
A— Age-Related Shifts in Neural Circuit Characteristics and Their Impact on Age-Related Cognitive Impairments
John H. Morrison
INTRODUCTION
Increasing chronological age carries with it a heightened risk for diseases such as Alzheimer's disease, as well as functional decline associated with senescence in the absence of any specific neurologic disease, such as age-related memory impairment. Alzheimer's disease leads to a catastrophic decline in cognitive abilities and memory performance in the affected individual. Age-related memory impairment in the context of senescence is far less catastrophic than Alzheimer's disease with respect to the quality of life, but it has a surprisingly high incidence and thus also represents a significant health problem associated with aging. With the increased life expectancy that has already occurred over the last century, let alone the projected further increase, it has become clear that one of the most important goals for neuroscientists over the next several decades will be to develop means of maintaining a high level of cognitive and memory performance in the aged population. The focus of this paper is to outline and to illustrate a circuit-based approach aimed at both revealing the neurobiological basis of age-related memory impairment and identifying targets for intervention.
A Multitiered Neuroanatomic Approach to Aging
The Link between Neurochemistry and Neuroanatomy
Traditionally, a given cell class or circuit has been defined and/or categorized on the basis of its physiological and anatomic characteristics, i.e., the
information being transmitted and the origin and termination of its connections, respectively. More recently, both neuroanatomical and electrophysiological characterizations of neural circuits have incorporated biochemical and molecular biological information in order to develop a more comprehensive portrayal of the essential qualities of a circuit that relate to its role in brain function. The key biochemical attributes of a given circuit or cell class are largely a reflection of the particular gene expression patterns, dynamics of protein synthesis, degradation and distribution, and selective activation of signaling cascades that are predominant in the neurons that furnish the circuit. The resultant biochemical profile essentially represents the neurochemical phenotype of a circuit or cell class. Operationally, one might define neurochemical phenotype as the complement of specific molecules, particularly proteins and their enzymatic products, that are enriched in and utilized by a given class of neurons in a manner that is not shared by other cell classes. The neurochemical phenotype of a neuron includes molecules related to synaptic transmission (e.g., receptors, neurotransmitters, and related enzymes), structural attributes, metabolic processes, or any characteristic that is uniquely well developed in that neuron and critically important to its designated role in brain function.
For example, a cortical neuron that uses GABA, the major inhibitory neurotransmitter, has a neurochemical phenotype that differs in many fundamental ways from a cortical neuron that uses glutamate, the major excitatory neurotransmitter. As implied in the definition of neurochemical phenotype, gene expression and protein distribution patterns are not uniform across the brain or even across a single brain region. In fact, brain circuits are highly heterogeneous with respect to which genes are expressed over time and space, and the intracellular distribution of gene products (i.e., proteins) is highly regulated. Therefore, a comprehensive neuroanatomic analysis of a circuit must address its particular neurochemical profile as well as its anatomic connections, since both will impact that circuit's functional characteristics and role in behavior.
With respect to the task at hand, the goal is to link age-related shifts in neurochemical phenotype and neuroanatomic characteristics in key cortical and hippocampal circuits to functional decrements in memory that occur with aging. Why place the emphasis for studies of aging on cell classes and circuits rather than on brain regions? More specifically, if memory is the issue and the hippocampus is critical to memory, why not address the issue of age-related pathology at the level of the entire hippocampus, rather than isolated cells, circuits, and synapses?
As described below, there are important reasons to identify and consider the brain region of interest as an important step in such analyses. However, the main rationale for bringing the analysis to a higher level of resolution and focusing on distinct cell classes and circuits is that it more accurately reflects
neuronal vulnerability to aging and Alzheimer's disease. A given brain region does not generally suffer as a whole; it is more likely that vulnerable circuits in a given region tend to suffer while other circuits are unaffected. Even a region as vulnerable to Alzheimer's disease as the hippocampus has cell classes and circuits that are highly resistant to degeneration as well as those that are vulnerable.
We suspect that normal aging will exhibit an even higher degree of selectivity in affected circuits than does Alzheimer's disease, thus it is even more important to address the question of age-related neuronal vulnerability at the highest possible level of cellular and circuit resolution. In turn, as interventions are developed to prevent age-related decline, they should be targeted to the vulnerable cell classes and circuits, not to a given region. Ideally, the goal should be to sustain the health of vulnerable circuits without impacting those that are resistant to age-related decrements, and in order to do this we must first clarify the neurobiological underpinnings of age-related functional decline at the level of the selectively vulnerable cells, circuits, and synapses.
In addition, it is critical that the cellular and neuroanatomic analyses be quantitative. Qualitative impressions of age-related cell loss, morphologic aberrations, or down-regulation of a particular receptor will not allow for a sufficiently precise or accurate depiction of age-related changes, nor will qualitative judgments possess the requisite statistical power to test many hypotheses. In contrast, quantitative descriptions of structural and biochemical attributes can be readily correlated with quantitative assessments of physiological and behavioral output, allowing for direct links to be established between neurobiological indices and function. This is perhaps most important when testing the effectiveness of an intervention, since one would like to be able to equate any improvement in functional output with a measurable reflection of increased neuronal viability. Throughout this article, the emphasis will be on microscopic approaches to cell and circuit analysis rather than electrophysiological approaches; however, the electrophysiological approach to the hippocampus, aging, and memory is a critical partner to the microscopic data, particularly with respect to delineating complex properties that emerge from the activity of hippocampal neurons.
Targeting Multiple Levels of Resolution: From the Brain Region to the Synapse
The Brain Region
In any such analysis, the first task is to define the brain region(s) and circuits to be analyzed, by virtue of their hypothesized role in the neural function or behavior that is compromised by aging. In addition, the process of neural circuitry analysis becomes more focused as the behavioral measures
are improved and refined in their sensitivity and quantitative power. The great advances over the last few decades in the analysis of memory offer an excellent example of the manner in which behavioral data can guide neuroanatomic analysis toward a given brain region, e.g., the hippocampus, and in turn certain circuits, e.g., the perforant path (discussed below). Defining the brain region(s) of interest is particularly important if one of the experimental goals is to obtain estimates of neuron number, glial cell number, or synapse number, since such estimates are not useful if the region of interest cannot be clearly and precisely defined or its boundaries recognized. In fact, the accurate determination of neuron number in key hippocampal fields such as CA1 and the adjacent entorhinal cortex is what led to the realization that neuron death is unlikely to be at the root of age-related impairment in memory and cognition (Rapp and Gallagher, 1996; Morrison and Hof, 1997; Peters et al., 1998a), challenging the accepted dogma that people inevitably ''lose nerve cells" as they age. The use of modern stereological techniques for determining neuron number in a defined brain region has been invaluable for establishing the sustained viability of neurons during aging. One of the many attributes of the stereological techniques that make them particularly valuable for analyses of aging and neurodegenerative disorders is that they generate estimates of neuron or synapse number that are not confounded by tissue or cellular shrinkage, two major potential confounds in studies of aging (West, 1993a, 1993b). The stereologic approach (West, 1993a, 1993b, 1999; Geinisman et al., 1996; Coggeshall and Lekan, 1996) to estimates of neuron number, synapse number, axon length, etc., will continue to be powerful for such studies, particularly as a quantitative database of neuroanatomic information relevant to aging develops.
Cell Classes and Circuits
The next level of resolution beyond that of brain region is the analysis of specific circuits and related cell classes. With respect to the hippocampus and memory, attention is drawn to the neurons in layer II of the entorhinal cortex, which provide the perforant path that connects the entorhinal cortex to the dentate gyrus (Van Hoesen and Pandya, 1975). This circuit is highly vulnerable in aging and Alzheimer's disease (Hyman et al., 1984; Morrison and Hof, 1997; Hof et al., 1999). The entorhinal cortex receives convergent inputs from multiple neocortical association areas and in turn provides the major cortical input to the hippocampus, and thus along with associated parahippocampal and perirhinal areas, it is positioned for a critical role in memory (Squire and Zola-Morgan, 1991; Zola-Morgan and Squire, 1993). The analysis of any key circuit such as the perforant path moves quickly into the issue of cell classes or cell types with respect to the cells of origin of such a projection. While cell type has been traditionally defined exclusively by morphologic
criteria (size, shape, extent of neuritic arborization), a more comprehensive definition of cell type might be a designated class of neurons that share key characteristics with regard to morphology, location, connectivity, and neurochemical phenotype. In fact, the concepts of cell type and neurochemical phenotype bear a critical relationship to selective vulnerability in aging or neurodegenerative diseases.
Selective vulnerability is most readily appreciated in the context of neurodegenerative disorders such as amyotrophic lateral sclerosis, Parkinson's disease, and Alzheimer's disease. Each disease is characterized by its own, unique pattern of degeneration, with amyotrophic lateral sclerosis involving the loss of upper and lower motor neurons, Parkinson's disease noted for the selective degeneration of dopaminergic neuRons in substantia nigra, and Alzheimer's disease characterized primarily by the degeneration of key cortical circuits. In many cases, the vulnerable neurons in a given disorder share particular neurochemical characteristics that can be linked to their selective vulnerability. For example, both the neurons that provide the perforant path from the entorhinal cortex and the neurons that provide the long corticocortical interconnections are highly vulnerable in Alzheimer's disease, and both are marked by a particular cytoskeletal profile that can be linked to their vulnerability (Morrison et al., 1987; Hof et al., 1990: Hof and Morrison, 1990; Hof et al., 1999).
In order to define and understand the determinants of selective vulnerability in neurodegenerative diseases as well as in normal aging, it will be important to determine neuron number according to specific classes of neurons wherein class is based on morphology, connectivity, and neurochemical phenotype. In this way, hypotheses can be tested at a finer level of cell-type resolution, and links can be drawn between a particular element of the neurochemical phenotype and vulnerability. Quantitative analyses using chemically specific approaches (e.g., immunohistochemistry, in situ hybridization) allow for direct investigation of the molecular determinants of selective vulnerability, as reflected, for example, in intracellular biochemical changes in neurons as they change with age or begin to degenerate. This approach is particularly important with respect to links to gene expression, with the goal being a quantitative dataset that links gene expression patterns with circuits, vulnerability, and, potentially, with age-related changes in cognition.
Neuronal Compartments
Beyond the analysis of cell class(es), it is important to determine the degree to which biochemical characteristics of neuronal compartments in individual neurons are affected by age. Given that neuron death is unlikely to be the substrate for age-related memory impairment, it has become increasingly important to investigate more subtle changes in cellular morphology
and neurochemical phenotype that would impact function, but not be lethal. This step will require careful quantitative analyses of individual neurons, both in terms of morphometric measures as well as in terms of subcellular analyses of protein distribution patterns. For example, age-related changes in dendritic spine number have been described that could impact neuronal function (Coleman and Flood, 1987), and in fact, in primate neocortex it appears that spine density decreases with age with no change in dendritic length or branching patterns (Duan et al., 1999), and such changes may predominate in the most distal portion of the dendrite (Peters et al., 1998b). In addition, changes in receptor distribution can affect a single portion of the dendritic tree that receives a particular input, leaving the rest of the neuron unaffected (Gazzaley et al., 1996a). Moreover, some age-related changes might preferentially affect the nerve terminal rather than the dendrite, or vice versa. Such subtle changes could profoundly affect circuits and neural transmission with no evidence of generalized cell death.
The Synapse
The highest-resolution morphologic analysis is directed at the synapse and represents a search for potential changes in synapse structure or molecular constituents of the synapse that are related to age and might impact circuit function and behavior. Neocortical synapse loss clearly occurs in Alzheimer's disease and correlates well with degree of cognitive impairment (DeKosky and Scheff, 1990; Terry et al., 1991). High-resolution structural analyses of the synapse suggest that there are synaptic changes in the hippocampus with normal aging in the rat (Geinisman et al., 1995), although such changes may not occur in aged primate (Peters et al., 1996). In addition, the dendritic spine is more plastic than previously thought, and structural changes in the dendritic spine may occur on a time course consistent with induction of changes in synaptic function underlying memory, such as long-term potentiation (Toni et al., 1999). Thus, age-related spine loss or loss of spine plasticity could lead to age-related decline in memory and/or learning.
While purely structural analyses of the aging synapse have been and will continue to be illuminating (Geinisman et al., 1995; Tigges et al., 1996; Peters et al., 1996), perhaps the most exciting applications of electron microscopy to studies of aging will emerge from the use of immunogold postembedding electron microscopy. This immunohistochemical technique has extremely high resolution, in that the molecule of interest is identified by the presence of a discrete gold particle that is 10–25 nanometers in diameter and is highly quantifiable (Chaudhry et al., 1995). In addition, multiple antibodies bound to gold particles of different sizes can be used simultaneously to localize multiple synaptic molecules in a single synapse (He et al., 2000). The number of gold particles can be equated to the number of molecules of the targeted
protein in a very small, discrete region, such as a single synapse (see Figure A-1). Such an approach allows for a very high-resolution analysis of the molecular constituents of identified synapses and presumably will be able to reveal shifts in the molecular constituents with experimental manipulations and/or aging at a quantitative level. When such studies are done in behaviorally characterized aged animals, we will be able to draw direct correlations between the distribution of key synaptic molecules and age-related behavioral impairment. No such studies have been completed to date, but they are now under way.
As is apparent from the above discussion, key studies of aging have been and will continue to be directed at questions requiring different levels of resolution in the analysis. The illustrations cited below will focus on the microscopic delineation of vulnerable circuits and cell classes, as well as changes in protein distribution in specific cell classes, circuits, and neuronal compartments. While these examples will hopefully highlight the power of a circuit-based approach, they will also illuminate the present shortcomings of the data, such as a paucity of chemically specific synaptic data and the need for more interdisciplinary analyses. Electrophysiological data will be crucial in the interdisciplinary context, since they are particularly powerful at revealing the emergent properties and information content of hippocampal circuits coding for memory (Barnes et al., 1997a, 1997b; Eichenbaum et al., 1999; Shapiro and Eichenbaum, 1999; Wood et al., 1999). As the data emerge, it will be crucial to develop a set of models and/or databases that link data across studies, such that the synaptic and cellular data will be in a context that is easily linked to the functional role of a known circuit, in a region that is clearly implicated in a function that is compromised in aging, such as memory.
DIFFERENTIATING ALZHEIMER'S DISEASE FROM SENESCENCE: THE CRITICAL ROLE OF THE ENTORHINAL CORTEX AND ITS PROJECTION TO DENTATE GYRUS
At the outset, it is important to draw a distinction between the neurobiological events underlying the dementia of Alzheimer's disease and those that underlie age-related memory impairment (Morrison and Hof, 1997). In Alzheimer's disease and neurodegenerative disorders in general, neuron death occurs that results in circuit disruption and profound impairment of the neural functions dependent on the degenerating circuits (see Hof et al., 1999, for a review). As mentioned above, neuron death is not ubiquitous in neurodegenerative disorders, in that neurons display a particular pattern of selective vulnerability in each disorder. In Alzheimer's disease, the highly vulnerable circuits are: (1) neurons that interconnect functionally linked neocortical areas (Pearson et al., 1985; Rogers and Morrison, 1985; Lewis et al., 1987); (2) the projection from the entorhinal cortex to the hippocampus, referred to
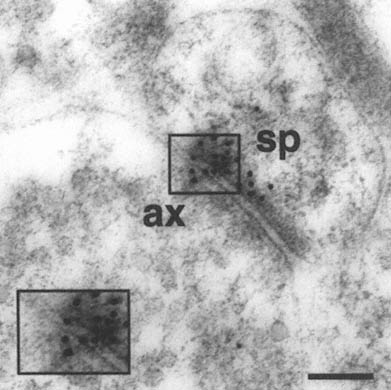
FIGURE A-1 This is a photograph of an electron microscope image that illustrates the postembedding immunogold method. The tissue has been treated with a primary antibody directed against the NMDA receptor subunit NR2A, followed by a secondary antibody that is both conjugated to a gold particle of 10 nanometer diameter, and binds to the primary antibody, thereby forming a bridge and revealing the location of the receptor protein NR2A.
This electron micrograph demonstrates intense NR2A immunogold localization between an axon terminal(ax) and dendritic spine(sp) forming an asymmetric (Type 1) synapse on the dendrites of pyramidal cells in CA1 of monkey hippocampus. The majority of the gold particles, each of which probably represents an individual receptor, are associated with the postsynaptic specialization of the dendritic spine and synaptic cleft, and thus are in a position to mediate NMDA receptor activity at this particular synapse. In addition, two particles are clearly associated with the presynaptic axon terminal, suggesting that NR2A may also participate in autoreceptors that modulate glutamate release presynaptically. Thus, with this approach we can resolve the molecular constituents of the synapse and quantify their relative distribution in specific regions of the synapse. The boxed area is represented at higher magnification in lower left corner (Scale bar = 0.13µm). The author thanks William Janssen and Prabhakar Vissavajjhalla for providing this illustration.
as the perforant path, as well as certain circuits intrinsic to the hippocampus (Hyman et al., 1984); and (3) key diffuse projections to the cerebral cortex, such as the cholinergic projection from nucleus basalis (Whitehouse et al., 1982; for a review, see Kemper, 1999). In contrast to the selective but extensive neuron loss reflective of Alzheimer's disease, neuron death is minimal in the regions classically associated with cognition and memory in the normal course of aging (Peters et al., 1998a). The lack of significant hippocampal and neocortical neuron death in normal aging has now been demonstrated in humans, monkeys, and rats (West et al., 1993b; Gomez-Isla et al., 1996; Rapp and Gallagher, 1996; Gazzaley et al., 1997; Peters et al., 1998a), although some neuron loss appears to occur in humans in the hilus of the dentate and in the subiculum (West, 1993b).
However, a lack of quantifiable neuron loss does not necessarily mean that no degenerative changes are occurring in a given brain region, and it does not rule out more subtle changes that lead to compromised function without cell loss. The entorhinal cortex is a particularly instructive case in this regard. It appears that the neurons within layer II of entorhinal cortex, which serve as a neocortical conduit to the hippocampus through the perforant path, are likely to be the single most vulnerable class of neurons in the brain with respect to both aging and Alzheimer's disease. While these neurons are clearly devastated early in Alzheimer's disease, their status in cognitively normal, aged individuals and those with mild cognitive impairment has been more difficult to pinpoint. Neuron counts in neurologically normal individuals suggest that there is no neuron loss in the entorhinal cortex (Gomez-Isla et al., 1996; West, 1993b). However, analyses of neurofibrillary tangles (NFT), the classic reflection of a degenerating neuron in Alzheimer's disease, suggest that virtually all humans over the age of 55 have some NFT in layer II of entorhinal cortex (Vickers et al., 1992; Bouras et al., 1994).
How does one reconcile these two findings and draw a distinction between age-related degenerative events in the entorhinal cortex that are progressive and those that are not? While the answer to this question continues to be elusive, one approach that appears promising is the use of a comprehensive panel of antibodies in a quantitative experimental design in order to distinguish and quantify transitional events in the neurons within the entorhinal cortex that can be correlated with the clinical dementia rating scale. This approach has led to a focus on patients with a rating of 0.5 that have mild cognitive impairment, yet it is unclear whether their condition represents early Alzheimer's disease or a more stable condition that might be referred to as age-related memory impairment.
The key to reconciling the presence of NFT in this region with the fact that there does not appear to be neuron loss is that the various neuronal profile counts that have been done have not taken into consideration "transitional neurons," i.e., neurons that are still intact and included in an analysis
of total neuron counts, yet have transitional intraneuronal pathology resembling an NFT. Such neurons are confusing in that they could be misconstrued as "normal neurons" in a Nissl stain or NFTs in a tau immunohistochemical stain, the two stains that are most commonly used to quantify total "healthy" neurons and NFT, respectively. But these neurons may actually be the key to understanding the difference between early Alzheimer's disease and senescence.
For example, when neurons in layer II of the entorhinal cortex are counted in three categories—ghost tangles, transitional neurons, and healthy neurons—the data are far more revealing with respect to early pathologic events in layer II of entorhinal cortex, and it is quite clear that there might be significant "transitional" pathology in neurologically normal individuals or individuals with a clinical dementia rating of 0.5 in the absence of quantifiable neuron death and in the absence of massive NFT formation (Gimmel et al., 1998; Bussière et al., 1999; see Figure A-2).
These estimates of neuron number in three classes can also be related to each other as ratios in a given case, establishing a case-by-case "index of neurodegeneration" that is not hampered by the individual variability in raw number of neurons that invariably occurs in studies of neuron number. It will be very important to continue to study patients with a clinical dementia rating of 0.5 to try and determine whether this condition is a precursor to Alzheimer's disease or whether it is a condition that can be sustained and stabilized over a long period of time without cascading to Alzheimer's disease. It will be even more enlightening to do these kinds of neuropathologic analyses in patients with prospective neuropsychological assessment. In addition, other molecules linked to degeneration (e.g., neurofilament, presenilin) can be incorporated into the analyses of transitional neurons to obtain even more discrete molecular information on the changes that occur in these neurons in aging and the nature of their selective vulnerability. However, even a more precise delineation of the events surrounding degeneration will not provide a full understanding of the vulnerability of this circuit, since, as described below, the entorhinal-hippocampal connections display age-related changes short of degeneration that could also impact function.
Age-Related Neurochemical Shifts in Identified Circuits and Cell Classes
Introduction and Technical Considerations
Modern neuroanatomy is often centered on circuit analysis within the context of biochemical attributes, as described earlier. The most common methods used to link gene expression patterns with cell classes and circuits are immunohistochemistry and in situ hybridization, the localization of proteins
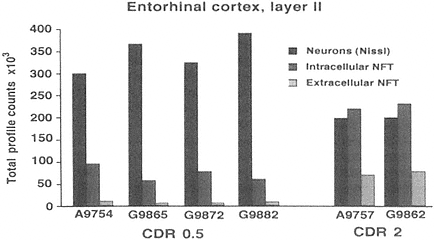
FIGURE A-2 This histogram represents a stereological analysis of the number of neuronal profiles in three different categories in layer II of the human entorhinal cortex, in one hemisphere. These are the neurons that form the perforant path, projecting to the dentate gyrus in the hippocampus proper. The three categories represent three different stages across the spectrum from healthy to degenerated. "Neurons (Nissl)" represent the number of neurons that are intact and appear healthy in a Nissl stain, without a trace of tau-accumulation. "Intracellular NFT" refers to neurons that are still intact as reflected by the Nissl stain, but are also immunoreactive for hyperphosphorylated tau, suggesting that a degenerative process is occurring, leading to tau accumulation and early stages of neurofibrillary tangle (NFT) formation. Extracellular NFTs, often called ghost NFTs, refer to a fully formed NFT with essentially no associated Nissl staining, suggesting that nothing remains of the neuron except the remnants of degeneration. These are end-stage NFTs that can be viewed as a dead neuron.
Six individual cases are shown, four with a clinical dementia rating (CDR) of 0.5, representing very mild cognitive impairment, and two with a CDR of 2, representing early Alzheimer's disease. Note the rather strong correlation between the CDR scores and the increase in transitional, intracellular NFT and extracellular NFT. In the CDR 0.5 cases, at least 75 percent of the layer II entorhinal cortex neurons remain free of pathology, and there are virtually no end-stage extracellular NFTs. However, the 25 percent that are transitional clearly suggest that a degenerative process is well under way in a significant proportion of these neurons. In CDR 2 cases, there is the beginning of an inversion in total numbers of NFT-bearing and NFT-free neurons, with the largest category being transitional intracellular NFTs, and 20 percent having prog ressed to end-stage NFTs. This may represent a pivotal stage in the development of dementia. How this pattern may be related to other aspects of neurochemical pheno type that may be linked to selective vulnerability will be investigated further in multiple labeling stereologic studies using other antibodies. The author thanks Patrick Hof for providing this illustration.
and mRNA, respectively. This is a crucial level of understanding of brain organization, since it is estimated that over half of the 100,000 genes in the mammalian genome are expressed exclusively or predominantly within the brain, however, many of them are not expressed ubiquitously or in a uniform pattern. In fact, the selective and nonuniform expression of genes is a critical element in each brain region, cell type, or neural circuit's particular neurochemical phenotype, i.e., the set of proteins that are required for that region, cell class, or circuit to perform its unique role in brain function. The regional and cellular delineation of gene expression patterns is thus important as a reflection of function, but it is also increasingly critical as a background for analysis of genetic manipulations, particularly in mouse transgenic models.
While in situ hybridization and several biochemical approaches have been applied in a quantitative fashion on the regional level to reveal relative mRNA levels in one brain region compared with another, the analysis of mRNA or protein levels on a quantitative cellular level has presented special problems with respect to obtaining quantitative data. Just as we need quantitative databases that define neuronal structure, we need quantitative data on gene expression patterns, otherwise we will not be able to measure the effect of a genetic manipulation in the context of neural circuits. This will require precise measurements of protein concentration and/or mRNA levels in specific regions, identified circuits, cell classes, neuronal compartments, and at the level of the synapse. There is little doubt that the emerging gene chip technology will augment efforts to obtain regional data on gene expression patterns, and the single-cell mRNA amplification approaches pioneered by Eberwine and colleagues have already been useful to obtain mRNA expression data on the single-cell level (Eberwine et al., 1992; Kacharmina et al., 1999). In addition, quantitative immunocytochemical analyses of protein levels on a cellular level haVe been fruiTful, although the measurements generally are interpreted as relative protein levels rather than absolute molar measurements (Gazzaley et al., 1996a, 1996b). These quantitative cellular measurements are crucial if we are to obtain data at the desired level of resolution (see earlier discussion) that can be linked to individual cell classes and circuits. The analyses of putative age-related or experimentally induced shifts in glutamate receptors (GluRs) offer an excellent example of this approach, particularly in the hippocampus, where information flow through the trisynaptic circuit is highly ordered anatomically and mediated through glutamate receptors, with the NMDA receptor in particular strongly implicated in memory and age-related changes in memory (Caramanos and Shapiro, 1994; Barnes, 1994; Barnes et al., 1997a, 1997b).
NMDA Receptors, Hippocampal Circuits, and Aging
Both the available data and the missing data on changes in NMDA receptor distribution with aging offer an excellent example of the importance of
microscopic analyses at several levels of resolution, as outlined in an earlier section. At the regional level, receptor binding studies have been used to study potential age-related changes in hippocampal GluRs in several species. While several studies have reported decreases in NMDA binding in the hippocampus of mice, rats, and monkeys (Wenk and Walker, 1991; Clark et al., 1992; Magnusson and Cotman, 1993; Le Jeune et al., 1996), other studies suggested that there is no change in NMDA receptors with aging (Nicolle et al., 1996) or perhaps even an increase in humans without Alzheimer's disease (Johnson et al., 1996).
In studying age-related changes in receptors or any of the key synaptic molecules, it is particularly important to be able to take the analysis from the regional level to that of cell classes, circuits, individual neuronal compartments, and synapses, since the changes are very likely to be cell, circuit, and synapse specific and therefore difficult to resolve at the regional level. In the case of multisubunit receptors like the NMDA and AMPA GluRs, it is optimal for the immunohistochemical data to be available at high anatomic resolution and with the highest molecular specificity, since receptor composition at the synapse is a crucial determinant of the functional characteristics of synaptic transmission.
In the same vein, it is best if shifts in GluR expression are detected at the level of GluR families (e.g., AMPA, kainate, NMDA receptors) and subunits within a family (e.g., for the AMPA receptor, GluRs1-4; for NMDA receptors, NRI and NR2A-D), given that the subunit composition profoundly impacts function (see Hollman and Heinemann, 1994, for a review). Thus, for multisubunit receptor studies in aging, the demands are particularly high in that we want the localization to: (1) have the highest level of molecular specificity, (2) be linked to identified cells and circuits, (3) be available at a quantitative level, and (4) precisely describe the receptor composition at the synaptic level. Furthermore, if the analysis is to be truly comprehensive, then a given circuit needs to be characterized beyond the receptor subunits themselves to the associated proteins that modulate receptor function (e.g., PSD-95).
We have been able to get a start on such a comprehensive analysis, although it is still in its infancy. Motivated by the importance of both the entorhinal cortex projection to the dentate gyrus and the NMDA receptor in age-related changes in memory, we investigated the GluR distribution and immunofluorescence intensity within the dentate gyrus of juvenile, adult, and aged macaque monkeys with the combined use of subunit-specific antibodies and quantitative confocal microscopy (Gazzaley et al., 1996a).
This circuit has great advantages for such an analysis because the projection from the entorhinal cortex to the dentate gyrus is strictly confined to the outer molecular layer, i.e., the distal dendrites of granule cells, whereas other excitatory inputs terminate in a nonoverlapping fashion in the inner molecular layer, the proximal dendrites (Rosene and Van Hoesen, 1987; Witter and
Amaral, 1991). This strict laminar organization allows for putative changes in GluR distribution at the laminar level to be interpreted at the level of intraneuronal compartmentalization (i.e., distal versus proximal dendrites) and with reference to an isolated excitatory circuit (i.e., the perforant path).
This quantitative analysis demonstrated that aged monkeys, compared with young adult monkeys, exhibit a decrease in the fluorescence intensity for the subunit NR1 in the outer molecular layer of the dentate gyrus compared with the inner molecular layer. Given the tight laminar organization of these circuits, this suggests that the decreased NR1 levels impact the input from the entorhinal cortex, but not the other excitatory inputs to the dentate gyrus, again, pointing to the entorhinal input to the hippocampus as a key element in age-related changes. Since NR1 is the obligatory subunit for the NMDA receptor, this shift probably represents a general shift in NMDA receptor localization. Parallel qualitative and quantitative studies with antibodies to AMPA and kainate subunits demonstrated that the intradendritic alteration in NRI occurs without a similar alteration of non-NMDA receptor subunits, even though all three classes of GluRs are colocalized within these dendrites (Siegel et al., 1995). Further analyses, using markers for presynaptic terminals and dendritic markers, demonstrated that these elements were structurally intact in these aged animals.
These findings suggested that, in aged monkeys, a circuit-specific alteration in the intradendritic concentration of NR1 occurs without concomitant gross structural changes in dendritic morphology or a significant change in the total synaptic density across the molecular layer, suggesting that the intradendritic distribution of a neurotransmitter receptor is modifiable in an age-related and circuit-specific manner. Such a shift would lead to compromised NMDA receptor-mediated transmission, which could explain age-related shifts in long-term potentiation and spatial memory (Barnes et al., 1997a, 1997b) in the absence of any purely structural damage.
While these results are compelling, in that they represent a particularly high level of both molecular and anatomic specificity for age-related shifts in circuit attributes, they also are limited in two important ways. First, the animals were not behaviorally characterized, so these neurobiological changes cannot be directly linked to functional change. Second, the aged animals were all females, and their endocrine status was not carefully monitored, although they were presumably all postmenopausal. Given that estrogen is known to affect circuit characteristics in the hippocampus (McEwen and Alves, 1999), perhaps the endocrine status of the aged animals contributed to the receptor changes as much or more than chronological age. Presently, the data are not available from aged primates to clarify this potential confound, but we investigated the cellular mechanisms of estrogen-induced NMDA receptor regulation at the protein and mRNA levels in ovariectomized (OVX) rats with and without estrogen replacement therapy (ERT), using immunocytochemical and
in situ hybridization techniques (Gazzaley et al., 1996b). Quantitative confocal microscopy was used to quantify alterations in immunofluorescence intensity levels of NR1 subunit proteins within neuronal cell bodies and dendrites of discrete hippocampal fields. In parallel, in situ hybridization was used to examine NR1 mRNA levels in corresponding hippocampal regions.
The data indicate that ERT in OVX rats significantly increases immunofluorescence intensity levels in comparison to nonsteroid-treated OVX rats within the cell bodies and dendrites of CA1 pyramidal cells and to a lesser extent within the granule cells of the dentate gyrus, without affecting CA3. In contrast, such alterations in immunofluorescence intensity occur without concomitant changes in mRNA hybridization levels. These data demonstrate a substantial degree of GluR plasticity in response to ERT over a fairly short period of time, since the ERT was initiated only one week after OVX, and the animals were sacrificed after only two days of ERT. These data are consistent with the estrogen-induced augmentation of NMDA receptor-mediated transmission by electrophysiological measurements (Woolley, 1999).
As compelling as these data are with respect to E-induced plasticity in the NMDA receptor, they are incomplete, in that we cannot comment on the effects on other related proteins (e.g., other NMDA receptor subunits, other GluRs.). In addition, we do not know how an increase in NR1 at the level of the soma and dendritic shafts translates into a change at the synapse. Moreover, these experiments were performed on young rats. Thus, while the receptor analyses on aged monkeys and estrogen-manipulated young rats suggest that both aging and endocrine status can alter NMDA receptors in a profound and circuit-dependent manner, the appropriate multidisciplinary analyses have yet to be carried out to determine if such alterations directly impact age-related memory decline. Studies of young and aged primates that are closely monitored both behaviorally and endocrinologically will need to be done to properly extend these studies. In addition, these studies will have to be extended to the ultrastructural level, in order to determine whether or not such dendritic shifts in NR1 are manifested at the synapse. Thus, while certain NMDA receptor-mediated hippocampal circuits are excellent targets for gene/circuit/behavioral links to be established in the context of age-related cognitive decline, most of the data needed to solidify such links are still missing.
INTERDISCIPLINARY APPROACHES
While neuroanatomic datasets and behavioral datasets can be compared across experiments, it is most powerful when the neuroanatomic and cellular analyses are done in the same animals that have been behaviorally characterized. This has been particularly powerful in the hands of investigators that behaviorally screen aged animals so that the behaviorally impaired aged ani-
mals can be considered as a distinct group from those that are not behaviorally impaired (Rapp and Gallagher, 1996; Gallagher and Rapp, 1997; Peters et al., 1998b). In addition, when the neurobiological data are quantitative and derived from behaviorally characterized animals, direct correlations can be drawn in individual subjects between a given neurobiological index (e.g., synapse number) and behavioral performance (Rapp and Gallagher, 1996; Peters et al., 1998b). Such studies should be encouraged in animal models as well as in human studies; large interdisciplinary teams have begun to develop brain banks from subjects that have had extensive premortem longitudinal assessment.
Recently, an interdisciplinary team carried out a comprehensive analysis of several neurochemical indices in the hippocampus of behaviorally characterized young and aged rats, using quantitative immunohistochemical procedures to examine the hypothesis that changes in the connectional organization of the hippocampus contribute to age-related learning impairment (Smith et al., 1999b). Immunohistochemical markers were used for key pre-and postsynaptic proteins as well as structural proteins that would reveal the degree to which the circuits were affected by shifts in protein distribution as differentiated from frank degeneration. Young and aged rats were tested on a hippocampal-dependent version of the Morris water maze, which revealed substantial variability in spatial learning ability among aged rats (Gallagher and Rapp, 1997). A quantitative confocal method was used to quantify changes in immunofluorescence staining for the presynaptic vesicle glycoprotein, synaptophysin (SYN), which is an established marker for presynaptic terminals and is required for synaptic release. The intensity of specific immunoreactivity was measured in inner (IML), middle (MML) and outer (OML) portions of the dentate gyrus molecular layer, stratum lucidum (SL) and stratum laconosum-moleculare (LM) of CA3, and CA1 stratum radiatum (SR) and LM.
This approach allowed us to link neurochemical changes with specific cell classes and circuits in a very comprehensive fashion involving all three elements of the trisynaptic circuit through the hippocampus, as well as multiple sites of termination of the entorhinal input to the hippocampus. Comparisons based on chronological age alone failed to reveal a reliable difference in SYN staining intensity in any region examined. In contrast, aged subjects with robust spatial learning deficits displayed significant reductions in SYN immunoreactivity in CA3-LM relative to either young controls or age-matched rats with preserved learning. In addition, across all aged rats, individual differences in spatial learning capacity correlated with levels of SYN staining in three of the regions examined: the OML and MML of the dentate gyrus and CA3-LM. These changes in relative SYN levels occurred in the absence of any evidence of structural degeneration of the innervated dendrites, and thus would impact synaptic transmission, perhaps though com-
promised glutamate release rather than degeneration of pre-or postsynaptic elements.
Most importantly, all three of the regions displaying decreased levels of SYN receive a major projection from layer II of entorhinal cortex, offering further evidence that this circuit is exquisitely sensitive to aging. These findings suggest that circuit-specific alterations in glutamate release in the hippocampus may contribute to the effects of aging on learning and memory, in the absence of frank degeneration. This is a compelling example of the power of using quantitative, chemically specific approaches in behaviorally characterized animals in order to pinpoint the subtle circuit-specific neurobiological substrates of age-related memory impairment.
In these same animals, we investigated the AMPA receptor subunit, GluR2, and the NMDA receptor subunit NR1 to determine whether or not postsynaptic shifts in receptors might also be occurring in the context of aging that would further impact the functional status of the entorhinal inputs to dentate gyrus and CA3 (Adams et al., 1999). Interestingly, there was no statistically significant decrease in NR1 directly associated with age-related memory impairment. However, there was a positive correlation between performance on the Morris water maze and NR1 fluorescence intensity levels regardless of age, and this correlation was present only in CA3. AMPA receptors did not show such a correlation.
Could performance on a memory task be so clearly linked to one particular GluR in a small subset of hippocampal circuits? Clearly, it is too early to draw any causal inference from these data; however, recent transgenic mouse experiments support the notion of a direct relationship between the NMDA receptor proteins and memory performance. First, mice that have the NR1 gene knocked out in a manner that is confined to the hippocampus have impaired learning and memory performance (Tsien et al., 1996). In addition, mice that have a different NMDA receptor subunit, NR2B, overexpressed in the forebrain display enhanced memory and learning in several behavioral paradigms (Tang et al., 1999).
Clearly, these data represent a powerful example of gene/circuit/behavior links that will help to illuminate the role of the NMDA receptor in the hippocampus in age-related memory decline, and they further reinforce the power of multidisciplinary approaches as we move forward in our investigations of the neurobiology of aging. These latter experiments in mice also demonstrate the power of genetically manipulated mice as models for the investigation of memory and, potentially, age-related memory impairment. The required mouse genetics is sufficiently advanced; however, mouse neurophysiology, neuropsychology, and neuroanatomy lag far behind, making detailed interdisciplinary analyses difficult. With respect to mouse neuroanatomy, an important goal for the future will be to develop the quantitative datasets that will link gene products with specific circuits so that genetic manipulations that
affect learning and memory can be linked to brain structure in a precise manner. It will also be crucial to avoid the syndrome of ''looking for your keys under the street lamp." If one is manipulating gene X to affect behavior Y, one cannot assume that targeted circuits will react in a simple predictable fashion. Analyses of genetically manipulated mice must be comprehensive and quantitative, casting a wide net with respect to affected circuits and behaviors.
The nonhuman primate is also an increasingly important animal model, particularly for studies of cognition (Rapp and Amaral, 1992; Moss et al., 1999) and studies of hormonal effects (Abel et al., 1999). The issues of estrogen, menopause, and the effects of ERT on the hippocampus will be particularly important to analyze in a primate model, given the similarities between nonhuman primate reproductive physiology and that of humans, as well as the advanced cognitive abilities of the nonhuman primate compared with rodent models. In addition, given the extraordinary resource that aged primates represent, these studies should be multidisciplinary whenever possible in order to ensure that interactive datasets are obtained and that the animals are used as fully as possible.
Restoring Circuits
Given that both normal aging and neurodegenerative disorders disrupt selectively vulnerable circuits, it would appear that the most successful interventions will be those that have a sufficient degree of circuit selectivity. Is there any evidence that damaged or degenerating circuits can be restored in the brain, and if so how successful and selective is the restoration? There is a long history of such attempts and a large resultant literature that is beyond the scope of this review; however, it is informative to the present discussion on aging and neurodegenerative disorders to highlight several of the key approaches that have been attempted, such as tissue transplantation, transplanting engineered cells or viral vectors for gene therapy, the promise of stem cells, and the potential for exploiting the natural neurogenesis that occurs in the adult brain.
Transplantation Strategies
Transplantation of embryonic brain tissue into a damaged adult brain was one of the first strategies developed for circuit repair. Generally, select tissue from the donor embryo brain that contains the neurons destined to provide a replacement for the damaged circuit is surgically implanted in the deafferented region. The most successful animal models have involved the initial surgical destruction of the dopaminergic innervation of caudate followed by the transplantation of fetal substantia nigra from a donor brain in
the hopes of developing a therapy for Parkinson's disease. As early as the 1970s and 1980s, this strategy was demonstrated to be successful with respect to survival of the transplanted neurons, formation of the appropriate connections, and functional recovery of the animal (Bjorklund et al., 1981; Stromberg et al., 1985). Based on a large body of animal literature, the prospects for transplantation in such human neurodegenerative diseases as Parkinson's disease were considered in the 1980s, and there was limited success as a therapeutic intervention (see Lindvall, 1991, for a review).
However, in recent years the success of this strategy has improved, and in fact, there are now clinical trials with relatively long-term evaluation that demonstrate success with respect to the survival of the transplant, a degree of functional recovery, and even demonstrated release of dopamine from the transplanted axons into the newly innervated striaturn (Olanow et al., 1996; Hauser et al., 1999; Piccini et al., 1999). The strategy of transplanting fetal tissue has been modified and improved through the use of genetically modified cells that secrete doparnine into the striaturn, and these cells can be injected directly into the striaturn (Martinez-Serrano et al., 1995; Kordower et al., 1994, 1995). In addition, a gene delivery system employing a viral vector to deliver a growth factor into the septum successfully protected experimentally damaged cholinergic neurons innervating the hippocampus (Blomer et al., 1998).
While these approaches are potentially very promising for certain neurodegenerative disorders such as Parkinson's disease, such transplant approaches will clearly be most successful with a target circuit that has relatively little synaptic specificity in its target region, which is the case with the nigrostriatal doparninergic circuit. For example, such a strategy would seem to have very limited feasibility with respect to replacing complex, highly specific, corticocortical circuits.
Perhaps most relevant to aging are two recent primate studies that have employed gene therapy to reverse naturally occurring age-related compromise in two vulnerable circuits. One study targeted the cholinergic projection emanating from nucleus basalis that is known to be vulnerable in Alzheimer's disease (Smith et al.,1999a), and the other study used a viral vector approach to deliver a growth factor to reverse the natural age-related decline in dopa-mine function in the nigrostriatal projection (Kordower et al., 2000). Smith et al. demonstrated in primates that atrophic cell shrinkage and loss of cholinergic markers in nucleus basalis neurons could be reversed with human NGF (nerve growth factor) therapy. Interestingly, the neurons had not fully degenerated, and in that sense this is not analogous to late-stage Alzheimer's disease. The neurons had shrunk and their gene expression had shifted in a manner that impaired cholinergic function, but they had not degenerated. Both the morphologic and biochemical age-related shifts were reversible with NGF therapy.
In the analogous study of the dopamine neurons (Kordower et al., 2000), a viral vector was used to deliver GDNF, a growth factor that is known to be crucial for the sustained viability of the doparninergic neurons that furnish the nigrostriatal projection. Several morphologic, neurochemical, and physiologic parameters of the nigrostriatal system were augmented in the GDNF-treated aged animals, and a healthy youthful phenotype of these neurons was restored.
The aspect of these two studies that is perhaps most interesting and relevant to the present discussion is that these circuits had not degenerated, they were simply in poor health and had lost their robust high-functioning phenotype. Thus, these experiments may be particularly relevant to the circumstances that occur in normal aging, in which circuits have suffered with respect to gene expression and subtle morphologic attributes that impact function, but they are still intact and still able to be rescued with no need to replace the circuit.
The Promise of Stem Cells
In all of the transplant approaches outlined above, it was crucial to provide either neurons, modified cells, or viral vectors that replaced a missing neurotransmitter or growth factor that was required at a high level in a particular target region. This approach will have limited application to many other kinds of age-related problems that involve more complex circuits with high synaptic specificity. While the recent discoveries regarding stem cells are still in their infancy with respect to relevance to brain aging, they may in fact have broad application to the brain in the near future.
Stem cells are cells within the body that continue to proliferate in a state that has not differentiated into any particular organ, and they therefore retain what is referred to as a "pluripotent" capacity. The fact that these cells can be coaxed into a particular path of differentiation has generated enormous excitement about their potential for restoration of damaged cells and related functions in multiple organ systems, including the brain. In fact, stem cells and their potential application was chosen as the breakthrough of the year for 1999 by the journal Science. The excitement in this area was fueled particularly by two reports in late 1998 that demonstrated the potential to keep undifferentiated embryonic stem cells alive for long periods of time, and then promote their differentiation into various target tissues after a sustained period of remaining undifferentiated (Thomson et al., 1998; Shamblott et al., 1998). However, reliance on human embryonic stem cells with an eye toward therapeutics presents both technical problems as well as political and ethical issues that revolve around the use of embryonic human tissue.
Soon after the initial discovery regarding embryonic stem cells it was shown that adult stem cells from one particular organ retained the capacity to
survive, differentiate, and presumably function in a different organ; an example is the conversion of neural stem cells into a variety of blood cells that circulate and reside in the bone marrow (Bjornson et al., 1999; Scheffler et al., 1999). The reverse appears to be true as well, in that bone marrow stem cells can survive and migrate within the brain and differentiate into cells that developed a phenotype for astrocytes and, in some cases, neurons (Kopen et al., 1999). Also, embryonic and neural stem cells can replace oligodendrocytes in a brain deficient in such cells and the newly derived oligodendrocytes successfully myelinate axons (Brustle et al., 1999; Yandava et al., 1999).
These studies serve as a powerful animal model for testing potential therapies for such diseases as multiple sclerosis. It may also be possible to replace damaged neural circuits with such an approach. In such a study, neural embryonic stem cells from a healthy mouse were transplanted into a rat's spinal cord several days after traumatic injury (McDonald et al., 1999). His-tological analysis showed that the transplant-derived cells from the mouse had not only survived but had differentiated into astrocytes as well as neurons, and the investigators were able to demonstrate some functional recovery in these animals. The potential of such approaches that utilize neural stem cells for therapies directed at age-related pathology and functional decline is profound, and while this research arena is still in its infancy, the progress that has already occurred is very impressive (see Shihabuddin et al., 1999, for a review).
The most likely limitation of the stem cell approaches will be the need for a high degree of circuit specificity in any devised therapy. The use of neural stem cells to achieve some recovery of function after spinal cord injury suggests that, at least in some cases, an adequate level of circuit specificity may emerge with little direct coaxing. However, it is not clear at this point whether or not this will occur in the key brain circuits affected in aging. For example, if we were to try and use stem cell therapy to replace the entorhinal neurons that provide the perforant path, how would we guide the neural stem cells and the differentiated neurons into becoming the particularly highly differentiated neurons that reside in layer II of entorhinal cortex with the appropriate afferents and efferents? Perhaps even more difficult, how would we replace the neurons that furnish the corticocortical circuits that interconnect frontal and temporal regions that are so damaged in Alzheimer's disease, while leaving the intact circuits unaffected? As described below, in some cases the brain may solve this problem itself by continuing to generate neurons that can replace certain circuits throughout life.
Neurogenesis in the Adult Hippocampus
The recent data on neurogenesis in the adult brain have demonstrated clearly that we need to reevaluate the accepted dogma that when neurons die
they cannot be replaced by the generation of new neurons. While it has been known for a long time from work in rodents that some neurogenesis occurs in the adult dentate gyrus (Altman and Das, 1965), it was demonstrated only recently that this also occurs in the nonhuman primate (Gould et al., 1999b) and in humans as well (Eriksson et al., 1998). In addition, while the functional status of the new neurons in the dentate gyrus is unclear, it has been demonstrated that they project to CA3 and thus may form normal connections (Hastings and Gould, 1999). Furthermore, the prevailing notion that the hippocampus is the only telencephalic region in which neurogenesis occurs has now been challenged by a report that neurogenesis occurs in the nonhuman primate neocortex, particularly in cortical regions that would presumably play a dominant role in learning, memory, and cognition (Gould et al., 1999c); however, these data on neocortex are presently controversial and will need to be expanded and replicated.
How are we to integrate the notion of new neurons into our prevailing attitudes regarding age-related functional decline and neurodegenerative disorders? While much work lies ahead, several interesting recent reports demonstrate the potential importance of neurogenesis to aging, at least with respect to the dentate gyrus. For one thing, neurogenesis in the dentate gyrus is decreased in aging (Kuhn et al., 1996). There are also positive influences on neurogenesis that may be exploited in preventing the age-related decrease in neurogenesis. The simple process of training animals on a learning task that requires the hippocampus enhanced neurogenesis and the viability of the new neurons in the dentate gyrus of rats (Gould et al., 1999a). Similarly adult mice living in an enriched environment have increased neurogenesis in the hippocampus (Kempermann et al., 1997). Moreover, increased experience and social interaction led to an enhancement of neurogenesis in the dentate gyrus of aged animals (Kempermann et al., 1998). Finally, simple physical exercise (e.g., running) increased cell proliferation in the adult mouse dentate gyrus (van Praag et al., 1999).
The potential for hormonal impact on these processes makes this issue even more relevant to aging. For example, estrogen has been demonstrated to stimulate a transient increase in neurogenesis in the dentate gyrus of the adult female rat (Tanapat et al., 1999). Furthermore, it was recently demonstrated that the level of neurogenesis typical of a young animal could be restored in an aged animal by decreasing the high levels of circulating corticosteroids that commonly occur in aged animals (Cameron and McKay, 1999). Thus, while it is not yet possible to fit these data on neurogenesis into the present context of aging and the neurobiological substrate for age-related functional decline, it is clear that this will be an area of intense investigation in the future and an area of paramount importance in aging research. It will be especially important to determine the quantitative extent of neurogenesis and the functional implications of adding neurons to the aged brain. Do the new neurons par-
ticipate in the appropriate circuits? To what degree does adult neurogenesis occur in areas other than the dentate gyrus? In a related fashion, to what degree can the natural process of neurogenesis in the hippocampus be used to replace neurons in a neurodegenerative disorder such as Alzheimer's disease? Obviously it will take time to obtain answers to these questions, but the field is moving rapidly, and we must now incorporate the potential for new neurons into our thinking about malfunctioning neurons and degenerating neurons in the aging brain.
CONCLUDING REMARKS
This report outlines a microscopic and neuroanatomic approach to understanding neurobiological events that underlie memory decline with aging. The available data suggest that the focus of such analyses should be the vulnerable circuit, and the delineation of phenotypic characteristics that render a cell class or circuit selectively vulnerable to aging. With respect to Alzheimer's disease, the key reflection of vulnerability is degeneration, whereas memory decline in the context of normal aging is likely due to subtle neurochemical and morphologic alterations that lead to functional impairment in the absence of frank neuronal degeneration. This differentiation is not absolute, however, in that degenerative events are clearly under way in the entorhinal cortex of neurologically normal elderly people. Many of these neurons appear as "transitional' with respect to degenerative profiles typical of Alzheimer's disease, and it will be critical to focus more attention on these transitional events if we are to adequately differentiate a stable, relatively high-functioning state from the early stages of a progression toward Alzheimer's disease. In addition, much of the work on animal models suggests that functional decline in the context of normal aging or senescence (e.g., age-related memory impairment) is unlikely to be due primarily to neuron loss and is more likely to be a reflection of shifts in gene expression or key neurochemical attributes that impair function in an intact circuit. This suggests that therapy targeted at restoring a youthful phenotype to vulnerable circuits may be particularly effective, and data exist demonstrating the rescue of age-impaired cholinergic and dopaminergic circuits. In addition, replacing dead neurons or impaired circuits through the use of stem cells may be more realistic than previously thought, although obtaining the requisite circuit specificity from such an approach may be problematic. Finally, naturally occurring neurogenesis, particularly in the adult dentate gyrus, offers another avenue for restoration of function, and data exist showing that the generation of new hippocampal neurons and their continued viability are responsive to behavioral and endocrine intervention.
Thus, while conditions such as Alzheimer's disease clearly involve devastating neuron loss, the scenario for normal aging is far more dynamic and
adaptable than we once thought, and even neuron loss might not be as irreversible as we assumed just a few years ago. However, the development of helpful interventions will have to be carefully guided by a healthy respect for the specificity and heterogeneity of neural circuits, and the fact that even in regions considered vulnerable (e.g., the hippocampus), individual circuits vary enormously in their vulnerability to aging. Therapies that are not adequately specific with respect to targeting vulnerable circuits are likely to have unanticipated behavioral and functional consequences that will compromise their effectiveness.
ACKNOWLEDGMENTS
The author thanks Patrick Hof, Michelle Adams, Peter Rapp, and Bill Janssen for helpful discussions and comments regarding this manuscript.
REFERENCES
Abel, T.W., M.L. Voytko, and N.E. Rance 1999 The effects of hormone replacement therapy on hypothalamic neuropeptide gene expression in a primate model of menopause. Journal of Clinical Endocrinology and Metabolism 84:2111–2118.
Adams, M.M., T.D. Smith, P.R. Rapp, M. Gallagher, and J.H. Morrison 1999 Immunofluorescence intensity in CA3 of hippocampus predicts spatial learning in young and aged rats. Society for Neuroscience Abstracts 25:2163.
Altman, J., and G.D. Das 1965 Autoradiographic and histological evidence of postnatal neurogenesis in rats. Journal of Comparative Neurology 124:319–335.
Barnes, C.A. 1994 Normal aging: Regionally specific changes in hippocampal synaptic transmission. Trends in Neurosciences 17:13–18.
Barnes, C.A., G. Rao, and J. Shen 1997a Age-related decrease in the N-methyl-D-aspartateR-mediated excitatory postsynaptic potential in hippocampal region CAI. Neurobiology of Aging 18:445–452.
Barnes, C.A., M.S. Suster, J. Shen, and B.L. McNaughton 1997b Multistability of cognitive maps in the hippocampus of old rats. Nature 388:272–275.
Bjorklund, A., U. Stenevi, S.B. Dunnett, and S.D. Iversen 1981 Functional reactivation of the deafferented neostriaturn by nigral transplants. Nature 289:497–499.
Bjornson, C.R., R.L. Rietze, B.A. Reynolds, M.C. Magli, and A.L. Vescovi 1999 Turning brain into blood: A hematopoietic fate adopted by adult neural stem cells in vivo. Science 283:534–537.
Blomer, U., T. Kafri, L. Randolph-Moore, I.M. Verma, and F.H. Gage 1998 Bcl-xL, protects adult septal cholinergic neurons from axotomized cell death. Proceedings of the National Academy of Sciences of the United States of America 95:2603–2608.
Bouras, C., P.R. Hof, P. Giannakopoulos, J. Michel, and J.H. Morrison 1994 Regional distribution of neurofibrillary tangles and senile plaques in the cerebral cortex of elderly patients: A quantitative evaluation of a one-year autopsy population from a geriatric hospital. Cerebral Cortex 4:138–150.
Brustle, O., K.N. Jones, R.D. Learish, K. Karram, K. Choudhary, O.D. Wiestler, I.D. Duncan, and R.D. McKay 1999 Embryonic stem cell-derived glial precursors: A source of myelinating transplants. Science 285:754–756.
Bussière, T., B. Wicinsky, G.I. Lin, D.P. Perl, P. Davies, R. Nixon, J.H. Morrison, and P.R. Hof 1999 Early neurodegenerative alterations in the cerebral cortex during normal aging and Alzheimer's disease. Society for Neuroscience Abstracts 25:593.
Cameron, H.A., and R.D.G. McKay 1999 Restoring production of hippocampal neurons in old age. Nature Neuroscience 2:894–897.
Caramanos, Z., and M.L. Shapiro 1994 Spatial memory and N-methyl-D-aspartate receptor antagonists APV and MK-801: Memory impairments depend on familiarity with the environment, drug dose, and training duration. Behavioral Neuroscience 108:30–43.
Chaudhry, F.A., K.P. Lehre, M. van Lookeren Campagne, O.P. Ottersen, N.C. Danbolt, and J. Storm Mathisen 1995 Glutamate transporters in glial plasma membranes: High differentiated localizations revealed by quantitative ultrastructual immunocytochemistry. Neuron 15:711–720.
Clark, A.S., K.R. Magnusson, and C.W. Cotman 1992 In vitro autoradiography of hippocampal excitatory amino acid binding in aged Fischer 344 rats: relationship to performance on Morris water maze. Behavioral Neuroscience 106(2):324–335.
Coggeshall, R.E., and H.A. Lekan 1996 Methods for determining numbers of cells and synapses: A case for more uniform standards of review. Journal of Comparative Neurology 364:6–15.
Coleman, P.D., and D.G. Flood 1987 Neuron numbers and dendritic extent in normal aging and Alzheimer's disease. Neurobiology of Aging 8:521–545.
DeKosky, S.T., and S.W. Scheff 1990 Synapse loss in frontal cortex biopsies in Alzheimer's disease: Correlation with cognitive severity. Annals of Neurology 27:457–464.
Duan, H., Y. He, B. Wicinsky, G. Yeung, T.L. Page, W.G.M. Janssen, J.H. Morrison, and P.R. Hof 1999 Age-related changes in cortical projection neurons in macaque monkey: Dendrite morphology, spine density, and neurochemical features. Society for Neuroscience Abstracts 25:362.
Eberwine, J., H. Yeh, K. Miyashiro, Y. Cao, S. Nair, R. Finnell, M. Zettel, and P. Coleman 1992 Analysis of gene expression in single live neurons. Proceedings of the National Academy of Sciences of the United States of America 89:3010–3014.
Eichenbaum, H., P. Dudchenko, E. Wood, M.L. Shapiro, and H. Tanila 1999 The hippocampus, memory, and place cells: Is it spatial memory or memory space? Neuron 23:209–226.
Eriksson, P.S., E. Perfilieva, T. Bjork-Eriksson, A.M. Alborn, C. Nordborg, D.A. Peterson, and F.H. Gage 1998 Neurogenesis in the adult human hippocampus. Nature Medicine 4:1313–1317.
Gallagher, M., and P.R. Rapp 1997 The use of animal models to study the effects of aging on cognition. Annual Review of Psychology 48:339–370.
Gazzaley, A.H., S.J. Siegel, J.H. Kordower, E.J. Mufson, and J.H. Morrison 1996a Circuit-specific alterations of N-methyl-D-aspartate subunit 1 in the dentate gyrus of aged monkeys. Proceedings of the National Academy of Sciences of the United States of America 93(7):3121–3125.
Gazzaley, A.H., M.M. Thakker, P.R. Hof, and J.H. Morrison 1997 Preserved number of entorhinal cortex layer II neurons in aged macaque monkeys. Neuroblology of Aging 18:549–553.
Gazzaley, A.H., N.G. Weiland, B.S. McEwen, and J.H. Morrison 1996b Differential regulation of NMDARI mRNA and protein by estradiol in the rat hip-pocampus. Journal of Neuroscience 16:6830–6838.
Geinisman, Y., L. Detoledo-Morrell, F. Morrell, and R.E. Heller 1995 Hippocampal markers of age-related memory dysfunction: Behavioral, electrophysiological and morphological perspective. Progress in Neurobiology 45:223–252.
Geinisman, Y., H.J. Gunderson, E. van der Zee, and M.J. West 1996 Unbiased stereological estimation of the total number of synapses in a brain region. Journal of Neurocytology 25:805–819.
Gimmel, D., J.H. Morrison, D.P. Perl, C. Bouras, and P.R. Hof 1998 Development of stereologic indices of neurodegeneration in the cerebral cortex during normal aging and Alzheimer's disease. Society for Neuroscience Abstracts 24:960.
Gomez-Isla, T., J.L. Price, D.W. McKeel, Jr., J.C. Morris, J.H. Growdon, and B.T. Hyman 1996 Profound loss of layer II entorhinal cortex neurons occurs in very mild Alzheimer's disease. Journal of Neuroscience 16:4491–4500.
Gould, E., A. Beylin, P. Tanapat, A. Reeves, and T.J. Shors 1999a Learning enhances adult neurogenesis in the hippocampal formation. Nature Neu-roscience 2:260–265.
Gould, E., A.J. Reeves, M. Fallah, P. Tanapat, C.G. Gross, and E. Fuchs 1999b Hippocampal neurogenesis in adult Old World primates. Proceedings of the National Academy of Sciences of the United States of America 96:5263–5267.
Gould, E., A.J. Reeves, M.S. Graziano, and C.G. Gross 1999c Neurogenesis in the neocortex of adult primates. Science 286:548–552.
Hastings, N.B., and E. Gould 1999 Rapid extension of axons into the CA3 region by adult-generated granule cells. Journal of Comparative Neurology 413:146–154.
Hauser, R.A., T.B. Freeman, B.J. Snow, M. Nauert, L. Gauger, J.H. Kordower, and C.W. Olanow 1999 Long-term evaluation of bilateral fetal nigral transplantation in Parkinson's disease. Archives of Neurology 56:179–187.
He, Y., W.G.M. Janssen, J.D. Rothstein, and J.H. Morrison 2000 Differential synaptic localization of the glutamate transporter EAAC I and glutamate receptor subunit GluR2 in the rat hippocampus. Journal of Comparative Neurology 418:255–269.
Hof, P.R., C. Bouras, and J.H. Morrison 1999 Cortical neuropathology in aging and dementing disorders: Neuronal typology, connectivity, and selective vulnerability. Pp. 175–311 in Cerebral Cortex, Vol. 14, Neurodegenerative and Age-Related Changes in Structure and Function of Cerebral Cortex, A. Peters and J.H. Morrison, eds. New York: Kluwer Academic/Plenum Publishers.
Hof, P.R., K. Cox, and J.H. Morrison 1990 Quantitative analysis of a vulnerable subset of pyramidal neurons in Alzheimer's disease: 1. Superior frontal and inferior temporal cortex. Journal of Comparative Neurology 301:44–54.
Hof, P.R., and J.H. Morrison 1990 Quantitative analysis of a vulnerable subset of pyramidal neurons in Alzheimer's disease: II. Primary and secondary visual cortex. Journal of Comparative Neurology 301:55–64.
Hollmann, M., and S. Heinemann 1994 Cloned glutamate receptors. Annual Review of Neuroscience 17:31–108.
Hyman, B.T., A.R. Damasio, G.W. Van Hoesen, and C.L. Barnes 1984 Alzheimer's disease: Cell pecific pathology isolates the hippocampal formation. Science 225:1168–1170.
Johnson, M., R.H. Perry, M.A. Piggott, J.A. Court, D. Spurden, S. Lloyd, P.G. Ince, and E.K. Perry 1996 Glutamate receptor binding in the human hippocampus and adjacent cortex during development and aging. Neurobiology of Aging 17:639–651.
Kacharmina, J.E., P.B. Crino, and J. Eberwine 1999 Preparation of cDNA from single cells and subcellular regions. Methods in Enzymology 303:3–18.
Kemper, T.L. 1999 Age-related changes in subcortical nuclei that project to the cerebral cortex. Pp. 365–397 in Cerebral Cortex, Vol. 14, Neurodegenerative and Age-Related Changes in Sructure and Function of Cerebral Cortex, A. Peters and J.H. Morrison, eds. New York: Kluwer Academic/Plenum Publishers.
Kempermann, G., H.G. Kuhn, and F.H. Gage 1997 More hippocampal neurons in adult mice living in an enriched environment. Nature 386:493–495.
1998 Experience-induced neurogenesis in the senescent dentate gyrus. Journal of Neuroscience 18:3206–3212,
Kopen, G.C., D.J. Prockop, and D.G. Phinney 1999 Marrow stromal cells migrate throughout forebrain and cerebellum, and they differentiate into astrocytes after injection into neonatal mouse brains. Proceedings of the National Academy of Sciences of the United States of America 96:10711–10716.
Kordower, J.H., J. Blich, M.E. Emborg, S.Y. Ma, Y. Chu, E.-Y. Chen, S. Palfi, L. Leventhal, B.Z. Roitberg, W.D. Brown, J.E. Holden, R. Pyzalski, M.D. Taylor, P. Carvey, D. Trono, P. Hantraye, N. Deglon, and P. Aebischer 2000 A lentivirus encoding for GDNF reverses doparnine insufficiency in an aged monkey model of progressive nigrostriatal degeneration. (Submitted)
Kordower, J.H., Y.T. Liu, S. Winn, and D.F. Emerich 1995 Encapsulated PC12 cell transplants into hemiparkinsonian monkeys: A behavioral, neuroanatomical, and neurochemical analysis. Cell Transplant 4:155–171.
Kordower J.H., S.R. Winn, Y.T. Liu, E.J. Mufson, J.R. Sladek, Jr., J.P. Hammang, E.E. Baetge, and D.F. Emerich 1994 The aged monkey basal forebrain: Rescue and sprouting of axotomized basal fore-brain neurons after grafts of encapsulated cells secreting human nerve growth factor. Proceedings of the National Academy of Sciences of the United States of America 91:10898–10902.
Kuhn, H.G., H. Dickinson-Anson, and F.H. Gage 1996 Neurogenesis in the dentate gyrus of the adult rat: Age-related decrease of neuronal progenitor proliferation. Journal of Neuroscience 16:2027–2033.
Le Jeune, H., D. Cecyre, M.J. Meaney, and R. Quirion 1996 Ionotropic glutamate receptor subtypes in the aged memory-impaired and unimpaired Long-Evans rat. Neuroscience 74:349–363.
Lewis, D.A., M.J. Campbell, R.D. Terry, and J.H. Morrison 1987 Laminar and regional distributions of neurofibrillary tangles and neuritic plaques in Alzheimer's disease: A quantitative study of visual and auditory cortices. Journal of Neuroscience 7:1799–1808.
Lindvall, O. 1991 Prospects of transplantation in human neurodegenerative diseases. Trends in Neuro-sciences 14:376–384.
Magnusson, K.R., and C.W. Cotman 1993 Age-related changes in excitatory amino acid receptors in two mouse strains . Neurobiology of Aging 14:197–206.
Martinez-Serrano, A., W. Fischer, and A. Bjorklund 1995 Reversal of age-dependent cognitive impairments and cholinergic neuron atrophy by NGF-secreting neural progenitors grafted to the basal forebrain. Neuron 15:473–484.
McDonald, J.W., X.Z. Liu, Y. Qu, S. Liu, S.K. Mickey, D. Turetsky, D.I. Gottlieb, and D.W.Choi 1999 Transplanted embryonic stem cells survive, differentiate and promote recovery in injured rat spinal cord. Nature Medicine 5:1410–1412.
McEwen, B.S., and S.E. Alves 1999 Estrogen actions in the central nervous system. Endocrine Reviews 20:279–307.
Morrison, J.H., and P.R. Hof 1997 Life and death of neurons in the aging brain. Science 278:412–419.
Morrison, J.H., D.A. Lewis, M.J. Campbell, G.W. Huntley, D.L. Benson, and C. Bouras 1987 A monoclonal antibody to non-phosphorylated neurofilament protein marks the vulnerable cortical neurons in Alzheimer's disease. Brain Research 416:331–336.
Moss, M.B., R.J. Killiany, and J.G. Herndon 1999 Age-related cognitive decline in the rhesus monkey. Pp. 21–48 in Cerebral Cortex, Vol. 14, Neurodegenerative and Age-Related Changes in Structure and Function of Cerebral Cortex, A. Peters and J.H. Morrison, eds. New York: Kluwer Academic/Plenum Publishers.
Nicolle, M.M., J.L. Bizon, and M. Gallagher 1996 In vitro autoradiography of ionotropic glutamate receptors in hippocampus and striaturn of aged Long-Evans rats: Relationship to spatial learning. Neuroscience 74:741–756.
Olanow, C.W., J.H. Kordower, and T.B. Freeman 1996 Fetal nigral transplantation as a therapy for Parkinson's disease. Trends in Neuro-sciences 19:102–109.
Pearson, R.C.A., M.M. Esiri, R.W. Hiorns, G.K. Wilcock, and T.P.S. Powell 1985 Anatomical correlates of the distribution of the pathological changes in the neocortex in Alzheimer's disease. Proceedings of the National Academy of Sciences of the United States of America 82:4531–4534.
Peters, A., J.H. Morrison, D.L. Rosene, and B.T. Hyman 1998a Are neurons lost from the primate cerebral cortex during aging? Cerebral Cortex 8:295–300.
Peters, A., D.L. Rosene, M.B. Moss, T.L. Kemper, C.R. Abraham, J. Tigges, and M.S. Albert 1996 Neurobiological bases of age-related cognitive decline in the rhesus monkey. Journal of Neuropathology and Experimental Neurology 55:861–874.
Peters, A., C. Sethares, and M.B. Moss 1998b The effects of aging on layer 1 in area 46 of prefrontal cortex in the rhesus monkey. Cerebral Cortex 8:671–684.
Piccini, P., D.J. Brooks, A. Bjorklund, R.N. Gunn, P.M. Grasby, O. Rimoldi, P. Brundin, P. Hagell, S. Rehncrona, H. Widner, and O. Lindvall 1999 Dopamine release from nigral transplants visualized in vivo in a Parkinson's patient. Nature Neuroscience 2(12):1137–1140.
Rapp, P.R., and D.G. Amaral 1992 Individual differences in the cognitive and neurobiological consequences of normal aging. Trends in Neurosciences 15:340–345.
Rapp, P.R., and M. Gallagher 1996 Preserved neuron number in the hippocampus of aged rats with spatial learning deficits. Proceedings of the National Academy of Sciences of the United States of America 93:9926–9930.
Rogers, J., and J.H. Morrison 1985 Quantitative morphology and regional and laminar distribution of senile plaques in Alzheimer's disease. Journal of Neuroscience 5:2801–2808.
Rosene, D.L., and G.W. Van Hoesen 1987 The hippocampal formation of the primate brain—A review of some comparative aspects of cytoarchitecture and connections. Pp. 345–356 in Cerebral Cortex, Vol. 6, Further Aspects of Cortical Function, Including Hippocampus, E.G. Jones and A. Peters, eds. New York: Plenum Press.
Scheffler, B., M. Horn, 1. Blumcke, E.D. Laywell, D. Coomes, V.G. Kukekov, and D.A. Steindler 1999 Narrow-mindedness: A perspective on neuropoiesis. Trends in Neurosciences 22:348–357.
Shamblott, M.J., J. Axelman, S. Wang, E.M. Bugg, J.W. Littlefield, P.J. Donovan, P.D. Blumenthal, G.R. Huggins, and J.D. Gearhart 1998 Derivation of pluripotent stem cells from cultured human primordial germ cells. Proceedings of the National Academy of Sciences of the United States of America 95(23):13726–13731.
Shapiro, M.L., and H. Eichenbaum 1999 Hippocampus as a memory map: Synaptic plasticity and memory encoding by hippocampal neurons. Hippocampus 9(4):365–384.
Shihabuddin, L.S., T.D. Palmer, and F.H. Gage 1999 The search for neural progenitor cells: Prospects for the therapy of neurodegenerative disease. Molecular Medicine Today 5:474–480.
Siegel, S.J., W.G.M. Janssen., J.W. Tullai, S.W. Rogers, T. Moran, S.F. Heinemann, and J.H.Morrison 1995 Distribution of the excitatory amino acid receptor subunits GluR2(4) in monkey hippocampus and colocalization with subunits GluR5-7 and NMDARI. Journal of Neuroscience 15:2707–2719.
Smith D.E., J. Roberts, F.H. Gage, and M.H. Tuszynski 1999a Age-associated neuronal atrophy occurs in the primate brain and is reversible by growth factor gene therapy. Proceedings of the National Academy of Sciences of the United States of America 96:10893–10898.
Smith, T.D., M.M. Adams, J.H. Morrison, M. Gallagher, and P.R. Rapp 1999b Alterations in hippocampal synaptophysin immunoreactivity predict spatial learning impairment in the aged rat. Society for Neuroscience Abstracts 25:2163.
Squire, L.R., and S. Zola-Morgan 1991 The medial temporal lobe memory system. Science 253:1380–1386.
Stromberg, L, S. Johnson, B. Hoffer, and L. Olson 1985 Reinnervation of dopamine-denervated striaturn by substantia nigra transplants: Immunohistochemical and electrophysiological correlates. Neuroscience 14:981–990.
Tanapat, P., N.B. Hastings, A.J. Reeves, and E. Gould 1999 Estrogen stimulates a transient increase in the number of new neurons in the dentate gyrus of the adult female rat. Journal of Neuroscience 19:5792–5801.
Tang, Y.P., E. Shimizu, G.R. Dube, C. Rampon, G.A. Kerchner, M. Zhuo, G. Liu, and J.Z. Tsien 1999 Genetic enhancement of learning and memory in mice. Nature 401:63–69.
Terry, R.D., E. Masliah, D.P. Salmon, N. Butters, R. DeTeresa, R. Hill, L.A. Hansen, and R. Katzman 1991 Physical basis of cognitive alterations in Alzheimer's disease: Synapse loss is the major correlate of cognitive impairment. Annals of Neurology 30:572–580.
Thomson, J.A., J. Itskovitz-Eldor, S.S. Shapiro, M.A. Waknitz, J.J. Swiergiel, V.S. Marshall, and J.M. Jones 1998 Embryonic stem cell lines derived from human blastocysts. Science 282:1145–1147.
Tigges, J., J.G. Herndon, and D.L. Rosene 1996 Preservation into old age of synaptic number and size in the supragranular layer of the dentate gyrus in rhesus monkeys. Acta Anatomica 157:63–72.
Toni, N., P.A. Buchs, 1. Nikonenko, C.R. Bron, and D. Muller 1999 LTP promotes formation of multiple spine synapses between a single axon terminal and a dendrite. Nature 402:421–425.
Tsien, J.Z., P.T. Herta, and S. Tonegawa 1996 The essential role of hippocampal CAI NMDA receptor-dependent synaptic plasticity in spatial memory. Cell 87:1327–1338.
Van Hoesen, G.W., and D.N. Pandya 1975 Some connections of the entorhinal (area 28) and perirhinal (area 35) cortices of the rhesus monkey. III. Efferent connections. Brain Research 95:39–59.
van Praag, H., G. Kempermann, and F.H. Gage 1999 Running increases cell proliferation and neurogenesis in the adult mouse dentate gyrus. Nature Neuroscience 2:266–270.
Vickers, J.C., A. Delacourte, and J.H. Morrison 1992 Progressive transformation of the cytoskeletal associated with normal aging and Alzheimer's disease. Brain Research 594:273–278.
Wenk, G.W., and L.C. Walker 1991 Loss of NMDA, but not GABA-A, binding in the brains of aged rats and monkeys. Neurobiology of Aging 12:93–98.
West, M.J. 1993a New stereological methods for counting neurons. Neurobiology of Aging 14:275–285.
1993b Regionally specific loss of neurons in the aging human hippocampus. Neurobiology of Aging 14:287–293.
1999 Stereological methods for estimating the total number of neurons and synapses: Issues of precision and bias. Trends in Neurosciences 22:51–61.
Whitehouse, P.J., D.L. Price, R.G. Struble, A.W. Clark, J.T. Coyle, and M.R. DeLong 1982 Alzheimer's disease and senile dementia: Loss of neurons in basal forebrain. Science 215:1237–1239.
Witter, M.P., and D.G. Amaral 1991 Entorhinal cortex of the monkey: V. Projections to dentate gyrus, hippocampus, and subicular complex. Journal of Comparative Neurology 307:437–459.
Wood, E.R., P.A. Dudchenko, and H. Eichenbaum 1999 The global record of memory in hippocampal neuronal activity. Nature 397:613–616.
Woolley, C.S. 1999 Electrophysiological and cellular effects of estrogen on neuronal function. Critical Reviews in Neurobiology 13:1–20.
Yandava, B.D., L.L. Billinhurst, and E.Y. Snyder 1999 ''Global" cell replacement is feasible via neural stem cell transplantation: Evidence from the dysmyelinated shiverer mouse brain. Proceedings of the National Academy of Sciences of the United States of America 96:7029–7034.
Zola-Morgan, S., and L.R. Squire 1993 Neuroanatomy of memory. Annual Review of Neuroscience 16:547–563.

































