
7
New and Emerging Analytical Techniques for the Detection of Organic Contaminants in Water
Martin Reinhard and Jean-François Debroux
Detailed chemical analysis of water is a prerequisite for assuring the safety of water supplies. Limitations in our ability to identify contaminants in water also limit our ability to assure water quality and to assess environmental impacts. Recent improvements in analytical techniques have expanded the "analytical window," that is, the compound range that is amenable to specific identification. Using these improved tools, much has been learned about the occurrence, fate, and transport of organic chemicals in the environment. The boundaries of the analytical window represent the "analytical frontiers" and are continuously expanded. These boundaries are defined loosely by the contaminant concentration and chemical properties, such as molecular weight, polarity, chemical lability, and structural complexity.
In typical natural water samples, the mass of compounds that is within the analytical window (i.e., that can be specified in terms of both a specific structure and concentration) is small compared to the total organic carbon. Using coupled gas chromatography/mass spectroscopy (GC/MS) in conjunction with derivatization, the specifically identified compounds accounted for less than 12 percent (or < 1 mg/L) of the organic carbon in different groundwater samples (Reinhard et al., 1994). For 80 percent (or 2 to 10 mg/L) of the total organic carbon, this fraction typically remains uncharacterized or only in aggregate form (e.g., in terms of average chemical properties, functional group content, or size distribution) (Fujita et al., 1996).
Even though good mass spectra are obtained from a majority of the contaminants that are detected, most remain unidentified and/or unquantified because of a lack of reference spectra and/or reference compounds. The information that can be deduced from the mass spectra may be sufficient to propose structures using spectral determinations. However, verification and quantification of the proposed structures are often impossible because of a lack of reference spectra.
Since the majority of the organic carbon in environmental samples is not amenable to specific identification, aggregate properties (or group parameters) are often used as indicators of general water quality. Included in this category are such parameters as elemental composition, functionality, acidity, metal binding properties, total halogen content, ultraviolet (UV) absorption,
fluorescence properties, and molecular size distribution. Although very important, analysis of these parameters is not discussed in detail here.
Concern about the presence of potentially harmful contaminants is particularly relevant in cases where drinking water sources are impacted by point sources, such as wastewater effluents, landfills, and industrial dump sites. Other threats to water quality stem from the use of agricultural pesticides, leaking fuel and solvent storage tanks, and byproduct formation during disinfection with oxidants. To deal with these concerns, regulatory agencies have responded by establishing maximum contaminant limits (MCLs) for hazardous organic compounds (priority pollutants) known to have significant potential to contaminate drinking water, including pesticides, aromatic hydrocarbon compounds, disinfection byproducts, chlorinated solvents, and a number of other synthetic chemicals. MCLs are as low as 50 ng/L for ethylene dibromide and 200 ng/L for 1,2-dibromo-3-bromopropane. The European Union Commission has set a maximum limit for pesticide residues of 100 ng/L in drinking water.
Specifying MCLs for selected compounds addresses the potential threat of chemicals known to be hazardous. However, water quality can be threatened by numerous unidentified compounds that may be of anthropogenic origin or that may originate from natural sources. Compounds of anthropogenic origin include byproducts in consumer products, additives to fuels, waste products of manufacturing processes; natural sources for organics include detritus and exudates of plant and animal matter. The lowest MCLs are in the low nanogram per liter range, and it is evident that target concentrations for detailed characterizations of individual compounds should be in the low nanogram per liter range as well. This paper reviews new and emerging analytical approaches for the analysis of aqueous environmental samples.
Examples of compounds that have emerged recently as particularly relevant are indicated in Table 7-1. The new and emerging analytical techniques are discussed in the context of these compound classes.
Analytical Procedures
Basic Analytical Approaches
Analytical procedures consist of several interdependent operations carefully tuned to provide maximum efficiency. Some of the analytical operations that may apply are shown in Figure 7-1. The sensitivity of the method defines the lower limit of the sample size that must be processed. The sample is processed to preserve, recover, isolate, and/or concentrate the analytes to produce extracts that are compatible with subsequent instrumental analyses. The processes that lead to a concentrate that can be analyzed using instrumental methods are called sample preparation. Derivatization is a microanalytical technique that serves one or several of the following purposes: increase recovery from the aquatic matrix, facilitate separation from other organic constituents,
TABLE 7-1
Emerging Compound Classes
|
Compound Category |
Examples |
|
Pharmaceuticals |
Antibiotics, hormones and contraceptives |
|
Pesticides, herbicides, and their transformation products |
Triazines, phenoxy acids |
|
Surfactants and surfactant residues |
Aliphatic alcohol polyethoxylates, octyl-and nonylphenol polyethoxylates, octyl-and nonylphenol ethoxycarboxylates, linear alkyl benzene sulfonates |
|
Industrial additives and agents |
Aromatic sulfonates, chelating agents, amino carboxylic acids |
|
Taste and odor compounds |
Xylenes, aldehydes, disulfides, chlorophenol |
|
Gasoline additives |
Dialkyl ether, alcohol, methyl tertiary butyl ether |
|
Disinfection byproducts |
Trihalomethanes, haloacetic acids |
and/or improve identification or detection. Because reference compounds for most contaminants are not available, synthesis is often necessary for structure verification and the development of rapid detection and quantification procedures.
Methods used to characterize aggregate dissolved organic carbon (DOC) reflect the physical-chemical properties of the heterogeneous mixture of organic molecules present. The molecular size distribution (selective membranes or gel permeation chromatography), the absorbance and fluorescence of UV and visible light, the bonding configurations (13Carbon, 1Hydrogen nuclear magnetic resonance (NMR), infrared spectroscopy), and acidity (potentiometric titrations) are all commonly investigated properties. A thorough review of these techniques has been prepared by members of the International Humic Substances Society and can be found in Aiken et al. (1985) and Hayes et al. (1989).
Sampling And Sample Preparation Methods
Sampling Considerations
Sample size is one of the most important considerations in designing analytical protocols. If the needed sample size exceeds one liter many studies are not feasible. For instance, the sampling and shipping costs in large-scale field investigations may become prohibitive if the sample size is large and samples are
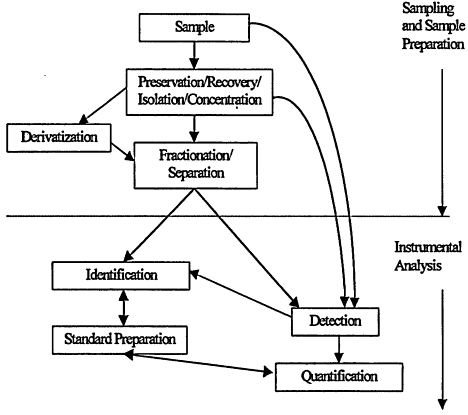
Figure 7-1
General analytical protocol for the analysis of organic trace compounds in water.
shipped by air. Preservation depends on the biodegradability of the analyte. For biolabile compounds, acidification to pH < 2, refrigeration at 4°C, physical separation of microbes by filtration, and the exclusion of light are often sufficient to preserve the sample for a few days. Chemically and biologically stable analytes such as the triazine pesticides are stable under refrigeration in dark conditions for a few years (Bucheli et al., 1997).
Some contaminants, such as chelating agents or some steroid alcohols, are strongly bound to the matrix or to co-contaminants. For the analysis of total testosterone, for example, the sample may have to be subjected to enzymatic hydrolysis prior to extraction to make the testosterone extractable. For ethyl-enediaminetetraacetic acid (EDTA) analysis the speciation (i.e., type of central ion) is an important analytical factor since it influences the stability of the complex and its spectroscopic properties.
Liquid-Liquid and Solid-Phase Extraction
Liquid-liquid extraction (LLE) is a commonly used method of removing organic analytes from an aqueous matrix. LLE methods are used in most official pesticide analysis methods. As LLE methods can be laborious, may cause emulsification problems, involve the evaporation of large solvent volumes, and the disposal of large volumes of hazardous wastes, ways are sought to replace LLE. One increasingly popular alternative is solid-phase extraction (SPE; Altenbach and Giger, 1995; Crescenzi et al., 1997; Pan et al., 1997; Suter et al., 1997). SPE uses hydrophobic sorbents, such as cartridges or discs. Solid-phase microextraction (SPME) utilizes small volumes of sorbents that are extracted using thermal desorption or a small volume of solvent after contacting the sample with the solvent containing the analyte(s) of interest. An elegant form of SPME involves the sorption by a sorbent phase that is coated onto a silica rod mounted into a syringe (Zhang et al., 1994). The compounds can be introduced into the GC by simply injecting the needle and pushing the silica rod into the hot injector.
Another alternative to conventional LLE is supercritical fluid extraction (SFE), which typically uses carbon dioxide as the extractant. SFE can be used indirectly by first concentrating solutes onto solid sorbents, such as SPE cartridges or SPME discs.
As LLE methods are successful at isolating relatively nonpolar organics from water, resin chromatography and membranes are currently used to increase the recovery of organic matter when isolation and concentration are necessary for analysis. A series of ionic and nonionic resins are utilized to exploit the low solubility, and hence absorption, of organic molecules onto resin surfaces. These isolation procedures can be carried out at neutral as well as depressed pH values, relying on the natural or modified (by pH) polarity of organic compounds. Desorption is performed by aqueous eluents possessing different pH values, altering the polarity, or by less polar organic solvents. The resulting fractions of the organic matter pool possess operational definitions that pertain to similar chemical properties.
Concentration by Semipermeable Membranes
Membrane filtration (i.e., reverse osmosis, nanofiltration, and ultrafiltration) is used to separate larger organic molecules from smaller water molecules, increasing the concentration of organic matter in the retentate. As this method has been shown to be successful in retaining a substantial fraction of the larger organic molecules present, retention of smaller organic molecules as well as inorganic components is dependent on membrane pore size and charge. A tradeoff emerges, with smaller pore sizes increasing the fraction of organic matter isolated corresponds to increasing inorganics retention. A comprehensive review of resin chromatography and membrane isolation is presented by Aiken et al. (1985).
Derivatization
Derivatization techniques are used to modify the properties of polar analytes such that they can be processed with a given analytical technique. Typical analytes that must be derivatized prior to analysis include relatively large compounds with thermolabile functional groups (hydroxyl, carboxyl, aminogroups) or small molecules with multiple functional groups. The derivatization chemistry utilizes an agent that reacts selectively with the functional group to form a stable product that can be separated from the water matrix by extraction or sorption, from other analytes by chromatography, detected by chromatographic detectors, or identified by mass spectrometry. Selected examples relevant to water analysis are indicated in Table 7-2.
Most derivatization agents are water reactive and can only be used in water-free extracts. Recent research has been directed toward the development of procedures that allow derivatization directly in water (Minero et al., 1994; Vincenti et al., 1995; Angelino et al., 1997). One approach is to react amino alcohols, amino phenols, polyhydroxy polycarboxylic acids, glycols, and polyhydroxybenzenes with n-hexyl chloroformate. The products hydrolyze slowly owing to their increased hydrophobicity. When fluorinated with an n-hexyl chloroformate, the detection limit could be lowered from the low µg/L to the low ng/L range.
Owing to public health concerns, there is a strong interest in the analysis of some low molecular weight carbonyl compounds, such as glyoxal and methyl glyoxal. One approach to analyzing such compounds involves a reaction with 2,4-dinitrophenylhydrazine and subsequent HPLC analysis. A method for GC analysis has been presented by Glaze and Weinberg (1997), who modified the method by Yamada and Somiya for the analysis of C1 to C3 carbonyls in water. A
TABLE 7-2
Examples of Derivatization Reactions
|
Derivatization Reaction |
Common Derivatizing Agent |
|
Methylation of carboxylic acids |
Diazomethane, methanol/sulfuric acid |
|
Oxime formation of carbonyl functionality |
PFBHA |
|
N-hexyl carbonate, carbamate, and ester formation from hydroxylic, aminic, and carboxylic functionality |
N-hexyl chloroformate |
|
Heptafluorobutyramide formation from aromatic amines |
Heptafluorobutyramide |
review of aqueous carbonyl derivatization by the use of O-(2,3,4,5,6-pentafluorophenyl) methylhydroxylamine hydrochloride (PFBHA) is presented by Cancilla and Que Hee (1992). PFBHA makes an excellent derivatizing agent Since this reaction can be performed in water, resulting oximes are readily extracted into an organic solvent and separate well during GC, and ECD detectors am particularly sensitive to multifluorinated aromatics.
Although diazomethane is a commonly used carboxylic acid methylating reagent, it is very toxic and unstable. Alternative methylating reagents, such as the combination of methanol and sulfuric acid, can be advantageous as they can be time saving and safer to use in the laboratory (Xie et al., 1998). Methylation is also performed in combination with other derivatizing reactions. Multiple derivatizations can enhance the separation of desired compounds from aqueous matrix.
There are numerous derivatizing reagents in use to produce numerous products that can then be separated and quantified. The above text discusses only a select few. Articles or books that solely discuss derivatization chemistry present a more thorough review of the topic (Knapp, 1979; Drozd, 1981; Lingeman and Underberg, 1990). A current review of the various derivatizations utilized to quantify herbicide residues by gas chromatography is presented by Tadeo et al. (1996).
Instrumental Analysis
Fractionation and Separation
Conventional fractionation and separation involve preparative column chromatography, followed by GC or high-performance liquid chromatography. Combinations of derivatization and extraction techniques with either liquid chromatography (LC) or GC allows for the separation of numerous families of compounds. The objective of recent research is to circumvent these laborious techniques by using highly selective MS techniques.
Mass Spectrometry
Mass spectrometry is the method of choice for the identification of trace contaminants in water. State-of-the-art MS has been reviewed elsewhere (Burlingame et al., 1996). The spectrometry of biomacromolecules represents one of the major breakthroughs of the past decade. This breakthrough became possible because of the discovery that polar labile substances could be ionized by sputtering from viscous liquid surfaces using energetic atom and ion beams. Over the past five years, soft ionization techniques with high ion yields have become available, such as electrospray ionization (ESI) and matrix-assisted laser desorption (MALD) ionization. ESI involves the formation of low-energy, even-electron ions by polyprotonation or deprotonation solutions (Burlingame et al., 1996). To achieve a unimolecular compound-specific fragmentation of these low-energy (cold) species, energy is added in tandem mass spectrometers. ESI is applicable for compounds up to 200 kDa.
MALD ionization involves laser-induced ablation as it is protonated or deprotonated from a cocrystalline solid in the matrix chosen. The laser energy is deposited selectively into the solids. Burlingame et al. (1996) have predicted that these technologies will "become as commonplace as HPLC did in the early 1980s." If one looks at the recent environmental literature, this prediction is clearly becoming a reality, as research papers that are using ESI are becoming more and more frequent.
The high selectivity of MS/MS offers the possibility for direct injection of water samples without chromatographic separation and derivatization. An increasingly important tool is molecular modeling techniques for the interpretation of mass spectral fragmentations.
Identification of Chiral Compounds
Many environmental contaminants occur as enantiomeric mixtures. The widely used pesticide mecoprop (2-(4-)chloro-2-methyl-phenoxy propionic acid) is an example. Because different enantiomers often respond selectively in biochemical systems, there is a need for enantioselective determination.
Examples
Pharmaceuticals
Pharmaceuticals are used in large quantities in human and veterinary medicine or as a food additive in animal production. In a series of studies, Stan and Heberer (1997) detected 29 different pharmaceutical compounds. Methods for the analysis of drugs are typically adapted from methods used in pharmacology. Pharmaceuticals have long been known to occur in sewage effluents (Reinhard and McCarty, 1980), but the widespread occurrence of these compounds has been documented only recently, mainly by researchers in Germany (Heberer and Stan, 1997).
Antibiotics
In a bioassay-directed study using umuC genotoxicity, Hartman et al. (1998) analyzed the occurrence of fluoroquinone antibiotics in the sewage of a Swiss hospital.
Hormones and Contraceptives
Ramsey et al. (1997) analyzed for estrone, hexestrol, and zeranol by interfacing an aqueous SFE vessel to an LC/MS instrument equipped with an amino column and a UV/vis diode-array detector. The analyses progressed in
two steps: first, the sample was extracted with super critical carbon dioxide and then the analytes were concentrated on the octadecasylane (ODS) column. Second, by means of a 10 port valve the ODS column inlet was connected to an HPLC pump, and the outlet was connected in series to the chromatographic amine column and the LC/MS. Hexane is used as the extractant for the ODC column and the moile phase for the amine column. The method is suitable for the detection of analytes at the 200 ng/L level.
Stumpf et al. (1996) developed a method for trace analysis of steroids in water with a detection limit of 1 ng/L. The method is suitable for the detection of natural and synthetic estrogen as well as the phytoestrogen β-sitosterol in matrices containing high organic contents (such as secondary effluents). One-liter samples are filtered using 0.45-mm glass fiber filters and then acidified to pH 3. Analytes are concentrated using SPE with a mixture of Lichrolut-EN and Lichrolut C18 (Merck). The extracted materials are eluted with acetone and cleaned up using slica gel chromatography with hexane/acetone as the eluent. The analytes are silylated and then analyzed using GC/MS. Recovery rates at 10 ng/L were 76 to 97 percent. The detection limit ranges from 1 ng/L for estrone, estradiol, estriol, mestranol, 17-ethinylestradiol to 5 ng/L for estradiol-17-valerate and 10 ng/L for β-sitosterol. In river water the observed concentrations for synthetic steroids exceeded the detection limit only occasionally; for β-sitosterol, 50 ng/L was observed in most cases.
Schlett and Pfeifer (1996) report a method for the detection of steroidal hormones that is similar to that of Stumpf et al. (1996). The sample is not acidified prior to SPE, and cleanup of the e tract is not necessary. For derivatization a 1,000:2:2 mixture of MSTFA/TMIS/DTE is used.
Nichols et al. (1997) evaluated the hypothesis that poultry litter applied to pasture contributes to the estrogen 17-β-estradiol in runoff using an enzyme-linked immunoassay. Environmental concentrations in poultry litter (water-extractable fraction) were found to be 133 μg/kg. The observed concentrations in municipal waterwater were 0.03 μg/kg, and in runoff 0.3 to 1.3 μg/kg. The detection limit was found to be 0.02 μg/L. The reported limitation of the study was that only one of many hormones found in poultry waste was measured. Other hormones, including testosterone and estrone, were not measured.
Drug and Drug Metabolites
Heberer and Stan (1997) reported on a procedure for analyzing clofibric acid and N-(phenylsufonyl)-sarcosine. Clofibiric acid (2-(4-)-chlorophenoxy-2-methyl propionic acid) is the metabolite of several blood lipid-regulating drugs (clofibrate, etofyllincolfibrate, etofibrate). Clofibiric acid was detected in wastewater effluents in the 1970s by several investigators in raw and treated sewage. Cartridges containing 1 g of C18 reversed-phase adsorbent were used to extract 1L samples after acidification at pH < 2. Two and one-half milliliters of ethanol were used to elute the analytes from the SPE cartridge. For derivatization, pentafluorobenzoyl bromide is used with triethylamine as the catalyst. The extracts were analyzed using a GC/MS system in the multiple ion detection mode. Concentrations of clofibric acid artificially recharged
groundwater ranged from below the detection limit to 170 ng/L. N-(phenyl)-sarcosine was found at concentrations up to 105 ng/L. In drinking water the concentrations of clofibric acid and N-(phenyl)-sarcosine were as high as 270 and 105 ng/L, respectively; near wastewater infiltration ponds, concentrations were 4 and 150 μg/L, respectively. Occurrence of these compounds in European rivers is consistent with the widespread use of these compounds.
In another series of studies, Stem and Heberer (1997) screened sewage, surface, groundwater, and drinking water for drug and drug metabolites. The observed concentrations in sewage and surface concentrations were as high as 4.5 and 0.5 μg/L, respectively. In a groundwater well used by a drinking water treatment plant, for example, maximum concentrations of clofibiric acid where found to be 7.3 μg/L, diclofenac, 0.4 μg/L; ibuprofen, 0.2 mg/L; and phenazone, 1.2 μg/L.
Buser et al. (1998) report the occurrence of diclofenac, a nonsteroidal antiinflammatory drug, in Swiss rivers and streams, and its photodegradative removal. The analytical procedure involved adsorption at pH < 2 using a macroporous polystyrene adsorbent, extraction, methylation with diazomethane, and GC/MS analysis using selected-ion monitoring.
Pesticides
Environmental contamination with pesticides remains an important topic in environmental chemistry (Barbash and Resek, 1996). The classes of chemicals used as pesticides are expanding, and the chemical structures of the chemicals are increasing in complexity, especially when compared to the chlorinated hydrocarbon pesticides used during the 1950s and 1960s.
Banoub et al. (1997) describe an ESI MS/MS method for the detection of tebufenozide (N'-t-butyl-N'-(3,5-dimethylbenzoyl)-N-(4-ethylbenzoyl)hydrazine) in lakewater samples without either derivatization or further purification. These authors used an ESI with collision-activated decomposition MS/MS for detection of the analyte. The detection limit is approximately 5 ng/L.
Bucheli and coworkers (1997) investigated the occurrence and behavior of three groups groups of herbicides (triazines, acetamides, and phenoxy acids) in roof runoff. Their analytical approach involved concentration on GCB, followed by sequential elution of the neutral and acidic fractions and derivatization of the acidic fraction with diazomethane. 13C-labeled internal standards were used for quantification. Recoveries were 84 percent or better. Mecoprop, which occurs in two enantiomeric forms, was separated with a fused silica capillary column. The detection limits in rainwater are approximately 1 ng/L.
Crescenzi et al. (1997) reported that 45 pesticides, representing 15 compound classes and possessing a wide range of polarity, were quantified using SPE followed by LC/ESP/MS. All compounds but One were greater than 80 percent recovered during SPE, with the majority of the compounds > 95 percent. Detection limits ranged between 1 and 9 ng/L. This method was also used to
quantify polyethylene glycols (ethoxy chain lengths >3) in environmental samples (Crescenzi et al., 1997). Mangiapan and coworkers (1997) report quantification of arachlor and its degradation products by using SPE followed by GC/MS. Select samples were also analyzed by LC/MS.
Surfactants
Although most high-volume surfactants are classified as biodegradable, residual concentrations of the parent product and/or metabolites are frequently found in effluents and surface waters impacted by effluents. Of greatest interest have been the linear alcohol polyethoxylates (AE), alkyl phenol polyethoxylates, and the linear alkyl sulfonates.
Aliphatic Alcohol Polyethoxylates
Marcomini and Pojana (1997) describe an approach to simultaneously characterize both neutral and carboxylated AE biointermediates using liquid chromatography. Biointermediates include linear alcohols, polyethylene glycols (PEGs) of varying lengths, AE carboxylated at the hydrophilic end or on the hydrophobic end, polyethylene glycol monocarboxylates (PEGC), polyethylene glycol dicarboxylates (PEGDC), and short-chain dicarboxylated PEGs. Depending on the type of sample, 10- to 1,000-ml aliquots are used. The approach is based on SPE using 0.3 to 1.0 g GCB as the sorbent. The reported advantage of GCB is that it exhibits properties of reversed-phase sorbents and cation exchange resins. Using elutions of increasing strength, analyte fractions of increasing acidity are obtained. Because of the lack of chromophores, the analytes were converted into derivatives that could be analyzed using HPLC. To take advantage of the great sensitivity and specificity of the fluorescence detector, Marcomini and Pojana (1997) utilize a fluorescent derivatizing agents. For the two neutral compound classes AE and PEG, 1-naphthoyl chloride (NC) and 1-naphtyl isocyanate (NIC) are used, respectively. The authors note that the two derivatizing agents are highly complementary in terms of the attainable separations by reversed-phase HPLC.
The AE and PEG derivatives of NIC can be separated by homologues and total ethoxymer elution, whereas the NC derivatives allow the separation of each AE ethoxymer and the oligomer-by-oligomer separation of PEGs. Hence, the choice of NC or NIC depends on the information red. The reported detection limit for the neutrals is 100 ng/L.
The carboxylated metabolites (carboxylated PEG and carboxylated AE) are derivatized using 9-chloromethyl anthracene and separated using normal-phase and reversed-phase HPLC. Normal-phase chromatography using an amine column separates PEGC and PEGDC, proving a full separation of all intermediates. Reversed-phase chromatography separates the PEGC and the DPEGDC from the AEC but not from each other. The reported recoveries from environmental matrices were better than 90 percent for AEC and CAEC and 67 to 84 percent for PEGC and PEGDC (Macromini and Pojana, 1997) possibly because of matrix effects.
Linear Alkylbenzene Sulfonates (LAS) and Alkylphenol Polyethoxylates (APEO)
LAS and their associated degradation products sulfophenyl carboxylates (SPC), were measured by Gonzales-Mazo et al. (1997) using SPE followed by HPLC. Multiple detection (fluorescence and diode array) was used prior to definitive identification with ISP/MS (operated in the negative ion mode). Detectable levels of C7 to C13 SPCs in environmental samples were reported.
Di Corcia et al. (1994) present a method wherein LAS and SPC can be isolated and quantified in conjuction with APEO, specifically nonylphenol polyethoxylate and its degradation products. These compounds were initially captured by utilizing cartridge SPE and then separated and quantified using liquid chromatography.
Industrial Additives and Chelating Agents
This compound class includes chemically diverse compounds that are used as additives in plastic or concrete, foundry mold, as tanning agents. Of recent interest here are the aromatic sulfonates (Suter et al., 1997). Altenbach and Giger (1995) used SPE and ion-pair chromatography combined with UV absorption and fluorescence detection to detect aromatic sulfonates in a landfill leachate. Compound identification is based on UV/vis and fluorescence spectral libraries. Surer et al. (1997) used SPE in conjunction with LC/MS for the quantitative analysis of aromatic sulfonates (p-toluenesulfonic acid and naphthalene mono and disulfonic acids) from the same site. The LC/MS system utilized was equipped with a single quadrupole mass filter and employed ESI in the negative ion mode.
Taste and Odor Compounds
Taste and odor compounds include relatively simple compounds such as toluene, the xylenes, geosmin, disulfides, and low molecular weight aldehydes (Bruchet and Hochereau, 1997). Odor-thresholds are typically at the nanogram per liter level or below. Most odorous compounds are volatile and although there are exceptions, including chlorophenol and triiodomethane. Bruchet and Hochereau summarize the analytical approaches used for taste and odor compounds: closed-loop and open-loop stripping analysis; purge-and-trap, liquid-liquid, and liquid-solid extraction coupled to high-resolution GC/MS. Solvent-less procedures are important for the detection of highly volatile compounds that would elute within the window of the solvent. Bruchet and Hochereau note that SPE methods have found limited applications owing to background problems,
although they see a potential for SPME. The human nose is very sensitive, and for many taste and odor compounds the sensitivity of available analytical techniques is insufficient to identify the odor-causing compound. Efforts are under way to increase the sensitivity of analytical techniques. New approaches aim to directly inject water volumes up to 100 μl using an ion-column injector.
Disinfection Byproducts
Disinfection byproducts (DBPs) are primarily a drinking water issue. However, because effluents are often chlorinated before discharge, halogenated disinfection byproducts can reach surface waters and groundwater on discharge. Over 100 different reaction products were identified by GC/MS when natural organic matter was chlorinated (Christensen et al., 1983), but Krasner et al. (1989) state that the most abundant DBPs found in chlorinated drinking waters are trihalomethanes, haloacetic acids, haloacetonitriles, haloketones, chlorophenols, chloral hydrate, chloropicrin, and cyanogen chloride.
Gasoline Additives
Church et al. (1997) describe a method that quantifies various gasoline oxygenates: methyl tert-butyl ether (MTBE), ethyl tert-butyl ether (ETBE), and tert-amyl methyl ether (TAME) and also characterizes their most characteristic degradation products. Direct aqueous injection (DAI) was utilized in a GC/MS system. Although purge-and-trap methods have been successful at measuring MTBE, ETBE, and TAME, the DAI/GC/MS method was used to simultaneously measure these compounds and their common metabolites. Detection limits were reported as 0.1 μg/L for all compounds except one metabolite (5 μg/L).
Summary and Conclusions
The size of the analytical window is expanding but still limits our ability to characterize organics in water. The second major limitation is the lack of reference spectra and reference compounds. Even if detected, contaminants often cannot be identified.
The target concentrations for trace organics are being pushed from the microgram to the nanograms per liter range. The trend to lower detection limits is driven by the low MCLs and the potency of some endocrin disruptors. As a consequence, sample preparation techniques require new levels of refinement.
The contaminants that are analyzed include more and more reactive contaminants, contaminants that interact with matrix or sediment components by complexation or other chemical-bonding mechanisms. Chemical speciation is an increasingly important consideration in water quality analysis.
HPLC/MS in conjunction with ESI is becoming more and more a standard method for organic trace analysis. HPLC/MS offers the possibility to circumvent costly and time-consuming derivatization procedures.
Acknowledgements
Funding for this research was provided by the Orange County Water District, Fountain Valley, California, and by the American Water Works Association Research Foundation through a grant to Arizona State University.
References
Aiken, G. R., D. M. McKnight, R. L. Wershaw, and P. MacCarthy, eds. 1985. Humic Substances in Soil, Sediment, and Water: Geochemistry, Isolation, and Characterization. New York: John Wiley & Sons.
Altenbach, B. and W. Giger. 1995. Determination of benzenesulfonates and naphthalenesulfonates in wastewater by solid phase extraction with graphitized carbon-black and ion pair liquid chromatography with UV detection. Analytical Chemistry 67:2325-2333.
Angelino, S., V. Maurino, C. Minero, E. Pelizzetti, and M. Vincenti. 1997. Improved procedure for n-hexyl hydrophilic compounds in water by direct derivatization of highly hydrophilic substances directly in water-hydroxyaminic compounds. Journal of Chromatography 793:307-316.
Banoub, J. H., R C. Martin, H. Hodder, G. Sheppard, and S. Sharpe. 1997. Determination by electrospray tandem mass spectrometry of the insecticide mimic® (tebufenozide) dollowing an areal overspray of a small lentic pond. Analusis Magazine 25:15-19.
Barbash, J. E., and E. A. Resek. 1996. Pesticides in Ground Water, Distribution, Trends, and Governing Factors Vol. 2. Ann Arbor, Mich.: Chelsea.
Bruchet, A., and C. Hochereau. 1997. Analysis of trace compounds responsible for tastes and odors in drinking water. Analusis Maganzine 25:32-34.
Bucheli, T. D., F. C. Gruebler, S. R. Mueller, and R. P. Schwarzenbach. 1997. Simultaneous determination of neutral and acidic pesticides in natural water at the low nanogram per liter range. Analytical Chemistry 69:1569-1576.
Burlingame, A. L., R. K. Boyd, and S. J. Gaskell. 1996. Mass spectrometry. Analytical Chemistry 68:599R-651R.
Buser, H. R., T. Poiger, and M.D. Müller. 1998. Occurrence and fate of the pharmaceutical drug diclophenac in surface waters: Rapid photodegradation in a lake. Environmental Science and Technology 32:3449-3456.
Cancilla, D. A. and S. S. Que Hee. 1992. O-(2,3,4,5,6-Pentafluorophenyl) methylhydroxylamine hydrochloride: A versatile reagent for the determination of carbonyl-containing compounds. Journal of Chromatography 627:1-16.
Christman, R. F., D. L. Norwood, D. S. Millington, J. D. Johnson, and A. A. Stevens. 1983. Identity and yields of major halogenated products of aquatic fulvic acid chlorination. Environmental Science and Technology 17:625-628.
Church, C. D., and L. M. Isabelle, J. F. Pankcow, D. L. Rose, and P. G. Tratnyek. 1997. Method for determination of methyl tert-butyl ether and its degradation products in water. Environmental Science and Technology 31:3723-3726.
Crescenzi, C., A. Di Corcia, E. Guerriero, and R. Samperi. 1997a. Development of a multiresidue method for analyzing pesticide traces in water based on solid-phase
extraction and electrospray liquid chromatography mass spectrometry. Environmental Science and Technology 31:479-488.
Crescenzi, C., A. Di Corcia, A. Marcomini, and R. Samperi. 1997b. Detection of poly(ethylene glycols) and related acidic forms in environmental waters by liquid chromatography/electrospray/mass spectrometry. Environmental Science and Technology 31:2679-2685.
Di Corcia, A., R. Samperi, and A. Marcomini. 1994. Monitoring aromatic surfactants and their aromatic surfactants and their biodegradation intermediates in raw and treated sewage by solid phase extraction and liquid chromatography. Environmental Science and Technology 28:850.
Drozd, J. 1981. Chemical Derivatization in Gas Chromatography. Amsterdam: Elsevier.
Fujita, Y., W. H. Ding, and M. Reinhard. 1996. Identification of wastewater DOC characteristics in reclaimed wastewater and recharged groundwater. Water Environment Research 68:867-876.
Glaze, W. H. and H. S. Weinberg. 1997. A unified approach to the analysis of polar organic by-products of oxidation in aqueous matrices. Water Research 31:1555-1572.
Gonzalez-Maze, E., M. Honing, D. Barcelo, and A. Gomez-Parra. 1997. Monitoring long-chain intermediate products from the degradation of linear alkylbenzene sulfonates in the marine environment by solid-phase extraction followed by liquid chromatography/ionspray mass spectrometry 31:504-510.
Hartman, A., A. C. Alder, T. Keller, and R. M. Widmer. 1998. Identification of fluoroquinoline antibiotics as the main source of umuC genotoxicity in native hospital wastewater. Environmental Toxicology and Chemistry 17:337-382.
Hayes, M. H. B., P. MacCarthy, R. L. Malcolm, and R. S. Swift, eds. 1989. Humic Substances II: In Search of Structure. New York: John Wiley & Sons.
Heberer, T., and H. J. Stan. 1997. Determination of clofibric acid and N-(phenylsufonyl)-sarcosine in sewage, river and drinking water. International Journal of Environmental Analytical Chemistry. 67:113-124.
Knapp, D. R 1979. Handbook of Analytical Derivatization Reactions. New York: John Wiley & Sons.
Krasner, S. W., M. J. McGuire, J. G. Jacangelo, N. L. Patania, K. M. Reagan, and E. M. Aita. 1989. The occurrence of disinfection by-products in U.S. drinking water. Journal of the American Water Works Association 81:41-53.
Lingeman, H., and W. J. M. Underberg, eds. 1990. Detection-Oriented Derivatization Techniques in Liquid Chromatography. New York: Marcel Dekker, Inc.
Mangiapan, S., E. Benfenati, P. Grasso, M. Terreni, M. Pregnolato, G. Pagani, and D. Barcelo. 1997. Metabolites of arachlor in water. Identification by mass spectrometry and chemical synthesis. Environmental Science and Technology 31:3637-3646.
Marcomini, A., and G. Pojana. 1997. Surfactant metabolites in environmental aqueous samples: A liquid chromatographic approach to the routine analysis of alcohol polyethoxylate biointermediates. Analusis Magazine 25:35-37.
Minero, C., M. Vincenti, S. Lago, and E. Pelizatti. 1994. Determination of trace amounts of highly hydrophilic compounds in water by direct derivatization and gas-chromatography: Mass-spectrometry . Fresenius' Journal of Analytical Chemistry 350:403-409.
Nichols, D. J., T. C. Daniel, P. A. Moore, D. R. Edwards, and D. H. Pote. 1997. Runoff of estrogen 17β-estradiol from poultry litter applied to pasture. Journal of Environmental Quality 26:1002-1006.
Pan, L., J. M. Chong, and J. Pawliszyn. 1997. Determinaion of amines and water using derivatization combined with solid-phase microextraction. Journal of Chromatography 773:249-260.
Ramsey, E. D., B. Minty, and A. T. Rees. 1997. Drugs in water: Analysis at the part-per-billion level using direct supercritical fluid extraction of aqueous samples coupled on-line with ultraviolet-visible diode-array liquid chromatography-mass spectrometry. Analytical Communications 34:261-264.
Reinhard, M, and P. L. McCatry. 1980. Pp. 33-52 in Chemistry of Water Reuse Vol. 2, W. J. Cooper, ed. Ann Arbor, Mich.: Ann Arbor Science.
Reinhard, M., W. H. Ding, and Y. Fujita. 1994 Organic Carbon Characterization of Advanced Treated Wastewater at Water Factory 21, Orange County Department of Civil Engineering, Stanford University , Stanford, Calif.
Schlett, C, and B. Pfeifer. 1996. Determination of steroidal hormones in drinking and surface water samples. Vom Wasser 87:327-333.
Stan, H. J., and T. Heberer. 1997. Pharmaceuticals in the aquatic environment. Analusis Magazine 25:20-23.
Stumpf, M, T. A. Ternes, K. Haberer, and W. Baumann. 1996. Nachweis von natürlichen und synthetischen Östogenen in Kläranlagen und Fliessgewässern (in German). Vom Wasser 87:251-261.
Suter, M. J. F, S. Riediker, C. Zipper, H. P. E. Kohler, and W. Giger. 1997. Polar organic compounds in landfill leachates. Analusis Magazine 25:23-25.
Tadeo, J. L., C. Sanchez-Brunete, A. I. Garcia-Valcarcel, L. Martinez, and R. A. Perez. 1996. Determination of cereal herbicide residues in environmental samples by gas-chromatography. Journal of Chromatography 754:347-365.
Vincenti, M., C. Minero, S. Lago, and C. Rovida. 1995. Determination of hydroxycarbamates in aqueous matrices by direct derivatizaton and GC-MS analysis in chemical-ionization. Journal of High Resolution Chromatography 18:359-362.
Xie, Y., D. A. Reckhow, and D. C. Springborg. 1998. Analyzing HAAs and ketoacids without diazomethane. Journal of the American Water Works Association 90:131-138.
Zhang, Z., M. S. Yang, and J. Pawliszyn. 1994. Solid-phase microextraction. Analytical Chemistry 66:A844-A853.
















