
1
Introduction
The level of trust that has characterized science and its relationship with society has contributed to a period of unparalleled scientific productivity. But this trust will endure only if the scientific community devotes itself to exemplifying and transmitting the values associated with ethical scientific conduct.
National Academy of Sciences (NAS, 1995, p. v)
The scientific community today recognizes how crucial it is to understand and to honor ethical research conduct as well as scientific progress if it is to sustain the trust placed in it by policymakers and the public, including parents who are considering whether to enroll their child in clinical research. This report examines how this recognition has been demonstrated in the development of policies and practices to protect the safety and well-being of the children who participate today in research that advances the future prevention, diagnosis, and treatment of child health problems. It also describes continuing problems and concerns and makes recommendations for further action by policymakers and those who sponsor, conduct, review, and monitor research.
The benefits that biomedical research has brought to infants, children, and adolescents are remarkable. In recent decades, research has helped change medical care and public health practices in ways that, each year, save or lengthen the lives of tens of thousands of children around the world, prevent or reduce illness or disability in many more, and improve the
quality of life for countless others. Beyond the infants, children, and adolescents directly affected, the benefits of research extend to the families, friends, and communities who love and care for them.
Since the 1950s, research has led to polio, measles, and other vaccines that have dramatically cut child deaths, disability, and discomfort from communicable diseases (CDC, 1999). Similarly, many premature babies with underdeveloped lungs who once would have died now survive with the use of mechanical ventilators and surfactants (substances that make breathing easier). Statistical analyses of clinical trial data have suggested a 30 to 40 percent absolute decrease in the number of deaths among affected infants after the adoption of surfactant therapy (Jobe, 1993). With improved therapies, the rate of mortality from acute lymphocytic leukemia (formerly called acute lymphoblastic leukemia) dropped by 65 percent between 1975 and 1999 for children under age 20 years (Ries et al., 2003).
Children and their families have also benefited from research identifying the unanticipated harms or ineffectiveness of what were once standard therapies. For example, in the 1940s and early 1950s, an epidemic of blindness occurred among premature newborns who were routinely treated with high-dose oxygen, which at that time was almost universally viewed as reducing the risk of anoxic brain injury (Silverman, 1977). Three controlled clinical trials demonstrated oxygen’s toxic effects on the developing retina (James and Lanman, 1976). Another once widely used practice that long-term follow-up studies showed to be dangerous was irradiation for purported thymus enlargement in young children (see, e.g., Shore et al., 1985, 1993).
Despite many advances, pediatricians have argued that infants, young children, and adolescents have not shared equally with adults in the achievements of biomedicine (see, e.g., AAP, 1977, 1995). Most attention has focused on pharmaceutical research. Surveys of the Physician’s Desk Reference (a comprehensive guide to pharmaceuticals that includes prescribing information) found in 1973 and again in 1991 that approximately 80 percent of the medications listed had no prescription information for children (Wilson, 1975; Gilman and Gal, 1992; both cited in AAP, 1995). These analyses did not assess which drugs were realistically candidates for use with children, but they nonetheless suggested an information gap for clinicians and families who were searching for safe and effective medications for sick children. This information gap leaves physicians with the choice of not prescribing such medications for children (and thus potentially undertreating them) or using the medications based on their or their colleagues’ experience and judgment about whether and how data from studies with adults might apply to children of different ages.
In fact, children differ physiologically from adults in myriad ways that can affect how drugs work in the body. Extrapolation based on adult drug
doses can be dangerous and lead to underdosing, overdosing, or specific adverse effects that do not occur in adults. Such extrapolation and unsystematic “experimentation” thus may expose children to risk while simultaneously failing to generate a trustworthy knowledge base for future care. For example, the drug cyclosporine was approved for adults in 1982 to counter immune system rejection of transplanted organs. The drug was then used in children without testing in clinical trials and without the same degree of success as achieved in adults. Eventually, researchers discovered that young children metabolize cyclosporine much more quickly than adults and thus need more frequent dosing to maintain therapeutic levels of the drug. For more recent immunosuppressive agents, the National Institutes of Health (NIH) and pharmaceutical companies have sponsored clinical trials to test the agents’ action and effectiveness in children prospectively (Hoppu et al., 1991; Harmon, 2003; Schachter et al., 2004).
Another example of problems created by lack of pediatric studies is the undertreatment of children with schizophrenia because many drugs that have helped adults have not been tested in studies with children (Quintana and Keshavan, 1995; Findling et al., 2000). Additional examples of research shortfalls are cited in Chapter 2.1
Laboratory experiments, animal studies, and research involving adults helped lay the foundation for many of the research advances cited above, but most ultimately required studies involving children. Some advances, for example, the use of surfactants to treat hyaline membrane disease, required studies that could not be done initially with adults because only infants have the disease. Other advances (e.g., those involving chloramphenicol) required participation in research by children in several age groups to identify different developmental effects. Often, the research involved ill children, including premature babies. Sometimes, it depended on participation by healthy children, for example, in vaccine studies.
In recent years, both NIH and the Food and Drug Administration (FDA) have adopted policies to increase the amount of clinical research involving children. These policies are discussed in Chapter 2.
Notwithstanding the expected benefits of policies to increase the amount of research involving infants, children, and adolescents, some cau-
tion is appropriate. Unlike most adults, children usually lack the legal right and the intellectual and emotional maturity to consent to research participation on their own behalf. Their vulnerability demands special consideration from researchers and policymakers and additional protections beyond those provided to mentally competent adult participants in research.
As discussed later in this chapter, instances of unethical research practices involving children have prompted public criticism and concern that has contributed to the development of current federal regulations to protect both child and adult participants in research. Since the 1960s, policymakers, researchers, research institutions, and research sponsors have taken a number of steps to strengthen ethical standards and policies for human research and to create formal programs, including institutional review boards (IRBs), to approve and monitor research. Clinical studies funded, conducted, or regulated by the government are now subject to a (mostly) common set of provisions for the protection of human participants in research, including special protections for children. One result is that some potentially important clinical studies that would be approved for adult participation cannot be approved for participation by children.
At the same time, the challenges in implementing human research protection policies consistently and effectively have multiplied as clinical research has increased in size, scope, and complexity. For example, multisite studies are now the norm for much research involving children, with a consequent increase in opportunities for delays and variations in protocol reviews and approvals across different sites.
Scientific advances, such as those emerging from the Human Genome Project, have created new challenges for the assessment of risk and benefit in research involving children. As new knowledge about genetic risk emerges, the psychological consequences of knowledge may become more or less serious for children and families, as may the social and economic harms that could follow a breach of confidentiality.
Despite the strengthening of human research protection policies and programs and in the face of highly complex advances in biomedical science, deficiencies in the conduct of research—some resulting in deaths or serious injuries—continue to be exposed. The 1999 death of 18-year-old Jesse Gelsinger, legally an adult, in a gene transfer trial at the University of Pennsylvania led to widely publicized investigations and discoveries of numerous deficiencies in gene transfer trials (see, e.g., Thompson, 2000a and Weiss and Nelson, 2000). These deficiencies included the substantial underreporting of serious health problems involving participants in the trials. As one recent report concluded, “the system intended to protect [Jesse Gelsinger] from unacceptable risks in research instead failed him” (IOM, 2001, p. 4).
Less dramatic examples of deficiencies in the conduct or review of
research, some involving children, have also been identified. For example, the federal Office for Human Research Protections (OHRP) has cited several major research universities for deficiencies in their oversight of studies involving children. (The letters of determination for the years since 2000 can be viewed at http://ohrp.osophs.dhhs.gov/detrm_letrs/lindex.htm.)
These and other problems make clear that the design of standards, policies, and formal programs to protect research participants must be matched by consistent, effective implementation. As a consequence, recent years have seen more efforts to monitor policy implementation, to match responsibilities with adequate resources, and to hold investigators and institutions accountable for fulfilling their responsibilities. Still, concerns persist about the adequacy, interpretation, and application of standards and policies for research involving humans, including infants, children, and adolescents. Another area of concern is whether the various administrative and other burdens or costs imposed by protective regulations are, in all cases, justified by the contribution that they make to the goal of protecting children from unethical or harmful research.
These concerns, combined with the public commitment to expanding clinical research to benefit children, provided the impetus for this study. The major themes of this report are
-
Well-designed and well-executed clinical research involving children is essential to improve the health of future children—and future adults—in the United States and worldwide. Failure to undertake such research can deny children timely access to effective new therapies and expose them to harm from therapies not specifically demonstrated to be safe and effective for children, including infants and adolescents. Children should not be routinely excluded from potentially beneficial clinical studies, and no subgroup of children should be either unduly burdened as research participants or unduly excluded from involvement.
-
A robust system for protecting human participants in research in general is a necessary foundation for protecting child research participants in particular. An efficiently administered, effectively performing system with adequate resources must, however, commit additional resources and attention to meet ethical and legal standards for protecting infants, children, and adolescents who participate in research. All investigators conducting studies that include infants, children, and adolescents should work under the umbrella of a formal program for the protection of human research participants.
-
Effective implementation of policies to protect child participants in research requires appropriate expertise in child health at all stages in the design, review, and conduct of such research. This expertise includes knowledge of infant, child, and adolescent physiology and development as well as
-
awareness of the unique requirements and challenges of pediatric clinical care and research. It also includes understanding of ethical principles and regulatory requirements specific to child participants in research and appreciation of the family systems in which decisions about children’s clinical care and research participation are made.
ORIGINS OF STUDY AND OVERVIEW OF REPORT
This report was provided for in the Best Pharmaceuticals for Children Act of 2002 (P.L. 107-109). The broad purpose of the legislation was to improve the safety and efficacy of drugs for children. One key provision renewed incentives for pharmaceutical manufacturers to test drugs in studies with children to establish the safe doses of medications that had been approved as safe and effective in adults. The legislation also called for a study by the Institute of Medicine (IOM) of research involving children.
The IOM, which is the health policy arm of the National Academy of Sciences, was to prepare a report that reviewed federal regulations, reports, and research and made recommendations about desirable practices in ethical research involving children. Appendix A describes the specific topics to be considered and the information strategies used in developing the report. The IOM appointed an expert committee of 14 members to prepare this report, which covers children of all ages, including infants and adolescents. In its work, the committee focused primarily on research that involved preventive, diagnostic, treatment, or similar interventions and direct interactions with children. It examined, but less intensively, research that involved only observation, questionnaires, medical records, or stored samples of blood or other biologic material. The committee also did not consider in depth other important questions, including financial and other conflicts of interest, standards for pediatric research in developing countries, priorities for pediatric research, scientific methods, scientific misconduct, and appropriate review of low-risk social science research. Past reports from the IOM and the National Research Council have examined a number of these issues as well as the topics that are the primary focus of this report.2 As far as the
committee could determine, this is one of very few comprehensive reports on ethical issues in research involving children since the first major report on this topic in 1977 (National Commission, 1977).
This report presents the committee’s analysis and recommendations. It is written for a broad audience that may not be familiar with the technical aspects of clinical research nor the intricacies of federal regulations.
The remainder of this chapter offers arguments for a systems perspective on human research protection, summarizes core principles for ethical research involving humans, and reviews the evolution of policies based on these principles. Chapter 2 examines the necessity for clinical research involving children, the challenges in undertaking such research, and initiatives to encourage pediatric research. It also discusses how different government agencies and private groups or individuals conceptualize the periods of infancy, childhood, and adolescence. Chapter 3 reviews the regulations governing human research generally and pediatric research specifically.
In Chapter 4, the focus is on the interpretation and application of ethical principles and federal regulations relating to the assessment of risks and potential benefits in pediatric research. The chapter includes several recommendations intended to encourage greater consistency in the interpretation of the regulations. Chapter 5 turns to the question of what children and parents understand about participation in research; it makes recommendations about processes for seeking parent’s permission and children’s assent to research participation. Chapter 6 examines the question of paying for children’s participation in research and makes recommendations for IRBs.
Chapter 7 considers compliance with the regulations governing pediatric research. The chapter also discusses accreditation and quality improvement as strategies for improving performance. The final chapter discusses the roles and responsibilities of IRBs as well as investigators, regulators, and others whose actions affect the safety and well-being of child participants in research.
Appendix B presents an in-depth review of state laws relating to children’s agreement to medical care and research participation. Appendix C briefly considers other protections for research participants beyond those emphasized in the text of the report. Appendix D includes a glossary, and Appendix E contains short biographies of committee members.
DEFINITIONS: RESEARCH, CLINICAL RESEARCH, AND HUMAN SUBJECTS OR RESEARCH PARTICIPANTS
As defined in federal regulations, research is “a systematic investigation, including research development, testing and evaluation, designed to develop or contribute to generalizable knowledge” (45 CFR 46.102(d)).
The regulations also refer more specifically to research that either generates data through intervention or interaction with the individual or obtains identifiable private information about an individual (45 CFR 46.102(f)).
What constitutes an effort to develop generalizable knowledge is the subject of some disagreement and confusion (see, e.g., NBAC, 2001b). For example, student “research” projects involving questionnaires or observation are often intended to teach students about research design and techniques, statistical analysis, scientific methods, and, more broadly, scientific thinking. Some may qualify as research, whereas others are clearly learning exercises that hold no promise of creating generalizable knowledge.
Questions have also arisen about certain kinds of institutional projects to improve the quality of their medical care by systematically assessing the link between processes of care and health or other outcomes (see, e.g., Brett and Grodin, 1991; Casarett et al., 2000; Bellin and Dubler, 2001; and NBAC, 2001b). These quality improvement projects use systematic planning, control, assessment, and intervention methods that rely on many scientific precepts, methods, and analytic strategies that are also used in health services and other kinds of research (see, e.g., Berwick et al., 1990; Batalden et al., 1994; Nelson et al., 1998; and IOM, 2000a). Some projects are undertaken from the outset with the intent to generalize and publish findings and so qualify as research. Many other projects have only internal, institutional goals and do not constitute research. Like other routine management decisions and actions that are clearly not research, quality improvement activities may cause harm, be monitored for consequences, and even be described in trade publications. Drawing the line between research and certain health care management strategies continues to be a challenge and suggests the need for better communication between human research protection programs and institutional quality improvement activities (see, e.g., Bellin and Dubler, 2001).
Some questions also arise about the boundary between clinical research and clinical practice innovations by individual physicians. Typical examples of such innovations include a surgeon’s modification of an existing surgical technique or trying different mechanical ventilation strategies for patients with respiratory distress. The general view is that radically new procedures should “be made the object of formal research at an early stage in order to determine whether they are safe and effective” (National Commission, 1978a, p. 3). The definitions of “radically new” and “early stage” are, however, controversial.
For purposes of regulatory oversight, the National Bioethics Advisory Commission (NBAC) recommended that research should be considered to involve human participants “when individuals (1) are exposed to manipulations, interventions, observations, or other types of interactions with investigators or (2) are identifiable through research using biological materials,
medical and other records, or databases” (NBAC, 2001b, p. 40). Thus, research involving human biological materials, medical record data, or other information that cannot be linked to identifiable individuals is not human research in this context. NBAC also recommended that federal policy explicitly identify research activities that are not subject to federal regulations.
Clinical research is commonly viewed as research that uses human participants to test the safety or effectiveness of medical interventions (e.g., drugs or diagnostic tests) or to study the diagnosis or pathophysiology of diseases, disorders, or injuries. Synonyms include clinical study and clinical investigation. A clinical experiment is one kind of clinical research. Consistent with the FDA’s statutory mandate, agency regulations on the protection of human subjects define clinical investigation as “any experiment that involves a test article [e.g., a drug or medical device] and one or more human subjects and that either is subject to requirements for prior submission to the Food and Drug Administration… [or] … the results of which are intended to be submitted later to, or held for inspection by, the Food and Drug Administration as part of an application for a research or marketing permit” (21 CFR 50.3(c)).3 More broadly conceived, “clinical investigation … includes all studies intended to produce knowledge valuable to the prevention, diagnosis, prognosis, treatment, or cure of human disease” (IOM, 1994a, p. 35). Disease, in this context, can be interpreted to include disorders and injuries. This broad definition encompasses biomedical research and certain kinds of psychosocial, health services, and epidemiological studies, as well as laboratory research involving, for example, tissues, cells, and genes. As explained earlier, the emphasis in this report is on clinical research that involves direct interactions with child participants in research.
Some statements of ethical principles for research have made an implicit or explicit distinction between therapeutic and nontherapeutic research.4 The former category of research would, for example, include the
administration of a new combination of chemotherapeutic agents for the treatment of leukemia to test the hypothesis that the experimental agents will provide a benefit over standard therapy. In contrast, a study involving various tests intended solely to increase knowledge of the pathophysiology of a disease would be nontherapeutic, although the knowledge gained might contribute to the development of a therapy that might subsequently benefit those who had participated in the study. Federal regulations on protection of human subjects in research do not use therapeutic-nontherapeutic distinction but refer to interventions with the prospect of direct benefit or with no prospect of direct benefit to participants (45 CFR 46.405 and 46.406). This wording, which is also adopted in this report, puts the focus not on the research as a whole but rather on the characteristics of the specific interventions that are included in a study. Some of the interventions may have the prospect of direct benefit whereas others may not. Under federal regulations, these distinctions can affect what aspects of a research protocol are approvable by an IRB. Another problem with the characterization of studies as therapeutic or nontherapeutic is that such labeling may contribute to the common confusion between clinical care and clinical research. Chapters 4 and 5 discuss these distinctions further.
Federal regulations define a human subject of research as “a living individual about whom an investigator (whether professional or student) conducting research obtains (1) data through intervention or interaction with the individual, or (2) identifiable private information. Intervention includes both physical procedures by which data are gathered (for example, venipuncture) and manipulations of the subject or the subject’s environment that are performed for research purposes)” (45 CFR 46.102(f)). This report generally follows the practice of recent IOM and other reports in referring to research participants rather than subjects (see, e.g., IOM, 2001; 2003a; and NBAC, 2001b). This usage recognizes the subjects of research as members of a research project who may, depending on their maturity and capacities, have their own special responsibilities, for example, adhering to drug, diet, exercise, or other intervention protocols. It also conveys a more respectful stance. Although the 2001 NBAC report also supported the use of the term participants, it noted that the term subject portrays more accurately than any other “the relationship and the unequal balance of power between the investigator and the individual in the research” (NBAC, 2001b, p. 33).
Parents sometimes participate with their children in clinical studies, for instance, when a study assesses the health knowledge, beliefs, or practices of both. Even when parents are not research participants in this direct way, they may be “surrogate” participants in certain respects; for example, when outcome measurements rely in whole or in part on parental assessments of aspects of the child’s quality of life.
A SYSTEMS PERSPECTIVE ON PROTECTING HUMAN PARTICIPANTS IN RESEARCH
Although many have accepted the wisdom of Henry Beecher’s observation more than three decades ago that in addition to informed consent, “there is the more reliable safeguard provided by the presence of an intelligent, informed, conscientious, compassionate, responsible investigator,” it would be unfair and unrealistic to expect individual clinicians and researchers, who often face multiple conflicts of interest, to both recognize and resolve by themselves the complex moral problems arising from the use of human subjects in research trials. It is not adequate to focus these ethical responsibilities only on the individual investigator who, in fact, functions within a much broader research and clinical environment.
National Bioethics Advisory Commission (NBAC, 1998, p. 15)
Clinical Research as a Complex, High-Stakes Enterprise
Clinical research today is a complex, high-stakes enterprise. A clinical trial may cost many millions of dollars, and one recent estimate put the cost of developing a new drug at nearly $900 million (including postmarketing studies) (Kaitin, 2003). The challenges of accommodating the physical, intellectual, social, and emotional characteristics and needs of infants, children, and adolescents may make pediatric research even more costly than studies that involve only adults.
For commercial sponsors, the financial rewards of positive research findings can be substantial, particularly when the population of potential users is large. Increasingly, research institutions and investigators too can reap substantial economic rewards from research. In addition, the careers of investigators and the stature of research institutions often hinge on success in the competition for research funding and the publication of findings in prestigious journals.
Nonfinancial conflicts of interest related to professional advancement or stature may be as potent as financial conflicts (see, e.g., NBAC, 2001b; Levinsky, 2002; and IOM, 2003a). An important rationale for requiring the inclusion and, indeed, increasing the proportion of nonscientists and community members on IRBs is to provide balance by involving individuals who are independent of research institutions and sponsors (IOM, 2003a). Another concern about conflicting interests arises when physician investigators recruit their own patients. In these situations, patients’ decisions may be influenced by feelings of obligation, worry about antagonizing someone on whom they depend, or confusion about the goals of the physician as a
researcher versus the goals of the physician as a clinician. Given the pressures on trial enrollment created by the often small numbers of eligible children, the potential for physician role conflict must be taken seriously.
Clinical research is often organizationally and socially complicated, reaching far beyond the boundaries of single institutions. In many clinical trials, investigators work in teams that must develop and negotiate topics and protocols with multiple additional participants. These participants are likely to include government or private sponsors (or both), at least one and sometimes several research review boards, and possibly legal advisers for different parties. Depending on the study, sites might include sophisticated medical centers, community hospitals, nursing homes, private physicians’ offices, research participants’ homes, schools, or other locations or combinations of locations. For many pediatric studies in particular, recruitment of sufficient numbers of research participants may take years and require several, even dozens, of study sites.
Value of a Systems Perspective
Given the complexity of modern clinical research and the stakes involved, an effective program for protecting human participants in research cannot focus narrowly on individuals or organizations (IOM, 2001, 2003a). Rather, a broader perspective is needed that envisions a system of interrelated structures, policies, procedures, and resources that function successfully across institutional boundaries to protect adult and child participants in research. Relevant structures include staff positions and organizational units (e.g., university offices of research administration, institutional and freestanding IRBs, and government regulatory offices). Policies include both public and private rules governing individual and organizational behavior (e.g., federal laws and regulations providing special protections for child participants in research, institutional policies relating to conflict-of-interest disclosures and determinations, and journal policies on conflict of interest and informed consent). Procedures are the mechanisms for carrying out policies (e.g., information collection and reporting arrangements and methods for collecting and analyzing data on adverse events in research). Resources include funding, laws, training in research ethics and methods, and leadership. The central objective of this system of interrelated elements is to protect research participants by encouraging and sustaining responsible behavior from all those involved in sponsoring, reviewing, monitoring, or regulating research and disseminating research findings.
This systems perspective can be applied to clinical research involving children by considering whether each component of the system is adequate to the specialized responsibilities of protecting child participants in re-
search. For example, does an IRB have sufficient expertise in child health to review the kinds of pediatric research protocols that come before it? Is sufficient expertise in child health present on the safety monitoring boards that monitor injuries and other adverse events that occur during the course of a study? Are information systems organized to report separately on protocols involving children?
The recent report Integrity in Scientific Research observed that the research environment, like any system, includes both “variables and constants” and that “the most unpredictable and influential variable” is the individual investigator (IOM/NRC, 2002, p. 26). Each investigator’s professional integrity is shaped by his or her education, culture, and ethical upbringing and is, inevitably, unique. This means that “the constants” operating in behalf of ethical conduct must come from the institutions and larger systems within which investigators work.
One advantage of considering human research protections in a systems framework of shared responsibilities is that it reduces the temptation to focus too narrowly on discrete individuals and organizations and, thereby, to underrate or ignore the diverse forces that powerfully shape their behavior. Figure 1.1 depicts, in highly simplified form, a program of human research protections operating within a larger social, economic, and political environment and a surrounding ethical culture and climate.
As shown in Figure 1.1, a significant system component is a human research participants protection program. A program in this sense is not a discrete IRB but, rather, a variable mix of individuals, organizational units, and organizations (see the discussions in IOM, 2001 and 2003a). The core functions of such a human research participants protection program include review of research protocols for ethical and scientific soundness, monitoring of participant safety appropriate to the risk presented by individual studies, ethical interactions between investigators and research participants, and arrangements for assessing compliance with rules and policies and improving program performance.
The specific components or modules of a human research protection program may differ depending on the characteristics of a particular study (e.g., the setting or the risks to participants), its sponsorship, and other factors. A program consists of the collection of organizational structures, policies, and procedures that apply to a particular research protocol or group of protocols. Thus, a program may include a body appointed to monitor data related to research participant safety if a study presents appreciable risk to participants, but such a body will not be part of a minimal risk study. (See discussion in Chapter 3 of data and safety monitoring boards and data monitoring committees.)
For complex multicenter clinical trials, the human research protection program may involve multiple research organizations, IRBs, research teams,
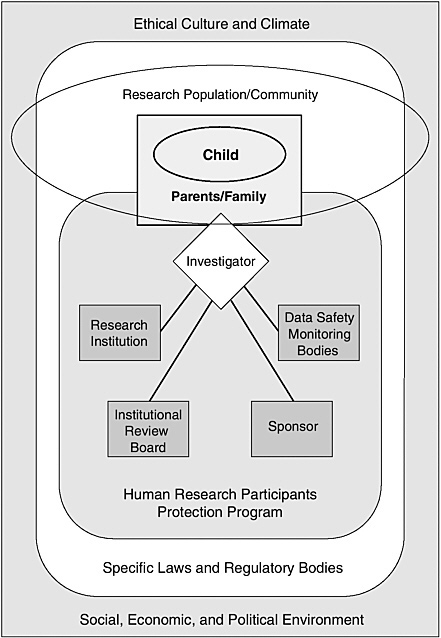
and even research sponsors. The investigators for such trials may have to cope with different and possibly conflicting institutional policies and practices, legal frameworks (e.g., different state policies on when minors make decisions in their own right), social and economic conditions, and community cultures and ethical norms. Even within a single community, investigators, research participants, and others involved may be immersed in or influenced by more than one culture.
The government has recognized the importance of a systems perspective in its creation of a quality improvement initiative that will “work together with all components of the human research community (e.g., subjects, institutions, IRBs, investigators, sponsors, and the public)” to strengthen programs for protecting human participants in research (OHRP, 2002b, p. 1). The statement announcing the initiative noted that “public trust in our nation’s human research enterprise is threatened” and that investigations have “too frequently discovered serious systemic deficiencies” in programs for protecting human participants in research (OHRP, 2002b, p. 1).
An important but underdeveloped part of a system for research protections for children is the prospective, rigorous evaluation of potential long-term benefits and harms of research and the identification of emerging or nontraditional research risks. For example, given the rapidly evolving state of knowledge of the human genome, it is important for investigators, IRBs, and sponsors of research involving children to develop methods to identify and evaluate risks that are unique to or specially evident in genetic research. Such methods should take into account the long-term nature of the potential psychological effects of such research on children who are developing cognitively, emotionally, and socially. They should also consider the risks to family members and family relationships. Although the risks of adverse drug reactions may be clear for all involved in a traditional drug study, such is not the case for the risks of learning (based on genetic investigations) that one will or may develop a debilitating or lethal disease. Long-term follow-up of child research participants and their families will help identify risks that are not now well understood and thereby provide a basis for better protecting children and families from future harm.
ETHICAL PRINCIPLES FOR A SYSTEM TO PROTECT HUMAN RESEARCH PARTICIPANTS
If a study is unethical to start with, it does not become ethical because it produces useful results.
Henry Beecher, 1970, p. 122
The core ethical principles for protecting the dignity and well-being of
human participants in research originate from a variety of historical and philosophical sources, some of which are discussed further in the next section of this chapter. Today, in the United States, the most widely cited statement of these ethical principles is the Belmont Report of the National Commission for the Protection of Human Subjects of Biomedical and Behavioral Research (hereafter referred to as the National Commission) (National Commission, 1978a).5 The U.S. Congress created the National Commission in 1974 and charged it with, among other tasks, identifying basic principles for ethical research involving human subjects and developing ethical guidelines for applying those principles to the conduct of research. The charge also called for the National Commission to examine issues in research involving fetuses, prisoners, children, and those with mental disabilities, which it did in a series of additional reports (National Commission, 1975, 1976, 1977, and 1978b). Although the principles laid out in the Commission’s reports are generally accepted, their interpretation or their application in specific cases may be unclear or contentious.
As summarized in Box 1.1, the Belmont Report presented three basic principles to guide ethical research involving humans: respect for persons, beneficence, and justice. In some formulations, respect for persons is labeled autonomy and beneficence is subdivided to distinguish a fourth principle, nonmaleficence (see, e.g., Beauchamp and Childress, 1994, and Jonsen et al., 1998). The latter division has ancient roots in the injunction of Hippocrates “to help or at least to do no harm” (Goold, 1923, p. 165). Although ethicists and others differ in their analyses of these principles, the following overview presents the committee’s perspectives. Later chapters offer additional discussion as indicated below.
The principle of respect for persons underlies the emphases on the confidentiality of personal information and the provision of voluntary, informed consent for both medical treatment and participation in research. Voluntary participation in research also entails the freedom to withdraw from a study. Respect for research participants further requires that participants not be asked to expose themselves to risks or invest their time and energy in studies that are directed at unimportant questions or that are not properly designed to answer the research question.
The Belmont Report emphasized protection of vulnerable individuals as an element of respect for persons. A somewhat different perspective not mentioned in the report stresses respect for children’s emerging autonomy (or respect for the capacities of other vulnerable individuals) as a basis for involving them in decisions, consistent with their capabilities. As discussed
|
BOX 1.1
SOURCE: National Commission, 1978a. |
in Chapters 3 and 5, the seeking (when appropriate) of children’s assent to research participation demonstrates such respect, but legal permission for participation must ordinarily come from parents.
It can be argued that the Belmont Report weakened the argument for respecting autonomy by joining it with the argument for protecting the vulnerable from undue influence or coercion (Kopelman, in press). For individuals who are capable and competent to make their own decisions and who are not harming others, the report’s formulation of the principle of respect for persons should not be interpreted to permit a balancing of autonomy against protection. For these competent individuals, respect for persons generally takes the form of noninterference within broad limits.
The Belmont Report observes that it is not possible to draw a precise line between justifiable persuasion and undue influence. It notes that unjustifiable pressure typically involves an individual with authority or commanding influence (e.g., a physician who could determine treatment options for a patient) over a prospective research participant. Although the report does not mention financial incentives for research participation, much attention has been devoted to payments to research participants as a potential source of undue influence (see Chapter 6 of this report).
In clinical research, particularly research that entails some risk but holds no prospect of benefiting the research participant, respect for indi-
viduals and their right to self-determination may conflict with values that focus on the potential benefits of research to the larger society. As discussed further in Chapter 4, some of the thorniest debates about research ethics involve pediatric studies that present some risk to children and that offer no prospect of direct benefit but promise to build knowledge beneficial to children in the future. Studies of the mechanisms of disease typically fall into this category.
The real-world application of the moral principle of respect for persons faces a number of practical difficulties in clinical care and research. These include imbalances in information and power between clinicians and patients and between investigators and research participants or their parents. For parents, the physical and emotional stresses associated with a child’s illness or injury and, frequently, the time constraints on decision making also may compromise their ability to obtain, absorb, and evaluate information, weigh options, and then provide truly informed permission for their child to be treated or enrolled in research. How to move from the general principle of respect for persons and the abstract concept of informed consent to effective implementation and desired outcomes is a major, open question for clinicians, investigators, administrators, ethicists, regulators, and others concerned with clinical care and research. Chapter 5 returns to this question as it examines what is known about children’s and parent’s comprehension of research and about parent’s permission and children’s assent to research participation.
The principles of beneficence and nonmaleficence specifically direct attention to the potential benefits and harms of participation in research. From these principles derive the responsibilities of investigators to maximize potential or expected benefits in research, minimize risks (i.e., potential harms), and balance or weigh the potential harms to an individual of participating in research against the potential benefits of participation.6 Also, because children usually do not have the intellectual capacity to assess and weigh the potential harms and benefits of research, parents and others have a duty to do this for them.
In the context of research, the principle of justice primarily involves the fair distribution of the potential harms and benefits of participating in research. Children and other vulnerable groups, including prisoners, residents of mental institutions, and the economically disadvantaged (in the United States and other countries), should not be disproportionately used in research or exploited because they are convenient, readily controlled or
coerced, or unduly susceptible to economic or other inducements to research participation. Overuse of vulnerable groups is a special concern when they are unlikely to benefit from the knowledge gained from research. Research in resource-poor countries has been particularly criticized as unjust when it is not responsive to needs of those of countries, for example, when the aim is to develop medical treatments that will not be practical or affordable except in wealthier countries (see, e.g., Edejer, 1999 and NBAC, 2001a).
Underuse as well as overuse of vulnerable groups in research also raises problems of justice when it limits the extent to which a group can experience the potential benefits of research. The principle of justice has been central in successful arguments for the expanded involvement in research of women (as exemplified in the congressional mandate for the Women’s Health Initiative announced by NIH in 1991 [NIH, 1994; IOM, 1994b]), children (see Chapter 2), elderly adults (ASCP, 1991; FDA, 1994b), and minorities (NIH, 1994)—and, generally, for not limiting clinical studies to nonelderly white adult males.
The Belmont Report emphasized the societal benefits “that serve to justify research involving children—even when individual research subjects are not direct beneficiaries” (National Commission, 1978a, p. 7). It did not review the debate on this point. The earlier National Commission report on children, however, included a lengthy discussion of the arguments for and against subjecting children to research involving some risk but no prospect of direct benefit (National Commission, 1977). In particular, that report reviewed the arguments of theologian Paul Ramsey that only potentially beneficial research was ethically permissible with children or others who could not provide informed consent (Ramsey, 1970). To engage children in research that could not benefit them was to treat them as “means to others’ ends” (National Commission, 1977, p. 93).
The 1977 report presented an extended analysis of Ramsey’s arguments and those who had offered various alternative views, including Richard McCormick, Stephen Toulmin, Victor Worsfold, Stanley Hauerwas, William Bartholome, Tristram Engelhardt, and others. The Commission eventually proposed that “nonbeneficial” research was acceptable but only under conditions more limited than those applicable to adults. The report pointed to the lack of alternative populations for studying certain conditions affecting children, the limitations of extrapolation from adult studies, and the serious consequences for children of prohibiting all child research that did not have the prospect of benefiting the participants.
The complexity and difficulty of the moral arguments about children’s participation in research are reflected in the multiple statements of views from members of the Commission in the report’s final chapter. In essence, the Commission (with two dissents) adopted what may be seen as a utilitar-
ian rationale—albeit a significantly limited one—that “foreseeable benefit to an identifiable class of children may justify a minor increment of risk” to child participants in research in certain restricted situations (National Commission, 1977, p. 125). Chapter 4 of the current report discusses the complexities and controversies in (1) identifying the types, probabilities, and magnitude of potential harms and benefits to which child research participants may be exposed; (2) judging whether the potential harms to the child are reasonable in relation to potential benefits; and (3) assessing whether the potential harms have been minimized.
HISTORICAL EVOLUTION OF POLICIES FOR PROTECTING HUMAN PARTICIPANTS IN RESEARCH
There is a long history of research on children … but a relatively short history of legal control of this activity.
Leonard Glantz, 1994, p. 103
Systematic attention to research ethics largely postdates World War II. For much of the period since the war, policymakers, ethicists, and others have focused on the articulation and refinement of general principles, guidelines, and regulations for research involving humans. Intensive attention to the special ethical issues related to research with children developed rather slowly. Policymakers then took longer to adopt special proposals to increase protections for children than to accept proposals affecting pregnant women, fetuses, and prisoners. Nonetheless, controversies about the ethics of research involving children have frequently served as a stimulus for proposals—if not action—to adopt or strengthen human research protection policies.
Before 1947
In 1945, ’50, the doctor … was king or queen. It never occurred to a doctor to ask for consent for anything … People say, oh, injection with plutonium, why didn’t the doctor tell the patient? Doctors weren’t in the habit of telling the patients anything. They were in charge and nobody questioned their authority. Now that seems egregious. But at the time, that’s the way the world was.
Leonard Sagan (radiologist), 1994
(as cited in ACHRE, 1995, p. 83)
Broadly viewed, research involving children is not an innovation of the twentieth century. Instances of experimentation with children date back centuries. Lederer and Grodin (1994) observed that physicians often used
their own children, children of their servants or slaves, and institutionalized children as subjects for early infectious disease and immunization “experiments” because the children were convenient and lacked experience with the diseases being investigated (p. 4). One widely cited example from the 1790s is Edward Jenner’s experimental injection of his gardener’s son and his own son with cowpox material to vaccinate them against smallpox (NLM, 2002). In the 1700s and later, physicians also used children in experiments with measles, pertussis, syphilis, gonorrhea and other infectious diseases.
The nineteenth and early twentieth centuries saw scattered or passing comments on ethical research conduct.7 In Prussia at the turn of the last century, public controversy over research practices (including the inoculation of healthy children with syphilis serum) led to appointment of a committee that issued recommendations for ethical research practices (Grodin, 1992; Vollmann and Winau, 1996). Prussian authorities subsequently issued the first known governmental directives on research practices in 1900. They advised medical directors of hospitals and clinics that research interventions should not go forward if “the human subject was a minor or not competent for other reasons.” If competent, subjects should provide “unambiguous consent” after a “proper explanation of the possible negative consequences.” The consent was also to be “documented in the medical history” (quoted in Vollmann and Winau, 1996, p. 1446). It is not evident that the nonbinding directive or the ethical analysis supporting it had any effect on research practices (Vollman and Winau, 1996).
At about the same time in the United States, legislative proposals were made after controversies arose about experiments with healthy children in hospitals, orphanages, and schools.8 As recounted by Lederer (1992, 1995) and Lederer and Grodin (1994), experiments with children in the late
nineteenth and early twentieth centuries included investigations of digestive processes (including the use of stomach tubes in infants), deliberate efforts to induce various sexually transmitted diseases to identify their causes and natural history, lumbar punctures, and studies of scurvy in orphaned infants that involved withholding of orange juice. A Swedish physician’s admission that he had used children provided by a foundling home rather than calves in a smallpox experiment because calves were costly prompted a U.S. pamphlet “Foundlings Cheaper than Animals” (Lederer, 1995, p. 50).
One American researcher who used institutionalized children in various studies commented in a 1914 publication that conditions in these institutions were similar to the “conditions which are insisted on in … [infection experiments] among laboratory animals, but which can rarely be controlled in a study of infection in man (Alfred F. Hess, quoted in Lederer and Grodin, 1994, p. 6). Despite considerable controversy, neither public action nor voluntary standards for human research won acceptance in the United States before World War II.
In 1931, the German government issued extensive new regulations protecting human participants in research after controversies over the use of healthy children in harmful studies on tuberculosis vaccines (Grodin, 1992). Among other provisions, the regulations stated that the potential adverse effects of research should be proportionate to the anticipated benefits and that disadvantaged individuals should not be exploited as research participants. The 1931 policies were both unprecedented for the era and profoundly ineffectual under the Nazi regime that took power in 1933.
Most current discussions of research ethics start with the Nuremberg Code’s Directives for Human Experimentation, which were announced by an American military tribunal in 1947 before the verdict in the trial of several Nazi physicians and others for atrocities in medical experiments (Annas and Grodin, 1992).9 (The tribunal convicted 16 of 23 defendants, most of them physicians, of war crimes and crimes against humanity and sentenced 7 of them to be executed.) These directives were the first internationally accepted statement of ethical principles in research. The lead principle stated that “the voluntary consent of the human subject is absolutely essential” meaning that “the person involved should have the legal capacity
to give consent” (Nuremberg Code, 1949, p. 181–182). The directives did not mention children. Strictly construed, they would have precluded research involving children or mentally or legally incapacitated adults.10
Although ethicists, investigators, and policymakers have considerably refined and extended the principles of ethical research (e.g., to cover children), many of the basic tenets in current national and international statements on the conduct of research are similar to those set forth by the Nuremberg judges in 1947. In addition to voluntary, informed, and competent consent, these tenets provided that the research should be necessary and that its risks should be balanced by its social importance and potential benefits. Research should also be designed and conducted by scientifically qualified investigators to produce valid results and minimize risk to participants.
1948 to 1974
It was just that we were so ethically insensitive that it never occurred to us that you ought to level with people that they were in an experiment.
Louis Lasagna on research in 1950s, 1994
(quoted in ACHRE, 1995)
By mid-1960, NIH officials were concerned about the agency’s traditional practice of relying exclusively on the moral character of investigators to safeguard humans in research. Moreover, NIH had no way to monitor the conduct of the investigators it was funding.
Irene Stith-Coleman, 1994, p. 7
As medical research accelerated in the 1950s and 1960s, the Nuremberg principles were both increasingly recognized and increasingly questioned in certain of their specifics (Faden and Beauchamp, 1986; ACHRE, 1995). Some of the questions highlighted the lack of provision for research involving children and others not competent to consent to research in their own right.
Early in the 1950s, the new Clinical Center at NIH developed explicit policies for the protection of human participants in research (which applied to studies conducted at the facility). Among other elements, the policies
provided for peer review of certain kinds of research (e.g., high-risk research, nontherapeutic research involving patients, and research involving healthy volunteers). They also directed attention to ethical questions in the review of research. According to Faden and Beauchamp, “[o]fficials at the Center expected these procedures to set the standard for other institutions … but [this] pioneering venture was an isolated and largely ignored event” (1986, p. 202). For example, as Faden and Beauchamp reported, a survey by researchers at Boston University, which was supported by a federal grant and published in 1962, suggested that few research centers had guidelines for clinical research or even accepted the concept of committee review of protocols.
With respect to research involving children, the 1995 report of the Advisory Committee on Human Radiation Experiments stated that “in the 1940s and 1950s there were apparently no written rules of professional ethics for pediatric research in general” (ACHRE, 1995, p. 203).11 In summarizing the discussion during a 1961 conference on Social Responsibility in Pediatric Research, the same report observed that it was not uncommon for “pediatric patients to be used as subjects of nontherapeutic research without the permission of their parents” (ACHRE, 1995, p. 202). The report also noted that some researchers, including researchers who failed to get parental permission, recognized that this was unethical (see Box 1.2).
In 1962, the U.S. Congress passed legislation that expanded the scope of FDA’s authority by passing amendments to the Federal Food, Drug, and Cosmetic Act (P.L. 87-781). The legislation included provisions that required investigators to obtain a subject’s consent to the use of an experimental drug unless it was not feasible or was not in the subject’s best interest (at Section 501(i)(4)). Four years later, the FDA commissioner issued explicit regulations providing for consent to participation in research, “at least partially in recognition of the widespread failure of the industry to obtain [it]” (Glantz, 1992, pp. 183–200; see also the discussion in Faden and Beauchamp, 1986).
After years of debate within NIH about the balance between ethical principles and scientific inquiry, the U.S. Surgeon General issued policy statements in 1966 that significantly expanded the conceptualization and application of informed consent for external clinical research funded by
|
BOX 1.2 In the opening minutes of the meeting, this researcher reminded his colleagues that “the question for us to discuss here today is how we operate on a daily basis.” He offered for discussion a provocative case from his personal experience in which he and his associates “wanted [to do] lumbar punctures on newborns.” He explicitly noted that “this study [was] not of benefit to the individual; it was an attempt to learn about normal physiology.” One of the other conferees asked, “Did you ask [parental] permission?” The researcher responded, “No. We were afraid we would not get volunteers.” The case prompted a great deal of discussion at the conference, but perhaps most tellingly this researcher frankly acknowledged toward the end of the discussion—in a meeting that had begun with an assurance of confidentiality from the organizers—that he had “sinned” in carrying out these lumbar punctures in “normal infants” without parental permission. SOURCE: Closed meeting on Social Responsibility in Pediatric Research, Boston University, 1961 (as described in ACHRE, 1995, p. 202). |
U.S. Public Health Services grants (U.S. Surgeon General, 1966). The policy statements also required research institutions to establish committees (IRBs) to review proposed human research (Faden and Beauchamp, 1986). This institution-level review was to consider the methods for obtaining informed consent, the balance of risks and benefits in proposed research, and the welfare of the research participants.
The 1966 NIH policies were shaped in part by the 1964 Declaration of Helsinki, published 2 years previously by the World Medical Association (WMA, 1964).12 The Declaration, which has been revised several times, set forth principles for ethical research that enlarged the Nuremberg directives and went beyond the Association’s first statement in 1954 (Annas et al., 1977). The 1964 Declaration distinguished research with aims seen as “essentially therapeutic for a patient” from research with only “scientific aims.” It specified looser standards of consent for the former, recommending that consent be obtained “consistent with patient psychology.” The 1964 document did not specifically mention children or minors, but it did provide for
the consent of legal guardians to the participation in “nontherapeutic” research of those not legally able to provide consent. Subsequent revisions to the Declaration added specific references to children and included provisions for children’s agreement to participate in research (for those children who are capable of providing it). More recent revisions refer to minor’s “assent” rather than “consent,” recommend committee review of research, and call for journals not to accept reports of research that are inconsistent with the Declaration.
During the 1960s, criticisms of unethical research practices in research involving children gained new attention (see, e.g., ACHRE, 1995 and Lowen, 1995). In an often cited 1966 article in the New England Journal of Medicine, Henry Beecher reviewed 22 studies, most of which involved “experimentation on a patient not for his benefit but for that, at least in theory, of patients in general” (Beecher, 1966, p. 367). Four of the studies discussed in the article included children. As described by Beecher, one used multiple spot X-rays to study bladder filling and voiding in babies; another involved the suturing of adult skin grafts to the chest wall of a subset of children being treated for congenital heart disease to examine the effect of thymectomy on growth and development; and a third included some children with mental retardation who were given an antibiotic (for the treatment of acne) to determine whether it caused liver dysfunction (which it did) (Beecher, 1966).
The fourth study involved children at New York’s Willowbrook State School. Researchers infected some of the child participants with a mild form of hepatitis during the initial stages of a study of the natural history, prevention, and treatment of viral hepatitis that extended from 1956 to 1972 and that eventually contributed to the development of a successful hepatitis vaccine. The Willowbrook research also contributed to the public debate over research ethics and the impetus for regulation (see, e.g., Goldman, 1971, 1973; President’s Commission, 1981; Faden and Beauchamp, 1986; Lederer and Grodin, 1994; ACHRE, 1995; and NBAC, 1998). Among the major points of discussion were the infecting of healthy institutionalized children and the adequacy or appropriateness of the process for securing parental consent (permission) during some stages of the study. In the 1970s, controversy over the ethics of this study reached medical journals, major newspapers, and Congressional hearings (see Goldman, 1971, 1973 generally and, e.g., Ramsey, 1970; Edsall, 1971; and Goldby, 1971 who criticized the research and, e.g., Krugman and Shapiro, 1971 and Ingelfinger, 1973 who defended it).
Many of the researchers in the studies cited by Beecher were well regarded, and research oversight committees had reviewed some of the study proposals. In the Willowbrook research, parents had been asked for and had provided consent in an era when that was not uniform practice.
Beyond public controversy about particular studies, problems with the Surgeon General’s 1966 policy statement became a concern. As described in a later report, site visits “to randomly selected institutions revealed a wide range of compliance … [and] widespread confusion about how to assess risks and benefits, refusal by some researchers to cooperate with the policy, and in many cases, indifference by those charged with administering research and its rules at local institutions” (ACHRE, 1995, pp. 100–101). The report also noted widespread complaints about overworked review committees and requests for policy clarification and guidance.
In 1971, what was then the U.S. Department of Health, Education, and Welfare (DHEW) further formalized its policies on protecting human research participants in the Institutional Guide to DHEW Policy for the Protection of Human Subjects. Faden and Beauchamp (1986, p. 212) describe this as a “major monograph on the subject of ethics and regulation of research.” The guide set forth six basic conditions for informed consent, including the condition that the discussion of participation describes risks and discomforts, expected benefits, alternatives to research participation, and freedom to discontinue participation at any time.
The 1971 Guide also required the consent of research participants or their authorized representatives. The Guide stated that review committees “should consider the validity of consent by next of kin, legal guardians, or by other qualified third parties representative of the subjects’ interests … [and] whether these third parties can be presumed to have the necessary depth of interest and concern with the subjects’ rights and welfare … [and are] legally authorized to expose the subjects to the risks involved” (quoted in National Commission, 1977, p. 93).
In 1973, DHEW issued a working document on experimentation with children that proposed several special protections for children (DHEW, 1973b; see also Glantz, 1994). The draft provided that children would be excluded from participation in research under several conditions, one of which was if they were age 6 or over and had not consented to participation—unless the agency waived the requirement.13 The draft also proposed
that an “ethical review board” should review research protocols involving children and that a “protection committee” should monitor aspects of research once it was initiated. This intensity of review does not appear to have been seriously considered in later assessments or policy deliberations.
DHEW did not include special provisions for children in the general regulations that it issued in 1974 (DHEW, 1974). In July 1975, however, the NIH Clinical Center is said to have required for its intramural research program that investigators obtain a child’s agreement to participate in research (National Commission, 1977).
During the 1970s, policymakers and the public were shocked to learn about the Tuskegee Syphilis Study. For more than 30 years, health researchers had followed black men diagnosed with syphilis but had neither informed them of their condition nor treated them for it (Heller, 1972; DHEW, 1973a; Jones, 1992; ACHRE, 1995). Revelations about this study contributed significantly to the passage in 1974 of the National Research Act (P.L. 93-348). That Act explicitly provided for the creation of IRBs to review biomedical and behavioral research that involved humans and was funded by DHEW. As noted earlier, it also established the National Commission and directed it to identify ethical principles for research involving humans with additional attention to research involving vulnerable individuals, including children, prisoners, and those with mental disabilities.
1975 to 1995
By the time that its mandate expired in 1978, the National Commission had produced 17 reports and supplementary documents. The best known is the Belmont Report, which was discussed earlier in this chapter, but other reports were also influential. DHEW revised its 1974 regulations following reports by the National Commission on research involving fetuses (National Commission, 1975) and prisoners (National Commission, 1976). In 1975, the agency added to the general regulations on human research protections (which became Subpart A of 45 CFR 46) a set of special regulations for pregnant women and fetuses and in vitro fertilization (Subpart B). In 1978, it added regulations relating to prisoners (Subpart C).
The U.S. Department of Health and Human Services (DHHS, formerly DHEW) did not adopt specific regulations on research involving children until 1983 (see below), 6 years after the National Commission produced the report, Research Involving Children (National Commission, 1977). That report laid out the case for involving children in research, described the extent of such research, surveyed institutional practices regarding consent for research involving children, and reviewed legal and ethical issues in pediatric research. In contrast to the Belmont Report, which has links from many IRB and other websites related to human research protection pro-
grams,14 neither the 1977 report on children nor its summary appear to be available online for easy reference.15
Two years later, DHHS issued revised general regulations governing human research, but these still did not include special protections for children (DHHS, 1981). The new rules expanded provisions related to informed consent. For example, they included requirements that the process of securing informed consent include descriptions of the extent, if any, to which confidentiality will be maintained, explanations that refusal to participate will not result in a penalty or a loss of benefits, and information about whom to contact with questions or in the event of a research-related injury.
The 1981 regulations also allowed IRBs to exempt or expedite certain categories of minimal-risk research involving, for example, many kinds of educational and survey research. The exempted research did not have to meet the requirements for informed consent, although IRBs might still impose the requirements. These provisions for exempt and expedited research review responded to some of the concerns from the social and behavioral science research communities that the 1974 regulations inappropriately imposed on their fields regulations that had been devised for the often different circumstances and risks of biomedical research.
Finally, in 1983, 10 years after its first proposals and 6 years after the National Commission report on children, DHHS issued special regulations for research involving children (Subpart D). The recommendations of the National Commission formed the foundation for these rules. In 2000, the Children’s Health Act (P.L. 106-310) required FDA to bring its regulations into conformity with the DHHS regulations providing additional protections for children participating in research (FDA, 2001b). The DHHS and FDA regulations are discussed further in Chapter 3 and subsequent chapters of this report.
In 1986, the government published proposed rules to extend the general regulations governing research conducted or supported by DHHS to all federal agencies and all federally supported research. This step followed earlier recommendations by the President’s Commission for the Study of Ethical Problems in Medicine and Biomedical and Behavioral Research
|
14 |
Availability does not guarantee familiarity. In a 1998 discussion with another federal commission, ethicist Albert Jonsen observed that he had recently spoken to group of IRB participants. “What they knew [were] the federal regulations. They didn’t know Belmont” (Jonsen, 1998b). |
|
15 |
During the course of this IOM study, a scanned copy of the report—obtained from committee member Robert Nelson—was made available for viewing on the study’s website. Chapter 8 encourages the Office for Human Research Protections (OHRP) to make the report available as a resource on its website. |
(President’s Commission, 1981). Not until 1991 did the government officially extend the rules to 15 other federal agencies (USDA et al., 1991).16 This step standardized a variety of different agency policies under what is termed the Common Rule. The Common Rule does not include Subpart B, C, or D, although the Department of Education, which also funds considerable research involving children, has adopted Subpart D.
Although FDA has been part of what is now DHHS for many decades (FDA, 2002a), it has separate rules applicable to the research that it oversees based on its specific statutory authority and associated legal codification. The regulations related to protection of human participants in research are found at 21 CFR 50 and 56. The FDA has not adopted the Common Rule as such but has revised its regulations to bring them into general conformity (FDA, 1991a). Interestingly, in 1979, FDA solicited comments on a proposed rule that would apply the principles set forth in the 1973 DHEW regulations to all pediatric research that was subject to FDA jurisdiction (FDA, 1979b). The proposed rules were never adopted and were formally withdrawn in 1991 (FDA, 1991b). In 2001, following directives in the Children’s Health Act of 2000, FDA brought its regulations largely into line with the provisions of Subpart D (FDA, 2001b).
Recent Years
Except for some limited revisions, the regulations issued in the 1980s still govern research conducted, supported, or regulated by DHHS, including research involving children. These regulations are discussed further in Chapter 3. Recent years have been marked by various critical reports on the performance of IRBs and DHHS in implementing these regulations. Chapter 7 reviews some of these reports as well as DHHS responses.
Arguably, the most attention-getting recent developments related to the protection of human participants in research have involved the temporary suspension or restriction of federally funded research at more than a dozen
institutions (NBAC, 2001b). Problems cited by the agency included inappropriate enrollment of patients in research, poor documentation of protocol approvals and continuing review of approved proposals, and deficiencies related to the design, approval, and conduct of research, including a study in which a volunteer died (NBAC, 2001b; McNeilly, 2001). In addition, after the death of Jesse Gelsinger, FDA shut down gene transfer trials at the University of Pennsylvania. Further investigation continues.
Responding to criticisms of its own performance, DHHS has recently taken a number of steps to underscore the importance of human research protections. In 2000, it moved the lead responsibility for issues related to the protection of human participants in research out of an office within NIH and into the Office of Public Health and Science within the Secretary’s Office (DHHS, 2000). To underscore program goals, the Office for Protection from Research Risks became the Office for Human Research Protections (OHRP). FDA took a similar step in March 2001 when it created the Good Clinical Practices Program within the Office of the Commissioner to take the lead on policy issues related to the protection of human participants in research. Individual centers, for example, the Center for Drug Evaluation Research still maintain research monitoring units relevant to their jurisdictions (David Lepay, M.D., Ph.D., Food and Drug Administration, personal communication, October 4, 2003). FDA and NIH have also taken steps to improve oversight and public disclosure of safety programs in gene transfer trials and, in the words of the FDA Commissioner, “restore the confidence in the trials’ integrity that is essential if gene [transfer] studies are to be able to fulfill their potential” (FDA, 2000c, online, unpaged).
Another recent development involves an increasing number of referrals to DHHS of proposed research protocols involving children that IRBs have determined they cannot approve under the federal regulations (sections 404, 405, and 406 of 45 CFR 46) but which can be approved by the Secretary of DHHS under 45 CFR 46.407. For all such proposals, DHHS has created a public review and comment process, which is described further in Chapters 3 and 8. The referred protocols have, to cite a few examples, proposed to test a diluted smallpox vaccine in children, study sleep mechanisms with children, and investigate precursors to diabetes in Japanese youth (DHHS, 2002a,b; DHHS, 2003b).
As noted earlier, OHRP has created a voluntary quality improvement program that includes institutional self-assessment tools and various opportunities for obtaining guidance and counsel from agency staff or through written materials (OHRP, 2002b; see also the discussion in Chapter 7). The initiative also will promote interactions and idea sharing among research institutions and review boards.
Another major development in recent years has been the growing
amount of research funded by American companies that is being conducted in other countries. Recent data indicate that approximately 25 percent of investigational new drug applications include critical data from studies conducted outside the United States (Lepay, 2003). In 2001, the DHHS Office of the Inspector General (OIG) reported that the number of foreign investigators conducting research under an FDA Investigational New Drug filing rose from 41 in 1980 to 271 in 1990 to 4,458 in 1999. The OIG concluded that FDA “receives minimal information on the performance of foreign institutional review boards … [and] has an inadequate database on the people and entities involved in foreign research” (OIG, 2001, p. ii). Furthermore, it “cannot necessarily depend on foreign investigators signing attestations that they will uphold human subject protections” (OIG, 2001, p. ii).
In a report that explored the ethical and practical complexities of overseeing foreign research in more depth, the National Bioethics Advisory Commission also expressed concern about current DHHS policies and procedures (NBAC, 2001a). Chapter 3 discusses federal regulations on human participants in research as they apply to research conducted in other countries.
The globalization of clinical research has encouraged international efforts to “harmonise” (to use accepted international spelling) national regulations and practices relating to human research. Examples include the Guideline for Good Clinical Practice: E6 (ICH, 1996) and Clinical Investigation of Medicinal Products in the Pediatric Population: E11 (ICH, 2000b) of the International Conference on Harmonisation (ICH) and the International Ethical Guidelines for Biomedical Research Involving Human Subjects of the Council for International Organizations of Medical Sciences (CIOMS, 2002, updating guidelines first issued in 1982).17
The movement of clinical research overseas reflects an array of economic, social, and political forces related to such factors as the cost of doing research and the rigors of regulations governing the conduct of research (and the ease of on-site inspection of research sites). It also reflects the higher prevalence of many serious medical problems in less developed countries and, thus, the larger pool of potential research participants. This is not a trivial attraction, given that most children in developed countries are healthy, which means that recruiting sufficient numbers of children for clinical studies is often difficult.
As noted earlier in this chapter, international research can raise problems of justice, particularly when sponsors of research in resource-poor countries largely ignore the needs of those of countries; for example, when knowledge derived from research in those countries will mainly benefit wealthier countries. This may occur when diseases that are rare in wealthy nations but common in poorer countries are neglected or when the prices or costs of new preventive or therapeutic measures are beyond the resources of poorer countries.
A major impetus for the CIOMS guidelines has been concerns about ethics in international biomedical research and the challenges of applying universal ethical principles “in a multicultural world with a multiplicity of health-care systems and considerable variation in standards of health care” (CIOMS, 2002, online, unpaged). The Declaration of Helsinki, last revised in 2000 and 2002, also reflects concerns about justice in research involving resource-poor countries (WMA, 2002).
The next chapter considers many of the challenges in undertaking research involving infants, children, and adolescents. It also outlines the necessity and rationale for this research.

































