
This paper was presented at a colloquium entitled “Genetic Engineering of Viruses and of Virus Vectors,” organized by Bernard Roizman and Peter Palese (Co-chairs), held June 9–11, 1996, at the National Academy of Sciences in Irvine, CA.
Early events in poliovirus infection: Virus-receptor interactions
VINCENT R. RACANIELLO *
Department of Microbiology, Columbia University College of Physicians and Surgeons, 701 West 168th Street, New York, NY 10032
ABSTRACT The interaction of poliovirus with its cell receptor initiates conformational changes that lead to uncoating of the viral RNA. Three types of genetic analyses have been used to study the poliovirus-receptor interaction: (i) mutagenesis of the poliovirus receptor (PVR), (ii) selection of viral mutants resistant to neutralization with soluble PVR, and (iii) selection of viral variants adapted to use mutant PVRs. The results of these studies show that a small portion of the first immunoglobulin-like domain of PVR contacts viral residues within a deep depression on the surface of the capsid that encircles the fivefold axis of symmetry. Viral capsid residues that influence the interaction with PVR are also found in locations such as the capsid interior that cannot directly contact PVR. These mutations might influence the ability of the capsid to undergo receptor-mediated conformational transitions that are necessary for high-affinity interactions with PVR.
All viruses initiate infection of susceptible cells by first binding to a cell surface receptor. For some viruses, the cell receptor plays an active role in the uncoating of the viral genome, whereas for others the receptor is nothing more than a tether that concentrates virus particles on the cell surface and directs them toward disassembly pathways. For example, the uncoating of influenza viruses is triggered by the acidification of the endocytic vesicles that bring the virus into the cells (1). Adenovirus is brought into the endocytic pathway by its cell receptor, where it is dismantled in a process that does not appear to require the receptor (2). Some enveloped viruses fuse with cell membranes at neutral pH; the interaction with the cell receptor may trigger conformational changes in viral glycoproteins that convert them to fusogenic forms (3). The interaction of poliovirus with receptor-bearing cells leads to the production of the conformationally altered A particle (4), which is believed to be an intermediate in cell entry (5). The determination of the three-dimensional structure of the viral capsid (6) and identification of the cell receptor for poliovirus (7) have lead to studies aimed at understanding how virus-receptor interactions lead to uncoating of the viral RNA.
Poliovirus and Its Cell Receptor
The poliovirus capsid consists of 60 copies of each of the four viral polypeptides VP1, VP2, VP3, and VP4, arranged with icosahedral symmetry. All three serotypes of poliovirus utilize a cell surface receptor called the poliovirus receptor (PVR), which is a novel member of the immunoglobulin superfamily, to initiate infection of cells (7). The PVR polypeptide contains an N-terminal signal sequence, three extracellular immunoglobulin (Ig)-like domains, a transmembrane domain, and a cytoplasmic tail. Alternative splicing produces two mRNAs encoding polypeptides of 392 and 417 amino acids that differ in the lengths of their cytoplasmic domains. Both forms of PVR function as receptors for poliovirus. The predicted molecular size of the two polypeptides is 43 or 45 kDa, although posttranslational modification in HeLa cells produces a predominant species of 80 kDa (8).
Two human genes related to PVR, PRR1 and PRR2, have been identified, although it is not known whether the encoded polypeptides function as poliovirus receptors (9, 10). A mouse homolog, MPH, does not bind poliovirus (11), including strains that are adapted to grow in mice (Y.Dong and V.R.R., unpublished data). The cellular functions of PVR, PRR1, PRR2, and MPH are unknown, although like many members of the Ig superfamily, they may play a role in cell adhesion and recognition. The cytoplasmic domain of one isoform of PVR is phosphorylated at serine, possibly by calcium/calmodulin kinase II (12), and several protein kinases bind to and phosphorylate the cytoplasmic domain of MPH (Y.Dong and V.R.R., unpublished data). Identification of these protein kinases should provide clues about the normal role of PVR and MPH in the cell.
PVR and the Uncoating of Viral RNA
Shortly after poliovirus binds to cell surface PVR, it releases its RNA genome into the cytoplasm. PVR is likely to play a role in the uncoating step, as suggested by its ability to induce dramatic structural changes in the virus particle. When poliovirus is bound to cells at 37°C, a large proportion of the virus is eluted as a conformationally altered form known as the A particle (4). These particles contain infectious RNA, but they differ from native virus in their sedimentation coefficient (135 S compared with 160 S for native virions), their increased sensitivity to detergent and proteinases, and the absence of VP4 (5). The N terminus of VP1, normally on the interior of the virion, has been translocated to the surface, making the capsid hydrophobic. Conversion of poliovirus to 135S particles can also be accomplished in solution by incubation with detergent extracts of insect cells expressing PVR (13) or with soluble PVR released into the culture medium from expressing cells (14). It is likely that PVR is sufficient for 135S particle formation, although this possibility has not yet been tested with the purified protein.
The A particle has been proposed to be an essential intermediate in the entry of poliovirus into cells (5). The N terminus of VP1 may form an amphipathic helix that inserts into the cell membrane, producing a pore through which the viral RNA may leave the capsid. To determine the role of 135S particle formation in poliovirus replication, we took advantage of the observation that A particles are not formed at temperatures below 33°C (15) and determined whether poliovirus could replicate at 25°C (A.Dove and V.R.R., unpublished data). Our findings indicate that wild-type Mahoney strain of
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. §1734 solely to indicate this fact.
Abbreviations: PVR, poliovirus receptor; ca, cold adapted.
|
* |
e-mail: racaniello@cuccfa.ccc.columbia.edu. |
poliovirus type 1 (P1/Mahoney) is unable to grow at 25°C, but cold-adapted (ca) mutants are readily selected at this temperature. Ca mutants replicate efficiently at 25°C without forming 135S particles (they do form 135S particles at 37°C). The Ca phenotype maps to a central region of the viral RNA encoding nonstructural proteins, suggesting that the block to replication in wild-type P1/Mahoney is past the stage of cell entry— possibly RNA replication, proteolytic processing, or even assembly. In support of this hypothesis, when the entry steps are bypassed by transfection of viral RNA into cells, ca viral RNA replicates at 25°C, but wt RNA does not.
These results suggest that the formation of 135S particles is not required for poliovirus replication. The altered particle might be a stable end product that is readily detected; the true intermediate in RNA uncoating might be an earlier particle, perhaps less stable than 135S particles, that represents a less-drastic PVR-induced conformational change. The ability to study a productive poliovirus infection at 25°C, in the absence of 135S particle formation, should enable the identification of such structural changes. We are left with the question of why so many nonfunctional 135S particles are formed. The answer is not known, but 135S particle formation has been studied mainly in cultured cells; in vivo, where the accessibility and/or level of cell receptors might be limited, fewer A particles may be generated.
PVR Sequences That Control Virus Binding
To fully understand how the poliovirus-PVR interaction initiates cell entry, a detailed picture of how the virus and receptor combine is required. Ultimately, resolution of a virus-receptor complex will be needed, but the results of genetic analyses have provided some insight into the interaction. The binding site for poliovirus appears to be contained within domain 1, which can bind poliovirus when expressed on the cell surface either alone or linked to other domains from CD4, the intracellular cell adhesion molecule ICAM-1, or MPH (for review, see ref. 16). Virus does not bind as well to domain 1 as it does to native PVR, suggesting that domains 2 and 3 contribute to the interaction, either directly or by influencing the structure of domain 1. Several laboratories have mutagenized PVR domain 1 to identify the putative contact point, and the results show that three main sites are important for poliovirus binding (Fig. 1): (i) the C-C′ loop through the C″ strand, (ii) the border of the D strand and the D-E loop, and (iii) the G strand. A mutation at the beginning of the F strand also reduces virus binding, probably by altering domain structure. Mutagenesis of other loops and strands has not revealed other regions that are important for binding.
These studies indicate that the C′-C″ ridge is likely to be the main part of PVR that contacts poliovirus. The homologous part of CD4 plays a major role in the interaction with human immunodeficiency virus type 1 (for review, see ref. 18). The D-E loop of domain 1 may also contact poliovirus, but the G strand is more distant and not likely to be directly involved with the binding site. Consistent with this hypothesis is the observation that substitution of PVR residues 70–100, which contains the C′-C″ ridge (Fig. 1) into the corresponding region of MPH produces a chimeric receptor that can be recognized by type 1 but not types 2 and 3 poliovirus (Y.Lin and V.R.R., unpublished data). This result suggests that the poliovirus binding site on PVR is contained with this 30-amino acid segment, although contribution of conserved MPH residues to poliovirus binding cannot be excluded. The three serotypes of poliovirus contact PVR slightly differently, a conclusion also drawn from studies of a G-strand mutation (Fig. 1) that abrogates binding of types 1 and 2 but not type 3 poliovirus (19).
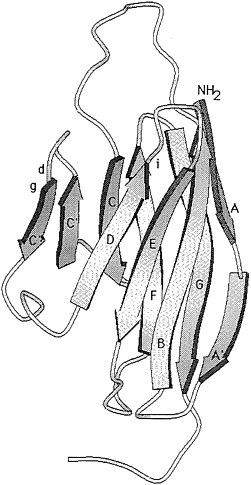
FIG. 1. Structural model of PVR domain 1 (17). The locations of three mutations that influence poliovirus binding are shown as letters: d (amino acid 82, Gln replaced with Phe), g (amino acid 56, insertion of Val-Asp-Phe), and i (amino acid 99, Leu-Gly replaced with ProGlu-Thr-Asn). The β-strands are lettered A-G.
Viral Capsid Sequences That Regulate Receptor Binding
When the three-dimensional structures of rhinovirus and poliovirus were solved, a 1.2-nm-deep 1.5-nm-wide channel was noted surrounding the prominent peak at the fivefold axis of symmetry of the particle (6, 20). This channel was called the canyon and was proposed to be the receptor binding site for rhinovirus 14 (20). A model of the interaction of HRV-16 with its soluble receptor, ICAM-1, indicates that ICAM-1 does bind in the canyon (21). Evidence that the canyon is the receptor binding site in poliovirus comes from the study of two types of viral mutants: soluble receptor resistant (srr) mutants and viruses adapted to utilize mutant PVRs. Detergent-solubilized PVR expressed in insect cells converts poliovirus to 135S particles, neutralizing its infectivity (13). Poliovirus mutants resistant to neutralization with soluble PVR have been selected that possess a range of binding defects to PVR (22, 23). Each srr mutant contains a single mutation, located on the surface or the interior of the capsid. The surface mutations (Fig. 2) are located in the canyon and may form part of the contact site for PVR. Mutation at any one of eight residues decreases the binding affinity of poliovirus for PVR, indicating that multiple points in the virus-receptor interface contribute to binding. Mutations at internal capsid residues also reduce binding affinity. These residues are not likely to contact the receptor directly but may affect the ability of the virus to bind to PVR with high affinity by altering the flexibility of the capsid. The proximity of several of the internal mutations near a hydrocarbon binding pocket that appears to contain sphingosine (24) is consistent with this hypothesis. This pocket is believed to regulate the ability of the capsid to undergo receptor-mediated structural transitions (24).
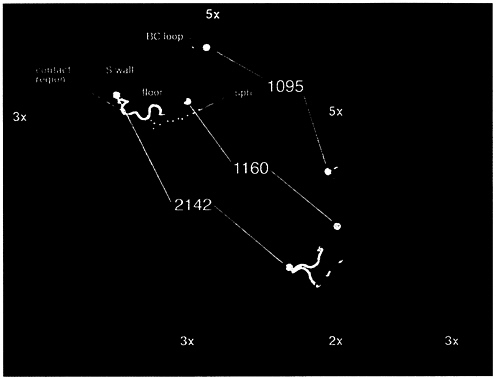
FIG. 2. α-Carbon trace of the poliovirus type 1 Mahoney protomer (6). Upper left, a single protomer viewed from the side; lower right, two neighboring protomers shown in different colors, viewed from the outside of the particle. The fivefold, twofold, and threefold axes of symmetry, the VP1 BC loop, the canyon floor, and canyon wall are labeled. Sphingosine (sph) in the hydrocarbon-binding pocket is shown as blue spheres, srr mutations are shown as a white line labeled contact region, and adapting mutations are shown as yellow spheres and their amino acid residues are given (e.g., 1095, residue 95 of VP1).
Additional information on capsid sequences that control receptor interaction comes from the analysis of viral variants that are adapted to grow on cells expressing mutant forms of PVR that do not bind wild-type 1 poliovirus (25). PVR mutants d, g, and i (Fig. 1) were constructed by substituting residues of PVR with corresponding amino acids from MPH (17). Stable mouse L-cell lines expressing d, g, or i mutants cannot bind poliovirus, but viral variants were isolated that can utilize the mutant PVRs to infect cells. These adapted viruses can still use wild-type PVR to infect cells and, therefore, possess an expanded receptor recognition. Sequence analyses and site-directed mutagenesis identified three sites of mutation that are responsible for the adapted virus phenotype (Fig. 2). Every adapted mutant contained a change at VP1 position 95 from Pro to Ser or Thr; when either amino acid is introduced into wild-type virus by site-directed mutagenesis, viruses are produced that can use all three mutant receptors. Position 95 of VP1 is located in the B–C loop at the fivefold axis of symmetry, distant from the putative receptor contact site defined by srr mutations (Fig. 2). Although it is possible that this portion of the capsid also contacts PVR, the lack of allele specificity of the VP1–95 adapting mutation suggests that this residue is not likely to contact the mutated portions of PVR. This sequence might instead modulate the flexibility of the capsid and its ability to accommodate mutant receptors, a mechanism consistent with the absence of allele specificity. Substitution of the entire VP1 B–C loop with the sequence from the mouse-adapted type 2 Lansing strain (P2/Lansing) enables P1/Mahoney to recognize an unidentified receptor in mice that cannot be used by the wild-type virus (26, 27). In this case, the VP1 B–C loop of P2/Lansing loop may directly contact the mouse receptor, or it may impart to the capsid the flexibility to recognize a new receptor.
A second adapting mutation is a change from Val to Ile at VP1 amino acid 160. This amino acid is located at the interface between protomers (the capsid subunit consisting of one copy each of VP1, VP2, VP3, and VP4), near the hydrophobic binding pocket of VP1. This mutation is not allele specific and might also act by influencing the flexibility of the capsid. The VP1–160 mutation also allows P1/Mahoney to recognize a receptor in mice, thereby causing disease in that host. The mouse-adapted P2/Lansing strain contains an Ile at amino acid 1160; curiously, it can use the g receptor but not the d and i receptors. A third adapting mutation, a change from His to Tyr at VP2 amino acid 142, is located on the canyon wall near the receptor binding site defined by the srr mutations. This mutation is allele-specific and will only correct the defect conferred by the d and g mutations, which are adjacent in PVR (Fig. 1). The nature of the amino acid at this location in the capsid may influence the contact point with PVR. The type 3 Leon strain of poliovirus Y at VP2–142 can only bind the d receptor. These studies emphasize the serotype-specific differences in the interaction of poliovirus with PVR.
Does PVR Require a Cofactor?
A mAb directed against HeLa cells that blocks the binding of poliovirus to HeLa cells in a serotype-specific manner (28) recognizes an isoform of the lymphocyte homing receptor CD44H (29). This cell surface molecule is not a receptor for poliovirus, because expression of CD44H cDNA in PVR-negative mouse L cells does not confer the ability to bind poliovirus. Because the protein recognized by this mAb is restricted to certain tissues that are susceptible to poliovirus infection, it was suggested that CD44 might be a determinant of poliovirus tissue tropism (28). However, the results of growth curve analyses indicate that all three poliovirus serotypes multiply normally in cells that express PVR but not CD44, and the addition of CD44 by stable transformation has no effect on virus multiplication. Furthermore, the binding affinity constant for all three poliovirus serotypes is identical in the presence or absence of CD44 (M.Bouchard and V.R.R., unpublished data). We conclude that CD44 is not required for poliovirus replication in cell culture. CD44H and PVR may be associated in the cell membrane, and the anti-CD44 mAb may block poliovirus binding by its proximity to the virus binding site on PVR.
Summary
The results discussed herein suggest an hypothesis for how the interaction of poliovirus with PVR might initiate uncoating of
the viral RNA. Contact between the virus and receptor occurs through capsid residues in the canyon and the C′-C″ ridge on domain 1 of PVR. High-affinity binding is probably dependent on the nature of the contact residues in the virus and the receptor and on capsid residues at the protomer interface and in the interior that allow the capsid to conform to the receptor. Because the contact points in the canyon are located at the protomer interface, above the hydrocarbon-binding pocket, the interaction with PVR may destabilize the interface and weaken the affinity of sphingosine for the pocket. As additional PVR molecules bind to the capsid, sphingosine may be released, leading to complete destabilization of the capsid. The RNA might then emerge from a portal at the protomer interface. Crystallographic resolution of the virus-receptor complex will be required to demonstrate precisely how the virus and receptor interact. Whether or not PVR, either in soluble form or associated with membranes, is sufficient to drive RNA uncoating can also be determined experimentally. Finally, the location in the cell at which the uncoating event occurs must be identified. These experiments will provide clues about how cell receptors participate in the uncoating of an icosahedral virus.
Work cited from my laboratory has been supported by the National Institutes of Health and the American Cancer Society.
1. White, J. (1994) in Cellular Receptors for Animal Viruses, ed. Wimmer, E. (Cold Spring Harbor Lab. Press, Plainview, NY), pp. 281–301.
2. White, J. (1990) Annu. Rev. Physiol. 52, 675–697.
3. Greber, U., Willetts, M., Webster, P. & Helenius, A. (1993) Cell 75, 477–486.
4. Joklik, W.K. & Darnell, J.E. (1961) Virology 13, 439–447.
5. Fricks, C.E. & Hogle, J.M. (1990) J. Virol. 64, 1934–1945.
6. Hogle, J.M., Chow, M. & Filman, D.J. (1985) Science 229, 1358–1365.
7. Mendelsohn, C., Wimmer, E. & Racaniello, V.R. (1989) Cell 56, 855–865.
8. Bernhardt, G., Bibb, J.A., Bradley, J. & Wimmer, E. (1994) Virology 199, 105–113.
9. Eberl, F., Dubreuil, P., Mattei, M.G., Devilard, E. & Lopez, M. (1995) Gene 159, 267–272.
10. Lopez, M., Eberl, F., Mattei, M.G., Gabert, J., Birg, F., Bardin, F., Maroc, C. & Dubreuil, P. (1995) Gene 155, 261–265.
11. Morrison, M.E. & Racaniello, V.R. (1992) J. Virol. 66, 2807– 2813.
12. Bibb, J.A., Bernhardt, G. & Wimmer, E. (1994) J. Virol. 68, 6111–6115.
13. Kaplan, G., Freistadt, M.S. & Racaniello, V.R. (1990) J. Virol. 64, 4697–4702.
14. Zibert, A., Selinka, H.-C., Elroy-Stein, O. & Wimmer, E. (1992) Virus Res. 25, 51–61.
15. Gómez Yafal, A., Kaplan, G., Racaniello, V.R. & Hogle, J.M. (1993) Virology 197, 501–505.
16. Racaniello, V.R. (1995) in Human Enterovirus Infections, ed. Rotbart, H.A. (Am. Soc. Microbiol., Washington, DC), pp. 73–93.
17. Morrison, M.E., Yuan-Jing, H., Wien, M.W., Hogle, J.W. & Racaniello, V.R. (1994) J. Virol. 68, 2578–2588.
18. Ryu, S., Kwong, P.D., Truneh, A., Porter, T.G., Arthos, J., Rosenberg, M., Dai, X., Xuong, N., Axel, R., Sweet, R.W. & Hendrickson, W.A. (1990) Nature (London) 348, 419–426.
19. Harber, J., Bernhardt, G., Lu, H.-H., Sgro, J. & Wimmer, E. (1995) Virology 214, 559–570.
20. Rossmann, M.G., Arnold, E., Erickson, J.W., Frankenberger, E.A., Griffith, J.P., Hecht, H.-J., Johnson, J.E. & Kamer, G. (1985) Nature (London) 317, 145–153.
21. Olson, N.H., Kolatkar, P.R., Oliveira, M.A., Cheng, R.H., Greve, J.M., McClelland, A., Baker, T.S. & Rossmann, M.G. (1993) Proc. Natl. Acad. Sci. USA 90, 507–511.
22. Kaplan, G., Peters, D. & Racaniello, V.R. (1990) Science 250, 1596–1599.
23. Colston, E. & Racaniello, V.R. (1994) EMBO J. 13, 5855–5862.
24. Filman, D.J., Syed, R., Chow, M., Macadam, A.J., Minor, P.D. & Hogle, J.M. (1989) EMBO J. 8, 1567–1579.
25. Colston, E.M. & Racaniello, V.R. (1995) J. Virol. 69, 4823–4829.
26. Martin, A., Wychowski, C., Couderc, T., Crainic, R., Hogle, J. & Girard, M. (1988) EMBO J. 7, 2839–2847.
27. Murray, M.G., Bradley, J., Yang, X.F., Wimmer, E., Moss, E.G. & Racaniello, V.R. (1988) Science 241, 213–215.
28. Shepley, M.P., Sherry, B. & Weiner, H.L. (1988) Proc. Natl. Acad. Sci. USA 85, 7743–7747.
29. Shepley, M.P. & Racaniello, V.R. (1994) J. Virol. 68, 1301–1308.




