

PAUL DOUGHTY BARTLETT
August 14, 1907–October 11, 1997
BY F. H. WESTHEIMER
PAUL D. BARTLETT was one of the great chemists of the twentieth century. His research and teaching were in the area of physical-organic chemistry, and he dominated that field for perhaps four decades. He wasn't old enough to be among the earliest practitioners; that honor is shared among Arthur Lapworth, A. Hantzsch, C. K. Ingold, and L. P. Hammett. But Bartlett created a school of physical-organic chemistry that sparked a revolution in the way organic chemistry is taught and practiced throughout the world. Physical-organic chemistry is concerned with the mechanisms of reaction in organic chemistry and with the properties of organic chemicals. Today, syntheses are designed on the basis of theory, and the theoretical implications and stereochemistry of each step in any complete synthesis are carefully considered, so that the dichotomy of theory and practice is happily fading into the past. No one can contemplate the elegant chemistry of any of the great masters of synthesis today without realizing that the enormous effi-
Reprinted with permission from the Proceedings of the American Philosophical Society 142(3):September 1998.
ciency of modern practice is based on the findings of physical-organic chemistry.
Bartlett himself is responsible for more than a few of these findings, but he is responsible for much more. Only two great schools of physical-organic chemistry have thrived: that of Sir Christopher Ingold at University College, London, and that of Paul Bartlett at Harvard. Both schools have had an enormous impact on organic chemistry, but certainly Bartlett attracted many more, and more productive, collaborators and has had a greater influence on the practice of chemistry.
COLLABORATORS
More than 270 graduate students and postdoctoral fellows worked in Bartlett's laboratory. Before discussing his scientific discoveries, a few more words about these collaborators and his teaching are warranted. Bartlett instituted a course in organic chemistry at Harvard based primarily on reaction mechanisms (Chemistry 105, formerly Chemistry 5) for first-year graduate students and advanced undergraduates. He revised his course year after year, so it developed as the field advanced. Many of the best young chemists throughout America were attracted to Harvard to work with Bartlett, and his graduate students and postdoctorals occupy prominent professorships in universities and major positions in industry throughout the country.
The list of scientists who worked with him and learned from him is long and impressive, and the chemistry they have created is truly remarkable. To avoid, as far as possible, invidious comparisons among Bartlett's many distinguished collaborators, I will name here only those who have been elected to the National Academy of Sciences or the American Academy of Arts and Sciences, or both: Norman Allinger (University of Georgia), Ned Arnett (Duke), Myron
Bender (Northwestern), Saul Cohen (Brandeis), Fred Greene II (MIT), George Hammond (California Institute of Technology and then industry), Daniel Koshland, Jr. (University of California, Berkeley), John D. Roberts (California Institute of Technology), C. Gardner Swain (MIT), D. Stanley Tarbell (Vanderbilt), Nick Turro (Columbia), and Saul Winstein (University of California, Los Angeles). Although this list is extraordinary, the full list of his productive scientific offspring is even more so.
Koji Nakanishi, one of the great practitioners of chemical synthesis, spent a few years at Harvard, and although he didn't work directly with Bartlett, he reports in his autobiography the revelation of learning mechanism, and of hearing such terms as Ingold's SN1 for the first time. He carried Bartlett's teaching around the world with him, specifically to Japan. Many of Bartlett's distinguished collaborators gave courses in advanced organic chemistry based on his “105” and spread his ideas far and wide. Actually, his teaching went farther, for many of his students and postdoctorals themselves had distinguished students and postdoctorals. These scientific grandchildren—his grandstudents, so to speak—continued, and continue, his tradition in physical-organic chemistry.
RESEARCH
BRIDGE-HEAD HALOGENS
Bartlett published around 300 papers in chemistry, several of which broke exciting new ground, and many supplied critical support for ongoing theory. In 1939 he and Lawrence Knox published their epic research on bridgehead halogens. In particular, they synthesized 1-bromonorbornane and showed that halogen is essentially inert to hot strong alkali or to hot silver nitrate solution.
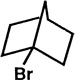
One can readily understand that a nucleophilic attack by hydroxide ion, with displacement of the halogen and Walden inversion, is impossible, since the back side of the carbon bearing the halogen is thoroughly blocked by the bicyclic structure. But it wasn't obvious in 1939 why a tertiary halide shouldn't readily ionize, as t-butyl halides do, and then participate in SN1 reactions. Knox's halide didn't, and Bartlett postulated that carbonium ions, with only six electrons around the central carbon, are not stable unless they are essentially planar, and of course the bicyclic ion, were it to exist, would be constrained to a quite different, non-planar geometry.
A few years later, he and Saul Cohen confirmed Knox's finding, and extended it, by synthesizing triptycene, and (with E. S. Lewis and Josephine Ryan, and later with Fred Greene) the corresponding bridge-head bromide and iodide.
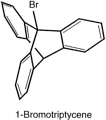
These two bridge-head halides, too, proved inert; the reaction with silver nitrate occurs at least 1015 times as fast with t-butyl bromide as with 1-iodotriptycene. The contrast of the behavior of these bridge-head halides with that of
triphenylmethyl halides is even more extraordinary than the contrast between the behavior of bromonorbornane and t-butyl halides. Triphenyl methyl cation is quite stable; whereas the corresponding cation from triptycene could not be formed. By way of contrast, Bartlett and his coworkers showed that the 1-triptycyl radical is readily formed, but it reacts like an aliphatic radical, rather than as a triaryl one.
J. D. Roberts defined a physical-organic chemist as a scientist who carries out physical-chemical experiments on new organic compounds that he has designed to establish a specific theoretical point, and then synthesized; it is the combination of design, synthesis, and physical measurement that characterizes the field. No better illustration of this definition exists than Bartlett's investigations of bridge-head halides.
REACTIVE CARBOCATIONS
Bartlett made a second highly original discovery with two of his graduate students, F. E. Condon and Abraham Schneider. In 1944 they discovered the hydrogen-halogen exchange reaction. In the presence of catalytic amounts of the Lewis acid, aluminum tribromide, t-butyl chloride reacts with isopentane to yield t-amyl bromide and isobutane; the reaction required only about 0.001 seconds. Presumably the t-alkyl halide reacts with the aluminum halide to yield the t-butyl cation, and this in turn plucks a t-hydrogen atom from an alkane. The alleged inert character of paraffins is illusory; they react readily enough with carbocations. Bartlett postulated a sequence of reactions, such as:
(CH3)3CCl + AlBr3 ? AlBr3Cl- + (CH3)3C+
(CH3)3C+ + CH3CH2CH(CH3)2 ? CH3CH2C+(CH3)2 + (CH3)3CH
CH3CH2C+(CH3)2 + AlBr3Cl- ? CH3CH2CBr(CH3)2 + AlBr2Cl
Prior to the paper by Bartlett, Condon, and Schneider, the field of reactions of petroleum hydrocarbons was in confusion; subsequently it became science. This major discovery from Bartlett's lab in 1944 was an important part of the background for George Olah's beautiful work four decades later on the reactions of paraffins in superacid solutions, work for which he was awarded the Nobel Prize in chemistry in 1994.
NON-CLASSICALIONS
Bartlett and his students made a number of other contributions to carbonium ion chemistry, crowned by the monograph entitled “Nonclassical Ions” (Benjamin, 1965) that Bartlett edited. With characteristic modesty, he presented many papers by Winstein, by Roberts, and by other researchers, along with only one of his own. But he interpreted all the papers he selected and combined them with his commentaries into a useful whole.
FREE RADICALS
A third major field of investigation in Bartlett's lab was free radical chemistry. He and C. Gardner Swain conducted the first research, using the rotating sector method, to find rate constants for a free radical polymerization in solution. Subsequently, he and F. A. Tate illuminated the free radical pathway for the polymerization of allyl acetate by carrying out the reaction with a selectively deuterated substrate, CH2=CH-CD2-O-COCH3. This deuterated compound polymerized two to three times as fast to produce a polymer more than twice as long as did ordinary allyl acetate. The polymerization of allyl acetate, like other free radical polymerizations, is a chain reaction, and Bartlett explained the highly unusual deuterium isotope effect that he and Tate discovered (where the deuterated substrate reacts faster,
rather than more slowly, than its hydrogen counterpart) as the result of attack at a deuterium atom in the chain-breaking step of the polymerization.
SINGLET OXYGEN
Oxidations with atmospheric oxygen are also free radical reactions. Ordinary oxygen exists in the triplet state 3O2, (i.e., as a diradical). Reversing the usual, where the ground state of a molecule (i.e., the state of least energy) has only paired electrons, the ground state of oxygen is a triplet with two unpaired electrons; singlet oxygen (1O2) with nothing but paired electrons is a high-energy species. Triplet oxygen reacts rapidly with radicals (that is, with other molecules that present unpaired electrons), but fortunately reacts relatively slowly with the larger world of compounds with paired electrons. Bartlett prepared singlet oxygen by photoactivation and by the decomposition of phosphite ozonides. He showed that trimethylphosphite ozonide is stable at very low temperatures, but it readily yields singlet oxygen on warming to room temperature.
(CH3O)3P + O3 ? (CH3O)3PO3
(CH3O)3PO3 ? (CH3O)3PO + 1O2
a-LACTONES
A number of investigators had postulated that a-lactones participated in chemical reactions as transitory intermediates, but such species had never been observed. Bartlett and Robert Wheland synthesized and identified di-t-butyl a-lactone by the oxidation of di-t-butyl ketene with ozone. The a-lactone was stable in solution at -78°, and could be identified by its spectra. This bold initiative, which demonstrated the reality of alpha-lactones, admitted them to the category of high-energy intermediates.
This sampling of the nearly 300 papers from Bartlett's laboratory gives some feeling for the range and originality of his projects, and explains at least in part the awards he received, and the high regard in which he was held—and is held.
PERSONAL
Paul D. Bartlett was born in Ann Arbor, Michigan, on August 14, 1907, and grew up in Indianapolis, Indiana, where he attended public schools. He went on to Amherst College and in 1928 graduated summa cum laude, acquiring an honor that Amherst gave most sparingly. He then went on to graduate work in chemistry with James Bryant Conant at Harvard. In 1932 he and Conant published a study of the rates and equilibria of semicarbazone formation in aqueous solution, a study that made the distinction clear between rates and equilibria. That distinction, so obvious now, was not previously well established for organic chemistry.
Bartlett was plainly marked as Conant's successor in theoretical organic chemistry. But, at that time, the Harvard chemistry department held tightly to the doctrine that it would not appoint one of its own graduate students or postdoctorals until he or she (this was just at the time when Mary Fieser came to Harvard and made that second pronoun a reality) had gone elsewhere and by publication had demonstrated his or her originality. The idea was to avoid allowing a dominant professor (as Conant certainly was) to fill the department with his students unless and until they had shown independent creativity. Bartlett served a year as a postdoctoral fellow at the Rockefeller Institute (as it was then) in New York and subsequently served for two years as an instructor at the University of Minnesota. With his research at Minnesota, Bartlett more than satisfied the department's requirement of independent accomplishment;
he was sufficiently decontaminated, and could safely be added to the staff of the chemistry department at Harvard. He rapidly ascended through the ranks of instructor, assistant professor, associate professor, and Erving professor of chemistry, while he taught his famous course, trained his co-workers, and made his seminal discoveries.
Several of these discoveries have been outlined above. The first, however, was his work with Stanley Tarbell. They examined the kinetics and product formation in the bromination of stilbene in methanol as solvent and established that this is a two-step process, where a cationic intermediate is formed from stilbene and bromine, followed by reaction of that intermediate with either methanol or bromide ion. Some years later George Kimball and Irving Roberts made the imaginative suggestion that Bartlett and Tarbell's cation was a cyclic bromonium ion.
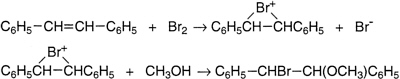
In 1972, when he was eligible either to retire or to remain on the staff at Harvard for another four years (this was long before the Pepper law outlawed retirement), Bartlett elected to retire from Harvard and to accept a Welch Professorship at Texas Christian University in Fort Worth. There he continued to attract postdoctorals to work with him and launched on a second career. At TCU he frequently collaborated with W. H. Watson, an X-ray crystallographer, to determine the structure of the products of carbonium-ion rearrangements, effectively enlarging the set of tools with which he faced the problems of modern physical-organic chemistry. When he retired a second time (this time from
TCU in 1985) he returned to Harvard, where he was welcomed by the friends he had left in 1972.
HONORS
Inevitably, with his record of accomplishment, Bartlett received many honors and prizes. He obtained the American Chemical Society's Young Chemists Award in 1938. He was elected to the National Academy of Sciences in 1947, and he received the A. W. von Hofmann Gold Medal of the German Chemical Society in 1962. He received the National Medal of Science from President Johnson in 1968, and was elected to the American Philosophical Society in 1978. In 1969 he was elected an honorary member of the Chemical Society of London and an honorary member of the Swiss Chemical Society. In 1981 he received the Robert A. Welch Award in chemistry. He received honorary degrees from Amherst and from the Universities of Chicago, Montpellier, Paris, and Munich. He received the James Flack Norris Awards, both in physical-organic chemistry and in teaching, as well as a dozen or so other prizes and awards.
In 1931 Bartlett joined Mary Lula Court in a wonderfully successful marriage that was only terminated fifty-eight years later by Lou Bartlett's death in 1989. Lou took care of Paul's graduate students and postdoctorals almost as if they had been her children, inviting them to the Bartlett's lovely home and gardens in Weston, Massachusetts, and again entertaining a new generation of students in Fort Worth during Paul Bartlett's second career.
Paul was a large, ruggedly handsome man who enjoyed the outdoors. He took his research group on skiing and hiking trips with great and evident pleasure. He had a ready smile, a hearty laugh, and a fine sense of humor, which was always evident. He wrote verse of all kinds for all occasions, ranging from carefully rhymed and metered poems to dog-
gerel that was marvelously entertaining. He was a splendid colleague both in and out of the chemical laboratory. Regrettably, his last years in a retirement home in Lexington, Massachusetts, were less than ideal, as he lost his independence, but none of his resolve. The chemistry department gave a reception for him for his ninetieth birthday in which he happily participated, but following the party his health steadily deteriorated.
He is survived by daughters Joanna Bartlett Kennedy and Sara W. Bartlett, son Geoffrey Bartlett, and seven grandchildren, to say nothing of his numerous scientific offspring down to the second and third generation. It will be hard to find another colleague of his quality.
THE AUTHOR GRATEFULLY acknowledges J. M. McBride, Therese Wilson, and Lily Birladeanu, who helped to correct the first draft of this paper.
SELECTED BIBLIOGRAPHY
1932
With J. B. Conant. A quantitative study of semicarbazone formation. J. Am. Chem. Soc.54:2881.
1936
With S. Tarbell. A kinetic study of the addition of methyl hypobromite to stilbene J. Am Chem. Soc.58:466.
1939
With L. H. Knox. Bicyclic structures prohibiting the Walden inversion. Replacement reactions in 1-substituted apocamphanes. J. Am. Chem. Soc.61:3184.
1942
With M. J. Ryan and S. G. Cohen. Triptycene (9,10-o-benzenoanthracene). J. Am. Chem. Soc.64:2647.
1944
With F. E. Condon and A. Schneider. Exchanges of halogen and hydrogen between organic halides and isoparaffins in the presence of aluminum halides. J. Am. Chem. Soc.66:1531.
1946
With C. G. Swain. Rate constants of the steps in addition polymerization. Use of the rotating sector method on liquid vinyl acetate. J. Am. Chem. Soc.68:2381.
1950
With S. G. Cohen, J. D. Cotman, Jr., N. Kornblum, J. R. Landry, and E. S. Lewis. The synthesis of 1-bromotriptycene. J. Am. Chem. Soc.72:1003.
With E. S. Lewis. Bicyclo structures prohibiting the Walden inversion. Further studies of triptycene and its derivatives, including 1-bromotriptycene. J. Am. Chem. Soc.72:1005.
1953
With F. A. Tate. The polymerizatio of allyl compounds. VI. The
polymerization of Allyl-d2 acetate and the mechanism of its chain termination. J. Am. Chem. Soc.75:91.
1954
With F. D. Greene. Triptycene 1-carboxylic acid and related compounds. The decomposition of ditriptycyl peroxide. J. Am. Chem. Soc.76:2349.
1960
With T. G. Traylor. O18 tracer evidence of the termination mechanism in the autoxidation of cumene. Tetrahedron Lett.24:30.
1967
With G. Guaraldi. Di-t-butyl trioxide and di-t-butyl tetroxide. J. Am. Chem. Soc.89:4799.
1970
With R. Wheland. a-lactones from diphenylketene and di-t-butylketene. J. Am. Chem. Soc.92:6057.
With G. D. Mendenhall and A. P. Schaap. Competitive modes of reaction of singlet oxygen. Ann. N. Y. Acad. Sci.171:79.
1980
With H.-K. Chu. Mechanism of the direct reaction of phosphite ozonide with olefins J. Org. Chem.45:3000.
With G. D. Mendenhall and D. L. Durham. Controlled generation of singlet oxygen at low temperatures from triphenyl phosphite ozonide. J. Org. Chem.45:4269.














