
3
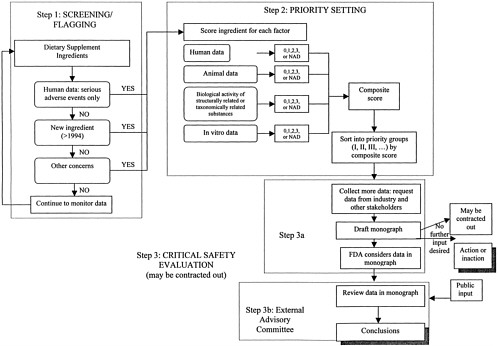
Outline of the Overall Process for Evaluation of Dietary Supplement Ingredients
PROPOSED FRAMEWORK FOR EVALUATION OF DIETARY SUPPLEMENT INGREDIENTS
The Food and Drug Administration (FDA) asked the Institute of Medicine to propose a safety framework that would assist it in overseeing the safety of dietary supplement ingredients. In an ideal situation, FDA would be able to immediately undertake a full safety evaluation for every supplement ingredient. Since this is not possible, a process must be implemented to determine which supplement ingredients warrant the highest priority for review.
This report outlines a three-step framework for considering the safety of dietary supplement ingredients. The first two steps in the process, “screening/flagging” and “priority setting,” are designed to categorize dietary supplement ingredients based on theoretical or possible concern, and therefore the immediacy of the need for in-depth evaluation of safety, the third step of the process.
For the purposes of describing the proposed process, it is convenient to think of the overall framework as three distinct phases, separating the screening and priority-setting phases into two distinct steps in the framework, with the third step being a critical in-depth review. The first step in the process, screening/flagging, is essentially a beginning point that incorporates the factors that will bring an ingredient to the attention of FDA and will indicate whether the material should immediately be examined more closely. This step is important because it is not feasible initially to research the information necessary for priority setting for the entire universe of dietary supplement ingredients. Over time, it is likely that steps one and two will be viewed as one ongoing system. The priority-setting system will determine which ingredients require a full safety evaluation first and can be done on an on-going basis once an ingredient is flagged in the screening step. After dividing flagged ingredients into priority categories for further evaluation, the third step—in-depth safety evaluations of highest priority ingredients—can be completed.
Ingredients versus Products
It is important to note that although dietary supplement products are the substances sold on the market, this framework is designed to consider the safety of dietary supplement ingredients. For the purpose of this framework, botanical ingredients are defined as the plant parts (e.g., seed, root, leaf) rather than the many individual chemical compounds contained in a plant. Although this framework focuses on identifying and reviewing ingredients with inherent safety concerns, it is very important to remember that all products containing a particular ingredient are not likely to
have equivalent safety profiles. As discussed in the following paragraphs, differences in safety profiles may exist because the products contain different amounts of an ingredient, there are differences in bioavailability (the degree to which a substance becomes available to the target tissue after administration), there are differences in the amounts or presence of other substances (including contaminants), or the products are sold in combination with different ingredients.
Substantial variation can exist among the different brands of products purportedly containing a given dietary supplement ingredient (Foreman, 2000; Howe, 2000) due to lack of standardization. Products labels may claim that products are standardized to contain a particular amount of a substance. Several reports of product analyses, however, suggest that product labels may be inaccurate—that products may contain significantly higher or lower amounts of substances than indicated on the label (Green et al., 2001; Hamilton-Miller et al., 1999; Kamber et al., 2001). While a number of reports have suggested that substances do not contain the substances purported on the label, reports of labeling discrepancies with several botanicals have been disputed on the basis that laboratories used different analytical methods or measured different chemical markers that may not be relevant (Betz et al., 1995; Marrone, 1999). The eventual development of standards may address this problem.
In addition to differences in the amount of a substance contained in a product, significant variation may exist in bioavailability. Variability in bioavailability may result from differences in manufacturing and formulation that affect how much of a substance is absorbed. For example, dissolution maybe incomplete, or even if the ingredient’s dissolution is complete, absorption may be incomplete if it is degraded in the intestinal fluid or it does not undergo active or passive transportation out of the intestinal mucosa. Dietary supplements may be delivered in matrices (tablets, capsules, etc.) that impact dissolution and absorption, or they may contain ingredients that change absorption and other aspects of bioavailability (Chambliss, 2001).
Quality control guidelines for many ingested substances are described by current Good Manufacturing Practices (GMPs). As described in Chapter 1, FDA has not yet published proposed or final GMPs for the dietary supplement industry. Quality control variables that can impact the safety of dietary supplement products include, but are not limited to, contamination by heavy metals, contamination by harmful microorganisms, contamination by pesticides, misidentification of raw plant ingredients, and improper storage. GMPs should provide guidance in a number of these areas (CFSAN, 2000; FDA, 1997).
In addition to variations in products containing a particular supplement ingredient, many products sold today are “combination products” that are mixtures of more than one dietary supplement ingredient. These products raise another set of safety concerns because mixtures can have safety profiles different than the summed effect of discrete ingredients. There is a potential for interaction among ingredients, and even mixtures containing the same dietary supplements may differ in dose and ratio of components. Although combination products can produce effects distinct from the individual ingredients, a first approximation of a combination product’s safety can come from examining the safety of the component ingredients. A combination product containing ingredients that individually are not considered safe is likely to demonstrate some of the same safety concerns as the individual ingredients.
Even if no single ingredient raises safety concerns, MedWatch and other information sources should be monitored for clusters of serious adverse events and other possible indicators of problems related to specific combination products. For example, when data suggest that interactions between individual supplement ingredients may be associated with adverse effects, combination products that contain these interacting ingredients warrant particular attention. If
potential interactions or clusters of serious adverse events from particular combinations come to FDA’s attention, then the combination product itself should undergo the screening/flagging and priority-setting steps.
In the case of most combinations, it is expected that more data will be available about the safety of the individual ingredients than about the safety of the combination. If a review raised questions about the combination of kava kava, Saint John’s wort, and passionflower, for example, the initial approach would be to consider the safety of kava kava, Saint John’s wort, and passionflower individually to determine if any of these three ingredients were individually thought to be of potential concern from a safety perspective. If any of these individual botanicals were considered unsafe, then the combination product should also be considered unsafe. If none of the three ingredients alone had raised safety concerns, then it would be appropriate to monitor the literature, the MedWatch database, and other sources of information for clusters of adverse events or other indications that harmful effects might be associated with the combination. In the case of popular combinations of substances, it is possible that a significant amount of data about the safety of the combination will be available, perhaps even more than is available on the safety of the individual substances (the combination of glucosamine and chondroitin sulfate maybe such an example). In this case, it may be appropriate to consider the safety of the combination itself, in addition to the safety of the individual ingredients. If safety concerns have been raised for an individual ingredient, then these concerns should generally not be considered as mitigated when the ingredient is combined or used in combination with another ingredient.
In summary, this framework focuses on ingredients rather than products, but the variability among products necessitates that FDA consider the safety of products as well. Regular monitoring of MedWatch and other information sources will be necessary to detect indications of serious adverse events possibly related to specific brands of products.
Initiation of the Process
The three steps of the proposed framework are outlined in Table 3–1 and Figure 3–1. Step One is the initial screening/flagging process; Step Two, the priority-setting process; and Step Three, the evaluation process. The stepwise framework is important, but it is recognized that before FDA can get started with the framework, it needs an initiation process to start screening the large list of dietary supplement ingredients currently on the market. Several options for this initiation point were considered. It is possible to start by examining:
-
The number of serious adverse events reported. MedWatch is a system for collecting adverse events. While anyone is free to file a MedWatch report, a sizeable portion of contributors to MedWatch are members of the medical community. The MedWatch system could therefore form the basis for concern about the safety of particular ingredients (or products). That is, the framework process could be initiated by first considering ingredients with a greater number of reported serious adverse events. The major disadvantage of this approach is the incomplete nature of the reports and the limited number of reports to MedWatch (GAO, 1997).
-
A priority list prepared by experts. A group of several experts in the field could, within a day, provide a list of the top ingredients causing the most concern in their opinion. It is proposed that one expert would have a background in botanicals (e.g., a pharmacognosist), one would be a physician practicing alternative medicine, and one would be a nutritional pharmacologist or toxicologist. A key advantage of this method is that it would have a high probability of identifying most ingredients with any significant concern prior to collection and review of
TABLE 3–1 Overall Framework
|
Step in the Process |
Step One: Screening/Flagging |
Step Two: Priority Setting |
Step Three, Part A: Draft Monograph Preparation and Monograph Review by the Food and Drug Administration (FDA) |
Step Three, Part B: Critical Safety Evaluation |
|
Which ingredients |
All ingredients are considered “New” ingredients are automatically flagged |
Ingredients flagged in screening step |
Ingredients with highest priority based on Step Two ranking |
Monographed ingredients for which a decision is not clear cut or for which further input is desired |
|
Completed by |
FDA |
FDA |
FDA or contractor |
External advisory committee |
|
Factors and modifiers used |
Human data: serious adverse events only Other concerns,a as they come to FDA’s attention |
Human data Animal data Biological activity of structurally related and taxonomically related substances In vitro data Vulnerable group use (modifies other factors) Prevalence of use (modifies priority ranking) |
Human data Animal data Biological activity of structurally related and taxonomically related substances In vitro data Vulnerable group use considered with other factors |
Human data Animal data Biological activity of structurally related and taxonomically related substances In vitro data Vulnerable group use (considered with other factors) |
|
Level of information search |
Easily obtainable information (see Table 4–1) |
Literature search is more comprehensive |
Comprehensive Request industry data and data from other stakeholders |
Comprehensive Public input |
|
Depth of evaluation |
Low level evaluation: is there evidence suggesting a concern may exist? |
Weighting based on evidence of possible risk, potential seriousness of harm, and relative importance of factor |
Comprehensive: totality of evidence is considered, including data requested from industry and other stakeholders |
Totality of evidence; monograph reviewed and revised |
|
Goal |
Ingredients warranting further investigation are flagged |
Table of ingredients sorted into priority groups for further evaluation |
Monograph AND FDA decision for action/inaction OR Referral to external advisory committee |
Monograph with conclusions of external advisory committee |
|
a The term, “other concerns,” as described in Chapter 3, encompasses concerns FDA becomes aware of without extensive information searching. These may include concerns expressed by other regulatory agencies, concerns expressed in secondary literature, or concerns expressed by other organizations. |
||||
-
available data. However, this method is not transparent, could be biased, and the exact ranking of the ingredients may tend to be somewhat arbitrary.
-
Sales volume. On the basis (that an unsafe product would do the most harm if it reaches a large number of people, ingredients could be sorted by sales volume and those with the highest sales volume sent first through the screening/flagging and priority-setting steps. The major disadvantage of this system is that sales volumes are not static and sales figures serve only as proxy estimates of use. Another problem with this system is that as a class, vitamins will have the highest sales volume although their safety has been more thoroughly monitored than other classes of dietary supplements. It might therefore be appropriate to begin with the top sellers in the different classes of substances (e.g., the top 10 to 20 percent of hormones, of botanicals, of animal products, of vitamins, and so on).
-
Randomly, in no particular order. The advantage of a random approach, rather than a systematic one, is that initiation of the process will not require additional resources and time investment.
One of these methods, or a combination of them, may be an effective way for FDA to initiate the screening process of ingredients described in this report.
Description of the Process
The first two steps in the process, screening/flagging and priority setting, are organized to categorize dietary supplement ingredients based on concern and therefore immediacy of need for subsequent in-depth evaluation of safety, the third step of the process. All three steps of this proposed framework are described in detail in Chapter 5 and briefly below.
Information or Factors Used to Identify or Flag Ingredients for Further Evaluation
Several types of readily available information, or “factors,” are used to identify ingredients warranting farther consideration, as outlined in Table 3–1 and Figure 3–1, and described in more detail in Chapters 4 and 5. The first factor considered is the ingredient status; ingredients introduced to the market as dietary supplements after 1994 are automatically flagged in Step One to be reviewed in Step Two, priority setting. For pre-1994 ingredients, evidence of serious adverse events in humans is considered. If there is evidence of serious adverse events that may be related to a specific dietary supplement ingredient, that ingredient is flagged. The next consideration is “other information” available to FDA. This other information factor encompasses a broad range of potential concerns. Information that may make FDA aware of potential problems ranges from materials prepared by other groups that have evaluated the safety of dietary supplements, to expressions of concern from consumer protection or advocacy organizations. All flagged ingredients enter the priority-setting step of the framework (Step Two).
Evaluation and Weighting of Available Information
In Step Two, the priority-setting step, several key types of information about each flagged ingredient are considered. These types of information, or factors, are described in Chapter 4. They include human data, animal data, bioactivity of related substances, and in vitro data.
Information about each of these primary factors is retrieved and four key aspects of these data are considered:
-
the relevance of the information to safe use by consumers;
-
the potential seriousness of the harm reported;
-
the methodological quality of the evidence; and
-
the quantity of the evidence.
These aspects are used to give each ingredient a score for each of the four factors. The information for each factor in also considered in terms of how it might suggest possible susceptibility of particular subpopulations. After the scores are tabulated, they are sorted into several priority groups based on the scores, the hierarchy of the different types of data, and the estimated prevalence of use of the ingredient. This sorting process allows FDA to consider the different factors independently and individually for each ingredient.
Monograph Preparation, Internal Review, and In-Depth Safety Evaluation
As high priority ingredients are identified, safety review monographs will be compiled (Step Three) as described in Chapter 6. In this step, industry and other stakeholders are invited to bring forward information relevant to the safety of the ingredients being reviewed. Monographs will then be drafted. Monographs will include comprehensive reviews of human data, animal data, other different types of evidence about the ingredients’ safety, and notations of where information is lacking. The monograph preparation may be done by FDA or may be contracted out. Once the monographs are prepared, FDA will decide whether to turn them over to an external advisory committee for further attention, to take action without further input from an external advisory committee, or to take no action at the current time. If FDA decides not to take immediate action, it could choose to make the monographs available as draft monographs. FDA may later decide to take action or to refer the ingredient to an external advisory committee, especially if additional information becomes available. A decision not to take further action does not indicate that the product is safe, and FDA may choose not to make a statement about the safety of the product.
If the data do not lead to a clear-cut FDA decision, then the ingredient and its draft monograph are referred to an external advisory committee for analysis and advice, and the draft monograph is made public. The external advisory committee will review the draft monograph, collect additional information as needed, and provide opportunity for public input on the ingredient’s inherent safety. The external advisory committee will review the science base for the monograph and revise it as necessary. Finally, the committee will summarize the science relevant to assessing safety of the ingredient and provide opinions on the possible risks associated with the ingredient, including risks that may be specific to particularly vulnerable subpopulations. The committee will also outline where additional research may help resolve safety questions. The revised monograph and the committee’s comments will be made available to the general public, so that the expert opinions of scientists are known even if no action is taken.
Ongoing Review and Reassessment
As new information becomes available to FDA, re-evaluation of internal draft monographs and monographs revised by an external advisory committee may be necessary. Such new information should be considered as described in the priority-setting step to determine if there is sufficient substantive new information to review and possibly revise the monograph.
REFERENCES
Betz JM, White KD, der Marderosian AH. 1995. Gas chromatographic determination of yohimbine in commercial yohimbe products. J AOAC Int 78:1189–1194.
CFSAN (Center for Food Safety and Applied Nutrition). 2000. Dietary Supplement Strategy (Ten Year Plan). Online. Food and Drug Administration. Available at http://vm.cfsan.fda.gov/~dms/ds-strat.html. Accessed March 5, 2002.
Chambliss WG. 2001. Nutraceutical formulations: Impact on bioavailability. In: Examining the Science Behind Nutraceuticals: Proceedings of the AAPS Dietary Supplements Forum. Arlington, VA: AAPS Press. Pp. 367–380.
FDA (Food and Drug Administration). 1997. Current good manufacturing practice in manufacturing, packaging, or holding dietary supplements; Proposed rule. Fed Regis 62:5699–5709.
Foreman J. 2000. St. John’s wort: Less than meets the eye. Online. The Boston Globe. Available at http://www.boston.com/globe/search/stories/health/health_sense/011000.htm. Accessed March 5, 2002.
GAO (General Accounting Office). 1997. Medical Device Reporting: Improvements Needed in FDA’s System for Monitoring Problems with Approved Devices. HEHS-97–21. Washington, DC: U.S. Government Printing Office.
Green GA, Catlin DH, Starcevic B. 2001. Analysis of over-the-counter dietary supplements. Clin J Sport Med 11:254–259.
Hamilton-Miller JM, Shah S, Winkler JT. 1999. Public health issues arising from microbiological and labeling quality of foods and supplements containing probiotic microorganisms. Public Health Nutr 2:223–229.
Howe K. 2000. Herb Remedies: Panacea or Problem? Online. San Francisco Chronicle. Available at http://www.sfgate.com/cgibin/article.cgi?file=/chronicle/archive/2000/06/02/MN75760.DTL. Accessed March 5, 2002.
Kamber M, Baume N, Saugy M, Rivier L. 2001. Nutritional supplements as a source for positive doping cases? Int J Sport Nutr Exer Metab 11:25 8–263.
Marrone CM. 1999. Safety issues with herbal products. Ann Pharmacother 33:1359–1362.








