
3
Microbiology, Ecology, and Natural History of Coronaviruses
OVERVIEW
Coronaviruses cause a substantial fraction of human colds and a host of common respiratory infections in many other animals, including economically important diseases of livestock, poultry, and laboratory rodents. Moreover, although these viruses were not known for producing more than mild infections in humans prior to the SARS epidemic, veterinary coronavirologists have long been aware of their potential for producing lethal infections in animals, as Linda Saif describes in this chapter’s first paper. For this reason, there is already an extensive amount of research on animal coronaviruses that can be drawn from for understanding the life cycle and pathogenicity of the SARS virus, and veterinary scientists are now being called on to join the research response to the epidemic and share their knowledge of coronaviruses with a broader audience. Mark Denison’s paper describes the current state of research on animal coronaviruses and discusses how results from these animal models suggest promising directions for future research on SARS and other emerging zoonoses.
Animal coronaviruses tend to follow one of two basic pathogenic models, producing either enteric or respiratory infections. Both models show parallels to the clinical features of SARS patients, the majority of whom presented with respiratory infections but in some cases also suffered from enteric complications. In adult animals, coronavirus infections of a respiratory nature have shown increased severity in the presence of several factors, including high exposure doses, respiratory coinfections, stress related to shipping or commingling with animals from different farms, and treatment with corticosteroids. In young, seronegative animals, enteric coronaviruses can cause fatal infections. Although coronaviruses
generally cause disease in a single animal species, some have been demonstrated to cross species barriers.
Considerable effort has already been applied toward uncovering an animal source of the SARS virus. This has been sought primarily through the genetic characterization of viral isolates from suspected animal sources and comparison with human SARS coronavirus samples. In the past, however, epidemiological detective work has identified the source of many outbreaks of infectious disease, and one workshop participant suggested that a case control study of the first 50 to 100 SARS patients from China’s Guangdong Province, where the earliest cases of the disease were detected, might prove similarly fruitful. While a natural reservoir for the SARS virus has not yet been identified, the combination of such genomic and epidemiological techniques is already yielding suggestive results. For example, the last paper in this chapter by Yi Guan et al. describes the presence of coronaviruses closely related to SARS among live animals sold in Guangdong markets. Similar epidemiological principles may yet provide valuable direction for further laboratory surveys of animal viruses aimed at finding the original source and reservoir of the SARS coronavirus.
Coronaviruses have been classified into three major categories based on their genetic characteristics. While the SARS virus has been linked with Group II coronaviruses, whose members include human and bovine respiratory viruses and the mouse hepatitis virus, there is still some debate over whether its genetic features might be sufficiently distinct to warrant classification within a separate, fourth class of coronaviruses. Studies of coronavirus replication at the molecular level reveal several mechanisms that account for the repeated, persistent infections typical of coronaviral disease. High rates of mutation and RNA-RNA recombination produce viruses that are able to adapt to acquire and regain virulence. Although researchers have identified several potential targets for antiviral therapies, the ability of the virus to mutate and recombine represents a major challenge to vaccine development. A vaccine that can provide highly effective, long-term protection against respiratory coronavirus infections has not yet been developed, nor have appropriate animal models been developed to test potential vaccines against SARS. It was noted by several workshop participants that a coordinated, multidisciplinary research effort, drawing on expertise in both the veterinary and biomedical sciences, will likely be needed to meet these goals.
ANIMAL CORONAVIRUSES: LESSONS FOR SARS
Linda J. Saif
Department of Food Animal Health Research Program, Ohio Agricultural Research and Development Center
The emergence of severe acute respiratory syndrome (SARS) illustrates that coronaviruses (CoVs) may quiescently emerge from possible animal reservoirs and
can cause potentially fatal disease in humans, as previously recognized for animals. Consequently the focus of this review will be on the emergence of new CoV strains and the comparative pathogenesis of SARS CoV with those CoVs that cause enteric and respiratory infections of various animal hosts. A review of animal CoV vaccines recently has been compiled (Saif, in press), so this topic will not be addressed.
Emergence of New Coronaviruses
The medical community was amazed by the emergence of a new coronavirus associated with SARS in healthy adults in 2003 (Drosten et al., 2003; Ksiazek et al., 2003; Peiris et al., 2003b; Poutanen et al., 2003). Historically human CoV infections (229E and OC43 CoV strains) were mild and associated with only common cold symptoms although reinfections, even with the same strain, occur (Callow et al, 1990; Holmes, 2001). However, veterinary coronavirologists had previously recognized the potential for coronaviruses to cause fatal enteric or respiratory infections in animals and for new CoV strains to emerge from unknown reservoirs, often evoking fatal disease in naïve populations. For example, the porcine epidemic diarrhea CoV (PEDV) first appeared from an unknown source in Europe and Asia in the 1970s and 1980s, causing severe diarrhea and widespread deaths in baby pigs before becoming endemic in swine (Pensaert, 1999). The PEDV is absent in U.S. swine. Interestingly, PEDV is genetically more closely related to human CoV 229E than to the other animal group I CoV (Duarte et al., 1994), and unlike the other group I CoV, it grows in Vero cells like SARS CoV (Hoffman and Wyler, 1988). These observations raise intriguing, but unanswered, questions about its origin.
Alternatively new CoV strains differing in tissue tropism and virulence may arise from existing strains. The less virulent porcine respiratory coronavirus (PRCV) evolved as a spike (S) gene deletion mutant of the highly virulent enteric CoV, transmissible gastroenteritis virus (TGEV) ( reviewed in Laude et al., 1993; Saif and Wesley, 1999). Curiously, differences in the sizes of the 5′ end S gene deletion region (621–681 nucleotides) between European and U.S. PRCV strains provided evidence for their independent origin on two continents within a similar time frame (1980s). Deletion of this region (or in combination with deletions in ORF 3a) presumably accounted for altered tissue tropism from enteric to respiratory and reduced virulence of the PRCV strains (Ballesteros et al., 1997; Sanchez et al., 1999). The ability of certain CoVs to persist in their host also provides a longer opportunity for new mutants to be selected with altered tissue tropisms and virulence from among the viral RNA quasispecies (or swarm of viruses). An example is the virulent systemic variant, the feline infectious peritonitis virus (FIPV), which likely arises from persistent infection of cats with the less virulent feline enteric CoV (Herrewegh et al., 1997; Vennema et al., 1995).
Furthermore, animal CoVs may acquire new genes via recombination, as exemplified by the acquisition of an influenza C-like hemagglutinin by bovine
CoV or its ancestor CoV (Brian et al., 1995). Recombination events among CoVs may also generate new strains with altered tissue or host tropisms. For example, targeted recombination between feline and mouse S proteins enables feline CoV to infect mice (Haijema et al., 2003). Recent phylogenetic analysis suggests that SARS CoV may have evolved from a past recombination event between mammalian-like and avian-like parent strains with the S gene representing a mammalian (group 1)–avian origin mosaic (Stavrinides and Guttman, 2004). This recognition that CoVs can further evolve in a host population to acquire new tissue tropisms or virulence via mutations or recombination suggests that similar events may occur if SARS CoV persists in humans.
Interspecies Transmission of Coronaviruses
The genus coronavirus is composed of at least three genetically and autigenically distinct groups of CoV that cause mild to severe enteric, respiratory, or systemic disease in domestic and wild animals, poultry, rodents, and carnivores and mild colds in humans (Table 3-1) The SARS CoV is genetically distantly related to known CoVs and comprises a provisional new group (IV) (Drosten et al., 2003; Marra et al., 2003; Rota et al., 2003) or alternatively, using rooted tree phylogenetic analysis, belongs to a subgroup of group II (Snijder et al., 2003). Coronaviruses from two wild animal species (civet cats and raccoon dogs) recently have been characterized genetically as members of the SARS CoV group (Guan et al., 2003). Coronaviruses within each group share various levels of genetic and antigenic relatedness and several show cross-species transmission. Thus the likelihood that SARS CoV is a zoonotic infection potentially transmitted from wild animals to humans is not unprecedented based on previous research on interspecies transmission of animal CoV and wildlife reservoirs for CoV. As examples, the porcine CoV, TGEV, and canine and feline CoVs can cross-infect pigs, dogs, and cats with variable disease expression and levels of cross-protection in the heterologous host (Saif and Wesley, 1999; Saif and Heckert, 1990). These three related CoVs appear to be host range mutants of an ancestral CoV. Wildlife reservoirs for CoVs were recognized prior to SARS. Captive wild ruminants harbor CoVs antigenically closely related to bovine CoV and CoV isolates from the wild ruminants experimentally infected domestic calves (Tsunemitsu et al., 1995; Majhdi et al., 1997). The promiscuousness of bovine CoV is evident by infection of dogs and also humans by genetically similar (>97 percent identity) CoV strains (Erles et al., 2003; Zhang et al., 1994). Even more dramatic than infection of mammalian hosts by bovine CoV is the finding that bovine CoV can experimentally infect and cause disease (diarrhea) in phylogenetically diverse species such as avian hosts, including baby turkeys, but not baby chicks (Ismail et al., 2001b). It is notable that in the latter study, the bovine CoV-infected baby turkeys also transmitted the viruses to unexposed contact control birds. The reasons for the broad host range of bovine CoV are unknown, but may relate to the
TABLE 3-1 Coronavirus Groups, Target Tissues, and Diseases
|
|
|
|
Disease/Infection Site |
||
|
Genetic Group |
Virus |
Host |
Respiratory |
Enterica |
Otherb |
|
I |
HCoV-229E |
Human |
X upper |
|
|
|
|
TGEV |
Pig |
X upper |
X S1 |
|
|
|
PRCV |
Pig |
X upper/lung |
|
Vitremia |
|
|
PEDV |
Pig |
|
X SL, colon |
|
|
|
F1PV |
Cat |
X upper |
X |
Systemic |
|
|
FCoV |
Cat |
|
X S1 |
|
|
|
CCOV |
Dog |
|
X S1 |
|
|
|
RaCoV |
Rabbit |
|
|
Systemic |
|
11 |
HCoV-OC43 |
Human |
X upper |
?? (BCoV?) |
|
|
|
NUN |
Mouse |
|
X |
Hepatitis, CNS, systemic |
|
|
RcoV |
Rat |
X |
|
|
|
|
(sialodocryadenitis) |
Pig |
X |
|
Eye, salivary glands CNS |
|
|
BEV |
Cattle |
X upper/lung |
X S1, colon |
|
|
|
BCoV |
|
|
|
|
|
III |
IBV |
Chicken |
X upper |
X |
Kidney, oviduct |
|
|
TCoV (TECoV) |
Turkey |
|
X S1 |
|
|
IV?? |
SARS |
Human |
X lung |
X (?) |
Viremia, kidney? |
|
IIA? |
Civet cat CoV |
Himalayan palm civet |
X |
X |
Subclinical? |
|
|
|
Raccoon dog |
|
|
|
|
|
Raccoon dog CoV |
|
? |
X |
Subclinical? |
|
aSl = small intestine; ?? = BCoV-Iike CoV from a child, Zhang et al. (1994); ? = unknown. bCNS = central nervous system. |
|||||
presence of a hemagglutinin on bovine CoV and its possible role in binding to diverse cell types.
Recent data suggest that SARS CoV may also have a broad host range besides humans. Genetically similar CoVs were isolated from civet cats and raccoon dogs (Guan et al., 2003). In experimental studies, the SARS CoV infected and caused disease in macaques and ferrets and infected cats subclinically (Fouchier et al., 2003; Martina et al., 2003). In the latter two species, the SARS CoV was further transmitted to exposed contacts, documenting transmission within the new host species. Consequently, although previous data document the emergence of new animal CoV strains and the broad host range of several CoVs, the determinants for host range specificity among CoVs are undefined. In addition, we understand little about CoVs circulating in wildlife and relatively few animal CoV strains have been fully sequenced for comparative phylogenetic analysis to trace their evolutionary origins.
Pathogenesis of Animal Enteric and Respiratory Coronaviruses
Pathogenesis of Group I TGEV and PRCV CoV: Models of Enteric and Respiratory Infections
Because both pneumonia and diarrhea occur in SARS patients, an understanding of the tissue tropisms and pathogenesis of respiratory and enteric animal CoVs should contribute to our understanding of similar parameters for SARS. The TGEV targets the small intestinal epithelial cells leading to severe villous atrophy, malabsorptive diarrhea, and a potentially fatal gastroenteritis (Table 3-1). The virus also infects the upper respiratory tract with transient nasal shedding (Van Cott et al., 1993), but infection or lesions in the lung are less common. In adults, TGEV is mild with transient diarrhea or inappetence, but pregnant or lactating animals develop more severe clinical signs and agalactia (Saif and Wesley, 1999).
The PRCV, an S gene deletion mutant of TGEV, has an altered tissue tropism (respiratory) and reduced virulence (Laude et al., 1993; Saif and Wesley, 1999). Like SARS, PRCV spreads by droplets and has a pronounced tropism for the lung, replicating to titers of 107-108 TCID50 and producing interstitial pneumonia affecting 5 to 60 percent of the lung (Cox et al., 1990; Halbur et al., 1993; Laude et al., 1993; Saif and Wesley, 1999). Although many uncomplicated PRCV infections are mild or subclinical, lung lesions are invariably present. Like SARS, clinical signs of PRCV include fever with variable degrees of dyspnea, polypnea, anorexia, and lethargy, and less coughing and rhinitis (Cox et al., 1990; Halbur et al., 1993; Hayes, 2000; Laude et al., 1993; Saif and Wesley, 1999). Further resembling SARS, PRCV replicates in lung epithelial cells, although viral antigen is also detected in type I and II pneumocytes and alveolar macrophages. In lungs, bronchiolar infiltration of mononuclear cells, lymphohistiocytic exudates, and epithelial cell necrosis leads to interstitial pneumonia. PRCV induces transient
viremia with virus also detected from nasal swabs and in tonsils and trachea, similar to SARS (Drosten et al., 2003; Ksiazek et al., 2003; Peiris et al., 2003b). The PRCV further replicates in undefined cells in the gut lamina propria, but without inducing villous atrophy or diarrhea and with limited fecal shedding (Cox et al., 1990; Saif and Wesley, 1999). Recently, however, fecal isolates of PRCV were detected with consistent, minor point mutations in the S gene compared to the nasal isolates from the same pig (Costantini et al., in press). Such observations suggest the presence of CoV quasispecies in the host with some strains more adapted to the intestine, a potential corollary for the fecal shedding of SARS CoV (Drosten et al., 2003; Ksiazek et al., 2003; Peiris et al., 2003a). Of further relevance to SARS was the displacement of the virulent TGEV infections by the widespread dissemination of PRCV in Europe and the disappearance of PRCV from swine herds in summer with its reemergence in older pigs in winter (Laude et al., 1993; Saif and Wesley, 1999).
Group II Bovine CoV (BCoV): Models of Pneumoenteric Infections
The shedding of SARS in feces of many patients and the occurrence of diarrhea in 10 to 27 percent of patients (Peiris et al., 2003a), but with a higher percentage (73 percent) in the Amoy Gardens, Hong Kong, outbreak (Chim et al., 2003) suggests that SARS may be pneumoenteric like BCoV. BCoV causes three distinct clinical syndromes in cattle: calf diarrhea; winter dysentery with hemorrhagic diarrhea in adults; and respiratory infections in cattle of various ages, including cattle with shipping fever (Table 3-1) (Clark, 1993; Lathrop et al., 2000a; Lathrop et al., 2000b; Saif and Heckert, 1990; Storz et al., 1996, 2000a, Tsunemitsu et al., 1995). Based on BCoV antibody seroprevalence, the virus is ubiquitous in cattle worldwide. All BCoV isolates from both enteric and respiratory infections are antigenically similar in virus neutralization (VN) tests, comprising a single serotype, but with two to three subtypes identified by VN or using monoclonal antibodies (MAbs) (Clark, 1993; Hasoksuz et al., 1999a; Hasoksuz et al., 1999b; Saif and Heckert, 1990; Tsunemitsu and Saif, 1995). In addition, genetic differences (point mutations but not deletions) have been detected in the S gene between enteric and respiratory isolates, including ones from the same animal (Chouljenko et al., 2001; Hasoksuz et al., 2002b). Nevertheless, inoculation of gnotobiotic or colostrum-deprived calves with calf diarrhea, winter dysentery, or respiratory BCoV strains led to both nasal and fecal CoV shedding and cross-protection against diarrhea after challenge with a calf diarrhea strain (Cho et al., 2001b; El-Kanawati et al., 1996). However, subclinical nasal and fecal virus shedding detected in calves challenged with the heterologous BCoV strains (Cho et al., 2001b; El-Kanawati et al., 1996) confirmed field studies showing that subclinically infected animals may be a reservoir for BCoV (Heckert et al., 1990, 1991). Cross-protection against BcoV-induced respiratory disease has not been evaluated.
Calf Diarrhea and Calf Respiratory BCoV Infections
Calf diarrhea BCoV strains infect the epithelial cells of the distal small and large intestine and superficial and crypt enterocytes of the colon, leading to villous atrophy and crypt hyperplasia (Saif and Heckert, 1990; Van Kruiningen et al., 1987). One- to 4-week-old calves develop a severe, malabsorptive diarrhea, resulting in dehydration and often death. Concurrent fecal and nasal shedding often occur. BCoV are also implicated as a cause of mild respiratory disease (coughing, rhinitis) or pneumonia in 2- to 24-month-old calves and are detected in nasal secretions, lungs, and often the intestines (Clark, 1993; Heckert et al., 1990; Heckert et al., 1991; Saif and Heckert, 1990). In studies of calves from birth to 20 weeks of age, Heckert and colleagues (1990, 1991) documented both fecal and nasal shedding of BCoV, with repeated respiratory shedding episodes in the same animal with or without respiratory disease, and subsequent increases in their serum antibody titers consistent with these reinfections. These findings suggest a lack of long-term mucosal immunity in the upper respiratory tract after natural CoV infection, confirming similar observations for human respiratory CoV (Callow et al., 1990; Holmes, 2001).
Winter Dysentery BCoV Infections
Winter dysentery (WD) occurs in adult cattle during the winter months and is characterized by hemorrhagic diarrhea, frequent respiratory signs, and a marked reduction in milk production in dairy cattle (Saif, 1990; Saif and Heckert, 1990; Van Kruiningen et al., 1987). Intestinal lesions and BCoV-infected cells in the colonic crypts resemble those described for calf diarrhea. The BCoV isolates from WD outbreaks at least partially reproduced the disease in BCoV seropositive nonlactating cows (Tsunemitsu et al., 1999) and in BCoV seronegative lactating cows (Traven et al., 2001). Interestingly, in the later study, the older cattle were more severely affected than similarly exposed calves, mimicking the milder SARS cases seen in children versus adults (Kamps and Hoffmann, 2003a).
Shipping Fever BCoV Infections
More recent studies done in 1995 have implicated BCoV in association with respiratory disease (shipping fever) in feedlot cattle (Lathrop et al., 2000a, Storz et al., 1996). BCoV was isolated from nasal secretions and lungs of cattle with pneumonia and from feces (Hasoksuz et al., 1999a, 2002a; Storz et al., 2000a, b). In a subsequent study, a high percentage of feedlot cattle (45 percent) shed BCoV both nasally and in feces by ELISA (Cho et al., 2001a). Application of nested RT-PCR detected higher BCoV nasal and fecal shedding rates of 84 percent and 96 percent, respectively (Hasoksuz et al., 2002a).
Cofactors That Exacerbate CoV Infections, Disease, or Shedding
Underlying disease or respiratory coinfections, dose and route of infection, and immunosuppression (corticosteroids) are all potential cofactors related to the severity of SARS. These cofactors can also exacerbate the severity of BCoV, TGEV, or PRCV infections. In addition, these cofactors may play a role in the superspreader cases seen in the SARS epidemic (Kamps and Hoffmann, 2003b) by enhancing virus transmission.
Impact of Respiratory Co-Infections on CoV Infections, Disease, and Shedding
Shipping fever is recognized as a multifactorial, polymicrobial respiratory disease complex in young adult feedlot cattle with several factors exacerbating respiratory disease, including BCoV infections (Lathrop et al., 2000a,b; Storz et al., 1996; Storz et al., 2000a; Storz et al., 2000b). Shipping fever can be precipitated by several viruses, alone or in combination, including viruses similar to common human respiratory viruses (BCoV, bovine resiratory syncytial virus, parainfluenza-3 virus), bovine herpesvirus, and viruses capable of mediating immunosuppression (bovine viral diarrhea virus, etc.). The shipping of cattle long distances to feedlots and the commingling of cattle from multiple farms creates physical stresses that overwhelm the animal’s defense mechanisms and provides close contact for exposure to new pathogens or strains not previously encountered. Such factors are analogous to the physical stress of long airplane trips with close contact among individuals from diverse regions of the world, both of which may play a role in enhancing an individual’s susceptibility to SARS. For shipping fever, various predisposing factors (viruses, stress) allow commensal bacteria of the nasal cavity (Mannheimia haemolytica, Pasteurella spp., Mycoplasma spp., etc.) to infect the lungs, leading to fatal fibrinous pneumonia (Lathrop et al., 2000a,b; Storz et al., 1996, 2000a,b). Like PRCV or SARS infections, it is possible that antibiotic treatment of such individuals with massive release of bacterial lipopolysaccharides (LPS) could precipitate induction of proinflammatory cytokines, which may further enhance lung damage. For example, Van Reeth et al. (2000) showed that pigs infected with PRCV followed by a subclinical dose of E. coli LPS within 24 hours developed enhanced fever and more severe respiratory disease compared to each agent alone. They concluded that the effects were likely mediated by the significantly enhanced levels of proinflammatory cytokines induced by the bacterial LPS. Thus there is a need to examine both LPS and lung cytokine levels in SARS patients as possible mediators of the severity of SARS. Bacteria (Chlamydia spp.) have been isolated from SARS patients, but their role in enhancing the severity of SARS is undefined (Poutanen et al., 2003).
Interactions between PRCV and other respiratory viruses may also parallel the potential for concurrent or preexisting respiratory viral infections to interact with SARS CoV (such as metapneumoviruses, influenza, reoviruses, respiratory
syncytial virus [RSV], OC43 or 229E CoV). Hayes (2000) showed that sequential dual infections of pigs with the arterivirus (order Nidovirales, like CoV) PRRSV followed in 10 days by PRCV significantly enhanced lung lesions and reduced weight gains compared to each virus alone. The dual infections also led to more pigs shedding PRCV nasally for a prolonged period and surprisingly, to fecal shedding of PRCV. The lung lesions observed resembled those in SARS victims (Nicholls et al., 2003).
In another study, Van Reeth and Pensaert (1994) inoculated pigs with PRCV followed in 2 to 3 days by swine influenza A virus (SIV). They found that SIV lung titers were reduced in the dually compared to the singly infected pigs, but paradoxically the lung lesions were more severe in the dually infected pigs. They postulated that the high levels of IFN-alpha induced by PRCV may mediate interference with SIV replication but may also contribute to the enhanced lung lesions. Such studies are highly relevant to potential dual infections with SARS CoV and influenza virus and potential treatments of SARS patients with IFN alpha.
Impact of Route (Aerosols) and Dose on CoV Infections
Experimental inoculation of pigs with PRCV strains showed that administration of PRCV by aerosol compared to the oronasal route, or in higher doses, resulted in higher virus titers shed and longer shedding (Van Cott et al., 1993). In other studies, high PRCV doses induced more severe respiratory disease. Pigs given 108.5 TCID50 of PRCV had more severe pneumonia and deaths than pigs exposed by contact (Jabrane et al., 1994), and higher intranasal doses of another PRCV strain (AR310) induced moderate respiratory disease whereas lower doses produced subclinical infections (Halbur et al., 1993). By analogy, hospital procedures that could potentially generate aerosols or exposure to higher initial doses of SARS CoV may enhance SARS transmission or lead to enhanced respiratory disease (Kamps and Hoffman, 2003a,b).
Impact of Treatment with Corticosteroids on CoV Infections of Animals
Corticosteroids are known to induce immunosuppression and reduce the numbers of CD4 and CD8 T cells and certain cytokine levels (Giomarelli et al., 2003). Many hospitalized SARS patients were treated with steroids to reduce lung inflammation, but there are no data to assess the outcome of this treatment on virus shedding or respiratory disease. A recrudescence of BCoV fecal shedding was observed in one of four winter dysentery BCoV infected cows treated with dexamethasone (Tsunemitsu et al., 1999). Similarly, treatment of older pigs with dexamethasone prior to TGEV challenge led to profuse diarrhea and reduced lymphoproliferative responses in the treated pigs (Shimizu and Shimizu, 1979). These data raise issues for corticosteroid treatment of SARS patients re-
lated to possible transient immunosuppression leading to enhanced respiratory disease or increased and prolonged CoV shedding (superspreaders). Alternatively, corticosteroid treatment may be beneficial in reducing proinflammatory cytokines if found to play a major role in lung immunopathology (Giomarelli et al., 2003).
Group I Feline CoV (FCoV): Model for Systemic and Persistent CoV Infection
The spectrum of disease evident for FCoV (feline infectious peritonitis virus) exemplifies the impact of viral persistence and macrophage tropism on CoV disease progression and severity. Historically, two types of FCoVs have been recognized: feline enteric CoV (FECoV) and FIPV. Current information suggests that the FECoV that causes acute enteric infections in cats establishes persistent infections in some cats, evolving into the systemic virulent FIPV in 5 to 10 percent of cats (deGroot and Horzinek, 1995; Herrewegh et al., 1997; Vennema et al., 1995). The relevance of this model to SARS is whether similar persistent CoV infections might occur in some patients, leading to the emergence of macrophage-tropic mutants of enhanced virulence and precipitating systemic or immune-mediated disease. The initial site of FCoV replication is in the pharyngeal, respiratory, or intestinal epithelial cells (deGroot and Horzinek, 1995; Olsen, 1993), and clinical signs include anorexia, lethargy, and mild diarrhea. The prolonged incubation period for FIPV and its reactivation upon exposure to immunosuppressive viruses or corticosteroids suggested that FCoVs could cause chronic enteric infections in cats (deGroot and Horzinek, 1995; Olsen, 1993). Recent reports of chronic fecal shedding and persistence of FCoV mRNA or antigen in infected cats confirm this scenario (Herrewegh et al., 1997).
A key pathogenetic event for development of FIPV is productive infection of macrophages followed by cell-associated viremia and systemic dissemination of virus (deGroot and Horzinek, 1995; Olsen, 1993). Stress (immunosuppressive infections, transport to new environments, cat density) leading to immune suppression may trigger FIP in chronically infected cats, similar to its role in shipping fever CoV infections of cattle. Two major forms of FIP occur: (1) effusive, with a fulminant course and death within weeks to months, and (2) noneffusive, progressing more slowly (deGroot and Horzinek, 1995; Olsen, 1993). The effusive form is characterized by fibrin-rich fluid accumulation in peritoneal, pleural, pericardial, or renal spaces, with fever, anorexia, and weight loss. Noneffusive FIP involves pyogranulomatous lesions with thrombosis, central nervous system, or ocular involvement. Fulminant FIP with accelerated early deaths appears to be immune mediated in FCoV seropositive cats. At least two mechanisms implicating IgG antibodies to FCoV S protein in FIP immunopathogenesis have been described. In the first, circulating immune complexes (IC) with C’ depletion in sera and IC in lesions are evident in cats with terminal FIP (deGroot and Horzinek, 1995). In the second, antibody dependent enhancement (ADE) of FCoV infection of macrophages in vitro is mediated by neutralizing IgG MAbs to the S protein of
FIPV, or of interest, to the antigenically-related CoV, TGEV (Olsen et al., 1993). Similar accelerated disease was seen in vivo in cats inoculated with recombinant vaccinia virus expressing the S protein (but not the M or N proteins) of FIPV (deGroot and Horzinek, 1995; Olsen et al., 1993). Thus the FIPV model provides a frightening glimpse of the severity and potential complications associated with a persistent, systemic CoV infection.
Group III CoVs: Infectious Bronchitis Virus (IBV): Model for Respiratory CoV Infection with Other Target Tissues
The IBV is a highly contagious respiratory disease of chickens, like SARS, spread by aerosol or possibly fecal-oral transmission, and distributed worldwide (Cavanagh and Naqi, 2003; Cook and Mockett, 1995). Genetically and antigenically closely related CoV have been isolated from pheasants and turkeys (Guy et al., 1997; Ismail et al., 2001a), but in young turkeys, they cause mainly enteritis. Respiratory infections of chickens are characterized by tracheal rales, coughing, and sneezing, with the disease most severe in chicks (Cavanagh and Naqi, 2003; Cook and Mockett, 1995). The IBV also replicates in the oviduct, causing decreased egg production. Nephropathogenic strains can cause mortality in young birds. In broilers, severe disease or death ensues from systemic E. coli co-infections after IBV damage to the respiratory tract or Mycoplasma sp. co-infections with IBV. The IBV is recovered intermittently from the respiratory tract for about 28 days after infection and from the feces after clinical recovery, with the cecal tonsil being a possible reservoir for IBV persistence, similar to the persistence of FCoV in the intestine of cats (Herrewegh et al., 1997). The IBV was recovered from both tracheal and cloacal swabs in chickens at onset of egg production, suggesting re-excretion of IBV from chronically infected birds, as also demonstrated for fecal shedding of FCoV or BCoV after induction of immunosuppression (Olsen, 1993; Tsunemitsu et al., 1999).
The IBV replicates in epithelial cells of the trachea and bronchi, intestinal tract, oviduct, and kidney, causing necrosis and edema with small areas of pneumonia near large bronchi in the respiratory tract and interstitial nephritis in the kidney (Cavanagh and Naqi, 2003; Cook and Mockett, 1995). Of interest for SARS is the persistence of IBV in the kidney and its prolonged fecal shedding because SARS CoV is detected in urine and shed longer term in feces. However, it is unclear if SARS CoV shedding in urine is a consequence of viremia or a kidney infection like IBV. Both diagnosis and control of IBV are complicated by the existence of multiple serotypes and the occurrence of IBV recombinants (Cavanagh and Naqi, 2003; Cook and Mockett, 1995). This is unlike the scenario for most group 1 or 2 respiratory CoVs in which only one or two (FCoV) serotypes are known. Also relevant to SARS CoV is the finding that IBV strains also replicate in Vero cells, but only after passage in chicken embryo kidney cells (Cavanagh and Nagi, 2003).
In summary, studies of animal CoV infections in the natural host provide enteric and respiratory disease models that enhance our understanding of both the similarities and divergence of CoV disease pathogenesis and targets for control. Unanswered questions for SARS pathogenesis, but highly relevant to the design of strategies for prevention and control, include the following: What is the initial site of viral replication? Is SARS CoV pneumoenteric like BCoV, with variable degrees of infection of the intestinal and respiratory tracts and disease precipitated by the co-factors discussed or unknown variables? Alternatively, is SARS primarily targeted to the lung like PRCV, with fecal shedding of swallowed virus and with undefined sequelae contributing to the diarrhea cases? Does SARS CoV infect the lung directly or via viremia after initial replication in another site (oral cavity, tonsils, upper respiratory tract) and does it productively infect secondary target organs (intestine, kidney) via viremia after replication in the lung?
Finally, the persistent, macrophage tropic, systemic FIPV CoV infection of cats presents yet another CoV disease model and a dilemma for attempted control strategies. In this disease scenario, induction of neutralizing IgG antibodies to the FIPV S protein not only fails to prevent FIPV infections, but actually potentiates the immunopathogenesis of FIPV (Olsen, 1993).
The suspected zoonotic origin of SARS CoV (Guan et al., 2003) and the recognized propensity of several CoV to cross species barriers illustrate the need for additional animal studies of the mechanisms of interspecies transmission of CoVs and adaptation to new hosts. The possible animal reservoir for SARS remains undefined. At present we understand very little about CoVs or other viruses circulating in wildlife or their potential to emerge or recombine with existing CoVs (Stavrinides and Guttman, 2004) as public or animal health threats. Hopefully the SARS epidemic will generate new interest and funding for these fundamental research questions applicable not only to SARS CoV, but also to the estimated 75 percent of newly emerging human diseases arising as zoonoses (Taylor, 2001).
CORONAVIRUS RESEARCH: KEYS TO DIAGNOSIS, TREATMENT, AND PREVENTION OF SARS
Mark R. Denison, M.D.
Department of Pediatrics, Department of Microbiology & Immunology, Elizabeth B. Lamb Center for Pediatric Research, Vanderbilt University Medical Center, Nashville, TN
For coronavirus investigators, the recognition of a new coronavirus as the cause of severe acute respiratory syndrome (SARS) was certainly remarkable, yet perhaps not surprising (Baric et al., 1995). The cadre of investigators who have worked with this intriguing family of viruses over the past 30 years are familiar with many of the features of coronavirus biology, pathogenesis, and disease that
manifested so dramatically in the worldwide SARS epidemic. Advances in the biology of coronaviruses have resulted in greater understanding of their capacity for adaptation to new environments, transspecies infection, and emergence of new diseases. New tools of cell and molecular biology have led to increased understanding of intracellular replication and viral cell biology, and the advent in the past five years of reverse genetic approaches to study coronaviruses has made it possible to begin to define the determinants of viral replication, transpecies adaptation, and human disease. This summary will discuss the basic life cycle and replication of the well-studied coronavirus, mouse hepatitis virus (MHV), identifying the unique characteristics of coronavirus biology and highlighting critical points where research has made significant advances, and which might represent targets for antivirals or vaccines. Areas where rapid progress has been made in SCoV research will be described. Finally, areas of need for research in coronavirus replication, genetics, and pathogenesis will be summarized.
Coronavirus Life Cycle
The best studied model for coronavirus replication and pathogenesis has been the group 2 murine coronavirus, mouse hepatitis virus, and much of what is known of the stages of the coronavirus life cycle has been determined in animals and in culture using this virus. Thus this discussion will focus on MHV with comparisons to SCoV and other coronaviruses. This is appropriate because bioinformatics analyses suggest that SCoV, while a distinct virus, has significant similarities in organization, putative protein functions, and replication to the group II coronaviruses, particularly within the replicase gene (Snijder et al., 2003). Excellent, detailed reviews of MHV and coronavirus replication are available elsewhere (Holmes and Lai, 1996; Lai and Cavanagh, 1997).
The coronavirus virion is an enveloped particle containing the spike (S), membrane (M), and envelope (E) proteins. In addition, some strains of coronaviruses, but not SCoV, express a hemagglutinin protein (HE) that is also incorporated in the virion. The genome of coronaviruses is a linear, single-stranded RNA molecule of positive (mRNA) polarity, and from 28 to 32 kb in length (Bonilla et al., 1994; Drosten et al., 2003; Lee et al., 1991). Within the virion, the genome is encapsidated by multiple copies of the nucleocapsid protein (N), and has the conformation of a helical RNA/nucleocapsid structure. The S protein has been a focus of pathogenesis studies in mice because it appears to be the critical determinant of cell tropism, species specificity, host selection, cell tropism, and disease (Navas and Weiss, 2003; Navas et al., 2001; Rao and Gallagher, 1998).
Virus replication is initiated by binding of the S protein to specific receptors on the host cell surface. For MHV, the primary receptor has been shown to be the carcino-embryonic antigen–cell adhesion molecule (CEACAM) (Dveksler et al., 1991; 1996; Holmes and Lai, 1996; Yokomuri and Lai, 1992), and for the human
coronavirus, HCoV-229E, and other group 1 coronaviruses, the receptor is aminopeptidase N (Yeager et al., 1992). The precise mechanisms of entry and uncoating have yet to be defined, but likely occur by either fusion from without or viroplexis through endocytic vesicles. For wildtype MHV, entry and uncoating constitute a pH independent process that is probably direct fusion mediated by a fusion peptide in the S protein (Gallagher et al., 1991). The understanding of the region of the S1 component of coronavirus that binds to receptors was the basis for studies leading to the very recent and very rapid identification of angiotensin converting enzyme 2 (ACE 2) as a receptor for SCoV (Li et al., 2003).
The next discrete stage in the life cycle is translation and proteolytic processing of viral replicase proteins from the input genome RNA, followed by formation of cytoplasmic replication complexes in association with cellular membranes (Denison et al., 1999; Gosert et al., 2002; Shi et al., 1999; van der Meer et al., 1999). Replication complexes are thought to be sites of all stages of viral RNA transcription and replication, and possibly assembly of nascent viral nucleo-capsids. Viral assembly occurs both temporally and physically distinct from viral replication complexes in the endoplasmic-reticulum-Golgi-intermediate compartment (ERGIC), a transitional zone between late ER and Golgi (deVries et al., 1997; Klumperman et al., 1994; Krijnse-Locker et al., 1994; Rottier and Rose, 1987). Although the mechanisms by which replication products are delivered to sites of assembly remain to be determined, it has been shown that subpopulations of replicase proteins and the structural nucleocapsid (N) translocate from replication complexes to sites of assembly and may mediate the process in association with cellular membrane/protein trafficking pathways (Bost et al., 2000). Virus assembly in the ERGIC involves interactions of genome RNA, N, the membrane protein (M), and the small membrane protein (E), resulting in budding of virions into the lumen of ER/Golgi virosomes (Opstetten et al., 1995). Further maturation of virus particles occurs during movement through the Golgi, resulting in virosomes filled with mature particles (Salamuera et al., 1999). Trafficking of the virosomes to the cell surface has not been well characterized, but is presumed to occur via normal vesicle maturation and exocytic processes. The outcome is the nonlytic release of the vast majority of mature virions into the extracellular space. For MHV and several other coronaviruses that can directly fuse with cells, there is a characteristic and rapidly detectable cytopathic effect of cell-cell fusion into multinucleated syncytia. Production of infectious virus continues even after the majority of cells are fused. Syncytia were recently reported as a readout of SCoV receptor expression and cell infection (Li et al., 2003).
Viral Replication Complex Formation and Function
Following entry and uncoating, the 5′ most replicase gene of the input positive strand RNA genome is translated into two co-amino terminal replicase polyproteins that are co- and post-translationally processed by viral proteinases
to yield 15 to 16 mature replicase proteins, as well as intermediate precursors. The nascent replicase polyproteins and intermediate precursors likely mediate the formation of viral replication complexes in the host cell cytoplasm. Interestingly, coronavirus replication requires continuous replicase gene translation and processing throughout the life cycle to maintain productive infection (Kim et al., 1995; Perlman et al., 1987; Sawicki and Sawicki, 1986). Replication complexes of MHV are associated with double-membrane vesicles (Gosert et al., 2002), and all tested MHV replicase proteins have been shown to colocalize to replication complexes at the earliest time of detection, likely both by membrane integration and by protein-protein and protein-RNA interactions (Bost et al., 2000; Denison et al., 1999; Prentice and Denison, in press; Shi et al., 1999; Sims et al., 2000; van der Meer et al., 1999). Further, replicase proteins likely mediate the process of double-membrane vesicle formation, likely by induction of cellular autophagy pathways (E. Prentice, unpublished results).
Coronavirus replication complexes are sites for replicase gene translation and replicase polyprotein processing, and also for viral RNA synthesis. Replicase gene proteins likely mediate positive-strand, negative-strand, subgenomic, and genomic RNA synthesis, as well as processes of capping, polyadenylation, RNA unwinding, template switching during viral RNA synthesis, and discontinuous transcription and transcription attenuation. The coronavirus replicase polyproteins and mature replicase proteins represent the largest and most diverse repertoire of known and predicted distinct enzymatic functions of any positive-strand RNA virus family. Until recently, of the 15 or more mature replicase proteins, only the proteinase, RNA helicase, and RNA-dependent RNA polymerase activities had been predicted or experimentally confirmed (Brockway et al., 2003; Heusipp et al., 1997; Lee et al., 1991; Ziebuhr et al., 2000). With the advent of SARS, more extensive bioinformatics analyses have resulted in predictions of several additional functions involved in RNA processing, including methyltransferase and exonuclease activities (Snijder et al., 2003; Thiel et al., 2003). Even with inclusion of distant predicted relationships, up to eight of the replicase proteins remain without predicted or confirmed functions. In summary, it is likely that coronaviruses have exploited their genetic capacity to encode proteins in the replicase gene with distinct functions in RNA synthesis and processing, as well as proteins with specific roles in induction or modification in host cellular membrane biogenesis and trafficking, delivery of replication products to sites of assembly, and possibly virus assembly. Thus replicase translation, replicase polyprotein processing, and mature replicase proteins constitute important targets for interference with coronavirus replication, virus-cell interactions, or viral pathology.
Coronavirus Replicase Protein Expression and Processing
The proteinase activities for all coronaviruses include both papain-like proteinase (PLP) and picornavirus 3C-like proteinase activities that are encoded
within the replicase polyproteins and mediate both cis and trans cleavage events (Ziebuhr et al., 2000). Because of the parallel evolution of the proteinases, their cleavage sites, and the hierarchical cleavage processes, the proteolytic processing of the coronavirus replicase proteins may serve as distinct regulatory and genetic elements (Ziebuhr et al., 2001). Specifically, there are both conserved and divergent regions of the replicase polyproteins by amino acid identity and similarity, with the sequences and predicted mature proteins beginning with the 3C-like proteinases through the carboxy terminus of the replicase polyprotein retaining higher identity and similarity across the predicted proteins. In contrast, the amino-terminal third of the replicase demonstrates the most variation in proteins, cleavage site locations, and the number of proteinases that mediate maturation processing. SCoV appears to have the general organization of, and similar protein sizes to, the group 2 coronaviruses such as MHV in this part of the genome (Snijder et al., 2003). However, SCoV likely uses only one PLP to mediate the cleavages, similar to the group 3 coronavirus infectious bronchitis virus (IBV). Thus this region of the replicase may experience the most variability, suggesting either the encoding of accessory functions that are flexible and tolerant of changes, or conversely group or host-specific roles that are subject to pressure for more rapid change.
Expression of Structural and Accessory Genes
Only the 5' most replicase gene is translated from the input positive-strand genome RNA. The genome contains multiple other genes for the known structural proteins S, E, M, and N, as well as other genes for expression of proteins that have been labeled as “nonstructural” or “accessory” because they have been presumed to not be required for replication, and are not thought to be incorporated into virions. MHV encodes six of these genes, while SCoV encodes possibly up to 11 structural and accessory genes, which are expressed from subgenomic mRNAs (Snijder et al., 2003). Subgenomic RNA transcription occurs during minus-strand RNA synthesis by acquisition of the antileader RNA sequences from the 5′ end of the genome via homology to a transcriptional regulatory sequence (TRS, also known as an intergenic sequence), and requiring a discontinuous activity of the nascent minus-strand template and polymerase complex to acquire the leader (Sawicki and Sawicki, 1998). The outcome of transcription is the generation of a “nested set” of subgenomic negative-strand RNAs that all contain the antileader sequences that serve as templates for similar size subgenomic mRNAs. This transcriptional strategy exposes different genes as the 5′ ORF in different mRNAs, all of which also contain the 3′ sequence downstream of the gene, including the 3′ nontranslated region of the genome.
For MHV, genes 3, 5b, 6, and 7 encode S, E, M, and N, respectively. Genes 2, 4, and 5a are not required for replication in culture, and have been mutated to block expression, deleted, or substituted with noncoronavirus genes such as GFP (de Haan et al., 2002; Ortego et al., 2003; Sarma et al., 2002). Because all
coronaviruses retain these genes in various combinations in the face of presumed pressure for genetic economy and apparent lack of functions in RNA synthesis, it is presumed that these genes serve roles in modification of host cells, pathogenesis, or interactions with the immune system. SCoV encodes a larger and more complex array of these genes than MHV or other coronaviruses, which may reflect its evolution in its original animal host (Ksiazek et al., 2003; Marra et al., 2003; Snijder et al., 2003; Thiel et al., 2003). In addition, the report of a deletion within one of the accessory genes in human isolates of SCoV suggests that this may be a gene involved in host range or adaptation for replication and transmission in humans (Guan et al., 2003).
Coronavirus Genetics
Until recently, the genetics of coronavirus replication and pathogenesis have largely been studied using natural variants, host range mutants, passaged virus, and mutagenized viruses selected for temperature sensitivity and specific phenotypes. Classical complementation of functions made it possible to define at least eight genetic groups for MHV, with most of the complementation groups localized to the replicase gene (Stalcup et al., 1998). Taking advantage of naturally high rates of homologous RNA-RNA recombination and of host range determinants in the S protein, the development of targeted recombination has allowed more defined and detailed studies of the accessory and structural genes of MHV, transmissible gastroenteritis virus (TGEV), and feline infectious peritonitis virus (FIPV) (Haijema et al., 2003; Kuo et al., 2000; Masters et al., 1994). Studies with natural variants and targeted recombination genetic studies have demonstrated that the S protein is the major determinant of host range, tropism, and pathogenesis; other genetic elements, possibly in the replicase, may influence these characteristics of different coronaviruses (Navas and Weiss, 2003). The capacity of coronaviruses to change host range, transmission, pathogenesis, and disease has been established in the laboratory using cell adaptation and virus passage (Baric et al., 1997, 1999; Chen and Baric, 1995, 1996), and has been demonstrated in nature by natural variants of MHV, TGEV, and bovine coronavirus (BCoV), as well as by studies using heterologous viruses such as canine coronavirus (CcoV) to immunize cats against FIPV (Enjuanes et al., 1995; Tresnan et al., 1996). Further, targeted recombination studies have confirmed the genetic flexibility of the coronavirus genome and the ability of coronaviruses to recover wild-type replication following deletions, mutations, substitutions, and gene order rearrangements in the structural and accessory genes (de Haan et al., 2002).
Challenges for genetic studies using natural variants and mutants, particularly in defining the precise changes responsible for altered phenotypes, has limited progress in genetic studies. Targeted recombination, while a robust system with powerful selection, has been limited to studies of the 3′ 10 kb of the MHV genome, and is limited to selection of viable recombinants. Recently, the estab-
lishment of “infectious clone” reverse genetic strategies for the coronaviruses TGEV (Transmissible Gastroenteritis Coronavirus), HCoV-229E, IBV, and MHV has made it theoretically possible to study the genetics of the entire genome and all of the structural, accessory, and replicase genes. Approaches to “infectious cloning” have included full-length cDNA clones of TGEV genome in bacterial artificial chromosomes (Gonzalez et al., 2001), recombinant vaccinia viruses containing full-length cDNA clones of HCoV-229E and IBV genomes (Casais et al., 2001; Thiel et al., 2001), and in vitro assembly strategies for TGEV, MHV, and most recently, SCoV (Yount, 2000; Yount et al., 2002, 2003).
The in vitro assembly approach was developed to overcome the challenge of full-length cDNA cloning of the TGEV and MHV genomes, which contained “toxic” regions in the replicase gene, resulting in unstable or toxic clones in E. coli (Yount, 2000; Yount et al., 2002). Subcloning of the regions required splitting the toxic domains into separate clones. The result of this strategy was the cloning of the MHV genome into seven fragments (A through G). To recover viable virus, the following strategy is pursued: (1) cloned cDNA fragments are excised from plasmid using class 2 restriction enzymes that remove the recognition site and leave overhanging genomic sequence; (2) excised fragments are ligated (assembled) in vitro; (3) transcription of full-length genomic RNA is driven in vitro using a T7 promoter on the 5′ fragment A; (4) full-length genome RNA is electroporated into competent cells that are then plated on a monolayer of naturally permissive cells; and (5) cells are monitored for cytopathic effect or plaques, and virus is recovered from plaques or media supernatant.
In vitro assembly has many advantages for genetic studies of such a large and complex genome RNA. First, genetic changes can be introduced and confirmed in stable small fragments without the need for a ~30kb genomic clone. Second, the cloned fragments make it possible to develop libraries of mutations that can rapidly be tested in different combinations. Furthermore, identification of putative second-site reversion mutations for deleterious introduced changes can be introduced with the original mutation to confirm their reversion potential. In combination with biochemical and cell imaging approaches, it also is possible to study highly defective or lethal mutations in electroporated cells, in order to define critical determinants of replication. The in vitro assembly approach has been used to introduce marker mutations that are silent for replication in culture (Yount et al., 2003). In addition, we have engineered mutations in the MHV replicase gene to define the requirements for polyprotein processing and to determine the role of specific replicase proteins in replication in culture and in pathogenesis in animals. Using this approach we have recovered viruses with mutations at polyprotein cleavage sites and proteinase catalytic residues, all of which have distinct phenotypes in protein processing, viral growth, and viral RNA synthesis (unpublished results). Thus, direct reverse genetic studies of the critical replicase gene functions can be performed using in vitro assembly of infectious clones.
Advances in SCoV Research
The rapid progress in the identification and characterization of SCoV as the etiologic agent of SARS was made possible by the fact that the virus grows well in culture, and by the foundational research in coronaviruses that has been supported by the National Institutes of Health, the Multiple Sclerosis Foundation, the U.S. Department of Agriculture, and other organizations over the past two decades. The application of knowledge concerning virus structure, genetics, receptor binding, virus entry, and viral pathogenesis has made it possible to target the spike protein for studies of SCoV replication, pathogenesis, and immune response (Xiao et al., 2003). The remarkably rapid identification of ACE 2 as a receptor for SARS has demonstrated the foundational importance of studies of other coronaviruses (Li et al., 2003). Similarly, understanding of replicase gene expression, processing, and predicted functions has identified possible targets for structure/function studies and possible therapeutic intervention. The studies of coronavirus proteinase activities, cleavage site, and structures were the basis for studies leading to the rapid determination of SCoV replicase polyprotein cleavage sites and 3CLpro crystal structure (Anand et al., 2003; Campanacci et al., 2003; Snijder et al., 2003; Thiel et al., 2003).
Application of Reverse Genetics to Studies of SCoV
Because of the potential for reemergence of SARS, it is important to move forward with research in diagnostics, vaccines, and therapeutics for SCoV. Experience with the development and use of reverse genetics to study other coronaviruses resulted in establishment of reverse genetics for SCoV within months of the onset of the worldwide epidemic (Yount et al., 2003). How should the understanding of other coronaviruses, the rapid advances in research with SCoV, and the development of reverse genetics for SCoV be harnesssed to achieve these goals and attack these critical questions in SCoV replication, pathogenesis, and disease? Certainly, the use of SCoV reverse genetics, along with robust tissue culture systems and emerging animal models, creates the potential to rapidly answer questions concerning: (1) determinants of virus growth in culture; (2) potential mechanisms of transpecies adaptation; (3) sensitivity to and escape from biochemical and immune interference with replication; (4) determinants of virulence and pathogenesis; (5) mechanisms of genome recombination and mutation; (6) functions of and requirements for replicase, structural, and accessory proteins; and (7) development of stably attenuated viruses for use as seed stocks for inactivated vaccine or testing as live-attenuated vaccines.
How then should these critical issues be investigated while recognizing the potential of SCoV to cause severe disease, as well as the potential for rapid spread? First, there is significant experience with other coronaviruses in attenuation of virus replication and pathogenesis, both using virus passage and by direct engineering of changes. Although coronavirus genome organization, proteins, and replication appear more tolerant of changes then previously thought, all changes
of gene order, gene deletion, insertion, or mutagenesis so far reported have led to viruses impaired in replication, pathogenesis, or both. Many of the attenuating changes in MHV and other coronaviruses are conserved in SCoV and thus could be tested for likely attenuation in SCoV culture and animal models. Second, where there is clear conservation of sequences, motifs, proteins, or putative functions between SCoV and model viruses such as MHV, new or untested changes might be most rapidly analyzed under BSL2 conditions in those model viruses, and then directly applied to SARS once their phenotypes are determined. Third, all work with SCoV will be performed only under BSL3 conditions. This would also apply to chimeric viruses, whether engineered by introduction into the SCoV background, or by introducing SCoV proteins or sequences with known or predicted pathogenic consequences into other coronavirus backgrounds. Finally, it is important to develop strains of SCoV that are attenuated and stabilized against reversion and recombination, to be used as the basis for studies of other replication and pathogenesis determinants and construction of virus chimeras. Such attenuated variants would provide additional safeguards while allowing application of powerful genetic tools to the study of SCoV emergence, biology, disease, treatment, and prevention. Overall, newly invigorated programs in other human and animal coronaviruses, combined with the new research in SCoV, will shed important new light on this important virus family and perhaps lead to better understanding of the potential for resurgence of SCoV or the emergence of other coronaviruses into human populations.
ISOLATION AND CHARACTERIZATION OF VIRUSES RELATED TO THE SARS CORONAVIRUS FROM ANIMALS IN SOUTHERN CHINA
Y. Guan,1 B. J. Zheng,1 Y. Q. He,2 X. L. Liu,2 Z. X. Zhuang,2 C. L. Cheung,1 S. W. Luo,1 P. H. Li,1 L. J. Zhang,1 Y. J. Guan,1 K. M. Butt,1 K. L. Wong,1 K. W. Chan,3 W. Lim,4 K. F. Shortridge,1 K. Y. Yuen,1 J. S. M. Peiris,1 and L. L. M. Poon1,5
Reprinted with permission from Guan et al., 2003. Copyright 2003 AAAS.
A novel coronavirus (SCoV) is the etiological agent of severe acute respiratory syndrome (SARS). SCoV-like viruses were isolated from Himalayan palm civets
found in a live-animal market in Guangdong, China. Evidence of virus infection was also detected in other animals (including a raccoon dog, Nyctereutes procyonoides) and in humans working at the same market. All the animal isolates retain a 29-nucleotide sequence that is not found in most human isolates. The detection of SCoV-like viruses in small, live wild mammals in a retail market indicates a route of interspecies transmission, although the natural reservoir is not known.
Severe acute respiratory syndrome (SARS) recently emerged as a human disease associated with pneumonia (WHO, 2003c). This disease was first recognized in Guangdong Province, China, in November 2002. Subsequent to its introduction to Hong Kong in mid-February 2003, the virus spread to more than 30 countries and caused disease in more than 7900 patients across five continents (WHO, 2003d). A novel coronavirus (SCoV) was identified as the etiological agent of SARS (Ksiazek et al., 2003; Peiris et al., 2003a), and the virus causes a similar disease in cynomolgous macaques (Fouchier et al., 2003). Human SCoV appears to be an animal virus that crossed to humans relatively recently. Thus, identifying animals carrying the virus is of major scientific interest and public health importance. This prompted us to examine a range of domestic and wild mammals in Guangdong Province.
Because the early cases of SARS in Guangdong reportedly occurred in restaurant workers handling wild mammals as exotic food (Zhong et al., 2003), our attention focused on wild animals recently captured and marketed for culinary purposes. We investigated a live-animal retail market in Shenzhen. Animals were held, one per cage, in small wire cages. The animals sampled included seven wild, and one domestic, animal species (see Table 3-2). They originated from different regions of southern China and had been kept in separate storehouses before arrival to the market. The animals remained in the markets for a variable period of time, and each stall holder had only a few animals of a given species. Animals from different stalls within the market were sampled. Nasal and fecal samples were collected with swabs and stored in medium 199 with bovine serum albumin and antibiotics. Where possible, blood samples were collected for serology. Before sampling, all animals were examined by a veterinary surgeon and confirmed to be free of overt disease. Serum samples were also obtained, after informed consent, from traders in animals (n = 35) and vegetables (n = 20) within the market. Sera (n = 60) submitted for routine laboratory tests from patients
|
|
ogy, The University of Hong Kong, for the excellent technical assistance. We also thank C.C. Hon and F.C. Leung from the Department of Zoology, The University of Hong Kong, and Richard Webby from St. Jude Children’s Research Hospital (Memphis, TN) for assistance in the phylogenetic analysis. We thank K.V. Holmes’ laboratory from the Department of Microbiology, University of Colorado Health Sciences Center (Denver, CO) for validating the animal viral sequences. Supported by research funding from Public Health Research (Grant A195357), the U.S. National Institute of Allergy and Infectious Diseases, the Wellcome Trust (067072/D/02/Z), and SARS research funds from The University of Hong Kong. |
TABLE 3-2 Animal Species Tested for Coronavirus Detection
hospitalized for nonrespiratory disease in Guangdong were made anonymous and used for comparison.
Nasal and fecal swabs from 25 animals were tested for SCoV viral nucleic acid by using reverse transcription–polymerase chain reaction (RT-PCR) for the N gene of the human SCoV. Swabs from four of six Himalayan palm civets were positive in the RT-PCR assay (see Table 3-2). All specimens were inoculated into FRhK-4 cells as previously described for virus isolation (Peiris et al., 2003a). A cytopathic effect was observed in cells inoculated with specimens from four Himalayan palm civets (Paguma larvata), two of which also positive for coronavirus in the original specimen by RT-PCR. A virus was also detected by virus isolation
and direct RT-PCR from the fecal swab of a raccoon dog (Nyctereutes procyonoides). No virus was detectable in six other species sampled. Electron microscopy of one infected cell supernatant (SZ16) showed viral particles with a morphology compatible with coronavirus (see Figure S-1)6. Sera from five animals had neutralizing antibody to the animal coronavirus; these were from three palm civets, a raccoon dog, and a Chinese ferret badger, respectively (see Table 3-2).
To further validate the results from the neutralization test, a Western blot assay was used to detect SCoV-specific antibodies from these animal serum samples (see Figure 3-1). Indications of positive antibodies were observed from samples SZ2, SZ3, SZ11, and SZ17 (which were also positive in the neutralization assay) and from the positive control human serum. No positive signal was observed from those serum samples that were negative in the neutralization test. There was insufficient serum left over from the raccoon dog (SZ13) to be analyzed by this assay.
Sera from humans working in the market were tested for antibody to SZ16 virus by neutralization and indirect immunofluorescence assays. Although 8 out of 20 (40 percent) of the wild-animal traders and 3 of 15 (20 percent) of those who slaughter these animals had evidence of antibody, only 1 (5 percent) of 20 vegetable traders was seropositive. None of these workers reported SARS-like symptoms in the past 6 months. In comparison, none of 60 control sera from patients admitted to a Guangdong hospital for nonrespiratory diseases was seropositive (see Table 3-3).
Two of the virus isolates (SZ3 and SZ16) isolated from the nasal swabs of palm civets were completely sequenced, and the amino acid sequence was deduced. Two other viruses were partially sequenced, from the S gene to the 3' end of the virus (GenBank accession numbers AY304486 to AY304489). Viral RNA sequences from these original swab samples from animal were confirmed in an independent laboratory (Holmes K., unpublished observations). The full-length genome sequences had 99.8 percent homology to the human SCoV, which indicates that the human and animal SCoV-like viruses were closely related. Phylogenetic analysis of the S gene of both human and animal SCoV-like viruses indicated that the animal viruses are separate from the human virus cluster (see Figure 3-2 and Figure S-2)7. However, the viruses SZ1, SZ3, and SZ16 from palm civets were phylogenetically distinct. The viruses SZ3 and SZ16 had 18 nucleotide differences between them over the 29,709–base pair (bp) genome, whereas the human SCoV isolated from five geographically separate sites (GZ50, CUHK-W1,
|
6 |
Supporting online material, www.sciencemag.org/cgi/content/full/1087139/DC1, Materials and Methods, Figures S1 and S2, and References and Notes. |
|
7 |
Supporting online material, www.sciencemag.org/cgi/content/full/1087139/DC1, Materials and Methods, Figures S1 and S2, and References and Notes. |
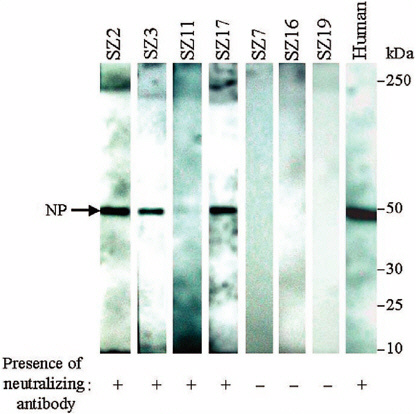
FIGURE 3-1 Detection of antibodies against recombinant nucleocapsid protein of SCoV in animal sera by Western blot assay. Recombinant nuleocapsid protein (NP, 49.6 kD) was used as an antigen to detect anti-ScoV antibodies in animal sera. Protein A-HRP was used as a secondary antibody, and reactive bands were visualized by the enhanced chemiluminesence Western blotting system. A serum sample from a convalescent SARS patient was used as a positive control. Blots reacted with animal (SZ2, SZ3, SZ11, SZ17, SZ7, SZ16, or SZ19) or human sera are indicated. Results from the neutralization test for ScoV-specific antibodies in these serum samples are also shown.
TABLE 3-3 Prevalence of Antibody to Animal SCoV SZ16 in Humans. Controls Are Serum Specimens from Patients Hospitalized for Nonrespiratory Diseases in Guangdong Made Anonymous
|
Occupation |
Sample numbers |
Antibody positive (%) |
|
Wild-animal trader |
20 |
8 (40) |
|
Slaughterer of animals |
15 |
3 (20) |
|
Vegetable trader |
20 |
1 (5) |
|
Control |
60 |
0 (0) |
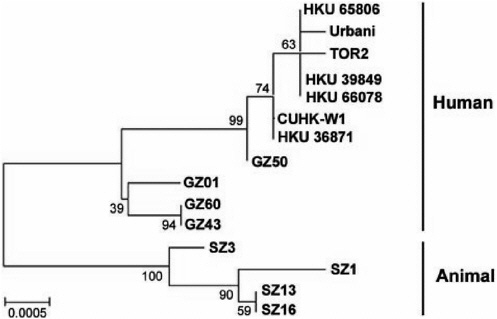
FIGURE 3-2 Phylogenetic analysis of the nucleotide acid sequence of the spike gene of SCoV-like viruses. Nucleotide sequences of representative SCoV Sgenes (Sgene coding region 21477 to 25244, 3768 bp) were analyzed. The phylogenetic tree was constructed by the neighbor-joining method with bootstrap analysis (1000 replicates) using MEGA 2 (Kumar et al., 2001). Number at the nodes indicates bootstrap values in percentage. The scale bar shows genetic distance estimated using Kimura’s two-parameter substitution model (Kimura, 1980). In addition to viruses sequenced in the present study, the other sequences used in the analysis could be found in GenBank with accession number: from AY304490 to AY304495, AY278741, AY278554, AY278491, AY274119, and AY278489.
Tor-2, HKU-39848, and Urbani) differed by only 14 nucleotides (nt). Nevertheless, animal virus SZ13 (raccoon dog) and SZ16 (palm civet) were genetically almost identical, and transmission or contamination from one host to the other within the market cannot be excluded.
When the full genome of the animal (n = 2) and human (n = 5, see above) virus groups were compared, the most striking difference was that these human viruses have a 29-nt deletion (5'-CCTACTGGTTACCAACCTGAATGGAATAT-3', residue 27869 to 27897) that is 246 nt upstream of the start codon of the N gene (see Figure 3-3). Of human SCoV sequences currently available in GenBank, there was only one (GZ01) with this additional 29-nt sequence. In addition to that, there were 43 to 57 nucleotide differences observed over the rest of the genome. Most of these differences were found in the S gene coding region. The existence of the additional 29-nt sequence in the animal viruses results in demolishing the open reading frames (ORFs) 10 and 11 (Marra et al., 2003) and merging these two ORFs into a new ORF encoding a putative protein of 122
amino acids (see Figure 3-3). This putative peptide has a high homology to the putative proteins encoded by ORF10 and ORF11. Because ORF11 does not have a typical transcription regulatory sequence for SCoV (Marra et al., 2003), the putative ORF11 reported by others may just be the direct result of the deletion of the 29-nt sequence. BLAST search of this peptide yields no significant match to any other known peptide. Further investigation is required to elucidate the biological significance of this finding.
When the S-gene sequences of the four animal viruses were compared with 11 human SCoV viruses, 38 nucleotide polymorphisms were noted, and 26 of them were nonsynonymous changes (see Table 3-4). The S genes among the four
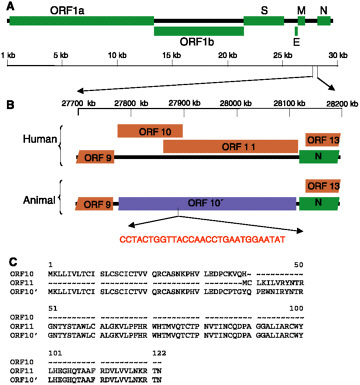
FIGURE 3-3 A 29-nt deletion in the human SCoV genome. (A) Genetic organization of SCoV-like viruses found in humans and animals. ORFs 1a and 1b, encoding the nonstructural polyproteins, and those encoding the S, E, M, and N structural proteins are indicated (green boxes). (B) Expanded view of the SCoV genomic sequence (27700 nt to 28200 nt, based on AY278554 numbering). ORFs for putative proteins and for N in human isolates are indicated as brown and green boxes, respectively (Marra et al., 2003). An extra 29-nt sequence is present downstream of the nucleotide of 27868 of the animal SCoV (based on AY278554 numbering). The presence of this 29-nt sequence in animals isolates results in fusing the ORFs 10 and 11 (top) into a new ORF (bottom; ORF10', light blue box). (C) Protein sequence alignment of ORF10 and 11 from human isolates and ORF 10' from animal isolates.
TABLE 3-4 Nucleotide Sequence Variation of the S Gene of Animal and Human SCoV
animal viruses had eight nucleotide differences, whereas there were 20 nucleotide differences among 11 human viruses. Thus, the animal viruses, although isolated from one market, are no less divergent than the human viruses isolated from Hong Kong, Guangdong, Canada, and Vietnam. However, whereas 14 (70 percent) of the 20 polymorphisms among the human viruses were nonsynonymous mutations, only two (25 percent) of the eight nucleotide substitutions within the animal viruses were. An amino acid deletion (nucleotide positions 21690 to 21692) was observed in two of the human viruses (GZ43 and GZ60). Of the 38 polymorphisms, there were 11 consistent nucleotide signatures that appeared to distinguish animal and human viruses. The observation that the human and animal viruses are phylogenetically distinct (see Figure 3-2) makes it highly unlikely that the SCoV-like viruses isolated in these wild animals is due to the transmission of SCoV from human to animals.
Our findings suggest that the markets provide a venue for the animal SCoV-like viruses to amplify and to be transmitted to new hosts, including humans, and this is critically important from the point of view of public health. However, it is not clear whether any one or more of these animals are the natural reservoir in the wild. It is conceivable that civets, raccoon dog, and ferret badgers were all infected from another, as yet unknown, animal source, which is in fact the true reservoir in nature. However, because of the culinary practices of southern China, these market animals may be intermediate hosts that increase the opportunity for transmission of infection to humans. Further extensive surveillance on animals will help to better understand the animal reservoir in nature and the interspecies transmission events that led to the origin of the SARS outbreak.
REFERENCES
Anand K, Ziebuhr J, Wadhwani P, Mesters JR, Hilgenfeld R. 2003. Coronavirus main proteinase (3CLpro) structure: basis for design of anti-SARS drugs. Science 300(5626):1763-7.
Anonymous. 2003. Severe acute respiratory syndrome (SARS). Weekly Epidemiological Record78:81-3.
Ballesteros ML, Sanchez CM, Enjuanes L. 1997. Two amino acid changes at the N-terminus of transmissible gastroenteritis coronavirus spike protein result in the loss of enteric tropism. Virology 227(2):378-88.
Baric RS, Fu K, Chen W, Yount B. 1995. High recombination and mutation rates in mouse hepatitis virus suggest that coronaviruses may be potentially important emerging viruses. Advances in Experimental Medicine and Biology 380:571-6.
Baric RS, Sullivan E, Hensley L, Yount B, Chen W. 1999. Persistent infection promotes cross-species transmissibility of mouse hepatitis virus. Journal of Virology 73(1):638-49.
Baric RS, Yount B, Hensley L, Peel SA, Chen W. 1997. Episodic evolution mediates interspecies transfer of a murine coronavirus. Journal of Virology 71(3):1946-55.
Bonilla PJ, Gorbalenya AE, Weiss SR. 1994. Mouse hepatitis virus strain A59 RNA polymerase gene ORF 1a: heterogeneity among MHV strains. Virology 198(2):736-40.
Bost AG, Carnahan RH, Lu XT, Denison MR. 2000. Four proteins processed from the replicase gene polyprotein of mouse hepatitis virus colocalize in the cell periphery and adjacent to sites of virion assembly. Journal of Virology 74(7):3379-87.
Bost AG, Prentice E, Denison MR. 2001. Mouse hepatitis virus replicase protein complexes are translocated to sites of M protein accumulation in the ERGIC at late times of infection. Virology 285(1):21-9.
Brian DA, Hogue BG, Kienzle TE. 1995. The Coronavirus Hemagluttinin Esterase Clycoprotein. In: Siddell SG, ed., The Coronaviridae. New York: Plenum Press. Pp. 165-79.
Brockway SM, Clay CT, Lu XT, Denison MR. 2003. Characterization of the expression, intracellular localization, and replication complex association of the putative mouse hepatitis virus RNA-dependent RNA polymerase. Journal of Virology 77(19):10515-27.
Callow KA, Parry HF, Sergeant M, Tyrrell DA. 1990. The time course of the immune response to experimental coronavirus infection of man. Epidemiology and Infection 105:435-46.
Campanacci V, Egloff MP, Longhi S, Ferron F, Rancurel C, Salomoni A, Durousseau C, Tocque F, Bremond N, Dobbe JC, Snijder EJ, Canard B, Cambillau C. 2003. Structural genomics of the SARS coronavirus: cloning, expression, crystallization and preliminary crystallographic study of the Nsp9 protein. Acta Crystallographica. Section D, Biological Crystallography 59(Pt 9):1628-31.
Casais R, Thiel V, Siddell SG, Cavanagh D, Britton P. 2001. Reverse genetics system for the avian coronavirus infectious bronchitis virus. Journal of Virology 75(24):12359-69.
CDC. 2003. Update: outbreak of severe acute respiratory syndrome-Worldwide, 2003. MMWR52:241-8.
Chen W, Baric RS. 1995. Evolution and persistence mechanisms of mouse hepatitis virus. Advances in Experimental Medicine & Biology 380:63-71.
Chen W, Baric RS. 1996. Molecular anatomy of mouse hepatitis virus persistence: coevolution of increased host cell resistance and virus virulence. Journal of Virology 70(6):3947-60.
Chim SS, Tsui SK, Chan KC, Au TC, Hung EC, Tong YK, Chiu RW, Ng EK, Chan PK, Chu CM, Sung JJ, Tam JS, Fung KP, Waye MM, Lee CY, Yuen KY, Lo YM. 2003. Genomic characterisation of the severe acute respiratory syndrome coronavirus of Amoy Gardens outbreak in Hong Kong. Lancet 362(9398):1807-8.
Cho KO, Hasoksuz M, Nielsen PR, Chang KO, Lathrop S, Saif LJ. 2001. Cross-protection studies between respiratory and calf diarrhea and winter dysentery coronavirus strains in calves and Rt-Pcr and nested Pcr for their detection. Archives of Virology 146(12):2401-19.
Cho KO, Hoet AE, Loerch SC, Wittum TE, Saif LJ. 2001. Evaluation of concurrent shedding of bovine coronavirus via the respiratory tract and enteric route in feedlot cattle. American Journal of Veterinary Research 62(9):1436-41.
Chouljenko VN, Lin XQ, Storz J, Kousoulas KG, Gorbalenya AE. 2001. Comparison of genomic and predicted amino acid sequences of respiratory and enteric bovine coronaviruses isolated from the same animal with fatal shipping pneumonia. Journal of General Virology 82(12):2927-2933.
Clark MA. 1993. Bovine coronavirus. British Veterinary Journal. 149(1):51-70.
Cook J, Mockett APA. 1995. Epidemiology of infectious bronchitis virus. In: Siddell SG, ed., The Coronaviridae. New York: Plenum Press. Pp. 317-35.
Costantini V, Lewis P, Alsop J, Templeton C, Saif LJ. In press. Respiratory and enteric shedding of porcine respiratory coronavirus (PRCV) in sentinel weaned pigs and sequence of the partial S gene of the PRCV isolates. Archives of Virology.
Cox E, Hooyberghs J, Pensaert MB. 1990. Sites of replication of a porcine respiratory coronavirus related to transmissible gastroenteritis virus. Research in Veterinary Science 48(2):165-9.
deGroot RJ, Horzinek MC. 1995. Feline infectious peritonitis. In: Siddell SG, ed., The Coronaviridae. New York: Plenum Press. Pp. 293-315.
de Haan CA, Masters PS, Shen X, Weiss S, Rottier PJ. 2002. The group-specific murine coronavirus genes are not essential, but their deletion, by reverse genetics, is attenuating in the natural host. Virology 296(1):177-89.
de Jong JC, Claas EC, Osterhaus AD, Webster RG, Lim WL. 1997. A pandemic warning? Nature 389:544.
de Vries AAF, Horzinek MC, Rottier PJM, de Groot RJ. 1997. The genome organization of the nidovirales: similarities and differences between arteri-, toro-, and coronaviruses. Seminars in Virology 8(1):33-47.
Denison MR, Spaan WJ, van der Meer Y, Gibson CA, Sims AC, Prentice E, Lu XT. 1999. The putative helicase of the coronavirus mouse hepatitis virus is processed from the replicase gene polyprotein and localizes in complexes that are active in viral RNA synthesis. Journal of Virology 73(8):6862-71.
Domingo E, Holland JJ. 1997. RNA virus mutations and fitness for survival. Annual Review of Microbiology 51:151-78.
Drosten C, Gunther S, Preiser W, van der Werf S, Brodt HR, Becker S, Rabenau H, Panning M, Kolesnikova L, Fouchier RA, Berger A, Burguiere AM, Cinatl J, Eickmann M, Escriou N, Grywna K, Kramme S, Manuguerra JC, Muller S, Rickerts V, Sturmer M, Vieth S, Klenk HD, Osterhaus AD, Schmitz H, Doerr HW. 2003. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. New England Journal of Medicine 348(20):1967-76.
Duarte M, Tobler K, Bridgen A, Rasschaert D, Ackermann M, Laude H. 1994. Sequence analysis of the porcine epidemic diarrhea virus genome between the nucleocapsid and spike protein genes reveals a polymorphic ORF. Virology 198(1):466-476.
Dveksler GS, Gagneten SE, Scanga CA, Cardellichio CB, Holmes KV. 1996. Expression of the recombinant anchorless N-terminal domain of mouse hepatitis virus (MHV) receptor makes hamster of human cells susceptible to MHV infection. Journal of Virology 70(6):4142-5.
Dveksler GS, Pensiero MN, Cardellichio CB, Williams RK, Jiang GS, Holmes KV, Dieffenbach CW. 1991. Cloning of the mouse hepatitis virus (MHV) receptor: expression in human and hamster cell lines confers susceptibility to MHV. Journal of Virology 65(12):6881-91.
El-Kanawati ZR, Tsunemitsu H, Smith DR, Saif LJ. 1996. Infection and cross-protection studies of winter dysentery and calf diarrhea bovine coronavirus strains in colostrum-deprived and gnotobiotic calves. American Journal of Veterinary Research 57(1):48-53.
Enjuanes L, Smerdou C, Castilla J, Anton IM, Torres JM, Sola I, Golvano J, Sanchez JM, Pintado B. 1995. Development of protection against coronavirus induced diseases: a review. Advances in Experimental Medicine and Biology 380:197-211.
Erles K, Toomey C, Brooks HW, Brownlie J. 2003. Detection of a group 2 coronavirus in dogs with canine infectious respiratory disease. Virology 310(2):216-23.
Fouchier RA, Kuiken T, Schutten M, van Amerongen G, van Doornum GJ, van den Hoogen BG, Peiris M, Lim W, Stohr K, Osterhaus AD. 2003. Aetiology: Koch’s Postulates Fulfilled for Sars Virus. Nature 423(6937):240.
Gallagher TM, Escarmis C, Buchmeier MJ. 1991. Alteration of the pH dependence of coronavirus-induced cell fusion: effect of mutations in the spike glycoprotein. Journal of Virology 65(4):1916-28.
Gonzalez JM, Almazan F, Penzes Z, Calvo E, Enjuanes L. 2001. Cloning of a transmissible gastroenteritis coronavirus full-length cDNA. Advances in Experimental Medicine and Biology 494:533-6.
Gosert R, Kanjanahaluethai A, Egger D, Bienz K, Baker SC. 2002. RNA replication of mouse hepatitis virus takes place at double-membrane vesicles. Journal of Virology 76(8):3697-708.
Guan Y, Peiris JM, Lipatov AS, Ellis TM, Dyrting KC, Krauss S, Zhang LJ, Webster RG, Shortridge KF. 2002. Emergence of multiple genotypes of H5N1 avian influenza viruses in Hong Kong SAR. Proceedings of the National Academy of Sciences 99:8950-5.
Guan Y, Zheng BJ, He YQ, Liu XL, Zhuang ZX, Cheung CLLSW, Li PH, Zhang LJ, Guan YJ, Butt KM, Wong KLCKW, Lim W, Shortridge KF, Yuen KY, Peiris JSM, Poon LLM. 2003. Isolation and characterization of viruses related to the SARS coronavirus from animals in southern China. Science 302(5643):276-8.
Guangdong Public Health Office. January 21, 2003. Summary report of investigating an atypical pneumonia outbreak in Zhongshan, Document No 2.
Haijema BJ, Volders H, Rottier PJM. 2003. Switching species tropism: an effective way to manipulate the feline coronavirus genome. Journal of Virology 77(8):4528-38.
Hayes JR. 2000. Evaluation of dual infection of nursery pigs with U.S. strains of porcine reproductive and respiratory syndrome virus and porcine respiratory coronavirus. Master’s Thesis, Food Animal Health Research Program, OARDC/The Ohio State University.
Herrewegh AA, Mahler M, Hedrich HJ, Haagmans BL, Egberink HF, Horzinek MC, Rottier PJ, de Groot RJ. 1997. Persistence and evolution of feline coronavirus in a closed cat-breeding colony. Virology 234(2):349-63.
Heusipp G, Harms U, Siddell SG, Ziebuhr J. 1997. Identification of an ATPase activity associated with a 71-kilodalton polypeptide encoded in gene 1 of the human coronavirus 229E. Journal of Virology 71(7):5631-4.
Hoffmann C, Kamps BS. 2003. Clinical presentation and diagnosis. In: Kamps BS, Hoffmann C, eds., SARS Reference. 3rd ed. Pp. 124-43. [Online] Available: http://www.SARSreference.com.
Hoffmann M and Wyler R. 1988. Propagation of the virus of porcine epidemic diarrhea in cell culture. Journal of Clinical Microbiology (26):2235-9.
Holland JJ, de la Torre JC, Clarke DK, Duarte E. 1991. Quantitation of relative fitness and great adaptability of clonal populations of RNA viruses. Journal of Virology 65(6):1960-2967.
Holmes KV. 2001. Coronaviruses. In: Knipe DM, Howley PM, eds. Field Virology, 4th ed. Philadelphia: Lippincott Williams and Wilkins. Pp. 1187-203.
Holmes KV. 2003. SARS coronavirus: a new challenge for prevention and therapy. Journal of Clinical Investigation 111(11):1605-9.
Holmes KV, Lai MMC. 1996. Coronaviridae: the viruses and their replication. In: Fields BN, Knipe DM, Howley PM, eds., Virology. Vol. 1. 3rd ed. Philadelphia: Lippincott-Raven. Pp. 1075-93.
Ismail MM, Cho KO, Hasoksuz M, Saif LJ, Saif YM. 2001. Antigenic and genomic relatedness of turkey-origin coronaviruses, bovine coronaviruses, and infectious bronchitis virus of chickens. Avian Diseases 45(4):978-84.
Ismail MM, Cho KO, Ward LA, Saif LJ, Saif YM. 2001. Experimental bovine coronavirus in turkey poults and young chickens. Avian Diseases 45(1):157-63.
Kamps BS, Hoffmann C. 2003a. Pediatric SARS. In: Kamps BS, Hoffmann C, eds., SARS Reference. 3rd ed. Pp. 49-60. [Online] Available: http://www.SARSreference.com.
Kamps BS, Hoffmann C. 2003b. Transmission. In: Kamps BS, Hoffmann C, eds., SARS Reference. 3rd ed. Pp. 49-60. [Online] Available: http://www.SARSreference.com.
Kim JC, Spence RA, Currier PF, Lu X, Denison MR. 1995. Coronavirus protein processing and RNA synthesis is inhibited by the cysteine proteinase inhibitor E64d. Virology 208(1):1-8.
Kimura M. 1980. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. Journal of Molecular Evolution 16(2):111-20.
Klumperman J, Locker JK, Meijer A, Horzinek MC, Geuze HJ, Rottier PJ. 1994. Coronavirus M proteins accumulate in the Golgi complex beyond the site of virion budding. Journal of Virology 68(10):6523-34.
Krijnse-Locker J, Ericsson M, Rottier PJ, Griffiths G. 1994. Characterization of the budding compartment of mouse hepatitis virus: evidence that transport from the RER to the Golgi complex requires only one vesicular transport step. Journal of Cell Biology 124(1-2):55-70.
Ksiazek TG, Erdman D, Goldsmith CS, Zaki SR, Peret T, Emery S, Tong S, Urbani C, Comer JA, Lim W, Rollin PE, Dowell SF, Ling AE, Humphrey CD, Shieh WJ, Guarner J, Paddock CD, Rota P, Fields B, DeRisi J, Yang JY, Cox N, Hughes JM, LeDuc JW, Bellini WJ, Anderson LJ, SARS Working Group. 2003. A novel coronavirus associated with severe acute respiratory syndrome. New England Journal of Medicine 348(20):1953-66.
Kumar S, Tarnura K, Jakobsen IB, Nei M. 2001. MEGA2: molecular evolutionary genetics analysis software. Bioinformatics 17:1244-5.
Kuo L, Godeke GJ, Raamsman MJ, Masters PS, Rottier PJ. 2000. Retargeting of coronavirus by substitution of the spike glycoprotein ectodomain: crossing the host cell species barrier. Journal of Virology 74(3):1393-406.
Lai MM, Cavanagh D. 1997. The molecular biology of coronaviruses. Advances in Virus Research 48:1-100.
Laude H, Van Reeth K, Pensaert M. 1993. Porcine respiratory coronavirus: molecular features and virus-host interactions. Veterinary Research 24(2):125-50.
Lee N, Hui D, Wu A, Chan P, Cameron P, Joynt GM, Ahuja A, Yung MY, Leung CB, To K, Lui SF, Szeto CC, Chung S, Sung JJ. 2003. A major outbreak of severe acute respiratory syndrome in Hong Kong. New England Journal of Medicine 348:1986-94.
Lee HJ, Shieh CK, Gorbalenya AE, Koonin EV, La Monica N, Tuler J, Bagdzhadzhyan A, Lai MM. 1991. The complete sequence (22 kilobases) of murine coronavirus gene 1 encoding the putative proteases and RNA polymerase. Virology 180(2):567-82.
Li W, Moore MJ, Vasilieva N, Sui J, Wong SK, Berne MA, Somasundaran M, Sullivan JL, Luzuriaga K, Greenough TC, Choe H, Farzan M. 2003. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 426(6965):450-4.
Lu Y, Lu X, Denison MR. 1995. Identification and characterization of a serine-like proteinase of the murine coronavirus MHV-A59. Journal of Virology 69(6):3554-9.
Majhdi F, Minocha HC, Kapil S. 1997. Isolation and characterization of a coronavirus from elk calves with diarrhea. Journal of Clinical Microbiology 35(11):2937-42.
Marra MA, Jones SJ, Astell CR, Holt RA, Brooks-Wilson A, Butterfield YS, Khattra J, Asano JK, Barber SA, Chan SY, Cloutier A, Coughlin SM, Freeman D, Girn N, Griffith OL, Leach SR, Mayo M, McDonald H, Montgomery SB, Pandoh PK, Petrescu AS, Robertson AG, Schein JE, Siddiqui A, Smailus DE, Stott JM, Yang GS, Plummer F, Andonov A, Artsob H, Bastien N, Bernard K, Booth TF, Bowness D, Czub M, Drebot M, Fernando L, Flick R, Garbutt M, Gray M, Grolla A, Jones S, Feldmann H, Meyers A, Kabani A, Li Y, Normand S, Stroher U, Tipples GA, Tyler S, Vogrig R, Ward D, Watson B, Brunham RC, Krajden M, Petric M, Skowronski DM, Upton C, Roper RL. 2003. The genome sequence of the SARS-associated coronavirus. Science 300(5624):1399-404.
Masters PS, Koetzner CA, Kerr CA, Heo Y. 1994. Optimization of targeted RNA recombination and mapping of a novel nucleocapsid gene mutation in the coronavirus mouse hepatitis virus. Journal of Virology 68(1):328-37.
Navas S, Seo SH, Chua MM, Das Sarma J, Hingley ST, Lavi E, Weiss SR. 2001. Role of the spike protein in murine coronavirus induced hepatitis: an in vivo study using targeted RNA recombination. Advances in Experimental Medicine and Biology 494:139-44.
Navas S, Weiss SR. 2003. Murine coronavirus-induced hepatitis: JHM genetic background eliminates A59 spike-determined hepatotropism. Journal of Virology 77(8):4972-8.
Opstelten DJ, Raamsman MJ, Wolfs K, Horzinek MC, Rottier PJ. 1995. Envelope glycoprotein interactions in coronavirus assembly. Journal of Cell Biology 131(2):339-49.
Ortego J, Sola I, Almazan F, Ceriani JE, Riquelme C, Balasch M, Plana J, Enjuanes L. 2003. Transmissible gastroenteritis coronavirus gene 7 is not essential but influences in vivo virus replication and virulence. Virology 308(1):13-22.
Peiris JS, Lai ST, Poon LL, Guan Y, Yam LY, Lim W, Nicholls J, Yee WK, Yan WW, Cheung MT, Cheng VC, Chan KH, Tsang DN, Yung RW, Ng TK, Yuen KY, SARS study group. 2003a. Coronavirus as a possible cause of severe acute respiratory syndrome. Lancet 361(9366):1319-25.
Peiris JS, Chu CM, Cheng VC, Chan KS, Hung IF, Poon LL, Law KI, Tang BS, Hon TY, Chan CS, Chan KH, Ng JS, Zheng BJ, Ng WL, Lai RW, Guan Y, Yuen KY, HKU/UCH SARS Study Group. 2003b. Clinical progression and viral load in a community outbreak of coronavirus-associated SARS pneumonia: a prospective study. Lancet 361(9371):1767-72.
Peng GW, He JF, Lin JY, Zhou DH, Yu DW, Liang WJ, Li LH, Guo RN, Luo HM, Xu RH. 2003. Epidemiological study on severe acute respiratory syndrome in Guangdong province. Chinese Journal of Epidemiology 24:350-2.
Pensaert MB. 1999. Porcine epidemic diarrhea. In: Straw BE, D'Allaire S, Mengeling WL, Taylor D. eds., Diseases of Swine. 8th ed. Ames, IA: Iowa State Press. Pp. 179-85.
Perlman S, Ries D, Bolger E, Chang LJ, Stoltzfus CM. 1986. MHV nucleocapsid synthesis in the presence of cycloheximide and accumulation of negative strand MHV RNA. Virus Research 6(3):261-72.
Poutanen SM, Low DE, Henry B, Finkelstein S, Rose D, Green K, Tellier R, Draker R, Adachi D, Ayers M, Chan AK, Skowronski DM, Salit I, Simor AE, Slutsky AS, Doyle PW, Krajden M, Petric M, Brunham RC, McGeer AJ, National Microbiology Laboratory Canada, Canadian Severe Acute Respiratory Syndrome Study Team. 2003. Identification of severe acute respiratory syndrome in Canada. New England Journal of Medicine 348(20):1995-2005.
Prentice E, Denison MR. 2001. The cell biology of coronavirus infection. In: Lavi E, Weiss SR, Hingley S, eds., The Nidoviruses. Philadelphia, PA: Plenum.
Rao PV, Gallagher TM. 1998. Mouse hepatitis virus receptor levels influence virus-induced cytopathology. Advances in Experimental Medicine and Biology 440:549-55.
Rota PA, Oberste MS, Monroe SS, Nix WA, Campagnoli R, Icenogle JP, Penaranda S, Bankamp B, Maher K, Chen MH, Tong S, Tamin A, Lowe L, Frace M, DeRisi JL, Chen Q, Wang D, Erdman DD, Peret TC, Burns C, Ksiazek TG, Rollin PE, Sanchez A, Liffick S, Holloway B, Limor J, McCaustland K, Olsen-Rasmussen M, Fouchier R, Gunther S, Osterhaus AD, Drosten C, Pallansch MA, Anderson LJ, Bellini WJ. 2003. Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science 300(5624):1394-9.
Rottier PJ, Rose JK. 1987. Coronavirus E1 glycoprotein expressed from cloned cDNA localizes in the Golgi region. Journal of Virology 61(6):2042-5.
Saif LJ. In press. Comparative biology of coronaviruses: Lessons for SARS. In: Peiris M, ed., SARS: The First New Plague of the 21st Century. Oxford, UK: Blackwell.
Saif L, Wesley R. 1999. Transmissible gastroenteritis virus. In: Straw BE, D'Allaire S, Mengeling WL, Taylor D. eds., Diseases of Swine. 8th ed. Ames, IA: Iowa State University Press.
Saif LJ, Heckert RA. 1990. Enteric coronaviruses. In: Saif LJ, Thiel KW. Viral Diarrheas of Man and Animals. Boca Raton, FL: CRC Press. Pp. 185-252.
Salanueva IJ, Carrascosa JL, Risco C. 1999. Structural maturation of the transmissible gastroenteritis coronavirus. Journal of Virology 73(10):7952-64.
Sanchez CM, Izeta A, Sanchez-Morgado JM, Alonso S, Sola I, Balasch M, Plana-Duran J, Enjuanes L. 1999. Targeted recombination demonstrates that the spike gene of transmissible gastroenteritis coronavirus is a determinant of its enteric tropism and virulence. Journal of Virology 73 (9):7607-18.
Sarma JD, Scheen E, Seo SH, Koval M, Weiss SR. 2002. Enhanced green fluorescent protein expression may be used to monitor murine coronavirus spread in vitro and in the mouse central nervous system. Journal of Neurovirology 8(5):381-91.
Sawicki SG, Sawicki DL. 1986. Coronavirus minus-strand RNA synthesis and effect of cycloheximide on coronavirus RNA synthesis. Journal of Virology 57(1):328-34.
Sawicki SG, Sawicki DL. 1998. A new model for coronavirus transcription. Advances in Experimental Medicine and Biology 440:215-9.
Shi ST, Schiller JJ, Kanjanahaluethai A, Baker SC, Oh JW, Lai MM. 1999. Colocalization and membrane association of murine hepatitis virus gene 1 products and De novo-synthesized viral RNA in infected cells. Journal of Virology 73(7):5957-69.
Shortridge KF, Stuart-Harris CH. 1982. An influenza epicentre? Lancet 2:812-13.
Sims AC, Ostermann J, Denison MR. 2000. Mouse hepatitis virus replicase proteins associate with two distinct populations of intracellular membranes. Journal of Virology 74(12):5647-54.
Snijder EJ, Bredenbeek PJ, Dobbe JC, Thiel V, Ziebuhr J, Poon LL, Guan Y, Rozanov M, Spaan WJ, Gorbalenya AE. 2003. Unique and conserved features of genome and proteome of SARS-coronavirus, an early split-off from the coronavirus group 2 lineage. Journal of Molecular Biology 331(5):991-1004.
Stalcup RP, Baric RS, Leibowitz JL. 1998. Genetic complementation among three panels of mouse hepatitis virus gene 1 mutants. Virology 241(1):112-21.
Stavrinides J, Guttman DS. 2004. Mosaic evolution of the severe acute respiratory syndrome coronavirus. Journal of Virology 78(1):76-82.
Subbarao K, Klimov A, Katz J, Regnery H, Lim W, Hall H, Perdue M, Swayne D, Bender C, Huang J, Hemphill M, Rowe T, Shaw M, Xu X, Fukuda K, Cox N. 1998. Characterisation of an avian influenza A (H5N1) virus isolated from a child with a fatal respiratory illness. Science 279:393-6.
Thiel V, Herold J, Schelle B, Siddell SG. 2001. Infectious RNA transcribed in vitro from a cDNA copy of the human coronavirus genome cloned in vaccinia virus. Journal of General Virology 82(Pt 6):1273-81.
Thiel V, Ivanov KA, Putics A, Hertzig T, Schelle B, Bayer S, Weissbrich B, Snijder EJ, Rabenau H, Doerr HW, Gorbalenya AE, Ziebuhr J. 2003. Mechanisms and enzymes involved in SARS coronavirus genome expression. Journal of General Virology 84(Pt 9):2305-15.
Tresnan DB, Levis R, Holmes KV. 1996. Feline aminopeptidase N serves as a receptor for feline, canine, porcine, and human coronaviruses in serogroup I. Journal of Virology 70(12):8669-74.
Tsang KW, Ho PL, Ooi GC, et al. 2003. A cluster of cases of severe acute respiratory syndrome in Hong Kong. New England Journal of Medicine 348:1977-85.
Tsunemitsu H, El-Kanawati ZR, Smith DR, Reed HH, Saif LJ. 1995. Isolation of coronaviruses antigenically indistinguishable from bovine coronavirus from wild ruminants with diarrhea. Journal of Clinical Microbiology 33(12):3264-9.
Tsunemitsu H, Saif LJ. 1995. Antigenic and biological comparisons of bovine coronaviruses derived from neonatal calf diarrhea and winter dysentery of adult cattle. Archives of Virology 140(7):1303-11.
van der Meer Y, Snijder EJ, Dobbe JC, Schleich S, Denison MR, Spaan WJ, Locker JK. 1999. Localization of mouse hepatitis virus nonstructural proteins and RNA synthesis indicates a role for late endosomes in viral replication. Journal of Virology 73(9):7641-57.
Vennema H, Poland A, Floyd Hawkins K, Pedersen NC. 1995. A comparison of the genomes of FECVs and FIPVs and what they tell us about the relationships between feline coronaviruses and their evolution. Feline Practice 23:40-4.
WHO (World Health Organization). 2003a. Cumulative number of reported probable cases of severe acute respiratory syndrome (SARS). [Online] Available: http://www.who.int/csr/sarscountry/2003_07_11/en [accessed July 16, 2003].
WHO. 2003b. Case definitions for surveillance of severe acute respiratory syndrome (SARS) [Online] Available: http://www.who.int/csr/sars/casedefinition/en/ [accessed May 1, 2003].
WHO [Online] Available: www.who.int/csr/sars/en/.
WHO. Cumulative Number of reported probable cases of severe acute respiratory syndrome (SARS). [Online] Available: www.who.int/csr/sars/country/2003_05_20/en/.
Xiao X, Chakraborti S, Dimitrov AS, Gramatikoff K, Dimitrov DS. 2003. The SARS-CoV S glycoprotein: expression and functional characterization. Biochemincal and Biophysical Research Communucations 312(4):1159-64.
Yeager CL, Ashmun RA, Williams RK, Cardellichio CB, Shapiro LH, Look AT, Holmes KV. 1992. Human aminopeptidase N is a receptor for human coronavirus 229E. Nature 357(6377):420-2.
Yokomori K, Lai MM. 1992. Mouse hepatitis virus utilizes two carcinoembryonic antigens as alternative receptors. Journal of Virology 66(10):6194-9.
Yount B, Curtis KM, Baric RS. 2000. Strategy for systematic assembly of large RNA and DNA genomes: transmissible gastroenteritis virus model. Journal of Virology 74(22):10600-11.
Yount B, Curtis KM, Fritz EA, Hensley LE, Jahrling PB, Prentice E, Denison MR, Geisbert TW, Baric RS. 2003. Reverse genetics with a full-length infectious cDNA of severe acute respiratory syndrome coronavirus. Proceedings of the National Academy of Sciences 100(22):12995-3000.
Yount B, Denison MR, Weiss SR, Baric RS. 2002. Systematic assembly of a full-length infectious cDNA of mouse hepatitis virus strain A59. Journal of Virology 76(21):11065-78.
Zhang XM, Herbst W, Kousoulas KG, Storz J. 1994. Biological and genetic characterization of a hemagglutinating coronavirus isolated from a diarrhoeic child. Journal of Medical Virology 44 (2):152-61.
Zhong NS, Zheng BJ, Li YM, Poon, Xie ZH, Chan KH, Li PH, Tan SY, Chang Q, Xie JP, Liu XQ, Xu J, Li DX, Yuen KY, Peiris, Guan Y. 2003. Epidemiology and cause of severe acute respiratory syndrome (SARS) in Guangdong, People’s Republic of China. Lancet 362(9393):1353-8.
Ziebuhr J, Snijder EJ, Gorbalenya AE. 2000. Virus-encoded proteinases and proteolytic processing in the Nidovirales. Journal of General Virology 81(Pt 4):853-79.
Ziebuhr J, Thiel V, Gorbalenya AE. 2001. The autocatalytic release of a putative RNA virus transcription factor from its polyprotein precursor involves two paralogous papain-like proteases that cleave the same peptide bond. Journal of Biological Chemistry 276(35):33220-32.




































