers alone is an adequate solution to the accuracy and size problems (indeed, in the case of increased accuracy, it is manifestly not adequate). What is needed is to improve or to develop new methodologies so that they can handle the problems of interest in the way we wish to treat them, taking advantage of the increased computing power of ASCI-class machines to achieve that end. The first steps must be in methodology, not in porting existing approaches to new architectures.
What of other components of computational chemistry, such as reaction dynamics or molecular dynamics simulations? A chat with practitioners in these areas is likely to indicate a need to perform more accurate dynamics studies, on larger systems, including the effects of bulk environment, etc., plus an additional concern about studying phenomena at longer time scales (that is, how best to simulate phenomena that take place in nature on a time scale of microseconds to milliseconds when normal simulation time steps are at the femtosecond level). In the case of increased accuracy and larger systems, the issues are of course similar in spirit to those discussed above in the context of quantum chemistry. Indeed, increased accuracy in reaction dynamics and simulations would probably mandate increased accuracy (for larger systems) in the quantum-chemical calculations used to provide potentials
for the dynamics. Our attention is again focused on the need to develop new methods rather than to simply look at ways to implement existing methods or to port existing programs to new architectures.
Development of new computational methods will lead to new implementations for those methods, which brings additional advantages. First, there is the opportunity to consider explicitly the nature of new computer architectures when developing the methods. For example, methods that might have seemed inappropriate in the past because their implementation would have enormous memory requirements may be perfectly feasible in the large memory environment provided by ASCI-class machines. Second, new implementations can take advantage of modern software engineering practices and modern computer languages, leading to increased ease of maintenance compared to the traditional "dusty decks." Third, new methods and implementations can take advantage of modern technologies, such as the ability to store, retrieve, and manipulate large data sets, or interaction environments via which users can not only visualize their data but also steer simulations.
As we noted at the beginning of this essay, computational chemistry has been a remarkably successful subdiscipline. Nevertheless, we repeat also that success and maturity should not be confused with completion; our efforts have been very successful, but much remains to be done. A primary need is to encourage the development of new methods, and their implementation, rather than just seeking to reimplement existing methods on new hardware. Efforts are needed to increase the accuracy of our results and to achieve that accuracy for larger and larger systems, to model experiments more realistically, and to develop software appropriate to new hardware so as to fulfill the opportunities offered by ASCI-class machines. These efforts, in turn, require strong support from agencies and institutions, not only to support research and development activities, but also to provide a cadre of trained computational scientists through support of education and training.
Acknowledgments: The author was supported by the National Science Foundation through Cooperative Agreement DACI-9619020 and by Grant No. CHE-9700627.
Discussion
Sam Kounaves, Tufts University: Considering the amount of investment that has been put into these types of supercomputing centers and initiatives, one of the final outcomes is, of course, the ability to transfer some of this technology to other areas. In my own particular specialty, for example, we would love to have more in terms of modeling electrode position nucleation for the fusion phenomena. Microsoft last year spent $3 billion just to develop software, and that may not be significant. But are there any plans for transferring? What type of systems do you foresee, for example, to make it more generalized, to make it accessible to other communities in the chemistry world?
Peter Taylor: There are a couple of efforts, I think. Some of the work done at EMSL (Environmental Molecular Sciences Laboratory) at PNNL, in Northwest Chemistry Codes, and some of the activities done under our own support in San Diego and our larger partnership, National Partnership for Advanced Computational Infrastructure, attempt to do just this. And I think there has been some success with some of these things already. Both of these projects really are just getting rolling in terms of dealing with the outside community. But we have plans to do this and I would expect to see the fruits of this over the next couple of years.
That, I think, is something we can do in cases where we already have the methods that we need. If new methods have to be developed, then, of course, one has to be less specific about the lead time. We simply do not know how long it will take to develop some of the new methodology, and so forth. But in
a number of communities, certainly those supported by the Department of Energy and in the NSF program, there is a desire to harden these things and move them out so that use can be made of them in a much larger community.
Peter Cummings, University of Tennessee and Oak Ridge National Laboratory : I just wanted to comment, Peter, about the things that you said were coming from the molecular dynamics community, which I will just generalize as the classical simulation community. I think it is not simply an issue of larger systems. It is also longer simulation times; more complex systems, like proteins and polymers, all have very much longer relaxation times. And I think that one of the challenges facing parallel computing is how to get beyond these much longer time scales, because parallelization does not solve that problem. And, in fact, I think there may be a lot of potential in non-dynamical—that is, Monte Carlo-type—techniques that subvert the time-scale problem. Rethinking those on parallel architectures, I think, can lead to a lot of progress.
Peter Taylor: I think that is a very important point. I certainly would not want anybody to go away with the assumption that I felt I had listed all of the issues here. The one of time scale is important enough that I should have said something about it. It is also another useful illustration of one of the points I was making, because that is an area, clearly, where effort needs to be invested in developing new methods. Getting beyond longer time scales is not something we know how to do today. We need to try to find novel ways, as you say, to subvert, in essence, the time-scale problem faced otherwise in traditional dynamics.
Judith Hempel, University of California, San Francisco: I would like to raise a question that I cannot answer. What is the impact, really, on computational chemistry of the increase in the computing power of the ASCI initiative, and what if that were only the beginning? So, in a sort of visionary sense, if computing power were to increase at this same rate or even at an accelerated rate, what would be the impact of computational chemistry on the world as we know it?
Peter Taylor: Well, I think the issue is once again related to some of these questions about accuracy and about the size of the calculations. If you look at, for example, small-molecule chemistry, then typically the accurate methods we have scale roughly on the order of the fifth or sixth power of the size of the molecule. So with a computer 100,000 times faster than what you have today, you would still only be able to study a system 10 times larger. Now, we would all like to be able to do systems 10 times larger, but that capability does not necessarily seem that much of a reward for a five-orders-of-magnitude gain in computing power. If we really want to see dramatic qualitative changes in the type of work we can do with computational chemistry, we need to put a lot more effort into figuring out how to make the calculations scale better with the size of the system.
Judith Hempel: Do you see a time in the future when quantum mechanics will take over for the empirical methods, like the force-field methods that now use quantum mechanics to extract parameters?
Peter Taylor: What I see is an increasing use of, in essence, hybrid methods. At some level, even with something very large like an enzyme, I do not think it should be necessary—I would like to think we are more ingenious than this—to treat the entire enzyme and the medium in which it is found quantum mechanically. But I suspect that in order to be able to do a good job in predicting the catalysis, the actual
details of what happens at the bottom of the active site in the enzyme, we will need to use quantum mechanics.
The question is, If you start using quantum mechanics there, where do you switch over as you move out in space to a point where you can go over to a lower-level treatment, a semi-empirical or empirical method? There has been a lot of effort in that recently. Some efforts have been more successful than others. I think this is something we will work out over the next few years, and I think this is the real future. Ideally there should be no need to treat a system like that entirely quantum mechanically. If we could do it cheaply enough, well, then we could do it that way. But in practice I do not think that should be necessary.
But I think we do not completely understand yet how to splice these different levels of treatment together. How do we go, for example, from a very accurate quantum chemistry calculation for an area where we are really interested in the details to a lower-level quantum chemistry calculation, say a density functional calculation or something in a larger region, and then eventually to some completely empirical type of molecular mechanics approach in the next layer out?
Thom Dunning: Let me make one addition to that statement. One of the surprises that came out of the ASCI program was that the increased computing power makes it possible—even though, as Peter pointed out, the techniques that we use right now do not scale very well with the size of the system—to actually compute all of the thermodynamics for all of the species and the reactions involved in combusting both gasoline and diesel fuel. And, in gasoline there are some 1,000 species that are thought to be involved, and some 2,000 reactions that the modelers say they need to have kinetic data for.
For diesel fuel you are talking about 2,000 species and 4,000 reactions. And some of those species have not even been seen or characterized in the laboratory yet, so even with the techniques as they are right now, one can, without increasing computing power, actually make substantial contributions to very practical problems that the country faces. But, I would agree entirely with Peter in that we need techniques that scale better with the size of the system.






