
4
Behavior and Fate of Oil
|
HIGHLIGHTS This chapter points out the following:
|
In Chapter 3, annual loadings of petroleum hydrocarbons are estimated for each of the 16 coastal zones around North America and for the world’s oceans. These loading rates, in units of mass per time, are useful in comparing the relative importance of various types of loadings and in exploring the spatial distribution of these loadings. Mass loadings, however, are not a direct indicator of the potential effects of petroleum hydrocarbons in the oceans. Ecological and human health risks generally scale to the magnitude and duration of exposure, and these mass loading rates must be translated for all hydrocarbon sources into temporally and spatially variable concentrations in the sea. Equal mass loadings to different parts of the ocean may have substantially different consequences. For example, 1,000 tonnes per year of crude oil discharged to a low-energy mangrove swamp will certainly have a much different impact than the same 1,000 tonnes per year released into the deep water of the North Sea. Similarly, equal mass loadings of different types of petroleum may result in different concentrations and potential risks. Quantitative geochemical models are used to estimate ambient hydrocarbon concentrations from mass loading estimates. This chapter discusses the many physical, chemical, and biological mechanisms that process hydrocarbon loadings once they enter the ocean. Models provide both the conceptual framework to evaluate these loadings and the deterministic tools to translate loadings into ambient concentrations and, ultimately, effects. These models combine the inherent properties of petroleum components (e.g., solubility, volatility, reactivity) with the interacting water to predict petroleum hydrocarbon concentrations. The minimum information required to translate loads into concentrations, therefore, includes knowledge of the chemical composition of the loadings and the hydrodynamics of the interacting water.
A full understanding of the impact of petroleum loadings into the ocean requires an accurate assessment of the magnitude, spatial extent, and duration of exposure. Because of the incredible diversity of physical environments within the world’s oceans, it is not possible to derive simple generic relationships between petroleum mass loadings and ambient concentrations that can be applied universally. The “fate” (where it goes) and “persistence” (how long it remains in the system) of petroleum in sea water are controlled by processes that vary considerably in space and time. The processes that
control petroleum transport (movement) in surface waters are reasonably well understood, and conceptual models exist to build deterministic models for specific loadings in a specific area for periods of time (less than one week).
Figure 4-1 shows the interrelationships among the physical, chemical, and biological processes that crude oil undergoes when introduced into the marine environment, subsequently weathers, and is then transported away from the source. Processes involved in the weathering of crude oil include evaporation, emulsification, and dissolution, whereas chemical processes focus on oxidation, particularly photooxidation. The principal biological process that affects crude oil in the marine environment is microbial oxidation. As crude oil weathers, it may also undergo various transport processes including advection and spreading, dispersion and entrainment, sinking and sedimentation, partitioning and bioavailability, and stranding which leads in some cases to tarball formation. These processes are all discussed briefly, along with special considerations of oil and ice, and oil from deepwater releases. This chapter concludes with a discussion of conceptual and computer models and a summary of fates of oil inputs to the ocean from seeps, surface spills, deepwater releases, and diffuse sources such as the atmosphere, land run off, and recreation.
PROCESSES THAT AFFECT THE IMPACT OF OIL RELEASES
Weathering
Following an oil spill or any other event that releases crude oil or crude oil products into the marine environment, weathering processes begin immediately to transform the materials into substances with physical and chemical characteristics that differ from the original source material.
Evaporation
In many oil spills, evaporation is the most important process in terms of mass balance. Within a few days following a spill, light crude oils can lose up to 75 percent of their initial volume and medium crudes up to 40 percent. In contrast, heavy or residual oils will lose no more than 10 percent of their volume in the first few days following a spill. Most oil spill behavior models include evaporation as a process and as a factor in the output of the model.
Despite the importance of the process, relatively little work has been conducted on the basic physics and chemistry of oil spill evaporation (Fingas, 1995). The particular difficulty with oil evaporation is that oil is a mixture of hundreds of compounds, and this mixture varies from source to source and over time. Much of the work described in the literature focuses on “calibrating” equations developed for water evaporation (Fingas, 1995). Initial prediction of oil evaporation was carried our by using water evaporation equations such as the one developed by Sutton (1934).
Later work of Mackay and colleagues (Mackay and Matsugu, 1973; Stiver and Mackay, 1984) was applied to describe the evaporation of crude oil through the use of mass-transfer coefficients as a function of wind speed and spill area. Stiver and Mackay (1984) further developed relationships between evaporative molar flux, mass transfer coefficient at prevailing wind speed, area of spill, vapor pressure of the bulk liquid, gas constant, and temperature.
In all of this previous work, boundary-layer regulation was assumed to be the primary mechanism for petroleum evaporation. This assumption was never tested by experimentation. Subsequently, Fingas (1995) showed that boundary regulation is slight for petroleum evaporation in the thin layers typically found on surface oil slicks, and a simple equation can be used to model evaporation:
Percentage evaporated = C (T)ln (t), (1)
where C is a constant that can be empirically-determined or predicted on the basis of distillation data, T is temperature, and t is time. Empirical equations for many oils have been determined, and the equation parameters found experimentally for the evaporation of oils can be related to commonly available distillation data for the oil (Fingas, 1999). For example,
Percentage evaporated = 0.165 (percent D)ln(t), (2)
where percent D is the percentage (by weight) distilled at 180ºC and t is time in minutes, can be used for oil evaporation prediction. Figure 4-2 shows typical evaporation rates of different oils, the values of which were obtained from experiments under controlled conditions.
Emulsification
Emulsification is the process of formation of various states of water in oil, often called “chocolate mousse” or “mousse” among oil spill workers. These emulsions significantly change the properties and characteristics of spilled oil. Stable emulsions contain between 60 and 85 percent water thus expanding the volume by three to five times the original volume of spilled material. The density of the resulting emulsion can be as great as 1.03 g/mL compared to a starting density ranging from about 0.95 g/mL to as low as 0.80 g/mL. Most significantly, the viscosity of the oil typically changes from a few hundred to a few hundred thousand milli Pascal-seconds, a typical increase of three orders of magnitude. This increase in viscosity can change a liquid petroleum product into a heavy, semi-solid material. Emulsification, if it occurs, has a great effect on the behavior of oil spills at sea. As a result of emulsification, evaporation slows spreading by orders of magnitude, and the oil rides
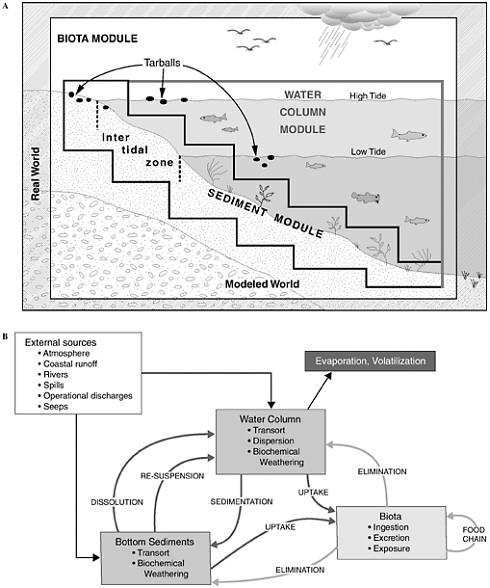
FIGURE 4-1 Graphic representation (A) and detailed interactions (B) of a conceptual model for the fate of petroleum in the marine environment. Various modules depicted are often included as significant components of computer models attempting to simulate or predict behavior and fate of petroleum compounds.
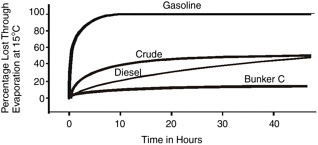
FIGURE 4-2 Evaporation rates of different types of oil at 15ºC (adapted from Fingas, 2000).
lower in the water column, showing different drag with respect to the wind. Oils will generally take up water once spilled at sea, but emulsions may not always form. Water can be simply “entrained” by the oil due to viscous forces, without forming a more stable emulsion. Thus, emulsification also has significant effects on the choice of oil spill recovery methods.
In the late 1960s, Berridge et al. (1968) were the first to describe emulsification in detail. They measured several physical properties and described the emulsions as forming because of the presence of asphaltenes and resins. After these studies, there was little emphasis on the causes of emulsion formation. Mackay et al. (1982) hypothesized that emulsion stability was due to the formation of a film in oil that resisted the coalescence of water droplets; however, this work was used largely for modeling, and not for understanding, the process. Several studies have shown that water is stabilized in oil by two forces: viscous and elastic forces resulting from the interfacial action of resins and asphaltenes. This stabilization was noted as early as the 1970s when formation of emulsion correlated with the oil (Fingas et al., 1996). Only in the 1990s did studies show the effects of composition and propose clear reasons for water-in-oil emulsions. A significant factor in defining mechanisms and other characteristics of emulsions has been the development of analysis techniques for them. Sjöblom et al. (1999) have been instrumental in conducting studies on the formation of emulsions, focusing on the emulsions associated with oil production. Methods were developed to use radio-frequency conductivity to study emulsions. This group also used a Langmuir trough to show that asphaltenes formed barriers of greater strength than those formed by resins.
|
BOX 4-1 T/B North Cape Barge Spill, Rhode Island On January 19, 1996, the tug Scandia caught fire while towing the tank barge North Cape. The tug and tow were abandoned and storm-force winds pushed the Scandia ashore at the Trustom Pond National Wildlife Refuge, 3 miles west of Pt. Judith, Rhode Island. Approximately 2,850 tonnes (828,000 gallons) of home heating oil were released from the barge over a two-day period (Michel et al., 1997). Oil spread over a large area and throughout the water column, resulting in a fishing closure for approximately 250 square miles of Block Island Sound and seven coastal ponds that lasted nearly five months (Mauseth et al., 1997). This spill highlighted the conditions that occur when a light oil is released under high-wave-energy conditions, resulting in very high loading of a refined oil directly into the water column immediately after release. Thus, there was little time for traditional oil weathering processes to occur, whereby the toxicity of the oil is reduced by evaporation of the lighter, more toxic components. Two types of home heating oil were spilled, containing 3 and 6 percent polynuclear aromatic hydrocarbons (PAH), dominated by the 2- and 3-ringed PAH. An estimated 80 percent of the initial release of 700,000 gallons during the storm was physically dispersed into the water column and 12 percent evaporated in the first eight hours after each discharge. Only 10 percent of the oil was estimated to remain on the water surface in the form of sheens after the first 24 hours. Dissolved and dispersed oil concentrations in the water column reached 1-6 parts per million (ppm) total PAH. The dispersed oil droplets resurfaced during calm periods, leaving the dissolved fraction behind. The plume of dissolved oil moved along and offshore, significantly affecting benthic resources. In contrast, very little oil stranded on the shoreline, with relatively small impacts on marshes and intertidal communities, and no shoreline cleanup was necessary. Nearshore benthic resources were greatly impacted, with estimated mortality of 9 million lobsters (mostly juveniles), 19.4 million surf clams, 7.6 million rock and hermit crabs, 4.2 million fish, and 2.8 million kilograms of amphipods and worms (NOAA et al., 1998). The extent of impacts to benthic resources was a function of the richness of the nearshore habitat, particularly for juvenile lobsters and surf clams, as well as the very cold conditions during the spill (water temperatures were 4ºC). Acute mortality of benthic organisms in the salt ponds, particularly amphipods, was also estimated to be high. There were no population-level impacts on winter flounder adults who were present and spawning in the ponds at the time of maximum exposures, nor were there any growth or survival impacts for young-of-the-year winter flounder in the ponds that year. Wildlife directly affected by the spill were seabirds and waterfowl in wintering grounds in nearshore marine waters. Of the 114 live birds collected, all but 9 died or were euthanized. The very cold conditions during the spill decreased the survival rate. Total bird mortality was estimated to be 2,300 birds (Sperduto et al., 1998). Piping plovers showed reduced productivity the breeding season after the spill (NOAA et al., 1998). The rapid weathering of the light refined oil and the absence of mixing with fine-grained sediment limited ecological impacts to short-term toxic effects on both water column and benthic resources. |
McLean et al. (1998) studied water-in-crude-oil emulsions and found that there were two stabilizating factors, viscosity and surface-active agents. Systems were studied using model emulsions with the addition of resins and asphaltenes. They found that resins and asphaltenes accumulate at the oil-water interface and form a barrier to recoalescence. Asphaltenes form more stable emulsions than those stabilized by resins alone. The state of asphaltene solubilization influences the stability of the emulsion. If aromatic solvents are in abundance, the emulsions are not as readily formed. The amounts of asphaltenes and resins were very important as are the ratios between these compound mixtures. These findings have been confirmed by a number of researchers including Sjöblom et al. (1999) and Fingas et al. (2000).
Stability is an important characteristic of a water-in-oil emulsion. Characterization of an emulsion as stable or unstable is required before other properties can be considered, because properties change significantly for each type of emulsion. Emulsion stability and four water-in-oil states: stable emulsions, meso-stable emulsions, unstable emulsions (or simply water and oil), and entrained water (Fingus, 2000; Schramm, 2000). These four states are distinguished by perseverance through time, visual appearance, and by rheological measurements. Meso-stable emulsions, which can be red to black in appearance, have properties between stable and unstable emulsions. Meso-stable emulsions lack sufficient asphaltenes to render them completely stable, although the viscosity of the oil may be high enough to stabilize some water droplets for a period of time. Meso-stable emulsions may degrade to form layers of oil and stable emulsions. Unstable emulsions are those that largely decompose to water and oil after mixing, generally within a few hours. Some water, usually less than about 10 percent, may be retained by the oil, especially if the oil is viscous. This entrained state has a short life span, but residual water, typically about 10 percent, may persist for a long time.
An important measurement to characterize water-in-oil states is forced oscillation rheometry (Fingas et al., 2000). From this measurement the presence of significant elasticity clearly defines whether a stable emulsion has been formed. Viscosity by itself can be an indicator, under some conditions, of the stability of the emulsion. Color is also used as an indicator, but it may not be definitive. All stable emulsions are usually reddish, although some meso-emulsions also have a reddish color, but unstable emulsions are always the color of the starting oil. Water content is not an indicator of stability because excess water may be present. Stable emulsions often have water contents greater than about 60 percent, whereas unstable emulsions or entrained water in oil generally have water contents less than 50 percent. Table 4-1 illustrates water-in-oil states formed from various oils under controlled laboratory conditions.
Dissolution
Dissolution is the chemical stabilization of oil components in water. Dissolution accounts for only a small portion of oil loss, but it is still considered an important behavior parameter because the soluble components of oil, particularly the smaller aromatic compounds, are more toxic to aquatic species than the aliphatic components. Modeling interest in dissolution is directed at predicting the concentrations of dissolved components in the water column. Most models in existence do not separate the dissolution component. The entrainment model is sometimes used but fails to distinguish between dispersion and dissolution.
Brookman et al. (1985) reviewed the solubility of oil and oil components in water. Most solubility data were obtained for distilled water at 25ºC, using various schemes. The solubility of oil components in water varies widely depending on composition. Table 4-2 shows the solubility of very common aromatic hydrocarbons typically found in crude oils (Mackay et al., 1992). Solubility decreases very rapidly with increasing size and increasing substitution. In contrast, the solubility of the aliphatic oil components is very low relative to that of aromatic hydrocarbons and is considered to be negligible. The solubility of crude oils and petroleum products was investigated by Shiu et al. (1990) using several methods in two different laboratories and under a variety of conditions. Table 4-3 contains examples of whole oil solubilities.
The kinetics of dissolution have remained largely unstud
TABLE 4-1 Examples of Water-in-Oil States
|
Oil Type |
Water-In-Oil State Formed |
Starting mPa.sa |
After Formation mPa.sa |
After One Week mPa.sa |
Ratio of Starting/Formation |
|
Arabian Light Crude |
Stable |
14 |
23000 |
23000 |
1640 |
|
Bunker C (15% evaporated) |
Entrained |
8700 |
28000 |
150000 |
3 |
|
Carpenteria (15% evaporated) |
Meso-stable |
3400 |
29000 |
20000 |
9 |
|
Carpenteria Crude (20% evaporated) |
Unstable |
160 |
~200 |
~250 |
~1 |
|
Dos Cuadras Crude (20% evaporated) |
Meso-stable |
740 |
9800 |
2500 |
13 |
|
Port Hueneme Crude |
Entrained |
4100 |
1600 |
8700 |
4 |
|
Sockeye |
Stable |
45 |
6900 |
2800000 |
1533 |
|
aViscosity |
|||||
TABLE 4-2 Solubility of Some Aromatic Oil Components
|
Compound |
Solubility (mg/L) |
|
Benzene |
1700 |
|
Toluene |
530 |
|
Ethylbenzene |
170 |
|
p-Xylene |
150 |
|
Naphthalene |
30 |
|
1-Methyl naphthalene |
28 |
|
1.3-Dimethyl naphthalene |
8 |
|
1,3,6-Trimethyl naphthalene |
2 |
|
Phenanthrene |
1 |
|
Fluorene |
2 |
|
Dibenzothiophene |
1.1 |
|
Chrysene |
0.002 |
ied. In oil spill models, dissolution is often assumed to occur immediately (Hibbs et al., 1999). Some models have incorporated the effect of oil droplet size in the water column and used this parameter to create a kinetic behavior model (Mackay and Leinonen, 1977). In groundwater, kinetics of dissolution are often modeled using a depletion concept and based on the rate of water flow (Mackay et al., 1991).
Oxidation
Crude oil is a complex mixture of organic compounds, mostly hydrocarbons. Oxidation alters these mixtures by creating new compounds and by rearranging the distribution of residual compounds, based on their susceptibility to the oxidative process. The ultimate oxidative fate of all of the organic compounds, given an unrestricted supply of oxygen and time, is conversion to carbon dioxide and water, as expressed in the following equation:
CH2O + O2 <—> CO2 + H2O, (3)
where CH2O is a symbol for all organic compounds. Not only is this the basic reaction for oxidation (to the right), it is the reaction known as “respiration” in animals and “com
TABLE 4-3 Examples of Whole Oil Solubility Data
|
Oil Type |
Solubility mg/L |
Temperature ºC |
Salinity % |
|
Prudhoe Bay |
29 |
22 |
distilled |
|
Lago Media |
24 |
22 |
distilled |
|
Lago Media |
16.5 |
22 |
33 |
|
Diesel fuel |
3 |
20 |
distilled |
|
Diesel fuel |
2.5 |
25 |
33 |
|
Bunker C |
6 |
22 |
distilled |
|
Automotive gasoline |
98 |
22 |
distilled |
bustion” when high temperatures are involved, and the reverse reaction (to the left) is the basic equation for photosynthesis. Hence, this reaction is fundamental to life on earth.
In the oxidation of crude oil, hydrocarbons are oxidized to alcohols, ketones, and organic acids. Oxidized products are more water soluble than the hydrocarbon compounds from which they are derived. The order in which hydrocarbons are oxidized depends on a variety of factors, but in general, small molecules up to C20 (molecules with 20 carbon atoms or less) are oxidized before larger ones. Within the same molecular weight range, the order is the aliphatic n-paraffins (n-alkanes) first, followed by branched and cyclic alkanes (naphthalenes) and then the polycyclic aliphatic and aromatic hydrocarbons. Thus, the degree of oxidation can be ascertained on the residue, based on the type and distribution of the residual compounds. In addition, preferential oxidation of low molecular weight compounds increases the density of the unoxidized residue.
Oxidation of crude oil is mediated by two processes, photooxidation and microbial oxidation, that provide the energy to drive the oxidative reactions. Where crude oil is exposed to sunlight and oxygen in the environment, both photooxidation and aerobic microbial oxidation take place. Where oxygen and sunlight are excluded in anoxic environments, anaerobic microbial oxidation takes place.
Photooxidation in Sea Water
Photooxidation is a family of light-catalyzed reactions that oxidize the reduced carbon in petroleum hydrocarbons. These reactions include both direct photoreactions, where the reactant absorbs light energy, to form a less stable intermediate, and indirect photoreactions, where other chemical species in solution absorb light energy. Both produce reactive intermediates (e.g., solvated electrons, hydroxy radicals) that attack the hydrocarbon molecule or transfer energy directly to the reactant hydrocarbon. The necessary ingredients for photooxidation are radiation and light-absorbing molecules (chromophores). Because few petroleum hydrocarbons absorb sunlight efficiently, most photooxidation occurs via indirect photoreactions. The formation of singlet oxygen from the energy transfer of the triplet excited state of natural organic matter in sea water provides the dominant oxidant for this reaction. Heterogeneous photooxidation, in which reactions occur at the liquid-solid and liquid-liquid interfaces, may also be important. Heterogeneous photolysis of adsorbed species on natural particulate matter may result from direct photochemistry, surface semiconductor redox reactions, or photosensitized reactions on the surfaces of algal cells. Heterogeneous photolysis at the oil-water interface (i.e., surface petroleum slicks) is complex due to the large number of chromophores and reactants that change in absolute and relative abundance during photooxidation (Larsen et al., 1977; Patton et al., 1981; Payne and McNabb, 1984; Payne and Phillips, 1985). (Parker et al., 1971, cited in

PHOTO 15 The construction of new roads and parking lots to support larger number of trucks and automobiles leads to increased runoff, runoff that often contains elevated levels of petroleum hydrocarbons. (Photo courtesy of Larry Roesner.)
Malins, 1977) Photooxidation is unimportant from a mass-balance consideration; however, products of photooxidation of petroleum slicks may be more toxic than those in the parent material (Lacaze and Villedon de NeVde, 1976). Photooxidation also plays an important role in the removal of dissolved petroleum hydrocarbons. Aliphatic and aromatic fractions of petroleum are oxidized photochemically in sunlight to more polar ketones, aldehydes, carboxylic acids, and esters. Because these products are more soluble in seawater, photooxidation enhances the overall solubilization of intact petroleum. These dissolved products can undergo further oxidative processes by either direct or indirect photolysis. In contrast, photooxidation may also result in higher-molecular-weight products through the condensation of peroxide intermediates, ultimately leading to tar and gum residues. Photochemical processes are described in detail in Zitka and Cooper (1987) and Schwarzenbach et al. (1993) and were summarized previously (NRC, 1985).
The aromatic and unsaturated fractions of dissolved petroleum hydrocarbons undergo both direct and indirect photolysis in seawater. Polycyclic aromatic hydrocarbons (PAH) degrade to relatively stable quinones via reactions initiated by electron transfer from singlet state PAH to molecular oxygen (Sigman et al., 1998). Colored natural organic matter (humic and fulvic acids) may play a role in catalyzing the indirect photolysis of PAH, both by capturing light energy and by concentrating hydrophobic PAH, within relatively nonpolar micellular environments of the macromolecules.
The extent of photooxidation depends upon (1) the spectrum and intensity of incident light, (2) the optical properties of the surface water as modified by the petroleum hydrocarbons and other dissolved and particulate constituents, (3) the optical properties of the hydrocarbons themselves, and (4) the presence of photo-quenchers and activator compounds. Petroleum photooxidation occurs faster under short-wavelength light (<300 nm) than in broad-spectrum natural sunlight. Modeling the photooxidation of petroleum hydrocarbons is complex because the surface film alters the intensity and spectrum of the incident sunlight. As weathering proceeds, individual components of the petroleum hydrocarbon mixture degrade by photooxidation at different rates and to different products, further altering the spectral environment.
Microbial Oxidation
There are generally two biological fates of petroleum in marine systems. Both utilize the same metabolic pathway, respiration, but have different end points (Figure 4-3). The first process utilizes hydrocarbons as a carbon source to produce energy, while subsequently degrading the long-chained
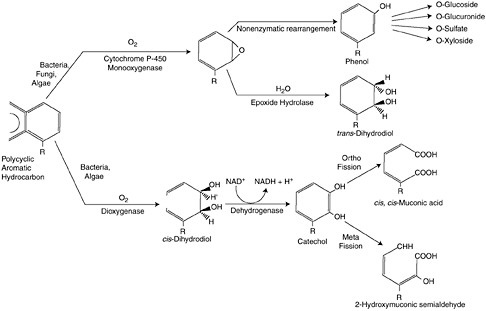
FIGURE 4-3 Two pathways for degrading PAH in oxygenated environments. Pathway one (upper) utilizes the detoxification enzyme system, cytochrome P450 1A, whereby hydrophobic PAH are oxidized to a smaller, and more water soluble molecule that can be excreted by cells. Pathway two (lower) is aerobic respiration, whereby energy in the form of adenosine triphosphate (ATP) is generated from the NADH (reduced form of nicotinamide adenine dinucleotide) produced in the breakdown of hydrocarbons. (American Insititution of Biological Sciences.)
molecules. Microorganisms, primarily bacteria and fungi, and to a lesser extent, heterotrophic phytoplankton, are responsible for these processes. The metabolic process is called oxidative phosphorylation, or respiration, and is the most energetically favorable metabolic pathway to make energy. The second biological process is primarily a detoxification mechanism in response to exposure to oil whereby an organism metabolizes the hydrocarbons to more water-soluble products that can be excreted from the body.
Biodegradation of hydrocarbons has been considered one of the principal removal mechanisms in the aquatic environment. Much of the earlier research was based on laboratory experiments that elucidated the metabolic pathways for degradation, environmental influences on oil degradation rates, and rates of oxidation. Since 1990, biodegradation of hydrocarbons has become a premiere research area as noted by at least five reviews on the topic (Leahy and Colwell, 1990; Atlas and Barth, 1992; Prince, 1993; Swannell et al., 1996; Heider et al., 1999).
There are several energetically favorable metabolic pathways to degrade hydrocarbons that are utilized by different types of microorganisms, including oxidative phosphorylation or respiration (heterotrophic bacteria, fungi, and heterotrophic phytoplankton), nitrate reduction (denitrifiers), and sulfate reduction. Complete microbial oxidation of oil produces CO2 and H2O often from the breakdown of smaller alkanes and cyclic hydrocarbons. More complex hydrocarbons, such as branched alkanes and multicyclic compounds (polycyclic aromatic and aliphatic hydrocarbons), require multiple metabolic pathways for degradation and likely involve a consortium of bacterial strains (Sugiura et al., 1997). Figure 4-3 summarizes two pathways for degrading PAH in oxygenated environments. Pathway one utilizes the detoxification enzyme system, cytochrome P450 1A, whereby hydrophobic PAH are oxidized to a smaller, and more water soluble molecule that can be excreted by cells. Pathway two is aerobic respiration, whereby energy in the form of adenosine triphosphate (ATP) is generated from the NADH (reduced form of nicotinamide adenine dinucleotide) produced in the breakdown of hydrocarbons.
Anaerobic degradation of hydrocarbons by sulfate- and iron-reducing bacteria has recently been measured in marine environments (Loveley et al., 1995, Coates et al., 1996). Because energy yield is relatively low, anaerobic degrada
tion of hydrocarbons in marine environments is limited to low-oxygen areas where heterotrophic bacteria cannot out-compete the anaerobes for carbon substrate.
Rates of biodegradation are dependent on the ability of microbes to contact hydrocarbons as well as on the bacterial metabolic processes operating within the cell. Rates of biodegradation in a natural experiment range from 50 to 100 g/ m3 per day (Lee and Levy, 1987). In the environment, rates of degradation have been reported to be between 0.001 and 60 g/m3 per day (Atlas and Bartha, 1992). Rates for anaerobic degradation of hydrocarbons have not been measured but are generally thought to be a fraction of aerobic respiration rates. In marine environments subject to oil spills (e.g., harbors), prior exposure to hydrocarbons decreases the response times for biodegradation to occur but does not increase the rate. In more pristine environments, there is a longer time lag between the oil spill and biodegradation because the natural populations must adapt to a new carbon substrate and produce the necessary enzymes.
There is a general hierarchy for rates of biodegradation of hydrocarbons: saturated alkanes are more quickly degraded by microorganisms than aromatic compounds; alkanes and smaller-sized aromatics are degraded before branched alkanes, multi-ring and substituted aromatics, and cyclic compounds (Leahy and Colwell, 1990; Atlas and Bartha, 1992). Polar petroleum compounds such as sulfur- and nitrogen-containing species are the most resistant to microbial degradation (Prince, 1993). There are several reasons for this, including water solubility and surface area that affect their availability for bacterial adhesion and metabolism. Increasingly complex structures (e.g., branched methyl groups) and the stability of hydrocarbons decrease the rates of mineralization, which are likely a consequence of the greater stability of carbon-carbon bonds in aromatic rings than in straight-chain compounds. Emulsification (formation of small droplets) provides greater surface area for microorganisms to attach. This implies that only a certain percentage of an oil can be readily biodegraded, typically a few percent for a crude oil (over and above the percentage evaporated) and very little for a heavy oil.
Environmental factors such as oxygen concentrations, nutrients, temperature, salinity, and pressure, as well as the physical properties of oil (including surface-to-volume ratios) and the energy level of the environment, can greatly influence biodegradation rates. In addition, energy levels in marine systems, such as the physical mixing of water as well as wind and wave action, can impede biodegradation by aiding in the formation of large oil globules that have a low surface area-to-volume ratio and impede microbial cell attachment and decomposition processes. In marine systems, microorganism growth is controlled by oxygen activity, nutrient concentrations, light, temperature, salinity, and pressure. Oxygen is required for metabolism by heterotrophic bacteria and phytoplankton, as well as fungi, and is prevalent in high-energy environments where the oil-water interface is constantly aerated by the atmosphere. In addition, the marine environment is generally limited by the scarcity of the nitrogen macronutrients, and secondarily, phosphate, although site-specific and seasonal exceptions to this nutrient hierarchy abound.
Temperature can influence biodegradation. In low-temperature environments, oil viscosity increases and water solubility decreases, thereby limiting microbial attachment. In addition, volatilization of toxic, short-chained hydrocarbons is decreased and may be detrimental to microorganisms. Finally, temperature can affect cellular enzymatic activity, where the rate of enzymatic activity approximately doubles up or down with every 10ºC change in temperature (the Q10 principle). The net effect is a decrease of biodegradation with decreased temperatures that has important implications in assessing oil spills in colder environments.
Transport
Horizontal Transport
Horizontal transport and horizontal dispersion are separate processes that stand apart from, but may enhance, spreading and Langmuir circulation. Horizontal transport means displacement along a horizontal axis, whereas, horizontal dispersion or diffusion is movement about a defined point and does not necessarily involve net movement.
Spreading
The most used models for spreading are based on the work by Fay (1969). Fay suggested that spreading is best described in three phases—inertial, viscous, and surface tension. The inertial phase is dominated by gravity forces, the viscous phase by gravity and viscosity forces, and the surface tension phase by surface tension spreading. Other models, often involving constants, have never been used extensively (Fallah and Stark, 1976). The Fay model has been subject to criticism for several reasons. First the viscosity of the water, not the oil, is used as a primary driving mechanism. Second, the model generally under-predicts spreading when tested. This observation may be explained in part as a consequence of horizontal diffusion resulting from shear diffusion of waves (Elliott, 1986).
Several tests of the Fay spreading model have been conducted. Flores et al. (1998) found that the Fay model under-predicted the spread of oil under quiescent conditions. Lehr et al. (1984) studied spreading using a series of test spills in the Arabian Gulf. They also found that the Fay model grossly under-predicted and proposed amendments to the model, suggesting that the sheen and thicker portions of the spill be modeled separately. No new formulations of the Fay spreading model have found wide acceptance; however, the formulation is often adjusted in models to account for the under-predictions shown in tests.
Other, less common models based on work by Mackay, divide the slick into thick and thin segments that spread separately (Garcia-Martinez et al., 1996). Elliot et al. (1986) develop a spreading formula based on the shear processes cited earlier. Many models combine processes when computing their oil spread rates (Plutchak and Kolpak, 1981). The impact of these approaches in terms of modeling spill trajectories is unclear.
Advection
Few studies have been conducted on the subsurface advection of oil (Spaulding, 1995). The potential for mixing petroleum with water due to evaporation and cooling of surface waters seems limited as the buoyant forces working on the droplet tends to overcome these mechanisms. Limited modeling and observation suggest that the dissolved and particulate oil move as the bulk water moves and that the water moves in concert with mass circulation including the influence of currents and tides (Spaulding, 1995). Additional influences in the subsurface movement include vertical mixing by Langmuir circulation (McWilliams and Sullivan, 2000).
Empirical studies in the 1960s established that oil slicks on a sea surface are transported with the surface current (top centimeter of water) at 2.5 to 4 percent of the wind speed (Fallah and Stark, 1976; Reed, 1992). Furthermore, it was established that a deflection angle was appropriate to account for the Coriolis effect during slick transport. The drift velocity has largely been taken as 3.5 percent, which is the mean of the range shown above but also is a result of several carefully measured experiments (Audunson et al., 1984; Youssef and Spaulding, 1993; Reed et al., 1994). The deflection angle has been sometimes taken as 3 percent; however, Youssef and Spaulding (1993) have provided calculated values that vary with wind speed.
Langmuir Circulation
Langmuir cells (LC), often expressed as windrows, are a common feature in the sea and are generated by a wind-driven shear instability in combination with the mean Lagrangian motion from surface waves (so-called Stokes drift) as depicted in Figure 4-4. The so-called cells that compose LC have time scales of minutes and length of tens of meters. LC creates convergence and divergence zones on the sea surface running parallel to the wind vector. In the vertical, LC cause local downwelling regions that can drag surface pollutants such as oil down into the water column.
LC can potentially have many effects on surface oil. First, it enhances movement of the slick. Second, LC can create convergence and divergence zones on the surface that affect oil thickness, which in turn can affect biota, weathering rates, and cleanup strategies. Finally, LC enhances vertical dispersion of oil droplets. By pushing the droplets down into the
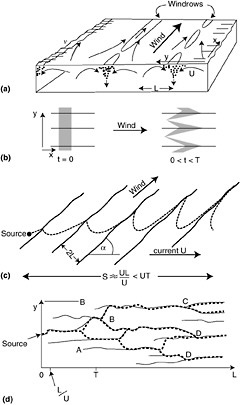
FIGURE 4-4 Diagram of Langmuir Circulation (LC) showing (a) perspective sketch of some of the features of LC (Note regions of convergence on the surface and characteristic length scale of the cells is “L” in the vertical and horizontal); (b) longitudinal dispersion and advection of floating particles by Langmuir circulation. A rectangular cloud of particles is shown at the start of LC motion (t=0) and after some time but before LC breaks us (t=T); (c) advective dispersion of a plume of seed particles when the wind and current angles are inclined at angle, a; and, (d) dispersion of seed particles at LC break-up or instability. (Modified from Thorpe, 2001, Elsevier Science, Inc.)
water column, LC can indirectly affect horizontal advection and dispersion, and increase the amount of hydrocarbon that dissolves into the water column.
McWilliams and Sullivan (2001) compare the LC enhancement of vertical and horizontal dispersion and argue persuasively that vertical dispersion is the most important. They argue that since the characteristic mixing length of LC

PHOTO 16 A spill of roughly 320,000 gallons of south Louisiana crude in May 1997, streaming across the broken marshes of Lake Barre. There is very little substrate exposed thus the oil is being pushed through the submerged vegetation by wind. (Photo courtesy of Jacqui Michel, Research Planning, Inc.)
is the same in both the vertical and the horizontal (order of tens of meters), the vertical component is more important because it is strong compared to other normal vertical mixing processes in the ocean. In contrast, LC-induced horizontal dispersion is weak compared to other horizontal mixing processes. Rye (2001) shows aircraft observations from numerous spills that indicate LC horizontal dispersion, but the effects are relatively small scale. It is interesting to note that Rye’s (2001) comparison focused solely on horizontal scales and not vertical, presumably because of the lack of good data in the vertical.
Lehr and Simecek-Beatty (2001) point out that LC may well be as important at enhancing vertical dispersion as wave breaking. Theory suggests that wave breaking will drive oil droplets roughly one wave height into the water column, whereas LC could drive smaller near-neutrally buoyant droplets tens of meters down, perhaps as far as the base of the mixed layer. Given this, it is an apparent paradox that state-of-the-art oil spill trajectory models include vertical dispersion due to wave breaking but not LC. The primary reason for this is that there is presently no relatively simple verified algorithm to include LC in a spill model. A realistic model would have to not only include a physical model of the Langmuir cell hydrodynamics but also to consider the buoyancy of the oil droplets and hence the droplet-sized distribution. None of these are well understood.
In summary, LC is a potentially important mechanism whose effects have been seen in real spills but are not presently well understood. Further measurements are clearly needed especially with regard to the efficiency of LC in enhancing vertical dispersion and subsequent hydrocarbon dissolution. If further research demonstrates the importance of LC compared to other processes, then a relatively simple LC algorithm should be developed and incorporated into oil spill trajectory models. LC effects on cleanup strategies are another potentially fruitful topic of research.
Horizontal Dispersion
Dispersion is a mixing process caused by the turbulence field in the ocean. It is the process that would cause a liter of instantaneously released dyed water to expand over time and eventually dissipate in the ocean. Without dispersion, advection would move that liter downstream, but the volume of dyed water would not change over time. Dispersion occurs in both the horizontal and the vertical directions, but because the hydrodynamic processes in the vertical and horizontal are often quite different, a distinction is usually made.
In oil spill modeling, horizontal dispersion is often combined with “spreading,” but they are fundamentally different processes characterized by different length and time scales. A liter of oil dumped on a tabletop will spread but it will not
disperse. Although spreading and horizontal dispersion start to work immediately after a spill occurs, spreading is nearly complete within a day while dispersion continues to increase. For most offshore spills, dispersion will move more oil around than will spreading.
Dispersion originates from ocean eddies of various scales, Langmuir circulation, boundary-layer shear (e.g., wind gusts blowing on the sea surface), and other seemingly random turbulence. Dispersion is typically modeled using a Fickian law that assumes a neutrally buoyant, noncohesive substance. Clearly oil is different, so at the very least the dispersion coefficients used in a Fickian model will likely be different from those determined for miscible substances. Some composite oil slick models simply ignore horizontal dispersion and focus on the “center of mass” of the slicks. The National Oceanic and Atmospheric Administration’s GNOME model uses a Fickian law. Others have developed heuristic methods with coefficients tuned to observed slick data. Examples include Morales et al. (1997) who have developed a random-walk method and Howlett et al. (1993) who break the spill into parcels called “spillets” and disperse them numerically.
Vertical Dispersion and Entrainment
Vertical dispersion and entrainment are the movements of oil droplets of sizes less than about 100 μm into the water column. Typically droplets that display a residence time of minutes to hours have droplet sizes less than about 20 μm (Reed, 1992). Larger droplets will rise quickly to the surface. MacKay developed an early model of entrainment based on the square of wind speed, the viscosity of oil, slick thickness, and surface tension (Reed, 1992; ASCE, 1996). Tests of this model showed that it provided reasonable results at moderate wind speeds, but otherwise deviated from experimental values.
Delvigne et al. (1987) and Delvigne (1993) developed a series of models based on a number of different flume tests, tank tests, and at-sea measurements. These commonly used models are empirical and are based on breaking wave energy, film thickness, oil type, and temperature. Energy is included as turbulent energy dissipation by the waves per unit area. Later models were developed to account for energy applied by other than breaking waves and included movement around obstacles and hydraulic jumps. The models have been applied successfully under a variety of circumstances. They do not, however, account for the stability of the droplets in the water column, a factor that largely depends on droplet size and has been modeled based on empirical data (Delvigne et al., 1987). Few tests of models have been done at sea because of the analytical difficulties of measuring the many factors involved. The tests conducted thus far have been mainly an extrapolation of the fate of oil to the Delvigne model (ASCE, 1996). The depth of mixing was found to conform largely to the rule of thumb that states that the depth of mixing is 1.5 times the wave height (Delvigne et al., 1987).
Sinking and Sedimentation
Sinking is the mechanism by which oil masses that are denser than the receiving water are transported to the bottom. The oil itself may be denser than water, or it may have incorporated enough sediment to become denser than water. Sedimentation is the sorption of oil to suspended sediments that eventually settle out of the water column and accumulate on the seafloor. There is a significant difference in the relative amount of oil incorporated by the two processes; sinking oil may contain a few percent sediment, whereas contaminated sediments accumulating on the seafloor will contain at most a few percent oil (McCourt and Shier, 2001). Sedimentation requires a mechanism for oil to become attached to sediments. One mechanism is ingestion of small oil droplets dispersed in the water column by zooplankton and excretion of oil in fecal pellets that then sink to the seafloor. This process has been documented only during the Arrow spill in Chedabucto Bay (Conover, 1971).
The National Research Council (NRC, 1999) developed conceptual behavior models for nonfloating oils that described the factors determining whether spilled oil will sink. Figure 4-5 shows the interaction of these factors. Because most nonfloating oils are only slightly denser than water, the presence of currents can keep the oil in suspension and prevent its accumulation as a coherent mass on the bottom. For example, little or no oil accumulation on the bottom was reported after heavy-oil spills in the Mississippi River (Weems et al., 1997) and Puget Sound (Yaroch and Reiter, 1989). In very few spills of oil that was heavier than water, the oil sank directly to the bottom, and these kinds of spills occurred only in sheltered settings (e.g., from the vessels Sansinena and Mobiloil). In contrast, a buoyant oil can pick up enough sediment, either after stranding onshore or mixing with sediment suspended by wave action, to become an oil-sediment mixture that is denser than sea water. If the sediment separates from the oil mass, the still-buoyant oil can then re-float, as was observed at the Morris J. Berman spill in 1991 in Puerto Rico.
Recent studies on sedimentation of spilled oil have focused on the interaction of fine particles (clay) and oil stranded on the shoreline as a mechanism that speeds natural removal of residual stranded oil (Bragg and Owens, 1995). This process involves oil-fine interaction of micron-sized mineral fines with oil droplets in the presence of water containing ions. Once processed, the oil droplets do not coalesce, and the oil is readily removed from the shoreline by tidal flushing and wave action. The oil-clay oil particle clusters are of near-neutral buoyancy and are easily kept in suspension. The oil attached to the fine particles is more available for biodegradation. This process was first described during the Exxon Valdez oil spill and has been proposed as occurring at other recent spills (Bragg and Owens, 1995). No field or laboratory measurement techniques, however, have been developed that enable immediate identification of
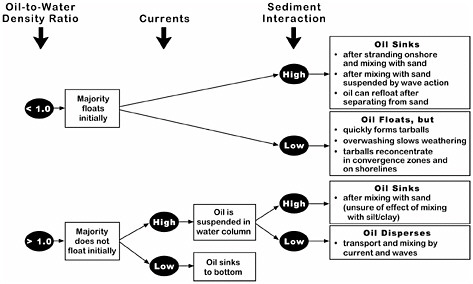
FIGURE 4-5 Factors determining whether spilled oil will float or sink (NRC, 1999).
the process or the potential for the process to occur in a spill situation. Recent work on oil-fines interaction stimulated new research on the interaction of oil and suspended solids in glacier-fed rivers in Alaska. In laboratory mixing tests with water from eight rivers collected over three seasons in Alaska, oil-sediment interactions occurred, but the factors controlling the amount of oil loading could not be identified (McCourt and Shier, 2001).
Overwashing
Overwashing is the temporary submergence of oil below the water surface. The oil can be described as “floating” just below the water surface. Environment Canada conducted several studies in the 1980s to investigate the factors influencing oil submergence as part of an overall program on the behavior of spilled oil (Wilson et al., 1986; Clark et al., 1987; Lee et al., 1989). Equations for overwashing were developed by Mackay et al. (1986) and have been used in some models to predict this process. There have been no significant advances in the theory of overwashing since this work.
The principal cause of overwashing is the action of waves and near-surface turbulence (Clark et al., 1987). Two other factors are also very important: the density of the oil must be close to that of water and the oil must become viscous enough so that the slick breaks up into discrete masses such as tarballs (Buist and Potter, 1987).
Overwashing is particularly important because submerged oil is difficult to see visually or with remote sensors, making it difficult to detect the oil, track its path, and make accurate trajectory predictions. During the Nestucca spill, the oil broke into tarballs that became overwashed and could not be visually tracked. Two weeks later, the oil showed up along 150 km of shoreline on Vancouver Island, Canada (NOAA, 1992). Submerged oil also weathers more slowly because there is almost always a thin water layer on top of the oil (Clark et al., 1987). Thus, relatively fresh oil can travel hundreds of kilometers from the release site. Finally, it is very difficult to recover submerged oil using standard skimming equipment.
There are three mechanisms by which submerged oil can resurface: (1) the density of the water increases, as in an estuary where the oil moves from fresh water to salt water; (2) the turbulence of the water surface ceases, when the wind dies down or a river plume enters a bay; and (3) the oil becomes stranded on a shoreline.
Partitioning and Bioavailability
Partitioning
Petroleum hydrocarbons come in many chemical forms or species and partition among the myriad particulate and
dissolved phases of surface sea water including minerals (e.g., iron oxides) and live and dead cells. These partitioning processes include adsorption where the hydrocarbon attaches to the two-dimensional surface of a solid or other interface and absorption where the chemical partitions into the interior of a cell or detrital particle. Understanding the distribution of petroleum hydrocarbons between the dissolved phase and the variety of aquatic particles is important for determining the fate of hydrocarbons in the sea and the bioavailability of these chemicals to marine biota.
For nonpolar organic chemicals, including most of the components of petroleum, the science of partitioning is based on early studies of pesticide retention by soils, which formed the basis for later work with sediments. This field has been extensively reviewed and synthesized (e.g., Karickhoff, 1984; Schwartzenbach et al., 1993; Chiou et al., 1998; Stangroom et al., 2000). For these compounds, associations with solids result primarily from nonspecific interactions with the solids, often driven by hydrophobic exclusion from the dissolved-phase. The simplest model of dissolved-solid distributions is the equilibrium isotherm. The concentration of a solid-associated chemical is related to the dissolved phase concentration by an equilibrium constant K = Cp/Cd, where Cp is the solid-associated concentration of a chemical (mass of chemical per unit mass of solid) and Cd is the thermodynamically dissolved concentration. (In practice, “dissolved” is an operational definition corresponding to the water fraction that can pass through a 0.2 or 0.45 μm membrane filter and includes solids such as minerals, bacteria cells, and colloids; in these cases, the true equilibrium partition coefficient, K, is approximated by a distribution coefficient, Kp.) For nonpolar compounds, the magnitude of K is inversely proportional to the compound’s aqueous solubility and is directly proportional to the octanol-water partition coefficient (Kow). Furthermore, K varies with the nature of the solid phase, especially the fractional organic carbon content and grain size.
Since the NRC (1985) report, there have been great advances in analytical measurements of the different fractions and in predicting the dissolved-solid distributions and bioavailability of hydrocarbons, especially PAH. Initially, partitioning of PAH was modeled as an equilibrium process, based on laboratory observations that the distribution of hydrocarbons apparently reached constant conditions after a few hours. Subsequent work established that desorption may be much slower that adsorption, especially after the sorbed chemical has been allowed to age within the solid (Karickhoff, 1984; Wu and Gschwend, 1988; Ball and Roberts, 1991; Huang et al., 1998; Kan et al., 1998, 2000). Slow desorption has important implications for the fate and transport of hydrocarbons, both in surface waters and in the subsurface environment (Mackay et al., 1986). For example, concentrations of PAH attached to particles from land-based sources may be supersaturated with respect to the corresponding dissolved phase in coastal waters, resulting in a desorption gradient driven by diffusion. The extent to which this disequilibrium will persist depends on the relative rate of desorption compared to the residence time of the particle in the coastal waters As another example, the amount of hydrocarbon released from contaminated sediments that are resuspended into the water column as a result of storms, tides, or dredging depends directly on the desorption rate. Also, the surface to volume ratios (S:V) of laboratory studies should be considered when evaluating field conditions, and compared with the S:V of oils occupying different environments. During the past 15 years, kinetic adsorption-desorption algorithms that describe rate processes have begun to find their way into newer hydrocarbon fate and transport models. Most commonly used “off-the-shelf” modeling packages continue to employ equilibrium partitioning.
Another significant improvement in the description of dissolved-solid partitioning is the recognition that highly sorbing phases within aquatic particle populations can greatly reduce hydrocarbon bioavailability and reactivity in the marine environment. The presence of soot particles in coastal marine sediments significantly alters the partitioning of PAH between sediments and porewater (McGroddy and Farrington, 1995; Gustaffson et al., 1997a; Naes et al., 1998). Whether this alteration is due to highly energetic adsorption sites on the soot particle surfaces or to correspondingly slow desorption kinetics from these particles (or, more likely, both) is not yet clear. This strong binding within the sediments likely decreases the availability of PAH to benthic organisms (Maruya et al., 1996; Naes et al., 1998; Lamoureux and Brownawell, 1999; Krauss et al., 2000). The composition of sedimentary organic matter also affects the efficiency with which benthic organisms extract PAH from sediments (Landrum et al., 1997; Standley, 1997; Weston and Mayer, 1998; Baumard et al., 1999).
Bioavailability
Organisms are exposed to petroleum hydrocarbons in the marine environment. They are not exposed to the total amount of hydrocarbons in the water and sediment, however, because some portions of the chemical occur in forms not accessible to the organisms. The processes controlling bioavailability have been reviewed by. Partitioning strongly affects the mechanisms and magnitude of exposure of aquatic organisms to hydrocarbons. Dissolved hydrocarbons can diffuse across gill and cell membrane surfaces, and those associated with particles can be ingested during feeding. If oil droplets are present in the water column, marine filter feeders are exposed to PAH by direct uptake of the oil (Menon and Menon, 1999). Unlike other nonpolar compounds such as polychlorinated biphenols (PCBs) and certain pesticides, PAH sometimes bioaccumulate in the food chain depending on the metabolic rate of the organism.
Single-cell organisms, such as phytoplankton, are exposed to hydrocarbons primarily through partitioning of dis

PHOTO 17 Oil on intertidal flat in Saudi Arabia in May 1991. The heavy oil slicks persisted long enough (for months) in protected bays to coat the entire intertidal zone. (Photo courtesy of Jacqui Michel, Research Planning, Inc.)
solved hydrocarbons, operationally defined as passing through a 0.2-μm filter and including colloids and small particles. Any geochemical process that decreases the dissolved hydrocarbon concentration, including partitioning onto larger solids, will reduce hydrocarbon exposure of phytoplankton. Conversely, partitioning of hydrocarbons onto organic-rich particles, including plankton and detritus, results in transfer of hydrocarbons to higher trophic levels, including filter feeders (e.g. mussels, oysters), fish, and mammals. The extent of hydrocarbon accumulation in organisms is controlled by the desorption rate of hydrocarbons from particles in the gut (for higher trophic organisms) and metabolic rates that degrade or transfer hydrocarbons outside the cell.
Shoreline Stranding and Tarball Formation
Persistent oil residues have two major fates: shoreline stranding for spills near to shore and tarball formation for releases in offshore waters. Oil loading on a shoreline can be highly variable, and the amount of oil and the rate of natural removal drive the decision to conduct shoreline cleanup. The sensitivity of shorelines to oil has been embodied in a ranking system called the Environmental Sensitivity Index (ESI) that has been widely applied for oil spill planning and cleanup decision-making (Halls et al., 1997; Hayes et al., 1980); (see Box 2-2). The ESI classifies and ranks shorelines according to the factors that influence oil persistence and impacts, such as degree of exposure, substrate permeability, and shoreline slope. Highest on the scale are the sheltered habitats, such as muddy tidal flats, marshes, and mangroves. These shoreline types are usually priority areas for protection because of their sensitivity and the difficulty of cleanup. Gravel beaches have the highest ranking for beaches because their high permeability allows deep penetration, complex patterns for sediment reworking during storms, low rates of natural replenishment, and the presence of localized, sheltered areas where oil can persist for years (Hayes and Michel, 2001). Decision-making for shoreline cleanup must evaluate the trade-offs between the impact of the oil and the impact of the cleanup. The objective is oil removal to the point that allows recovery without causing more harm than leaving the oil in place (NOAA, 2000). There are well-established guidelines for shoreline assessment, cleanup methods, and cleanup end points (NOAA, 2000; Environment Canada, 2000).
A coastal zone oil spill model (COZOIL) was developed to predict the behavior, loading, and fate of oil stranded on different shoreline types (Reed and Gundlach, 1989). It considers the oil density, viscosity, wave energy, grain size of
shoreline sediments, and empirical holding thicknesses, penetration depths, and removal coefficients for each type of shoreline. This work provided the algorithms needed to improve the accuracy of oil fate models that previously did not fully consider the amount of oil stranded on a shoreline.
As a result of the various physical and chemical processes that affect floating oil from seeps, spills, and operational discharges (e.g., discharge of ballast water), oil can eventually coagulate into residues called tarballs. While some tarballs may be as large as pancakes, most are coin sized. Spills of heavy oils often quickly break into patches of tarballs, making them difficult to track. Tarballs are very persistent in the marine environment and can travel hundreds of miles, sometimes reconcentrating in convergence zones far from the original spill site. Tarballs are problematic because of their long-term persistence and ubiquitous nature along shipping routes (see Plummer, 1996; Butler et al., 1998; Gabche et al., 1998).
Natural oil seeps, which release small amounts of oil over long periods of time (thousands of years) have also caused contamination of beaches, particularly in southern California (Landes, 1973), and naturally occurring tarballs in the Gulf of Mexico and the Caribbean have been described in detail (Geyer and Giammona, 1980). Anthropogenic pollution of beaches began in the twentieth century with the increased shipment of crude oil and refined petroleum products by sea. Thus, coastal locations near natural oil seeps and/or tanker traffic routes are likely candidates for impact by tarballs that continue to contaminate many coastlines worldwide (Butler and Morris, 1974; Hiffe and Knap, 1979; Knap et al., 1980; IOC, 1984; Smith and Knap, 1985; Atwood et al., 1987a,b; GESAMP, 1993; Butler et al., 1998).
Biomarkers (organic compounds whose structure reflects the biological source) and isotope geochemistry (mainly 13C) have been widely applied to characterize tarballs and to differentiate anthropogenic from natural sources, as well as to discriminate among the many possible subsources of anthropogenic contamination. For example, oil pollution in the Straits of Malacca was determined by biomarkers to be from Middle East and South Asia crude oils (Zakaria et al., 2000). In the eastern Mediterranean, biomarkers differentiated four different oil types in tar residues on the coast at Sidi Heneish, 240 km west of Alexandria, Egypt (Barakat et al., 1999). Biomarkers and carbon isotopic compositions were also used to show that crude oil had been spilled in Prince William Sound before the Exxon Valdez spill in 1989 (Box 1-1). In fact, the spill likely occurred in 1964 as a result of the Great Alaska Earthquake during which stored oil, originally shipped from California, was inadvertently released into Prince William Sound when storage facilities were destroyed (Kvenvolden et al., 1995).
The sources of tarballs remain a global coastal contamination issue; thus, continued efforts should be made to develop analytical tools and parameters to enhance the identification of sources. Systematic monitoring of ocean-facing beaches, as recommended by Butler et al. (1998), should continue and be augmented as an inexpensive means to verify changes in pollution input from shipping and the effectiveness of international laws. In addition, efforts to distinguish anthropogenic and natural crude oil contamination of coastlines should continue so that the role of natural seepages, over which humankind has little control, and anthropogenic spills of crude oil and refined products, which humankind is able to minimize, can be readily and accurately distinguished.
Oil and Ice
The behavior and fate of oil in the Arctic, and in other ice-containing marine environments, is modified by the presence of the ice and by the lower temperature (Fingas and Hollebone, 2001). The different ice conditions largely dictate the fate and behavior of oil in a specific situation. The relevant ice conditions can be considered as the following: pack and other ice on water, in leads, with frazil/grease/brash ice, under ice, encapsulated in ice, on ice, and on snow. Several of these have subconditions that significantly change the way oil behaves in the Arctic environment. The amount of ice present on water influences the behavior of oil significantly; generally more than three-tenths (i.e., 30 percent) ice coverage is taken as a condition in which the behavior of oil changes significantly over an open water condition. The behavior of oil under ice differs significantly if the ice is first-year or multiyear ice. First-year ice contains micro-channels with concentrated brine formed by the exclusion of the salt during ice formation. During spring, these brine channels drain and allow oil under or in the ice to rise to the surface. Table 4-4 summarizes the important behavior and fate processes that are modified by the presence of ice and cold.
Several behavioral modes are affected by the presence of ice or the lower temperature in the Arctic. The evaporation rate is reduced by the lower temperature. The extent of evaporation of the oil is not the same as that of warmer climates and is, in addition, slowed by adsorption to snow or ice. Ice-encapsulated oil does not evaporate. Emulsification of oil in Arctic environments has not been fully studied; however, it is believed to occur as readily as it would in other environments. The increased viscosity of the oil at lower temperatures would, in many cases, enhance the formation of water-in-oil emulsions. Although ice on the water damps waves, there is often sufficient sea energy of water interaction with ice to form emulsions. An important aspect of the behavior of oil in the Arctic is adhesion to snow and ice. The effect of adhesion is to remove the oil from other immediate processes to which it might be subjected. For example, oil that adheres to ice edges will no longer spread, nor is it likely to emulsify. Another result of adhesion is to contain the oil initially, but later to spread it out over a larger area. Because the outer edge of the ice pack is very dynamic as is the first-year ice upon breakup, oiled ice can spread over a long dis
TABLE 4-4 Behavior Modes Influenced by the Presence of Ice
|
Location |
Sublocation |
Behavior mode |
|
On water |
<3/10 ice cover |
as open water |
|
>3/10 ice cover |
spreading evaporation adhesion to ice |
|
|
In leads |
spreading and evaporation as above pumping to ice surface |
|
|
With frazil/grease/ brash ice |
spreading adhesion to ice |
|
|
Under ice |
Under first year ice |
spreading evaporation rise to surface through brine channels |
|
|
Under multiyear ice |
spreading and evaporation as above rise to surface through channels |
|
In ice |
Encapsulated |
evaporation rise to surface |
|
On ice |
On ice |
spreading evaporation pooling |
|
|
Under snow |
spreading evaporation pooling adhesion to snow |
|
On snow |
|
spreading adhesion to snow |
tance (perhaps as much as 100 km), and as the ice melts to release the oil, oil is spread over a wide area. Spreading is also a factor that changes very much with ice conditions. Spreading on open water is slower with increasing ice coverage. Spreading on water covered with frazil or grease ice (new crystalline ice in a slush form) or brash ice (ice in chunks up to 2 meters in diameter) is very slow. Studies have been conducted on the spread of oil under ice, and models have been developed for this process. Oil will fill the cavities under ice and be swept along with the current. Studies have shown that spreading and transport by currents will spread oil very widely under ice. Oil spilled on top of ice surfaces will also spread, depending on ice roughness and snow cover. Snow will absorb the oil and retard its spreading. As this oil is heated by the sun, it quickly sinks below the snow/ice line and thus does not affect the bulk albedo for that area (Fingas, 2000).
Oil trapped under first-year ice will rise through the ice to the surface through brine channels during spring. The oil will form pools on the surface of the “rotting” ice and then enter the water as the ice completely melts. Oil trapped under multiyear ice will remain there until it rises through cracks. There are no brine channels in multiyear ice. If there are no cracks, the oil may rise through the natural ablation of ice over a multiyear period, perhaps 7 to 10 years. Less is known about the fate of oil under multiyear ice. Oil caught in leads (cracks in pack ice) may be forced under the ice as well or, if the lead closes rapidly, it can be forced to the ice surface. Often leads will freeze over and thus freeze the oil at the same time as would occur on open water.
The ultimate fate of oil in the Arctic depends largely on the location and ice conditions; however, the effect of the ice is generally to slow the spreading and contain the oil when it is first spilled, compared to open water conditions. Oil caught in ice and snow over winter will generally be released in spring when the ice melts. Adherence to snow and ice may ultimately result in widespread distribution of the oil in the spring or summer. Table 4-5 summarizes the fate of oil in an ice environment.
Fates of Subsurface Releases
Oil spills sometimes are not released at the surface but further down in the water column. Examples include natural seeps, blowouts during drilling of exploratory wells, pipeline leaks, and shipwrecks. Subsurface releases differ from surface releases in several important ways. First, oil can move substantial distances beneath the surface before it finally floats to the surface. This is especially true in deeper water where currents are strong. Such behavior makes tracking difficult, but more importantly it potentially provides time for the more soluble oil fractions to dissolve. Dissolution is enhanced because of mixing and higher pressure, and other aspects.
The special nature of subsurface spills was not been addressed in previous NRC (1975, 1985) reports. The latter report did discuss the Ixtoc spill, which was a subsurface spill, but it did not examine in any detail the subsurface nature of that spill. To facilitate this discussion, subsurface releases are separated into deepwater and shallow water, corresponding to a break at roughly 200 m. The separation is due to a number of physical and chemical complications that arise in deeper water.
Shallow-Water Releases
Considerable research has been conducted and measurements have been made on subsea shallow releases in the last 25 years. Initial efforts at modeling started with Topham (1975), followed by McDougall (1978), Fannelop and Sjoen (1980), and Milgram (1983). Through these efforts, the so-called integral plume models evolved based on an Eulerian reference frame. Zheng and Yapa (1998) and Yapa and Zheng (1997) improved the efficiency of the integral plume model using a Lagrangian scheme and incorporated more realistic ambient currents. Arguably their biggest contribution has been in verifying the integral plume model through extensive comparisons with analytical solutions and through laboratory and field experiments. Of most interest are their
TABLE 4-5 Fate of Oil in Ice Environments
|
Location |
Sublocation |
Fate during freeze-up |
Oil fate after thaw |
|
On water |
<3/10 ice cover |
as open water |
melt out onto open water |
|
|
>3/10 ice cover |
largely trapped among ice |
melt out onto open water |
|
|
In leads |
frozen into leads or on edges |
melt out onto open water |
|
|
With frazil.grease/brash ice |
frozen into brash ice |
melt out onto open water |
|
Under ice |
Under first year ice |
encapsulated |
rise through brine channels to surface |
|
|
Under multiyear ice |
encapsulated |
rise through cracks to surface or remains encapsulated |
|
In ice |
Encapsulated |
remains encapsulated |
melt out onto open water |
|
On ice |
On ice |
will remain on ice |
melt out onto open water |
|
|
Under snow |
absorbed into snow |
melt out onto open water |
|
On snow |
|
absorbed into snow |
melt out onto open water |
comparisons with two major field experiments conducted in 1995 and 1996 and described in Rye et al. (1996) and Rye and Brandvik (1997), respectively. The experiments released roughly 25 m3 of oil in about 100 m of water in the North Sea. The Zheng and Yapas model compared well with the time it took the oil to reach the surface as well as the center location of the surface slick. However, no comparisons of hydrocarbon concentrations in the water outside the plume were made because of limited measurements in the field experiment.
The basic physics of a shallow-water release are as follows. If the oil or gas leaves the source under considerable pressure (i.e., blowout of an exploration well), there is a jet phase that consists of high-velocity fluid or gas (up to the order of 10 m/s) confined to a narrow but expanding cone. The initial momentum in the jet phase is dissipated rapidly within about 1 m of the release point. By this time, distinct droplets (and bubbles if gas is present) form with sizes that are a function of the details of the release but typically with a size distribution of the order of 3 mm (5 percent non-exceedence probability) although it can be much less in the case of very high exit velocities. Subsequently, the hydrocarbons start to rise as a “plume”—a collection of bubbles or droplets that act in concert to drag significant volumes of the adjacent sea water upward in the water column. Plumes rise at a different rate than individual bubbles or droplets, and this is accounted for in the existing models with a so-called slip velocity (the difference between the rise velocity of the mean bubbles or droplets and the plume). If gas is involved, the oil-gas plume will reach the surface in a matter of minutes driven by the large buoyancy of the gas bubbles. The resulting surface slick will spread into a thin film due to the radial outflow of entrained water near the surface. Radial velocities the same order of magnitude as the plume rise velocity (1 m/s) can be expected near the source (Milgram and Burgess, 1984). The typical film thickness will be in the order of 50-100 μm in slicks that are several kilometers wide. Initially, very heavy oil slicks can be as thick as 1mm or more. If the release consists of oil only, the rise time will be longer but still on the order of 10 minutes. If a substantial quantity of oil reaches the surface, spreading will occur, driven by gravity forces and restrained by surface tension and viscous forces in a manner similar to a surface slick.
If there is insufficient integrated buoyancy to set up the plume dynamics described above, the droplets or bubbles rise as individual elements at the rise velocity dictated by their diameter and shape. The threshold for plume dynamics is controlled largely by the volume of oil and gas released, and to a lesser degree, the details of the release orifice (which can control bubble and droplet size), the density of the fluids involved, and the ratio of oil to gas.
Strong cross-flowing currents can complicate the above picture in several ways. First, the plume will tend to bend over much as a plume of smoke is bent by the wind, resulting in a horizontal offset in the surfaced oil slick. Second, as identified initially by Hugi (1993), the rising bubbles or droplets can be sieved downstream according to size, with the largest bubbles rising on the upstream side of the plume and the smallest rising on the downstream side. If the cross-flow current is strong enough, the sieving process will disrupt the establishment of the plume, in which case the oil or bubbles will rise individually. Both these effects of cross-currents will influence how long the oil/gas takes to rise to the surface and where it surfaces. However, the differences in time are perhaps a factor of two and, in space, on the order of a hundred meters.
Another complication is that the oil droplets may entrain water and become emulsified much more quickly than they would in a surface release. Emulsification was observed at the Ixtoc blowout (Boehm and Fiest, 1982) and was due to the intense mixing and turbulence set up by this massive blowout. The presence of emulsified oil significantly impacts the weathering of the oil as discussed earlier. It also greatly lessens the buoyancy of the oil, thus increasing the time it takes for oil to reach the surface.
Deepwater Releases
Substantial efforts to study petroleum releases in deeper water have only recently begun, although there were a few
early efforts—most notably studies of deepwater gas hydrate formation by Bishnoi and Maini (1979) and Topham (1984). Most recent efforts began in 1997 and were triggered by the expansion of hydrocarbon exploration efforts into deeper water. Results are starting to appear: Johansen (2000), Masutani and Adams (2000), Spaulding et al. (2000), Johansen et al. (2001), Chen and Yapa (2001), and Socolofsky and Adams (2001).
The present understanding of deepwater releases suggests they are much more complicated than those in shallow water. Figure 4-6 shows a schematic of the important processes, which are discussed in the subsections below along with some further comments on hydrates and overall hydrocarbon fates. Note that the water depth shown in Figure 4-6 is 1,500 m. In fact, all of the processes discussed below apply to water depths of 200 m or greater with the exception of hydrate formation.
Jet phase
This is the same as in the shallow water case except that there is potential for hydrate formation in deep water.
Plume phase
For most substantial releases, the discharged hydrocarbon will form a rising plume shortly after release, which is analogous to the previously described plume that forms in a shallow-water release. However, in deep water, the plume eventually entrains so much dense water that the aggregate density of the oil-gas-hydrate-seawater suspension is no longer buoyant. Once the plume sheds some of its heavier components, it may re-form as the plume ceases to rise, and “mushrooms.” This process can occur numerous times (known as peeling). Whether or not a given plume reaches a terminal level will depend on the depth of discharge, the plume buoyancy (flow rate and composition of oil, gas, and hydrates), and the strength of the ambient stratification. Most substantial releases will reach a terminal depth within an order of 100 meters of the release orifice although it can be much higher especially if the stratification is weak.
As in the case of the shallow release, a crosscurrent may add the complication of bending and/or sieving (see earlier discussion of shallow releases), although the effect will be even stronger. Also, like the shallow-water case, there must be sufficient integrated buoyancy to set up a plume. If the volume is too small or the bubbles or droplets are too small, then the plume is not established and movement will look like the post-terminal phase described below.
Post-terminal phase
Once the plume reaches its final terminal layer the rise of the oil-gas-hydrates is driven purely by the balance between the buoyancy of the individual droplets, bubbles, and hydrate flakes, and their hydrodynamic drag.
Hydrates
Natural gas released deeper than about 300 m can theoretically form hydrates based on thermodynamic equilibrium calculations. However, the recent laboratory and experimental studies cited above suggest that these calculations predict hydrate formation prematurely. For example, in a field experiment, Johansen et al. (2001) found that the thermodynamic equilibrium suggested for hydrate formation should have occurred below 450 m, but no hydrate was observed at the release depth of 840 m. The laboratory experiments of Masutani and Adams (2000) suggest that hydrate formation requires saturation of the water with the gas.
Hydrate formation is an important factor in determining how quickly oil rises to the surface. Hydrate has a specific
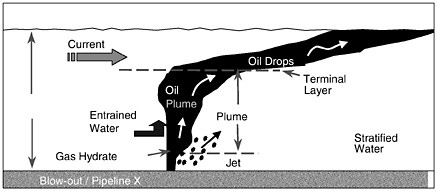
FIGURE 4-6 Schematic diagram depicting the basic physical processes involved in a deepwater subsurface oil and gas release.
gravity of roughly 0.9, so if it forms, much of the buoyancy driver disappears from the plume, thus increasing the time it takes the hydrocarbon to reach the surface.
At some very great depths or very high volumes of gas release, hydrates will form. As the hydrate rises above the critical depth it will start to decompose. If it is a flake, it will decompose into small bubbles that will almost certainly dissolve in the water before reaching this surface. If the hydrate consists of a thin skin as observed by Masutani and Adams (2000), then the skin will decompose and the bubble dissolve.
Deepwater release fates
Based on the work cited above, several important aspects of the fates of deepwater releases have become apparent:
-
The majority of the oil in most deepwater releases will rise to the surface although it may take several hours. Theoretical calculations of a light-weight oil suggests that, at most, 10 percent might dissolve under perfect conditions. For a diesel and a light medium crude, Johansen et al. (2001) found that it took about an hour for hydrocarbons to appear at the surface when released from 800 m of water.
-
The surface slick formed once the oil reaches the surface will be thinner than that seen during a shallow-water release or a surface release. This is due in part to the fractionation of oil droplets that results in a staged arrival of the oil at the surface and in part to diffusion or dispersion of the oil as it rises.
-
Much if not all of the natural gas (85 percent methane or more) associated with the oil will likely be dissolved in the water column, regardless of whether hydrate forms or not. This is due to the high solubility of methane in sea water at the high pressures and cold temperatures found in deeper water.
-
Integral plume models like those of Johansen (2000) and Chen and Yapa (2001) do appear to capture much of the major features observed in the field including the terminal layer and time to surface.
Despite the fact that surface slicks capture the public attention, there are a substantial percentage of accidental spills that occur beneath the surface, typically from the seafloor. Examples include blowouts of exploration wells, pipeline ruptures, and shipwrecks.
The release of oil beneath the surface introduces a number of complications compared to oil released at the surface. From the standpoint of fate the most important complications are enhanced dissolution in the water column and, perhaps, emulsification. If natural gas is present it will tend to dissolve rapidly during the rise through the water column.
The state of modeling is well advanced in shallow water in many respects. Several integral plume models compare well to field data, which include the time the oil takes to reach the surface, the size and shape of the rising plume, and the surface slick. In deeper water, the models are just now appearing. Predicting hydrate formation and emulsification appears to be problematic. The most serious, in terms of fate problems, for both shallow and deepwater appears to be the limited validation of the dissolved component. Without this, it is difficult to assess some potentially important fates.
MODELS AND MODELING
Conceptual Model
In order to better understand the interplay of the various processes discussed earlier, it may be worthwhile to examine the potential fate of a petroleum spill through the development of a conceptual model. (A conceptual model in this sense, is not a software application, but a conceptual tool that might lead to the development of a computer model or, more likely, a group of models—many of which are already available.) A conceptual model for the fate of oil in the environment should start with the most basic sources of crude oil. The model should then trace the products and by-products of oil refining and their ultimate fate in the environment. That is, the model should perform a mass balance on the oil beginning with its extraction from below the surface to its ultimate receptor (e.g., sediment, animal tissue, atmospheric gas). In this case the basic sources would be:
-
natural seeps
-
land-based oil production facilities
-
offshore platforms
The conceptual model should then trace the transport and fate of this material as it moves from source => refinery => use => recycling => waste => environment. For the present study, whose the emphasis is on the fate of oil and its by-products in the marine environment and the subsequent effects on biota, only inputs of petroleum hydrocarbons to the marine environment in time and space have to be taken into account. The fate of these inputs is tracked using three-dimensional transport and weathering models within the water column, the sediments, and the biota. Figure 4-1 illustrates this conceptual model, which partitions the marine environment into three interdependent environmental compartments or modules—the water column, bottom sediments, and biota. The various mechanisms whereby petroleum hydrocarbons move from one module to another are identified on the figure. Petroleum hydrocarbons are introduced into either the water column or the bottom sediments from one or more of the external sources shown in the rectangular boxes. Describing the fate of petroleum hydrocarbons in the ocean requires tracking their movement from the external source through each environmental compartment. The effects of oil in the sea are described in the bottom sediments and biota modules. The approach to developing a quantitative expression of the fate and/or effects within each of the modules is discussed below.
External Sources
The conceptual model is driven by external sources of petroleum hydrocarbons as shown on Figure 4-1, including:
-
atmosphere
-
coastal runoff
-
rivers
-
spills
-
operational discharges
-
seeps
In Chapter 3 of this report, each of these sources is quantified by coastal zone within North America. To quantitatively describe the fate and effects of these external sources, they must be redefined as point sources or diffuse sources, and then further quantified in time and space. The division of the external sources into point and diffuse sources can be done as follows:
-
Point sources:
-
Spills (vessels, platforms, pipelines, facilities)
-
Rivers
-
Produced water
-
Coastal refinery wastewater
-
-
Diffuse sources:
-
Natural seeps
-
Atmosphere on open seas
-
Coastal urban runoff
-
Marine transport operations
-
Recreational boating
-
The time and space scales required to characterize these inputs depend on the degree of time and space definition desired for examining fate and effects. For example, to track the fate and effects of a medium-sized spill where the initial concern is toxic effects and physical adsorption on animals, the time scale would be on the order of days and the space scale meters. Each of the environmental modules (with the possible exception of bottom sediments) would also have to describe fates and effects within the compartment in similar time and space scales until the short-term effects of the spill were no longer discernible. Then the long-term effects of the spill might be calculated at seasonal time steps, with space scales in kilometers.
For minor spills that occur frequently in a geographic area, the effects of these discharges would most likely exhibit themselves as chronic effects. Thus, these minor spills could be categorized as diffuse sources, constant in time (or season) and space over a specified geographic area.
Most of the other inputs can be considered constant in time (or seasonal), and fixed in space. Time scales for the environmental compartments can thus be seasonal, with space scales in kilometers.
Transfer of Materials Within and Between Modules
Within each module, equations governing the processes that occur in the module can be developed to whatever extent is desirable, (i.e., simple models to complex models; single compound models to multiple-compound models). Most likely, a single physical oceanographic circulation model could be used as the basis for the transport calculations performed in the water column and the bottom sediments. The macro biological part of the biotic module is the ultimate system of interest in terms of effects, and the other two modules are required only to the extent necessary to accurately define the transfer paths between them and the biotic module.This model is probably good for all animals, including birds with respect to oil impacts due to ingestion, but it is not suitable for physical effects such as coating of the animals with oil, which has occurred during significant oil spills. The approach to quantitatively describing the fate and effect processes that occur in each environmental module is discussed further below.
The Water-Column Module
The water-column module does not describe any effects of oil in the ocean, but rather describes the fate of petroleum hydrocarbon compounds within the water column. The processes include interrelating transfers to and from the water column from external sources and the other two environmental modules and calculating internal biochemical transformations of petroleum hydrocarbon compounds (weathering). The water-column fate model can be expressed using a mass-balance model in the form of differential equations.
The Bottom-Sediment Module
The bottom sediments module shown on Figure 4-1 is also a fates module describing (1) chemical weathering of petroleum hydrocarbons within the sediment, (2) the transfer across the sediment-water interface of petroleum hydrocarbon compounds between the sediment module and the water column, and (3) transfers of petroleum hydrocarbon compounds between the sediment and biota modules by benthic organisms. It would be possible to include the benthic organisms in the sediment module, but it is conceptually easier to have the biological process confined to one module (the biota module). A simple two-dimensional (horizontal) mass-balance model can be written for the sediment module and also can be put in the form of a differential equation. More complex models can be envisioned involving, for example, aerobic and anaerobic processes that take place in the sediment and also, the water column.
Note that transport is included on Figure 4-1 in the bottom-sediment module. Transport of bottom sediment would occur in river deltas during times of high flow (such as seasonal high flows or flood flows) or anywhere in the intertidal
zone due to episodic events such as upwelling or hurricanes. Quantifying this phenomenon would be very difficult and most likely would not result in any increase in the accuracy of tracing the fate and effects of oil in the ocean.
The Biota Module
The biota module is the most difficult of the modules to describe quantitatively. It includes both fates and effects. To better illustrate the processes that must be quantified in this complex module, the biota module shown in Figure 4-1 is illustrated in more detail in Figure 4-7.
Basically the fate of petroleum hydrocarbons in the biota module results from (1) ingestion (uptake) of one suite of petroleum hydrocarbons by an organism and subsequent excretion to the sediment or the water column of a different suite after digestion and metabolism; (2) transfer of petroleum hydrocarbons up the food chain through the predatorprey relationship; and (3) sorption or ingestion of petroleum hydrocarbons by a marine organism followed by death of the organism and return of the petroleum hydrocarbon to the water column or sediment column through biochemical decay of the organism. In addition, certain petroleum hydrocarbons will be carried away from the sea by birds, and land-based animals, when oil adsorbs on their bodies and they carry the oil away from the sea or ingest seafood and then excrete products or die onshore.
Perhaps the best way to quantitatively describe the biota module would be through the application of a trophic-level food web model that describes the movement up the food chain of petroleum hydrocarbons ingested at any given level. Writing an ingestion-growth-excretion relationship for each trophic level and coupling trophic levels with a predator-prey relationship would enable one to track the migration of petroleum hydrocarbons through the food chain and to determine the concentration of the petroleum hydrocarbons per gram of biomass at each trophic level. Ingestion into the food chain would be a sink for petroleum hydrocarbons in the water-column or bottom-sediment modules, and petroleum hydrocarbons excreted in the biota module would become sources for the water-column or bottom-sediment modules.
Computer Models
The advent of fast personal computers has enabled the development and wide distribution of a number of fairly comprehensive composite fates models that include many of the elements in the ideal conceptual model described in the pre
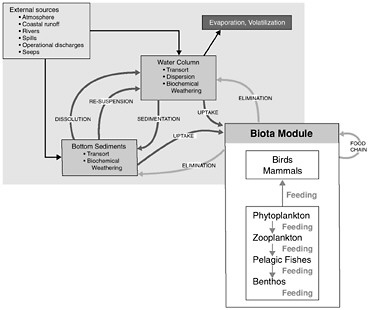
FIGURE 4-7 Detailed interactions among components of the biota module of conceptual model for the fate of petroleum in the marine environment shown in Figure 4-1.
vious section. Most of the models have focused largely on surface oil slicks. Several of these models have graphical user interfaces and integrated Geographical Information Systems (GIS) and are widely used by agencies and industry for:
-
Tactical (emergency) spill response. In the event of an accidental spill, a composite model is often used to predict where the spill will go and how it will weather. This kind of information plays a key role in determining protection priorities.
-
Strategic (contingency) planning. The models are run in a stochastic manner (multiple-runs) to develop maps of the probability a spill would impact a region given that a spill occurs.
-
Post-Spill assessment. The models are used after a spill to fill observation gaps and estimate damage. Another use is to determine the source of unknown oil. In this model, one knows where the spill ended up but needs to determine the most likely origin to identify the responsible party.
There have been two reviews of composite oil slick models published recently: ASCE (1996) and Reed et al. (1999). ASCE (1996) cites more than 50 composite slick models in existence at the time of its study. Clearly a complete review of all these models would be a major publication in itself and is beyond the scope of this report. Nevertheless a closer look at a few representative models does provide valuable insight into the present state of the art. Table 4-6 summarizes and differentiates among four widely used composite models. Some of the differences in Table 4-6 are a result of the fact that the models are intended for different purposes. SIMAP is primarily for strategic planning and perhaps post-spill assessment. OSIS and GNOME/ADIOS2 can be used for all three purposes: tactical planning, strategic planning, and post-spill assessment. OSRA is focused on strategic planning. The intent of Table 4-6 is to provide an overview of the range of models. The rows in the table indicate how each model accounts for the major processes identified in the previous sections. Two of the models are used by U.S. government agencies, while the other two are commercially available and widely used by private industry.
The National Oceanic and Atmospheric Administration has a primary responsibility in providing spill modeling expertise to the U.S. Coast Guard during major spills in U.S. waters. NOAA also works in other parts of the world on an invitation basis. NOAA actually uses two models: GNOME to account for advection, spreading, and first-order evaporation and ADIOS2 to estimate detailed weathering, dispersion, and so forth. The Minerals Management Service (MMS) model known as OSRA (Smith et al., 1982) is used to estimate spill impact probability associated with offshore oil development in the Outer Continental Shelf (OCS). This model is a key consideration in determining the potential environmental impact of future oil and gas developments. OSIS (Leech et al., 1993) is a commercial product frequently used in Europe. SIMAP (French, 1998) is another commercial product that is used in the United States especially for post-spill Natural Resources Damage Assessments
TABLE 4-6 Summary of Processes Included in Four Composite Models
|
Process |
GNOME-ADIOS2 (NOAA) |
OSIS (BMT) |
OSRA (MMS) |
SIMAP (ASA) |
|
Dimensions |
Near-surface |
Near-surface |
Near-surface |
Entire water column |
|
Advection |
Wind factor + background + stochastic uncertainty |
Wind factor + background current + wave (Elliot and Wallace, 1989) |
Wind factor + background |
External hydrodynamic model + wind factor (if not in hydro model) |
|
Horizontal dispersion |
Fickian |
Random walk based by Morales et al. (1997) |
Optional |
Heuristic method + spillets |
|
Spreading |
Modified Fay + wind component |
None |
None |
Modified Mackay et al. (1980) |
|
Emulsification |
Eley (1988) |
Mackay et al. (1980) |
None |
Mackay et al. (1982) |
|
Vertical dispersion (entrainment) |
Modified Delvigne and Sweeney (1988). Includes wave break and Langmuirs |
CONCAWE (Van Oudenhaven et al., 1983) |
None |
Delvigne and Sweeney (1988) |
|
Dissolution |
None |
None |
None |
Mackay and Leinonen (1977) |
|
Evaporation |
Modified Payne (Jones, 1997) |
Stiver and Mackay (1984) |
None |
Stiver and Mackay (1984) |
|
Oxidation |
None |
None |
None |
First order decay with heuristic components |
|
Sediment & settling |
Payne (1987) |
None |
None |
French et al. (1999) |
|
Subsurface release |
None |
None |
None |
Simple passive point source |
|
Coastal-interaction |
All oil sticks |
All oil sticks |
All oil sticks |
COZOIL (Reed and Gundlach, 1989) |
(NRDAs). SIMAP calculates effects as well as fates, but only the fates component is discussed here. OilMap is a widely used relative of SIMAP that considers only near-surface fates, but does provide a choice of process algorithms. As indicated in Table 4-6, three of the four models consider only surface waters. SIMAP is apparently the only widely available model that considers the entire water column. In addition it tracks four oil components separately:
-
monoaromatics: aromatics with molecular weights (MW) less than 100 g/mole;
-
polynuclear aromatic hydrocarbons (PAH): volatile aromatics with MW between 100 and 200 grams/mole;
-
non-aromatic volatiles (<200 g/mole MW); and
-
a residual fraction that is neither volatile nor soluble (>200 g/mole MW).
A comparison between SIMAP and the Conceptual Model of Figure 4-1 shows that SIMAP accounts for all the processes, although obviously each process submodel is often far less than ideal. The simplest model in Table 4-6 is the OSRA model, although it is more complex than can be described in a table. A multiple-step process is actually involved. The first step is to run the basic OSRA model in a Monte Carlo fashion to establish contact probability and time-to-impact contour maps. The second step is to look at weathering and dispersion of some specific spill scenarios impacting critical resources. The first step gives the probability that the spill will hit that resource along with the time to impact. Weathering for these specific scenarios is calculated using NOAA’s ADIOS2, described in part in Table 4-6.
Composite models compare reasonably well with observations. ASCE (1996) briefly compares hindcast results from several two-dimensional models with data from two actual spills (simulations for three historical spills are given, but observations are included for just two of the three). The focus is on time periods on the order of one week after a sudden surface point source spill, with observations consisting of time series locations of major patches of oil. (Comparisons are only for surface oil. No modeling was attempted for overwashed oil.) In general, the ASCE (1996) study concluded that the models evaluated did well, but also noted several major limitations should be kept in mind:
-
The comparisons are largely qualitative, and no mass-balance comparisons are possible because of the lack of comprehensive field observations.
-
The models are run in “hindcast” mode, i.e., after the fact. Actual wind and especially current measurements are minimal; thus the current and wind inputs into the model are modified so that the modeled slicks track the observed one as closely as possible.
-
If the models are run in a “forecast” mode in which future winds and currents must be estimated, then model forecasts will often deviate substantially from observations.
The cases considered by ASCE (1996) are quite limited in the context of the greater problem of “Oil in the Sea.” For example, these cases did not consider subsurface releases, continuous point sources, non-point source releases, or long-term (greater than one week) fates (for more details see ASCE [1996]). French (1998) provided some comparisons between fates and effects calculated from SIMAP, as observed during the North Cape oil spill. Comparisons were generally good.
FATE OF OIL INPUT
Table 4-7 is a summary of the fate processes that affect petroleum hydrocarbons from the seven major input categories. Each input is ranked using a scale of high, medium, and low that indicates the relative importance of each fate process. Table 4-7 was developed by consensus of the committee and is based on many assumptions. It is intended to provide only a general idea of the relative importance of the fates processes. Clearly one of the biggest problems in developing a table such as this is that the importance of a particular fate process will depend on the details of the event. The committee has tried to account for this to a limited extent in the case of accidental spills by including subcategories for various oil types. With these caveats in mind, an explanation of each of the fates is as follows.
Evaporation-volatilization is ranked according to the relative volume of the release that would be lost by net transport from the sea surface to the atmosphere. For example, gasoline would have “high” evaporation whereas a heavy crude would be “low.” Evaporation has been ranked “high” for two-stroke engine inputs, which consist largely of unburned gasoline. Emulsification rankings are driven largely by the oil type whereby gasoline, which has no emulsification potential, would have a low ranking, while a medium, fresh-crude could have a high ranking, although this depends on the specific crude composition.
Dissolution rankings consider the total water-soluble fraction, the rate of dissolution, and the rate of volatilization from the water, reflecting the relative potential of releases to impact water-column resources.
Oxidation rankings reflect the relative rate and extent of oil removal by microbial and photooxidative degradation for those oils that have moderate persistence in the marine environment. Thus, releases of crude oils are ranked “medium” because microbial degradation is a significant weathering process for the intermediate-weight hydrocarbon components in crude oil, whereas releases of heavy, weathered oils are ranked “low” because they are recalcitrant to microbial and photodegradation. In contrast, light oils such as gasoline and light distillates are mostly lost by evaporation-volatilization (Figure 4-2) and not to oxidation, and are ranked as not relevant.
Horizontal transport is a combination of spreading, advection, and horizontal dispersion, and the rankings are
TABLE 4-7 Processes that Move Petroleum Hydrocarbons Away from Point of Origin
driven by the persistence of the release on the water surface as well as the areal extent of the input. Thus, even though atmospheric inputs are expected to have low persistence because they volatilize quickly, they are introduced over large areas compared to point sources such as spills. Heavy oils and seeps form residues that can persist at sea for long periods.
Vertical transport and movement include the processes of vertical dispersion, entrainment, Langmuir circulation, sinking, and overwashing. Rankings reflect the potential for mixing into the water column by natural processes, given that very light oils evaporate quickly and heavy oils are too viscous to disperse naturally. Sedimentation rankings consider the suspended sediment concentrations at the release sites and the potential for the oil to adhere to sediments after stranding onshore or by mixing with suspended sediments.
Shoreline stranding is ranked highest for those petroleum hydrocarbons that persist on the water surface and are likely to be released close to shore, increasing the potential for a significant amount of the released oil to strand. Tarball formation, like shoreline stranding, is ranked highest for releases of crudes and heavy oils that form persistent residues.
Seeps
Crude oil released into the marine environment through natural seepage undergoes most of the same physical and chemical process as crude oil released into the ocean at the seafloor. One main difference is in the rate of addition of oil to the environment. With natural oil seeps, the leakage rate is relatively low and chronic. On the other hand, oil spills result in a release that is a sudden, one-time event. The same basic processes act to degrade and remove oil with time, although in the case of seeps, crude oil is replenished as long as the seeps remain active. Thus, the persistence of natural oil seeps is reckoned in terms of years.
Ranking the fate processes that move petroleum hydrocarbons away from natural oil seeps is difficult because these seeps occur worldwide in numerous geographic settings. In the four major weathering processes of evaporation, emulsification, dissolution, and oxidation, they are ranked “medium.” The overall ranking is similar to that of crude oil spills. Also, comparison between natural seeps and crude oil spills show the similarities in rankings between the processes of transport, sedimentation, shoreline stranding, and tarball formation.
Spills
Spills range widely in oil type, spill size, location, and environmental conditions during the release.
Gasoline
With a very low viscosity, gasoline spills spread rapidly as thin sheens. Gasoline is also light, with a specific gravity of about 0.8, so it causes a slick on the sea surface, or rapidly re-floats if dispersed into the water column. Evaporation-volatilization is the dominant process affecting spills on the water surface and may eliminate nearly all of the spilled gasoline within a few hours to a day. As a result, gasoline spills in marine waters have low persistence. There is little potential for spreading, mixing into the water column, sedimentation, or stranding, and no risk of forming emulsions or
tarballs. Even though gasoline has the highest water solubility of all oil types, dissolved concentrations under slicks decrease rapidly by evaporation.
Light Distillates
Light refined products, such as diesel, No. 2 fuel oil, jet fuels, and kerosene, are narrow-cut fractions that have low viscosity and spread rapidly into thin sheens. They do not tend to form emulsions except under very cold conditions. They evaporate more slowly (compared to gasoline) and incompletely; therefore, they are ranked as “medium” in terms of their horizontal transport or movement. As low-viscosity, moderately persistent oils, light distillates tend to disperse readily into the water column by even gentle wave action. Thus, they have the highest potential of any oil type for vertical mixing. There is also a greater potential for dissolution to occur, from both surface sheens and droplets dispersed in the water column. The water-soluble fractions are dominated by two- and three-ringed PAH, which are moderately volatile and may affect aquatic biology. Thus, spills of light distillates have the greatest risk of impacting water-column resources. Light distillates are not very adhesive; therefore, they do not adhere strongly to sediments or shoreline habitats. Loading levels on the shoreline are relatively low because of the thinness of sheens on the water surface and the low adhesion of stranded oil. The constituents of these oils are light to intermediate in molecular weight and can be readily degraded by aerobic microbial oxidation. Long-term persistence in sediments is greatest under heavy loading and reducing conditions where biodegradation rates for anaerobic bacteria are low.
Crude Oils
Crude oils contain a wide range of compounds, from light to heavy; thus, they are affected by many fate processes. Evaporation can remove about one-third of the volume of a medium crude oil slick within the first day, but there will always be a significant residue. Many crudes will emulsify readily, a process that greatly reduces subsequent weathering rates. As a result, crude oil spills close to shore often strand and persist on shorelines, particularly on permeable substrates such as gravel beaches and sheltered habitats such as marshes. Crude oils tend to adsorb heavily onto intertidal sediments, with the risk of subsequent erosion of oiled sediments from the shoreline and deposition in nearshore habitats. Under high-energy, nearshore conditions, oil and sediments can mix and be transported to the bottom sediments. For spills that are transported offshore, the slicks eventually break up into fields of tarballs that can be transported long distances because they are so persistent. The water-soluble fraction of crude oils include a wide range of PAH. Dissolution from slicks and stranded oil can persist for weeks to years.
Heavy Distillates
These oil types, such as No. 6 fuel oil, bunker C, and heavy slurry oils, lose only up to 10 percent of their volume via evaporation. Some products are so viscous that they cannot form emulsions, but many emulsify shortly after release. They show low natural dispersion because the oil is too viscous to break into droplets. These oils have the lowest water-soluble fraction; thus, loadings to the water column are generally low under slicks. Spills of heavy distillate quickly break up into thick streamers and then fields of tarballs that are highly persistent. The heavy distillate can be transported hundreds of miles, eventually stranding on shorelines and posing significant impacts to birds and other marine animals such as turtles. Because of their high density, these releases are more likely to sink after picking up sediment, either by mixing with sand in the surf zone or after stranding on sandy shorelines. Some heavy distillates are so dense that they are heavier than brackish or sea water and will not float when spilled.
Produced Water
Large volumes of produced water are discharged in offshore oil-producing areas. Produced water is treated to remove most free oil prior to discharge; however, the water still contains a moderate amount of soluble and volatile petroleum hydrocarbons. The concentrations of benzene, toluene, ethylene and xylenes (BTEX) in produced water vary by almost a factor of 10, with an average concentration of about 5 mg/L. The polynuclear aromatic hydrocarbons (PAH) also vary by about an order of magnitude, with an average concentration of about 1 mg/L. Because these compounds are already in solution, their concentrations are reduced rapidly by volatilization and dilution, particularly when released to open, well-mixed waters. Elevated levels of contaminants in sediments typically extend 100-200 m from the discharge point. There are occasional events in the treatment process that result in light sheens, but they are expected to disperse rapidly with a low risk of stranding on shorelines and no risk of emulsification or tarball formation.
Vessel Operational Discharges
Operational discharges from vessels generally occur more than 50 miles from shore and are concentrated along shipping lanes. Discharges are composed of bilge water, cargo tank washings, fuel oil sludge, and other oily wastes, which are all considered moderate in terms of their loss by evaporation and dissolution, formation of tarballs, and potential for long-distance transport. Vessel discharges pose a low risk of vertical mixing because the releases are generally viscous. Because these discharges are released in offshore waters, there is low potential for contact with sediments and sedi
mentation, and shoreline stranding is a concern only where shipping lanes pass close to shore.
Recreational Marine Vessels
Fuel for two-stroke outboard engines is a mixture of gasoline and lube oil in volume ratios varying from 20:1 (5 percent) in older engines to 50:1 (2 percent) in newer models. The bulk of the fuel, gasoline, is comprised of the lighter, molecular-weight fraction (e.g., BTEX) and volatilizes from the surface water. The rate of volatilization is temperature dependent, but the product will remain for several minutes to hours at 15ºC, given the amount of gasoline that is emitted from two-stroke engines in coastal waters and the time of year that they are used (usually during warm periods, when biological productivity is highest). The potential effect from toxins, such as PAH, in unburned gasoline and lubricating oil, on the biota including larvae and phytoplankton is large (see Chapter 5 for greater discussion of potential environmental effects from the release of refined petroleum products). To date there are no published field studies evaluating the effect of gasoline released from the operation of two-stroke engines.
The lube oil mixed with gasoline forms the sheens and slicks that trail behind two-stroke engines during operation. Evaporation and dissolution are the most important fate mechanisms. After two days, nearly 75 percent of the lubricating oil can evaporate at 15ºC (Figure 4-2). A smaller portion of the light lube oil can remain on the surface marine microlayer for longer periods (days) depending on environmental conditions including physical, chemical, and biological processes.
Land-based Sources
Unlike the discharge of liquid petroleum, hydrocarbons that enter the coastal ocean from land-based sources via rivers have already undergone considerable biogeochemical weathering. Land-based sources result from petroleum inputs to streams and rivers and subsequent transport to surface coastal waters. This transport is selective, with more water-soluble and stable components of the petroleum mixture carried downstream. During riverine transport, the petroleum mixture can undergo further weathering, including evaporation-volatilization and microbial degradation, such that the material reaching the coastal ocean is likely more stable and recalcitrant than the original mixture.
In addition to weathering between the release point and the coastal ocean, the nature of the river transport will play a major role in the magnitude and fate of petroleum products reaching the ocean. A good example is the differential behavior of petroleum transport in the Columbia River and the Chesapeake Bay. The Columbia is a large, relatively fast-flowing river whose plume discharges directly into the coastal Pacific. Petroleum hydrocarbons entering the Columbia River are likely transported rapidly to the coastal ocean, with relatively little retention within the river basin. The Chesapeake Bay, on the other hand, is a shallow, productive, semi-enclosed estuary with a long water residence time and a well-characterized ability to trap eroded solids. Due to its large surface area-to-volume ratio and its relatively high sedimentation rates, the Chesapeake Bay is likely to efficiently transport petroleum hydrocarbons entering from the tributaries. While this trapping reduces the loadings of petroleum hydrocarbons to the coastal oceans, it may result in locally enriched hydrocarbon levels in estuaries and other embayments. (Note that in this report, tidal embayments are included as part of the coastal ocean, so these removal processes in estuaries would be counted as “losses” from the coastal ocean.)
Atmospheric Deposition
Petroleum hydrocarbons enter the coastal ocean from the atmosphere by wet deposition (scavenging of atmospheric hydrocarbons by precipitation), dry aerosol deposition (transport of marine aerosol particles to the sea surface), and gas exchange. Of these three, it is estimated that gas exchange dominates the total gross loading of hydrocarbons from the atmosphere. Since gas exchange results from the dissolution of gaseous hydrocarbons in sea water, the magnitude of its flux depends on the concentration in the gas phase and the solubility of the hydrocarbon in sea water. Unlike the other sources discussed in this report, atmospheric deposition supplies hydrocarbons somewhat uniformly to the coastal ocean at relatively low loading rates over large areas.
Analysis of the concentrations of petroleum hydrocarbons in the coastal ocean indicates that the surface waters are greatly oversaturated with n-alkanes with respect to the overlying atmosphere. All of the input sources discussed in this report lead in varying degrees to these ambient concentrations in the coastal ocean. Volatilization is the dominant fate process for petroleum hydrocarbons. Terrestrial hydrocarbon loadings (land-based sources) and other nearshore sources support dissolved hydrocarbon loadings in coastal waters that far exceed the loadings in equilibrium with the atmosphere. Hydrocarbon degassing to the atmosphere from coastal water is therefore a major geochemical process.
Summary
The behavior and fate of crude oil and refined products in the marine environment are controlled by many different processes that vary considerably in space and time. Physical, chemical, and biological processes all interact to (1) alter oil introduced into the oceans; (2) transport the resulting degradation (weathering) products away from the source; and (3) incorporate the residual substances into compartments of the earth’s surface system. These compartments involve disso

PHOTO 18 Refinery capacity is an important factor in gasoline prices. However, such facilities are also sources of petroleum spills and atmospheric releases of volatile organic compounds that can play a role in local air quality. In the United States most refineries are located near marine transportation terminals along the Gulf of Mexico and the northeastern Atlantic seaboard. (Photo courtesy of Environmental Research Consulting.)
lution in the hydrosphere, deposition in the lithosphere, volatilization into the atmosphere, and ingestion by organisms in the biosphere. Physical processes degrading oil include evaporation, emulsification, and dissolution, whereas chemical processes focus on photooxidation and biological processes emphasize microbial oxidation.
The transportation of oil in the marine environment occurs in two directions, horizontal and vertical. Horizontal transport involves spreading and surface advection, leading in some instances to shoreline stranding and tarball formation. Transport in the vertical direction includes dispersion, entrainment, Langmuir circulation, sinking, overwashing, and sedimentation. Consideration is also given to oil in icy conditions and oil released in deep water.
Conceptual models can be developed to build deterministic models for specific oil loadings for specific sources. The development and distribution of composite fate models, up to now, focus largely on surface oil slicks.
Oil entering the marine environment comes from natural sources (oil seeps) and from sources over which humankind has some control (oil spills, urban runoff, pollution resulting from oil transportation and production, and oil usage in ve
hicles, including boats). The ultimate fates of oil in the sea depend on the amount and rate of discharge, composition, source, and environmental setting and persistence.
The effect of petroleum hydrocarbon is not directly related to the volume released. It is instead a complex function of the rate of release, the nature of the released hydrocarbon, and the local physical and biological ecosystem. Some progress has been made in understanding the basic processes affecting fates such as evaporation. Much more needs to be learned about oil-sediment interaction, vertical dispersion and entrainment, dissolution, Langmuir cells, and hydrate formation (as related to deep subsurface releases of gas). Furthermore, the priorities for research into petroleum hydrocarbon fate and transport have historically been driven by large spills. Thus, resource allocation to support these efforts tends to wane in periods during which a large spill has not recently occurred. Federal agencies, especially NOAA, MMS, the U.S. Coast Guard, and the USGS, should work with industry to develop and support a systematic and sustained research effort to further basic understanding of the processes that govern the fate and transport of petroleum hydrocarbons released into the marine environment from a variety of sources (not just spills).
Response plans depend heavily on site-specific modeling predictions of the behavior of spills of various sizes and types, under a variety of environmental conditions. There is a need for both better baseline data, including ambient background levels of hydrocarbons in the sea, and better data for calibrating fate and behavior models. Because experimental release of petroleum is not feasible under most circumstances, comprehensive data on the fate of the oil must be collected during spills. Such efforts are generally neglected, because moving needed equipment and personnel to spill sites to collect data naturally is of lower priority than containing the spill and minimizing damage to the environment and property. Federal agencies, especially the U.S. Coast Guard, NOAA, and EPA should work with industry to develop a more comprehensive database of environmental information and ambient hydrocarbon levels, and should develop and implement a rapid response system to collect in situ information about spill behavior and impacts.
Natural seep systems and sites of historical spills offer good opportunities for field studies of the fate and effect of the release of crude oil and (in the case of spills) refined products, especially to understand dissolution and long-term weathering. Federal agencies, especially the USGS, NOAA, EPA, and MMS, should develop and support targeted research into the fate and behavior of hydrocarbons released to the environment naturally through seeps or past spills.






























