
2
Physiological Role of Copper
THIS chapter begins by discussing the basis of the essentiality of copper. The kinetics of copper in the body and select roles of copper at the cellular and molecular level are described. In addition, factors influencing the bioavailability of copper are presented.
ESSENTIALITY
Copper is both essential and toxic to living systems. As an essential metal, copper is required for adequate growth, cardiovascular integrity, lung elasticity, neovascularization, neuroendocrine function, and iron metabolism. An average adult human ingests about 1 mg of copper per day in the diet; about half of which is absorbed (Harris 1997). An expert committee of the World Health Organization recommends 30 micrograms (µg) per kilogram (kg) of body weight per day, which equates to about 2 mg per day for an average adult (WHO 1996). The Food and Nutrition Board (FNB) recommends dietary copper intake of 1.5–3.0 mg per day for adults (NRC 1989). Copper is obligatory for enzymes involved in aerobic metabolism, such as cytochrome c oxidase in the mitochondria, lysyl oxidase in connective tissue, dopamine monooxygenase in brain, and ceruloplasmin. As a cofactor for apo-copper-zinc superoxide dismutase (apoCuZnSOD), copper protects against free-radical damage to proteins, membrane lipids, and nucleic acids in a wide range of cells and organs. Severe copper deficiencies, either gene defects due to mutations or low dietary copper intakes, although relatively rare in humans, have been linked to mental re-
tardation, anemia, hypothermia, neutropenia, diarrhea, cardiac hypertrophy, bone fragility, impaired immune function, weak connective tissue, impaired central-nervous-system (CNS) functions, peripheral neuropathy, and loss of skin, fur (in animals), or hair color (Linder and Goode 1991; Uauy et al. 1998; Cordano 1998; Percival 1998).
BIOCHEMISTRY AND PHYSIOLOGY
Copper taken in through the diet might be absorbed partially in the stomach, where the highly acidic environment frees the bound copper ions from partially digested food particles. However, the largest portion of ingested copper passes into the duodenum and ileum, which are the major sites of absorption. As a result of complexing with amino acids, organic acids, or other chelators, a high fraction of copper is soluble in the intestinal tract. Studies on isolated segments of the duodenum suggest that copper ions enter into the mucosal cells lining the intestine by simple diffusion and exit at the basolateral surface by a different mode of transport (Bremner 1980). Recent reports indicate that there is a divalent transporter that might transport copper (Rolfs and Hedinger 1999).
Basolateral transport is markedly reduced in Menkes disease, which results in systemic copper deficiency. Studies of this disease led to the prediction of a copper-transporting adenosine triphosphatase (ATPase) in the basolateral membrane of mucosal cells. The copper-transporting ATPase presumably discharges the copper into the serosal capillaries where the copper binds to albumin and amino acids for transport to the liver via the portal circulation. From the liver, copper is transported to extrahepatic tissues by albumin, amino acids, and, to a lesser extent, ceruloplasmin (Dunn et al. 1991).
A large fraction of circulating copper is returned to the liver as ceruloplasmin-bound copper. Ceruloplasmin, a sialoglycoprotein, is constantly being secreted from the liver into the blood. When ceruloplasmin returns to the liver, the sialic acid can be removed by the outer endothelial cells followed by an endocytosis of the desialated protein via the asialoglycoprotein receptor in the liver parenchyma (Irie and Tavassoli 1986). Likewise, removal of copper from ceruloplasmin hastens its uptake by liver parenchymal cells (Holtzman and Gaumnitz 1970).
As discussed in Chapter 4, Wilson disease, a genetic disease characterized by accumulation of copper mainly in the liver and brain, attests to a potential role for ceruloplasmin biosynthesis in liver homeostasis of copper. Copper-containing fragments of ceruloplasmin are found in the bile of normal subjects and are generally absent from the bile of Wilson patients.
Although many Wilson patients do not synthesize sufficient amounts of ceruloplasmin, this decrease in biliary ceruloplasmin is observed even in Wilson patients who have reasonably normal blood concentrations of the protein. That observation suggests that biliary secretion of copper bound to ceruloplasmin accounts for most of the copper that is excreted (Iyengar et al. 1988). In aceruloplasminemia, a genetic defect in ceruloplasmin biosynthesis, ceruloplasmin in the circulation is totally absent (Harris et al. 1995). Yet, the aceruloplasminemic patient does not develop Wilson disease, nor does copper accumulate in the liver in aceruloplasminemia. Thus, factors besides ceruloplasmin are required for biliary copper excretion and for maintaining normal liver copper homeostasis in the liver. A clue to the identity of such factors has come from studies of lipofuschin-like granules that accumulate in the liver of patients who suffer from primary biliary cirrhosis and are unable to excrete copper. These granules apparently arise from primary lysosomes and might contain degradation fragments of metallothionein-bound copper (Humbert et al. 1982). Studies in Wistar rats suggest that biliary copper excretion occurs by a glutathione-dependent (rapid) phase and a glutathione-independent (slow) phase (Houwen et al. 1990). No identification has been made of which phase is associated with ceruloplasmin.
A second copper-transporting ATPase enzyme is required to transport the copper ions into the Golgi compartment for incorporation into apoceruloplasmin (Murata et al. 1995). Several types of cells have been shown to have receptors for ceruloplasmin. However, the above observation supports, but does not define a role for ceruloplasmin in copper delivery to tissues. The receptors have been found on many cell types. K562 cells are capable of engaging ceruloplasmin in vitro and transporting ceruloplasmin-bound copper to cellular enzymes (Percival and Harris 1991). Unlike transferrin, which delivers iron by a receptor-mediated endocytosis of the whole protein, ceruloplasmin protein is not endocytosed (except in hepatic cells) and delivers its bound copper to components at the cell surface. The role of the protein, therefore, stops at the cell membrane, and transport of copper to the interior requires transport proteins in the membrane. Ascorbate facilitates the release of copper, and chelators of copper prevent its absorption by the cell (Percival and Harris 1989, 1990).
When excess copper is fed to rats, it selectively accumulates in periportal and midzones of the liver lobules (Fuentealba et al. 1989), suggesting that portal blood flow determines copper disposition (Haywood 1981). In some patients with chronic cirrhosis, copper also accumulates in lysosomes (Humbert et al. 1982), suggesting that those organelles take part in copper storage or, more likely, in copper excretion when copper concentra-
tions are high (Gross et al. 1989).1 Metallothionein is a small cysteine-rich protein that tightly binds copper. This protein is thought to play roles in both copper storage and internal copper movement (Cousins 1985). At high concentrations, copper can stimulate metallothionein synthesis. Under normal conditions, copper exists at extremely low concentrations as the free ion in the cytosol. Copper-binding ligands, therefore, are key regulators in copper movement and adaptation to toxic effects. The ligands protect against toxicity and can help target the proteins for copper incorporation. Copper-binding ligands include glutathione (GSH), amino acids, ATP, and the recently identified copper metallochaperones. All of these ligands have been shown to transport copper within the cell from one location to another, and make copper available to intracellular enzymes (Sternlieb 1980; DiSilvestro and Cousins 1983; Holt et al. 1986; Harris 1995).
Numerous biochemical and nutritional studies have focused on the mechanisms of copper absorption and metabolism. The challenge is to explain how extremely small quantities of copper are able to pass through cell membranes and into enzymes that require copper for function. Some insight has been obtained from studies in yeast. Yeast mediate copper transport with separate low- and high-affinity systems for copper uptake (Eide 1998). The yeast gene CTR1 (copper transport 1) was the first copper transport gene to be discovered (Dancis et al. 1994a,b). CTR1 encodes a transmembrane protein that selectively transports Cu(I). A homologous human gene, hCTR1, that encodes a transporter protein was identified in HeLa cells (Zhou and Gitschier 1997). Because the above transporters recognize only Cu(I), a reductase enzyme must reduce the Cu2+ before transport into the cell. Two reductase genes that can reduce Cu2+ for transport were identified in yeast (Hassett and Kosman 1995; Georgatsou et al. 1997). A reductase system that uses NADH as an electron donor to reduce copper has been found in rat liver cells (van den Berg and McArdle 1994).
Cells in a defined culture medium take up copper. High-affinity membrane permeases allow copper ions at micromolar concentrations to move through the phospholipid bilayer and enter the cytoplasm. The rapid up-
take is not ATP dependent, suggesting a passive carrier system (Schmitt et al. 1983; Tong and McArdle 1995). In general, chemical form, valence state, and relative concentrations of competing metals influence the quantity of copper that is absorbed. Select amino acids such as histidine and glutamine (Harris and Sass-Kortsak 1967), and ions, such as sodium, chloride, and bicarbonate (Alda and Garay 1990), stimulate transmembrane copper movement in vitro, whereas zinc and copper chelators, such as phytate, reduce absorption.
Glutathione is a ubiquitous cysteine-containing tripeptide, is present in millimolar quantities in liver, brain, kidney, and other tissues (Deneke and Fanburg 1989). Glutathione avidly binds copper. There is a rapid turnover of a GSH-Cu(I) complex in hepatoma cells. That turnover is consistent with GSH involvement in the reduction of Cu(II) to Cu(I), potentially facilitating its binding to metallothionein (Freedman and Peisach 1989a; Freedman and Peisach 1989b).
Mammalian and yeast cells have small polypeptides that bind Cu(I) and transfer it to selected recipients. These proteins, called copper chaperones or metallochaperones, transiently bind Cu(I) at a cysteine-rich region in the peptide chain (Valentine and Gralla 1997). Chaperones move copper ions from one location in the cell to another, sometimes crossing through organelle membranes. The chaperone promotes a rapid exchange of copper with the target proteins (Portnoy et al. 1999). Targets include cytochrome c oxidase in the mitochondria, ATP7B in the trans-Golgi, and apoCuZnSOD in the cell cytosol. Three chaperones first described in yeast are known to have human counterparts. ATOX1 (formerly HAH1) is the human homolog of ATX1 and is abundantly expressed in all tissues. ATX1 and ATOX1 specifically deliver copper to the secretory pathway where the targets are membrane-bound copper ATPases that regulate the flow of copper into cell compartments. Examples include Ccc2p, which delivers copper to a multicopper oxidase (Fet3p) required for iron uptake in yeast, and ATP7B (the Wilson disease gene product), which delivers copper to apoceruloplasmin in the trans-Golgi of liver. Proper ATP7B function is essential for the excretion of copper in the bile. COX17 is a chaperone that transports copper to cytochrome oxidase in the mitochondria of yeast cells (Amaravadi et al. 1997). The human homolog is hCOX17. LYS7, a 27-kilodalton (kDa) protein that delivers copper to the apoCuZnSOD (Culotta et al. 1997, 1999), has a human counterpart designated CCS (copper chaperone for SOD). Yeast mutants that are defective in LYS7 are unable to incorporate copper into SOD 1 and hence are defective for SOD1 activity. CCS is comparable in size and has a 28% sequence identity with LYS7. Both proteins contain a single MHCXXC consensus copper-binding sequence.
FACTORS AFFECTING BIOAVAILABILITY
The amount of copper which is absorbed from the diet can vary considerably depending on other dietary constituents. However, in general approximately half the copper consumed in the diet is typically absorbed by the gastrointestinal (GI) tract. Approximately two-thirds of the copper that is absorbed is rapidly secreted into the bile (Lönnerdal 1998; Turnlund 1998; Wapnir 1998) (Figure 2-1). Thus, approximately 80–90% of dietary copper is typically excreted in the feces. Thus, copper homeostasis is primarily regulated at the GI level, through biliary excretion with the kidney excreting only small amounts of copper. Small amounts of copper are also excreted in hair and sweat. The bioavailability, or the fraction of copper absorbed from the GI tract, has been shown to be influenced by the age of the individual, the amount of copper in the GI tract, and various dietary components. With respect to dietary components, copper in meat has been reported to be more bioavailable than that in vegetables. The bioavailability of copper is also expected to depend on the form of copper present (Baker et al. 1991). Absorption is higher for soluble or ionic forms than for less soluble or insoluble mineral forms or copper deposited in soil.
In adults, absorption varies according to the amount of copper in the diet. Animal studies indicate that absorption rates can be as low as about 10% with very high copper intakes, and as high as around 70% with low copper intakes. The average for typical diets in animals and humans is 30-40% (Lönnerdal 1996, 1998; Turnlund et al. 1998; Wapnir 1998). Turnlund et al. (1998) measured the amount of copper excreted by humans in the feces for 12 days after an oral dose, or intravenous dose, of copper for four subjects and five subjects, respectively. The intravenous dose allows for an estimation of the endogenous excretion into the gut. Retention from oral intake as estimated from copper in the feces was shown to be highest when dietary copper concentrations were lowest: 67% at 0.38 mg per day, 54% at 0.66 mg per day, and 44% at 2.49 mg per day (Turnlund et al. 1998). However, the estimated total percentage that was actually absorbed by the GI tract before endogenous (or biliary) excretion back to the GI tract was 77%, 73%, and 66%, respectively, for the three doses. Thus, changes in endogenous excretion (in the bile), rather than GI absorption, is the primary mechanism of action in regulating total body copper. Specifically, the change in endogenous excretion between the lowest dose and the highest dose varied between 12% and 34% (Turnlund et al. 1998), although those data might not adequately reflect long-term homeostasis because of the short-term nature of the study. Thus, increases in copper excretion in the feces with increasing copper dose is a function of decreased absorption and increased biliary secretion, the latter having a greater effect.
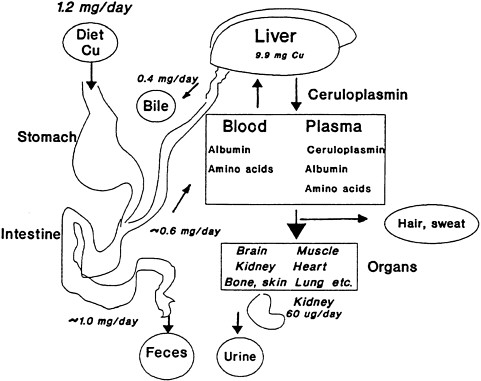
FIGURE 2-1 Overview of copper absorption, transport, and excretion. The liver receives copper from the intestine via the portal circulation and redistributes the copper to the tissue via ceruloplasmin, albumin, and amino acids. Nearly half of the copper consumed is not absorbed and passes into the feces. Another two-thirds of the daily intake is returned to the liver and released into the bile. Therefore, fecal excretion accounts quantitatively for nearly all of the copper consumed as the system endeavors to stay in balance. A small amount is excreted by the kidney via the urine, and still lesser amounts appear in hair and sweat. This interplay among the various systems maintains homeostasis and balance throughout the organism. The values in the figure are based on a dietary input of 1.2 mg per day.
Source: Harris 1997. Reprinted from Handbook of Nutritionally Essential Mineral Elements by courtesy of Marcel Dekker.
Effect of Age
Studies in rats indicate that the absorption and retention of copper can be particularly high in the neonatal period, and that it decreases by the time of weaning (summarized by Lönnerdal 1996, 1998). Data on absorption in human infants are sparse; however, studies on copper balance (i.e.,
the amount of copper in the body) show similar decreases in retention (a function of absorption and biliary excretion) of copper with age in full-term infants. Negative copper balance (a loss of copper from the body which exceeds dietary intake) can arise in preterm infants as a consequence of a reduced prenatal storage of copper and low dietary copper intake (Widdowson 1974; Sann et al. 1980; Hillman et al. 1981; Lönnerdal 1996, 1998; Cordano 1998). Both animal and human infant studies indicate that, within the intake range examined, copper absorption and retention increase linearly with intake amount, although the percentage of the dietary copper retained decreases (Lönnerdal et al. 1985; Lönnerdal 1996). For example, in 14-day-old suckling rat pups, the percentage of copper retained from a meal (0.5 mL) containing 0.2 mg of copper per liter was approximately 30%, and it was closer to 20% when the meal contained 2 mg of copper per liter. However, it is important to note that while the percentage of copper retained from the high-copper meal was lower than that from the low-copper meal, the total amount of copper retained from the high-copper meal was 7 times higher than that from the low-copper meal (Lönnerdal et al. 1985).
Due to tissue growth, and an increased expression of some copper proteins during the postnatal period, the percentage of absorbed copper retained by the body is higher in infants than in adults. Little information is available on dietary copper absorption, or copper retention, in toddlers and young children.
Dietary and Other Interactions
Early research with whole animals showed that the rate and amount of copper ions transferred across intestinal epithelia were influenced positively by dietary amino acids, but negatively by ascorbate and competing metal ions (Bremner 1980; Hogan and Rauser 1981; L'Abbe and Fischer 1984; Oestreicher and Cousins 1985; Fields et al. 1986). Chloride ions and bicarbonate appeared to stimulate cellular absorption (Alda and Garay 1990). The transport across the membrane was considered a property of transport proteins, themselves subject to antagonistic effects of competing metals, principally zinc and ferrous iron. Studies in yeast and bacteria have led to the discovery of membrane proteins that transport copper ions across cell membranes.
Copper absorption is reported to be greater in infants who ingest human milk rather than cows milk (Lönnerdal 1996 1998). That increased bioavailability might be attributed to the greater association of copper with lipids and whey in human milk, than in cow's milk, where much of the copper is bound to casein (Wapnir 1998).
The literature indicates that copper absorption is greater when diets are animal protein rather than plant protein (i.e., vegetarian) (Srikumar et al. 1992; Lönnerdal 1996; Wapnir 1998). Studies in experimental animals found that phytates and dietary fiber generally reduce copper bioavailability; however, effects on bioavailability are less clear in humans (Turnlund 1988; Lönnerdal 1996). Declines in serum copper give some indication that phytates or α-cellulose added to the diet might alter copper utilization or distribution (Turnlund 1988). In general, the effect of phytates and dietary fiber on absorption of copper appears to be less than the effect on absorption of other divalent cations, such as zinc (Wapnir 1998).
Dietary differences have been found in patients with Wilson disease. Observations of two Wilson patients on lactovegetarian diets suggest copper bioavailability is reduced by about 25% (Brewer et al. 1993b). The first of two patients on that diet was asymptomatic (i.e., normal liver function and normal slit lamp examination for Kayser-Fleischer rings) for 12 years despite having a typical average daily copper intake and no anticopper therapy. The second patient stopped using the anticopper therapy for 2 years and then switched to a lactovegetarian diet. After switching, her serum transaminase and transpeptidase activity (alanine aminotransaminase, asparate aminotransaminase, and liver γ-glutamyltranferase), which were previously elevated, showed improvement over the succeeding year. When last observed about 2 years after starting the diet, the patient remained clinically well. Other Wilson-disease patients who discontinued their anticopper therapy had serious difficulty after 3 to 4 months and serious degeneration in their condition after 1.5 years on average (Brewer and Yuzbasiyan-Gurkan 1992).
Carbohydrates, such as fructose, have been studied for their effect on copper absorption and metabolism. Fructose, or the fructose moiety of sucrose, fed to rats increased fecal and urinary excretion of copper (Lönnerdal 1996). The influence of fructose on copper balance in humans has not been well defined.
Based on studies in rats, ascorbic acid is thought to lower plasma and liver copper levels by reducing copper absorption, and the reduced copper absorption later stimulates copper absorption and depresses biliary excretion (Van den Berg et al. 1994). The decrease in absorption is caused by a reduction of cupric (Cu2+) ions to cuprous ions (Cu1+), which are less well-absorbed (Lönnerdal 1996). High levels of ascorbic acid might also decrease ceruloplasmin oxidase activity, although the overall effect of ascorbic acid on absorption and metabolism of copper in humans may be less than in animals (Lönnerdal 1996; Turnlund 1988). Administration of ascorbic acid with zinc at 1 g per day in patients with Wilson disease showed no interaction of ascorbic acid and zinc, on copper balance, or 64Cu absorption (Brewer et al. 1993a). Ascorbic acid had no detectable effect on the efficacy of zinc for copper-balance control in those patients.
Amino acids such as histidine and cysteine reduce copper absorption by forming complexes that are not well absorbed (Lönnerdal 1996). Histidine also enhances the inhibitory effects of zinc on copper absorption in rats (Wapnir and Balkman 1991). On the other hand, complexes of copper with other amino acids and organic acids might result in similar bioavailability to that of soluble copper sulfate (Wapnir 1998).
Based on studies to date, zinc is the primary mineral, and dietary element, which affects copper absorption. The effect of excess zinc on reducing copper absorption is well documented in a number of species (see summaries in Turnlund 1998; Wapnir 1998), and zinc has been used effectively in the treatment of Wilson disease (Hoogenraad et al. 1978; Hoogenraad et al. 1978; Hoogenraad et al. 1987; Brewer and Yuzbasiyan-Gurkan 1992; Brewer et al. 1994; Brewer et al. 1998). In pregnant rats, the consumption of diets containing high levels of zinc can result in fetal copper deficiency (Reinstein et al. 1984). In humans, the consumption of zinc supplements (50 mg per day for 6–8 weeks) can result in reductions in erythrocyte copper-zinc superoxide dismutase activity (Fisher et al. 1984; Yadrick et al. 1989; Johnson et al. 1998), suggesting that the chronic consumption of this level of zinc supplement could result in a condition of marginal copper status. Given the above possibility, the Institute of Medicine recommended that, at least for pregnant women, copper supplements (2 mg) should be provided to women when zinc supplements (25 mg or more) are given (IOM 1990).
In addition to zinc, various other minerals, such as iron, tin, calcium, phosphorus, cadmium, and molybdenum, interact with copper absorption and metabolism, although their effect compared with that of zinc is relatively minor or debatable in humans (Lönnerdal 1996; Turnlund 1988; Wapnir 1998). These minerals are cations that might compete for uptake in the digestive tract, thereby reducing absorption. These minerals might also affect copper utilization and excretion. For example, molybdenum has long been known to result in copper deficiency in ruminants but has little effect in nonruminants. Along with zinc for maintenance therapy, tetrathiomolybdate is now being used in the initial treatment of patients with the neurological or psychiatric form of Wilson disease. Tetrathiomolybate acts by blocking absorption of copper when given with food and by complexing with serum copper when given separately from food (Brewer et al. 1996).
Similar to other metals, the bioavailability of copper in soils or suspended particulates in water is likely to be a function of its mineral or surface sorbed form, solubility, and particle size (Davis et al. 1993; Ruby et al. 1996). As demonstrated for lead, solubility and bioavailability can vary greatly, depending on chemical and physical form (Ruby et al. in press). Copper acetate and sulfate are considerably more soluble and thus more bioavailable than cupric oxide (Johnson et al. 1998; Wapnir 1998), copper
sulfides (e.g., chalcopyrite), and other less-soluble minerals. Copper in soil and sediments also adsorbs strongly to soil components, such as clay minerals, hydrous iron, and manganese oxides (Tyler and McBride 1982), resulting in reduced solubility and mobility.
CONCLUSIONS
-
Copper is an essential nutrient.
-
Studies of absorption, transport and metabolism of copper have provided insights into the biochemical mechanisms for coping with copper deficiency and excess.
-
The retention of copper from the diet is influenced by age, amount and form of copper in the diet, and genetic background.
-
Bioavailability of copper varies with age and diet composition.
-
The liver plays a central role in copper homeostasis by varying the excretion of copper into the bile for loss in the stool.
-
The newly discovered chaperones for copper have provided insight into how copper ions in cells are guided to their target proteins.
RECOMMENDATIONS
-
Studies are needed to elucidate mechanisms of copper absorption, distribution, and excretion in humans.
-
Studies are needed on age-related changes in copper absorption or retention.
-
Research should be conducted on the genetic basis of the absorption mechanism and on whether variation in absorption efficiency has a genetic basis.
-
Research is needed to define extrahepatic processes for uptake and distribution.
-
The ability of copper to induce proteins involved in its metabolism and transport should be investigated. Particular emphasis should be given to the investigation of metal response elements on copper transport proteins.
-
Research is needed to determine the ontogeny of copper transporters.
-
Research is needed to identify how the form of copper (i.e., valence state and complexed forms) influences absorption and distribution.
REFERENCES
Amaravadi, R., D.M. Glerum and A. Tzagoloff. 1997. Isolation of a cDNA encoding the human homolog of COX17, a yeast gene essential for mitochondrial copper recruitment. Hum. Genet. 99(3):329–333.
Alda, J.O. and R. Garay. 1990. Chloride (or bicarbonate)-dependent copper uptake through the anion exchanger in human blood cells. Am. J. Physiol. 259(4 Pt 1):C570–C576.
Baker, D.H., J. Odle, M.A. Funk, and T.M. Wieland. 1991. Research note: bioavailability of copper in cupric oxide, cuprous oxide, and in a copperlysine complex. Poult. Sci. 70(1):177–179.
Bremner, I. 1980. Absorption, transport and distribution of copper. Pp. 23–48 in Biological Roles of Copper. Ciba Foundation Symposium 79. Amsterdam: Excerpta Medica.
Brewer, G.J. and V. Yuzbasiyan-Gurkan. 1992. Wilson disease. Medicine 71(3):139–164.
Brewer, G.J., R.D. Dick, V. Yuzbasiyan-Gurkan, V. Johnson and Y. Wang. 1994. Treatment of Wilson's disease with zinc: XIII. Therapy with zinc in presymptomatic patients from the time of diagnosis. J. Lab. Clin. Med. 123(6):849–858.
Brewer, G.J., R.D. Dick, V.D. Johnson, J.A. Brunberg, K.J. Kluin, and J.K. Fink. 1998. Treatment of Wilson's disease with zinc: XV. Long-term follow-up studies. J. Lab. Clin. Med. 132(4):264–278.
Brewer, G.J., V. Johnson, R.D. Dick, K.J. Kluin, J.K. Fink, and J.A. Brunberg. 1996. Treatment of Wilson Disease with ammonium tetrathiomolybdate. II. Initial therapy in 33 neurologically affected patients and follow-up with zinc therapy. Arch. Neurol. 53(10):1017–1025.
Brewer, G.J., V. Yuzbasiyan-Gurkan, V. Johnson, R.D. Dick, and Y. Wang . 1993a. Treatment of Wilson's disease with zinc: XI. Interaction with other anticopper agents. J. Am. Coll. Nutr. 12(1):26–30.
Brewer, G.J., V. Yuzbasiyan-Gurkan, R. Dick, Y. Wang, and V. Johnson. 1993b. Does a vegetarian diet control Wilson's disease? J. Am. Coll. Nutr. 12(5):527–530.
Cordano, A. 1998. Clinical manifestations of nutritional copper deficiency in infants and children. Am. J. Clin. Nutr. 67(5 Suppl.): 1012S–1016S.
Cousins, R.J. 1985. Absorption, transport, and hepatic metabolism of copper and zinc: Special reference to metallothionein and ceruloplasmin. Physiol. Rev. 65(2): 238–309.
Culotta, V.C., L.W. Klomp, J. Strain, R.L. Casareno, B. Krems, and Gitlin J.D. 1997. The copper chaperone for superoxide dismutase. J. Biol. Chem. 272(38):23469–23472.
Culotta, V.C., S.J. Lin, P. Schmidt, L.W. Klomp, R.L. Casareno, and J. Gitlin. 1999. Intracellular pathways of copper trafficking in yeast and humans . Adv. Exp. Med. Biol. 448:247–254.
Dancis, A., D.S. Yuan, D. Haile, C. Askwith, D. Eide, C. Moehle, J. Kaplan, and R.D. Klausner. 1994a. Molecular characterization of a copper transport protein in S. cerevisiae: An unexpected role for copper in iron transport. Cell 76:393–402.
Dancis, A., D. Haile, D.S Yuan, and R.D. Klausner. 1994b. The Saccharomyces cerevisiae copper transport protein (Ctr1p). Biochemical characterization, regulation by copper, and physiologic role in copper uptake. J. Biol. Chem. 269(41):25660–25667.
Davis, A., J.W. Drexier JW, M.V. Ruby, and A. Nicholson. 1993. Micromineralogy of mine wastes in relation to lead bioavailability, Butte, Montana. Environ. Sci. Technol. 27(7):1415–1425.
Deneke, S.M. and B.L. Fanburg. 1989. Regulation of cellular glutathione. Am. J. Physiol. 257(4 Pt 1):L163–L173.
DiSilvestro, R.A., and R.J. Cousins. 1983. Physiological ligands for copper and zinc . Ann. Rev. Nutr. 3:261–288.
Dunn, M.A., M.H. Green and R.M. Leach, Jr. 1991. Kinetics of copper metabolism in rats: a compartmental model. Am. J. Physiol. 261(1 Pt 1):E115-25.
Eide, D.J. 1998. The molecular biology of metal ion transport in Saccharomyces cerevisiae. Ann. Rev. Nutr. 18:441–469.
Fields, M., J. Holbrook, D. Scholfield, J.C. Smith, Jr., and S. Reiser. 1986. Effect of fructose or starch on copper-67 absorption and excretion by the rat. J. Nutr. 116(4):625–632.
Fischer, P.W., A. Giroux, and M.R. L'Abbe. 1984. Effect of zinc supplementation on copper status in adult man. Am. J. Clin. Nutr. 40(4): 743–746.
Freedman, J.H., and J. Peisach. 1989a. Resistance of cultured hepatoma cells to copper toxicity. Purification and characterization of the hepatoma metallothionein. Biochim. Biophys. Acta 992(2):145–54.
Freedman, J.H. and J. Peisach. 1989b. Intracellular copper transport in cultured hepatoma cells. Blochem. Biophys. Res. Commun. 164(1):134–140.
Fuentealba, I., S. Haywood, and J. Trafford. 1989. Variations in the intralobular distribution of copper in the livers of copper-loaded rats in relation to the pathogenesis of copper storage diseases. J. Comp. Pathol. 100(1):1–11.
Georgatsou, E., L.A. Mavrogiannis, G.S. Fragiadakis, and D. Alexandraki. 1997. The yeast Fre1p/Fre2p cupric reductases facilitate copper uptake and are regulated by the copper-modulated Maclp activator. J. Biol. Chem. 272:13786–13792.
Gross, J.B., Jr., B.M. Myers, L.J. Kost, S.M. Kuntz, and N.F. LaRusso. 1989. Biliary copper excretion by hepatocyte lysosomes in the rat. Major excretory pathway in experimental copper overload. J. Clin. Invest. 83(1):30–39.
Harris, E.D. 1995. Role of ligands in the translocation of metals. Pp. 71–88 in Handbook of Metal-Ligand Interactions in Biological Fluids. Bioinorganic Medicine, Vol. 1, G. Berthon, ed. New York: Marcel Dekker.
Harris, E.D. 1997. Copper. Pp. 231–273 in Handbook of Nutritionally Essential Mineral Elements, B.L. O'Dell and R.A. Sunde, eds. New York: Marcel Dekker.
Harris, D.I. and A. Sass-Kortsak. 1967. The influence of amino acids on copper uptake by rat liver slices. J. Clin. Invest. 46(4):659–677.
Harris, Z.L., Y. Takahashi, H. Miyajima, M. Serizawa, R.T. MacGillivray and J.D. Gitlin. 1995. Aceruloplasminemia: molecular characterization of this disorder of iron metabolism. Proc. Natl. Acad. Sci. (USA) 92(7):2539–2543.
Hassett, R. and D.J. Kosman. 1995. Evidence for Cu(II) reduction as a component of copper uptake by Saccharomyces cerevisiae. J. Biol. Chem. 270(1):128–134.
Haywood, S. 1981. A non-random distribution of copper within the liver of rats. Br. J. Nutr. 45(2):295–300.
Hillman, L.S., L. Martin and B. Fiore. 1981. Effect of oral copper supplementation on serum copper and ceruloplasmin concentrations in premature infants. J. Pediatr. 98(2):311–313.
Hogan, G.D. and W.E. Rauser. 1981. Role of copper binding, absorption, and translocation in copper tolerance of agrostis gigantea roth. J. Exp. Bot. 32(126):27–36.
Holt, D., D. Dinsdale, and M. Webb. 1986. Intestinal uptake and retention of copper in the suckling rat, Rattus rattus. I. Distribution and binding. Comp. Biochem. Physiol. 83(2):313–316.
Holtzman, N.A. and B.M. Gaumnitz. 1970. Studies on the rate of release and turnover of ceruloplasmin and apoceruloplasmin in rat plasma. J. Biol. Chem. 245(9):2354–2358.
Hoogenraad, T.U., R. Koevoet, and E.G. de Ruyter Korver. 1979. Oral zinc sulphate as long-term treatment in Wilson's disease (hepatolenticular degeneration). Eur. Neurol. 18(3):205–211.
Hoogenraad, T.U., C.J. van den Hamer, R. Koevoet, and E.G. Korver. 1978. Oral zinc in Wilson's disease [letter]. Lancet 2(8102):1262–1263.
Hoogenraad, T.U., J. van Hattum, C.J. van den Hamer. 1987. Management of Wilson's disease with zinc sulfate. Experience in a series of 27 patients. J. Neurol. Sci. 77(2–3):137–146.
Houwen, R., M. Dijkstra, F. Kuipers, E.P. Smit, R. Havinga and R.J. Vonk. 1990. Two pathways for biliary copper excretion in the rat. The role of glutathione. Blochem. Pharmacol. 39(6):1039–1044.
Humbert W., M. Aprahamian, C. Stock, and J.F. Grenier. 1982. Copper accumulation in primary biliary cirrhosis. An electron and X-ray microanalytical study. Histochemistry 74(1):85–93.
Irie S. and M. Tavassoli. 1986. Liver endothelium desialates ceruloplasmin. Blochem. Biophys. Res. Commun. 140(1):94–100.
IOM (Institute of Medicine). 1990. Nutrition during pregnancy. Part I: Weight Gain. Part II: Nutrient Supplements. Washington, DC: National Academy Press.
Iyengar V., G.J. Brewer, R.D. Dick, and O.Y. Chung. 1988. Studies of cholescystokinin-stimulated biliary secretions reveal a high molecular weight copper-binding substance in normal subjects that is absent in patients with Wilson's disease. J. Lab. Clin. Med. 111(3):267–274.
Johnson, M.A., M.M. Smith, and J.T. Edmonds. 1998. Copper, iron, zinc, and manganese in dietary supplements, infant formulas, and ready-to-eat breakfast cereals. Am. J. Clin. Nutr. 67(5 Suppl.):1035S–1040S.
L'Abbe M.R., and P.W.R. Fischer. 1984. The effects of dietary zinc on the activity of copper-requiring metalloenzymes in the rat. J. Nutr. 114(5):823–828.
Linder M. and C.A. Goode. 1991. Biochemistry of Copper. New York: Plenum Press.
Lönnerdal, B. 1996. Bioavailability of copper. Am. J. Clin. Nutr. 63(5 Suppl.):821S–829S.
Lönnerdal, B. 1998. Copper nutrition during infancy and childhood. Am. J. Clin. Nutr. 67(5 Suppl.):1046S–1053S.
Lönnerdal, B., J.G. Bell, and C.L. Keen. 1985. Copper absorption from human milk, cow's milk, and infant formulas using a suckling rat model. Am. J. Clin. Nutr. 42(5):836–844.
Murata Y., E. Yamakawa, T. Lizuka, H. Kodama, T. Abe, Y. Seki, and M. Kodama. 1995. Failure of copper incorporation into ceruloplasmin in the Golgi apparatus of LEC rat hepatocytes. Biochem. Biophys. Res. Commun. 209:349–355.
NRC (National Research Council). 1989. Recommended Dietary Allowances, 10th Ed. Washington, D.C.: National Academy Press.
Oestreicher P. and R.J. Cousins. 1985. Copper and zinc absorption in the rat: Mechanism of mutual antagonism. J. Nutr. 115(2):159–166.
Percival, S.S. 1998. Copper and immunity. Am. J. Clin. Nutr. 67(5 Suppl.): 1064S–1068S.
Percival S.S., and E.D. Harris. 1989. Ascorbate enhances copper transport from ceruloplasmin into human K562 cells. J. Nutr. 119(5):779–784.
Percival S.S., and E.D. Harris. 1990. Copper transport from ceruloplasmin: Characterization of the cellular uptake mechanism. Am. J. Physiol. 258(1 Pt 1):C140–C146.
Percival S.S., and E.D. Harris. 1991. Regulation of Cu, Zn superoxide dismutase with copper. Caeruloplasmin maintains levels of functional enzyme activity during differentiation of K562 cells. Biochem. J. 274(Pt 1):153–158.
Portnoy M.E., A.C. Rosenzweig, T. Rae, D.L. Huffman, T.V. O'Halloran,
and V.C. Culotta. 1999. Structure-function analyses of the ATX1 metallochaperone. J. Biol. Chem. 274(21):15041–15045.
Reinstein, N.H., B. Lonnerdal, C.L. Keen and L.S. Hurley. 1984. Zinc-copper interactions in the pregnant rat: fetal outcome and maternal and fetal zinc, copper and iron. J. Nutr. 114(7):1266–1279.
Rolfs A. and M.A. Hediger. 1999. Metal ion transporters in mammals: structure, function and pathological implications. J. Physiol. 518 (Pt 1):1–12.
Ruby M.V., A. Davis, R. Schoof, S. Eberle, and C.M. Sellstone. 1996. Estimation of lead and arsenic bioavailability using a physiologically based extraction test. Environ. Sci. Technol. 30(2):422–430.
Ruby M.V., R. Schoof, J. Drexier, W. Brattin, M. Goldade, G. Post, M. Harnois, W. Berti, M. Carpenter, D. Edwards, D. Cragin, and W. Chappell. In press. Advances in evaluating the oral bioavailability of inorganics in soil for use in human health risk assessment. Environ. Sci. Technol.
Sann, L., D. Rigal, G. Galy, F. Benvenu and J. Bourgeois. 1980. Serum copper and zinc concentration in premature and small-for-date infants. Pediatr. Res. 14(9):1040–1046.
Schmitt R.C., H.M. Darwish, J.C. Cheney, and M.J. Ettinger. 1983. Copper transport kinetics by isolated rat hepatocytes. Am. J. Physiol. 244:G183–G191.
Srikumar T.S., G.K. Johansson, P.A. Ockerman, J.A. Gustafsson, and B. Akesson. 1992. Trace element status in health subjects switching from mixed to a lactovegetarian diet for 12 mo. Am. J. Clin. Nutr. 55(4):885–890.
Sternlieb, I. 1980. Copper and the liver. Gastroenterology 78(6):1615–1628.
Tong, K.K., and H.J. McArdle. 1995. Copper uptake by cultured trophoblast cells isolated from human term placenta. Biochim. Biophys. Acta 1269(3):233–236.
Turnlund, J.R. 1988. Copper nutriture, bioavailability, and the influence of dietary factors. J. Am. Diet. Assoc. 88(3):303–308.
Turnlund, J.R. 1998. Human whole-body copper metabolism. Am. J. Clin. Nutr. 67(5 Suppl.):960S–964S
Turnlund, J.R., W.R. Keyes, G.L. Pfeiffer, and K.C. Scott. 1998. Copper absorption, excretion, and retention by young men consuming low dietary copper determined by using the stable isotope 65Cu1,2. Am. J. Clin. Nutr. 67(6):1219–1225.
Tyler L.D., and M.B. McBride. 1982. Mobility and extractability of cadmium, copper, nickel, and zinc in organic and mineral soil columns. Soil Sci. 134(3):198–205.
Uauy, R., M. Olivares and M. Gonzalez. 1998. Essentiality of copper in humans. Am. J. Clin. Nutr. 67(5 Suppl.):952S–959S.
Valentine, J.S. and E.B. Gralla. 1997. Delivering copper inside yeast and human cells. Science 278(5339):817–818.
van den Berg, G.J., and H.J. McArdle. 1994. A plasma membrane NADH oxidase is involved in copper uptake by plasma membrane vesicles isolated from rat liver. Biochim. Biophys. Acta 1195(2):276–280.
van den Berg, G.J., S. Yu, A.G. Lemmens, and A.C. Beynen. 1994. Ascorbic acid feeding of rats reduces copper absorption, causing impaired copper status and depressed biliary copper excretion. Biol. Trace Elem. Res. 41(1–2):47–58.
Wapnir, R.A. 1998. Copper absorption and bioavailability. Am. J. Clin. Nutr. 67(5 Suppl.):1054S–1060S.
Wapnir R.A., and C. Balkman. 1991. Inhibition of copper absorption by zinc: Effect of histidine. Biol. Trace Elem. Res. 29(3):193–202.
WHO (World Health Organization). 1996. Trace Elements in Human Nutrition and Health. Geneva: World Health Organization.
Widdowson, E.M. 1974. Trace elements in foetal and early postnatal development. Proc. Nutr. Soc. 33(3):275–84.
Yadrick, M.K., M.A. Kenney, and E.A. Winterfeldt. 1989. Iron, copper, and zinc status: response to supplementation with zinc or zinc and iron in adult females. Am. J. Clin. Nutr. 49(1):145–50.
Zhou B., and O. Gitschier. 1997. hCTR1: A human gene for copper uptake identified by complementation in yeast. Proc. Natl. Acad. Sci. (USA) 94:7481–7486.

















