
This paper was presented at a colloquium entitled “Genetic Engineering of Viruses and of Virus Vectors,” organized by Bernard Roizman and Peter Palese (Co-chairs), held June 9–11, 1996, at the National Academy of Sciences in Irvine, CA.
Genetically engineered poxviruses for recombinant gene expression, vaccination, and safety
BERNARD MOSS*
Laboratory of Viral Diseases, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, MD 20892–0445
ABSTRACT Vaccinia virus, no longer required for immunization against smallpox, now serves as a unique vector for expressing genes within the cytoplasm of mammalian cells. As a research tool, recombinant vaccinia viruses are used to synthesize and analyze the structure-function relationships of proteins, determine the targets of humoral and cell-mediated immunity, and investigate the types of immune response needed for protection against specific infectious diseases and cancer. The vaccine potential of recombinant vaccinia virus has been realized in the form of an effective oral wild-life rabies vaccine, although no product for humans has been licensed. A genetically altered vaccinia virus that is unable to replicate in mammalian cells and produces diminished cytopathic effects retains the capacity for high-level gene expression and immunogenicity while promising exceptional safety for laboratory workers and potential vaccine recipients.
On May 14, 1776, an English country physician named Edward Jenner inoculated 8-year-old James Phipps with cowpox virus isolated from the infected hand of Sarah Nelmes, a milkmaid (1). Jenner demonstrated that the boy was resistant to smallpox and the new procedure became known as vaccination (L. vacca cow) to distinguish it from variolation, an older and far more risky procedure of inoculation with small amounts of the smallpox (variola) virus itself. Although vaccination was met with some initial skepticism (Fig. 1), it was widely adopted. Cowpox virus was later superseded by vaccinia virus, a closely related virus that may have been isolated from a horse (2) and that produced a milder vaccination reaction. Despite the profound differences in human virulence of variola, vaccinia, and cowpox viruses, they are now known to belong to the same Orthopoxvirus genus, accounting for their ability to cross protect. Jenner accurately predicted “that the annihilation of the Small Pox, the most dreadful scourge of the human species, must be the final result of this practice [vaccination]” (cited in ref. 3). The last endemic case of smallpox occurred in 1977, bringing to an end the ravages wrought by smallpox over the past 3000 or more years (3). In 1980, the Assembly of the World Health Organization declared smallpox eradicated and recommended the discontinuation of smallpox vaccination. Ironically, that year also marked the application of recombinant DNA technology to vaccinia virus (4, 5), providing the means for genetically engineering poxviruses and developing them as expression vectors and candidate vaccines against unrelated infectious diseases (6, 7).
Molecular Biology of Poxviruses. The study of poxviruses has been motivated by a desire to understand the unique replication program of these large complex genetic systems (8). An appreciation of the molecular biology of poxviruses was crucial for their rational development as expression vectors (9). The ability of poxviruses to replicate entirely within the cytoplasm of the infected cell, even though they have DNA genomes, is the most distinctive feature of the members of this family. Detailed information regarding poxviruses was derived mainly from studies with vaccinia virus, although the basic features apply to other family members as well.
Infectious vaccinia virus particles are brick-shaped, 300–400 nm in diameter with lipoprotein membranes that surround a complex core structure containing a linear and nearly 200,000-bp duplex DNA molecule. Numerous virus-encoded enzymes, including a multisubunit DNA-dependent RNA polymerase, a transcription factor, capping and methylating enzymes, and a poly(A) polymerase, are packaged within the virus core and enable particles to synthesize translatable mRNAs with typical eukaryotic features after entry into a cell (Fig. 2). Initially, only the early genes are transcribed: they encode proteins involved in stimulation of the growth of neighboring cells, defense against host immune responses, replication of the viral genome, and transcription of the intermediate class of viral genes.
The progeny viral DNA molecules serve as templates for the sucessive expression of intermediate and late genes. The three temporal classes of genes have promoters with distinctive sequence elements (10–12) that are recognized by specific viral proteins (13), providing the basis for a programmed cascade mechanism of gene regulation (Fig. 2). Upon synthesis of the late structural proteins, infectious virus particles are assembled and some are wrapped with an additional Golgiderived membrane (14, 15), are transported to the periphery of the cell (16, 17), bud through the plasma membrane, and either remain attached to the cell surface (18) or are released into the medium (19). The externalized forms of vaccinia virus mediate cell-to-cell spread.
Genetic Engineering of Poxviruses. The DNA molecules of many viruses are infectious, i.e., a complete round of replication occurs after transfection of the naked genome into the cell. Poxviral DNA is not infectious, however, because the viral genome is not transcribed by cellular enzymes, and therefore, viral proteins are not made. Most strategies for genetically engineering poxviruses have employed homologous DNA recombination in infected cells, a process that occurs naturally during the replication of poxviruses (20–22).
Genetic engineering of poxviruses has provided a way to study their biology and to express foreign genes. Early examples of the former include marker transfer for mapping the thymidine kinase (TK; ref. 23) and DNA polymerase (24) genes and temperature-sensitive mutations (25, 26), as well as the deletion of nonessential genes such as the TK (7) and
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. §1734 solely to indicate this fact.
Abbreviations: CTL, cytotoxic T lymphocytes; MHC, major histocompatability complex; MVA, modified vaccinia virus Ankara; TK, thymidine kinase; IL, interleukin.

FIG. 1. English engraving by James Gilray, 1802, suggesting the alarm that greeted the introduction of the smallpox vaccine (from the National Library of Medicine).
vaccinia virus growth factor (27). Techniques have been developed that permit precise gene sequence alterations (28), inducible regulation of gene expression (29–31), and the targeting of temperature-sensitive mutations (32). These technical developments have greatly facilitated the correlation of poxvirus biochemistry and genetics.
Because poxviruses replicate in the cytoplasm and use their own transcription systems, poxvirus promoters and continuous open reading frames are required for expression of foreign genes. The time and level of gene expression is regulated by the choice of an early, intermediate, or late promoter. A commonly used DNA sequence contains tandem early and late promoters allowing a continuous moderate level of gene expression (9). The highest levels of expression have been obtained with strong natural or synthetic late or early/late promoters (refs. 33 and 34; S.Chakrabarti, J.Sisler, and B.M., unpublished results). The precise sequence of thymidines TTTTTNT serves as a transcription termination signal specifically for vaccinia virus early genes (35); if such a sequence happens to be present within a foreign gene to be regulated by an early promoter, the run of thymidines can be altered by changing the codon usage (36).
General methods for the production of recombinant poxviruses employ plasmid transfer vectors that contain an expression casette, consisting of a poxvirus promoter with adjacent restriction endonuclease sites for foreign gene insertion, flanked by poxvirus sequences that direct recombination to the desired locus (9, 37). The relatively high frequency of homologous recombination (approximately 0.1%) makes it possible to screen virus plaques by DNA hybridization or for expression of the desired foreign gene product (38). Nevertheless, selection or general screening procedures are quite helpful. By targeting the foreign gene to the TK locus, recombinant viruses can be selected by their TK-negative phenotype in TK-deficient cells (9). Alternatively, the transfer vector may enable the cointegration of an antibiotic selection marker (39–42) or a reporter gene allowing color screening due to β-galactosidase (43, 44) or β-glucuronidase (45) synthesis. The reversal of host range restriction (46) or plaque phenotype (47) has also been used. A recently devised procedure involves the complementation of a defect in extracellular virus production so that all plaques contain recombinant virus (48).
Although the large size of poxvirus genomes would seem to make in vitro ligation of foreign genes technically daunting, this approach has been used successfully (49–51). The strategy involves (i) cutting the vaccinia virus genome at a unique restriction endonuclease site, (ii) religating the two halves of the genome with the recombinant gene between them, transfecting the ligated DNA into cells that have been infected with a helper virus: either a temperature-sensitive vaccinia virus mutant or an avian poxvirus. This approach does not require intermediate cloning in Escherichia coli and is useful in conjunction with polymerase chain reaction amplification of genes or screening large cDNA libraries or for very large segments of DNA. In this regard, the vaccinia virus genome can tolerate the addition of at least 25,000 bp of additional DNA (49, 52). Presumably, this quantity could be doubled by deleting DNA not required for replication in cultured cells.
Since vaccinia virus is the prototype poxvirus and infects a wide range of cells and experimental animals, it was the first member of the family employed for expression studies. Other poxviruses have been developed as vectors, primarily because of advantages associated with their restricted host ranges, and will be considered in subsequent sections.
Vaccinia Virus-Bacteriophage Hybrid Systems. Because of their high efficiency and stringent promoter specificity, the
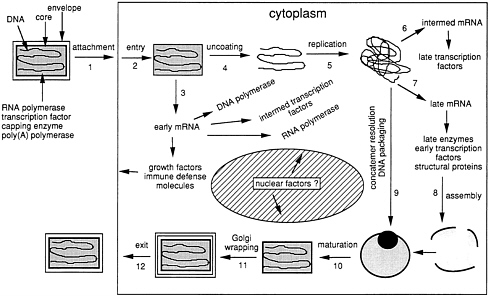
FIG. 2. Infectious cycle of vaccinia virus (from ref. 8, with permission).
single-subunit DNA-dependent RNA polymerase of bacteriophage T7 (53) and of related bacteriophages T3 (54) and SP6 (55) have been used for expression of selected genes in vaccinia virus expression vectors. Recombinant vaccinia viruses containing the gene encoding the bacteriophage polymerase, regulated by a vaccinia virus promoter, have been constructed. There are three basic versions of the system. In one, cells are infected with the recombinant vaccinia virus and then transfected with a plasmid containing the bacteriophage promoter regulating a foreign gene (53). In another version, the bacteriophage promoter regulated foreign gene is incorporated into a second recombinant vaccinia virus and expression obtained by coinfection with a recombinant vaccinia virus that encodes the T7 RNA polymerase gene (56) or by infecting cell lines that stably express the T7 RNA polymerase (57). Finally, there are inducible versions in which both the bacteriophage polymerase and expressed gene are in the same recombinant vaccinia virus allowing single infection protocols (58, 59).
Because of its simplicity and versatility, the transfection system has been widely used for analytical studies. In some cases, the same recombinant plasmid can be used for expression in both the vaccinia virus hybrid system and E. coli, as well as for in vitro transcription and translation. With liposome carriers, the transfection efficiency is very high and most cells express the desired gene (60). The level of expression can be enhanced by employing the untranslated leader sequence of encephalomyocarditis virus to provide cap-independent translation (61). This transient system is ideal for screening the effects of large numbers of mutations or for cDNA libraries as discussed below.
The double infection and the inducible systems are appropriate if large numbers of cells are used. In a recent configuration of the inducible system (59), the recombinant vaccinia virus contains (i) the E. coli lac repressor gene under control of a vaccinia virus early/late promoter for continuous repressor synthesis; (ii) the T7 RNA polymerase gene under control of a vaccinia virus late promoter and E. coli lac operator; and (iii) the gene to be expressed under control of a T7 promoter and lac operator. Because both the T7 polymerase and the T7 promoter-controlled genes are regulated by lac operators, control is very stringent providing an induction of more than 10,000-fold. Another modification of this system employs a thermolabile repressor, so that expression occurs upon temperature elevation rather than after addition of inducer, providing an advantage especially for fermentation technology.
Synthesis of Proteins in Cell Culture. Several characteristics of vaccinia virus contribute to its wide use as an expression system. These include (i) relatively simple methods of recombinant virus construction, (ii) a wide choice of cell types, (iii) cytoplasmic expression eliminating special requirements for nuclear processing and transport of RNA, and (iv) relatively high expression levels. As anticipated, proteins synthesized by vaccinia virus vectors are processed and transported in accord with their primary structure and the inherent capabilities of the host cell. Although vaccinia virus vectors have been primarily used for expression in fully permissive mammalian and avian cells, the system has also been extended to nonpermissive amphibian cells (62–64).
Transient expression, by DNA transfection of infected cells, can be accomplished either using vaccinia virus promoters (65) or the hybrid vaccinia virus-T7 system described above. Generally, the expression level is considerably higher than that of conventional eukaryotic transfection systems. Applications of this transfection system are very diverse and, for example, have included analysis of protein-protein interactions (66), epitope mapping of monoclonal antibodies (67), cell fusion (68), and expression of ion channels (69, 70). Complex structures, such as infectious RNA virus particles, have been assembled by transfecting several plasmids simultaneously (71–73). The transfection system also provides a useful method for functional screening of cDNA libraries, as shown by the recent identification of the human immunodeficiency virus coreceptor fusin gene (74).
To harvest protein from large quantities of cells, the gene of interest needs to be integrated into a recombinant vaccinia
virus (75). High levels of recombinant protein have been obtained using strong synthetic vaccinia virus promoters (34) or the hybrid vaccinia virus-T7 system (59). The latter is preferred if the expressed protein is toxic to either the virus or cells. The hybrid vaccinia virus-T7 system has recently been adapted to Chinese hamster ovary cells, which are normally not permissive for vaccinia virus (76).
Determination of Targets of Humoral and Cell-Mediated Immunity. If recombinant vaccinia viruses that express one or more genes of another microorganism are used to immunize animals, the antiserum can be tested for neutralizing activity. This approach eliminates the need to purify the protein in a native state, which may be difficult as in the case of membrane proteins (77, 78). Genetic engineering can be used to enhance the immune response, for example, by converting a secreted protein into one that is membrane-anchored (79). Immunization with recombinant vaccinia viruses also provides a convenient way to produce monoclonal antibodies to native gene products without having to purify them (80, 81).
Recombinant vaccinia viruses are extensively used to induce CD8-bearing cytotoxic T lymphocytes (CTLs) in animals or to determine the targets of CTLs in vitro. Intracellular expression allows the antigen to be processed and presented in association with matched major histocompatability complex (MHC) class I molecules for recognition. For the determination of targets, autologous or MHC-matched cells are infected with a recombinant vaccinia virus that expresses a protein of interest, preferably under control of an early or early/late promoter (82). Alternatively, the recombinant virus can also express the appropriate MHC molecule. The target cells are then loaded with chromium and chromium release is measured after incubation with effector lymphocytes from an experimental animal (83–85) or human (86, 87). B cells, immortalized with Epstein-Barr virus, have been used as autologous cells from individuals. CTL epitopes can be located by expressing truncated proteins, prior to fine mapping with small peptides (87, 88). The hybrid vaccinia virus-T7 transfection system can be used to express CTL target proteins, avoiding the task of constructing new recombinant viruses (89).
Vaccinia vectors are also used to induce CD4-bearing CTLs and analyze the presentation of endogenous antigens with MHC class II molecules (90–93).
Protection Against Experimental Infections. The numerous examples, in which immunization of experimental animals (from mouse to chimpanzee) with recombinant vaccinia viruses that express one or more genes of a DNA or RNA virus have provided partial or complete protection against disease caused by challenge, are referenced elsewhere (94–96). In many cases, protection was correlated with neutralizing antibody against viral envelope proteins expressed by the recombinant vector. In other cases, vaccination provided a priming effect and protection was associated with an anamnestic antibody response, as when chimpanzees were inoculated with a recombinant vaccinia virus that expresses hepatitis B surface antigen (97). In some cases, protection may be due to induction of CTLs (98–103).
Smallpox vaccine was usually administered intradermally and other routes were thought to be less immunogenic (104). Good immunogenicity also follows intradermal inoculation of recombinant vaccinia virues. However, when intradermal and intranasal inoculations of recombinant vaccinia viruses that express influenza and respiratory syncytial virus envelope glycoproteins were compared, the latter route provided better local immunity and protection against an upper respiratory infection (105–107). Mucosal immunity also has been achieved by intestinal inoculation with recombinant vaccinia virus (108, 109). Coexpression of interleukin (IL) 5 and IL-6 reportedly enhances mucosal IgA production (110, 111).
Tumor Immunization. The ability of poxviruses to induce strong CTL responses has led to consideration of their use as cancer vaccines. Recombinant vaccinia viruses that express viral antigens (101, 112) or cellular tumor-associated antigens (113–116) have provided prophylactic and therapeutic effects against experimental tumors. Cytokines (IL-2 and IL-12) and/or costimulatory molecules (B7–1 and B7–2) may enhance the immune effects in model systems (117, 118).
Attenuated and Nonreplicating Vectors. At the site of percutaneous inoculation with vaccinia virus, a nonimmune individual typically develops a papule that becomes vesicular and pustular reaching a maximum size in 8–10 days. The pustule dries and the scab separates within 14–21 days. The local lymph nodes may feel tender and there may be some fever. In young children, eczema vaccinatum and encephalitis are serious but infrequent complications; in adults disseminated or progressive vaccinia may occur if there is a severe immunodeficiency (119, 120). Safety issues have been raised because of the possibilities of accidental laboratory infections and side effects of vaccination. In the United States, the immunization practices advisory committee (121) recommended that laboratory work with vaccinia virus be carried out under biosafety level 2 conditions; those that have direct contact with the virus are advised to receive a smallpox vaccination at 10-year intervals. In the United Kingdom, however, vaccination of laboratory workers is recommended only under special circumstances (122).
Several approaches have been taken to enhance the safety of vaccinia virus. During the smallpox era, highly attenuated vaccinia virus strains were developed (3). One of these, modified vaccinia virus Ankara (MVA), was passaged more than 500 times in chicken embryo cells, lost the ability to replicate in mammalian cells, became apathogenic even for immunodeficient animals, and was administered without apparent incident to about 120,000 humans including many who were considered a poor risk for the conventional smallpox vaccine (123, 124). Further studies showed that multiple genomic deletions had occurred (125) and that virus replication was blocked at a late stage of morphogenesis in mammalian cells, importantly leaving synthesis of viral proteins unimpaired (126). Marker transfer experiments suggest that multiple gene defects need to be corrected for efficient replication of MVA in mammalian cells (M.Carroll and B.M., unpublished results). Consequently, MVA is useful as an expression vector while providing a high degree of safety. For this reason, recombinant MVAs that express T7 RNA polymerase have been constructed (127, 128). At the National Institutes of Health, work exclusively with MVA is permitted at biosafety level 1 without vaccination. Despite its inablity to replicate, recombinant MVA has proven to be highly immunogenic and effective in protecting against influenza, parainfluenza and simian immunodeficiency virus infections in mouse or monkey models (129–131).
Varied degrees of attenuation can be achieved by deletion of one or more genes not required for replication in tissue culture (132–139). These include viral genes involved in nucleotide metabolism, host interactions, and extracellular virus formation. NYVAC, a derivative of the Copenhagen strain of vaccinia virus with multiple deletions and impaired replication in human cells, has provided protective immunity againtst Japanese encephalitis virus in swine (140, 141).
Another attenuation approach involves the expression of genes designed to enhance the host response to vaccinia virus. Recombinant vaccinia viruses that encode IL-2 have diminished pathogenicity for immunodeficient mice (142, 143) and immunocompetent monkeys (144, 145). The mechanism involves the rapid clearance of virus by natural killer and other cells secreting interferon-γ (146, 147). Recombinant vaccinia viruses that express interferon-γ are also attenuated (148, 149). The stability of such viruses in vivo needs to be examined, since loss of the IL-2 or interferon-γ gene would restore virulence.
Some members of the poxvirus family have a naturally restricted host range, which can provide increased safety. Avian poxviruses were initially considered as vectors for birds (150–152) since they do not replicate in mammals. Subsequent studies, however, indicated that expression of recombinant genes occurs in mammalian cells and that immune responses are induced in mammals (153). For unknown reasons, canarypox virus appears to be superior to fowlpox virus in this regard (154, 155). Raccoon poxvirus (156), capripox virus (157), and swinepox virus (158) vectors also have been made.
Veterinary Vaccine Trials. Recombinant poxviruses are capable of protectively immunizing animals against diseases of veterinary importance. Examples include the use of vaccinia virus recombinants to protect cattle against vesicular stomatitis virus (159) and rinderpest (160, 161), chickens against influenza virus (162), and raccoons and foxes against rabies virus (163–166). The recombinant vaccinia virus expressing the rabies virus glycoprotein was administered in bait form for immunization of wild animals and protective immunization with reduced incidence of rabies has been demonstrated in large field tests in Belgium (167). Examples of the use of other poxvirus vectors for veterinary purposes include a raccoon poxvirus vector to protect raccoons against rabies virus (156); a capripoxvirus vector to protect cattle against rinderpest (168); swinepox vectors to protect pigs against Aujeszky disease (pseudorabies) (158); fowlpox vectors to protect chickens against influenza virus (169), Newcastle disease virus (152, 170–172), and infectious bursal disease virus (173); canarypox virus to protect dogs against canine distemper virus (154); and pigeonpox virus vectors to protect chickens against Newcastle disease virus (174).
Human Vaccine Trials. Phase 1 clinical trials, to test the safety and immunogenicity of a recombinant vaccinia virus expressing the human immunodeficiency virus 1 (HIV-1) envelope gene, were conducted in the United States (175– 177). No serious side effects were encountered in healthy subjects that received percutaneous inoculations. Humoral and cell-mediated immune responses were higher in previously unvaccinated individuals and were enhanced after a booster injection with gp160 protein. Although an improvement could be made with regard to the level of expression achieved with the first-generation vaccinia vector, the prime-boost strategy provided both humoral and cell-mediated immunity.
In China, a recombinant vaccinia virus that expresses the major Epstein-Barr virus membrane glycoprotein was immunogenic when administered to infants and young children and apparently delayed or prevented natural infection over a period of 16 months (178).
Clinical tests of two recombinant canarypox viruses have been reported. The first, conducted in France, described the inoculation of a canarypox virus that expressed the rabies glycoprotein. Antibody was detected, although the titer was not as high as that obtained with a licensed rabies vaccine (179). The second, conducted in the United States, involved a recombinant that expressed the HIV-1 glycoprotein. No antibody response was detected after two subcutaneous inoculations; however, both humoral and cell-mediated immune responses were found after a gp160 protein booster injection, suggesting that priming had occurred (180).
Present and Future Considerations. Poxviruses are transcription machines, encoding and packaging proteins required for synthesis of mRNA in the cytoplasm of an infected cell. For this reason, as well as the ability to integrate large amounts of DNA into the viral genome and to infect a wide variety of cells, recombinant vaccinia viruses have become valuable laboratory research tools. The popularity of the vector system exists despite the cytopathic effects of the virus and the special precautions required to work with a class 2 infectious agent. The adaptation of a highly attenuated vaccinia virus strain that has lost the ability to produce infectious progeny in mammalian cells addresses these concerns. Basic research on poxviruses and innovations, such as the vaccinia virus-bacteriophage T7 system, have greatly increased the level of recombinant gene expression; elucidation of the mechanisms that perturb host cell macromolecular synthesis and other intracellular processes may lead to further improvements in the vector system.
The role of vaccinia virus in the eradication of smallpox suggests that poxviruses could have important uses as recombinant vaccines. Excellent field results, with a wild-life recombinant vaccinia virus rabies vaccine, fully support this idea. Other veterinary vaccine applications also seem promising. The relatively few human clinical studies that have been carried out suggest that the immunogenicity of the vaccine may need to be enhanced in some cases. Safety is an important consideration leading to the goal of effective “nonreplicating” vectors. Avian poxviruses and genetically altered strains of vaccinia virus, which meet this stringent criterion, need to be directly compared for efficacy. Although existing immunity to vaccinia virus may restrict the replication of vaccinia virus vectors and decrease the immune response to the recombinant protein, smallpox vaccination was largely stopped more than 15 years ago so this is becoming less of a concern with the passage of time. Should more than one poxvirus-based vaccine be developed, then their simultaneous administration as a mixture or a polyvalent virus would avoid the problem associated with immunity to the vector.
1. Jenner, E. (1798) in An Inquiry Into the Causes and Effects of the Variolae Vaccinae, A Disease Discovered in Some of the Western Countries of England, Particularly Near Gloucestershire, and Known by the Name of the Cow Pox, London, ed. Camac, C.N. B. (Dover, New York), pp. 213–240.
2. Baxby, D. (1977) J. Infect. Dis. 136, 453–455.
3. Fenner, F., Henderson, D.A., Arita, I., Jezek, Z. & Ladnyi, I.D. (1988) Smallpox and Its Eradication (W.H.O., Geneva).
4. Wittek, R. & Moss, B. (1980) Cell 21, 277–284.
5. Wittek, R., Cooper, J., Barbosa, E. & Moss, B. (1980) Cell 21, 487–493.
6. Panicali, D. & Paoletti, E. (1982) Proc. Natl. Acad. Sci. USA 79, 4927–4931.
7. Mackett, M., Smith, G.L. & Moss, B. (1982) Proc. Natl. Acad. Sci. USA 79, 7415–7419.
8. Moss, B. (1996) in Poxviridae: The Viruses and Their Replication, eds. Fields, B.N., Knipe, D.M. & Howley, P.M. (Lippincott-Raven, Philadelphia), Vol. 2, pp. 2637–2671.
9. Mackett, M., Smith, G.L. & Moss, B. (1984) J. Virol. 49, 857–864.
10. Davison, A.J. & Moss, B. (1989) J. Mol. Biol. 210, 749–769.
11. Davison, A.J. & Moss, B. (1989) J. Mol. Biol. 210, 771–784.
12. Baldick, C.J., Keck, J.G. & Moss, B. (1992) J. Virol. 66, 4710–4719.
13. Moss, B. (1993) in Vaccinia Virus Transcription, eds. Conaway, R. & Conaway, J. (Raven, New York), pp. 185–205.
14. Hiller, G. & Weber, K. (1985) J. Virol. 55, 651–659.
15. Schmelz, M., Sodeik, B., Ericsson, M., Wolffe, E.J., Shida, H., Hiller, G. & Griffiths, G. (1994) J. Virol. 68, 130–147.
16. Stokes, G.V. (1976) J. Virol. 18, 636–643.
17. Cudmore, S., Cossart, P., Griffiths, G. & Way, M. (1995) Nature (London) 378, 636–638.
18. Blasco, R. & Moss, B. (1992) J. Virol. 66, 4170–4179.
19. Payne, L.G. (1980) J. Gen. Virol. 50, 89–100.
20. Fenner, F. & Comben, B.M. (1958) Virology 5, 530–548.
21. Sam, C.K. & Dumbell, K.R. (1981) Ann. Virol. 132E, 135–150.
22. Nakano, E., Panicali, D. & Paoletti, E. (1982) Proc. Natl. Acad. Sci. USA 79, 1593–1596.
23. Weir, J.P., Bajszar, G. & Moss, B. (1982) Proc. Natl. Acad. Sci. USA 79, 1210–1214.
24. Jones, E.V. & Moss, B. (1984) J. Virol. 49, 72–77.
25. Condit, R.C., Motyczka, A. & Spizz, G. (1983) Virology 128, 429–443.
26. Ensinger, M.J. & Rovinsky, M. (1983) J. Virol. 48, 419–428.
27. Buller, R.M.L., Chakrabarti, S., Moss, B. & Frederickson, T. (1988) Virology 164, 182–192.
28. Falkner, F.G. & Moss, B. (1990) J. Virol. 64, 3108–3111.
29. Fuerst, T.R., Fernandez, M.P. & Moss, B. (1989) Proc. Natl. Acad. Sci. USA 86, 2549–2553.
30. Rodriguez, J.F. & Smith, G.L. (1990) Virology 177, 239–250.
31. Zhang, Y. & Moss, B. (1991) Proc. Natl. Acad. Sci. USA 88, 1511–1515.
32. Hassett, D.E. & Condit, D.E. (1994) Proc. Natl. Acad. Sci. USA 91, 4554–4558.
33. Patel, D.D., Ray, C.A., Drucker, R.P. & Pickup, D.J. (1988) Proc. Natl. Acad. Sci. USA 85, 9431–9435.
34. Davison, A.J. & Moss, B. (1990) Nucleic Acids Res. 18, 4285– 4286.
35. Yuen, L. & Moss, B. (1987) Proc. Natl. Acad. Sci. USA 84, 6417–6421.
36. Earl, P.L., Hügin, A.W. & Moss, B. (1990) J. Virol. 64, 2448–2451.
37. Earl, P.L. & Moss, B. (1991) in Generation of Recombinant Vaccinia Viruses, eds. Ausubel, F.M., Brent, R., Kingston, R.E., Moore, D.D., Seidman, J.G., Smith, J.A. & Struhl, K. (Greene & Wiley, New York), Vol. 2, pp. 16.17.1–16.17.16.
38. Piccini, A., Perkus, M.E. & Paoletti, E. (1987) Methods Enzymol. 153, 545–563.
39. Franke, C.A., Rice, C.M., Strauss, J.H. & Hruby, D.E. (1985) Mol. Cell. Biol. 5, 1918–1924.
40. Falkner, F.G. & Moss, B. (1988) J. Virol. 62, 1849–1854.
41. Boyle, D.B. & Coupar, B.E.H. (1988) Gene 65, 123–128.
42. Isaacs, S.N., Kotwal, G.J. & Moss, B. (1990) Virology 178, 626–630.
43. Chakrabarti, S., Brechling, K. & Moss, B. (1985) Mol. Cell. Biol. 5, 3403–3409.
44. Panicali, D., Grzelecki, A. & Huang, C. (1986) Gene 47, 193–199.
45. Carroll, M.W. & Moss, B. (1995) BioTechniques 19, 352–355.
46. Perkus, M.E., Limbach, K. & Paoletti, E. (1989) J. Virol. 63, 3829–3836.
47. Rodriguez, J.F. & Esteban, M. (1989) J. Virol. 63, 997–1001.
48. Blasco, R. & Moss, B. (1995) Gene 158, 157–162.
49. Merchlinsky, M. & Moss, B. (1992) Virology 190, 522–526.
50. Scheiflinger, F., Dorner, F. & Falkner, F.G. (1992) Proc. Natl. Acad. Sci. USA 89, 9977–9981.
51. Pfleiderer, M., Falkner, F.G. & Dorner, F. (1995) J. Gen. Virol. 76, 2957–2962.
52. Smith, G.L. & Moss, B. (1983) Gene 25, 21–28.
53. Fuerst, T.R., Niles, E.G., Studier, F.W. & Moss, B. (1986) Proc. Natl. Acad. Sci. USA 83, 8122–8126.
54. Rodriguez, D., Zhou, Y., Rodriguez, J.-R., Durbin, R.K., Jiminez, V., McAllister, W.T. & Esteban, M. (1990) J. Virol. 64, 4851–4857.
55. Usdin, T.B., Brownstein, M.J., Moss, B. & Isaacs, S.N. (1993) BioTechniques 14, 222–224.
56. Fuerst, T.R., Earl, P.L. & Moss, B. (1987) Mol. Cell. Biol. 7, 2538–2544.
57. Elroy-Stein, O. & Moss, B. (1990) Proc. Natl. Acad. Sci USA 87, 6743–6747.
58. Alexander, W.A., Moss, B. & Fuerst, T.R. (1992) J. Virol. 66, 2934–2942.
59. Ward, G.A., Stover, C.K., Moss, B. & Fuerst, T.R. (1995) Proc. Natl. Acad. Sci. USA 92, 6773–6777.
60. Rose, J.K., Buonocore, L. & Whitt, M.A. (1991) BioTechniques 10, 520–525.
61. Elroy-Stein, O., Fuerst, T.R. & Moss, B. (1989) Proc. Natl. Acad. Sci. USA 86, 6126–6130.
62. Yang, X.-C, Karschin, A., Labarca, C., Elroy-Stein, O., Moss, B., Davidson, N. & Lester, H.A. (1991) FASEB J. 5, 2209–2216.
63. Petit, d. L., Koothan, T., Liao, D. & Malinow, R. (1995) Neuron 14, 685–688.
64. Wu, G.-Y., Zou, D.-J. & Cline, H.T. (1995) Neuron 14, 681–684.
65. Cochran, M.A., Mackett, M. & Moss, B. (1985) Proc. Natl. Acad. Sci. USA 82, 19–23.
66. Mizukami, T., Fuerst, T.R., Berger, E.A. & Moss, B. (1988) Proc. Natl. Acad. Sci. USA 85, 9273–9277.
67. Keil, W. & Wagner, R.R. (1989) Virology 170, 392–407.
68. Ashorn, P., Berger, E.A. & Moss, B. (1990) J. Virol. 64, 2149–2156.
69. Leonard, R.J., Karschin, A., Jayashree-Aiyar, S., Davidson, N., Tanouye, M.A., Thomas, L., Thomas, G. & Lester, H.A. (1989) Proc. Natl. Acad. Sci. USA 86, 7629–7633.
70. Rich, D.P., Anderson, M.P., Gregory, R.J., Cheng, S.H., Paul, S., Jefferson, D.M., McCann, J.D., Klinger, K.W., Smith, A.E. & Welsh, M.J. (1990) Nature (London) 347, 358–363.
71. Schnell, M.J., Mebatsion, T. & Conzelmann, K.-K. (1994) EMBO J. 13, 4195–4204.
72. Whelan, S.P.J., Ball, L.A., Bar, J.N. & Wertz, G.T.W. (1995) Proc. Natl. Acad. Sci. USA 92, 8388–8392.
73. Collins, P.L., Hill, M.G., Camargo, E., Grossfeld, H., Chanock, R.M. & Murphy, B.R. (1995) Virology 92, 11563–11567.
74. Feng, Y., Broder, C.C., Kennedy, P.E. & Berger, E.A. (1996) Science 272, 872–877.
75. Barrett, N., Mitterer, A., Mundt, W., Eibl, J., Eibl, M., Gallo, R.C., Moss, B. & Dorner, F. (1989) AIDS Res. Hum. Retroviruses 5, 159–171.
76. Ramsey-Ewing, A. & Moss, B. (1996) J. Biol. Chem. 206, 984–993.
77. Elango, N., Prince, G.A., Murphy, B.R., Venkatesan, S., Chanock, R.M. & Moss, B. (1986) Proc. Natl. Acad. Sci. USA 83, 1906–1911.
78. Earl, P.L., Robert-Guroff, M., Matthews, T.J., Krohn, K., London, W.T. & Moss, B. (1989) AIDS Res. Hum. Retroviruses 5, 23–32.
79. Langford, D.J., Edwards, S.J., Smith, G.L., Moss, B., Kemp, D.J., Anders, R.F. & Mitchell, G.F. (1986) Mol. Cell. Biol. 6, 3191–3199.
80. Yilma, T., Ristow, S.S., Moss, B. & Jones, L. (1987) Hybridoma 6, 329–335.
81. Malvoisin, E. & Wild, F. (1990) J. Virol. 64, 5160–5162.
82. Townsend, A., Bastin, J., Gould, K., Brownlee, G., Andrew, M., Coupar, B., Boyle, D., Chan, S. & Smith, G. (1988) J. Exp. Med. 168, 1211–1224.
83. Bennink, J.R., Yewdell, J.W., Smith, J.W., Moller, C. & Moss, B. (1984) Nature (London) 311, 578–579.
84. Yewdell, J.W., Bennink, J.R., Smith, G.L. & Moss, B. (1985) Proc. Natl. Acad. Sci. USA 82, 1785–1789.
85. Bennink, J.R. & Yewdell, J.W. (1990) Curr. Topics Microbiol. Immunol. 163, 153–184.
86. McMichael, A.J., Michie, C.A., Gotch, F.M., Smith, G.L. & Moss, B. (1986) J. Gen. Virol. 67, 719–726.
87. Walker, B.D., Flexner, C., Paradis, T.J., Fuller, T.C., Hirsch, M.S., Schooley, R.T. & Moss, B. (1988) Science 240, 64–66.
88. Earl, P., Koenig, S. & Moss, B. (1991) J. Virol. 65, 31–41.
89. Eisenlohr, L.C., Yewdell, J.W. & Bennink, J.R. (1992) J. Immunol. Methods 154, 131–138.
90. Zarling, J.M., Moran, P.A., Lasky, L.A. & Moss, B. (1986) J. Virol. 59, 506–509.
91. Morrison, L.A., Lukacher, A.E., Braciale, V.L., Fan, D. & Braciale, T.J. (1986) J. Exp. Med. 163, 903–921.
92. Polydefkis, M., Koenig, S., Flexner, C., Obah, E., Gebo, K., Chakrabarti, S., Earl, P.L., Moss, B. & Siliciano, R.F. (1990) J. Exp. Med. 171, 875–887.
93. Jaraquamada, D., Marti, M. & Long, E.O. (1990) J. Exp. Med. 172, 947–954.
94. Moss, B. (1991) Science 252, 1662–1667.
95. Cox, W.I., Tartaglia, J. & Paoletti, E. (1992) in Poxvirus Recombinants as Live Vaccines, eds. Binns, M.M. & Smith, G.L. (CRC, Boca Raton, FL), pp. 123–162.
96. Flexner, C. & Moss, B. (1996) in Vaccinia Virus as a Live Vector for Expression of Immunogens, eds. Woodrow, G.C. & Levine, M.M. (Dekker, New York), in press.
97. Moss, B., Smith, G.L., Gerin, J.L. & Purcell, R.H. (1984) Nature (London) 311, 67–69.
98. Jonjic, S., del Val, M., Keil, G.M., Reddehase, M.J. & Kozinowski, U.H. (1988) J. Virol. 62, 1653–1658.
99. Hany, M., Oehen, S., Schulz, M., Hengartner, H., Mackett, M., Bishop, D.H.L., Overton, H. & Zinkernagel, R.M. (1989) Eur. J. Immunol. 19, 417–424.
100. Miyazawa, M., Nishio, J. & Chesebro, B. (1992) J. Virol. 66, 4497–4507.
101. Earl, P.L., Moss, B., Wehrly, K., Nishio, J. & Chesebro, B. (1986) Science 234, 728–731.
102. Kulkarni, A.B., Connors, M., Firestone, C.Y., Morse, H.C. & Murphy, B.R. (1993) J. Virol. 67, 1044–1049.
103. Kulkarni, A.B., Morse, H.C., III, Bennink, J.R., Yewdell, J.W. & Murphy, B.R. (1993) J Virol. 67, 4086–4092.
104. Galasso, G.J., Karzon, D.T., Katz, S.L., Krugman, S., Neff, J. & Robbins, F.C. (1977) J. Infect. Dis. 135, 131–186.
105. Small, P.A., Smith, G.L. & Moss, B. (1985) Vaccines 85, 175–176.
106. Renegar, K.B. & Small, P.A. (1991) J. Virol. 65, 2146–2148.
107. Meitin, C.A., Bender, B.S. & Small, P.A. (1991) Vaccine 9, 751–756.
108. Kanesaki, T., Murphy, B.R., Collins, P.L. & Ogra, P.L. (1991) J. Virol. 65, 657–663.
109. Meitin, C., Bender, B. & Small, P., Jr (1994) Proc. Natl. Acad. Sci. USA 91, 11187–11191.
110. Ramsay, A.J., Husband, A.J., Ramshaw, I.A., Bao, S., Matthaei, K.I., Koehler, G. & Kopf, M. (1994) Science 264, 561–563.
111. Ramsay, A.J. & Kohonen-Corish, M. (1993) Eur. J. Immunol. 23, 3141–3145.
112. Lathe, R., Kieny, M.P., Gerlinger, P., Clertant, P., Guizani, I., Cuzin, F. & Chambon, P. (1987) Nature (London) 326, 878–880.
113. Bernards, R., Destree, A., McKenzie, S., Gordon, E., Weinberg, R.A. & Panicali, D. (1987) Proc. Natl. Acad. Sci. USA 84, 6854–6858.
114. Estin, C.D., Stevenson, U.S., Plowman, G.D., Hu, S.L., Sridhar, P. & Hellstrom, I. (1988) Proc. Natl. Acad. Sci. USA 85, 1052–1056.
115. Kantor, J., Irvine, K., Abrams, S., Kaufman, H., Dipietro, J. & Schlom, J. (1992) J. Natl. Cancer Inst. 84, 1084–1091.
116. Roth, J., Dittmer, D., Rea, D., Tartaglia, J., Paoletti, E. & Levine, A.J. (1996) Proc. Natl. Acad. Sci. USA 93, 4781–4786.
117. Rao, J.B., Chamberlain, R.S., Bronte, V., Carroll, M.W., Irvine, K.R., Moss, B., Rosenberg, S.A. & Restifo, N.P. (1996) J. Immunol. 156, 3357–3365.
118. Chamberlain, R.S., Carroll, M.W., Bronte, V., Hwu, P., Warren, S., Yang, J.C., Nishimura, M., Moss, B., Rosenberg, S.A. & Restifo, N.P. (1996) Cancer Res. 56, 2832–2836.
119. Lane, J.M., Ruben, F.L., Neff, J.M. & Millar, J.D. (1969) N. Engl. J. Med. 281, 1201–1208.
120. Redfield, R.R., Wright, D.C., James, W.D., Jones, T.S., Brown, C. & Burke, D.S. (1987) N. Engl. J. Med. 316, 673–676.
121. Katz, S.L. & Broome, C.V. (1991) Morb. Mortal. Wkly. Rep. 40, 1–10.
122. Advisory Committee on Dangerous Pathogens and Advisory Committee on Genetic Modifications (1990) Vaccination of Laboratory Workers Handling Vaccinia and Related Poxviruses Infectious for Humans (HMSO Publications Center, London).
123. Mayr, A., Hochstein-Mintzel, V. & Stickl, H. (1975) Infection 3, 6–14.
124. Hochstein-Mintzel, V., Hänichen, T., Huber, H.C. & Stickl, H. (1975) Zbl. Bakt. Hyg. 230, 283–297.
125. Meyer, H., Sutter, G. & Mayr, A. (1991) J. Gen. Virol. 72, 1031–1038.
126. Sutter, G. & Moss, B. (1992) Proc. Natl. Acad. Sci. USA 89, 10847–10851.
127. Wyatt, L.S., Moss, B. & Rozenblatt, S. (1995) Virology 210, 202–205.
128. Sutter, G., Ohlmann, M. & Erfle, V. (1995) FEBS Lett. 371, 9–12.
129. Sutter, G., Wyatt, L.S., Foley, P.L., Bennink, J.R. & Moss, B. (1994) Vaccine 12, 1032–1040.
130. Wyatt, L.S., Shors, S.T., Murphy, B.R. & Moss, B. (1996) Vaccine, in press.
131. Hirsch, V.M., Fuerst, T.R., Sutter, G., Carroll, M.W., Yang, L.C., Goldstein, S., Piatak, M., Jr., Elkins, W.R., Alvord, W.G., Montefiori, D.C., Moss, B. & Lifson, J.D. (1996) J. Virol. 70, 3741–3752.
132. Buller, R.M.L., Smith, G.L., Cremer, K., Notkins, A.L. & Moss, B. (1985) Nature (London) 317, 813–815.
133. Buller, R.M., Chakrabarti, S., Cooper, J.A., Twardzik, D.R. & Moss, B. (1988) J. Virol. 62, 866–877.
134. Edwards, K.M., Andrews, T.C., Van Savage, J., Palmer, P.S. & Moyer, R.W. (1988) Microb. Pathog. 4, 325–333.
135. Kotwal, G.J., Hügin, A.W. & Moss, B. (1989) Virology 171, 579–587.
136. Child, S.J., Palumbo, G., Buller, R.M. & Hruby, D. (1990) Virology 174, 625–629.
137. Lee, S.L., Roos, J.M., McGuigan, L.C., Smith, K.A., Cormier, N., Cohen, L.K., Roberts, B.E. & Payne, L.G. (1992) J. Virol. 66, 2617–2630.
138. Isaacs, S.N., Kotwal, G.J. & Moss, B. (1992) Proc. Natl. Acad. Sci. USA 89, 628–632.
139. Moore, J.B. & Smith, G.L. (1992) EMBO J. 11, 1973–1980.
140. Tartaglia, J., Perkus, M.E., Taylor, J., Norton, E.K., Audonnet, J.C., Cox, W.I., Davis, S.W., Vanderhoeven, J., Meignier, B., Riviere, M., Languet, B. & Paoletti, E. (1992) Virology 188, 217–232.
141. Konishi, E., Pincus, S., Paoletti, E., Laegreid, W.W., Shope, R.E. & Mason, P.W. (1992) Virology 190, 454–458.
142. Flexner, C., Hügin, A. & Moss, B. (1987) Nature (London) 330, 259–262.
143. Ramshaw, A., Andrew, M.E., Phillips, S.M., Boyle, D.B. & Coupar, B.E.H. (1987) Nature (London) 329, 545–546.
144. Flexner, C., Moss, B., London, W.T. & Murphy, B.R. (1990) Vaccine 8, 17–22.
145. Ruby, J., Brinkman, C., Jones, S. & Ramshaw, I. (1990) Immunol. Cell Biol. 68, 113–117.
146. Karupiah, G., Blanden, R.V. & Ramshaw, I.A. (1990) J. Exp. Med. 172, 1495–1503.
147. Karupiah, G., Coupar, B.E.H., Andrew, M.E., Boyle, D.B., Phillips, S.M., Müllbacher, A., Blanden, R.V. & Ramshaw, I.A. (1990) J. Immunol. 144, 290–298.
148. Yilma, T., Anderson, K., Brechling, K. & Moss, B. (1987) Vaccines 87, 393–396.
149. Kohonen-Corish, M.R.J., Long, N.J.C., Woodhams, C.E. & Ramshaw, I.A. (1990) Eur. J. Immunol. 20, 157–161.
150. Taylor, J., Weinberg, R., Kawaoka, Y., Webster, R.G. & Paoletti, E. (1988) Vaccine 6, 504–508.
151. Boyle, D.B. & Coupar, B.E.H. (1988) Virus Res. 10, 343–356.
152. Taylor, J., Edbauer, C., Rey-Senelonge, A., Bouquet, J.-F., Norton, E., Goebel, S., Desmettre, P. & Paoletti, E. (1990) J. Virol. 64, 1441–1450.
153. Taylor, J., Weinberg, R., Languet, B., Desmettre, P. & Paoletti, E. (1988) Vaccine 6, 497–503.
154. Taylor, J., Trimarchi, C., Weinberg, R., Languet, B., Guillemin, F., Desmettre, P. & Paoletti, E. (1991) Vaccine 9, 190–193.
155. Taylor, J., Weinberg, R., Tartaglia, J., Richardson, C., Alkhatib, G., Breidis, D., Appel, M., Norton, E. & Paoletti, E. (1992) Virology 187, 321–328.
156. Esposito, J.J., Knight, J.C., Shaddock, J.H., Novembre, F.J. & Bauer, G.M. (1988) Virology 167, 313–316.
157. Romero, C.H., Barrett, T., Evans, S.A., Kitching, R.P., Gershon, P.D., Bostock, C. & Black, D.N. (1993) Vaccine 11, 737–742.
158. van der Leek, M.L., Feller, J.A., Sorenson, G., Isaacson, W., Adams, C.L., Borde, D.J., Pfeiffer, N., Tran, T., Moyer, R.W. & Gibbs, E.P.J. (1994) Vet. Rec. 134, 13–18.
159. Mackett, M., Yilma, T., Rose, J.K. & Moss, B. (1985) Science 227, 433–435.
160. Yilma, T., Hsu, D., Jones, L., Owens, S., Grubman, M., Mebus, C., Yamanaka, M. & Dale, B. (1988) Science 242, 1058–1061.
161. Giavedoni, L., Jones, L., Mebus, C. & Yilma, T. (1991) Proc. Natl. Acad. Sci. USA 88, 8011–8015.
162. Chambers, T.M., Kawaoka, Y. & Webster, R.G. (1988) Virology 167, 414–421.
163. Wiktor, T.J., Macfarlan, R.I., Reagan, K.J., Dietzschold, B., Curtis, P., Wunner, W.H., Kieny, M.P., Lathe, R., Lecocq, J.P., Mackett, M., Moss, B. & Koprowski, H. (1984) Proc. Natl. Acad. Sci. USA 81, 7194–7198.
164. Blancou, J., Kieny, M.P., Lathe, R., Lecocq, J.P., Pastoret, P.P., Soulebot, J.P. & Desmettre, P. (1986) Nature (London) 322, 373–375.
165. Rupprecht, C.E., Wiktor, T.J.A., Johnston, D.H., Hamir, A.N., Dietzschold, B., Wunner, W.H., Glickman, L.T. & Koprowski, H. (1986) Proc. Natl. Acad. Sci. USA 83, 7947–7950.
166. Brochier, B., Kieny, M.P., Costy, F., Coppens, P., Baudilin, B., Lecocq, J.P., Languet, B., Chappuis, G., Desmettres, P., Afiademanyo, K., Libois, R. & Pastoret, P.-P. (1991) Nature (London) 354, 520–522.
167. Brochier, B., Costy, F. & Pastoret, P.P. (1995) Vet. Microbiol. 46, 269–279.
168. Romero, C.H., Barrett, T., Chamberlain, R.W., Kitching, R.P., Fleming, M. & Black, D.N. (1994) Virology 204, 425–429.
169. Webster, R.G., Kawaoka, Y., Taylor, J., Weinberg, R. & Paoletti, E. (1991) Vaccine 9, 303–308.
170. Boursnell, M.E., Green, P.F., Samson, A.C., Campbell, J.I., Deuter, A., Peters, R.W., Millar, N.S., Emmerson, P.T. & Binns, M.M. (1990) Virology 178, 297–300.
171. Boursnell, M.E., Green, P.F., Campbell, J.I., Deuter, A., Peters, R.W., Tomley, F.M., Samson, A.C., Chambers, P.,
Emmerson, P.T. & Binns, M.M. (1990) J. Gen. Virol. 71, 621–678.
172. Edbauer, C., Weinberg, R., Taylor, J., Rey-Senelonge, A., Bouquet, J.-F., Desmettre, P. & Paoletti, E. (1990) Virology 179, 901–904.
173. Bayliss, C.D., Peters, R.W., Cook, J.K., Reece, R.L., Howes, K., Binns, M.M. & Boursnell, M.E. (1991) Arch. Virol. 120, 193–205.
174. Letellier, C., Burny, A. & Meulemans, G. (1991) Arch. Virol. 118, 43–56.
175. Cooney, E.L., Collier, A.C., Greenberg, P.D., Coombs, R.W., Zarling, J., Arditti, D.E., Hoffman, M.C., Hu, S.L. & Corey, L. (1991) Lancet 337, 567–572.
176. Graham, B.S., Belshe, R.B., Clements, M.L., Dolin, R., Corey, L., Wright, P.F., Gorse, G.J., Midthun, K., Keefer, M.C., Roberts, N.J., Schwartz, D.H., Agosti, J.M., Fernie, B.F., Stablein, D.M., Montefiori, D.C., Lambert, J.S., Hu, S.L., Esterlitz, J.R., Lawrence, D.N. & Koff, W.C. (1992) J. Infect. Dis. 166, 244–252.
177. Cooney, E.L., McElrath, M.J., Corey, L., Hu, S.L., Collier, A.C., Arditti, D., Hoffman, M., Coombs, R.W., Smith, G.E. & Greenberg, P.D. (1993) Proc. Natl. Acad. Sci. USA 90, 1882– 1886.
178. Gu, S.Y., Huang, T.M., Ruan, L., Miao, Y.H., Lu, H., Chu, C.M., Motz, M. & Wolf, H. (1995) Dev. Biol. Stand. 84, 171–177.
179. Cadoz, M., Strady, A., Meignier, B., Taylor, J., Tartaglia, J., Paoletti, E. & Plotkin, S. (1992) Lancet 339, 1429–1432.
180. Pialoux, G., Excler, J.L., Riviere, Y., Gonzalez-Canali, G., Feuillie, V., Coulaud, P., Gluckman, J.C., Matthews, T.J., Meignier, B., Kieny, M.P. & et al. (1995) AIDS Res. Hum. Retroviruses 11, 373–381.








