
Transcription, Chromatin Assembly, and Homologous Recombination in Chromatin
James T. Kadonaga, Ph.D.
University of California, San Diego
Figure 1 is a picture that I downloaded from the human genome web site. It reflects the considerable effort that is devoted to the study of our genetic material, DNA. However, DNA in the eukaryotic nucleus is not this simple fiber, but instead is packaged into a nucleoprotein complex termed chromatin.
Figure 2 is an electron micrograph of chromatin. This picture displays the periodic nature of chromatin, as the unfolded chromatin filament resembles beads on a string. Figure 3 shows a schematic diagram of chromatin. The DNA (in black) is wound around the core histones in yellow. The linker histone H1 is shown in blue.
The monomeric unit of this repeated structure is called the nucleosome. Figure 4 shows the x-ray crystal structure of the nucleosome, which was solved by Tim Richmond and coworkers a few years ago. It depicts the nice balance between the DNA and the histones. In native chromatin, the DNA to protein mass ratio is approximately 1 to 1. So, it is probably most reasonable to think that the DNA and histones act as partners, rather than as one dominant over the other.
What happens in chromatin? Basically, whatever happens in DNA, happens in chromatin (Figure 5). Here we have things like transcription and DNA replication, repair, and recombination. But there are also chromatin-specific processes, such as chromatin assembly and the post-translational modification of histones. In addition, there is euchromatin in which most transcribed genes are located as well as heterochromatin, which includes centromeres and telomeres.



FIGURE 3 Schematic diagram of the 10 nm diameter chromatin fiber.

FIGURE 4 Crystal structure of the Nucleosome.
SOURCE: Luger, K., Mäder, A. W., Richmond, R. K., Sargent, D. F., and Richmond, T. J. (1997). Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature 389:251-260.

FIGURE 5 A variety of biological phenomena involve chromatin.
I am going to give you three short stories that describe some of the things that we do in my lab. The first story actually does not involve chromatin itself, but instead focuses on the basic transcription process. Second, I am going to talk about chromatin assembly, and then lastly, I am going to say a few words on homologous recombination in chromatin.
TRANSCRIPTION
Figure 6 is from before I used computers to make graphics. What we have here is a typical eukaryotic transcriptional control region. In our genome, we have several tens of thousands of genes, and each one has its own unique transcriptional program. Now, we are faced with the challenge of understanding how the activity of each of these tens of thousands of genes is regulated. One approach to this problem is the analysis of the fundamental mechanisms by which transcription is regulated—and that is the general question that we address.
One key DNA element that is involved in transcription is the core promoter, which contains the information that is needed for the RNA polymerase II transcriptional machinery to initiate transcription. The core promoter is typically about 40 bp in length and encompasses the transcription start site.
Immediately upstream of the core promoter, there is the proximal promoter region. In this region, there are recognition sites for sequence-specific DNA binding transcription factors, such as Sp1. Then, at variable distances from the core promoter—which could be either upstream or downstream of the start site—are transcriptional enhancers. Enhancers typically contain many recognition sites for the binding of a wide variety of sequence-specific DNA binding factors, and can be located as far as 80 kbp from the core promoter.
Figure 7 is a modernized version of Figure 6. Things have gotten more complicated over the years, but we still have the DNA, the core promoter, and the promoter- and enhancer-binding factors. And now, we also have nucleosomes in the picture. In addition, there are coactivators

FIGURE 6 A typical Eukarytotic transcription control region.

FIGURE 7 Many factors affect the regulation of transcription by RNA polymerase II.
that link the activators to the basal machinery as well as all sorts of his-tone-modifying enzymes and nucleosome-mobilizing factors. So, it is a fairly complicated picture.
I am going talk about the core promoter—just what is happening at the RNA start site, and why would anybody ever want to study that? I think that it is important to know how the basic transcription process works in eukaryotes. And second, when you look at the function of all of these factors (shown in Figure 7)—all of these coactivators, coregulators, histone-modifying enzymes, and nucleosome-remodeling factors—after they are done doing what they do, the transcriptional signals ultimately lead to the core promoter. In other words, all transcriptional pathways ultimately lead to the core promoter. Thus, the core promoter is the ultimate target of all of the factors that regulate the initiation of gene transcription.
Today, I am going to focus on the cis-acting DNA elements in the core promoter (Figure 8). Most of you are probably familiar with the TATA box motif that is located about 25-30 nucleotides upstream of the transcription start site. The TATA box is, by far, the most widely studied core promoter motif, and it is sometimes incorrectly thought that all core promoters contain a TATA box. But, in humans, it is estimated that perhaps only about 30 percent of core promoters have a TATA box. It then follows

FIGURE 8 TATA versus DPE-dependent core promoters.
that about 70 percent of core promoters in humans do not have a TATA. We therefore felt that it would be important to understand the core promoter elements that are involved in the transcription of the large proportion of genes that lack a TATA box.
These studies led Tom Burke to the discovery of a downstream core promoter motif called the DPE, which is located about 30 nucleotides downstream of the transcription start site. The DPE is conserved from Drosophila to humans, and is most commonly found in TATA-less promoters. In DPE-dependent promoters, the spacing between the DPE and Initiator (Inr) motifs appears to be invariant. Like the TATA box, the DPE is a sequence-specific recognition site for the TFIID basal transcription factor, which binds cooperatively to the Inr and DPE motifs. Also, if you inactivate a TATA-dependent promoter by mutation of the TATA box, you can restore the activity of the core promoter by the addition of a DPE motif at its appropriate downstream location.
How common is the DPE? Alan Kutach carried out a statistical analysis of about 200 core promoters in Drosophila and found that about 29 percent of the promoters have a TATA box and no DPE, about 26 percent have a DPE and no TATA, about 14 percent have both, and about 31 percent have neither a TATA nor a DPE (Figure 9). So, it appears that the DPE is about as common as the TATA box in Drosophila.
It seemed possible that the fundamental mechanism of basal transcription from TATA-dependent promoters is different from the mechanism of transcription from DPE-dependent promoters. In fact, that appears to be the case. For instance, Trish Willy identified and purified an activity that activates DPE transcription but not TATA transcription, and found that this activity is mediated by a protein termed NC2 (Dr1-Drap1)

FIGURE 9 Frequency of occurrence of TATA box and DPE motifs in Drosophila core promoters.
SOURCE: Kutach, A. K., and Kadonaga, J. T. (2000). The downstream promoter element DPE appears to be as widely used as the TATA box in Drosophila core promoters. Mol. Cell. Biol. 20:4754-4764. Fig. 4A.
(Figure 10). NC2 (Dr1-Drap1) was initially purified by several labs as a repressor of TATA transcription. What Trish found is that it is an activator of DPE transcription. Thus, NC2 (Dr1-Drap1) is a factor that can discriminate between TATA- versus DPE-dependent core promoters. Moreover, Trish identified a mutant form of NC2 (Dr1-Drap1) that is able to activate DPE transcription but unable to repress TATA transcription. This result indicates that the activation of DPE-dependent transcription by NC2 (Dr1-Drap1) is distinct from its repression of TATA-dependent transcription. Trish’s findings exemplify the differences in the basic mechanisms of transcription from DPE-dependent versus TATA-dependent core promoters.
One other question that we asked was—why would a gene have a DPE or a TATA box in its core promoter? One somewhat fanciful idea was that the presence of a TATA or DPE motif might be important for the proper functioning of some transcriptional enhancers. As shown in Figure 11, sometimes you have a cluster of genes in which the yellow enhancer needs to find the yellow promoter, the blue enhancer needs to find the blue promoter, and so on. Enhancer-core promoter interactions may be one means by which such enhancer-promoter specificity is achieved.
To test this idea, Jenny Butler used a Drosophila P-element transformation construct called the waffle vector Figure 12. She inserted two related reporter genes into the waffle vector: DPE-GFP and TATA-GFP. DPE-GFP has a DPE-dependent core promoter, and TATA-GFP has a

FIGURE 10 NC2 (Dr1-Drap1) is a bifuncational, core-pomoter-specific transcription factor.
SOURCE: Willy, P. J., Kobayashi, R., and Kadonaga, J. T. (2000). A basal transcription factor that activates or represses transcription. Science 290:982-984. Fig. 4B.

FIGURE 11 TATA or DPE motif might be important for the proper functioning of some transcriptional enhancers.
SOURCE: Butler, J. E. F., and Kadonaga, J. T. (2001). Enhancer-promoter specificity mediated by DPE or TATA core promoter motifs. Genes Dev. 15:2515-2519. Fig. 1.

FIGURE 12 The waffle vectorgeneates allelic transgenes.
TATA-dependent core promoter. These two genes are identical except for seven nucleotides in the TATA box region and seven nucleotides at the DPE region. Jenny transformed flies with her waffle construct, and then used enhancer-trapping techniques to mobilize the element transiently. The biallelic construct would occasionally land near an enhancer, which would activate transcription from either or both of the reporter genes. To compare the effect of each trapped enhancer upon transcription from DPE-GFP relative to that from TATA-GFP, Jenny created two daughter lines by the use of either FLP recombinase (in blue) or Cre recombinase (in red) in vivo. As shown at the bottom of Figure 12, this procedure results in the generation of two sister lines that have chromosomes that are identical except for the 14 nucleotides at the TATA and DPE regions of the core promoter. In this manner, Jenny was able to determine the effect of randomly-trapped enhancers upon DPE-versus TATA-dependent transcription.
These experiments showed that there are indeed DPE- and TATA-specific transcriptional enhancers Figure 13. Approximately 25 percent of the enhancers that we tested exhibit specificity for the TATA or DPE motifs. Thus, some enhancers function specifically with DPE or TATA elements in the core promoter. More generally, these results indicate that the core promoter is much more than a sequence that directs the proper site of initiation by RNA polymerase II. Rather, the core promoter is an important regulatory element.
A corollary to these conclusions is that if you are studying your favorite enhancer, you should do it in conjunction with its cognate core promoter. It is a common practice to map enhancers by fusing portions of the

FIGURE 13 Identification of a DPE-specific enhancer. SOURCE: Butler, J. E. F., and Kadonaga, J. T. (2001). Enhancer-promoter specificity mediated by DPE or TATA core promoter motifs. Genes Dev. 15:2515-2519. Fig. 3B.
enhancer region with a generic TATA-containing reporter gene. In such an assay, a DPE-specific enhancer would not be detected.
To summarize the studies of the core promoter—we have identified and characterized a new core promoter motif termed the DPE (Figure 8). The DPE is a downstream core promoter element that is located around +30 relative to the transcription start site. The DPE is conserved from Drosophila to humans, and is a binding site for TFIID. In Drosophila, the DPE is about as common as the TATA box. We found that NC2 (Dr1-Drap1), which had been previously studied as a repressor of transcription from TATA-dependent promoters, is an activator of transcription from DPE-dependent promoters. We have also found that the presence of a DPE or TATA motif in the core promoter can have a critical role in enhancer function.
CHROMATIN ASSEMBLY
Next, I will describe some of our studies in the area of chromatin assembly. So, why would anybody want to study chromatin assembly? In eukaryotes, genome replication is really chromosome replication, and chromosome replication requires the duplication of the DNA and the assembly of the newly-replicated DNA into chromatin. In fact, chromatin assembly is required whenever DNA is synthesized, such as during repli-

FIGURE 14 Chromatin assembly is important for chromosome structure.
cation or repair. In Figure 14 I have depicted chromatin assembly as something of a “light blue box” rather than a “black box.”
Our studies of chromatin assembly began with the development of a crude Drosophila embryo extract, termed the S-190 that mediates the ATP-dependent assembly of periodic nucleosome arrays. This was the work of Rohinton Kamakaka and Mike Bulger. (I should point out here that Abe Worcel, in the early 1980s, pioneered the development of an ATP-dependent chromatin assembly extract from Xenopus oocytes.) Our Drosophila S-190 extract was designed, in particular, to be scaled up for the subsequent fractionation and purification of the assembly factors. To this end, Mike Bulger, Takashi Ito, and Jessica Tyler had fractionated, purified, and cloned the factors that mediate the ATP-dependent assembly of periodic nucleosome arrays. These studies led to the discovery of the ATP-utilizing motor protein involved in chromatin assembly, which is termed ACF, as well as one novel histone chaperone, ASF1, and three previously-known histone chaperones, CAF-1, NAP-1, and nucleoplasmin (Figure 15). ACF is the source of the ATP-dependence of the chromatin assembly reaction, and it is needed for both histone deposition and periodic nucleosome spacing. ACF comprises the ISWI ATPase motor and an Acf1 large subunit, which appears to ‘program’ the ISWI motor. Chromatin assembly can be mediated by ACF in conjunction with a core histone chaperone.
Today, we now have a completely purified, recombinant system for the ATP-dependent assembly of periodic nucleosome arrays (Figure 16). This system was developed by Dmitry Fyodorov and Mark Levenstein. It consists of purified recombinant ACF, purified recombinant NAP-1 (as the histone chaperone), purified native (or recombinant) core histones, DNA, and ATP.
We typically monitor the assembly of chromatin by using the micrococcal nuclease digestion assay (Figure 17). In this assay, chromatin is

FIGURE 15 A simple working model for chromatin assembly.

FIGURE 16 Purified chromatin assembly factor and core histones.
SOURCE: Jiang, W., Nordeen, S. K., and Kadonaga, J. T. (2000). Transcriptional analysis of chromatin assembled with purified ACF and dNAP1 reveals that acetyl CoA is required for preinitiation complex assembly. J. Biol. Chem. 275:39819-39822. Fig. 1.

FIGURE 17 Micrococcal nucleas digestion assay.
SOURCE: Ito, T., Levenstein, M. E., Fyodorov, D. V., Kutach, A. K., Kobayashi, R., and Kadonaga, J. T. (1999). ACF consists of two subunits, Acf1 and ISWI, that function cooperatively in the ATP-dependent catalysis of chromatin assembly. Genes Dev. 13:1529-1539. Fig. 4C.
partially digested with micrococcal nuclease, which makes a double-stranded cleavage in the linker DNA between the nucleosomes. Then, the oligonucleosomal fragments are deproteinized, and the resulting DNAs are resolved by agarose gel electrophoresis. If you have a periodic nucleosome array, then the DNA fragments derived from each oligonucleosome population (i.e., 1-mers, 2-mers, 3-mers, 4-mers, etc.) would each be of approximately the same length, and a DNA “ladder” would be obtained.
When we assemble chromatin with our purified factors and analyze the products by the micrococcal nuclease digestion assay, we see that there is efficient assembly of periodic nucleosome arrays (Figure 18). Each protomer of ACF can assemble at least 50 nucleosomes.
To study how ACF works, Dmitry Fyodorov performed template-commitment experiments as well as other related mechanistic analyses. His results suggest that ACF is a processive, ATP-driven motor that translocates along DNA and mediates chromatin assembly (Figure 19). Dmitry also carried out a genetic analysis of ACF in vivo in Drosophila. These studies have revealed that ACF influences the cell cycle as well as gene expression.
To summarize this section on chromatin assembly—we have fractionated, purified, and cloned the factors that mediate chromatin assembly. These studies led, in particular, to the discovery of ACF. In this work, we have achieved a purified, recombinant chromatin assembly system with ACF and dNAP1. The only other factors needed are core histones, DNA, and ATP. We have also found that ACF appears to function as a processive, ATP-driven motor that assembles nucleosomes as it translocates along DNA.

FIGURE 18 Chromatin assembly with purified, recombinant ACF.

FIGURE 19 A processive model for chromatin assembly.
HOMOLOGOUS RECOMBINATION IN CHROMATIN
Lastly, I will describe some of our studies of homologous recombination in chromatin. This work was carried out by Vassili Alexiadis. These experiments were inspired by two things—first, homologous recombination is a fundamental and important biological process; and second, the Rad54 protein, which is involved in homologous recombination, is a member of the Snf2-like family of ATPases, which comprises the ATPase subunits of all known chromatin remodeling complexes (Figure 20). For instance, Rad54 is related to the ISWI ATPase subunit of ACF. Thus, it seemed likely that Rad54 would have a chromatin-related function in homologous recombination.
Rad54 is involved in homologous recombination during double-strand break repair as well as during meiosis. Rad54 and Rad51, which is related to bacterial RecA protein, participate in the strand pairing reaction that yields a D loop (Figure 21).


There are different ways by which this strand pairing reaction can be carried out in vitro. Vassili used an assay with double-stranded circular plasmid DNA and a homologous single-stranded oligonucleotide (Figure 22). In this reaction, Rad51 associates with the single-stranded oligonucleotide in an ATP-dependent manner, and then forms a D loop in a reaction that requires Rad54 and ATP. When he carries out this reaction with purified Rad54 and Rad51, he observes that the formation of D loops requires Rad51, Rad54, ATP, and homologous single-stranded DNA (Figure 23).
Next, Vassili sought to use chromatin instead of DNA in these reactions (Figure 24). These experiments revealed that Rad54 and Rad51 work at least as well with chromatin as with naked DNA (Figure 25). In contrast, the bacterial RecA protein can catalyze D loop formation with naked DNA but not with chromatin. Thus, Rad54 and Rad51 work well in chromatin.
Vassili then sought to investigate the effect of superhelical tension upon D loop formation by Rad51 and Rad54, because the bulk of the eukaryotic genome possesses little superhelical tension. He therefore tested the effect of relaxation of superhelical tension upon D loop formation by Rad51 and Rad54. These experiments revealed that relaxation of naked DNA by topoisomerase I results in a greater than 100-fold decrease
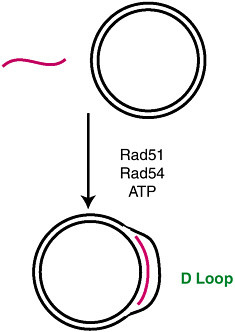
FIGURE 22 Catalysis of strand pairing by Rad51 and Rad54.

FIGURE 23 Formation of D loops with purified Rad51 and Rad54.
SOURCE: Alexiadis, V., and Kadonaga, J. T. (2002). Strand pairing by Rad54 and Rad51 is enhanced by chromatin. Genes Dev. 16:2767-2771. Fig. 1B.
in D loop formation by Rad54 and Rad51. In constrast, he found that topoisomerase I-mediated relaxation of chromatin has no effect upon the efficiency of D loop formation by Rad51 and Rad54 (Figure 26). Thus, the packaging of DNA into chromatin facilitates D loop formation by over 100-fold in the absence of superhelical tension.
To test further whether chromatin is important for D loop formation by Rad54 and Rad51, Vassili employed a different experimental approach. Instead of relaxing preassembled chromatin, he assembled relaxed DNA into chromatin and then performed D loop reactions. To this end, he assembled relaxed DNA into chromatin by using purified recombinant ACF, purified recombinant NAP-1, purified core histones, relaxed plasmid DNA, and ATP in the presence of purified topoisomerase I. These reactions were performed in the absence (no chromatin assembly) or presence (chromatin assembly) of core histones. As seen in this slide (Figure 27), the packaging of relaxed DNA into chromatin results in a greater than 100-fold stimulation of D loop formation by Rad54 and Rad51.
Hence, Rad51 and Rad54, but not E. coli RecA, can form D loops with chromatin. Notably, there is a greater than 100-fold enhancement of strand pairing by Rad54 and Rad51 upon assembly of relaxed DNA into chroma-

FIGURE 24 Strand pairing by Rad51 and Rad54 in chromatin.
tin. We have also found that Rad54 and Rad51 function cooperatively in the ATP-dependent remodeling of chromatin. It is generally thought that chromatin represses DNA-directed processes, such as transcription and replication, but here we have an example of a DNA-utilizing process that is facilitated by the packaging of DNA into chromatin. These results indicate that Rad54 and Rad51 have evolved to function optimally with chromatin (Figure 28). In addition, these findings may be applicable to studies of homologous recombination in vivo, such as for targeted knockouts and gene therapy.

FIGURE 25 Rad51 and Rad54, but not RecA, can form D loops with chromatin.
SOURCE: Alexiadis, V., and Kadonaga, J. T. (2002). Strand pairing by Rad54 and Rad51 is enhanced by chromatin. Genes Dev. 16:2767-2771. Fig. 2B.

FIGURE 26 Chromatin facilitates D loop formation in the absence of superhelical tension.
SOURCE: Alexiadis, V., and Kadonaga, J. T. (2002). Strand pairing by Rad54 and Rad51 is enhanced by chromatin. Genes Dev. 16:2767-2771. Fig. 3.

FIGURE 27 The assembly of relaxed DNA into chromatin facilitates strand pairing by Rad54 and Rad51.
SOURCE: Alexiadis, V., and Kadonaga, J. T. (2002). Strand pairing by Rad54 and Rad51 is enhanced by chromatin. Genes Dev. 16:2767-2771. Fig. 4.

FIGURE 28 Rad51 and Rad54 are optimized for strand exchange with chromatin.
SUMMARY
To summarize—I have described some of our studies in the area of basal transcription, chromatin assembly, and homologous recombination. In the area of basal transcription, we have identified and characterized the DPE downstream core promoter motif. The DPE is conserved from Drosophila to humans, and is something of a downstream analogue of the TATA box. Notably, some transcriptional enhancers act specifically with core promoters that contain a DPE or TATA motif. Thus, the core promoter can be an important component in the regulation of a gene.
In the area of chromatin assembly, we have fractionated, purified, and cloned the factors that mediate the ATP-dependent assembly of periodic nucleosome arrays. These studies led, in particular, to the discovery of ACF. In this work, we have achieved a purified, recombinant chromatin assembly system with ACF, NAP-1, core histones, DNA, and ATP. Mechanistic studies suggest that ACF functions as a processive, ATP-driven motor that translocates along DNA and mediates chromatin assembly.
We have also investigated the role of chromatin in homologous recombination in vitro. We observed a greater than 100-fold enhancement of strand pairing by Rad54 and Rad51 with chromatin in the absence of superhelical tension. In addition, we found that Rad54 and Rad51 function cooperatively in the remodeling of chromatin. These results indicate that Rad54 and Rad51 have evolved to function with chromatin, the natural substrate, rather than with naked DNA.
Lastly, and most importantly, I would like to mention the past and present coworkers who have contributed to this work (Figure 29). Tom Burke discovered the DPE, Alan Kutach studied the DPE consensus sequence, Trish Willy identified NC2 (Dr1-Drap1) as an activator of DPE-dependent but not TATA-dependent transcription, and Jenny Butler identified the DPE-specific transcriptional enhancers. Rohinton Kamakaka and Mike Bulger developed the S-190 chromatin assembly extract, Mike Bulger and Takashi Ito performed the fractionation of the S-190 and the purification of ACF, Jessica Tyler identified and purified the ASF1/RCAF assembly factor, Dmitry Fyodorov and Mark Levenstein established the purified recombinant chromatin assembly system, and Dmitry carried out the experiments suggesting that ACF is a processive DNA-translocating enzyme as well as the genetic analysis of ACF. Vassili Alexiadis performed the studies of homologous recombination with Rad54 and Rad51 in chromatin.
























