
5
Data Management and Bioinformatics Challenges of Metagenomics
Metagenomics studies are data-rich, rich both in the sheer amount of data and rich in complexity. Biologists now have over two decades of experience in handling and analyzing DNA sequence data, but these are mostly data on reasonably well understood structures—genes and complete genomes. We still do not comprehend the organizing principles of metagenomic data. The expected flood of sequence data from metagenomic studies therefore poses many new challenges that urgently need attention. Information from metagenomics studies will be fully exploited only if appropriate data-management and data-analysis methods are in place.
GENOMIC DATA
The rise of genomics has been characterized by technological and scientific innovations and by novel practices in data dissemination. With the coincident increase in computational power and electronic communication, they were critical for the success of the field of genomics. In the early 1980s the scientific community in Europe and the United States established archives of nucleic acid sequence data. This had several very important consequences. One was that the data were immediately accessible in a form suitable for computer analysis; another was that the data were freely available, without impediment to all researchers, be they in academe or industry. There are three nucleic acid sequence archives, all founded in the 1980s: GenBank, funded by the National Institutes of Health (NIH) through the National Library of Medicine; EMBL-Bank, funded by the European Molecular Biology Laboratory; and the DNA Databank of Japan
(DDBJ), funded by the Ministry of Education, Culture, Sports, Science and Technology of Japan. A formal collaboration between these bodies (the International Nucleotide Sequence Database Collaboration1) ensures that the contents of all three are effectively identical at any time. Major achievements of the INSDC have been to make the submission of nucleic acid sequence data to one of the three databases mandatory for publication of any scientific paper that reports new sequence data2 and to define standards for such submissions. Today, all DNA sequencing done in the public sector is captured in the archives. Their extraordinary growth is shown in Figure 5-1.
It is no exaggeration to state that without the INSDC and the sequences stored in and made available through the collaborating databases, the success of the Human Genome Project and similar genome projects would have been impossible. These databases and the analytical tools whose development was encouraged by the free availability of data allow researchers to access the totality of the world’s public DNA and protein sequence data. It is vital for the metagenomics community to continue to adhere to accepted standards with respect to the public deposition of data from community projects3 and continue to encourage and enable the development of analytical tools and agreed-upon data-management practices (see also Chapter 6).
DNA sequence data submitted to the international archives are processed sequences, they are not the “raw” sequences directly from the sequencing machines. In the late 1990s researchers recognized that public access to the raw sequence “traces” would also be of great value. The National Center for Biotechnology Information, the Wellcome Trust, and the European Bioinformatics Institute (EBI) therefore established the Trace Archive4 for these data. In December 2006, the Trace Archive contained over 1.4 billion traces from over 700 species. Despite the challenges arising from some of the new sequencing methods, timely deposition of raw sequence data to the Trace Archive by the metagenomics community will also be of great long-term community benefit.
The nucleic acid sequence data archives are a primary source of experimentally determined DNA and RNA sequences. Many types of analyses of genomes and individual genes, however, require protein sequences. Although historically these were experimentally determined, the great majority are now computationally predicted from DNA sequence data. This requires
|
1 |
See www.insdc.org. |
|
2 |
|
|
3 |
As accepted by the Fort Lauderdale Agreement of 2003: http://www.wellcome.ac.uk/assets/wtd003207.pdf. |
|
4 |
http://www.ncbi.nlm.nih.gov/Traces/trace.cgi?; http://trace.ensembl.org/. |
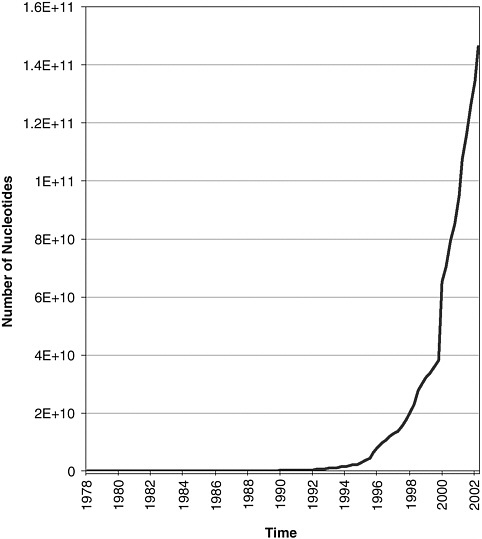
FIGURE 5-1 The growth in the size of the international sequence databases, since their inception in 1982. This graph shows the size of the databases at regular intervals, in numbers of nucleotides. (Data from the European Bioinformatics Institute.)
computational methods to predict which sequences constitute genes, that is, actually code for RNA and proteins. This is far from being a solved problem even for “complete” genomes. It will be even more difficult for the fragmentary sequences that will typically be obtained in metagenomics projects. Two databases, The Protein Information Resource and Swiss-Prot, were established as community resources for protein sequence data, in 1984
and 1986, respectively. They now collaborate closely to produce a common database of protein sequences—UniProt, a product of EBI, the Swiss Bioinformatics Institute, and the National Biomedical Research Foundation at Georgetown University. UniProt is not a primary database, but rather a highly curated database of protein sequences, the vast majority of which are derived computationally from gene models in the nucleic sequence data archive. Not surprisingly, the growth of UniProt has been slower than that of the nucleic acid sequence archive (see Figure 5-2).
METAGENOMIC DATA
In principle, there are no differences between DNA sequence data from “conventional” and metagenomics sequence projects. In both cases, the sequences are simply strings of the four bases A, T, C, and G. In practice, however, metagenomic sequence data require particular infrastructures for management and analysis (see Figure 5-3).
The growth of public DNA sequence data over the last 2 decades has been exponential, with a doubling time of about 14 months. Although predictions must be treated with some caution, early experience with metagenomics projects suggests that they will have a substantially shorter doubling time. Indeed, even the preliminary data from the Global Ocean Survey more than quadrupled the existing (predicted) protein-coding open reading frames (although this increase will be smaller when the GOS data are curated). The predicted growth of metagenomics sequences will result both from the new sequencing technologies now available (see below) and from the fact that metagenomic sequencing is not redundant. Conventional genomic sequencing projects are highly redundant. Multiple coverage of a genome is necessary for assembling a genomic sequence and ensuring accuracy. For example, the human genome was originally sequenced to a “depth,” that is, with a redundancy, of 4.5-fold. By contrast, metagenomic sequencing is far less redundant. A consequence is that many more DNA and (predicted) protein data are being generated for the same effort: rather than sequencing the same genome 10 times, 10 times as many data are being generated for different sequences. There are other consequences of the relative lack of redundancy of metagenomic sequences.
The major reasons for such high redundancy in conventional genomic sequencing were to achieve “complete” coverage of a genome and to ensure the accuracy of the sequence. For metagenomes, most sequence reads will be unique; instead of several overlapping fragments of the same underlying sequence, there will be on average only one fragment. Therefore, metagenomic sequence data have an intrinsically higher error rate than genomic sequencing data. Moreover, it is now clear that many metagenomics projects will rely on novel sequencing technologies for data collection (see
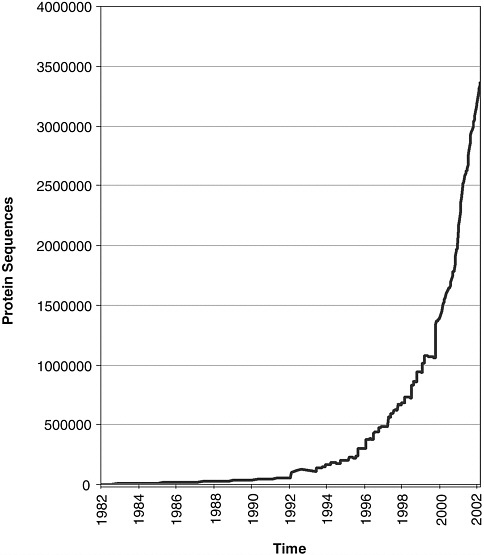
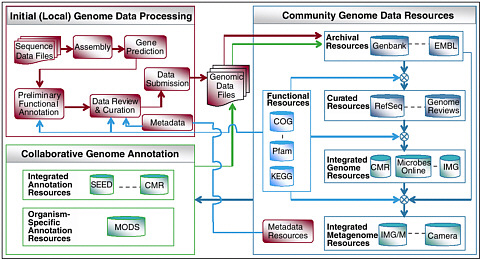
FIGURE 5-3 A typical life cycle of microbial genomic and metagenomic sequence data. This illustrates the pipelines of initial sequence data assembly and annotation, the integration of these data with annotation from other community resources, both integrative and species-specific the deposition of the annotated data into the archives of Genbank, EMBL-Bank and DDBJ; and the derivation of a number of specific integrated community data sets. With thanks to Victor Markowitz, whose original was modified.
Chapter 4). All the new-generation technologies produce sequence read lengths that are short—25-200 bases compared with 800-1000 bases for Sanger capillary sequencing technologies. Moreover, some of the new technologies are rather error-prone. These characteristics of the new technologies and the fact that within any study the sequences may be derived from many different organisms make the assembly of long sequences from the primary sequence data difficult, if not impossible.
THE IMPORTANCE OF METADATA
Metadata are data about data (Gray et al. 2005). They are also about biology. Metadata are the descriptions of sampling sites and habitats that provide the context for sequence information. Metadata are of great importance for metagenomic sequence data for two reasons. First, only by fully describing the samples from which metagenomics sequences have been obtained can one have any possibility of replicating a study. Samples from environmental or biological sources can never be fully replicated, but it is
important that samples be sufficiently well described for an independent researcher to have the possibility of resampling. Second, metadata are essential for the analysis of metagenomic sequence data. Metagenomic sequence data that lack an environmental context have no value.
There is an urgent need for the community to agree on what metadata must be included with the submission of any metagenomic sequence data. Appropriate metadata will depend on the type of metagenomics sample. For example, the metadata to be associated with a human gut sample will differ from that to be associated with an ocean sample. Without wishing to determine now what these metadata should be, some examples can be given:5
-
Detailed, three-dimensional geographic location of the sample, including depth (for water sampling) or height (for land and air samples).
-
The general features of the environment of the sample, such as ocean, soil, mine, human, or insect.
-
Specific features of the sample site, such as chemical data (pH, salinity, and so on), physical data (temperature, incident light, and so on), time when the sample was taken, and host condition, diet, and habitat.
-
Method of sampling, size of sample, and sample preparation.
Some of these data would be collected or recorded when the DNA sample was being collected, and others could be retrieved from other databases, including geospatial databases and weather or ocean-current databases.
The community must address these issues with a sense of urgency. If metagenomic sequence data are to be used to their fullest advantage, a metadata infrastructure, which defines the data that are to be collected and their semantics, is an urgent need. As indicated above, no single metadata standard will be appropriate for all samples. Nevertheless, close collaboration and coordination among the communities developing metadata standards, nationally and internationally, will be important. Much can be learned from standards initiatives in related communities, for example, those of the Microarray Gene Expression Data (MGED) Society.6 It is relevant that such standards are increasingly adopted and required by the major scientific journals.7
|
5 |
See, for example, the database developed by the International Census of Marine Microbes (http://icomm.mbl.edu/microbis) and the efforts of the Genomics Standards Consortium (http://darwin.nox.ac.uk/gsc/gcat). |
|
6 |
See http://www.mged.org/. |
|
7 |
See http://www.nature.com/nature/journal/v419/n6905/full/419323a.html. |
DATABASES FOR METAGENOMIC DATA
Absorbing the sequence that will be generated by metagenomics projects will be a challenge for the nucleic acid sequence data archive (GenBank, EMBL-Bank, and DDBJ) (see Box 5-1). But in addition to the archiving challenge, it is clear that the community will require new, secondary databases if the data are to be used effectively. Only a specialized database will be able to offer consistent storage and querying of the rich metadata that metagenomic sequences need. The analysis of metagenomic sequences will require computational programs that are best offered in the context of a specialized database, for example, programs for the clustering of metagenomic sequence reads or for time-series analysis. And metagenomic data must be integrated with data from different projects, such as satellite data on ocean temperatures and time-series data on changes in ocean salinity.
Two large projects have recently been initiated to build an infrastructure for metagenomic sequences and associated metadata. One is the CAMERA project,8 a joint venture of the University of California, San Diego and the J. Craig Venter Institute in Rockville, MD. CAMERA’s objective is to provide cyberinfrastructure tools and resources and bioinformatics expertise to enable the community to use metagenomic data. CAMERA will make raw environmental sequence data and their associated metadata accessible with pre-computed search results and access to high-performance computational resources.
At the Department of Energy’s Joint Genome Institute (Walnut Creek, CA), an existing microbial genome database project, Integrated Microbial Genomes, is being extended to cope with metagenomics data in a project called IMG/M.9 The objective of IMG/M is to integrate conventional microbial genomics data with data from metagenomics projects. In December 2006, it included data from over 680 microbial genomic projects, most of which were aimed at conventional complete genome sequencing.
Several smaller projects around the world have developed various specific data models and interfaces, often for specific metagenomic projects. Examples are the Micro-Mar10 and MICROBIS11 databases, in Alicante, Spain, and Woods Hole, MA, respectively.
There are interesting parallels between these projects and the genomic sequencing communities. In the latter, many of the major species being studied have special community genomics databases, for example, the Saccharomyces Genome Database for baker’s yeast,12 the Mouse Genome
|
BOX 5-1 The Metagenomic Data Deluge: Future Data Storage and Access Challenges From the perspective of sequence data repositories, projected data storage needs for archiving Sanger-based capillary sequence data might not seem overly formidable. Every year disk space gets cheaper, with storage density increasing steadily. Hard drives have experienced a 50-million-fold increase in storage density since their invention. So, is there cause for concern for future metagenomic data storage and retrieval? Projected future sequence DNA data storage challenges are more complex than simple extrapolation from today’s Sanger-based capillary sequence production rates. There are three central reasons why data accumulation is expected to accelerate dramatically, and soon:
|
Database for the laboratory mouse,13 FlyBase for the fruitfly Drosophila,14 and TAIR for Arabidopsis.15
These model organism databases are publicly funded (usually by the NIH or the National Science Foundation) and add value to the sequence data deposited in the GenBank, EMBL-Bank, and DDBJ archives. CAMERA, IMG/M, and similar projects promise to be “model organism databases” for metagenomes. Such databases will be essential if data are to be used to the greatest advantage by the scientific and biomedical communities. Different databases will doubtless be required for different needs. Cooperation and collaboration between them, especially in the development of standards for the description of data will be necessary. Not only will it be necessary for databases to include metadata about habitat and sample treatment, it will also be critical to document how the raw data has been processed, filtered, and analyzed. Maintenance and curation of metagenomics databases will greatly add to their value, but are expensive and will require consistent support. Funding for databases requires a different approach than that for research projects: there need to be mechanisms for long-term funding, coupled with community oversight and evaluation. The experiences of the National Center for Ecological Analysis and Synthesis16 and the National Evolutionary Synthesis Center17 in providing a community focus for data integration and analyses are examples the metagenomics community might wish to follow.
SOFTWARE
The analysis of genomic data depends on computer software. In general, grants for metagenomics projects will require an even higher percentage of funds for bioinformatic and statistical support than have conventional genomics studies or than may be typical of other kinds of biological research. It is important that appropriate new software be developed, conform to industry standards, and be well documented. The investment in the development of software needs to be timely. If the analytical needs of biologists are still uncertain, a major investment in robust software engineering is premature; but once an analytical technique becomes generally accepted and useful, investment in making the software more user-friendly and reliable is worthwhile. That pipeline is poorly supported by traditional grant-funding mechanisms. Funding agencies should consider a competi-
|
13 |
|
|
14 |
See http://flybase.org/. |
|
15 |
|
|
16 |
|
|
17 |
tive funding opportunity providing software engineering support to bring individually developed software programs that have found wide use in the community up to robust, engineered, documented form. Such a program would allow a variety of individual approaches to developing software, with community assessment of the software’s value.
ANALYSIS OF METAGENOMIC SEQUENCE DATA
Data from metagenomics projects share features that will require the development of novel computational tools and perhaps a new paradigm for the analysis of DNA data. In genome projects, the organization of the DNA in the organism was well known—a circular chromosome and plasmids in bacteria and multiple chromosomes in eukaryotes. The goal was to recover a sequence of a complete genome of a single organism. In metagenomics, we do not have a clear model of the organization of the DNA in the sample. We do know that each sample contains many different organisms, bacteria, viruses, and small eukaryotes. The different organisms will have different genome structures, linear or circular chromosomes, single or multiple chromosomes, and extrachromosomal elements, and these characteristics will be unknown at the time of data analysis. It is likely that even the most extensive sequencing of a specific sample will provide only partial sampling of the DNA in a given environment; therefore, the data in the sample may have to be used to predict features of the sample, rather than analyzing the features themselves. As an illustration of the complexity of metagenomic sequence analysis we illustrate, in Figure 5-3, an exemplar of a metagenomics project’s data “life cycle.”
There will be a need to analyze sequences in the context of their metadata, including independent environmental data, such as meteorological and oceanographic. Thus, the analyses that will be done on these sequences will be different from those done on conventional genome sequence data and will involve many questions that will combine data of various types (see Box 5-2).
Because of the unusual features of metagenomic data (fragmentation and high error rate), new computer algorithms and data models will be needed for the clustering of metagenomic sequences (both DNA and predicted peptide sequences) to characterize microbial communities from sequence data and to analyze changes in microbial communities over time. Novel techniques for the visualization of complex data will be needed.
Another major difference between data-management needs for metagenomics projects and those for conventional genomics problems will be the demand for continuing community input into data annotation. In conventional genomics, primary responsibility for annotating data falls on the authors, and this creates inconsistencies in the databases when old
|
BOX 5-2 Examples of Questions That Illustrate the Utility of Metadata
|
annotations are not updated and thus become inconsistent with new ones. Although there is now a mechanism (called third party annotation18) for the community to annotate genomic sequences in GenBank, EMBL-Bank, and DDBJ, the original authors’ annotations, even if outdated, remain as primary annotations seen in the database. For instance, annotations added through curation at the appropriate model organism database are only very slowly being incorporated into central databases. In metagenomics projects, where many types of annotations would become possible only after additional data (or metadata) are collected by other groups, an annotation database must be able to accept and integrate both individual and large-scale (computational) annotations of metagenomic data and able to integrate them in a transparent way for their user communities. The need for dynamic and flexible annotation will require ongoing, professional curation—another reason that long-term database funding will be important.
It will be seen that the scientific community will be presented with challenges by the generation of metagenomics data. Many of the challenges will require a high degree of community organization and collaboration. Given the wide array of microbial communities that will be studied—from toxic waste sites to agricultural soil to the human mouth—the interested scientific community will be extremely diverse, and coordination will be
more difficult. No existing body can take the lead to ensure that it occurs. However, the Microbe Project, a US government interagency group, has the appropriate broad membership to facilitate coordination and communication among the interested scientific communities (see Chapter 6).













