
This paper was presented at a colloquium entitled “Genetic Engineering of Viruses and of Virus Vectors,” organized by Bernard Roizman and Peter Palese (Co-chairs), held June 9–11, 1996, at the National Academy of Sciences in Irvine, CA.
Epstein-Barr virus vectors for gene delivery to B lymphocytes
ERLE S. ROBERTSON*, TADAMASA OOKA†, AND ELLIOTT D. KIEFF*‡
*Virology Program, Departments of Microbiology and Molecular Genetics and Medicine, Harvard Medical School, Boston, MA 02115; and †Laboratoire de Virologie, Moléculaire, Faculté de Médecine R.Laënnec, Université Lyon 1, C.N.R.S-UMR30, Rue Guillaume Paradin, 69372 Lyon, Cedex 08, France
ABSTRACT Basic research in Epstein-Barr virus (EBV) molecular genetics has provided means to maintain episomes in human cells, to efficiently deliver episomes with up to 150 kbp of heterologous DNA to human B lymphocytes, and to immortalize human B lymphocytes with EBV recombinants that can maintain up to 120 kbp of heterologous DNA. Episome maintenance requires an EBV nuclear protein, EBNA1, whereas immortalization of cells with EBV recombinants requires EBNA1, EBNA2, EBNA3A, EBNA3C, EBNALP, and LMP1. EBV-derived vectors are useful for experimental genetic reconstitution in B lymphocytes, a cell type frequently used in establishing repositories of human genetic deficiencies. The ability of EBV-infected cells to establish a balanced state of persistence in normal humans raises the possibility that cells infected with EBV recombinants could be useful for genetic reconstitution, in vivo.
The Epstein-Barr virus (EBV) genome has yielded reagents that have been useful in vector design. Genetic engineering of specifically mutated EBV recombinants has also had a major impact on basic EBV research and is useful for experimental genetic reconstitution in human cells. The purpose of this article is to review previous work, indicate potential utilities and liabilities, and describe recent experiments that should enable intensive investigation of an under explored and important part of the EBV genome.
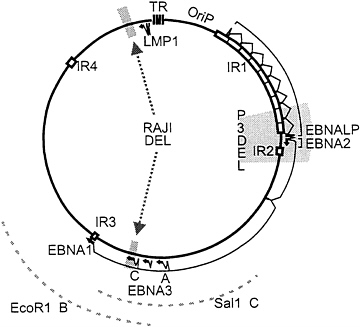
EBV is similar in its replication to other herpes viruses and many of the genetic approaches have substantial precedence in alpha herpes virus research. However, EBV also differs from alpha herpes viruses in that it establishes latency in and alters the growth of human B lymphocytes (for reviews, see refs. 1 and 2). These properties derive in large measure from a unique set of genes that encode nuclear and integral membrane proteins that have been given the acronyms, EBNAs (EBV nuclear antigens) and LMPs (latent membrane proteins). In initiation of latent infection in B lymphocytes, EBV first expresses EBNALP and EBNA2. EBNA2 upregulates the EBNA promoter leading to a longer primary transcript from which EBNALP, EBNA2, EBNA3A, EBNA3B, EBNA3C, and EBNA1 transcripts are derived (Fig. 1). EBNA2 also activates the LMP1 and LMP2 promoters. These proteins act in concert to alter B-lymphocyte growth and enable the maintenance of the EBV genome as a multicopy episome in a state of latent infection. Each of these proteins expressed in latently infected cells, with the exception of EBNA1, has epitopes that are presented on the B-cell surface in the context of common class I major histocompatibility complex (MHC) molecules and are recognized by immune human cytotoxic T lymphocytes. This high level of cytotoxic T-cell recognition and the ability of latently infected cells to shift between full latent gene expression with cell proliferation and an EBNA1 only type of latent infection that is immunologically privileged enables latently infected cells to achieve a balanced state of long-term persistence in humans. This state of long-term persistence could be useful for genetic reconstitution.
Orip/EBNA1-Based Vectors. The first EBV-derived reagents useful in vector development were EBNA1 and orip (5). Based on the knowledge that the EBV genome persists in latently infected cells as a multicopy episome (6), the EBV DNA cis-acting factor responsible for episome persistence was identified by screening for an EBV DNA sequence that would enable a heterologous plasmid with an expression cassette for toxic drug degradation to efficiently transform latently infected cells to toxic drug resistance. Plasmids containing an EBV DNA segment, designated plasmid origin or orip, transformed EBV-infected cells with higher efficiency than plasmids without the EBV DNA. Expression of EBNA1 is all that is required to enable an orip-containing plasmid to be maintained in cells without the rest of the EBV genome. Two molecules of EBNA1 bind to a 30-bp palindrome that is tandemly repeated 21 times and to a neighboring dyad repeat of the same sequence. In the presence of EBNA1, the dyad repeat is a bidirectional replication origin, while the tandem repeats enhance transcription from the episome and terminate DNA replication. The episome copy number varies and persistence in dividing cells over multiple divisions is dependent on continuous selection. Over time the episome DNA can integrate into chromosomal DNA (1).
Amplicon (Orip/EBNA1/orilyt/Terminal Repeat)-Based Vectors. Orip-containing plasmids have been further modified into amplicons that can be expanded and packaged into EBV in cells containing a replication-competent EBV genome that are permissive for EBV replication. The strategy is similar to that developed for herpes simplex virus (7). An EBV DNA lytic replication origin is all that is necessary for a plasmid to replicate when lytic EBV infection is induced. Linear head to tail concatemers are produced from the template plasmid DNA. The presence of the EBV terminal repeat sequence in the plasmid DNA enables the linear DNA concatemers to be packaged into virions and cleaved into head-full linear DNA molecules of 150–200 kbp. The addition of an EBNA1 expression cassette to the amplicon enables the packaged DNA to persist as an episome in infected human cells. The multiple tandem repeats of the original packaged plasmid DNA tend to persist in the infected cells as a head-full-size episome. Since all of the necessary components of the amplicon contain less than 10 kbp, the payload of heterologous DNA sequence(s) can be as large as 170 kbp. There is therefore ample space to include promoters for gene expression at the appropriate level in human B lymphocytes. Several genes or a large human gene in its own regulatory environment can be efficiently delivered
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. §1734 solely to indicate this fact.
Abbreviations: EBV, Epstein-Barr virus; EBNA, EBV nuclear antigen; BL, Burkitt lymphoma; LCL, lymphoblastoid cell line.
|
† |
To whom reprint requests should be addressed, e-mail: ekieff@bustoff.bwh.harvard.edu. |
to B lymphocytes. P3HR-1 cells are a suitable packaging cell lines since the endogenous EBV genome is transformation-incompetent and replication-competent. Although the assembly and transfection of very large amplicons into a latently infected lymphoblast packaging cell lines is inefficient, successful transfectants can be selected by using an amplicon-based positive selection marker and the transfected cells can be expanded in large amounts and fully characterized (1, 5). Such cell lines tend to be stable over several months of continuous positive selection. The induction of lytic replication results in amplification and packaging of linear concatemers of the amplicon. The endogenous EBV genome is also packaged into virus. Although the host range of EBV and of the amplicon packaged virus, in culture, is limited to B lymphocytes, EBV-transformed B lymphocytes are derived from genetically deficient humans to retain immortal cell lines with the specific genetic deficiency. Amplicon-based vectors can, therefore, be used for experimental reconstitution of genetic deficiencies in these cells, in vitro. EBV amplicons have recently been used to correct the TAP1 or 2 deficiency in B lymphoblasts derived from patients with type 1 diabetes (8). In a substantial fraction of cases, restoration of TAP1 expression corrected the abnormally low class I MHC expression that is characteristic of type 1 diabetes. An amplicon vector has also been used to correct an enzymatic defect (9).
The principal limitations of EBV-amplicon-based systems are their restricted host range, the production of replication competent endogenous EBV by the packaging cell line, and the possible effect of EBNA1 on cell growth. EBNA1 is a sequence-specific DNA binding protein that has transcriptional transactivating effects; an effect on transcription of cell genes could have important biological consequences. EBV amplicons packaged in P3HR-1 cells are mixed with P3HR-1 EBV. Although the P3HR-1 EBV cannot immortalize primary human lymphocytes, P3HR-1 is replication-competent and encodes all proteins that are important in EBV-mediated cell growth transformation except for EBNA2 and EBNALP. The EBV host range could likely be expanded by modification of the glycoprotein composition of the EBV outer envelope encoded by the packaging cell line. Gp350 is a highly specific ligand for CD21 that is abundantly expressed only on B lymphocytes. Modification of the ligand determinant or inclusion of other glycoproteins could broaden the host range. Gp350 can for example be incorporated into envelopes along with glycoproteins of varicella zoster virus (10).
Recombinant EBV-Based Vectors: Positive Selection Markers. Recent strategies for evaluating the effects of site-specific mutations in the viral genome have enabled a focused assessment of the role of specific genes and intragenic elements in primary B-lymphocyte growth transformation and in lytic EBV infection. The favored approach has been to introduce an EBV DNA fragment with a selectable marker into latently infected cells along with an expression vector for the Z immediate early transactivator of lytic EBV replication (1). Z induces lytic infection and the replicating EBV genome can recombine with the transfected EBV DNA fragment. The progeny virus could then be harvested and plated onto lymphocytes at a low multiplicity, and recombinant infected cells could be identified by genetic selection or marker-specific PCR (11–13). One strategy is to transfect a cell carrying a wild-type latent EBV genome with a mutated EBV DNA fragment carrying an expression vector for a gene that encodes an enzyme that inactivates a toxic drug (11, 12, 14–16). Progeny virus can then be used to infect primary B lymphocytes or a non-EBV-infected continuous B-cell line such as an EBV-negative Burkitt lymphoma (BL) cell line. Cells infected with the recombinant EBV can then be positively selected by growing the infected cells in the presence of the toxic drug (11, 12, 14–16). An advantage of this strategy is that mutations can be made in a gene that is essential for primary B-lymphocyte transformation and the mutant progeny genome can be positively identified by infection of BL cells and by plating of the infected cells under selective conditions (11, 12, 16). The weakness of the BL-cell approach is that BL cells are less susceptible to EBV infection than primary B lymphocytes. Further, reactivation of lytic infection from BL cells is more difficult than from primary B lymphocytes (11, 12, 16).
Recombinant EBV-Based Vectors: Marker Rescue for Transformation. The favored approach for most recombinant EBV molecular genetic analyses has been to use cells infected with the replication-competent but transformation-incompetent P3HR-1 EBV strain as the parent for positive selection by marker rescue of transformation. The P3HR-1 EBV genome is deleted for a DNA segment that includes the last two exons of EBNALP and the EBNA2 exon (16, 17). Recombination of P3HR-1 EBV with a transfected wild-type EBV DNA fragment spanning the EBNALP and EBNA2 deletion was, therefore, readily identified by the unique ability of the recombinant progeny virus to transform primary B lymphocytes into long-term lymphoblastoid cell lines (LCLs). The recombinant viral DNA can be characterized in the resultant LCL clones. Virus replication can be induced in these clones and the properties of the recombinant virus can be assayed on infection of new primary B lymphocytes. This has been a simple and relatively reproducible marker rescue strategy so that the efficiency of marker rescue of various specifically mutated EBNALP/ EBNA2 DNA fragments can be compared with wild-type DNA after the initial transfection into P3HR-1 cells. Null mutations in EBNALP/EBNA2 can be inferred by the consistent inability of several isogenic DNA fragments to marker rescue transforming virus in controlled experiments.
Recombinant EBV-Based Vectors: Second Site Homologous Recombination to Construct Specific Mutations at any Site in the EBV Genome Other Than EBNALP/EBNA2. Surprisingly, the same marker rescue of transformation strategy could be used to select for recombination at any other site in the EBV genome. A nonlinked second cotransfected EBV DNA fragment was incorporated into at least 10% of the P3HR-1 genomes that had recombined with the EBNA2 and EBNALP DNA fragment (18). EBV second-site recombinants that had acquired a mutation that did not interfere with transformation were readily derived since a total of 103 marker-rescued recombinants could be obtained from a single transfection and
more than 102 were recombinant at the site of the second cotransfected EBV DNA segment. Since P3HR-1 EBV is wild type at all sites other than the EBNALP/EBNA2 deletion, P3HR-1 EBV can also be used to transcomplement and rescue nontransforming mutations at second sites (18, 19). Thus, using this approach, specific mutations could be introduced anywhere in the genome except for nontransforming mutations within the EBNALP/EBNA2 DNA fragment (20).
Recombinant EBV-Based Vectors: EBV Cosmid or F-Factor-Derived Clones to Reconstruct Intact or Mini-Transforming EBV Genomes. Based on the observations that the pseudorabies virus genome can be reconstructed from overlapping clones of pseudorabies virus DNA, EBV genomes have been largely or completely reconstructed from overlapping cosmid clones of EBV DNA. After futile attempts to obtain lytic infection with overlapping EBV DNA cosmids in noninfected cells, P3HR-1 EBV-infected cells were used so that the endogenous P3HR-1 EBV could provide lytic replication and packaging functions in trans (17, 21, 22, 23). By checking seven sites characteristic of the transfected cosmid cloned EBV DNAs, it was found that approximately 10% of the resulting transforming recombinants were composed of only transfected cosmid DNA (20). The frequency of incorporation into recombinants of any single transfected EBV DNA fragment was twice as high with overlapping cosmid transfection as with second-site recombination (24). Most interestingly, a 12-kbp deletion in the transfected EBV DNA was much more frequently incorporated into recombinant genomes than occurred by second-site recombination (20). A much larger deletion was engineered by deleting from the transfected EBV DNA most of the 58 kbp of DNA between the EBNA1 and LMP1 open reading frames (Fig. 1). Only three overlapping cosmids were necessary for constituting this deleted EBV genome. More than 20% of the transforming recombinants that arose after transfection of P3HR-1 EBV-infected cells with the three cosmid EBV DNA clones had markers indicative of only the transfected EBV DNAs.
A further variation of this strategy is to assemble transformation competent, replication incompetent, mini-EBV genomes from several cosmid clones of EBV DNA cotransfected into cells carrying the P3HR-1 EBV or to assemble similar genomes in Escherichia coli as an F-factor-based plasmid that can then be packaged in cells carrying the P3HR-1 EBV (13, 23, 24). Both approaches have demonstrated that noncoding exons and introns of the long EBNA transcript and most of the lytic genes between EBNA1 and LMP1 can be deleted (13, 23, 25). An analysis of all the deletions made in the EBV genome demonstrates that the residual complexity of the genome required for transformation of a B lymphocyte in vitro is approximately 48 kbp, or 26% of the genome (14, 25–31). The construction of mini-transforming EBV genomes with large segment(s) of heterologous DNA could be independent of P3HR-1 since direct transformation of primary B lymphocyte with a cosmid DNA should be feasible given the small residual complexity of the transforming EBV genome. Progress will likely continue to be driven by the need for more efficient molecular genetic analyses in basic EBV research.
Mini-EBV genomes are potentially useful to make EBV recombinants that include 100–110 kbp of heterologous DNA; such mini-EBV genomes could ultimately have a place in genetic reconstitution. Mini-EBV genomes carrying a human gene or gene complex could be used to immortalize primary B cells of an individual suffering from a particular genetic defect. Transforming mini-EBV genomes would be unable to replicate and spread. Their particular utility as opposed to EBNA1/ orip-based approaches resides in the role of the EBNAs and LMPs in achieving a dynamic balance between renewal of latently infected cells and their destruction by immune T lymphocytes in normal humans. EBV-infected primary B lymphocytes would be expected to be long-lived latently infected lymphoblasts similar to those in natural EBV infection. EBV infection is largely nonpathogenic and almost all adults harbor EBV. In EBV-infected humans, latently infected cells constitute 1 in 10−5 to 10−6 circulating B lymphocytes and appear to be more prevalent in tonsils and other lymphoid organs. A substantial number of latently infected cells can persist over a long time. The persistence of latently infected cells in normal humans appears to be dependent on the ability of EBV-infected cells to be maintained in a dynamic balance between an EBNA1-only type of latency, which is nonproliferative but immunologically “privileged,” and an EBNALP, 2, 3A, 3B, 3C, LMP1, and LMP2 type of latency in which the cells are driven to proliferate but are subject to immune surveillance. The switch between the two types of latency appears to be dependent on the activity of the Wp/Cp promoter for EBNALP, 2, 3A, 3B, and 3C transcription, with the EBNA1-only promoter being a downstream default promoter. Regulation of the two types of latency could in theory be achieved by replacing the Wp/Cp EBNA promoter with a pharmaco-logically regulated promoter. Since each old world primate has an endogenous EBV-related agent, experimental studies, in vivo, are feasible. The relationship of EBV with malignancies is an important problem despite the very low frequency of EBV-related malignancy in normal people. Latently infected cells could be physically contained in a filter-bound unit, in vivo; this might assure an extra margin of safety against escape of these cells from immune surveillance. Specific hereditary defects that can be corrected through the release of wild-type protein into the peripheral circulation are the most likely targets for mini-EBV-transformed cell reconstitution. Correction of disorders in non-B lymphocytes is considerably more complicated since EBNA2 interacts with the B-cell-lineage-specific transcription factor PU.1 in achieving regulated EBV gene expression.
EBV Genes Essential for Primary B-Lymphocyte Growth Transformation. The central objective of most EBV-recombinant-based experimentation has been for genetic analysis of the role of EBV genes in latent growth transforming infection. The first marker rescue of transformation experiments from P3HR-1 cells demonstrated the importance of the EBNA2 open reading frame and of the last two exons of EBNALP in primary B-lymphocyte growth transformation (7, 21, 32). Subsequent extensive mutagenesis analyses of the EBNA2 open reading frame leave only seven prolines near the amino terminus, the domain that interacts with EBNA2 response element specific cellular DNA binding proteins RBPJkappa and PU.1, and the carboxyl-terminal acidic transactivating domain as being important for primary B-lymphocyte growth transformation (6, 7, 21, 33, 34). Thus, recombinant-EBV-based EBNA2 genetic analyses indicate that EBNA2 is essential for transformation because of its role as a transcriptional transactivator. EBNA2 binds to cellular transcription factors that recognize specific DNA sequences; EBNA2 then assembles basal and activated transcription components at nearby promoter sites.
Using second-site homologous recombination, the effect of a single nonsense mutation in EBNA3A, EBNA3B, or EBNA3C has been evaluated. LCLs infected with an EBV recombinant with a stop codon inserted at aa 109 in the EBNA3B reading frame arose at the expected frequency indicating from the start that EBNA3B is not critical for growth transformation of primary B lymphocytes in the context of an otherwise normal EBV genome. The LCLs infected with the EBNA3B stop codon recombinant were indistinguishable in their growth from wild-type-recombinant-infected LCLs and were similar in their response to activation of lytic EBV infection. These experiments demonstrate that the last 829 codons of EBNA3B are not important in primary B-lymphocyte infection or growth transformation in vitro (19). The dispensability of EBNA3B in tissue culture highlights a
limitation of these transformation assays. EBNA3B engenders strong cytotoxic T-cell responses in humans and is unlikely to have persisted in the viral genome unless it is important role in viral infection in vivo (1, 2).
In contrast to EBNA3B, stop codons inserted into the EBNA3A reading frame after codon 302 or into the EBNA3C reading frame after codon 365 only resulted in transforming recombinants when the recombinant-infected cells were coinfected with P3HR-1 EBV, which can provide wild-type EBNA3s in trans (24). Mutant EBNA3A recombinant EBV coinfected LCLs rapidly lost the mutant EBNA3A gene through secondary recombination between the mutant EBNA3A and wild-type EBNALP and EBNA2 EBV recombinant and the coinfecting P3HR-1 EBV genome (which is wild type for EBNA3A and deleted for EBNALP and EBNA2). These data suggest that EBNA3A is important in primary B-lymphocyte growth transformation and that expression of the first 302 amino acids has a negative effect on cells so that there is selection against its retention (24). Transfections of P3HR-1 infected cells with an F-factor-derived plasmid containing the known transforming regions of the EBV genome and a frameshift mutation in codon 304 of EBNA3A has provided additional evidence for a critical role in that almost all LCLs that arose were either coinfected with P3HR-1 EBV or were recombinant and wild type at EBNA3A (25). Two LCLs were isolated that lacked wild-type EBNA3A, indicating that the last 640 amino acids of EBNA3A are not absolutely essential for the maintenance of LCL growth.
An EBV recombinant with EBNA3C mutated by a nonsense codon insertion after codon 365 was extensively tested for its ability to transform primary B lymphocytes with or without added P3HR-1 to provide wild-type EBNA3C in trans. LCLs containing the mutant EBNA3C arose only when P3HR-1 was provided in trans (86 coinfected versus 0 singly infected). These results indicate that the last 627 codons of EBNA3C are critical for primary B-lymphocyte growth transformation (24). Biochemical data indicate that the EBNA3s are modulators of transcription of cell and viral genes that have RBPJkappa binding sites (35).
The integral membrane protein LMP1 is also essential for primary B-lymphocyte growth transformation. EBV recombinants with a stop codon after codon 9 or 84 could only transform primary B lymphocytes when wild-type LMP1 was provided in trans by P3HR-1 coinfection. The mutant open reading frame recombinants expressed N-terminally truncated LMP1s initiated at internal methionines. When virus was transferred from the open reading frame stop codon recombinant LCLs to primary B lymphocytes, mutant recombinant genomes only initiated growth transformation when wild-type LMP1 was encoded in trans by P3HR-1 coinfection (271 coinfected versus 0 singly infected). Thus, these results indicate a stringent requirement for wild-type LMP1 in primary B-lymphocyte growth transformation (29–31). In contrast, deletion of aa 2–7, 6–17, or 18–24 from the 20-aa amino-terminal cytoplasmic domain had at most (with the 6–17 deletion) a 10-fold reducing effect on primary B-lymphocyte growth transformation efficiency; the resultant LCLs were similar to wild-type LCLs in their growth. The 6–17 deletion removes all of the strongly basic amino acids from the amino-terminal cytoplasmic domain and adversely affects LMP1 patching in the plasma membrane. These results are most consistent with the important role for the amino-terminal cytoplasmic domain being to anchor the first transmembrane domain (29). The LMP1 open reading frame is 386 codons and the last transmembrane domain ends with aa 187. Insertion of a stop codon after codon 231 resulted in an EBV recombinant than can transform primary B lymphocytes with wild-type efficiency, at least as measured by growth on fibroblast feeders. In contrast, EBV recombinants with a stop codon inserted after codon 187 could only be recovered in LCLs when wild-type LMP1 was provided by coinfecting P3HR-1 genomes (0 singly infected versus 29 P3HR-1 EBV complemented clones on passage of five mutant recombinants). Similar results were obtained with or without fibroblast feeders (31). These results indicate that the first 44 aa of the carboxyl-terminal cytoplasmic domain may include an effector domain that is critical for primary B-lymphocyte growth transformation (31). More recent studies of this 44-aa domain using a yeast two-hybrid screen indicated that this region interacts with tumor necrosis factor receptor-associated factors, which appear to be a crucial link to B-lymphocyte growth transformation and NFkB activation (36).
Other than EBNA1, which has not been investigated because of its obvious importance for episome maintenance, most of the rest of the EBV genome has been shown to be unimportant for primary B-lymphocyte growth transformation. LMP2, EBERs, and BARF0, which are transcribed in latent EBV infection, as well as BHRF1 and BCRF1, which are transcribed early or late in EBV replication have been specifically mutated and found to be unimportant for primary B-lymphocyte growth transformation. BHRF1 was of interest because of its similarity to bcl2 and BCRF1, because of its near identity to IL10. The BCRF1 deletion interestingly included the Cp alternative EBNA promoter and the deletion had no adverse effect on EBV infection in primary B lymphocytes (1, 2, 22, 26, 28, 37, 38).
Derivation of a New Packaging Cell Line Useful for Specific Mutagenesis of the Central Part of the EBV Genome and for the Generation of Replication-Incompetent Recombinants. Raji, one of the earliest derived EBV-infected BL-derived cell lines, has an endogenous EBV genome that is defective in both transformation and lytic replication. Raji may be superior to P3HR-1 as a packaging cell line because of the inability of the endogenous virus to replicate in newly infected cells. The Raji EBV genome is deleted for a 3-kbp DNA segment that includes most of the EBNA3C open reading frame. Raji is also deleted for a 2.7-kbp DNA segment that encodes most of the major single-strand DNA binding protein that is an essential early lytic DNA replication protein. This second deletion also includes a second early open reading frame of unknown function and part of the LMP2 open reading frame. Surprisingly, transfection of Raji cells with an expression vector for the major single-stranded DNA binding protein has sufficed for productive virus replication after treatment of Raji cells with inducers of lytic infection. However, the resultant virus has not been previously tested in primary B-lymphocyte transformation assays (39). Based on the previous recombinant EBV molecular genetic experiment with a EBNA3C stop codon mutation, the Raji virus should be substantially more than 90% deficient in cell growth transformation. Depending on the amount of virus produced from Raji cells, there may be detectable residual transforming activity. Further, the Raji genome may have mutations in other essential transforming genes. However, if the nontransforming phenotype is due to the EBNA3C deletion, Raji cells could be quite useful for recombinant EBV-based genetic analyses of EBNA3C and surrounding genes in much the same way that the P3HR-1 EBV genome has been particularly useful for analyses of the EBNALP and EBNA2 genes. We therefore transfected Raji cells with EBV DNA fragments that included EBNA3C, induced lytic EBV infection, and assayed the resultant virus stocks for the ability to transform primary B lymphocytes.
MATERIALS AND METHODS
Cell Lines. B958 EBV is wild type for EBNA3C (1,3,4). Raji TK−, Raji R19, and Raji R30 are deleted for most of the EBNA3C open reading frame (3, 4). B-lymphocyte cell lines were cultured in RPMI 1640 medium containing 10% fetal bovine serum, Gentamicin (10 μg/ml), and 2 mM glutamine.
Primary B cells were processed from healthy adult donors (13, 23).
Cosmid Transfections. Cosmids SalIC and EcoRIB DNA was digested with restriction enzymes to release the EBV DNA from the vector and 25 μg of each DNA was phenol/ chloroform-extracted, ethanol-precipitated, and resuspended in 400 μl of RPMI 1640 medium containing 10% fetal bovine serum. pSVNaeZ (25 μg) and 15 million Raji cells were added for electroporation at 250 V and 960 μF. The transfected cells were transferred to 10 ml of complete medium and incubated at 37°C in 5% CO2/95% air for 5 days.
Infection of Primary B Lymphocytes. Human primary B lymphocytes were infected with 0.45-μm (pore size) filtered culture supernatant from Raji cells 5 days after electroporation. Virus was incubated with 10 million T-cell-depleted B cells for 2 h at 37°C. Infected cells were plated in complete medium with 15% fetal bovine serum and 2 mM glutamine in 96-well plates at 50,000 cells per well (13, 23). Each well was fed with 100 ml of complete medium after 7 days.
PCR Analyses. Primers flanking the BALF2 deletion (positions 163,978–166,635) corresponded to the B958 sequence of positions 163,739–163,761 (forward primer) and 166,758– 166,779 (reverse primer). The EBNA3C deletion (positions 99,126–102,118) primers were forward primer, positions 98,890–98,910, and reverse primer, positions 102,227–102,250 (3, 4). All primers were purchased from GIBCO/BRL. The primers that amplify across the BALF2 deletion amplified a 383-bp DNA fragment and the primers that amplify across the EBNA3C deletion amplified a 368-bp fragment (3, 4).
Immunoblot and Southern Blot Analyses. Expression of EBV antigens was evaluated by electrophoresis of denatured LCLs proteins in 8% SDS/PAGE gels and immunoblot analysis with a human polyclonal sera that recognizes latent and lytic antigens or with a monoclonal antibody that recognizes EBNA3C (35). For Southern blots, 15 μg of infected-cell DNA was digested with KpnI restriction enzyme and the fragments were size-fractionated by loading onto a 0.6% agarose gel and electrophoresed (13, 23). DNA was transferred to an activated nylon membrane (GeneScreenPlus; NEN) and was hybridized with a [32P]dCTP-labeled HindE probe.
RESULTS AND DISCUSSION
Marker Rescue of Transformation from Raji Cells Using Cosmid DNAs That Overlap the EBNA3C Deletion. To test whether Raji cells are highly deficient in primary B-lymphocyte transformation as a result of the EBNA3C deletion, Raji cells converted to stable expression of the single-strand DNA binding protein (BALF2) were transfected with SalIC and EcoRIB EBV DNA fragments. Two EBV DNA fragments were transfected into the converted Raji cells because SalIC ends only 3 kbp downstream of the EBNA3C gene and EcoRIB starts only 3 kbp upstream of the EBNA3C gene. In previous second-site homologous recombination experiments in P3HR-1 cells, SalIC was substantially less efficient in homologous replacement of EBNA3C than in replacement of EBNA3B or EBNA3A and this was attributed to the shorter flanking homology with EBNA3C (Fig. 1). The Z immediate early transactivator was cotransfected into the Raji cell to induce lytic EBV infection. The resultant virus was harvested and used to infect primary human B lymphocytes. Clones of transformed cells arose at a very low efficiency. From three Raji cell transfection and primary B-lymphocyte infection experiments, 3, 10, and 10 clones of transformed cells were obtained and most grew out into long-term lymphoblastoid cell lines. In contrast, no transformants were obtained from the same BALF2-converted Raji cells that were transfected with Z and no cosmid DNA. The Raji-derived LCLs were initially similar to other wild-type LCLs but their growth slowed over the first 6 months in culture compared with the wild-type LCLs and six of the LCLs did not survive for 6 months in culture. This could be due to the inability of the Raji virus to replicate and spread among the initially infected primary B lymphocytes. PCR analysis across the BALF2 deletion site amplified a 383-bp DNA fragment indicative of the specific Raji deletion (Fig. 2A). Thus, the LCLs are transformed with a Raji-derived EBV. The EBNA3C deletion was also analyzed by PCR and these analyses indicated that the LCLs did not have the deletion of EBNA3C that is characteristic of Raji cells. The wild-type fragment of 3.4-kbp fragment characteristic of wild-type DNA would not be amplified under these conditions but the 368-bp deletion DNA fragment was amplified from the parental Raji cell line and not from the LCLs (Fig. 2B). These data are compatible with replacement of the deletion with wild-type EBNA3C DNA in the Raji-EBV-derived LCLs.
Genomic Analysis of LCL DNAs Around EBNA3C. Southern blot analysis was done on a KpnI digest of the total cell DNA. The labeled EBV HindE DNA fragment that includes the EBNA3C open reading frame hybridized to 2.6-kbp and 0.7-kbp KpnI fragments that are characteristic of wild-type EBNA3C in the digest of wild-type B958 derived LCL DNA but not in the parental Raji EBV DNA. Each of the LCLs infected with the Raji recombinant virus had the 2.6-kbp and 0.7-kbp fragments characteristic of wild-type EBNA3C DNA (Fig. 3, compare lanes L1–L6 with lanes RTK, R19, and B95). The Southern blot and PCR data confirm that the LCL-infected marker rescued virus derived from Raji cells has wild-type and not mutant EBNA3C.
The Rescued LCLs Express EBNA3C but not BALF2. The LCLs were analyzed for expression of the 135-kDa major single-strand DNA binding protein by Western blot analysis with a human polyclonal serum. As expected, the LCLs and the RTK- Raji cells had no detectable p135 while p135 was readily detected in a B958 LCL and in the Raji cell lines that had been converted to BALF2 expression (39) (Fig. 4A). Also, EBNA3C was detected in each of the LCLs and in the B958-derived LCLs and was absent from RTK- Raji, from each of the BALF2-converted Raji cell lines, and from the EBV-negative BL cell line BJAB (Fig. 4B; compare lanes L1–L6 and lane B95). These experiments demonstrated that
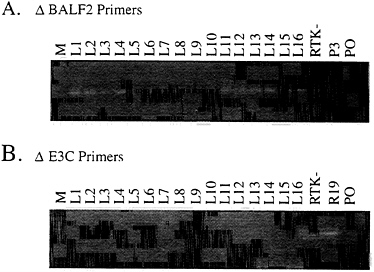
FIG. 2. PCR analysis of Raji LCLs showing the amplification of a DNA fragment across the BALF2 deletion and across the EBNA3C deletion in Raji cells. (A) Lanes: L1–L16, amplification from LCL DNAs; RTK-, from the parental Raji cell line DNA; P3, P3 DNA as a negative control; PO, primer-only control. The DNA fragment indicative of the BALF2 deletion 383 bp in size is seen in the representative lanes containing the Raji genome. (B) Amplification of the DNA fragment 368 bp in size representative of the EBNA3C Raji deletion junction (lanes were similar to those in A; R19, from the parental Raji cell line stably maintaining the BALF2 gene). Approximate sizes were determined by the ϕX174 RsaI DNA markers.
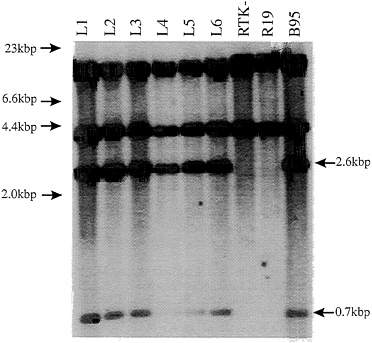
FIG. 3. Southern blot of LCL DNAs digested with KpnI restriction enzyme for comparison with the wild-type Raji TK- parental cell lines and the B958 DNA as control DNA from which the SalIC and EcoRIB cosmids were cloned (3, 4, 19). The HindE probe hybridizes to KpnI DNA fragments of 17.3 kbp, 2.6 kbp, 0.7 kbp, and 0.14 kbp across the EBNA3 region (3, 4). Fragments were fractionated on a 0.6% agarose gel, transferred to GeneScreenPlus nylon membrane, and then probed with [32P]dCTP-labeled HindE fragment. Markers used were λ DNA digested with HindIII as shown on the left. The arrows on the right indicate the appropriate KpnI fragments rescued in the LCLs.
the LCLs that arose after infection with virus from the EBV DNA SalIC- and EcoRIB-transfected Raji cells express levels of EBNA3C similar to a wild-type-EBV-infected LCLs. EBNA1 maps 7 kbp away from EBNA3C in the transfected EcoRIB fragment. Each of the six LCLs that was transformed by Raji recombinant virus had the EBNA1 encoded by the transfected EcoRIB fragment; EBNA1 encoded by the transfected EcoRIB fragment is identical in size to that in the B958-transformed LCL (from which the EcoRIB fragment was cloned) and is considerably larger than the Raji EBNA1 (compare lanes L1–L6 with lanes RTK-, R19, and B95; Fig. 4). These data indicate that a substantial part of the transfected EcoRIB DNA has recombined with the endogenous Raji genome.
These experiments describe a new packaging cell line for the construction of EBV recombinants. The cell line differs from previous packaging cell lines in that the endogenous EBV genome is not wild type for transformation or replication. The latently infected Raji cells have been rendered fully competent for lytic infection by complementation with a stably transfected expression vector for an essential early gene, BALF2, that is partially deleted from the endogenous Raji genome. Although the BALF2-converted Raji cells were previously shown to produce virions (39), to our knowledge, these experiments are the first proof that such cells can produce virions capable of infecting B lymphocytes.
The small numbers of virus recombinants that have so far been characterized are still deleted for BALF2, as is anticipated from previous experiments in which replicating herpes simplex virus genomes very rarely if ever recombined with chromosomally integrated viral genes. The frequency of recombination is less than 1 in 105 in situations in which the integrated DNA lacks a lytic replication origin and has no homology to the infecting viral DNA (40). The BALF2 DNA is presumed to be integrated and lacks a lytic replication origin; but, the endogenous Raji EBV genome has 2.6 kbp of the
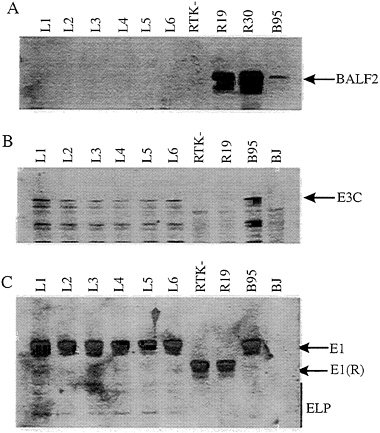
FIG. 4. Immunoblot analysis of cell lysate from LCLs compared with the BALF2 stably expressing cell lines R19 and R30 parental cell lines. Lanes, L1–L6, LCLs; R19 and R30, parental Raji cell lines; B95, cell lysate from a B958 LCL wild type for BALF2 (gp135); BJ, cell lysate from an EBV negative B lymphoma cell line. An 8% SDS/ PAGE gel was loaded, and samples were electrophoresed, then transferred to nitrocellulose membrane, and probed with a human polyclonal antiserum that recognizes the gp135 lytic antigen. (B) Similar immunoblot probed with monoclonal antibody A10 against the EBNA3C viral antigen (35). Bio-Rad broad-range prestained protein markers were used as standards. The arrows on the right indicate the appropriate protein. (C) Immunoblot of cell lysates from the LCLs (lanes L1–L6) for comparison of the EBV latent antigens to that of the parental strains RTK- and R19 (lanes RTK- and R19) and that of a B958 LCL (lane B95). The membrane was probed with a human polyclonal antiserum for detection of EBV latent antigens. The latent antigens are indicated by the arrows on the right. Prestained protein markers were used as standards.
BALF2 open reading frame. Recombination may therefore occur with a low frequency.
The Raji recombinant system should be particularly useful for the genetic analysis of EBNA3C and of surrounding genes. The data so far simply indicate that the endogenous Raji genome can be transcomplemented by BALF2 and will produce biologically active virus when the SalIC and EcoRIB EBV DNA fragments are transfected into Raji cells; the transforming virus that results has markers from the transfected DNA, including EBNA3C, which is known to be deleted from Raji and to be important for primary B-lymphocyte growth transformation. Thus, the simplest interpretation of the data is that the transfected DNA provides wild-type EBNA3C to the transformation-marker-rescued recombinants. However, the limited analyses that have been done indicate that the recombinants have much more than EBNA3C from the transfected EBV DNA. Transformation marker rescue from Raji has not yet been shown to be specifically dependent on an intact EBNA3C open reading frame. The power of the system in more precisely evaluating the importance of EBNA3C and of specific residues in EBNA3C will come from improvements in the efficiency of transformation marker rescue and from a reduction in the size of the marker rescuing DNA fragment. Now that the first recombinants have
been made in a Raji-EBV-genome-based packaging cell system, further experiments should specifically address these issues.
This work was supported by Grant CA47006 from the National Cancer Institute of the United States Public Health Service. E.S.R. is supported by a fellowship from the Leukemia Society of America.
1. Kieff, E.D. (1995) in Virology, eds. Fields, B.N., Howley, P., Knipe, D., Chanock, R., Melnick, J., Monath, T., Roizman, B. & Straus, S. (Raven, New York), 3rd Ed., pp. 2343–2396.
2. Rickinson, A.B. & Kieff, E.D. (1995) in Virology, eds. Fields, B.N., Howley, P., Knipe, D., Chanock, R., Melnick, J., Monath, T., Roizman, B. & Straus, S. (Raven, New York), 3rd Ed., pp. 2397–2446.
3. Baer, R., Bankier, A.T., Biggin, M.D., Deininger, P.L., Farrell, P.J., Gibson, T.J., Hatfull, G., Hudson, G.S., Satchwell, S.C., Séguin, C., Tuffnell, P.S. & Barrell, B.G. (1984) Nature (London) 310, 207–211.
4. Farrell, P.J. (1992) Genetic Maps ed. O’Brien, S.J. (Cold Spring Harbor Lab. Press, Plainview, NY), 6th Ed., pp. 1.120–1.133.
5. Sugden, B., Marsh, K. & Yates, J. (1985) Mol. Cell. Biol. 5, 410–413.
6. Lindahl, T., Adams, A., Bjursell, G., Bornkamm, G.W., Kascha-Dierich, C. & Jehn, U. (1976) J. Mol. Biol. 102, 511–530.
7. Kwong, A.D. & Frenkel, N. (1985) Virology 142, 421–425.
8. Wang, R, Li, X., Annis, B. & Faustman, D.L. (1995) Hum. Gene Ther. 6, 1005–1017.
9. Banerjee, S., Livanos, E. & Vos, J.H. (1995) Nat. Med. 1, 1303–1308.
10. Lowe, R.S., Keller, P.M., Ellis, R.W., Davison, A., Kieff, E. & Morgan, A. (1987) in Vaccines 87, eds. Chanock, R., Lerner, R., Brown, F. & Ginsberg, H. (Cold Spring Harbor Lab. Press, Plainview, NY), pp. 364–367.
11. Marchini, A., Longnecker, R. & Kieff, E. (1992) J. Virol. 66, 4972–4981.
12. Marchini, A., Kieff, E. & Longnecker, R. (1993) J. Virol. 67, 606–609.
13. Robertson, E.S., Tomkinson, B. & Kieff, E. (1994) J. Virol. 68, 1449–1458.
14. Lee, M.A., Kim, O.J. & Yates, J.L. (1992) Virology 189, 253–265.
15. Lee, M.A. & Yates, J.L. (1992) J. Virol. 66, 1899–1906.
16. Marchini, A., Cohen, J., Wang, F. & Kieff, E. (1992) J. Virol. 66, 3214–3219.
17. Cohen, J.I., Wang, F., Mannick, J. & Kieff, E. (1989) Proc. Natl. Acad. Sci. USA 86, 9558–9562.
18. Tomkinson, B. & Kieff, E. (1992) J. Virol. 66, 780–789.
19. Tomkinson, B. & Kieff, E. (1992) J. Virol. 66, 2893–2903.
20. Tomkinson, B., Robertson, E.S., Yalamanchili, R., Longnecker, R. & Kieff, E. (1993) J. Virol. 67, 7298–7306.
21. Hammerschmidt, W. & Sugden, B. (1989) Nature (London) 340, 393–397.
22. Miller, G., Robinson, J., Heston, L. & Lipman, M. (1994) Proc. Natl. Acad. Sci. USA 71, 383–387.
23. Robertson, E.S. & Kieff, E. (1995) J. Virol. 69, 983–993.
24. Tomkinson, B., Robertson, E.S. & Kieff, E. (1993) J. Virol. 65, 6765–6771.
25. Kempes, B., Pich, D., Zeidler, R., Sudgen, B. & Hammerschmidt, W. (1995) J. Virol. 69, 231–238.
26. Longnecker, R., Miller, C.L., Tomkinson, B. & Kieff, E. (1993) J. Virol. 67, 2006–2013.
27. Mannick, J.B., Cohen, J.L., Birkenbach, M., Marchini, A. & Kieff, E. (1991) J. Virol. 65, 6826–6837.
28. Marchini, A., Tomkinson, B., Cohen, J. & Kieff, E. (1991) J. Virol. 65, 5991–6000.
29. Izumi, K.M., Kaye, K.M. & Kieff, E.D. (1994) J. Virol. 68, 4369–4376.
30. Kaye, K.M., Izumi, K.M. & Kieff, E. (1993) Proc. Natl. Acad. Sci. USA 90, 9150–9154.
31. Kaye, K.M., Izumi, K.M. & Kieff, E. (1995) J. Virol. 69, 675–683.
32. Cohen, J.I. & Kieff, E. (1991) J. Virol. 65, 5880–5885.
33. Wang, F., Tsang, S., Kurilla, M.G., Cohen, J.I. & Kieff, E. (1990) J. Virol. 64, 3407–3416.
34. Yalamanchili, R., Tong, X., Grossman, S.R., Johannsen, E., Mosialos, G. & Kieff, E. (1994) Virology 204, 634–641.
35. Robertson, E., Grossman, S., Johannsen, E., Miller, C., Lin, J., Tomkinson, B. & Kieff, E. (1995) J. Virol. 69, 3108–3116.
36. Mosialos, G., Birkenbach, M., Yalamanchili, R., VanArsdale, T., Ware, C. & Kieff, E. (1995) Cell 80, 1–20.
37. Swaminathan, S., Tomkinson, B. & Kieff, E. (1991) Proc. Natl. Acad. Sci. USA 88, 1546–1550.
38. Swaminathan, S., Hesselton, R., Sullivan, J. & Kieff, E. (1993) J. Virol. 67, 7406–7413.
39. Decaussin, G., Leclerc, V. & Ooka, T. (1995) J. Virol. 69, 7309–7314.
40. Rice, S.A. & Knipe, D.M. (1990) J. Virol. 64, 1704–1715.







