
1 Phosgene1
Acute Exposure Guideline Levels
SUMMARY
Phosgene is a colorless gas at ambient temperature and pressure. Its odor has been described as similar to new-mown hay. Phosgene is manufactured from a reaction of carbon monoxide and chlorine gas in the presence of activated charcoal. The production of dyestuffs, isocyanates, carbonic acid esters (polycarbonates), acid chlorides, insecticides, and pharmaceutical chemicals requires phosgene. Manufacture of phosgene is approximately 1 million tons per year (y) in the United States, and more than 10,000 workers are involved in its manufacture and use. Manufacture of phosgene in the United States is
almost entirely captive—it is used in the manufacture of other chemicals within a plant boundary. Only one company sells phosgene on the U.S. merchant market.
Inhalation is the most important route of exposure for phosgene. Because of phosgene’s mild upper respiratory, eye, and skin irritancy and mildly pleasant odor, an exposed victim may not actively seek an avenue of escape before lower respiratory damage has occurred (Currie et al. 1987a; Lipsett et al. 1994). Pulmonary edema is the cause of death after a clinical latency period of ≤24 hours (h) (Franch and Hatch 1986).
Appropriate data were not available for deriving AEGL-1 values for phosgene. Odor cannot be used as a warning for potential exposure. The odor threshold is reported to be between 0.5 and 1.5 parts per million (ppm), a value above or approaching AEGL-2 and AEGL-3 values, and tolerance to the pleasant odor of phosgene occurs rapidly. Furthermore, following odor detection and minor irritation, serious effects may occur after a clinical latency period of ≤24 h.
AEGL-2 values were based on chemical pneumonia in rats (exposure at 2 ppm for 90 min) (Gross et al. 1965). An uncertainty factor (UF) of 3 was applied for interspecies extrapolation because little species variability is observed for lethal and nonlethal end points after exposure to phosgene. A UF of 3 was applied to account for sensitive human subpopulations due to the steep concentration-response curve and because the mechanism of phosgene toxicity (binding to macromolecules and causing irritation) is not expected to vary greatly among individuals. Therefore, the total UF is 10. The 1.5-h value was then scaled to the 30-min and 1-, 4-, and 8-h AEGL exposure periods using Cn×t=k, where n=1 (Haber’s law), because Haber’s law has been shown to be valid for phosgene within certain limits. Haber’s law was originally derived from phosgene data (Haber 1924). The 30-min value is also adopted as the 10-min value, because extrapolation would yield a 10-min AEGL-2 value approaching concentrations that produce alveolar edema in rats; Diller et al. (1985) observed alveolar pulmonary edema in rats exposed to phosgene at 5 ppm for 10 min. Applying a total UF of 10 to this data point yields a supporting 10-min AEGL-2 value of 0.5 ppm.
The 30-min and 1-, 4-, and 8-h AEGL-3 values were based on the highest concentration causing no mortality in the rat after a 30-min exposure (15 ppm) (Zwart et al. 1990). A UF of 3 was applied for interspecies extrapolation because little species variability is observed for lethal and nonlethal end points after exposure to phosgene. A UF of 3 was applied to account for sensitive human subpopulations due to the steep concentration-response curve and
because the mechanism of phosgene toxicity (binding to macromolecules and causing irritation) is not expected to vary greatly between individuals. Therefore, the total UF is 10. The value was then scaled to the 1-, 4-, and 8-h AEGL periods using Cn×t=k, where n=1 (Haber’s Law), because Haber’s Law has been shown to be valid for phosgene within certain limits. Haber’s Law was originally derived from phosgene data (Haber 1924). The 10-min AEGL-3 value was based on the highest concentration causing no mortality in the rat or mouse (36 ppm) after a 10-min exposure (Zwart et al. 1990). A UF of 3 was applied for interspecies extrapolation because little species variability is observed for lethal and nonlethal end points after exposure to phosgene. A UF of 3 was applied to account for sensitive human subpopulations due to the steep concentration-response curve and because the mechanism of phosgene toxicity (binding to macromolecules and causing irritation) is not expected to vary greatly between individuals (total UF, 10).
The calculated values are listed in Table 1–1.
1. INTRODUCTION
Phosgene is a colorless gas at ambient temperature and pressure. Its odor has been described as similar to new-mown hay (Leonardos et al. 1968). This mild odor and the weak acute irritant properties, however, provide little warning of its presence (Lipsett et al. 1994). The odor threshold has been established between 0.5 and 1.5 ppm (2.06 and 6.18 mg/m3) (Lipsett et al. 1994).
Phosgene is manufactured from a reaction of carbon monoxide and chlorine gas in the presence of activated charcoal. Manufacture of phosgene is approximately 1 million tons per year (y) in the United States, and more than 10,000 workers are involved in its manufacture and use (Currie et al. 1987a). Manufacture of phosgene in the United States is almost entirely captive (more than 99% is used in the manufacture of other chemicals within a plant boundary). Only one company sells phosgene on the U.S. merchant market. Over 80% of the phosgene used in the United States is involved in the manufacture of polyisocyanates in the polyurethane industry. The polycarbonate industry accounts for approximately 10% of phosgene used, and the remaining 10% is used in the production of aliphatic diisocyanates, monoisocyanates, chloroformates, agrochemicals, and intermediates for dyestuffs and pharmaceuticals. Phosgene can also be used in metal recovery operations (platinum, uranium, plutonium, and niobium) and has been used for manufacturing aluminum chloride, beryllium chloride, and boron trichloride. It has been pat-
TABLE 1–1 Summary of Proposed AEGL Values for Phosgene (ppm [mg/m3])
|
Classification |
10 min |
30 min |
1 h |
4 h |
8 h |
End Point (Reference) |
|
AEGL-1 |
NA |
NA |
NA |
NA |
NA |
NA |
|
(Nondisabling) |
|
|||||
|
AEGL-2 |
0.60 |
0.60 |
0.30 |
0.08 |
0.04 |
Chemical pneumonia rats (Gross et al. 1965) |
|
(Disabling) |
(2.5) |
(2.5) |
(1.2) |
(0.33) |
(0.16) |
|
|
AEGL-3 |
3.6 |
1.5 |
0.75 |
0.20 |
0.09 |
Highest concentration causing no mortality in the rat after a 30-min or 10-min exposure (Zwart et al. 1990) |
|
(Lethal) |
(15) |
(6.2) |
(3.1) |
(0.82) |
(0.34) |
|
ented as a stabilizer for liquid SO2. In addition, many pesticides have been produced by reaction of a thiol or dithiol with phosgene to produce thiol chloroformates (Kirk-Othmer 1991).
Inhalation is the most important route of exposure for phosgene. Because of phosgene’s mild upper respiratory, eye, and skin irritancy and mildly pleasant odor, an exposed victim may not actively seek an avenue of escape before lower respiratory damage has occurred (Currie et al. 1987a; Lipsett et al. 1994). Pulmonary edema is the cause of death after a clinical latency period of ≤24 h (Franch and Hatch 1986).
The chemical structure is depicted below, and the physicochemical properties of phosgene are presented in Table 1–2.
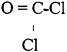
2. HUMAN TOXICITY DATA
2.1. Acute Lethality
Diller and Zante (1982) performed an extensive literature review concerning human phosgene exposure, and found that a great majority of data were
TABLE 1–2 Physical and Chemical Data
|
Parameter |
Data |
Reference |
|
Synonyms |
Carbonyl chloride, carbon oxychloride, carbonic dichloride, chloroformyl chloride |
Lipsett et al. 1994; EPA 1986 |
|
Chemical formula |
COCL2 |
Lipsett et al. 1994 |
|
Molecular weight |
98.92 |
Lipsett et al. 1994 |
|
CAS registry no. |
75–44–5 |
Lipsett et al. 1994 |
|
Physical state |
Gas |
Lipsett et al. 1994 |
|
Vapor pressure |
1215 mm Hg at 20°C |
EPA 1986 |
|
Vapor density |
3.4 (air=1) |
Lipsett et al. 1994 |
|
Specific gravity |
1.381 g/l at 20°C |
ACGIH 2000 |
|
Melting/boiling/ flash point |
−128°C/8.2°C/not applicable |
Lipsett et al. 1994; NIOSH 1994 |
|
Solubility |
Decomposes in water and alcohol; soluble in organic solvents |
EPA 1986 |
|
Conversion factors in air |
1 ppm=4.11 mg/m3 1 mg/m3=0.24 ppm |
Lipsett et al. 1994 |
|
Incompatibility |
Alkalis, ammonia, alcohols, copper |
NIOSH 1997 |
anecdotal or rough estimates and, thus, did not contain reliable exposure concentrations and/or durations. Information synthesized from this review is presented in Table 1–3. Based on observations during World War I, the 2 min LC50 value for humans was estimated to be 790 ppm (Chasis 1944).
Many case reports describe symptomology and postmortem results from human phosgene poisonings; however, exposure concentrations were not reported. Six men were occupationally exposed to phosgene when a pipe ruptured (Stavrakis 1971). A 24-y-old who had received the heaviest exposure arrived at the emergency room minutes after the accident. Upon admission, the patient was symptom-free; however, he was treated with methenamine intravenously and admitted for a 24-h observation. During this time, he remained symptom free and was discharged with no evidence of phosgene injury. The other five patients arrived at the emergency room between 6 and
TABLE 1–3 Effect of Phosgene Exposure in Healthy Humans
|
Effecta |
Cumulative Phosgene Exposure |
|
LCT1 |
~300 ppm·min |
|
LCT50 |
~500 ppm·min |
|
LCT100 |
~1,300 ppm·min |
|
aLethal concentration × time product. Source: Diller and Zante 1982. |
|
12 h after the accident, presenting with various degrees of phosgene intoxication. One 31-y-old who had been exposed “in almost the same degree as the previous patient” rapidly developed pulmonary edema. He also exhibited extreme hemoconcentration and leukocytosis. He did not respond to methenamine treatment and died 3.5 h after admission. The other four exposed workers were hospitalized for various periods of time and recovered satisfactorily.
A 23-y-old man (healthy nonsmoker) was exposed to phosgene at an estimated concentration of at least 5–10 ppm for 5 to 10 seconds (s) (Bradley and Unger 1982). He began coughing upon exposure to phosgene and experienced dyspnea and chest tightness within 30 min. Four hours after exposure, he was hospitalized with hypotension, tachycardia, tachypnea, cyanosis, and pulmonary edema. The patient was intubated and administered dopamine and methylprednisolone. From the second to the sixth day of hospitalization, he developed mediastinal and subcutaneous emphysema, bilateral pneumohydrothoraces, elevated white blood cell counts, fever, and hemiparesis on the right side. Death occurred after the patient developed ventricular fibrillation.
Misra et al. (1985) described another accidental occupational phosgene poisoning case. A 30-y-old male was exposed to phosgene at an undetermined concentration and immediately began coughing and experienced a sense of suffocation and burning eyes. After removal to fresh air and administration of oxygen, he felt better. However, approximately 7.5 h after the exposure, he was rushed to the emergency room with difficulty breathing. Despite oxygen administration and antibiotic therapy, his condition deteriorated. He died approximately 18 h after exposure. An autopsy showed pulmonary edema and bronchiolar necrosis, both of which were more severe in the lower lobes of the lungs than in the upper lobes.
Hegler (1928) reported the effects of a phosgene accident that occurred in Hamburg, Germany, on May 20,1928. Eleven metric tons of “pure phosgene”
were released from a storage tank on a warm, dry, slightly windy day. Within a few hours, people as far as six miles from the release site began reporting to hospitals. Three hundred people reported to hospitals within a few days of the accident. Effects ranged from mild or moderate illness to death; ten people were reported to have died. In general, exposed persons exhibited symptoms consistent with other reported phosgene poisonings (headache, dizziness, nausea and vomiting, irritant cough, and sickening-sweet taste, followed by a latency period and then pulmonary symptoms). Autopsies on six of the fatalities showed pulmonary effects in all cases. Fatty degeneration of the kidneys, liver, and heart were observed in a few cases and were thought to be secondary to the pulmonary damage. In an atypical case, damage in the gray matter of the brain and spinal cord, hyperemia, and signs of bleeding in the white matter were observed at autopsy. That patient died 11.5 days (d) postexposure from a blood clot lodged in the lung. It was uncertain if the extrapulmonary effects were due to phosgene exposure.
2.2. Nonlethal Toxicity
NIOSH (1976) performed two studies to determine the odor threshold of phosgene. In the first, 56 military personnel were exposed to phosgene at increasing concentrations until all subjects could detect odor. The lowest detectable concentration was 0.4 ppm. Thirty-nine percent of subjects could detect odor at 1.2 ppm, and 50% of subjects detected odor at 1.5 ppm. In the other study, four subjects identified 1.0 ppm as the lowest concentration at which the distinctive “new-mown hay” odor of phosgene could be detected.
In their literature review, Diller and Zante (1982) also identified nonlethal effects from phosgene exposure (lethal effects are described in Section 2.1). Nonlethal information synthesized from this review is presented in Table 1–4. From the above data and from animal data for “initial lung damage,” Diller and Zante (1982) synthesized information for nonlethal effects of phosgene in humans (Table 1–5).
2.2.1. Case Reports
A 30-y-old male was occupationally exposed to phosgene at an unknown concentration (Stavrakis 1971). After a short episode of coughing, he returned to work and completed the final 3 h of his shift. Approximately 4 h post-
TABLE 1–4 Acute Irritative Effects of Phosgene Exposure in Humans
|
Effect |
Phosgene Concentration |
|
Throat irritation |
3.1 ppm |
|
Ocular irritation |
4.0 ppm |
|
Cough |
4.8 ppm |
|
Severe eye and airway irritation |
10 ppm |
|
Source: Diller and Zante 1982 |
|
exposure, he presented at the emergency room with severe dyspnea, restlessness, chest pain, and persistent, productive cough. Chest x-rays confirmed acute pulmonary edema. He was treated and discharged free of symptoms 5 d after the phosgene exposure.
An investigator was exposed to phosgene at an undetermined concentration during an experiment (Delephine 1922). He entered the phosgene chamber “at frequent intervals” over a period of 45 min to take instrument readings. At first, he experienced only laryngeal and conjunctival irritation, but as the phosgene concentration increased, he was forced to hold his breath and not stay in the room for more than 1 min. Toward the end of the experiment, some phosgene escaped from the chamber. At this time, the investigator and a colleague experienced a violent cough and began to run away. During their escape, both men had to stop frequently due to the violent nature of their coughs. After exiting the contaminated area, both individuals continued to cough for approximately 20 min. They then improved for 3 or 4 h, after which they experienced a choking sensation that lasted approximately 24 h. Marked lassitude lasted for an additional few days, after which recovery appeared to be complete.
Everett and Overholt (1968) discussed a 40-y-old male who received a “massive” phosgene exposure. His initial symptoms included coughing and burning of the eyes, which subsided within 5 min. He was asymptomatic for the next 2 h, after which a hacking cough began. Three hours after exposure, mild dyspnea was present, and 6 h postexposure, severe dyspnea and moist rales were observed. He was admitted to an intensive care unit 8 h postexposure and presented with anxiety, agitation, cyanosis, thirst, constant cough, and severe pulmonary edema. By the fifth day in the hospital, he was asymptomatic, and by the seventh day, pulmonary function and chest x-ray were normal. A 2-y follow-up was unremarkable.
TABLE 1–5 Effect of Phosgene Exposure in Humans
|
Effect |
Phosgene Exposure |
|
Odor perception |
>0.4 ppm |
|
Odor recognition |
>1.5 ppm |
|
Ocular, nasal, throat, and bronchiolar irritation |
>3 ppm |
|
Initial lung damage |
>30 ppm·min |
|
Clinical pulmonary edema |
>150 ppm·min |
|
Source: Diller and Zante 1982. |
|
Regan (1985) described a phosgene release from a toluene diisocyanate plant. Fifteen employees were exposed to phosgene at an undetermined concentration, resulting in the hospitalization of four workers. Two of the four were released after an overnight observation. The other two were in more serious condition. One of them, a 31-y-old male, had pulmonary edema, rales in both lungs, and left chest pain 8 h postexposure. He was treated with oxygen, bronchodilators, steroids, and antibiotics and returned to work 6 d after the accident. His follow-up was unremarkable. The second man, a 47-y-old smoker, presented with dyspnea, bilateral rales, and pulmonary edema 11 h postexposure. He was also treated with oxygen, bronchodialators, steroids, and antibiotics but continued to deteriorate. He remained critical for 3 d with low right-side heart pressure, low arterial pressure, hemoconcentration, and leukocytosis. He was asymptomatic by 12 d postexposure. He had mild pulmonary obstruction four weeks after the accident; however, it is unclear if that was a result of phosgene exposure or of his smoking.
Longer-term effects from acute phosgene exposure have also been described. Galdston et al. (1947a) described the late effects of phosgene poisoning in six workers (two male, four female; ages 31–50). After an acute, accidental, occupational exposure to phosgene all of these workers experienced the typical effects of acute phosgene exposure. Chronic clinical findings present from 1 to 24 months (mo) postexposure included rapid, shallow breathing and changes in pulmonary function. However, no correlation was observed between the magnitude of phosgene exposure or the severity of acute effects and the severity of chronic symptoms. Galdston et al. (1947a) attributed the severity of chronic symptoms to the subjects’ psychological state. Smoking habits were not reported, and long-term follow-up was not performed.
Galdston et al. (1947b) also examined five males (ages 24–50) who had repeated occupational exposure to phosgene in “small amounts” during the course of 1.5 to 3.5 y. The subjects were examined with regard to pulmonary function and cardiac status. The subjects exhibited transitory effects such as cough, shortness of breath on exertion, and pain or tightness of the chest. These symptoms abated upon removal from phosgene exposure for several weeks. Results suggested that although symptoms of chronic exposure to low concentrations of phosgene are generally not as disabling as those from acute exposure, emphysema may develop after chronic exposure. Also, pulmonary function effects are more severe after chronic low-level exposure than after recovery from a serious acute exposure.
Diller et al. (1979) examined 12 originally healthy workers 3 to 9 y after intoxication with phosgene (ten workers), nitric oxide (one worker), or treflon smoke (one worker). Six of the 12 individuals complained of pulmonary symptoms for 3 y postexposure, and three of the 12 showed slight to severe lung function effects. The severity of lung function decrement correlated more closely with smoking habits than with the severity of chemical intoxication. Diller et al. (1979) concluded that originally healthy survivors of phosgene intoxication recover fairly well over a period of years. However, individuals with preexisting chronic bronchitis may suffer significant chronic deterioration of lung function after acute phosgene intoxication, as an additional observation over 25 y showed. The individual, a light smoker who had mild chronic bronchitis since childhood, was exposed to phosgene and smoke at age 35. He developed severe pulmonary edema and was hospitalized for 7 weeks (wk) following the exposure. During the months following the exposure, his general condition worsened and the bronchitis became more severe. After 2 y, pulmonary function (forced expiratory volume [FEV] and vital capacity [VC]) was decreased to 70% of normal. Ten years postexposure, he developed pulmonary emphysema, and VC and FEV were decreased to 50% of normal.
Herzog and Pletscher (1955) observed squamous metaplasia of the ciliated bronchial epithelium in two patients exposed to undetermined concentrations of phosgene. The metaplasia was observed 3 mo or 3 y postexposure, respectively.
In a phosgene processing factory, phosgene concentrations were measured over an 8-mo period with a device capable of detecting phosgene at 0–0.5 ppm (Henschler 1971). Positive values, of relatively short duration, were recorded on only 32 of 240 d:22 d, 0.05 ppm; 6 d, between 0.06 and 0.1 ppm; 3 d, between 0.1 and 0.5 ppm; 1 d, 0.5 ppm (of short duration). For longer time
periods, elevated concentrations were measured on three occasions: twice for 3 h, between 0.015 and 0.035 ppm, and once for 1.5 h, 0.35 ppm. The above described conditions produced no intoxication or adverse effects on lung function.
In another factory, use of a phosgene indicator badge revealed an average of 34 phosgene exposures per year during the period from 1978 to 1988 (Kaerkes 1992). The workforce contained approximately 200 individuals ranging in age from <20 to 60 y. Exposure concentrations ranged from <50 to 300 ppm·min. Below 50 ppm·min, no signs or symptoms of phosgene toxicity were observed in 75 of 88 individuals; however, three cases of temporary pulmonary edema were observed.
In another report, Sandall (1922) examined 83 British soldiers 3 y after phosgene exposure. Shortness of breath upon exertion (70%), cough with expectoration (54%), tight feeling in chest (25%), sporadic giddiness (14%), and nausea (12%) were the most frequently reported complaints. No physical lung abnormalities were noted in 53% of the men.
2.2.2. Epidemiologic Studies
Polednak (1980) studied a uranium processing plant where phosgene was produced as a by-product of a chemical process in which UO3 was combined with CCl4 to produce UCl4. Leaks and system failures resulted in the accidental release of phosgene into work areas. Although extensive monitoring data were not available, it was determined that the average exposure was below the detection limit of monitoring instruments used at that time. However, there were three to five episodes daily when concentrations exceeded the 1 ppm exposure limit recognized as the occupational standard of the time.
Approximately 30 y after exposure, there were no significant increases in mortality from overall cancer or cancers at specific anatomical sites, in diseases of the respiratory system, or in overall mortality noted in this cohort. However, the exposure period covered by the study was short, the exposed groups were small, and the exposure levels were not well documented. Consequently, evidence presented in this study is inadequate to assess the carcinogenicity of phosgene.
Polednak and Hollis (1985) reported a follow-up of the study discussed above. The study update reported the mortality experience of individuals through the end of 1978 and included 694 routinely exposed white male chemical workers, 97 acutely exposed chemical workers, and 9,280 controls.
Vital status ascertainment for the routinely exposed group and the controls was approximately 90% complete using Social Security Administration (SSA) records. For the acutely exposed group, SSA records as well as state death indexes were used to ascertain vital status, which was approximately 92% complete. Five individuals in the routinely exposed group, nine in the acutely exposed group, and 72 controls were lost to follow-up.
Approximately 35 y after exposure to phosgene, no increase in overall mortality or mortality from cancer or respiratory disease was noted in this cohort.
NIOSH (1976) compared the medical records of 326 workers exposed to phosgene with those of 6,288 unexposed workers from the same plant. Personal air sample measurements at this plant (20-min samples) showed phosgene concentrations ranging from undetectable to 0.02 ppm, and there was a 15 sample average of 0.003 ppm. Fixed-position air samples (20-min or 2-h collection) ranged from undectable to 0.13 ppm in 51 of 56 samples, and >0.14 ppm in 5 of 56 samples. There were no differences in pulmonary function or deaths attributable to respiratory disease between the exposed and control populations.
2.3. Developmental and Reproductive Toxicity
Developmental and reproductive studies regarding acute human exposure to phosgene were not available.
2.4. Genotoxicity
Genotoxic studies regarding acute human exposure to phosgene were not available.
2.5. Carcinogenicity
Polednak (1980) and Polednak and Hollis (1985) examined a cohort of chemical workers exposed to phosgene at chronic low levels as well as daily exposures above 1 ppm. Approximately 35 y after exposure to phosgene, no increase in overall mortality or mortality from cancer or respiratory disease
was noted. These studies are described in detail in Section 2.2.2 (Epidemiologic Studies).
2.6. Summary
Although there is a paucity of acute human data containing known exposure concentrations and times, reports of human phosgene poisonings present a relatively consistent set of clinical effects and sequelae. After acute phosgene exposure, brief (≤20 min) ocular and throat irritation, cough, nausea and vomiting, and dizziness are experienced, followed by a period (≤24 h) of apparent well-being. After this clinical latency phase, cough accompanied by expectoration, a sensation of pain or tightness of the chest, shortness of breath, and a choking sensation are experienced. Clinical findings may include hemoconcentration, leukocytosis, rales, and pulmonary edema. After recovery, rapid shallow breathing, shortness of breath on exertion, and a sense of decreased physical fitness may persist for months. Pulmonary emphysema may occur with repeated exposure to phosgene. Epidemiology studies have shown no increase in cancer in workers exposed to phosgene compared with controls. No information concerning reproductive and developmental toxicity or genotoxicity was available.
3. ANIMAL TOXICITY DATA
3.1. Acute Lethality
3.1.1. Mouse
Zwart et al. (1990) exposed groups of five male and five female Swiss mice to phosgene at varying concentrations for 5, 10, 30, or 60 min. The test atmosphere was monitored at both the inlet and outlet of the glass exposure chambers by gas chromatography and infrared analysis. Ten-minute LC50 values were 77 and 61 ppm for males and females, respectively. Thirty-minute LC50 values were 18 and 11 ppm for males and females, respectively, and 60-min LC50 values were determined to be 9 and 5 ppm for males and females, respectively.
Cameron et al. (1942) exposed 20 mice to phosgene at an average concentration of 0.86 ppm for 5 h. Twelve mice were dead the next morning. Sev-
eral other acute lethality studies of phosgene in mice have been reported. However, these studies do not contain experimental details such as strain or gender of mouse, number of animals exposed, or analytical methodology. These values, in addition to those of Zwart et al. (1990), are presented in Table 1–6.
3.1.2. Rats
Zwart et al. (1990) exposed groups of five male and five female Wistar rats to phosgene at varying concentrations for 5, 10, 30, or 60 min. The test atmosphere was monitored at both the inlet and outlet of the glass exposure chambers by gas chromatography and infrared analysis. The 10-min LC50 value was 80 ppm, and the 30- and 60-min LC50 values were 20 and 12 ppm, respectively.
A total of 118 male Wistar rats were exposed to phosgene at 0.5 to 4.0 ppm for 5 min to 8 h (Rinehart 1962; Rinehart and Hatch 1964). The exposures were varied to give CT products between 12 and 360 ppm-min and were carried out in 1,700-L wooden exposure chambers operating at a constant ventilation rate of 1,000 L/min. The chamber surfaces were lacquered, and thus, potential loss of phosgene by reaction with the wooden surface was minimized. This system provided for air “turnover” every 2 min and a 99% equilibrium time of 8 min. Air samples were taken frequently during exposures, and adjustments were made when necessary to maintain constant phosgene concentrations. An L(CT)0 of 180 ppm·min, a 75-min LC50 of 4 ppm, and a 125-min LC100 of 4 ppm were determined. The authors concluded that different combinations of concentration and time exposure giving equal products of C × T constitute equally effective doses.
Several other acute lethality studies of phosgene in rats have been reported. However, as is the case with the mice, these studies do not contain experimental details such as strain or gender, number of animals exposed, or analytical methodology. These studies are summarized in Table 1–7.
Box and Cullumbine (1947b) investigated phosgene-induced lethality in rats after the rats had experienced an exposure to phosgene at a nonlethal concentration. Rats were divided into two groups (12 per group). Half of each group was exposed to 19.2 ppm phosgene for 10 min and the other half served as a control group. Five days later, the preexposed and control rats were exposed to phosgene at 55.2, 60, 75.6, and 105.6 ppm for 10 min. The rats were then observed for the next 48 h for deaths. The pretreated rats had a reduced percentage of mortality (33%) compared with the control animals
TABLE 1–6 Acute Lethality of Phosgene in Mice
|
Time (min) |
LC50 (ppm) |
Reference |
|
1 |
850 |
Chasis 1944 |
|
1 |
3,300 |
Moor and Gates 1946 |
|
5 |
33 |
Kawai 1973 |
|
10 |
77 (male); 61 (female) |
Zwart et al. 1990 |
|
15 |
15 |
Cameron and Foss 1941 |
|
30 |
18 (male); 11 (female) |
Zwart et al. 1990 |
|
30 |
5.1 |
Kawai 1973 |
|
60 |
9 (male); 5 (female) |
Zwart et al. 1990 |
(74%). Thus, partial protection from phosgene-induced lethality was obtained by the phosgene pretreatment.
3.1.3. Guinea Pigs
The few acute lethality studies of phosgene in guinea pigs do not contain experimental details such as strain or gender, number of animals exposed, or analytical methodology. These less-than-adequate studies are summarized in Table 1–8.
3.1.4. Rabbits
As was the case with guinea pigs, the few acute lethality studies of phosgene in rabbits do not contain experimental details such as strain or gender, number of animals exposed, or analytical methodology. These less than adequate studies are summarized in Table 1–9.
3.1.5. Cats
Wirth (1936) reported no deaths in cats exposed to phosgene at 1.8 ppm for 470 min. A 1-min LC50 of 1,482 ppm was reported by Moor and Gates (1946), and a 15-min LC50 of 80 ppm was reported by Underhill (1920). No
TABLE 1–7 Acute Lethality of Phosgene in Rats
|
Strain |
Number/ Gender |
Exposure Time (min) |
Concentration (ppm) |
End Point |
Reference |
|
NR |
NR |
10 |
35 |
LC20 |
Shils 1943 |
|
NR |
NR |
10 |
60 |
LC40 |
Shils 1943 |
|
NR |
NR |
1 |
1,625 |
LC50 |
Chasis 1944 |
|
NR |
44/NR |
10 |
38–75 |
LC50 |
Box and Cullumbine 1947a |
|
NR |
NR |
12 |
30 |
LC50 |
Chasis 1944 |
|
NR |
NR |
15 |
35 |
LC50 |
Cameron and Foss 1941 |
|
NR |
NR |
20 |
15 |
LC50 |
Kimmerle and Diller 1977 |
|
Wistar |
40/NR |
30 |
10–15 |
LC75 |
Henschler and Laux 1960 |
|
NR |
NR |
20 |
25 |
LC50 |
Rothlin 1941 |
|
NR |
NR |
12 |
85 |
LC60 |
Shils 1943 |
|
NR |
NR |
10 |
40 |
LC70 |
Kimmerle and Diller 1977 |
|
NR |
32/NR |
10 |
39–103 |
LC75 |
Box and Cullumbine 1947a |
|
Wistar |
40/NR |
20 |
25 |
LC50 |
Henschler and Laux 1960 |
|
NR |
NR |
3 |
220 |
LC100 |
Winternitz et al. 1920 |
|
NR |
12/NR |
10 |
147 |
LC100 |
Box and Cullumbine 1947a |
|
NR |
10/NR |
13 |
73 |
LC100 |
Schultz 1945 |
|
Strain |
Number/ Gender |
Exposure Time (min) |
Concentration (ppm) |
End Point |
Reference |
|
NR |
NR |
20 |
37 |
LC100 |
Rothlin 1941 |
|
NR |
NR |
30 |
22 |
LC100 |
Winternitz et al. 1920 |
|
NR, not reported. |
|||||
details, such as number, gender, strain of cat, or methodology, were provided in any of these studies.
3.1.6. Dogs
Meek and Eyster (1920) exposed eight mongrel dogs (gender not specified) to phosgene at 80–100 ppm for 30 min. All eight died within 24 h postexposure. Pulmonary edema with some evidence of cardiac effects was observed at necropsy.
Additional dog lethality information is presented in Table 1–10.
3.1.7. Goats
Karel and Weston (1947) reported a 10-min LC50 of 250 ppm for a group of 30 female and 1 male goat, and Underhill (1920) reported a 15-min LC50 of 180 ppm for a group of 61 goats. All goats died when exposed to phosgene at 8,750 ppm for 1 min (Tobias 1945), 500 ppm for 3 min, or 110 ppm for 30 min (Winternitz et al. 1920). No further experimental details were available.
3.1.8. Sheep
A 10-min LC50 value of 333 ppm was determined from exposing groups of two Dorset crossbred wethers to phosgene at concentrations of 135, 240, 427, or 758 ppm for 10 min, followed by a 24-h observation period (Keeler et al. 1990b). Animals that died displayed a mixture of stringy mucous and frothy white material from the proximal trachea to the smaller bronchioles that
TABLE 1–8 Acute Lethality of Phosgene in Guinea Pigs
|
Exposure Time (min) |
Concentration (ppm) |
End Point |
Reference |
|
1 |
672 |
LC50 |
Chasis, 1944 |
|
15 |
32 |
LC50 |
Underhill, 1920 |
|
30 |
18 |
LC50 |
Chasis, 1944 |
|
30 |
141 |
LC50 |
Moor and Gates, 1946 |
|
9 |
85 |
LC99 |
Coman et al., 1947 |
|
3 |
220 |
LC100 |
Winternitz et al., 1920 |
|
30 |
20 |
LC100 |
Winternitz et al., 1920 |
|
20 |
77 |
LC100 |
Ong, 1972 |
filled the alveoli, perivascular spaces, and interlobular septa. Sheep that survived the 24-h postexposure period were noted to have airways with scant amounts of mucus and frothy white material and mild to moderate alveolar edema.
3.1.9. Nonhuman Primates
Chasis (1944) reported a 1-min LC50 of 240 ppm for a group of monkeys. The strain, gender, and number of animals were not reported. A 1-min LC50 of 500 ppm was reported for 19 male and 18 female Rhesus monkeys (Weston and Karel 1947). Moor and Gates (1946) found that all monkeys died when exposed to phosgene at a concentration of 1,087 ppm for 1 min. No other experimental details were available for either study.
3.2. Nonlethal Toxicity
3.2.1. Mice
Hatch et al. (1986) exposed Swiss albino mice (eight per group) to phosgene at 0, 0.1, 0.2, 0.5, or 1 ppm for 4 h in an 11.3 ft3 Rochester-type
TABLE 1–9 Acute Lethality of Phosgene in Rabbits
|
Exposure Time (min) |
Concentration (ppm) |
End Point |
Reference |
|
30 |
17 |
LC40 |
Frosolono 1977 |
|
1 |
3,200 |
LC50 |
Moor and Gates 1946 |
|
15 |
187 |
LC50 |
Underhill 1920 |
|
20 |
110 |
LC50 |
Cameron and Courtice 1946 |
|
20 |
20 |
LC50 |
Laquer and Magnus 1921 |
|
30 |
100–135 |
LC70 |
Halpern et al. 1950 |
|
30 |
93 |
LC75 |
Frosolono 1976 |
|
30 |
82 |
LC90 |
Shils 1943 |
|
35 |
151 |
LC99 |
Coman et al. 1947 |
|
15 |
220 |
LC100 |
Winternitz et al. 1920 |
|
30 |
110 |
LC100 |
Winternitz et al. 1920 |
chamber. The phosgene was mixed with filtered room air before the metering orifice of the chamber and introduced into the chamber airstream. The chamber airstream had a flow rate of 1 chamber volume per minute, and the chamber air was sampled every 10 min. Phosgene concentrations were first determined by gas chromatography, and an infrared analyzer was used for a second check. Actual chamber concentrations were within 2% to 6% of target concentrations. Eighteen to 20 h postexposure, the lungs were lavaged and analyzed for bronchiolar aveolar lavage fluid protein (LFP), an indicator of pulmonary edema. The LFP findings were 292±18, 302±21, 941±105, 1,302 ±149, or 2,168±167 (units not provided) for 0, 0.1, 0.2, 0.5, or 1 ppm, respectively. The lowest exposure concentration producing a statistically significantly (p<0.05) altered protein concentration was 0.2 ppm.
In another study, Illing et al. (1988) exposed groups of 37–39 female CD-1 mice to phosgene at 0.1 to 0.5 ppm for 4 h. Animals were exposed in stainless steel exposure chambers. Phosgene concentrations were monitored primarily by gas chromatography and double checked by infrared analysis. There was a significant (p<0.05) increase in pentobarbital-induced sleeping time in mice exposed at 0.15 to 0.5 ppm compared with air controls. No significant differences were observed in cytochrome P450 levels, body weights, or liver weights between phosgene-exposed mice and air controls.
TABLE 1–10 Acute Lethality of Phosgene in Dogs
|
Strain |
Number/ Gender |
Exposure Time (min) |
Concentration (ppm) |
End Point |
Reference |
|
NR |
12/NR |
10 |
110 |
LC25 |
Cameron and Courtice 1946 |
|
NR |
NR |
1 |
2,100 |
LC50 |
Chasis 1944 |
|
NR |
NR |
10 |
45 |
LC50 |
Kimmerle and Diller 1977 |
|
NR |
24/NR |
15 |
60–70 |
LC50 |
Underhill 1920 |
|
NR |
NR |
20 |
502 |
LC50 |
Chasis 1944 |
|
NR |
6/NR |
30 |
100–175 |
LC50 |
Patt et al. 1946 |
|
NR |
NR |
30 |
78 |
LC55 |
Postel and Swift 1945 |
|
Mongrel |
18/NR |
3 |
745–880 |
LC70 |
Coman et al. 1947 |
|
NR |
94/NR |
20 |
135 |
LC70 |
Freeman et al. 1945 |
|
NR |
42/NR |
30 |
98 |
LC70 |
Postel and Swift 1945 |
|
Mongrel |
15/M,F |
30 |
124 |
LC90 |
Schultz 1945 |
|
Mongrel |
32/NR |
10 |
39–103 |
LC75 |
Box and Collumbine 1947 |
|
Mongrel |
NR |
3 |
734 |
LC99 |
Coman et al. 1947 |
|
Mongrel |
NR |
30 |
90 |
LC99 |
Coman et al. 1947 |
|
NR, not reported. |
|||||
Box and Cullumbine (1947b) exposed a group of 37 mice to phosgene at 144 ppm for an unspecified period of time. Subgroups of four or five of these
mice were sacrificed on postexposure days 1, 2, 3, 4, 5, 7, 10, or 14. A histopathological examination was performed on the lungs of these animals. Varying degrees of edema, patches of leucocytic infiltration, and bronchial and bronchiolitic epithelia being lifted by edema were observed on postexposure day 1. More severe edema and collapsed lungs with leukocytic infiltration were observed on postexposure days 2–5. The lungs of the remaining mice were essentially normal by postexposure days 10 through 14.
3.2.2. Rats
A total of 118 male Wistar rats were exposed to phosgene at 0.5 to 4.0 ppm for 5 min to 8 h (Rinehart 1962; Rinehart and Hatch 1964). The exposures were varied to give CT products between 12 and 360 ppm·min and were carried out in 1,700-L wooden exposure chambers operating at a constant ventilation rate of 1,000 L/min. The chamber surfaces were lacquered, and thus, potential loss of phosgene by reaction with the wooden surface was minimized. This system provided for air “turnover” every 2 min and a 99% equilibrium time of 8 min. Air samples were taken frequently during exposures, and adjustments were made when necessary to maintain constant phosgene concentrations. In addition to the lethality data presented in Section 3.1.2, several conclusions were also drawn concerning nonlethal end points. First, the CT product appears to be a valid way to express pulmonary irritation due to phosgene exposure. This is based on the finding of equal degrees of respiratory response, as measured by reduction in pulmonary gas exchange capacity, from exposures to various combinations of C and T that yield the same CT product. Second, there is no decrease in pulmonary performance from exposures less than CT=30 ppm·min, but above this level, gas exchange capacity decreases directly with a log increase in CT. Finally, for low level exposures (below CT=100), the major site of action is the respiratory bronchioles, although above this level, the alveoli are involved.
In a later publication of the above experiment (Gross et al. 1965), pulmonary pathology from the phosgene-exposed rats was described. Exposure to phosgene at high concentrations (2 ppm for 90 min) produced chemical pneumonia, and exposure at lower concentrations produced “chronic pneumonitis.” The degree of pneumonitis produced by phosgene was rated as slight, moderate, or severe. Slight pneumonitis was defined as mural thickening of respiratory bronchioles with involvement of adjacent alveoli. Moderate pneumonitis was defined as alveolar involvement in a peribronchiolar zone that extends no
more than one-third of the distance to the next bronchiole. Severe pneumonitis was defined as mural thickening of the respiratory bronchioles accompanied by obliteration of adjoining alveoli and air spaces containing desquamated alveolar cells. The lowest 1-h phosgene concentration producing moderate pneumonitis was 0.8 ppm. The same effect occurred with 1-h exposures to phosgene at 0.9, 2.5, or 3 ppm. This occurred in two of three rats, the third rat displaying slight pneumonitis.
Hatch et al. (1986) exposed Sprague-Dawley rats (eight per group) to phosgene at 0, 0.1, 0.2, 0.5, or 1 ppm for 4 h. The exposure system and parameters were similar to those described in Section 3.2.1 (Hatch et al. 1986). Actual chamber concentrations were within 2% to 6% of target concentrations. Eighteen to 20 h postexposure, the lungs were lavaged and analyzed for bronchiolar aveolar lavage fluid protein (LFP). The LFP findings were 340 ±38, 258±18, 506±54, 1,642±116, or 2,471±125 (units not provided) for 0, 0.1, 0.2, 0.5, or 1 ppm, respectively. The lowest exposure concentration producing a significantly (p<0.05) increased protein concentration was 0.2 ppm.
Male Sprague-Dawley rats were exposed to phosgene at 0, 0.125, 0.25, 0.5, or 1 ppm for 4 h and sacrificed on day 0, 1, 2, or 3 postexposure (Currie et al. 1987a). Animals were exposed in 0.32 m3 Rochester-type inhalation chambers. The air flow was 0.32 m3/min, and the gas mixture passed downward through stainless steel wire cages holding the animals and was exhausted at the bottom through a water scrubber. Temperature in the chambers was 23.0±3.4°C, and humidity was 60±10% during exposures. The chambers were monitored by both infrared analysis and gas chromatography continuously during exposure, and even phosgene distribution was assured by sampling from various areas of the exposure chamber. Measured phosgene concentrations were within 2% to 6% of target concentrations. Exposure-related changes were observed in body weights, wet lung weights, LFP concentrations, and total cell count and differential cell count in lavage fluid. Body weights were decreased immediately after exposure through day 2 for the 0.5-and 1-ppm exposure groups. Wet lung weights were increased in the 0.5 ppm exposure group on postexposure days 1, 2, and 3 and in the 1-ppm exposure group immediately after exposure and on postexposure days 1, 2, and 3. The relative wet-lung-to-body-weight ratios were increased immediately after exposure and on days 1, 2, and 3 in the 0.5- and 1-ppm exposure groups. LFP concentrations were increased in the 0.25-ppm exposure group on day 1 and in the 0.5- and 1-ppm exposure groups immediately after exposure and on days 1, 2, and 3. Total cell counts in lavage fluid were elevated in the 1-ppm exposure group on days 2 and 3. Percentage of polymorphonuclear leukocytes
were increased in the 0.25-ppm exposure group on days 1 and 2 and in the 0.5-and 1-ppm exposure groups on days 1, 2, and 3. The LFP and cellular parameters had their peak effect on day 1 postexposure and had begun a return to control values by day 3 postexposure, suggesting that the pulmonary damage was reversible or rapidly repairable.
In another study, Currie et al. (1987b) exposed male Sprague-Dawley rats to phosgene at 0, 0.05, 0.125, 0.25, 0.5, or 1 ppm for 4 h. The exposure system and parameters were similar to those described in Currie et al. (1987a). Animals were sacrificed immediately after exposure and at days 1, 2, and 3 postexposure. The ATP concentrations in lungs were significantly (p<0.05) decreased immediately after exposure at all exposure concentrations. The 1-ppm exposure group also had decreased ATP concentrations on day 1 and increased ATP concentrations on days 2 and 3. All other exposure groups had ATP concentrations similar to control values on days 1, 2, and 3. LFP concentrations were only measured immediately after exposure and were linearly increased at 0.25, 0.5, and 1 ppm.
Frosolono and Pawlowski (1977) exposed anesthetized male CFE Carworth rats to phosgene at 0, 100, 200, or 430 ppm for approximately 10 min. Animals were exposed in a 364-L glass and stainless-steel chamber, and phosgene concentrations were estimated with a phosgene detector tube. The authors stated that “the analytical method for the determination of phosgene concentrations is not highly precise…and concentrations given should be considered putative or nominal.” Groups of six to eight rats per concentration were sacrificed 0, 30, 60, or 90 min after exposure. No percentage change of lung water (used as a measure of pulmonary edema) was observed in rats exposed to phosgene at 100 or 200 ppm and sacrificed 0 and 30 min after exposure. However, 60 min postexposure, the 100-ppm exposure group had a 6.7% increase in lung water and the 200-ppm exposure group had an 8.7% increase in lung water. Cytochrome C oxidase activities (specific and total) from the lungs were decreased, ranging from 30.1% to 79.8% after rats were exposed at 100 ppm and sacrificed 0, 30, and 60 min after exposure. After exposure at 200 ppm, relative serum lactic dehydrogenase (LDH) activities were increased from 1- to 3-fold over the postexposure time of 0 to 90 min. An exposure at 430 ppm resulted in decreased lung LDH activities (specific and total) ranging from 3.3% to 70.8% in the organelle fractions (homogenate and soluble). The authors suggested that the decreased LDH activities were indicative of cellular damage resulting in increased serum LDH activities.
In a different report, Pawlowski and Frosolono (1977) describe pulmonary ultrastructural alterations in male CFE Carworth rats exposed to phosgene in
a manner essentially identical to that described in Frosolono and Pawlowski (1977). Immediately after exposure, animals exposed to phosgene at 100 ppm exhibited vesiculation of ciliated and Clara cell cytoplasm in the bronchiolar epithelium, and interstitial edema was observed 30 min postexposure. In animals exposed to phosgene at 200 ppm, interstitial edema was observed immediately after exposure, and focal Type I cell discontinuities and interstitial and intracellular edema were present 30 min postexposure. At 48 min postexposure to phosgene at 200 ppm, interstitial cellular edema with general septal thickening and involvement of Type II cells was observed. At 430 ppm, cytoplasmic sequestration figures were observed in Type II cells 60 min postexposure.
In another study, lavage protein concentrations and histopathological assessments of the lungs were determined in male Wistar rats (10–15 per group) exposed to phosgene at 0, 0.1, 0.15, 1, 2.5, or 5 ppm for 10, 20, 50, 60, 250, 330, or 500 min (Diller et al. 1985). The exposure concentrations and durations were paired to provide different exposure scenarios corresponding to ≤50 ppm·min. A special 7-L plexiglass chamber was constructed to achieve reliable exposures at low concentrations of phosgene. Air exchange was 8-fold per minute, and consistency of phosgene concentration throughout the chamber was confirmed by a movable suction probe. Long-term mean phosgene concentrations were measured by collection of the gas samples in a glass bubbler and subsequent titration. Consistency throughout exposure was determined by a galvanometric analyzer, and concentration was checked using detector paper. Animals were sacrificed either 24 or 48 h after the phosgene exposure. A phosgene concentration at 50 ppm·min (5 ppm for 10 min) was required to produce alveolar edema. A concentration of 50 ppm·min was also required to produce an increase in LFP, and widening of pulmonary interstices was observed at 25 ppm·min. No phosgene threshold was observed for the latter two parameters down to 0.1 ppm, indicating that the CT=k relationship is valid for these two parameters. However, under the conditions of this study, Haber’s rule appears to be valid for pulmonary edema only down to 5 ppm.
Franch and Hatch (1986) exposed male Sprague-Dawley rats to phosgene at either 1 ppm for 4 h with the sacrifices occurring immediately after exposure and on recovery period days 1, 2, 7, or 14 or 1 ppm for 7 h with the sacrifices occurring hourly during the exposure period. The exposure system and parameters were similar to those described in Section 3.2.1 (Hatch et al. 1986). Actual chamber concentrations were 0.98±0.03 ppm for the 4-h exposure regimen and 1.03±0.02 ppm for the 7-h exposure regimen. Body weights of rats exposed at 1 ppm for 4 h were significantly (p≤0.01) decreased to 13% below controls on day 1 postexposure. Body weights in-
creased toward control values, achieving 3% below controls on day 14. Food consumption was also decreased compared with controls but was less than 10% below normal by day 4. Lung weights of exposed animals were 60% greater than controls immediately after exposure and remained elevated through day 7 postexposure. Lung nonprotein sulfhydryl (NPSH) content was similar in control and exposed rats on day 1, had increased to 80% above controls on day 2, and decreased back to control levels by day 7. Glucose-6-phosphate dehydrogenase (G6PD) activity was increased 40% in exposed animals compared with controls from days 1 through 14. Sequential examination every hour during a 7-h exposure revealed lung weights increasing 4 h into the exposure, reaching a maximum elevation of 60% above controls at 6 h. Both the NPSH levels and G6PD activities were decreased in this exposure regimen.
Frosolono and Currie (1985) investigated the effect of phosgene on the pulmonary surfactant-system (PSS) in groups of six to 14 rats exposed to phosgene at 1 ppm for 4 h. The exposure system and parameters were similar to those described in Section 3.2.1 (Hatch et al. 1986). The actual chamber concentration was 1.0±0.06 ppm. Animals were sacrificed immediately after exposure, or on postexposure days 1, 2, or 3. Pulmonary edema was present immediately after exposure and persisted through day 3. Phosphatidylinositol levels were significantly (p<0.05) decreased compared with controls immediately after exposure only. Phosphatidylserine and phosphatidylethanolamine levels were significantly increased compared with controls on days 1, 2, and 3 postexposure. Phosphatidylcholine levels were increased at all time points compared with controls.
Jaskot et al. (1989) exposed groups of male Sprague-Dawley rats (16 per group) to phosgene at 0 or 0.5 ppm for 4 h. The exposure system and parameters were similar to those described in Section 3.2.1 (Hatch et al. 1986). The actual chamber concentration was 0.54±0.05 ppm. The rats were sacrificed immediately or 24 h postexposure. Phosgene-exposed rats showed no changes in angiotensin-converting enzyme (ACE) activity in lavage fluid or serum compared with controls. Whole-lung ACE activity was significantly increased immediately after exposure and 24 h postexposure, with increases of 55% and 44% above controls, respectively. Phosgene-exposed rats also had increased ACE activity in lavage cell pellets, with increases of 50% and 54% above controls at 0 and 24 h, respectively.
Assessment of pulmonary immunocompetence was determined by exposing male Fischer 344 rats to phosgene at 0.1, 0.5, or 1 ppm for 4 h and measuring pulmonary natural killer cell activity on day 1, 2, 4, or 7 postexposure (Burleson and Keyes 1989). The animals were exposed in a Rochester cham-
her; temperature and humidity were maintained at 23.3±1.7°C and 60±10%, respectively. The chamber atmosphere was continuously monitored during exposures by both gas chromatography and infrared analysis. The actual chamber concentrations were 0.97, 0.49, and 0.10 ppm for the 1-, 0.5-, or 0.1-ppm groups, respectively. The pulmonary natural killer activities in the rats exposed at 1 ppm were significantly (p<0.05) decreased on days 1, 2, and 4. A decrease (p<0.05) in natural killer cell activity was also observed in the 0.5-ppm group. No effect was noted in the 0.1-ppm group.
Another immunological assessment, pulmonary cytotoxic T-lymphocyte (CTL) activity, was examined in male Fischer 344 rats exposed to phosgene at 0 or 1 ppm for 4 h. Animals were exposed in a Rochester exposure chamber, and the exposure atmosphere was monitored by gas chromatography and infrared analysis. The actual exposure concentration was 1.0±0.04 ppm. Twenty-four hours after phosgene exposure, subsets of both the control and phosgene-treated rats were infected with influenza virus. The remaining control and phosgene-exposed rats were sham infected with uninfected lung homogenate. Animals were sacrificed on day 2, 5, 7, 10, 15, or 20 postinfection (Ehrlich et al. 1989).
A significant suppression of CTL activity was noted in phosgene-exposed, influenza-infected rats compared with air-controls, air-infected, and phosgenetreated sham-infected animals. This effect was observed only on day 10 postinfection; however, this is a time when peak activity is normally detected in control rats. Body weights were significantly decreased (p<0.05) in phosgene-exposed, infected and uninfected rats on day 2 postinfection and in phosgene-exposed, infected animals at day 5. Lung weights were significantly increased (p<0.05) in phosgene-exposed, infected and uninfected rats on days 2 and 5 postinfection compared with air-infected and air-uninfected controls.
3.2.3. Guinea Pigs
Cameron et al. (1942) exposed ten guinea pigs to phosgene at an average concentration of 0.86 ppm for 5 h. All survived an apparent 24-h postexposure period.
Hatch et al. (1986) exposed Hartley guinea pigs (eight per group) to phosgene at 0, 0.1, 0.2, 0.5, or 1 ppm for 4 h. The exposure system and parameters are similar to those described in Section 3.2.1 (Hatch et al. 1986). Actual chamber concentrations were within 2% to 6% of target concentrations. Eighteen to 20 h postexposure, the lungs were lavaged and analyzed for bronchi-
olar alveolar lavage fluid protein (LFP). The LFP findings were 305±19, 228 ±47, 407±75, 524±47, or 1,212±149 (units not provided) for 0, 0.1, 0.2, 0.5, or 1 ppm, respectively. The lowest exposure concentration producing a significantly (p<0.05) altered protein concentration was 0.5 ppm.
In another study, Hartley guinea pigs (five per group) were exposed to phosgene at 0, 0.25, or 0.5 ppm for 4 h (Slade et al. 1989). The exposure chamber and atmosphere generation and measurement systems were similar to those used by Hatch et al. (1986). The LFP concentrations were measured 16 to 18 h after exposure. These investigators found that the LFP concentrations were elevated by 90% in animals exposed to phosgene at 0.25 ppm and 250% in animals exposed at 0.5 ppm, when compared with controls.
3.2.4. Hamsters
Hatch et al. (1986) exposed Syrian Golden hamsters (eight per group) to phosgene at 0, 0.1, 0.2, 0.5, or 1 ppm for 4 h. The exposure system and parameters were similar to those described in Section 3.2.1 (Hatch et al. 1986). Actual chamber concentrations were within 2% to 6% of target concentrations. Eighteen to 20 h postexposure, the lungs were lavaged and analyzed for bronchiolar alveolar lavage fluid protein (LFP). The LFP findings were 319 ±6, 347±14, 520±63, 1,289±92, or 3,035±111 (units not provided) for 0, 0.1, 0.2, 0.5, or 1 ppm, respectively. The lowest exposure concentration producing a significantly (p<0.05) altered protein concentration was 0.2 ppm.
3.2.5. Rabbits
Cameron et al. (1942) exposed ten rabbits to phosgene at an average concentration of 0.86 ppm for 5 h. All survived an apparent 24-h postexposure period.
Hatch et al. (1986) exposed New Zealand white rabbits (eight per group) to phosgene at 0, 0.1, 0.2, 0.5, or 1 ppm for 4 h. The exposure system and parameters were similar to those described in Section 3.2.1 (Hatch et al. 1986). Actual chamber concentrations were within 2% to 6% of target concentrations. Eighteen to 20 h postexposure, the lungs were lavaged and analyzed for bronchiolar alveolar lavage fluid protein (LFP). The LFP findings were 292±11, 309±20, 346±26, 517±68, and 855±71 (units not pro-
vided) for 0, 0.1, 0.2, 0.5, or 1 ppm, respectively. The lowest exposure concentration producing a significantly (p<0.05) altered protein concentration was 0.5 ppm.
3.2.6. Dogs
Coman et al. (1947) exposed adult mongrel dogs to phosgene at 108–197 ppm for 30 min and sacrificed the animals 0.63 to 8.58 h after exposure or exposed the dogs to phosgene at 71–80 ppm for 3 min and sacrificed the animals 0.13 to 5.32 h after exposure. Mild to severe emphysema was observed in all dogs, severity generally correlating to exposure concentration and time. Swelling and sloughing of the bronchiolar mucosa were then observed and were usually confined to bronchioles proximal to the respiratory bronchioles. Transient bronchiolar constriction, followed by dilatation of the bronchioles, was also observed. Congestion of the lung and alveolar edema usually followed in the lower exposure concentrations at the 30-min exposure time with the lung congestion preceding alveolar edema in the higher exposure concentrations at the 3-min exposure time. Lung-to-body weight ratios increased as both exposure concentration and/or sacrifice time after exposure increased.
In another study, adult mongrel dogs were exposed to phosgene for 30 min at concentrations that fluctuated between 24 and 40 ppm (Clay and Rossing 1964). The experimental design was as follows: (1) two dogs served as controls, (2) seven dogs were exposed one or two times and sacrificed 1 or 2 d after the last exposure, (3) seven dogs were exposed four to 10 times and sacrificed from 1 to 7 d after the last exposure, (4) five dogs were exposed 15 to 25 times and sacrificed immediately or from 1 to 14 d after the last exposure, and (5) four dogs were exposed 30 to 40 times and sacrificed immediately or from 1 to 12 wk after the last exposure. For all animals exposed one or two times, acute bronchiolitis or peribronchiolitis developed. Pulmonary emphysema was produced in dogs receiving more than two exposures.
3.2.7. Sheep
Groups often unanesthetized, adult sheep were exposed to phosgene at 0 or 767 ppm for 10 min (Assaad et al. 1990). Blood samples were collected
immediately before the exposure and 15, 30, 60, 120, 180, or 240 min after the exposure; also, plasma prostacyclin and thromboxane metabolites (6-keto-PGFla and TXB2) concentrations were measured. Levels of both metabolites were significantly (p<0.05) increased in the exposed animals compared with pre-exposure baseline values and air-control values. The 6-keto-PGFla returned to control values by 180 min, whereas the TXB2 did not. The authors concluded that (1) acute lung injury occurred immediately following exposure even though pulmonary edema and symptoms of pulmonary toxicity developed 4 to 8 h after exposure, (2) phosgene may induce pulmonary edema by injuring cell membranes, and (3) arachidonic acid metabolites may be useful as early, nonspecific markers for phosgene-induced lung injury.
In a subsequent study (Assaad et al. 1991), ten unanesthetized, adult sheep were exposed to phosgene at 0 or 767 ppm for 10 min and were sacrificed 4 h after exposure. Gross examination of the lungs revealed congestion and edema, and the light microscopic evaluation demonstrated alveolar and interstitial edema, fibrin and neutrophil exudation in the air spaces, and increased alveolar macrophages. The electron microscopic examination revealed that Type I pneumocytes had intracellular swelling, necrosis, and denuding of basement membrane with the preservation of the tight junctions. Type II pneumocytes showed loss of lamellar bodies, cytoplasmic swelling, and damage to the endoplasmic reticulum. Endothelial cells showed increased density and vesicular activity, cytoplasmic swelling, and displacement of the basement membrane.
Five Dorset-crossbred wether sheep underwent surgery in order to provide simultaneous information concerning phosgene exposure and pulmonary vascular and interstitial fluid dynamics (Keeler et al. 1990a). The efferent duct of the caudal mediastinal lymph node was cannulated to monitor pulmonary lymph flow. Additionally, a carotid atrial catheter, a pulmonary artery catheter, and a left atrial catheter were implanted to monitor systemic and pulmonary hemodynamics. After a 5- to 7-d recovery period, the sheep were exposed to phosgene at 480–600 ppm for 10 min. The control pulmonary lymph flow rate was 10.3±2.2 g/h, and the exposed sheep values were 19.5 ±6.0, 21.5±6.0, 22.5±6.0, 24.0±5.9, 26.5±5.3, 26.9±6.0, or 27.3±5.8 g/h for 1, 1.5, 2, 2.5, 3, 3.5, or 4 h postexposure, respectively. There was a small increase in mean pulmonary micro vascular pressure but no change in the ratio of lymph-to-plasma protein concentration. Sheep were sacrificed 4 h after exposure. Histopathological evaluations of the lungs revealed diffuse, moderate alveolar and interlobular edema.
3.2.8. Goats
Cameron et al. (1942) exposed two goats to an average phosgene concentration of 0.86 ppm for 5 h. Both survived an apparent 24-h postexposure period.
3.2.9. Cats
Cameron et al. (1942) exposed two cats to an average phosgene concentration of 0.86 ppm for 5 h. One cat became “very ill” with considerable labored breathing, but both survived an apparent 24-h postexposure period.
3.2.10. Monkeys
Cameron et al. (1942) exposed two monkeys to an average phosgene concentration of 0.86 ppm for 5 h. One of the monkeys became “very ill” with considerable labored breathing, but both survived an apparent 24-h postexposure period.
3.3. Developmental and Reproductive Toxicity
Developmental and reproductive toxicity studies regarding animal exposure to phosgene were not available.
3.4. Genotoxicity
Genotoxic studies regarding animal exposure to phosgene were not available. However, the two highly reactive chlorines of phosgene suggest that it could act on DNA in a similar manner to that of bifunctional alkylating agents (Shah et al. 1979).
3.5. Carcinogenicity
A study by Selgrade et al. (1989) showed that exposure to phosgene at very low levels enhances the susceptibility of mice to lung tumor formation.
Female C57BL/6 mice were exposed for 4 h to phosgene at 0.01 (N=13), 0.025 (N=28), or 0.05 ppm (N=35) and injected intravenously with syngeneic B16 melanoma cells on the following day. Controls were injected with tumor cells and exposed to air. The lungs were removed 2–3 wk after tumor cell injection and the tumors were counted. Compared with controls, there was a statistically significant (p<0.05) increase in the number of B16 melanoma tumors in the lungs of mice treated with phosgene at 0.025 or 0.05 ppm. The number of tumors per lung were 105, 110, or 185 in mice treated with 0.01, 0.025, or 0.05 ppm, respectively, compared with 90, 75, or 100 in the respective control groups. Exposure to 0.025 ppm was considered the lowest-observed-effect level. Extending the exposure time from 4 to 8 h did not alter the susceptibility to B16 tumors at 0.01 ppm.
In other experiments using a higher concentration, mice were exposed by inhalation to phosgene at 0.5 ppm for 4 h and injected intravenously with melanoma tumor cells on the following day or injected with tumors and then exposed at 0.5 ppm for 4 h/d for 4 consecutive days. There was a significant increase (p<0.05) in the number of lung tumors in the group exposed to phosgene prior to inoculation (96 tumors per lung compared with 38 tumors per lung for controls). Although the number of tumors in mice exposed to phosgene on 4 consecutive days beginning immediately after tumor injection was higher than in controls (65 tumors per lung compared with 48 tumors per lung for controls), the difference was not statistically significant. These experiments showed that exposure following tumor injection had little effect on tumor susceptibility compared with phosgene exposure prior to tumor injection.
3.6. Summary
Animal lethality studies are abundant; however, the studies are of varying quality and many are incompletely reported. Thus, the utility of the lethality studies must be considered on a case by case basis. Even though there are limitations concerning these studies, there appears to be little species variability between rats, mice, and guinea pigs, and the CT=k relationship appears to be generally valid. (Although at very high or very low concentrations or at exposure times so short that the animal can hold its breath, the CT=k relationship may not hold.)
Many nonlethal acute inhalation studies exist and are of generally good quality. These studies also suggest that there are few differences between species after acute exposure to phosgene and that the type and sequence of
effects are similar in humans and experimental animals. Many of the nonlethal toxicity studies describe biochemical changes in lung fluid, whose pathogenesis is likely due to acylation (see Section 4.2). Selected biochemical and other nonlethal effects are summarized in Table 1–11.
4. SPECIAL CONSIDERATIONS
4.1. Metabolism and Disposition
Following inhalation exposure, a small portion of phosgene hydrolyzes to hydrochloric acid (HCl) and carbon dioxide (CO2) in the mucous coating of the upper respiratory tract (Diller 1985), but in the moist atmosphere of the terminal spaces of the lungs, more extensive hydrolysis is thought to occur (Beard 1982). Although phosgene is only slightly soluble in water, once in solution it rapidly hydrolyzes to HCl and CO2. However, phosgene reacts even faster with other functional groups, such as amino, hydrazino, and sulfhydryl groups (Jaskot et al. 1991; Diller 1985).
4.2. Mechanism of Toxicity
The toxicity of phosgene is due to both acylation and hydrolysis. The acylation is most important and results from the reaction of phosgene with nucleophiles such as amino, hydroxyl, and sulfhydryl groups of macromolecules. The acylation causes lipid and protein denaturation, irreversible membrane changes, and disruption of enzymatic function. Phosgene depletes lung glutathione, and glutathione reductase and superoxide dismutase increase as a result of the lung’s response to injury. Cellular glycolysis and oxygen uptake are decreased following exposure to phosgene, and there is a decrease of intracellular ATP and cyclic AMP associated with increased permeability of pulmonary vessels and pulmonary edema. Phosgene exposure also causes increased lipid peroxidation and increased leukotriene synthesis but no change in cyclooxygenase metabolism (TEMIS 1997).
The hydrogen chloride formed by the hydrolysis of phosgene causes initial irritation to the eyes, nasopharynx, and respiratory tract. However, because of phosgene’s poor water solubility, a minimal amount of hydrogen chloride is formed (TEMIS 1997).
TABLE 1–11 Summary of Selected Nonlethal Effects of Phosgene
|
Phosgene Concentration (ppm) |
Exposure Time (h) |
Species |
Effect |
Reference |
|
0.05 |
4 |
Rat |
Decreased ATP |
Currie et al. 1987b |
|
0.1 |
1 |
Rat |
No edema; no histopathology |
Diller et al. 1985 |
|
0.1 |
4 |
Rat |
Lung histopathology |
Diller et al. 1985 |
|
0.1 |
4 |
Rat |
No decrease in PNKC activity |
Burleson and Keyes 1989 |
|
0.1 |
4 |
Rat |
No increase in LFP levels |
Hatch et al. 1986 |
|
0.1 |
4 |
Rat |
Decreased ATP; no changes in LFP level |
Currie et al. 1987b |
|
0.1 |
4 |
Mouse |
No changes in LFP levels |
Hatch et al. 1986 |
|
0.1 |
4 |
Hamster |
No changes in LFP levels |
Hatch et al. 1986 |
|
0.125 |
4 |
Rat |
No changes in LFP levels; no lung weight change |
Currie et al. 1987a |
|
0.2 |
4 |
Rat |
Increased levels of LFP |
Currie et al. 1987b |
|
0.2 |
4 |
Mouse |
Increased levels of LFP |
Hatch et al. 1986 |
|
0.2 |
4 |
Guinea Pig |
No changes in LFP levels |
Hatch et al. 1986 |
|
0.2 |
4 |
Rabbit |
No changes in LFP levels |
Hatch et al. 1986 |
|
0.2 |
4 |
Hamster |
Increased levels of LFP |
Hatch et al. 1986 |
|
0.25 |
4 |
Rat |
Increased levels of LFP and PMN; no lung weight change |
Currie et al. 1987a |
|
Phosgene Concentration (ppm) |
Exposure Time (h) |
Species |
Effect |
Reference |
|
0.25 |
4 |
Guinea Pig |
Increased levels of LFP |
Slade et al., 1989 |
|
0.5 |
4 |
Rat |
Decreased body weight; increased lung weight; increased levels of LFP |
Currie et al. 1987a |
|
0.5 |
4 |
Guinea Pig |
Increased levels of LFP |
Hatch et al. 1986 |
|
0.5 |
4 |
Rabbit |
Increased levels of LFP |
Hatch et al. 1986 |
|
0.5 |
4 |
Rat |
Decreased PNKC activity |
Burleson and Keyes 1989 |
|
0.5 |
4 |
Rat |
Increased ACE activity |
Jaskot et al., 1989 |
|
1 |
4 |
Rat |
Increased lung weight (14 d); decreased body weight; increased G6PD activity and NPSH content |
Franch and Hatch 1986 |
|
1 |
4 |
Rat |
Decreased PNKC activity |
Burleson and Keyes 1989 |
|
1 |
4 |
Rat |
Increased lung weight; decreased body weight; suppressed cytotoxic T-lymphocytes |
Ehrlich et al. 1989 |
4.3. Structure-Activity Relationships
Phosgene is a reactive intermediate of both chloroform and carbon tetrachloride metabolism. Chloroform is metabolized by oxidative dehydrochlorination of its carbon-hydrogen bond to form the highly unstable trichloro-methanol (Cl3COH), which is then spontaneously converted to phosgene. This reaction is catalyzed by cytochrome P-450 and occurs in both the liver and kidneys. The evidence for phosgene formation from chloroform was the isolation of 2-oxothioazolidine-4-carboxylic acid from the microsomal incubation of chloroform in the presence of cysteine. This compound is the expected product of the reaction of phosgene with cysteine (Pohl et al. 1977; Mansuy et al. 1977). The electrophilic phosgene further reacts with water to yield CO2 and Cl− (major end products of chloroform metabolism), but significant amounts of phosgene bind covalently with proteins and lipids or conjugate with cysteine or glutathione (GSH) (EPA 1985).
Covalent binding of phosgene with cellular macromolecules has been proposed as a mechanism of chloroform-induced hepatic and renal toxicity (Pohl et al. 1980a,b), and it is generally accepted that the carcinogenic activity of chloroform resides in its highly reactive intermediate metabolites such as phosgene. Irreversible binding of reactive chloroform metabolites to cellular macromolecules support several theoretical concepts as a mechanism for its carcinogenicity (EPA 1985).
Covalent macromolecular binding of phosgene may be prevented to some extent by endogenous GSH (Sipes et al. 1977). Phosgene reacts with two molecules of GSH to form diglutathionyl dithiocarbonate (GSCOSG), a compound identified as a metabolite of chloroform in rat liver microsomes and mouse kidney homogenates incubated with chloroform in the presence of GSH (Pohl et al. 1981; Branchflower et al. 1984). In mouse kidney homogenates, GSCOSG was shown to be further metabolized by kidney α-glutamyl transpeptidase to N-(2-oxothiazolidine-4-carbonyl)-glycine, which in turn is hydrolyzed, possibly in the presence of cysteinyl glycinase, to 2-oxothiazolidine-4-carboxylic acid (Branchflower et al. 1984).
The metabolism of carbon tetrachloride proceeds via cytochrome P-450-dependent dehalogenation (Sipes et al. 1977). The first step involves cleavage of one carbon-chlorine bond to yield Cl− and a trichloromethyl free radical, which is then oxidized to the unstable intermediate trichloromethanol, the precursor of phosgene. Hydrolytic dechlorination of phosgene yields CO2 and HCl (Shah et al. 1979). Although there are similarities in the metabolism of chloroform and carbon tetrachloride, metabolic activation of chloroform produces primarily phosgene, whereas the level of phosgene production from
carbon tetrachloride appears to be small. Pohl et al. (1981) compared the amount of phosgene (as diglutathionyl dithiocarbamate) produced by the aerobic metabolism of carbon tetrachloride and the amount produced from chloroform by liver microsomes from phenobarbital-treated rats. The results indicate that phosgene production from carbon tetrachloride is only 4% of that produced from chloroform. The reactive metabolites of both chloroform and carbon tetrachloride covalently bind to proteins and lipids but bind only minimally to DNA and nucleic acids. The failure of the reactive species (e.g. phosgene, trichloromethyl free radical, and other metabolites) to significantly bind to DNA has been ascribed to their short half-lives and to their lack of nuclear penetration (EPA 1985).
Given to intact rats, 14C-phosgene labeled liver proteins and to a smaller extent lipids (Reynolds 1967). The pattern of labeling was different from that of 14C-carbon tetrachloride and was similar to that of 14C-chloroform. It was also shown that 36Cl-carbon tetrachloride radioactivity was stably incorporated into liver lipid and protein, pointing to the trichloromethyl radical rather than phosgene as the reactive form of carbon tetrachloride. Cessi et al. (1966) reported that phosgene labeled the terminal amino groups of polypeptides in a manner similar to in vivo protein labeling produced by carbon tetrachloride. However, after inhalation, phosgene reacts completely with lavage fluid, lung tissue, and lung capillary blood so that it is unlikely that phosgene will reach tissue beyond the lung (Diller 1974).
4.4. Other Relevant Information
4.4.1. Haber’s Law and Time Scaling
The concept of a “death product” was introduced by Haber to explain the relationship between the extent of exposure to phosgene and death (Haber 1924). According to “Haber’s law,” the biological effect of phosgene is directly proportional to the exposure, expressed as the product of the atmospheric concentration (C) and the time of exposure (T), or CT=k, where k can be death, pulmonary edema, or other biological effects of phosgene exposure (EPA 1986). Haber’s law has subsequently been shown by other investigators to be valid for both nonlethal and lethal effects within certain limits.
For example, Rinehart (1962) and Rinehart and Hatch (1964) showed that the CT product appears to be a valid way to express pulmonary irritation due to phosgene exposure in rats. This is based on the finding of equal degrees of respiratory response, as measured by reduction in pulmonary gas exchange
capacity, from exposures to various combinations of C and T that yield the same CT product.
Rat and mouse lethality data from the well-conducted study of Zwart et al. (1990) also suggest that Haber’s law is valid for phosgene. The study by ten Berge et al. (1986) has shown that the concentration-exposure-time relationship for many irritant and systemically acting vapors and gasses can be described by the relationship Cn×t=k. When the 10- to 60-min rat LC50 data are utilized in a linear regression analysis a value of the exponent, n, of 0.93 is obtained. The mouse 10- to 60-min lethality data yield a value of 1.3 for n.
Thus, the fact that these empirically derived values for the exponent n approximate 1 is further support that Haber’s law is valid for phosgene.
5. RATIONALE AND PROPOSED AEGL-1
5.1. Human Data Relevant to AEGL-1
No human data were relevant for establishing AEGL-1 values.
5.2. Animal Data Relevant to AEGL-1
No animal data were relevant for establishing AEGL-1 values.
5.3. Derivation of AEGL-1
Appropriate data were not available for derivation of AEGL-1 values for phosgene. Odor cannot be used as a warning for potential exposure. The odor threshold is reported to be between 0.5 and 1.5 ppm, a value above or approaching AEGL-2 and AEGL-3 values, and tolerance to the pleasant odor of phosgene occurs rapidly. Furthermore, following odor detection and minor irritation, serious effects may occur after a clinical latency period of ≤24 h.
6. RATIONALE AND PROPOSED AEGL-2
6.1. Human Data Relevant to AEGL-2
No human data were relevant to establishing the AEGL-2 values.
6.2. Animal Data Relevant to AEGL-2
Chemical pneumonia was observed in rats exposed to phosgene at 2.0 ppm for 90 min (Gross et al. 1965). Biochemical markers of phosgene exposure, such as increased LFP, were observed in mice, rats, guinea pigs, hamsters, and rabbits exposed at up to 1 ppm for 4 h (Hatch et al. 1986; Diller et al. 1985). Other effects defined by AEGL-2 included “very ill” monkeys with labored breathing (Cameron et al. 1942) and acute bronchiolitis or peribronchiolitis in dogs (Clay and Rossing 1964). However, a lack of experimental details in the monkey and dog studies renders them inappropriate for AEGL derivation.
6.3. Derivation of AEGL-2
The chemical pneumonia observed in rats exposed to phosgene at 2 ppm for 90 min (Gross et al. 1965) will be used as the basis for deriving AEGL-2 values. This end point was chosen because at a C × t product of 180 ppm·min, approximately 60% of rats exhibited chemical pneumonia. Whereas, at C × t products ≤180 ppm·min, only 15% of exposed rats showed pneumonia or chemical pneumonitis. An uncertainty factor (UF) of 3 will be applied for interspecies extrapolation because little species variability is observed in lethal and nonlethal end points after exposure to phosgene. A UF of 3 will also be applied to account for sensitive human subpopulations due to the steep concentration-response curve and because the mechanism of phosgene toxicity (binding to macromolecules and irritation) is not expected to vary greatly between individuals. Thus, the total UF is 10. The concentration-time relationship for many irritant and systemically acting vapors and gases may be described by Cn × t=k, where the n ranges from 0.8 to 3.5 (ten Berge et al. 1986). Haber’s law (C×t=k; n=1) has been shown to be valid for phosgene within certain limits and will be used for scaling of the AEGL values for phosgene for the 30-min and 1-, 4-, and 8-h time points. The 30-min value is also adopted as the 10-min value, because extrapolation would yield a 10-min AEGL-2 value close to concentrations producing alveolar edema in rats. The AEGL-2 values for phosgene are presented in Table 1–12, and the calculations for these AEGL-2 values are presented in Appendix A.
These AEGL-2 values are supported by the nonlethal toxicity studies of Franch and Hatch (1986) and Ehrlich et al. (1989). In both of these studies, rats exposed to phosgene at 1 ppm for 4 h developed severe pulmonary edema and body-weight loss. If this exposure regimen and a total UF of 10 are uti-
TABLE 1–12 Proposed AEGL-2 Values for Phosgene (ppm [mg/m3])
|
Classification |
10 min |
30 min |
1 h |
4 h |
8 h |
|
AEGL-2 |
0.60 |
0.60 |
0.30 |
0.08 |
0.04 |
|
|
(2.5) |
(2.5) |
(1.2) |
(0.33) |
(0.16) |
lized to calculate AEGL-2 values, similar supporting values of 0.8, 0.4, 0.1, and 0.05 ppm are obtained for the 30-min and 1-, 4-, and 8-h time points, respectively. The 10-min value is supported by the fact that Diller et al. (1985) observed alveolar pulmonary edema in rats exposed at 5 ppm for 10 min. Applying a total UF of 10 to this data point yields a supporting 10-min value of 0.5 ppm.
7. RATIONALE AND PROPOSED AEGL-3
7.1. Human Data Relevant to AEGL-3
No human data were relevant to establishing the AEGL-3 values.
7.2. Animal Data Relevant to AEGL-3
Many lethality data exist for a variety of species (mouse, rat, guinea pig, rabbit, cat, dog, goat, sheep, and monkeys). However, in most cases, experimental parameters are poorly described, and the quality of the data is questionable for AEGL derivation. The mouse and rat LC50 studies of Zwart et al. (1990) are the exception and are appropriate for AEGL-3 derivation.
7.3. Derivation of AEGL-3
The highest concentration causing no mortality in the rat after a 30-min exposure is 15 ppm (Zwart et al. 1990). This value will be used as the basis for deriving 30-min and 1-, 4-, and 8-h AEGL-3 values. The highest concentration causing no mortality in the rat and mouse after a 10-min exposure is 36 ppm (Zwart et al. 1990); this value will be used as the basis for the 10-min AEGL-3 value. A UF of 3 will be applied for interspecies extrapolation
TABLE 1–13 Proposed AEGL-3 Values for Phosgene (ppm [mg/m3])
|
Classification |
10 min |
30 min |
1 h |
4 h |
8 h |
|
AEGL-3 |
3.6 |
1.5 |
0.75 |
0.20 |
0.09 |
|
|
(15) |
(6.2) |
(3.1) |
(0.82) |
(0.34) |
because little species variability is observed both in lethal and nonlethal end points after exposure to phosgene. A UF of 3 will also be applied to account for sensitive human subpopulations due to the steep concentration-response curve and because the mechanism of phosgene toxicity (binding to macromolecules and irritation) is not expected to vary greatly between individuals. Thus, the total UF is 10. The concentration-time relationship for many irritant and systemically acting vapors and gases may be described by Cn×t=k, where n ranges from 0.8 to 3.5 (ten Berge et al. 1986). Haber’s law (C×t=k; n=1) has been shown to be valid for phosgene within certain limits and will be used for scaling of the AEGL values for phosgene across time for the 1-, 4-, and 8-h values. The AEGL-3 values for phosgene are presented in Table 1–13 (above), and the calculations for these AEGL-3 values are presented in Appendix A.
8. SUMMARY OF PROPOSED AEGLS
8.1. AEGL Values and Toxicity End Points
The derived AEGL values for various levels of effects and durations of exposure are summarized in Table 1–14. Data were insufficient for deriving
TABLE 1–14 Summary of Proposed AEGL Values for Phosgene (ppm [mg/m3])
|
Classification |
10 min |
30 min |
1 h |
4 h |
8 h |
|
AEGL-1 |
NA |
NA |
NA |
NA |
NA |
|
(Nondisabling) |
|
||||
|
AEGL-2 |
0.60 |
0.60 |
0.30 |
0.08 |
0.04 |
|
(Disabling) |
(2.5) |
(2.5) |
(1.2) |
(0.33) |
(0.16) |
|
AEGL-3 |
3.6 |
1.5 |
0.75 |
0.20 |
0.09 |
|
(Lethal) |
(15) |
(6.2) |
(3.1) |
(0.82) |
(0.34) |
AEGL-1 values. Chemical pneumonia in rats was used as the basis for AEGL-2, and the highest concentration causing no mortality in the rat after a 10- or 30-min exposure (and mice, 10-min value only) was used for AEGL-3.
8.2. Comparison with Other Standards and Guidelines
Table 1–15 provides existing standards and guidelines for phosgene.
TABLE 1–15 Extant Standards and Guidelines for Phosgene
|
|
Exposure Duration |
||||
|
Guideline |
10 min |
30 min |
1 h |
4 h |
8 h |
|
AEGL-1 |
NA |
NA |
NA |
NA |
NA |
|
AEGL-2 |
0.60 ppm |
0.60 ppm |
0.30 ppm |
0.08 ppm |
0.04 ppm |
|
AEGL-3 |
3.6 ppm |
1.5 ppm |
0.75 ppm |
0.20 ppm |
0.09 ppm |
|
ERPG-la |
|
|
NA |
|
|
|
ERPG-2a |
|
|
0.2 ppm |
|
|
|
ERPG-3a |
|
|
1 ppm |
|
|
|
EEGL (NRC)b |
|
|
0.2 ppm |
|
0.02 ppm (24-h) |
|
NIOSH IDLHc |
2 ppm |
|
|
|
|
|
NIOSH STELd |
0.2 ppm (15-min ceilling) |
|
|
|
|
|
NIOSH RELd |
|
|
|
|
0.1 ppm (10-h) |
|
OSHA PEL-TWAe |
|
|
|
|
0.1 ppm |
|
ACGIH TLVf |
|
|
|
|
0.1 ppm |
|
MAK (Germany)g |
|
|
|
|
0.02 ppm |
|
MAC (Netherlands)h |
|
|
|
|
0.02 ppm |
8.3. Data Adequacy and Research Needs
No reliable, quantitative human data exist. Human data are limited to descriptive effects from accidental exposure and are thus inappropriate for derivation of AEGL values. There is, however, a plethora of acute inhalation data in many experimental species. The database is sufficient to have good confidence in AEGL-2 and AEGL-3 values.
9. REFERENCES
ACGIH (American Conference of Governmental Industrial Hygienists, Inc). 2000. Documentation of the Threshold Limit Values and Biological Exposure Indices. 6th Edition.
AIHA (American Industrial Hygiene Association). 2000. Emergency Response Planning Guidelines. AIHA, Akron, Ohio.
Assaad, A., Trotz, M., Moore, D., Nold, J, Corcoran, K., Hurt, H., Keeler, J., Phillips, K., and Said, S.I. 1990. Arachidonic acid metabolites as early markers of phosgene-induced acute lung injury and pulmonary edema. World Conference on Lung Health. Boston, MA. May 20–24. Am. Rev. Respir. Dis. 141:A420.
Assaad, A., Nold, J., Petrali, J., Moore, D., Mitcheltree, L., Corcoran, K., and Phillips, K. 1991. Pulmonary pathological alterations in sheep exposed to lethal dose of phosgene. 75th Annual Meeting of the American Societies for Experimental Biology, Atlanta, Georgia. April 21–25. FASEB J. 5:A484.
Beard, R.R. 1982. Inorganic compounds of oxygen, nitrogen, and carbon. In: Patty’s Industrial Hygiene and Toxicology, Vol 2C, Clayton, G.D. and Clayton, F.E., Eds. John Wiley & Sons, New York. Pp. 4126–4128.
Box, G.E.P. and Cullumbine, H. 1947a. The relationship between survival time and dosage with certain toxic agents. Br. J. Pharmacol. 2:27–37.
Box, G.E.P. and Cullumbine, H. 1947b. The effect of exposure to sub-lethal doses of phosgene on the subsequent L(Ct)50 for rats and mice. Brit. J. Pharmacol. 2:38– 55.
Bradley, B.L. and Unger, K.M. 1982. Phosgene inhalation: A case report. Texas Med. 78:51–53.
Branchflower, R.V., Nunn, D.S., Highet, R.J., et al. 1984. Nephrotoxicity of chloroform: metabolism to phosgene by the mouse kidney. Toxicol. Appl. Pharmacol. 72:159–168.
Burleson, G.R. and Keyes, L.L. 1989. Natural killer activity in Fischer 344 rat lungs as a method to assess pulmonary immunocompetence: Immunosuppression by phosgene inhalation. Immunopharmacol. Immunotoxicol. 11:421–443.
Cameron, G.R. and Courtice, F.C. 1946. The production and removal of oedema fluid in the lung after exposure to carbonyl chloride (phosgene). J. Physiol. 105:175–185. (Cited in EPA 1986)
Cameron, G.R. and Foss, G.L. 1941. Effect of low concentrations of phosgene for 5 hours on 5 consecutive days in groups of different animals. Washington, DC: British Embassy Defense Staff; Porton report no. 2316, serial no. 63. (Cited in EPA 1986)
Cameron, G.R., Courtice, F.C., and Foss, G.L. 1942. Effects of exposing different animals to a low concentration of phosgene 1:1,000,000 (4 mg/m3) for 5 hours. (Chapter IX in First Report on Phosgene Poisoning. Portion Report No. 2339.) Washington, D.C. British Defense Staff. British Embassy. (Cited in EPA 1986)
Cessi, C., Columbini, C., and Mameli, L. 1966. The reaction of liver proteins with a metabolite of carbon tetrachloride. Biochem. J. 101:46–47c.
Chasis, H. 1944. Phosgene: Review of Literature on the effect of exposure in man and experimental animals. Contract W–49–036–CWS–1.
Clay, J.R. and Rossing, R.G. 1964. Histopathology of exposure of phosgene: An attempt to produce pulmonary emphysema experimentally. Arch. Pathol. 78:544– 551.
Coman, D.R., Bruner, H.D., Horn, R.C., Friedman, M., Boche, R.D., McCarthy, M.D. Gibbon, M.H., and Schultz, J. 1947. Studies on experimental phosgene poisoning: I. The pathologic anatomy of phosgene poisoning with special reference to the early and late phases. Am. J. Pathol. 23:1037–1074.
Currie, W.D., Hatch, G.E., and Frosolono, M.F. 1987a. Pulmonary alterations in rats due to acute phosgene inhalation. Fundam. Appl. Toxicol. 8:107–114.
Currie, W.D., Hatch, G.E., and Frosolono, M.F. 1987b. Changes in lung ATP concentration in the rat after low-level phosgene exposure. J. Biol. Toxicol. 2:105–114.
Delephine, S. 1922. Summary notes on two fatalities due to inhaling phosgene. J. Ind. Hygiene. 4:433–440.
DHEW (U.S. Department of Health, Education, and Welfare). 1968. Eighth revision international classification of diseases, adapted for use in the United States. Public Health Services Publication No. 1693:1.
Diller, W.F. 1974. Klinik und Pathologie der phosgenvergiftung. Pneumonologie. 150:139–148.
Diller, W.F. 1978. Medical phosgene problems and their possible solution. J. Occup. Med. 20:189–193.
Diller, W.F., Bruch, J., and Dehnen, W. 1985. Pulmonary changes in the rats following low phosgene exposure. Arch. Toxicol. 57:184–190.
Diller, W.F. 1985. Pathogenesis of phosgene poisoning. Toxicol. Ind. Health. 1:7– 15.
Diller, W.F., Schnellbaecher, F., and Wuestefeld, E. 1979. Late pulmonary sequelae of phosgene poisoning or inhalation-toxic pulmonary edema. Arbeitsmed. Arbeitsschultz Prophyl. 29:5–16. (Cited in EPA 1986)
Diller, W.F. and Zante, R. 1982. Dosis-wirkungs-beziehungen bei phosgeneinwirkung auf mensch und tier. Zbl. Arbeitsmed. 32:360–368
Ehrlich, J.P., Gunnison, A.F., and Burleson, G.R. 1989. Influenza virus-specific cytotoxic T-lymphocyte activity in Fischer 344 rat lungs as a method to assess pulmonary immunocompetence: Effect of phosgene inhalation. Inhalation Toxicol. 1:129–138.
EPA (U.S. Environmental Protection Agency), 1985. Health Assessment Document for Chloroform. Prepared by the Office of Health and Environmental Assessment, Environmental Criteria and Assessment Office, Research Triangle Park, NC, for the Office of Air Quality Planning and Standards. EPA 600/8–84–004F.
EPA (U.S. Environmental Protection Agency). 1986. Health Assessment Document for Phosgene. Office of Health and Environmental Assessment. EPA/600/8– 86/022A. Everett, E.D. and Overholt, E.L. 1968. Phosgene poisoning. JAMA. 205:243–245.
Franch, S. and Hatch, G.E. 1986. Pulmonary biochemical effects of inhaled phosgene in rats. J. Toxicol. Environ. Health 19:413–423.
Freeman, S., Grodins, F.S., and Kosman, A.J. 1945. The use of helium in the treatment of phosgene poisoning. In: Fasciculus on chemical warfare medicine: v. II, respiratory tract. Washington, DC: National Research Council, Committee on Treatment of Gas Casualities; pp. 541–550. (Cited in EPA 1986)
Frosolono, M.F. 1976. Basic Mechanism, diagnostic procdures and therapeutic countermeasures for phosgene poisoning in animals. Final report, MCA. (Cited in EPA 1986)
Frosolono, M.F. 1977. Basic Mechanism, diagnostic procdures and therapeutic countermeasures for phosgene poisoning in animals. Final report, MCA. (Cited in EPA 1986)
Frosolono, M.F. and Currie, W.D. 1985. Response of the pulmonary surfactant system to phosgene. Toxicol. Ind. Health. 1:29–35.
Frosolono, M.F. and Pawlowski, R. 1977. Effect of phosgene on rat lungs after single high-level exposure: I. Biochemical alterations. Arch. Environment Health 32: 271–277.
Frosolono, M.F. and Currie, W.D. 1985. Effect of phosgene on the pulmonary surfactant system (PSS): Dose-response relationships . Biochemistry 24:3386.
Galdston, M., Leutscher, J.A., Longcope, W.T., et. al. 1947a. A study of the residual effects of phosgene poisoning in human subjects: I. After acute exposure. J. Clin. Invest. 26:145–168.
Galdston, M., Leutscher, J.A., Longcope, W.T., and Ballich, N.L. 1947b. A study of the residual effects of phosgene poisoning in human subjects: II. After chronic exposure. J. Clin. Invest. 26:169–181.
Gross, P., Rinehart, W.E., and Hatch, T. 1965. Chronic pneumonitis caused by phosgene. Arch. Environ. Health. 10:768–775.
Haber, F.R. 1924. Zur geschichte des gaskrieges [On the history of the gas war]. In: Fuenf Vortraege aus den Jahren 1920–23 [Five lectures from the years 1920-
1923]. Berlin, Germany: Verlag von Julius Springer; pp. 76–92. (Cited in EPA 1986)
Halpern, B.N., Cruchaud, S., Vermeil, G., and Roux, J.L. 1950. Experimental study on the pathogenesis and treatment of acute pulmonary edema. Arch. Int. Pharmacodyn. Ther. 82:425–476. (Cited in EPA 1986)
Hatch, G.E., Slade, R., Stead, A.G., and Graham, J.A. 1986. Species comparison of acute inhalation toxicity of ozone and phosgene. J. Toxicol. Environmental Health 19:43–53.
Hedlund, L.W., and Putman, C.E. 1985. Methods for detecting pulmonary edema. Toxicol. Ind. Health. 1:59–67.
Hegler, C. 1928. On the mass poisoning by phosgene in Hamburg. I. Clinical observations. 54:1551–1553.
Henschler, D. 1971. German MAK Criteria. (From Ehrlicher, H. 1971. Personal Communication, Department of Medicine, Bayer AG, Leverkusen).
Henschler, D. and Laux, W. 1960. On the specificity of a tolerance increase by repeated inhalation of pulmonary edema-producing gases. Naunyn-Schmiedbergs Arch. Exp. Pathol. Pharmakol. 239:433–441. (Cited in EPA 1986)
Herzog, H. And Pletscher, A. 1955. Die wirkung von industriellen reizgasen auf die bronchialschleimhaut des menschen. Schweizerische Medizinische Wochenschrift. 85:477–481.
Illing, J.W., Mole, M.L., Graham, J.A, et al. 1988. Influence of phosgene inhalation on extrapulmonary effects in mice . Inhalation Toxicol. 1:13–20.
Jaskot, R.H., Grose, E.C., and Stead, A.G. 1989. Increase in angiotensin-converting enzyme in rat lungs following inhalation of phosgene. Inhalation Toxicol. 1:71– 78.
Jaskot, R.H., Grose, E.C., Richards, J.H., and Doerfler, D.L. 1991. Effects of inhaled phosgene on rat lung antioxidant systems. Fund. Appl. Toxicol. 17:666–674.
Kaerkes, B. 1992. Experiences with a phosgene indicator badge during an elevenyear period. M.D. Dissertation, Heinrich-Heine University. Duesseldorf, Germany.
Karel, L. and Weston, R.E. 1947. The biological assay of inhaled substance by the dosimetric method: The retained median lethal dose and the respiratory response in unanesthetized, normal goats exposed to different concentrations of phosgene. J. Ind. Hyg. Toxicol. 29:23–28.
Kawai, M. 1973. [Inhalation toxicity of phosgene and trichloronitromethane (chloropicrin)]. J. Sangyo Igaku 15:406–407.
Keeler, J.R., Hurt, H.H., Nold, J.B., Corcoran, K.D., and Tezak-Reid, T.M. 1990a. Phosgene-induced lung injury in sheep. Inhalation Toxicol. 2:391–406.
Keeler, J.R., Hurt, H.H., Nold, J.B., and Lennox, W.J. 1990b. Estimation of the LCt50 of phosgene in sheep. Drug Chem Toxicol. 13:229–239.
Kimmerle, G. and Diller, W. 1977. Unveroffentilichte Untersuchungen. (Cited in EPA 1986)
Kirk-Othmer.. 1991. Encyclopedia of Chemical Technology. Fourth Edition. Volume 18. John Wiley & Sons. New York. Phosgene, pp. 645–655.
Laquer, E. and Magnus, R. 1921. Combat gas poisoning. III. Experimental pathology of phosgene poisoning. Z. Gesamte Exp. Med. 13:31–107.
Leonardos, G., Kendall, D.A., and Barnard, N.J. 1968. Odor threshold determinations of 53 odorant chemicals. Presented at the 61th Annual Meeting of the Air Pollution Control Association, St. Paul, Minn., June 23–27. [Requested. NIOSH, 1976, page 29]
Lipsett, M.J., Shusterman, D.J., and Beard, R.R. 1994. Phosgene. In: Industrial hygiene and toxicology, 4th ed. Vol. II, part F. Clayton, G.D. and Clayton, F.E. editors, J.Wiley & Sons, Inc. pp. 4557–4563.
MAC (Maximaal Aanvaaarde Concentratie [Maximal Accepted Concentration]). 2000. SDU Uitgevers (under the auspices of the Ministry of Social Affairs and Employment), The Hague, The Netherlands.
MAK (Maximale Argeitsplatzkonzentration [Maximum Workplace Concentration]) 2000. Deutsche Forschungsgemeinschaft (German Research Association).
Mansuy, D., Beaune, P., Crestell, T., Lange, M., and Leroux, M. 1977. Evidence for phosgene formation during liver microsomal oxidation of chloroform. Biochem. Biophys. Res. Comm. 79:513–517.
Meek, W.J. and Eyster, J.A.E. 1920. Experiments on the pathological physiology of acute phosgene poisoning. Am. J. Physiol. 51:303–320.
Misra, N.P., Manoria, P.C., and Saxena, K. 1985. Fatal pulmonary oedema with phosgene poisining. J. Assoc. Physicians India. 33:430–431.
Moor, S. and Gates, M. 1946. Technical report of division G/NDRC. (Cited in EPA 1986)
NIOSH (National Institute of Occupational Safety and Health). 1976. Criteria for a recommended standard: occupational exposure to phosgene. NIOSH-76–137.
NIOSH (National Institute of Occupational Safety and Health). 1997. Phosgene. NIOSH Pocket Guide to Chemical Hazards, pp. 252–253.
NRC (National Research Council). 1985. Emergency and Continuous Exposure Limits for Selected Airborne Contaminants. Phosgene, pp. 69–86. National Academy Press. Washington, D.C.
Ong, S.G. 1972. Treatment of phosgene poisoning with antiserum: anaphylactic shock by phosgene. Arch. Toxicol. 29:267–278. (Cited in EPA 1986)
OSHA (Occupational Safety and Health Administration). 1994. Limits for Air Contaminants. U.S. Department of Labor. 29 CFR 1910.1000.
Patt, H.M., Tobias, J.M., Swift, M.N., Postel, S., and Gerard, R.W. 1946. Hemodynamics in pulmonary irritant poisoning. Am. J. Physiol. 147:329–339.
Pawlowski, R. and Frosolono, M.F. 1977. Effect of phosgene on rat lungs after single high-level exposure. II. Ultrastructural alternations. Arch. Environ. Health 32:278–283.
Pohl, L.R., Bhooshan, B., Whittaker, N.F., and Krishna, G. 1977. Phosgene: a metabolite of chloroform. Biochem. Biophys. Res. Comm. 79:684–691.
Pohl, L.R., Martin, J.L., and George, J.W. 1980a. Mechanism of metabolic activation of chloroform by rat liver microsomes. Biochem. Pharmacol. 29:3271–3276.
Pohl, L.R., Martin, J.L., Taburet, A.M. and George, J.W. 1980b. Oxidative bioactivation of haloforms into hepatotoxins. In: Microsomes, Drug Oxidations, and Chemical Carcinogenesis, Vol. II. Coon, M.J., et al., Eds. Academic Press, New York. Pp. 881–884.
Pohl, L.R., Branchflower, R.V., Highet, R.J., et al. 1981. The formation of diglutathionyl dithiocarbonate as a metabolite of chloroform, bromotrichloromethane, and carbon tetrachloride. Drug. Metab. Dispos. 9:334–338.
Polednak, A.P. and Hollis, D.R. 1985. Mortality and causes of death among workers exposed to phosgene in 1943–1945. Toxicol. Ind. Health. 1:137–148.
Polednak, A.P. 1980. Mortality among men occupationally exposed to phosgene in 1943–1945. Environ. Res. 22:357–367.
Postel, S. and Swift, M. 1945. Evaluation of the bleeding-transfusion treatment of phosgene poisoning. In: Fasciculus on chemical warfare medicine: v. II, respiratory tract. Washington, DC: National Research Council, Committee on Treatment of Gas Casualties; pp. 664–690. (Cited in EPA 1986)
Regan, R.A. 1985. Review of clinical experience in handling phosgene exposure cases. Toxicol. Ind. Health. 1:69–71.
Reynolds, E.S. 1967. Liver parenchymal cell injury. IV. Pattern of incorporation of carbon and chlorine from carbon tetrachloride into chemical constituents of liver in vivo. J. Pharmacol. Exp. Ther. 155:126–127.
Rinehart, W.E. 1962. A study of the concentration × time (CT) relationship in sublethal exposures to phosgene (dissertation). Pittsburgh, PA. University of Pittsburgh. Publication No. 62–6675. University Microfilms, Inc. Ann Arbor, MI.
Rinehart, W.E. and Hatch, T. 1964. Concentration-time product (CT) as an expression of dose in sublethal exposures to phosgene. AIHA Journal. 25:545–553.
Rothlin, E. 1941. Pathogenesis and treatment for phosgene intoxication. Schweiz. Med. Wochenschr. 71:1526–1535. (Cited in EPA 1986)
Sandall, T.E. 1922. The later effects of gas poisoning. Lancet. 203:857–859. (Cited in EPA 1986)
Schultz, J. 1945. The prophylactic action of hexamethylenetetramine in phosgene poisoning. In: Fasciculus on chemical warfare medicine: v. II, Respiratory tract. Washington, DC: National Research Council, Committee on Treatment of Gas Casualties; pp. 691–712.
Selgrade, M.K., Starnes, D.M., Illing, J.W., Daniels, M.J., and Graham, J.A. 1989. Effects of phosgene exposure on bacterial, viral, and neoplastic lung disease susceptability in mice. Inhalation Toxicol. 1:243–259.
Shah, H., Hartman, S.P., and Weinhouse, S. 1979. Formation of carbonyl chloride in carbon tetrachloride metabolism by rat liver in vitro. Cancer Res. 39:3942– 3947.
Shils, S.E. 1943. The therapeutic and prophylactic value of hexamethylenetetramine in phosgene poisoning. MD (EA) Memorandum report Nr. 81 V. 19. (Cited in EPA 1986)
Sipes, I.G., Krishna, G., and Gillette, J.R. 1977. Bioactivation of carbon tetrachloride, chloroform, and bromotrichloromethane: role of cytochrome P-450. Life Sciences. 20:1541–1548.
Slade, R., Highfill, J.W., and Hatch, G.E. 1989. Effects of depletion of ascorbic acid or nonprotein sulfhydryls on the acute inhalation toxicity of nitrogen dioxide, ozone, and phosgene. Inhalation Toxicol. 1:261–271.
Stavrakis, P. 1971. The use of hexamethylenetetramine (HMT) in treatment of acute phosgene poisoning. Ind. Med. 40:30–31.
TEMIS (Trauma Emergency Medical Information System). 1997. Phosgene.
ten Berge, W.F., Zwart, A. and Appleman, L.M. 1986. Concentration-time mortality response relationship of irritant and systemically acting vapours and gases. J. Hazardous Materials 13:301–309.
Tobias, J.M. 1945. The pathological physiology of the lung after phosgene. In: Fasciculus on chemical warfare medicine: v. II, respiratory tract. Washington, DC: National Research Council, Committee on Treatment of Gas Casualties; pp. 331–391. (Cited in EPA 1986)
Underhill, F.P. 1920. The lethal war gases. Physiology and experimental treatment. New Haven Yale University Press.
Weston, R.E. and Karel, L. 1946. An application of the dosimetric method for biologically assaying inhaled substances: The determination of the retained median lethal dose, percentage retention, and respiratory response in dogs exposed to different concentrations of phosgene. J. Pharmacol. Exptl. Ther. 88:195.
Weston, R.E. and Karel, L. 1947. An adaptation of the dosimeteric method for use in smaller animals. The retained median lethal dose and the respiratory response in normal, unanesthetized, Rhesus monkeys (Macaca mulatta) exposed to phosgene. J. Ind. Hyg. Toxicol. 29:29–33.
Winternitz, M.C, Lambert, R.A., and Jackson, L. 1920. The pathology of phosgene poisoning. In: Collected studies on the pathology of was gas poisoning. New Haven, CT: Yale Univ. Press; pp. 35–66.
Wirth, W. 1936. Uber die wirkung kleinster phosgenmengen. Arch. Exp. Pathol. Pharmacol. 181:198–206.
Zwart, A., Arts, J.H.E., Klokman-Houweling, J.M., and Schoen, E.D. 1990. Determination of concentration-time-mortality relationships to replace LC50 values. Inhalation Toxicol. 2:105–117.
APPENDIX A
DERIVATION OF AEGL VALUES
Derivation of AEGL-1
Data were insufficient for derivation of AEGL-1 values for phosgene.
Derivation of AEGL-2
|
Key study: |
Gross et al. (1965) |
|
Toxicity end point: |
Chemical pneumonia in rats |
|
Scaling: |
C×t=k (2 ppm)×1.5 h=3 ppm·h |
|
Uncertainty factors: |
3 for interspecies variability 3 for intraspecies variability |
|
10-min AEGL-2: |
0.6 ppm (30-min value adopted as the 10-min value) |
|
30-min AEGL-2: |
C×0.5 hr=3 ppm·h C=6 ppm 30-min AEGL-2=6 ppm/10=0.6 ppm |
|
1-h AEGL-2: |
C×1 h=3 ppm·h C=3 ppm 1-h AEGL-2=3 ppm/10=0.3 ppm |
|
4-h AEGL-2: |
C×4 h=3 ppm·h C=0.75 ppm 4-h AEGL-2=0.75 ppm/10=0.075 ppm |
|
8-h AEGL-2: |
C×8 h=3 ppm·h C=0.375 ppm |
|
|
8-h AEGL-2=0.375 ppm/10=0.038 ppm |
Derivation of AEGL-3
|
Key study: |
Zwart et al. (1990) |
|
Toxicity end point: |
The highest concentration causing no mortality in the rat or mouse after a 10-min exposure (10-min). The highest concentration causing no mortality in the rat after a 30-min exposure (30-min, 1-, 4-, and 8-h). |
|
Scaling (30-min, 1-, 4-, and 8-h): |
C×t=k (15 ppm)×0.5 h=7.5 ppm·h |
|
Uncertainty factors: |
3 for interspecies variability 3 for intraspecies variability |
|
10-min AEGL-3: |
10-min AEGL-3=36 ppm/10=3.6 ppm |
|
30-min AEGL-3: |
C×0.5 h=7.5 ppm·h C=15 ppm 30-min AEGL-3=15 ppm/10=1.5 ppm |
|
1-h AEGL-3: |
C×1 h=7.5 ppm·h C=7.5 ppm 1-h AEGL-3=7.5 ppm/10=0.75 ppm |
|
4-hr AEGL-3: |
C×4 h=7.5 ppm·h C=1.875 ppm 4-h AEGL-3=1.875 ppm/10=0.19 ppm |
|
8-h AEGL-3: |
C×8 h=7.5 ppm·h C=0.94 ppm 8-h AEGL-3=0.94 ppm/10=0.094 ppm |
APPENDIX B
DERIVATION SUMMARY FOR ACUTE EXPOSURE GUIDELINE LEVELS FOR PHOSGENE (CAS No. 75–44–5)
|
AEGL-1 |
||||
|
10 min |
30 min |
1 h |
4 h |
8 h |
|
NA |
NA |
NA |
NA |
NA |
|
Key reference: NA |
||||
|
Test species/Strain/Number: NA |
||||
|
Exposure route/Concentrations/Durations: NA |
||||
|
Effects: NA |
||||
|
End point/Concentration/Rationale: NA |
||||
|
Uncertainty factors/Rationale: NA |
||||
|
Modifying factor: NA |
||||
|
Animal to human dosimetric adjustment: NA |
||||
|
Time scaling: NA |
||||
|
Confidence and Support for AEGL values: Data were insufficient for derivation of AEGL-1 values for phosgene. Odor cannot be used as a warning for potential exposure. The odor threshold is reported to be between 0.5 and 1.5 ppm, a value above or approaching AEGL-2 and AEGL-3 values, and tolerance to the pleasant odor of phosgene occurs rapidly. Furthermore, following odor detection and minor irritation, serious effects may occur after a clinical latency period of ≤24 h. |
||||
|
AEGL-2 |
||||
|
10 min |
30 min |
1 h |
4 h |
8 h |
|
0.60 ppm |
0.60 ppm |
0.30 ppm |
0.08 ppm |
0.04 ppm |
|
Key reference: |
Gross, P., Rinehart, W.E., and Hatch, T. 1965. Chronic pneumonitis caused by phosgene. Arch. Environ. Health. 10:768–775. |
|||
|
Test species/Strain/Number: Wistar rats/118 males |
||||
|
Exposure route/Concentrations/Durations: Rats/Inhalation: 0.5 to 4.0 ppm for 5 min to 8 h to give C×T products between 12 and 360 ppm·min (2 ppm for 1.5 h was determinant for AEGL-2) |
||||
|
Effects: 2 ppm for 1.5 h: chemical pneumonia; 0.9 ppm for 1 h: “chronic pneumonitis” |
||||
|
End point/Concentration/Rationale: Rats/2 ppm for 1.5 h/chemical pneumonia |
||||
|
Uncertainty factors/Rationale: Total uncertainty factor: 10 Interspecies: 3—little species variability is observed with both lethal and nonlethal end points in many studies after exposure to phosgene Intraspecies: 3—due to the steep concentration-response curve and effects appear to be due to irritation and binding to macromolecules are not expected to differ greatly among individuals. |
||||
|
Modifying factor: Not applicable |
||||
|
Animal to human dosimetric adjustment: Insufficient data |
||||
|
Time scaling: Cn×t=k where n=1. Haber’s Law (C×t=k) has been shown to be valid for phosgene within certain limits (EPA 1986). Haber’s Law was originally derived from phosgene data (Haber 1924). Reported 1.5 h data point used for AEGL-2 derivation. AEGL values for the 30-min and 1-, 4-, and 8-h exposure periods were based on extrapolation from the 1.5 h value. The 30-min value is also adopted as the 10-min value because Diller et al. (1985) observed alveolar pulmonary edema in rats exposed to 5 ppm phosgene for 10 min. Applying a total UF of 10 to this data point yields a supporting 10-min value of 0.5 ppm. |
||||
|
Data adequacy: The database is rich. The calculated AEGL-2 values are supported by rat studies where exposure of rats to 1 ppm phosgene for 4 h resulted in severe pulmonary edema and body weight loss. (Franch and Hatch 1986; Erlich et al. 1989). Use of these data (and application of a total UF of 10) results in supporting AEGL-2 values of 0.8, 0.4, 0.1, and 0.05 ppm for the 30 min, 1 h, 4 h, and 8 h time points, respectively. The 10-min value is supported by Diller et al. (1985) as described above in the time scaling section. |
||||
|
AEGL-3 |
||||
|
10 min |
30 min |
1 h |
4 h |
8 h |
|
3.6 ppm |
1.5 ppm |
0.75 ppm |
0.20 ppm |
0.09 ppm |
|
Reference: |
Zwart, A. et al. 1990. Determination of concentration-time-mortality relationships to replace LC50 values. Inhalation Toxicol. 2:105–117. |
|||
|
Test species/Strain/Sex/Number: Wistar rats/5 males and 5 females |
||||
|
Exposure route/Concentrations/Durations: Rats/Inhalation: 12, 15, 16, 17, or 24 ppm for 30 min (the highest concentration causing no mortality in the rat after a 30-min exposure of 15 ppm was determinant for AEGL-3) |
||||
|
Effects: |
Concentration |
Mortality |
||
|
|
12 ppm |
0/10 |
||
|
|
15 ppm |
0/10 |
||
|
|
16 ppm |
1/10 |
||
|
|
17 ppm |
5/10 |
||
|
|
24 ppm |
9/10 |
||
|
End point/Concentration/Rationale: The highest concentration causing no mortality in the rat after a 30-min exposure 30-min experimental no-effect-level for death (15 ppm) was used as a threshold for death in rats for the 30-min, 1-, 4-, and 8-h values. The highest concentration causing no mortality in the rat after a 10-min exposure (36 ppm) was utilized for the 10-min value. |
||||
|
Uncertainty Factors/Rationale: Total uncertainty factor: 10 Interspecies: 3—little species variability is observed with both lethal and nonlethal end points in many studies after exposure to phosgene Intraspecies: 3—due to the steep concentration-response curve and effects appear to be due to irritation and binding to macromolecules are not expected to differ greatly among individuals . |
||||
|
Modifying factor: Not applicable |
||||
|
Animal to human dosimetric adjustment: Insufficient data |
||||
|
Time scaling: Cn×t=k where n=1. Haber’s Law (C×t=k) has been shown to be valid for phosgene within certain limits (EPA 1986). Haber’s Law was originally derived from phosgene data (Haber 1924). Reported 30-min data point used to determine the 30-min AEGL value. AEGL-3 values for 1-, 4-, and 8-h were based on extrapolation from the 30 min value. The 10-min value was based on a reported 10-min data point. |
||||
|
Data adequacy: The AEGL-3 values are based on a well-conducted study in rats and the database is rich. |
||||


























































