
3
Current Practices for Assessing Risk for Developmental Defects and Their Limitations
Since the mid-1900s, various governmental agencies in the United States have taken responsibility for protecting the health of the public by regulating safe usage practices for drugs, food additives, pesticides, and environmental and occupational chemicals (Gallo 1996; Omenn and Faustman 1997). In the 1970s, risk assessment began as an organized activity of federal agencies to set acceptable exposure levels or tolerance levels. Earlier the American Conference of Governmental Industrial Hygienists had set threshold limit values for workers and the U.S. Food and Drug Administration (FDA) had established acceptable daily intakes for dietary pesticide residues and food additives. In 1983, the National Research Council published a report entitled Risk Assessment in the Federal Government: Managing the Process (often referred to as the “Red Book”), which provided a common framework for risk assessment (NRC 1983). In 1991, the U.S. Environmental Protection Agency (EPA) published risk assessment guidelines specific for developmental toxicity (EPA 1991).
In this chapter, the committee highlights risk assessment practices as they relate to the evaluation of chemicals as potential developmental toxicants and identifies limitations in the current risk assessment approaches.
THE DEVELOPMENTAL TOXICITY RISK ASSESSMENT PROCESS
“Human health risk assessment” refers to the process of systematically characterizing potential adverse health effects in humans that result from exposure to chemicals and physical agents (NRC 1983). For developmental toxicity, this assessment means evaluating the potential for chemical exposure to cause any of four types of adverse developmental end points: growth retardation; gross, skel-
etal, or visceral malformations; adverse functional outcomes; and lethality. Developmental toxicity risk assessment includes evaluating all available experimental animal and human toxicity data and the dose, route, duration, and timing of exposure to determine if an agent causes developmental toxicity (EPA 1991; Moore et al. 1995).
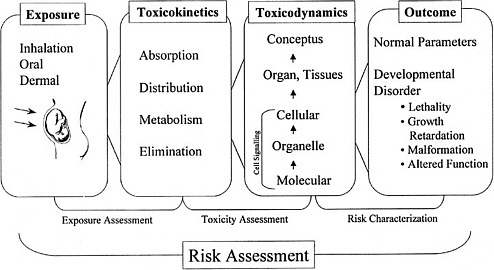
As discussed in the “Red Book,” risk management, in contrast to risk assessment, is the application of risk assessment information in policy and decision-making processes to balance risks and benefits (e.g., for therapeutic applications); set target levels of acceptable risk (e.g., for food contaminants and water pollutants); set priorities for the program activities of regulatory agencies, manufacturers, and environmental and consumer organizations; and estimate residual risks after a risk-reduction effort has been taken (e.g., folic acid supplementation in food). Figure 3-1 shows the NRC paradigm for risk assessment and risk management. As shown in this figure, risk characterization refers to the synthesis of qualitative and quantitative information for both toxicity and exposure assessments (EPA 1995). It also usually includes a discussion of the uncertainties in the analysis.
The following sections describe some of the specific approaches used for toxicity assessment. Four types of informational methods that can be used for

FIGURE 3-1 Risk assessment and risk management paradigm from the NRC modified for developmental toxicity risk assessments. In accordance with this committee’s deliberations, the research section now includes a two-way arrow and specifically highlights emerging research on gene-environment interaction and developmental cell-signaling pathways. The iterative feedback loop between research and risk assessment is necessary to translate new findings in biology into scientifically based risk assessments. Source: Adapted from NRC 1983.
these assessments are chemical structure-activity information, in vitro assessments, in vivo animal bioassays, and epidemiological studies. Two additional steps in risk assessment, dose-response assessment and exposure assessment, are described. Finally, the use of toxicokinetic information and biomarkers in developmental toxicity risk assessment is discussed.
Chemical Structure-Activity Information
Information on a chemical’s structure, stability, solubility, reactivity, and electrophilicity can provide useful clues to its potential to be absorbed and distributed throughout the body and to be reactive with biological tissues. In fact, despite early concepts of a true placental barrier, it is now appreciated that all lipid-soluble compounds have access to the developing cells of an embryo and fetus. Properties of lipid solubility and characteristics such as chemical size and pKa can be used to predict the potential for chemicals to cross the placenta and have access to conceptus tissues (Slikker and Miller 1994).
Structure-activity relationships (SARs) are developed to show the relationship between the specific chemical structures or moieties of agents and their capacity to produce certain toxic effects. For glycol ethers, retinoic acid, valproic acid, and their derivatives and for several other commercial products and therapeutics, good SAR data exist for developmental effects. Recently, SARs were reported for valproic acid derivatives that activate the peroxisomal proliferation pathway and cause developmental toxicity (Lampen et al. 1999).
Early research on receptor binding identified SARs for environmental agents such as benzopyrene and dioxin. Toxicity equivalency factors (TEFs) have been developed that relate the relative toxicity of each compound to a reference compound, such as benzo[a]pyrene (B[a]P) or 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) for pyrenes and dioxins, respectively (Van den Berg et al. 1998). Complexities arise when different toxicity end points have different SARs. To be useful for developmental toxicity risk assessments, SARs (and TEFs) must be evaluated for each of the end points of developmental toxicity.
In Vitro Assessments
Alternatives to pregnant experimental mammals in studies of developmental toxicology have often been grouped together as in vitro approaches, but that is misleading, because they include not only ex vivo mammalian embryos, tissues, cells, and subcellular preparations but also embryos of nonmammalian species. Broadly, such alternatives have had two applications: to test chemicals for potential effects and to analyze mechanisms of effect.
Mechanistic uses of ex vivo methods have much in common with investigative studies in other areas of biology. They have made major contributions to understanding developmental toxicity, because of the manipulations possible in
vitro, including the removal of the maternal environment, the ablation or transplantation of tissues and cells, labeling and tracking of cells or molecules, biochemical and gene manipulations by the use of inhibitors and anti-sense RNA, and real-time physiological monitoring of the embryo. The types of information generated include the identification of proximate developmental toxicants, exact tissue sites of accumulation, initial biochemical insults, gene expression changes, intrinsic SARs (of the parent compound), and identification of disrupted developmental pathways.
The search for alternatives for testing purposes is driven by the need to assess a larger number of chemicals than that allowed by using available resources for in vivo methods and also by the desire to reduce or replace the use of experimental mammals. Two levels of testing should be distinguished: secondary and primary. Secondary testing is the assessment of chemicals that have some known potential developmental toxicity. Most commonly, secondary testing involves analogs of prototype chemicals that have known in vivo developmental toxicity. The objective is to replicate the observed developmental toxicity in a simple system. The approach has been successful, especially for pharmaceuticals and particularly with the use of isolated mammalian embryos and embryonic cells in culture. For example, the approach has been used for testing retinoids (Kistler and Howard 1990) and triazoles (Flint and Boyle 1985). For that type of use, a universal validation of the method is not required. It is sufficient to show that the method replicates a specific in vivo effect for the particular chemicals under study.
Primary testing, in contrast, is the testing of chemicals that have no known potential toxicity, the aim being to predict in vivo actions. There must be confidence that the test outcome will accurately classify most chemicals by their potential to cause human developmental toxicity. Furthermore, the required sensitivity and selectivity will vary, depending on the purpose of the test. Sensitivity is the proportion of in vivo toxicants that are positive in the test, and selectivity is the proportion of inactive chemicals that are negative in the test. In some contexts, for example, in drug discovery by combinatorial chemistry, the aim is the early elimination of potential toxicants. False-positive results are not problematic, because there are many other chemicals from which to choose. Conversely, if the context is hazard identification and the aim is to set priorities for further in vivo testing, then a high rate of false-positive results would be inappropriate. Thus, there is a drive to validate tests for screening purposes by measuring their sensitivity and selectivity (Lave and Omenn 1986). Regardless of all the testing-related problems about to be discussed, it is worth bearing in mind that some countries have already banned the use of mammals for testing in certain situations, so there is an obligation to continue to refine in vitro approaches.
Alternative testing for developmental toxicity has a long history, encompassing regular international conferences (Ebert and Marois 1976; Kimmel et al. 1982; Schwetz 1993), comprehensive reviews (Brown and Freeman 1984; Faustman 1988; Welsch 1992; Brown et al. 1995), and much debate in print (Mirkes 1996;
Daston 1996). Alternative tests for development toxicity are not currently used by any regulatory agency.
Intrinsic Limitations
Alternatives to in vivo testing will never detect all the developmental toxicants that have actions in pregnant mammals. This is true for several reasons. First, some toxicants initiate their effects outside the embryo and in the maternal or placental compartments. Second, some effects are mediated by physiological changes only represented within the intact embryo (e.g., peripheral vascular perfusion). Third, known mechanisms of developmental toxicity are diverse, so it is unlikely that all targets will be present in a simple system. Fourth, some adverse outcomes are only observable as functional impairment postnatally. Finally, most alternative systems are static and have neither the dynamic changes in concentration associated with physiological disposition in vivo nor the metabolic transformation of the test agent.
Validation
Validation is complex and includes protocol standardization, interlaboratory consistency, and statistical prediction models, but the fundamental question remains: how well does the system mimic the susceptibility of human development? This has yet to be answered for any system, and there are a number of problems that are discussed below.
In Vitro Test for What Outcome? The type of adverse outcome induced by a chemical in vivo often varies between individuals, across species, and sometimes with routes or schedules of administration. Thus, although the initial aim of alternative tests was to predict the overall induction of congenital malformations, it is more appropriate to consider that in vitro tests can help to predict specified developmental toxicity and to identify potential mechanisms of disruption to particular cell-signaling and genetic regulatory pathways.
General Versus Specific Toxicity. Presumably, all chemicals would disturb development, if a high enough concentration were delivered to the embryo, even though such concentrations might be unattainable in mammals because of maternal toxicity. However, chemicals vary widely in their intrinsic hazard to development. For example, high-affinity ligands for some nuclear-hormone receptors cause irreversible developmental defects (see Chapter 4). It would be helpful to be able to discriminate such chemicals from those that affect development (D) only at exposures that are simultaneously toxic to the adult (A). The A/D ratio (the ratio of adult toxic dose to developmentally toxic dose) attempts to measure that specificity. However, use of that value has been tempered by the demonstra-
tion that A/D ratios are not necessarily consistent across species (Daston et al. 1991).
Which In Vivo Database? The database on humans is probably too heterogeneous to use for validation studies. For example, it is biased toward pharmaceuticals, and the exposure range is too small for most chemicals, so chemicals with reliable negative results are hard to identify. By default then, comparisons have been made with experimental mammal testing data. The information in this database is also heterogeneous in exposure times, routes, and doses; species; end points; and adverse outcomes. To avoid some of these problems but retain the use of existing data, perhaps the only option is to use data exclusively from orthodox segment II type tests in which animals are exposed during the period of major organogenesis. This approach eliminates many of the chemicals historically used in validation studies, because they have never been formally tested in vivo.
Chemicals for Validation. Much effort has been expended on the analysis of in vivo animal test data to produce a list of chemicals for use in validation studies. A prototype list produced by Smith et al. (1983) was subsequently found to be inadequate and an expert committee was set up to address that issue (Schwetz 1993). Because of the difficulty of the task, that committee was not able to complete its task. There has been considerable disagreement over what is and what is not developmentally toxic in vivo and over the severity of that action. There is currently no consensus on how to categorize, stratify, or quantify the developmental toxicity of chemicals. Most validation studies have used a binary classification: developmental toxicants or nontoxicants (Parsons et al. 1990; Uphill et al. 1990). This is a gross oversimplification of the richness of information available. More recently, chemicals have often been grouped informally into three or four categories: (1) toxic to development in all species, no maternal toxicity; (2) toxic to development in some species, no maternal toxicity; (3) toxic to development in some species, some maternal toxicity; (4) no evidence for developmental toxicity in any species tested. However, without formal definition of categories and consistent in vivo testing, there is disagreement in assigning chemicals to such groups (Wise et al. 1990; Daston et al. 1995; Newall and Beedles 1996; Spielmann et al. 1997). Many validation studies have been biased by the inherent toxicity inequality of chemicals selected (Brown 1987). It has been common to select chemicals of potent and general biological activity, such as antimetabolites, nucleotide or nucleoside analogs, and alkylating agents, as developmental toxicants. In contrast, the chosen nondevelopmental toxicants have frequently been endogenous intermediary biochemicals, such as acetate, glutamate, and lysine, or chemicals specifically designed to be nontoxic to mammalian cells, such as antibiotics, saccharin, and cyclamate. It comes as no surprise that developmental models respond differently to two such disparate groups. The proper strategy should be to select chemicals that are largely similar in their gen-
eral toxicity and potency, but different in specific developmental hazard (Schwetz and Harris 1993). This has never been achieved.
An additional problem in categorizing chemicals, even those tested according to standard protocols, is that toxicokinetics and metabolism are rarely investigated sufficiently to indicate whether a negative outcome in vivo is a reflection of a true lack of inherent developmental toxicity potential or a low embryonic exposure. This outcome can lead to a situation in which a chemical is correctly identified as a potential developmental toxicant from an in vitro test, but the effective exposure can never be achieved in vivo.
Existing and Extinct Alternative Tests for Primary Screening
Because there are no known common mechanisms of developmental toxicity on which to base a design for a primary screening test, three other approaches have been taken. These are the use of (1) mammalian embryos or parts of embryos in culture, (2) free-living nonmammalian embryos, and (3) cell cultures in which processes thought to be required for normal development are assayed (e.g., proliferation, adhesion, communication, and differentiation). More than 30 test systems have been devised and preliminarily assessed (see Table 3-1). All those test systems that use embryos monitor gross morphological end points. Few tests are actively used for screening purposes (Brown et al. 1995). Rodent embryo culture, micromass, and stem-cell assays are currently being validated in a European Union-sponsored trial (Spielmann et al. 1998). The validation of the frog embryo teratogenesis assay in Xenopus (FETAX) is being reviewed by the U.S. National Toxicology Program Interagency Center for the Evaluation of Alternative Toxicological Methods (NIEHS 1997; Fort et al. 1998).
Rather than having been eliminated by objective criteria, most other systems were simply not adopted by scientists and were not pursued by their originators. For example, there have been no studies comparing several systems for relative performance or using more sophisticated molecular end points. A few systems have been eliminated by poor performance. The mouse ovarian tumor (MOT) cell-attachment method and the human embryonic palatal mesenchymal (HEPM) cell-proliferation method were simultaneously assessed by the U.S. National Toxicology Program (Steele at al. 1988) and shown to have a combined specificity of only 50%. The hydra assay is novel in having been designed specifically to estimate the A/D ratio. Although transiently popular, usage diminished with the demonstration that the A/D ratio is not consistent across species (Daston et al. 1991) and with other concerns about comparability with mammalian responses.
New Receptor-Based Tests
Endocrine disruptions by chemicals are beyond the scope of this report but are relevant in terms of the overlap in receptors involved and in the in vitro ap-
TABLE 3-1 Systems Proposed as Alternatives to Pregnant Mammals to Test for Developmental Toxicitya
proaches being pursued. Interference with estrogen, androgen, glucocorticoid, or thyroxine receptor function can result in developmental and endocrine toxicities. Major efforts are under way to devise screening methods to assess interference with those receptors (EPA 1998b). This task is complex, because chemicals could be agonists, partial agonists, antagonists, or negative antagonists (Limbird and Taylor 1998) or interact with other steps in the pathway. Caution is needed, therefore, in extrapolating from simple tests. Nevertheless, a variety of tests have been devised to assess receptor binding, activation of response elements, and cellular responses (e.g., proliferation). Similar approaches could be devised for other signaling pathway receptors involved in developmental toxicity.
Animal Bioassays
In vivo animal bioassays are a critical component in human health risk assessment. A basic underlying premise of risk assessment is that mammalian animal bioassays are predictive of potential adverse human health impacts. This assumption, and the assumption that humans are the most sensitive mammalian species, have served as the basis for human health risk assessment.
Several study protocols to test for developmental toxicity in animals are accepted and used by regulatory agencies such as EPA and FDA. Describing the various protocols goes beyond the scope of this report and the reader is referred to the original guidelines (EPA 1991, 1996a, 1998c,d,e; FDA 1994; OECD 1998) for detailed descriptions. T.F. Collins et al. (1998) contains a discussion and a comparison of EPA, FDA, and OECD guidelines.
Information obtained from in vivo bioassays includes the identification of potentially sensitive target organ systems; maternal toxicity; embryonic and fetal lethality; specific types of malformations including gross, visceral, and skeletal malformations; and altered birth weight and growth retardation. These assays can also provide information on reproductive effects, multigenerational effects, and prenatal and postnatal function. In vivo bioassays determine critical effects that are used for quantitative assessments by taking the no-observed-adverse-effect level (NOAEL) for the most sensitive effects.
The focus of animal bioassays primarily has been toxicity assessment, including hazard identification and dose-response assessment. The aim of such studies is to identify qualitatively what spectrum of effects a test chemical can produce and to put those effects in the context of dose-response relationships. Because there is uncertainty in extrapolation from animal studies to humans, several assumptions are made, including the following: (1) an agent that causes an adverse developmental effect in experimental animals might cause an effect in humans; (2) all end points (i.e., death, structural abnormalities, growth alterations, and functional deficits) of developmental toxicity are of potential concern; and (3) specific types of developmental effects observed in experimental animals might not be manifested in the exact same manner as those observed in humans.
Much of the literature before 1975 concerning studies of in-utero-induced adverse developmental outcome is troubled by small sample sizes, inappropriate routes and modes of exposure, inconsistent methodology, and excessively high dose or concentration exposures. Many of those deficiencies have been corrected by the regulatory mandate of adhering to Good Laboratory Practices (OECD 1987; FDA 1987; EPA 1990).
Several studies of concordance between the perturbed developmental outcomes in experimental animal studies and the human clinical experience have been made (Nisbet and Karch 1983; Kimmel et al. 1984; Francis et al. 1990; Hemminki and Vineis 1985; Newman et al. 1993). The most rigorous and earliest of those was done in the early 1980s and is contained in a technical report for the National Center for Toxicological Research (NCTR) (C.A. Kimmel, EPA, unpublished report, 1984).1 In general, these studies concluded that there is concordance of developmental effects between animals and humans and that humans are as sensitive or more sensitive than the most sensitive animal species.
The NCTR study was notable because it employed criteria of acceptance for both human and experimental animal reports that included study design and statistical power considerations. Additionally, the authors held to the premise that adverse developmental effects represented a continuum of responses—or at least a number of interrelated effects—including in utero growth retardation, death of the products of conception, frank malformations, and functional deficits that manifest themselves in later stages in life. Hence, an effect on any one of these end points in experimental animals or human studies was considered a basis for concordance. Concordance did not require an exact mimicry of response among species. This was not required because exposure conditions (e.g., timing and duration of exposure and toxicokinetic differences) and tissue sensitivity (e.g., toxicodynamic differences) could differ enough between experimental animals and humans to result in a different type of effect.
Many different agents—mostly chemical agents but also physical agents—have been evaluated to determine their capacity to produce developmental toxicity in experimental animal models, such as the rat, mouse, and rabbit. Most of those studies have been conducted by private industry and federal government-funded research programs and involved test agents that had not yet entered the market. Schwetz and Harris (1993) provide a good review of 50 chemicals that the National Toxicology Program has evaluated for developmental toxicity using rodent bioassays.
As discussed in Chapter 2, humans were never exposed to many of the materials that have been evaluated in rodent bioassays and that have been shown to affect animal prenatal development adversely. Thus, it will never be known
whether comparable adverse effects would have been caused had similar human exposure occurred. Summary compilations from published data covering more than 4,000 different entities of exposure conditions indicate that more than 1,200 agents, predominantly chemical agents, have produced adverse developmental outcomes by the end point criteria stated above, often including congenital anomalies, in one or more species of experimental animals (Shepard 1998). Among this large number are about 50 agents (almost exclusively designated as drugs) that are known to cause adverse developmental effects in human beings. For most of the agents that were evaluated for developmental hazard potential in experimental animals, human exposures will never occur. Thus, public health was protected, but ascertainment of concordance of animal and human responses was undetermined for those agents. When exposures occur, rarely have human assessments been sufficient for definitive evaluation and establishment of cause-and-effect associations. Because of the background incidence of human developmental abnormalities (addressed in Chapter 2) and the difficulties in conducting epidemiology studies, such associations are extremely difficult to establish unless the outcome is unusual and striking, as was the case of thalidomide.
Among industrial chemicals and environmental contaminants that have been studied in pregnant animal models, often the estimated maximum tolerated dose (MTD) was repeatedly given in conformance with the testing guidelines. Internationally, regulatory authorities require in many instances that the MTD, even up to maternally toxic concentrations, be administered to ensure that no developmental toxicity occurs. Therefore, the underlying principle is that, if regulatory standards are set to protect against maternal toxicity, no adverse effects will occur in offspring. Unfortunately, all too frequently the focus of developmental toxicity testing has been to study the effects of an agent only at high doses that are most likely irrelevant to environmental and occupational exposures. For industrial and environmental chemicals, the dosing regimens at or even above MTDs, as applied in hazard identification studies, typically contrast sharply with anticipated human exposures that are commonly much lower in extent or magnitude, often uncertain, or even entirely unknown.
Because of the design of developmental hazard identification studies, the overwhelming majority of the more than 1,200 agents found to elicit adverse developmental outcomes in experimental animals were tested at doses many times higher than anticipated human exposures during pregnancy and have often elicited extreme maternal toxicity. Furthermore, exposure of the pregnant animals was sustained throughout all of organogenesis by daily repeated administrations, and minimal or no regard was taken for toxicokinetic considerations (see toxicokinetics section of this chapter for details).
Therefore, there are problems associated with the application of these assays for assessing human developmental toxicity potential. Repeated administration of an MTD might produce adverse results that are not indicative of risk from ambient exposure concentrations or intermittent exposures. It is a continuing
challenge for test design and interpretation to minimize the problems noted above and, thereby, improve the predictiveness of laboratory animal toxicology protocols.
Epidemiology
Four approaches have traditionally been used to evaluate human developmental toxicity: (1) case series, (2) randomized controlled trials, (3) cohort studies, and (4) case-control studies.
Case series comprise an important first step in assessing relationships between exposures and adverse pregnancy outcomes. Many developmental toxicants are first recognized by astute clinicians who correlate specific patterns of developmental defects or developmental disabilities with specific exposures during pregnancy. Most notable among agents first identified this way as causing developmental defects are rubella and thalidomide (Gregg 1941; Lenz and Knapp 1962; for a review, see Rosa 1992). Case series can be useful when the outcome is distinctive, the exposed population is large enough that numerous cases are recognized, and the dose and timing are well described. Case series should be interpreted with caution, however, because the association can be due entirely to chance. They rarely permit identification of a causal link between exposure and outcome due to their anecdotal nature and the high background of adverse pregnancy outcomes in humans (see Table 2-1). Their greatest value is in the generation of hypotheses for further investigation.
Randomized controlled trials are the most widely accepted type of epidemiological study for assessing the relationship between an intervention and an outcome. Subjects are enrolled into a randomized trial based on pre-established criteria. They are randomly assigned to a reference group (placebo or alternate treatment) or a test group and administered a test agent under controlled conditions. Because the agent of interest is deliberately administered, this type of study is not appropriate for assessing risk of chemical exposures on pregnancy, or of adverse effects of chemicals in general. Randomized control trials have their widest use in tests of the efficacy of pharmaceuticals and other medical interventions.
Cohort studies are observational epidemiological studies in which individuals are assigned to groups (cohorts) on the basis of pre-existing exposure status and are followed to determine pregnancy outcome. The cohort study approach is limited to the investigation of few exposures, but allows for the assessment of numerous developmental endpoints. Considering the rarity of congenital anomalies, large studies are needed to detect differences between cohorts. For example, spina bifida occurs in 0.1% of most American populations and might not be detected even in a cohort of 1,000 pregnancies. Even though cohort studies allow for the next best determination of a causal association (after randomized controlled trials), they are often not practical. An additional problem is that some
individuals from the cohort will no longer be available for follow-up of the pregnancy outcome. Postmarketing surveillance by the pharmaceutical industry can be viewed as a type of cohort study, although an unexposed cohort is often not studied concurrently.
The case-control study is the most common design used in assessing the association between exposure and pregnancy outcome. In this type of study, the pregnancy outcome is identified (usually congenital anomalies in live-born infants), and a retrospective evaluation is then conducted to determine the exposure pattern. In case-control studies, the number of developmental end points that can be assessed is small, but several exposures can be investigated. The case-control study is the most efficient study design for capturing rare events, such as congenital anomalies. Accurate ascertainment of exposure can be problematic for case-control studies. Recall bias can occur among women who deliver abnormal infants (i.e., exposures are recalled more extensively by women with abnormal infants than by those with normal infants). Selection of an appropriate control group, which ideally is identical to the case group except for the outcome of interest, can also be difficult.
There is no formula whereby a causal relationship can be established between an exposure and an adverse pregnancy outcome. Results from epidemiological studies should be interpreted with caution because associations found can be due to the following:
-
Unmeasured confounding, particularly confounding by indication.
-
Exposure misclassification (inability to pinpoint relevant dose and timing).
-
Outcome misclassification (related to the heterogeneity of birth defects).
-
Biological interactions (subgroups with differing genetic susceptibilities or presence of additional exposures).
-
Differential prenatal survival (in studies evaluating live-born infants, spontaneous abortion or elective termination of abnormal fetuses should be taken into account).
Evidence from a number of sources, including human and experimental animal data, must be collectively considered to determine the strength of the association (Rothman 1986; Khoury et al. 1992).
There are many problems in identifying associations between exposures and adverse pregnancy outcomes using conventional epidemiological approaches. Weak or moderate associations (relative risks or odds ratios ranging from 1 to 3) are typically found between environmental exposures and pregnancy outcomes (Khoury et al. 1992). For example, maternal smoking is weakly associated with oral clefts (odds ratios between 1 and 2) (Khoury et al. 1989). Insulin-dependent diabetes is associated somewhat more strongly with major malformations (a relative risk of 7) (Becerra et al. 1990), and potent developmental toxicants, such as isotretinoin and thalidomide, are very strongly associated with major malforma-
tions (relative risks in the range of 25 and 300-400, respectively) (Lenz and Knapp 1962; Lammer et al. 1985). Although conventional epidemiological studies have been useful in quantifying the magnitude of risk produced by those potent agents, they were first identified as human developmental toxicants through case reports. Conventional epidemiological studies can be influenced sufficiently with biases, uncertainties, and methodological weaknesses that they may not be useful to detect accurately and assign significance to weak associations—those with relative risks in the range of 1 to 3, the range in which many environmental toxicants can be expected to act (Taubes 1995). In the context of risk assessment, such methodological limits mean that a 2- or 3-fold increase of risk, which amounts to a major health problem, would go undetected.
Another concern with conventional epidemiological studies on chemicals is that studies frequently rely on occupationally exposed cohorts under conditions in which exposure patterns are higher and potentially more consistent than environmental exposures of the general public.
Another potential complication in interpreting data from conventional epidemiological studies is that the complexities and variabilities of human activities, such as life-style factors and diet, cannot be controlled in human studies in the same manner as animal studies. Thus, interpretation of epidemiological study results requires sophisticated experimental designs and analyses to ascertain true relationships.
The ability of an epidemiological study to identify chemically related effects is dependent on the size of the study population, the variability of population effects, the study design, and the background incidence of the adverse health effect being studied. Such information is especially important for risk assessors when they are evaluating epidemiological studies with widely varying results. Understanding how much power a study with negative results has to detect an adverse outcome strengthens the utility of these studies for risk assessments.
Dose-Response Assessment
As part of the evaluation of dose-response relationships, a quantitative evaluation is conducted (EPA 1991; Moore et al. 1995). Doses or concentrations are identified that have no or minimal associated adverse developmental effects. A NOAEL or a lowest-observed-adverse-effect level (LOAEL) is chosen from one of the experimental doses or concentrations tested. These levels are identified for each human and experimental animal study and manifestation of developmental toxicity (i.e., death, structural abnormalities, growth alterations, and functional deficits). Using the NOAEL or other most sensitive effect levels (i.e., end points adversely affected at the lowest doses tested), the reference dose (RfD) or reference concentration (RfC) is determined. These values are an estimate of a daily exposure to the human population that is assumed to be without appreciable risk of adverse developmental effects (EPA 1991).
An acceptable daily intake (ADI) can also be determined from a NOAEL. ADI values are used for pesticides and food additives to define the daily intake of chemicals, which appear to be without appreciable risk of harm during an entire lifetime.
In an alternative approach, a specific effect dose or concentration, such as the ED05 (EC05) or ED10 (EC10) (the best estimate of the dose at a 5% or a 10% level of response) is determined for the dose-response curve based on rodent or human epidemiological studies (Crump 1984; Allen et al. 1994 a,b). That dose, which, unlike the NOAEL or LOAEL, does not have to be one of the experimental doses, represents the dose that results in a 5% or 10% response in the study population. The dose is determined from the experimental results by using a dose-response curve fitting program. Studies have confirmed that these levels of response (5-10%) represent the minimal level of effect that can statistically be resolved in a relatively robust bioassay (with the current design for detecting developmental toxicants). The benchmark dose (BMD) is frequently calculated using the lower confidence limit of the dose that results in a 5-10% response and thus represents with 95% confidence the lowest dose giving an increased 5% or 10% response in exposed populations over unexposed populations. Continuous responses from developmental toxicity bioassays include percentage of fetuses malformed, percentage of litters having one or more malformed fetuses, and birth weights (Kavlock et al. 1995).
RfDs and ADI values are derived from NOAELs or BMDs by dividing by uncertainty factors. Uncertainty factors, which are derived from animal and human data, generally involve dividing by a default value of 10 to account for uncertainties (EPA 1991). Uncertainty factors for developmental effects are applied to the NOAEL or BMD to include a 10-fold factor for interspecies extrapolation and a 10-fold factor for intraspecies variation. Additional 10-fold factors might be used to account for insufficiency in the database, extrapolation from subchronic to chronic exposures, and extrapolation from a LOAEL to a NOAEL. In practice, the aggregate product of all the default values is most often a factor of 100 to 1,000 (i.e., the acceptable human exposure concentration is 100- to 1,000-fold lower than the dose in the animal study that had little or no observable developmental effects). Because of increasing concern for susceptibility of children, the Food Quality Protection Act (Public Law 104-170; August 3, 1996) specifies an additional 10-fold default factor be applied under specific conditions. The default values are used unless there are research results that support the use of a different value. The need for uncertainty factors could be reduced with better data on comparative toxicokinetics, susceptible populations, and mechanisms of action. For example, if a NOAEL in a rat developmental toxicity study is used to set a RfD, then, in general, the NOAEL would be divided by 10 to account for extrapolation from animal studies to humans (interspecies extrapolation) and by another 10-fold factor to account for sensitive versus average human responses (intraspecies differences). Modifying factors can be used to change these default
values of 10 and have been used to decrease the 10-fold factor used for interspecies extrapolation when there is sufficient knowledge about the similarities in toxicant kinetics in both rodents and humans (Moore et al. 1995).
Pharmaceutical agents almost always have a smaller difference between therapeutic and toxic dosages than is considered safe for environmental agents. A narrow therapeutic index is considered acceptable because pharmaceutical agents are given under the guidance of a health professional and because the therapeutic benefit has been determined to outweigh the risk. That is not the situation for agents such as environmental contaminants and food additives; hence, RfDs and ADIs are more conservative. It is worth stressing that the default uncertainty factors can be superceded when relevant exposure data are available to address key uncertainties. For example, the residual amounts of ethanol present in fruit juices, yeast breads, or vanilla ice cream are within a factor of 100-1,000 of the amount of alcohol (taken in alcoholic beverages) that results in fetal alcohol syndrome but do not constitute a risk to the fetus.
Renwick (1998) has discussed the concept of viewing uncertainty as being composed of kinetic and dynamic components and has proposed the concept of reducing the uncertainty associated with either or both of those components with additional mechanistic information. This structure has provided an initial framework by which mechanistic information can be used in the current risk assessment approaches. Chapters that follow will show how important new information on species differences and data on human variability can replace our reliance on such default approaches for developmental toxicity risk assessment.
Exposure Assessment
Exposure assessments are a critical component of the risk characterization process. Ideally, for developmental toxicity, one would like to have information about how much of the critical reactive species of the test chemical is present at the biological target throughout gestation. At the molecular level, that would mean knowing how much of and how long the toxic compound is bound to a specific target receptor. At the organism level, that would mean knowing how exposure occurred, when it occurred, how much of a compound was absorbed, what type of metabolism of the compound took place within the maternal compartment, and how that metabolism affected the exposure of the conceptus compartment. Information on how the conceptus metabolized and eliminated the compounds would also be important to know. Thus, an understanding of both time of exposure and dose is particularly important for characterizing the potential impacts of developmental toxicants. Numerous studies have shown dramatically different dose-response relationships when exposures occur even 8-12 hr apart due to the significant temporal differences in tissue susceptibility. Temporal differences make exposure assessments for developmental toxicants one of the most challenging of all exposure assessments. Although general toxicokinetic
models exist for pregnancy, a refined exposure-modeling tool is not available for most chemicals. The lack of that tool is probably a key factor behind efforts to use exposure biomarkers and direct tissue measurements to improve estimates of conceptus exposures. Biomarkers of exposure have proved to be especially useful in addressing the limitations of exposure data in epidemiological assessments. Such biomarkers allow for a more accurate and representative exposure assessment and, when linked temporarily with biomarkers of effect, can frequently enhance the ability of a human epidemiological study to estimate human risk. New advances in biomarkers of susceptibility are allowing investigators to understand more fully variability in human response and have been proposed as improvements for human risk characterization.
One approach for using exposure information is to calculate a margin of exposure (MOE). The MOE is a ratio of the dose judged to be without effect to the anticipated levels of human exposure (Moore et al. 1995). However, because the calculation does not include the use of any default values to account for sources of uncertainty, it can only give a general indication of different levels of effects versus exposures levels for a quick exposure-scenario comparison.
The challenge for human health risk assessment is to convert exposure assessment information into relevant information for humans. The subsequent section on toxicokinetics will expand upon these concepts and will explain what information is needed to conduct relevant human exposure assessments.
Toxicokinetics
Toxicokinetics is the description of the absorption, distribution, metabolism, and excretion of a toxic chemical into and from the body (commonly referred to as ADME). The importance of chemical toxicokinetics for risk assessment is demonstrated by several example documents (California Environmental Protection Agency 1991; Moore et al. 1995; EPA 1996a; O’Flaherty 1997). These combined documents show a consensus that toxicokinetic data provide key elements to understanding species differences in response to developmental toxicants. Figure 3-2 shows an illustration of how exposure to a compound is then evaluated by using ADME. ADME controls how much of, when, and in what form a toxicant comes in contact with target organs. For developmental toxicants, these key questions are related to the amount and form of the toxicant that reaches tissues of the conceptus. Such knowledge can reduce the uncertainty in the extrapolation of results collected from experimental animals for the prediction of hazard associated with exposure of pregnant women. This section provides a brief discussion of a number of issues that require further investigation, as work continues to improve the scientific basis for risk assessment.
Decisions about toxicity hazard and risk to human development based on toxicokinetics from pregnant animals can rarely provide an unequivocal answer
of yes or no. The use of retinoid creams for dermal application can serve as an example. Retinoids were already known to cause developmental toxicity in pregnant animals of every test species examined. The regulatory decision concerning retinoid creams for dermal application was based on barely detectable changes in the blood concentrations of endogenous retinoids after dermal application of the drug. However, there is still minimal marketing of retinoid creams for dermal application. A similar case has been noted for vitamin A pills (retinyl palmitate) administered at doses greater than 30,000 IU per day (R.K. Miller et al. 1998).
If data show that a chemical is absorbed into the systemic circulation, then the next most important piece of toxicokinetic information is a determination of whether the biologically active toxicant is the parent compound, a metabolite, or both. Without such knowledge, the usefulness of other toxicokinetic data is diminished. If the active toxicant is not clearly delineated, then the qualitative and quantitative metabolic patterns that often vary between species for the agent of interest cannot be constructively applied in the characterization of hazard and the management of potential risks.
A determination of species concordance in susceptibility, in both basic research studies and developmental toxicity hazard assessment testing of chemical agents, is often the key element that provides the foundation for a generalization of the findings and extrapolations relevant for humans. Toxicokinetics are therefore studied to compare absorption, distribution, metabolism, and elimination of the test agent and its relevant metabolites. Toxicokinetic measurements can be used to determine the internal dose delivered to target tissues rather than relying on the administered dose, thereby taking into account species differences and individual variations in the extent and duration of systemic exposure in maternal and conceptus compartments. These interspecies variations and the interindividual differences in the same species indicate that each individual has a specific “fingerprint” of unique alleles of genes encoding drug-metabolizing enzymes (DMEs) as well as receptor and transcription factors that regulate the expression of genes encoding those DMEs. All DMEs appear to have endogenous substrates and are used in the biological functions of the normal animal or human, yet these enzymes have specificities that are sufficiently broad to metabolize endogenous substrates and environmental agents (Nebert 1994). Extrapolating toxicokinetic results from laboratory animals to nonhuman primates and humans is further complicated by the fact that the DMEs in the embryo and fetus differ considerably from one another with species-specific temporal patterns of expression observed throughout gestation and postnatal periods (Miller et al. 1996; Cresteil 1998). Such differences can allow drug-metabolizing reactions to occur in primates that are not yet functional in the common laboratory animal conceptus. The significance of such differences is magnified when considered with the differences in the susceptibility between the conceptus and adult tissues.
Few studies have been conducted in pregnant animals that have compared species-specific toxicokinetics. The data collected make it apparent that inter-
species differences in susceptibility to developmentally toxic effects can frequently be due to differences in absorption, fate, or elimination of the agent of interest rather than to fundamental species-specific differences in biological response. Examples of embryotoxic drugs that have undergone detailed comparative evaluations are valproic acid (Nau 1986) and retinoids (Kraft et al. 1993; Kraft and Juchau 1993; Nau et al. 1994).
Regulatory authorities have required that the highest dose administered in animal toxicity studies in support of product registration be the estimated MTD. The administration of the MTD serves to maximize the likelihood of manifestation of a biological and possibly adverse response and ensure detection of all inherent toxicities. However, very large doses of environmental agents might be required in animals to reach the MTD, compared with anticipated human exposures at low concentrations. Such high doses of a test agent in animals can result in different kinetic and dynamic processes than those occurring at lower, more environmentally relevant exposures. For example, high doses can saturate elimination or repair processes or stimulate cell division or apoptosis, which might result in grossly exaggerated target organ concentrations and manifestations of toxicity. Indeed, toxicokinetic studies conducted in past decades have elucidated those phenomena for numerous therapeutic and environmental agents. The insights gained have led to the simplistic subdivision of linear and nonlinear classification of kinetics. Toxicokinetic data are essential to ascertain whether similar intervals or concentrations of the chemical or its metabolites result from different doses. High doses often result in nonlinear kinetics and subsequently elicit toxic effects that are not observed at low doses associated with linear toxicokinetics. Consideration of these kinetic and dose-response relationships is needed as new information is evaluated for use in risk assessment.
Toxicokinetic considerations are not only important in the interpretation of potential health effects and their relevance across species but they are also important in the determination of a developmental toxicity study design. For example, in one type of conventional developmental toxicology study design, studies initiate dosing at the beginning of organogenesis of the chosen test species. The study design might be flawed for compounds that have a long half-life, because steady-state concentrations are reached only after dosing for approximately four half-lives. Thus, a compound that has a half-life of 24 hr will not reach steady-state concentrations until 4 days after four consecutive daily administrations. Such a toxicokinetic property might miss the window of susceptibility to chemical perturbation of a specific developmental process in a long half-life test species. In animals with short gestation durations, such as the mouse or rat, the embryo might reach a developmental stage of decreased teratogenic sensitivity by the time the toxicologically critical steady-state concentration is achieved. For some agents, it may be possible to overcome this problem by starting exposure of the dam earlier in pregnancy. Toxicokinetic information can thus aid in the proper design of new studies or more accurately interpret results from inves-
tigations already completed. In this context, the committee needs to point out some serious shortcomings in the present approaches to interspecies extrapolations of toxicokinetic information, particularly those extrapolations that make the jump from laboratory animals to pregnant women. All too often, toxicokinetic data from pregnant animals are collected only for the maternal organism, and equally important aspects of the conceptus compartments are entirely lacking. As the examples of some chemicals studied in more detail have shown, the maternal-conceptus kinetics of chemicals and drugs change dynamically throughout gestation. Pharmacokinetic measurements in human pregnancy related to therapeutically used pharmaceutical agents are typically derived from blood and tissue samples collected from term deliveries and constitute only one or just a few time points after the drug’s administration. Many significant changes occur throughout gestation that can make such term kinetic assessments less directly applicable for evaluation of first-trimester exposures.
The present dosing regimens in safety evaluations do not always consider the profound differences in elimination half-lives of chemicals between animal test species and humans (Nau 1986). The studies on valproic acid revealed dramatic species differences in the toxicokinetics of this drug that correlate with the species-specific teratogenic response (Nau 1986). Technical means of dosing that overcome the toxicokinetic differences between humans and pregnant animals exist and have been shown to be applicable and useful. For example, the studies on valproic acid were conducted by subcutaneously implanting osmotic mini-pumps that can deliver a chemical at a constant rate and produce maternal serum pharmacokinetic profiles with concentrations of the test chemical that resemble those occurring in humans much more closely than single or even repeated bolus administrations (Nau et al. 1981, 1985). However, conventional developmental toxicity testing designs still do not use that methodology routinely. It was critical for the committee to consider these factors in their deliberations on how to use new biological information for human risk assessment. Toward that end, it will be helpful to understand whether a developmental toxicant acts by exceeding a certain threshold peak concentration (Cmax) for a brief period of time, or whether an extended exposure to a certain concentration of the chemical over some period of time, is required to induce abnormal development. Such toxicokinetic relationships are graphically expressed as a plot of the concentration of the chemical of concern in maternal plasma (and preferably also in the embryo) against time. This visual display of the chemical-analytical presence of the substance and time is commonly known as area under the curve (AUC), as shown in Figure 3-3. In developmental toxicity studies, both the Cmax and the AUC concepts have been all too uncritically applied in assessing the value of toxicokinetics in pregnancy. The maternal AUC has often been used to draw conclusions about the chemical exposure of the conceptus without the availability of any toxicokinetic information from the conceptus compartments. The assessment of conceptus toxicant levels can be invaluable for understanding animal species differences in develop-
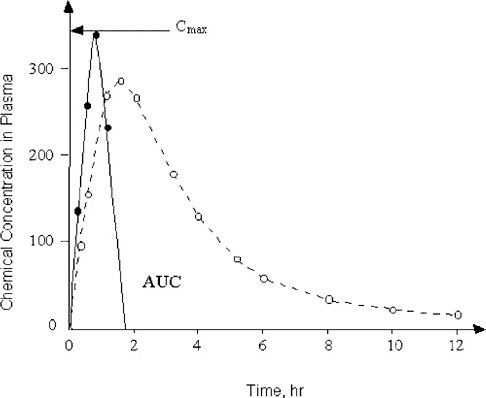
FIGURE 3-3 Two chemicals with different toxicokinetic properties are schematically illustrated. The concentration in maternal plasma of Chemical 1 (solid line) rises rapidly to reach its maximum (Cmax). Chemical 1 is then eliminated from the blood-plasma compartment in less than 2 hr after administration. In contrast, the plasma concentration of Chemical 2 (dashed line) rises more gradually and is slowly cleared from maternal plasma in an apparently biphasic fashion. The area under the curve (AUC) is defined by the plot of concentration of the chemical against time after administration.
mental toxicity and for assessing the validity of negative in vivo studies for compounds otherwise suspected to have high potential for developmental toxicity.
One chemical for which toxicokinetic data have been collected from maternal and conceptus compartments at two stages of pregnancy is 2-methoxyacetic acid, the proximate developmental toxicant derived from the maternal oxidation of 2-methoxyethanol, an ethylene glycol ether used as an industrial solvent. This chemical produces gross malformations in several test animal species examined, including nonhuman primates. Depending on the developmental age of an embryo at the time of exposure to sufficiently high concentrations of 2-methoxyacetic acid, the target tissues are either the developing anterior neuropore or
the differentiating paw skeleton of the limbs, and exposure causes exencephaly or digit malformations, respectively (Terry et al. 1994). In the case of 2-methoxyethanol, the maternal plasma AUC of 2-methoxyacetic acid was highly indicative of that in the embryo and might serve as a surrogate of separate conceptus toxicokinetic measurements (Welsch et al. 1995, 1996).
Toxicokinetic information could be helpful in judging the extent of the hazard to humans from exposures if human kinetics are known (Yacobi et al. 1993). For example, the anticonvulsant drug valproic acid given to pregnant mice induces exencephaly in their embryos when a certain maternal plasma threshold concentration is surpassed for a very brief duration (Nau 1986). Larger total exposure over time (larger AUC) achieved by constant maternal drug infusion causes a dramatically lower incidence of exencephaly, indicating that the peak concentration (Cmax) rather than total exposure over time (AUC) induces the teratogenic response in mice. In contrast, clinical use of valproic acid for antiepileptic therapy requires the maintenance of valproic acid concentrations in an effective therapeutic range at which the required human doses produce serum Cmax values that are 6-10-fold lower than the teratogenic concentrations in mice (Nau 1986). A similar inference regarding Cmax as a cause of embryotoxic effects was made for caffeine in mice. A large single dose (100 mg/kg) induced a teratogenic response, whereas the same total amount divided into four separate administrations did not cause any malformations (Sullivan et al. 1987).
The embryotoxicity of other agents appears to depend on the total exposure over time (AUC). For example, the developmental toxicity of all-trans retinoic acid and cyclophosphamide (a chemotherapeutic alkylating agent) in the rat correlates best with duration of exposure (Tzimas et al. 1997; Reiners et al. 1987).
Caution in the interpretation of maternal AUC information without concomitant conceptus toxicokinetics is necessary because a single agent might act through both toxicokinetic exposure patterns, depending on the stage of development. 2-Methoxyacetic acid seems to induce mouse digit malformations best correlated with maternal and conceptus AUC (Clarke et al. 1992, 1993; Welsch et al. 1995, 1996). However, additional toxicokinetic data from both the maternal and the conceptus compartments at an earlier stage of mouse embryogenesis indicate that the agent induces neural tube defects that correlate best with Cmax in the conceptus tissues (Terry et al. 1994; Welsch et al. 1996). What is still lacking in these data is information on the toxicodynamic interaction of 2-methoxyacetic acid with a specific and still unknown recognition site (receptor) in the conceptus. The significance of considering both AUC and Cmax measurements for developmental toxicity risk assessment is especially important because of known temporal differences in tissue susceptibility. In cancer risk assessment, Haber’s law (the product of concentration times time is equal to a constant) is used to normalize risk impacts. Such generalizing concepts cannot be applied in developmental toxicity risk assessment. A recent study by Weller et al. (1999) illustrated these differences for ethylene oxide developmental toxicity.
The toxicokinetic patterns that have been important in discriminating developmental toxicity are described here in terms of AUC and Cmax, and not in terms of metabolite profile (i.e., the qualitative similarities in a parent compound and its metabolites). Species are known to differ in the rates that they absorb, distribute, and excrete compounds (i.e., the metabolic rate manifested at AUC and Cmax). Pharmaceutical studies have demonstrated that metabolite profiles between species are often similar (Nau et al. 1994), and this similarity is one of the reasons that it is common practice to use various animal models to assess the potential toxicity of chemicals. The committee will later propose that human DME genes be introduced into model animals to further reduce differences in metabolism. These transgenic animals are likely to have similar metabolite profiles as humans but will be considerably different from humans in terms of metabolic rate.
In summary, the correct application of toxicokinetic information in the determination of hazard and in judgments concerning risk characterization requires a broad view of pharmacological, toxicological, and embryological principles. These principles have guided the committee in their considerations on how most effectively to incorporate recent advances in molecular and developmental biology in risk assessment.
BIOMARKERS
As the committee has outlined in the previous sections of this chapter, key challenges facing risk assessors include the need to understand critical initial events caused by toxicants (events that occur at low doses and early stages of toxicity) and to understand the implications of animal toxicity for human health. Ideally, appropriate biomarkers could serve as indicators to link exposure and early biological effects, and ultimately link those early effects with disease or pathogenesis. As numerous NRC reports have indicated, biomarkers of exposure, effects, and susceptibility are exactly the types of indicators that are needed to address these risk assessment challenges.
Specifically, biomarkers for developmental toxicity have been reviewed in the context of reproductive toxicology in a previous NRC (1989) report, Biologic Markers in Reproductive Toxicology. Three types of biomarkers have been defined (NRC 1989):
-
A biologic marker of exposure is an exogenous substance or its metabolite(s) or the product of an interaction between a foreign chemical and some target molecule or cell. The biomarker is measured in a compartment within an organism.
-
A biologic marker of effect represents a measurable biochemical, physiological, or other alteration within an organism that, depending on magnitude, can be recognized as causing an established or potential health impairment or disease.
-
A biologic marker of susceptibility is an indicator of an inherent or acquired limitation of an organism’s ability to respond to the challenge of exposure to a specific foreign chemical substance.
It is easy to see from those definitions that biomarkers of exposure and effect should be useful for linking early, low-dose exposures with pathogenesis and providing a platform for cross-species and cross-compound comparisons. Likewise, it is easy to see how biomarkers of susceptibility could be especially useful for assessing differences in temporal sensitivity and developing tissues and for cross-species and intraspecies comparisons.
The validity of a biomarker for risk assessment depends on a demonstration that it is highly associated with the outcome, such as in this context, a developmental defect. At present, few biomarkers meet the test. When evaluating biomarkers, one must investigate the mechanistic basis of the association between the biomarker and the adverse events and then determine the reliability of the comparison in a large and varied population for specificity, sensitivity, and reproducibility. A biomarker does not have to be the definitive end point for defining the problem, although that is preferable, but even having a tool to identify candidate individuals for more definitive testing can be helpful. For example, the maternal serum α-fetoprotein levels are useful in clinical screens for neural tube defects, but serum α-fetoprotein is not a definitive test for such defects.
Temporal considerations are important for using biomarkers for developmental toxicity. When considering the validity of a screening test, the gestational age at the time of assessment and, more important, the gestational age at the time of exposure to the toxicant must be considered. Such issues have growing importance as fetal therapeutic interventions are increasingly available for use (Miller 1991). Accessibility to the biological material of interest is temporally determined. For example, invasive (e.g., percutaneous umbilical blood sampling, PUBS) and noninvasive biomarker procedures (e.g., ultrasound and Doppler) for assessing the developmental state of the fetus have made possible the use of interventions that have revolutionized the clinical capabilities to treat the affected fetus. At the same time, physicians can now predict, based upon patterns of uterine blood flow, which pregnancies have a greater risk for a poor reproductive outcome (Jaffe 1998).
Biomarkers of Exposure
The pre-eminent example in this category is methylmercury (MeHg). Maternal hair concentrations, as well as blood concentrations, of MeHg correlate with adverse developmental outcomes in the children exposed in utero (Clarkson 1987). Different threshold exposures have been observed in the adult and fetus
for detrimental effects, based upon the hair analyses for MeHg. In fact, temporal records of MeHg exposure can be determined by measuring MeHg levels at various places in the human hair shaft.
Unfortunately, not all substances are comparable to MeHg in lending themselves to use as exposure biomarkers. For example, a debate continues concerning the dose of vitamin A, as retinyl esters versus retinol, that will produce malformations in humans. Of particular interest is the discussion about what dosages of vitamin A are needed to increase the blood concentrations of retinoic acid metabolites significantly above those seen in normal pregnant women. Doses of 30,000 international units of retinyl palmitate per day administered orally did not significantly increase the concentrations of retinoic acid in nonpregnant women above those concentrations already circulating in untreated pregnant women (R.K. Miller et al. 1998). Still, for many agents (e.g., ethanol, solvents, and retinoids) that cause developmental toxicity at or near adult toxic dosages, one might be able to monitor concentrations of the compound (or metabolites) in the exposed individual and thereby establish possible risk. Thus, biomarkers of exposure have the potential to be critical in establishing potential risk at a sensitive period during development.
For developmental toxicants that can produce developmental defects at dosages or concentrations not causing identifiable immediate adult toxicity (e.g., thalidomide and cigarette smoking), biomarkers of exposure that reveal actual concentrations of parent compounds or metabolites (e.g., cotinine as a nicotine-metabolite measure of cigarettes smoked) might be the only available indicators of risk.
It is believed that subtle changes in gene expression, as assayed by large-scale microassay analyses, are good examples of newly developing biomarkers of exposure. Those biomarkers still need to relate expression changes with early biological effects, occurring well before toxicity. In fact, there are extensive discussions to determine if these are truly “biomarkers of exposure” or “biomarkers of effect.” Current efforts are under way to improve the detection of differences in patterns of gene expression for various chemical classes (e.g., peroxisomal proliferators and oxidants), with the aim of improving use of patterns rather than single changes as exposure biomarkers. In cases in which maternal toxic effects occur, the patterns of expression changes might be especially useful biomarkers to improve detection of developmental versus maternal toxicity.
Biomarkers of exposure often are used in occupational and molecular epidemiology. Aniline-hemoglobin adducts, benzo[a]pyrene-DNA adducts, aflatoxin B1-DNA adducts, elevated metallothionein, and elevated urinary 8-hydroxy-deoxyguanosine levels have been useful biomarkers for specific exposures. The cancer risk of exposure to dangerous concentrations of foreign or endogenous chemicals is assessed by the activation of a proto-oncogene or the inactivation of a tumor-suppressor gene (e.g., p53), reflecting the mutation of these genes in
somatic tissues. In this usage, the biomarker of exposure also comes close to being a biomarker of effect, insofar as mutagenesis is thought to be an important step in carcinogenesis. Still other biomarkers, such as aryl hydrocarbon hydroxylase (AHH) and CYP1A1 at high concentrations, are taken to reflect induction of the enzymes by high internal concentrations of potentially toxic agents and are used to predict whether a population or individual might be at risk for perinatal morbidity or mortality.
Biomarkers of Effect
Biomarkers of effect at the molecular level are becoming as important as monitoring metabolites or a parent compound. Recently, Perera et al. (1998) confirmed an inverse relationship between concentrations of cotinine in plasma from newborns, a metabolite of nicotine, and birth weight and length. They also demonstrated a significant association between decreased body size at birth, body weight, and head circumference and increased concentrations of polycyclic aromatic hydrocarbon (PAH)-DNA adducts in umbilical cord blood above the median. This had previously been demonstrated for PAH-DNA adducts measured in the human placenta (Everson 1987; Everson et al. 1988). Such associations were related to cigarette smoking and environmental pollution. Those examples show that there can be a practical use of biomarkers of effect at the molecular level to assess exposure. Such measurements not only allow for epidemiological evaluations of environmental pollutants, such as cigarette smoke and air pollution, but they also allow those evaluations to help identify a subpopulation of individuals that might be at risk. Critical applications for such biomarkers in developmental toxicology are in the identification of those at risk, with hopes of reducing that risk by modifying exposure and by developing other intervention strategies to decrease the incidence of developmental defects. Other biomarkers include indicators of normal cell processes (e.g., cell proliferation that may occur at inappropriate times or at different levels of expression). Proliferation markers are often used for assessing immunological impacts where proliferation status is evaluated in the context of differentiation status. These immunological studies present similar issues to those in biomarker studies in developmental toxicology. Likewise, biomarkers of the apoptotic process (e.g., early biomarkers such as nexin, enzymatic changes in various caspase levels or types, and late biomarkers such as DNA fragmentation) can provide temporal, mechanistic biomarkers of effect that are also highly relevant for developmental toxicity assessments.
Other biomarkers of effect include increased concentrations of α-fetoprotein in amniotic fluid as indicative of neural tube defects, since delayed closure of the tube is thought to allow escape of this protein. Other biomarkers might be used in combination to enhance the collective ability to diagnosis or predict possible developmental anomalies (e.g., the triple assay of human chorionic gonadotropin, estriol, and α-fetoprotein for trisomy 21).
Biomarkers of Susceptibility
These biomarkers are used to identify either individuals or populations who might have a different risk based upon differences that are inherent (i.e., genetic) or acquired (i.e., from life history and conditions). The inherent category includes the polymorphisms for genes encoding DMEs and for genes for the receptor and transcription factors regulating the expression of the genes for DMEs, as discussed in a previous section of this chapter. The category also includes polymorphisms for genes encoding components of developmental processes, although the latter are still not well understood. The acquired category includes previous disease conditions, antibody immunity, nutrition, other chemical and pharmaceutical exposures, and various capacities for homeostasis.
As a monitor, the placenta has been a key test organ for identifying such sensitive populations and their responses to environmental exposures. For example, Welch et al. (1969) and Nebert et al. (1969) demonstrated that AHH is induced in the human placenta of cigarette smokers. With the ever-improving tools for investigation, biomarkers now have moved from proteins and enzyme activities induced by polycyclic aromatic hydrocarbons (e.g., benzo[a]pyrene) and dioxin (Manchester et al. 1984; Gurtoo et al. 1983) to biomarkers of combined effect and exposure, such as mRNAs (e.g., CYP1A1) plus DNA adducts (Everson et al. 1987,1988; Perera et al. 1998).
The molecular probes to identify such subpopulations are useful as biomarkers not only for identifying individuals at risk but also for exploring the underlying mechanisms by which those individuals or populations are at risk by demonstrating allelic polymorphisms in a particular gene. As discussed above, gene-environment interactions have been noted for the induction of cleft palate in humans (Hwang et al. 1995) through a combination of cigarette smoking and TGF genotype. Alone, neither variable demonstrates an association with cleft palate. Such an example demonstrates the possibility for understanding why only a small percentage of a population exposed to a developmental toxicant might be at risk, but more important, it identifies a biological association that might lead to a mechanistic understanding of how a particular developmental defect might occur.
There are serious concerns about using the term “biomarkers of susceptibility” to describe a person’s particular set of alleles because these have a hereditary basis (e.g., slow acetylator activity, low G6PD activity, low 5,10-methylene tetrahydrofolate reductase activity, or high CYP1A1 activity). The committee emphasizes the need for a distinction between biomarkers of susceptibility reflecting inherent limitations versus those reflecting acquired limitations. The former require a full understanding of the complex genetic implications before they can be used. An allele encoding an altered DME, for example, might put the individual at increased risk for toxicity caused by one environmental chemical but at decreased risk for toxicity caused by another drug or environmental
chemical. Combinations of alleles might also have exaggerating or compensating effects.
LIMITATIONS IN DEVELOPMENTAL TOXICITY RISK ASSESSMENTS
Although it can be argued that the current approach of risk assessment appears to have worked reasonably well for hazard identification, many assumptions must be made before it can be applied. One such default assumption is that outcomes for rodent tests are relevant for human risk prediction. Such assumptions are generically used because information on the mechanisms of action for specific developmental toxicants is inadequate and because the lack of mechanistic information results in the use of default uncertainty factors. The most important limitation is the paucity of human data, and the lack of methodology to adequately assess humans. Mechanism of action can be pursued in animal models, but it is also the lack of an understanding of human development that hampers risk assessment.
For risk characterization, the bioassays used for regulatory assessment have provided limited dose-response information. The information is limited because the focus is on the effects of high doses at or near maternal toxicity to emphasize identification of hazards. That focus has provided little quantitative information on the dose-response relationship in the low-dose region, the region of greatest importance for extrapolation in human risk assessment. The lack of useful dose-response data has had several impacts. As mentioned previously, conservative use of uncertainty factors predominates for converting NOAELs and BMDs to RfDs for determination of acceptable safe exposure levels. The dominance of animal testing at high doses has also had the unfortunate consequence of providing minimal useful mechanistic information, because assessments are frequently conducted at doses where homeostatic mechanisms are overwhelmed (Nebert 1994), and mechanistic clues about critical toxicant-induced changes are hidden.
The lack of mechanistic information has also resulted in assumptions about sensitivity among humans. Present practice in risk assessment almost always makes use of a default factor of 10 to take into account the variability in sensitivity (i.e., there is a 10-fold difference in susceptibility of the most sensitive individual and the average individual). This assumption has been experimentally addressed for relatively few chemicals (for a review, see Neumann and Kimmel 1998). However, the default assumption could change as researchers gain more information about the underlying basis for responses to toxicants. To date, the greatest progress in characterizing human variability is from research on DMEs. With time, there will also be data on other factors that influence susceptibility. For example, as discussed in detail in Chapter 5, a particular allele of transforming growth factor conveys more than a 10-fold increase in risk of oral clefts in infants whose mothers smoke cigarettes (Hwang et al. 1995; Shaw et al.1996).
As knowledge of human variation in responses increases from the results of the Human Genome Project, a risk-assessment framework is needed in which these default factors are replaced with mechanistic data on relevant toxicant-induced changes.
Molecular approaches should be useful in resolving the issue of extrapolation across species. There is general agreement that the molecular control of development is highly conserved, although the pattern of development of structures at higher levels of biological organization can be very dissimilar. The committee discusses such conservatism in Chapter 7 and suggests that models that assess a small number of those control points and pathways might be relevant for evaluating the potential for chemicals to impact the critical pathways in development, regardless of the species from which the model is derived. The same principle applies to extrapolation from rat or mouse to humans. The critical data to support predictions of ultimate effect in human embryos include a description of the pathogenetic steps that ensue from toxicant actions at the molecular level and lead to structural malformations. Pattern formation genes and signal transduction pathways are so highly conserved across groups of animals that actions of toxicants on those gene products and processes are likely to be comparable and have similar toxicodynamic impacts. The events that follow perturbations at the molecular level are likely to be more prone to interspecies variability, for example, the toxicokinetic differences observed with chemicals that require metabolic activation, as metabolic rates often vary markedly between species. At this point, the predictive value of hazard identification data in alternative models, particularly those that are phylogenetically removed from humans, becomes limited. Therefore, characterization of the pathogenetic events that result in dysmorphogenesis will lead to better prediction of (1) whether the critical events are present and when they are functional in humans, predisposing them to an adverse outcome; and (2) the kinds of adverse outcomes that are possible, based on the temporal and spatial locations of the critical events.
A better understanding of the molecular and cellular mechanisms involved in the pathogenesis of abnormal development might provide a method for answering such questions as to whether a residual level of risk exists at the RfD or ADI and what that level might be. A residual level of risk might also provide a method for determining what exposure concentration can be permitted before the probability of an adverse event begins to increase. The resolving power of animal studies to distinguish an increase in the rate of frank malformations is relatively weak. For example, in a study with 20 pregnant rats per dose group, an increase in the malformation rate must double from the background rate to be statistically significant. Mechanistic and pathogenetic events may prove to be much more sensitive and, therefore, provide a data-driven means to extend the dose-response curve below the NOAEL or BMD for malformations. Although these effects might not be adverse, they might be biomarkers or early indicators for the process of pathogenesis and might help to determine the shape and slope of the dose-
response curve at doses that approach relevance for human exposure. It then becomes a matter of conducting further research to understand the magnitude of response on a molecular or cellular end point that is needed to produce a structural defect with adverse physiological, structural, or functional developmental effects. A method has been proposed for constructing dose-response curves that combine data on frank malformations with data on less-severe effects that are not considered adverse. For example, Allen et al. (1996) combined data on rib malformations with those on rib variations in rats prenatally exposed to boric acid. This method could easily be adapted to include molecular events.
Although it can be postulated that many molecular and cellular events that are the precedents of abnormal development are unlikely to have strictly linear dose-response curves, there is minimal information on developmental specific processes. There have been extensive discussions on the shape of receptor-versus-nonreceptor-based responses that were initiated directly from recent advances in the understanding of molecular events; yet, little is known about actual events at low-dose exposures, as opposed to generation of hypothesized dose-response relationships at low doses. Hypothetical biologically based dose-response models have been proposed on a toxicant-specific basis (Shuey et al. 1995; Leroux et al. 1996). What appears to be most relevant for this report is a call for increased understanding of toxicant-induced molecular changes and an investigation of how these early events are linked to manifestations of adverse developmental outcomes. Empirical work will be needed to establish the magnitude of response at each level of organization required to provoke a response at the next level. Such investigations should be conducted to obtain quantitative information on the kinetics of the toxicant and the dynamics of the toxicological interaction in the temporal context of development (Faustman et al. 1999; Faustman et al. 2000).
Current practices in developmental toxicity risk assessment recognize the concept of “critical windows of sensitivity” in development, but a fundamental understanding of applying the molecular and developmental biological events that define those windows is lacking. This lack of understanding again results in the application of additional child-specific uncertainty factors in efforts to address the sensitivity of the developing conceptus rather than emphasizing the search for the biological understanding of critical windows of susceptibility.
A corollary to the problem of low-dose extrapolation is the assumption that effects observed at high-dose concentrations in experimental animals are relevant to the prediction of risk of adverse effects at ambient exposure concentrations. As discussed previously, testing of chemicals has the inherent dilemma of requiring exaggerated doses and concentrations to maximize the chances of detecting the potential for adverse effects and requiring understanding that the interpretability of the results might be limited because of the possibility that physiological processes in the pregnant animal have been so overwhelmed that the observed responses are qualitatively different from the responses at lower doses. The uneasy resolution of the dilemma has been to assume that the high dose and concen-
tration effects are predictive of all effects at low exposure doses and concentrations unless they are proved to be secondary to maternal toxicity. Some research has been conducted to demonstrate the existence of maternally mediated mechanisms of adverse development (Daston et al. 1991a, 1994). Examples are the induction of transitory zinc deficiencies in the dam by metallothionein inducers (Daston and Lehman-McKeeman 1996) and the overwhelming of acid-base buffering by acidic metabolites of ethylene glycol (Carney et al. 1996). Understanding the molecular processes that lead to specific developmental abnormalities will be useful in determining the low-dose relevance of high-dose effects. In those instances in which the high-dose effects are predictive of low-dose responses, the relevant molecular processes would be expected to increase with dose (i.e., to involve higher levels of gene expression or cellular response and involve more cells). For those instances in which the high-dose effects are the result of a secondary mechanism, the dose-response curve for the adverse effect and the underlying molecular perturbation would be expected to be steep, with an inflection at the dose where the maternal homeostasis was overwhelmed.
SUMMARY
This chapter has defined developmental toxicity risk assessment and outlined issues that regulators face as they strive to protect the human population from chemically induced birth defects. Each section also identified limitations in the current knowledge and methodologies. Biomarkers for developmental toxicity are also discussed. They hold great potential for epidemiological analysis of developmental defects, especially those defects due to complex gene-environment interactions. The information presented in this chapter, and in the next chapter on mechanisms of developmental toxicants (Chapter 4), will be used to define the current state of developmental toxicology and will provide a context for how advances in developmental biology and genomics can improve the approaches for protecting public health.
































