
9
Antimalarial Drugs and Drug Resistance
This chapter describes antimalarial drugs currently in use, with an emphasis on the artemisinins. It also reviews the way drug resistance develops and spreads, methods used to assess the presence and level of drug resistance, and the extent to which chloroquine and sulfadoxine/pyrimethamine (SP)—the two most widely used antimalarial drugs in the world today—have now lost efficacy.
CLINICAL MALARIA AND THE AIMS OF ANTIMALARIAL DRUG TREATMENT
Malaria sickens and kills people through several pathological mechanisms, understood to varying degrees. In addition to first- and second-line antimalarial drug treatments, adjunctive and supportive care measures (e.g., intravenous fluids, blood transfusions, supplemental oxygen, antiseizure medications) may be needed for severe manifestations. The aims of treatment are to prevent death or long-term deficits from malaria, to cut short the morbidity of an acute episode of illness, and to clear the infection entirely so that it does not recur.
Fever, sweating, and chills (or, in some cases, merely fever) triggered by the release of plasmodia into the bloodstream from mature blood schizonts, are the most common symptoms heralding the onset of a clinical case of uncomplicated falciparum malaria (see Chapter 6 for a description of the evolution of clinical symptoms). Without treatment—or an active immune
response primed by repeated previous malaria infections—the number of parasites will increase with every 2-day cycle of reproduction. A mature infection may involve up to 1012 circulating plasmodia.
At any time after the infection is established, the vast majority of plasmodia will be in some stage of asexual maturation leading to another round of multiplication within the patient’s bloodstream. However, a few parasites will have transformed into sexual stages (gametocytes) that, once ingested by mosquitoes, can perpetuate the transmission cycle. Because each stage of the malarial life cycle exhibits distinct biochemical and other characteristics (i.e., it expresses different proteins or locates in different sites within the body), a drug may kill one stage but have little effect on another. In other words, in each life-cycle stage the parasite manifests unique biological properties that can offer a target for the action of one or more antimalarial drugs.
ANTIMALARIAL DRUG CLASSES
Currently available antimalarials fall into three broad categories according to their chemical structure and mode of action (Appendix 9-A):
-
Aryl aminoalcohol compounds: quinine, quinidine, chloroquine, amodiaquine, mefloquine, halofantrine, lumefantrine, piperaquine, tafenoquine
-
Antifolate compounds (“antifols”): pyrimethamine, proguanil, chlorproguanil, trimethoprim
-
Artemisinin compounds (artemisinin, dihydroartemisinin, artemether, artesunate)
Atovaquone is an antimalarial in its own class with a unique mode of action; combined with proguanil it is sold under the trade name Malarone®. Several antibacterial drugs (e.g., tetracycline, clindamycin) also have antiplasmodial activity, although in general their action is slow for malaria treatment (as opposed to prophylaxis); they are recommended only in combination with other antimalarial drugs. Drugs active against Plasmodium falciparum also are active against the other three malaria species that affect humans—P. vivax, P. malariae, and P. ovale—with the exception of antifols, which work poorly against P. vivax.
Current treatment protocols for uncomplicated malaria and severe malaria are given in Tables 9-1 and 9-2.
TABLE 9-1 Treatment of Uncomplicated Malariaa
|
Malaria |
Drug Treatment |
|
P. vivax, P. malariae, P. ovale, known chloroquine-sensitive P. falciparuma |
Chloroquine 10 mg base/kg stat. Followed by:
|
|
Chloroquine-resistant P. falciparuma known to be sensitive to sulfadoxinepyrimethamine (SP) |
Pyrimethamine 1.25 mg/kg + sulfadoxine 25 mg/kg (single dose; 3 tablets in an adult) or Amodiaquine 10 mg base/kg/day for 3 days |
|
Chloroquine-resistant P. vivaxb and multidrug resistant P. falciparuma |
|
|
aFor acute treatment of falciparum malaria combinations containing an artemisinin-derivative are preferred. Artesunate (4 mg/kg/day for 3 days) has been combined successfully with chloroquine, amodiaquine, SP, mefloquine, and atovaquone-proguanil. bThis refers to truly resistant P. vivax infections, which are a significant problem only in Oceania and Indonesia and should not be confused with relapses. Amodiaquine is more effective than chloroquine for resistant P. vivax. |
|
Basic Properties of Antimalarials: Pharmacokinetics and Pharmacodynamics
Pharmacokinetics
The interactions of drugs with people who take them—how the compounds are absorbed, metabolized, distributed, and excreted—is referred to as pharmacokinetics. Antimalarial drugs differ considerably in their pharmacokinetics, which affect how well they work, how they are dosed, and how long they must be taken. People also vary in how they respond to drugs. Some of these responses are genetically determined, others by health status, others by dietary factors. In general, the pharmacokinetic properties of the antimalarials are similar in children and adults, although the metabo-
|
General points |
|
|
A. |
There are now few places in the world where chloroquine can be relied upon for falciparum malaria, and SP resistance is spreading rapidly—so recent information on drug susceptibility is required if these drugs are used. Amodiaquine is more effective than chloroquine against chloroquine-resistant P. falciparum, but highly amodiaquine-resistant parasites are prevalent in East Asia. |
|
B. |
Pregnancy: There are insufficient data on the safety of most antimalarial drugs in pregnancy. Artemisinin and its derivatives should not be given in the first trimester. Halofantrine, primaquine, and tetracycline should not be used at any time in pregnancy. There are theoretical concerns over inducing kernicterus when long-acting sulfonamides are used near term, but no evidence that this is a significant problem in practice. There are uncertainties over the safety of mefloquine in pregnancy. Quinine, chloroquine, proguanil, SP, and clindamycin are regarded as safe in the first trimester. Quinine may cause hypoglycemia, particularly in late pregnancy. |
|
C. |
Vomiting is less likely if the patient’s temperature is lowered before oral drug administration. |
|
D. |
Where possible, artesunate or quinine should be combined with a tetracycline or clindamycin. Short courses for artesunate or quinine (< 7 days) alone are not recommended. |
|
E. |
In renal failure. the dose of quinine should be reduced by one-third to one-half after 48 hours, and doxycycline but not tetracycline should be prescribed. |
|
F. |
The doses of all drugs are unchanged in children: however, several drugs including atovaquone, proguanil and artesunate have significantly altered kinetics in pregnancy. |
|
Specific points |
|
|
1. |
Patients with P. vivax and P. ovale infections also should be given primaquine 0.25 mg base/kg daily (0.375-0.5 mg base/kg in Oceania) for 14 days to prevent relapse. In mild G6PD deficiency 0.75 mg base/kg should be given once weekly for 6 weeks. In severe G6PD deficiency, primaquine should not be used. |
|
2. |
None of the tetracyclines should be given to pregnant women or children under 8 years of age. |
lism of several drugs is altered in pregnancy (e.g., atovaquone, mefloquine, cycloguanil).
A key pharmacokinetic property of antimalarials is how long they remain in the body. Artemisinin and its derivatives are absorbed and eliminated the most rapidly (half-life = 1 hour or less). Quinine also is absorbed and eliminated within one parasite life cycle (11 hours in healthy subjects to 18 hours in those with severe malaria). Other antimalarials are eliminated very slowly, remaining in significant concentrations for several days (pyrimethamine, halofantrine, lumefantrine, atovaquone), or even weeks (mefloquine, chloroquine, and piperaquine). In general, rapidly eliminated drugs (artemisinin, and quinine) must be taken over four asexual cycles (7 days) to ensure cure in nonimmune patients. In contrast, drugs that are
TABLE 9-2 Treatment of Severe Malaria
|
|
Health Clinic: |
|
|
|
|
Hospital Intensive Care Unit (Icu) |
No Intravenous Infusions Possible |
Rural Health Clinic: No Injection Facilities |
|
Chloroquine-resistant P. falciparum |
Quinine Quinine dihydrochloride 7 mg salt/kg infused over 30 minutes followed by immediately 10 mg/kg over 4 hours; or 20 mg salt/kg infused over 4 hours. Maintenance dose: 10 mg salt/kg infused over 2-8 hours at 8-hour intervalsa |
Quinine dihydrochloride 20 mg salt/kg diluted 1:2 with sterile water given by split injection into both anterior thighs. Maintenance dose: 10 mg/kg 8-hourlya |
|
|
|
Quindine 10 mg base/kg infused over 1-2 hours followed by 1.2 mg base/kg per hourb Electrocardiographic monitoring advisable |
|
|
eliminated slowly require fewer doses over shorter periods because they remain active in the body.
Halofantrine, lumefantrine, atovaquone and, to a much lesser extent, mefloquine, are hydrophobic and lipophilic (i.e., insoluble in water and capable of dissolving in fat); as a result, their absorption also varies according to the amount of dietary fat consumed. For this reason, blood concentrations of these drugs may vary considerably from one individual to another following the same dose.
Pharmacodynamics
The way drugs act on their target—in this case, plasmodia—is called pharmacodynamics. The principal effect of antimalarial drugs in uncomplicated malaria is to inhibit parasite multiplication by killing parasites. If an untreated infection progressed at maximum efficiency, with each life cycle, the total body parasite load would increase by a multiplication factor approximating the average number of viable parasites in a mature schizont (18-36) (White, 1997). Proliferation of parasites in nonimmune individuals often proceeds at multiplication rates of 6 to 20 per 2-day cycle (30-80 percent efficiency). Antimalarial drugs exerting maximum effect (Emax), on the other hand, reduce total parasite numbers 10- to 10,000-fold per cycle.
Individual antimalarial drugs differ in their Emax (i.e., the proportion of total plasmodia killed per treatment); for example, artemisinins often yield a 10,000-fold reduction per asexual cycle, whereas antimalarial antibiotics such as tetracycline or clindamycin may only achieve a 10-fold parasite reduction per cycle. The lowest blood or plasma concentration of an antimalarial drug that results in Emax can be considered a “minimum parasiticidal concentration” (MPC). Parasite reduction appears to be a first-order process throughout (Day et al., 1996), which means that a fixed fraction of the infecting malaria parasite population is removed with each successive cycle as long as the MPC is exceeded.
Clinical Pharmacodynamics
Patients with acute malaria may have up to 1012 parasites circulating in their blood. Even with killing rates of 99.99 percent per cycle, complete eradication of the parasite load requires at least three life cycles (6 days); therefore, therapeutic drug concentrations should be present for 4 cycles to effect a cure (White, 1997, 1998). Simply put, patients taking rapidly eliminated drugs must continue treatment for a full week. Treatment responses are always better in patients with some immunity (York and Macfie, 1924) because the immune response kills parasites in much the same way that a
drug does. In endemic areas, the worst treatment results are seen in young children who have little immunity. In contrast, although their degree of immune protection cannot be quantitated or assumed, older children or adults in high-transmission areas may do surprisingly well with failing drugs because much of their therapeutic response stems from immunity rather than antimalarial drug action.
In severe falciparum malaria, the stage at which an antimalarial drug acts is especially important since the ultimate goal of treatment is to halt parasite maturation to late-stage, cytoadherent parasites (i.e., mature schizonts that attach to endothelial cells lining small blood vessels), which are primarily responsible for life-threatening complications. The artemisinin derivatives are advantageous because they prevent parasites from maturing to these more pathological stages, whereas quinine and quinidine do not affect parasites until they have already cytoadhered. The antifols act even later in the cycle, and are not recommended for severe malaria (Yayon et al., 1983; ter Kuile et al., 1993). None of the drugs will prevent rupture of infected erythrocytes and reinvasion once a schizont has formed. Young ring forms (i.e., early asexual parasites) also are relatively drug resistant, especially to quinine and pyrimethamine.
Artemesinin derivatives offer the broadest antimalarial action against the range of developmental stages, and the most rapid in vivo activity (ter Kuile et al., 1993; White, 1997). These compounds (and, to a lesser extent, chloroquine) prevent ring stages from maturing, hastening their clearance, and preventing end-organ pathology that would otherwise occur if cytoadherence progressed unchecked (Chotivanich et al., 2000).
MECHANISMS OF ACTION AND DRUG RESISTANCE
Antifolate Drugs
Pyrimethamine, and biguanides such as cycloguanil interfere with folic acid synthesis, inhibiting the parasite enzyme known as dihydrofolate reductase-thymidilate synthase (DHFR). Sulfonamides act at the previous step in the folic acid pathway, inhibiting the parasite enzyme dihydropteroate synthase (DHPS). There is marked synergy between these two classes of drugs when they are taken together. However, resistance to pyrimethamine in P. falciparum developed within a few years of its introduction (Peters, 1987) due to point mutations in the DHFR gene, which cause 100- to 1,000-fold reduced affinity of the enzyme complex to the drug. Progressive mutations in the DHFR gene of P. falciparum further decreased efficacy. Triple mutant infections are relatively resistant to antifolate treatment; with a fourth mutation within the malaria parasite, antifolate drugs become completely ineffective.
Quadruple mutant P. falciparum strains are now prevalent in parts of Southeast Asia, and South America (Imwong et al., 2001). Resistance to partner antifols sulfonamide and sulfone results from progressive acquisition of mutations in the P. falciparum gene encoding the target enzyme DHPS.
Chloroquine
One of chloroquine’s most dramatic characteristics is its ability to concentrate itself from nanomolar (10-9) levels outside the parasite to levels one million times higher (millimolar levels, 10-3) in the acid food vacuole of the parasite inside a red blood cell (Krogstad and Schlesinger, 1987). This action in itself does not explain chloroquine’s antimalarial activity, however. Chloroquine works by interfering with heme dimerization, the detoxifying biochemical process within the malaria parasite that normally yields malaria pigment (hemozoin).
Reduced intracellular drug concentrations accompany chloroquine resistance because resistant parasites expel chloroquine from their acid food vacuoles 40-50 times faster than do drug-sensitive parasites (Bray et al., 1998). Such accumulation deficits were once attributed to changes in pH gradient, or to altered membrane permeability, or both (Le Bras and Durand, 2003). However, chloroquine resistance was then found to be reversible by verapamil, a drug which also modulates resistance in multidrug resistant (MDR) mammalian cancer cells. This discovery led to the identification of the protein Pgh1 (an analog to overexpressed glycoproteins that expel cytotoxic drugs in cancer cells) in the digestive vacuole membrane of P. falciparum. Genes encoding MDR proteins have been identified in P. falciparum (pfmdr1); amplification of these “wild type” MDR genes has recently been shown to cause mefloquine resistance (Price et al., 1999b).
Point mutations in the gene encoding a food vacuole transporter protein (pfcrt) have been linked to chloroquine resistance (Durand et al., 2001; Warhurst, 2001) and correlate with reduced in vivo chloroquine efficacy (Djimde et al., 2001). In the presence of pfcrt mutations, mutations in the second transport genes (Pfmdr1) further modulate resistance in vitro although the role of Pfmdr1 mutations in determining in vivo responses to chloroquine treatment is still unclear. Additional unlinked mutations are probably involved in the development of chloroquine resistance, some of which have not yet been discovered.
In the laboratory, the efflux mechanism seen in chloroquine-resistant P. falciparum parasites can be inhibited by several unrelated drugs (calcium channel blockers such as verapamil as well as tricyclic antidepressants, phenothiazines, and antihistamines), whereas mefloquine resistance can be
reversed by penfluridol (Martin et al., 1987; Oduola et al., 1993). Clinical applications of these findings are few to date, although chloroquine plus high doses of chlorpheniramine (an antihistamine) did show improved efficacy against chloroquine-resistant P. falciparum in Nigerian children (Sowunmi et al., 1997). Whether general use of resistance reversers will be safe and feasible in the future remains an open question (Personal communication, N. White, Mahidol University, March 2004).
Other Antimalarial Drugs
In general, antimalarial drug resistance to mefloquine, quinine, lumefantrine, and halofantrine is linked, whereas chloroquine, and mefloquine resistance are not. Cross-resistance between antimalarials is related to common aspects of their modes of action as well as their resistance mechanisms. Parasites with high-level chloroquine resistance (present in Southeast Asia), are generally resistant to amodiaquine as well; in residents of Southeast Asia, amodiaquine may thus fail as a back-up treatment (Le Bras and Durand, 2003). The same relationship holds true for halofantrine, and mefloquine. On the other hand, there may be an inverse correlation between chloroquine and mefloquine sensitivity: in Africa, for example, chloroquine-sensitive strains are substantially less sensitive to mefloquine or halofantrine, and vice versa (Oduola et al., 1987; Simon et al., 1988).
Atovaquone is a component of Malarone®, a new combination drug (consisting of atovaquone and proguanil) used for treatment and prevention of chloroquine-resistant P. falciparum. Atovaquone interferes with mitochondrial electron transport, and also blocks cellular respiration (Srivastava et al., 1997). High levels of atovaquone resistance result from single-point mutations in a gene encoding cytochrome b found on a small, extrachromosomal DNA-containing element in the parasite (Korsinczky et al., 2000).
ARTEMISININS
Of the available antimalarials, the artemisinins are effective at killing the broadest range of asexual stages of the parasite, ranging from medium-sized rings to early schizonts; they also produce the most rapid therapeutic responses by accelerating clearance of circulating ring-stage parasites (ter Kuile et al., 1993).
Qinghaosu, or artemisinin, is a sesquiterpene lactone peroxide extracted from the leaves of the shrub Artemisia annua (qinghao). Three derivatives are widely used: the oil-soluble methyl ether, artemether (artemotil [arteether] is a closely related compound); the water soluble hemi-succinate derivative, artesunate; and dihydroartemisinin (DHA).
Artesunate, artemether, and arteether are all synthesized from DHA, and they are converted back to it within the body. Artemisinin itself is available in a few countries in Asia. It is 5-10 times less active than the derivatives, and it is not metabolized to DHA.
Artemisinin is available as capsules of powder, or as suppositories. Artemether is formulated in peanut oil, and arteether in sesame seed oil, for intramuscular injection, and in capsules or tablets for oral use. Artesunate is formulated either as tablets, in a gel enclosed in gelatin for rectal administration (called a rectocap™), or as dry powder of artesunic acid for injection, supplied with an ampoule of 5 percent sodium bicarbonate. The powder is dissolved in the sodium bicarbonate to form sodium artesunate, and then diluted in 5 percent dextrose or normal saline for intravenous or intramuscular injection. Artelinic acid is a water-soluble second-generation compound under long-term development. It has not yet been used in treatment. The majority of clinical data pertain to the most widely used derivative, artesunate.
Botanical Properties
Artemisinin was first isolated from the stems, leaves, and flowers of Artemisia annua by Chinese scientists (Anonymous, 1982; Klayman et al., 1984), but details of the process were not released. Researchers at the Walter Reed Army Institute of Medical Research (WRAIR) successfully isolated artemisinin derivatives from air-dried parts of plants growing in the wild near Washington, D.C., using petroleum ether extraction (Klayman, 1985). The plant grows easily in temperate areas, and has become naturalized in many countries. It can attain a height of two meters or more, appearing as an erect specimen with a woody stem. Artemisinin accumulates in all parts of A. annua except for the roots (Abdin et al., 2003). Artemisinin content in flowers is 4-5 times higher than in leaves. Plant age correlates with artemisinin yield, presumably due to a progressive increase in leaf yield and artemisinin content with plant growth. In agricultural settings in Asia, artesunate production has varied from 5 kg/hectare to 50 kg/hectare (Personal communication, J-M. Kindermans, Médecins Sans Frontières, February 2004).
Mechanism of Action
Artemisinin’s chemical structure is unlike any other known antimalarial. It includes an endoperoxide bridge necessary for its antimalarial action (Brossi et al., 1988). Artemisinin treatment of membranes, especially in the presence of heme, causes lipid peroxidation (Scott et al., 1989;
Wei and Sadrzadeh, 1994; Berman and Adams, 1997); this event may occur as a result of the drug’s interaction with intracellular heme or iron (Meshnick et al., 1991). With respect to artemisinin’s direct effect on the malaria parasite, recent work suggests that artemisinin specifically inhibits PfATP6, the SERCA orthologue of Plasmodium falciparum, a calcium ATPase (Eckstein-Ludwig et al., 2003).
In vivo, artemisinins kill malaria parasites within host erythrocytes, after which dead parasites are culled by the spleen, leaving formerly infected red blood cells intact and circulating (Chotivanich et al., 2000). It is not yet clear which asexual parasite life-cycle stages are most sensitive to artemisinin derivatives: late rings and early trophozoites (ter Kuile et al., 1993) versus trophozoites (Geary et al., 1989). There is clear consensus, however, that artemisinin derivatives kill early-stage gametocytes and are more active over a broader range of the parasite life cycle than any other antimalarial drug currently in use.
General Clinical Experience with Artemisinins and ACTs
More randomized clinical trials have been published regarding the effects of artemisinins than any other individual or class of antimalarial drug (Myint et al., 2004). The artemisinins’ pharmacodynamic effects are due to their rapid absorption and activity against many stages of the malaria life cycle, from young asexual forms (rings) to early sexual forms (gametocytes) (Kumar and Zheng, 1990). Their half-lives are short (<1 hour for artesunate) which protects them from resistance. They reduce gametocyte carriage, thus decreasing infectiousness following treatment as witnessed by the dramatic fall in malaria transmission on the Thai-Burma border (Price et al., 1996; Nosten et al., 2000). Tolerability of the drugs is excellent (White and Olliaro, 1998).
Administered alone, artemisinin derivatives require at least 7 days of treatment. When combined with other drugs, however, artemisinin combination therapies (ACTs) given in 3-day regimens can eradicate parasites quickly and protect against the development of resistance to both drugs. In addition, artemisinin derivatives are the only first-line malaria treatments to act on gametocytes (early-stage).
On February 20, 2002, the World Health Organization (WHO) released a statement recommending that “Governments … rapidly adopt more effective treatments [for malaria]. The aim is to provide effective treatment against malaria and to slow the spread of drug resistance. … In particular, WHO recommends the use of artemisinin-based combination therapy” (WHO, 2001).
Current Evidence Base for ACTs1
Through the largest randomized controlled trials ever conducted on antimalarials in Africa, a considerable body of evidence has now been collected showing that artemisinin-based combinations improve cure rates, decrease gametocyte carriage, and are well tolerated with few serious adverse effects. In the first of these multicenter trials to be published, 941 children who had uncomplicated P. falciparum malaria were randomly assigned 3 days treatment with amodiaquine plus artesunate, or amodiaquine plus placebo. Both regimens were well tolerated. The combination of artesunate and amodiaquine significantly improved treatment efficacy in Gabon (85 versus 71 percent, p=0.02) and in Kenya (68 vs. 41 percent, p<0.0001). In Senegal, however, the two regimens were equivalent: day-28 cure rates for amodiaquine-artesunate versus amodiaquine were 82 versus 79 percent (p=0.5) (Adjuik et al., 2002).
The efficacy and safety of artesunate in combination with sulfadoxine-pyrimethamine (SP) has been evaluated in randomized controlled trials involving 2,865 patients in sub-Saharan Africa. Results from the first study published from The Gambia (von Seidlein et al., 2000) (where the cure rate with SP monotherapy was then 93 percent), showed that both cure rate and parasite clearance were significantly higher in patients who received 3 days of artesunate plus a single dose of SP compared with those who received SP alone. Gametocyte carriage was 68 percent following solo SP treatment in comparison with 21 percent following the artesunate-SP combination (p=0.001).
In contrast, underlying SP resistance in Uganda led to unacceptable rates of late recrudescence when artesunate-SP was used there (Dorsey et al., 2002). This finding underscores that combining an artemisinin with a longer-acting drug without any underlying resistance may prove the optimal regimen in many areas. On the other hand, in Thailand, where drug resistance is particularly severe, artesunate plus mefloquine was highly effective, even in areas where mefloquine resistance was previously quite common (Price et al., 1997). In the largest-ever series of therapeutic efficacy studies with ACTs, artesunate plus mefloquine produced a sustained, increased cure rate (almost 100 percent from 1998 onward) despite established resistance to high-dose mefloquine alone seen between 1990 and 1994 (Nosten et al., 2000). Cure rates with other ACTs (atovaquone-proguanil-artesunate, artemether-lumefantrine) in Asia have also been consistently above 90 percent (van Vugt et al., 1998, 1999).
Coartem (artemether-lumefantrine) is the first fixed-dose ACT whose
two antimalarial components were not widely used prior to marketing. When given in a six-dose regimen over 3 days, Coartem is effective against all P. falciparum and extremely well tolerated. Coartem’s principal drawback is its twice daily dosing and lumefantrine’s variable absorption linked to dietary fat intake (if patients are unable to eat, or consume a very low fat diet while taking the drug, low cure rates can result).
In randomized trials comparing a six-dose regimen of orally administered artemether-lumefantrine with mefloquine-artesunate for multidrug-resistant P. falciparum malaria, artemether-lumefantrine was as effective (94-100 percent) and overall better tolerated than mefloquine-artesunate (van Vugt et al., 1999, 2000; Lefevre et al., 2001).
Artesunate
Artesunate can be administered as a tablet, an injection, or as a rectal suppository. A comparison of intravenous artesunate and quinine in 113 adults with severe malaria reported mortality of 12 percent with artesunate, and 22 percent with quinine (p=0.22) (Newton et al., 2003). In patients with hyperparasitemia who had no other features of severe malaria but were at an increased risk of developing severe malaria, oral artesunate was found to be superior to intravenous quinine in reducing both the clinical symptoms and parasites (Luxemburger et al., 1995).
Rectally administered artesunate has been shown to be safe and highly effective in children and adults with uncomplicated or moderately severe falciparum malaria (Sabchareon et al., 1998; Karunajeewa et al., 2003; Barnes et al., 2004). A number of recent randomized controlled and open-label studies of rectal artesunate in Africa and Asia have demonstrated the rapid antimalarial efficacy of a single dose of rectal artesunate (10 mg/kg) in moderately severe falciparum malaria in both children and adults prior to referral for definitive treatment. All patients had evidence of adequate absorption of the drug. Clearance of malaria parasites from the peripheral blood was consistently more rapid with rectal artesunate than with quinine injection. There also are a number of small open label studies, some of which were randomized, demonstrating the clinical and parasitological efficacy of rectal artesunate in adults with severe P. falciparum infections (Awad et al., 2003) where rectal artesunate was administered repeatedly, and combined with a second oral antimalarial to prevent recrudescence.
These clinical and parasitological responses suggest that rectal artesunate could prove highly beneficial in the initial management of acute malaria in patients who cannot take medication by mouth (and for whom parenteral medication is not immediately available due to limited local resources). In Ghana, artesunate suppositories have already been given by trained village volunteers; in the future, traditional healers also could be
trained to use them as an emergency treatment for febrile seizures in malaria-endemic areas (Personal communication, B. Greenwood, London School of Hygiene and Tropical Medicine, April 2004). If used widely enough, rectal artesunate could have a major effect on malaria deaths in Africa.
Toxicity of Artemisinins2
In the early 1970s, Chinese scientists characterized the antimalarial properties of artemisinins and their excellent tolerability and safety (Anonymous, 1982; Luo and Shen, 1987). A subsequent review of published and unpublished studies of the arteminisinin derivatives confirmed the earlier Chinese findings (Pukrittayakamee et al., 2000). Those adverse events that were reported were not specifically associated with a single form of artemisinin or route of administration. Neutropenia (but not agranulocytosis) and asymptomatic ECG abnormalities occurred in 1.3 percent of patients; reduced reticulocyte count, anemia, eosinophilia, and elevated aspartate aminotransferase (a liver enzyme) occurred in ≤ 1.0 percent (Taylor and White, 2004).
In western Thailand, Price and colleagues (1999a) conducted a detailed study of oral artesunate or artemether used as monotherapy or in combination with mefloquine. The artemisinin derivatives were associated with substantially fewer adverse effects than the mefloquine-containing regimens: acute nausea (16 vs. 31 percent), vomiting (11 vs. 24 percent), anorexia (34 vs. 52 percent), and dizziness (15 vs. 47 percent). Oral artesunate or artemether alone were well tolerated. Since these data were published two cases of acute hives and anaphylaxis developed in the same population following artesunate monotherapy, raising the total number of allergic reactions to 6 patients out of roughly 17,000, or 1 in 2,833 (Leonardi et al., 2001).
Artemether-Lumefantrine (Coartem)
Experience with Coartem is increasing rapidly. In large-scale clinical trials the combination was very well tolerated, whether taken as a four- or six-dose regimen. Coartem has been better tolerated than either mefloquine alone or artesunate plus mefloquine (van Vugt et al., 1998; Looareesuwan et al., 1999). Reported adverse effects have generally been mild, including
gastrointestinal upset, headache, dizziness, fatigue, sleep disturbance, palpitations, maylgia, arthralgia, and rash (Bakshi et al., 2000). Detailed prospective studies have not demonstrated any cardiotoxicity (van Vugt et al., 1999; Bindschedler et al., 2000). Twenty severe adverse effects in 1,869 patients include 19 that could be explained on the basis of the malarial episode or a concurrent illness; artemether-lumefantrine may have contributed to the development of hemolytic anemia in one 35-year-old patient 13 days following discontinuation of the drug (Bakshi et al., 2000).
Neurotoxicity: Animal Studies
Dogs receiving high doses of intramuscular artemether or arteether have developed a peculiar selective pattern of brain stem damage, in particular involving the reticular formation, the vestibular system nuclei, and nuclei related to the auditory system. Clinical features included gait disturbances; loss of spinal and pain response reflexes; prominent loss of brain stem and eye reflexes; cardiorespiratory depression; and death. ECG changes included prolongation of QTc interval and bizarre ST-T segment changes (Brewer et al., 1994). A similar selective pattern of brain stem pathology also was found in mice, rats, and Rhesus monkeys given arteether or arthemether (Genovese et al., 1998; Petras et al., 2000). In mice, parenteral artemether was more neurotoxic than artesunate, resulting in escalating, irreversible neurological deficits involving balance, and death with increasing doses (Nontprasert et al., 1998). Recent studies have shown that neurotoxicity is determined by the pharmacokinetic properties of the drugs. Sustained CNS exposure from slowly absorbed or eliminated artemisinins is considerably more neurotoxic than intermittent brief exposure. Thus intramuscular artemether and arteether are more neurotoxic in experimental animals than the same drugs given orally, or artesunate given by any route.
Neurotoxicity in Humans
One case report to date has described acute cerebellar dysfunction including slurred speech, ataxia, impaired heel to shin movement, and dysdiadochokinesis after treatment of falciparum malaria with oral artesunate (Miller and Panosian, 1997). Detailed neurologic data from Price and colleagues (1999a) include neurological examinations conducted in nearly 2,000 children older than 5 at baseline, and on days 2, 7, and 28 posttreatment, with artemether alone, artesunate alone, or artesunate plus mefloquine. Short-course therapy with the artemisinins either alone or in combination with mefloquine was associated with self-limited, minor neurological deficits in a minority (<1 percent) of patients during the first few days of falciparum malaria.
In a recent case control study of 150 patients with uncomplicated malaria who received artemether-lumefantrine, a slight reduction in auditory acuity was noted compared with age-matched healthy controls (Toovey and Jamieson, 2004). In contrast, no differences in audiometry or auditory-evoked potentials were noted in 242 Vietnamese patients who received up to 21 treatment doses of artemisinin or artesunate compared with 108 age-and location-matched controls (Kissinger et al., 2000), or in 79 Karen patients who received two or more treatment doses of artemether or artesunate compared with 79 age- and location-matched controls (van Vugt et al., 2000).
Most recently, neuropathologic assessments of the brain stems of patients who died from severe falciparum malaria following treatment with high dose intramuscular artemether (the doses were higher than generally recommended) were performed (Hien et al., 2003), revealing no evidence for damage similar to that seen in experimental animals. Taken together with the cumulative body of negative clinical data—including detailed studies of audiometry, and auditory-evoked potentials in patients who received multiple courses of artemisinin derivatives in uncomplicated malaria as well as high dose artemether in severe malaria (Hien et al., 1996)—the weight of evidence suggests that neurotoxicity observed in animals does not occur in humans receiving current recommended treatment doses. The widespread use of water-soluble compounds (which are far less neurotoxic in animal models) as opposed to injected oil-based derivatives adds a further margin of safety.
Safety of Artemisinins During Pregnancy
Observations in Pregnant Women
Published data on treating malaria during pregnancy with artemisinin derivatives have come from China (Li et al., 1990), the Thai-Burmese border (where use has been greatest) (McGready et al., 1998, 2000, 2001), and The Gambia (Deen et al., 2001). The data from China describe 23 women treated with artemether or artemisinin between 17 and 38 weeks of gestation who developed no evidence of fetal or maternal toxicity. The published experience of artemisinin use during pregnancy in Thailand includes observational data on 461 women treated with artesunate (n=528) or artemether (n=11) for 539 episodes of acute P. falciparum malaria, including 44 episodes during the first trimester. Birth outcomes in the artemisinin-treated Thai women were comparable to community rates for abortion, stillbirth, congenital abnormality, and/or mean gestation at delivery. All newborns who were followed for 1 year developed normally, including those who had been exposed to an artemisinin in the first trimester of pregnancy
(McGready et al., 2001). Another study of 287 pregnant Gambian women inadvertently treated (during a mass drug administration) with a single dose of artesunate plus SP during their first, second, or third trimesters of pregnancy found no evidence of obstetric or fetal toxicity compared with women who were not exposed to artesunate plus SP (Deen et al., 2001). There are no published data on the use of artemether-lumefantrine in pregnant or breast-feeding women.
Evidence from Experimental Animals
Studies investigating the possible risks of artemisinins in during the first trimester of pregnancy (when birth defects are most likely to occur), also have been conducted in laboratory animals. In 2001, a WHO report concluded: “Preclinical studies have consistently shown that artemisinin and its derivatives do not exhibit mutagenic or teratogenic activity, but all of these drugs caused fetal resorption in rodents at relatively low doses” (WHO, 2001). More recently, in some experiments in rats and rabbits, but not in others, cardiovascular and limb abnormalities occurred when pregnant animals were given artemisinin doses similar to those used in man. For this reason, in 2002, WHO convened two further meetings of experts who reviewed the animal and human evidence relevant to the use of artemisinins by pregnant women, concluding:
Presently, artemisinin compounds cannot be recommended for treatment of malaria in the first trimester. However, they should not be withheld if treatment is considered to be lifesaving for the mother and other antimalarials are considered to be unsuitable. Because the safety data are limited, artemisinin compounds should only be used in the second and third trimesters when other treatments are considered unsuitable.
There is a need for further evidence of the safety of artemisinin compounds in pregnancy. All pregnant women treated with artemisinin compounds should be carefully followed up to document the pregnancy outcomes and subsequent development of the child and reported to the appropriate authorities. (WHO/RBM/UNDP/World Bank, 2003).
In summary, current data are encouraging but more safety data for the artemisinin derivatives are needed to support use in pregnant women with uncomplicated malaria. In contrast, although there are no published data regarding the treatment of severe malaria in pregnancy with artesunate or artemether, these drugs have been used widely and found effective. In particular, artesunate and artemether are often preferred over quinine and quinidine during pregnancy because they do not induce hypoglycemia, and they are easier to administer (Tran et al., 1996). Nonetheless, if ACTs are widely introduced, tens of thousands of pregnant African women—includ-
ing some of whom do not have malaria at all—will receive them. The use of artemisinins by pregnant women, especially during the first trimester, is an issue that is, appropriately, receiving continued attention by international monitoring bodies. On the country level, pharmacovigilance programs will be needed to track a variety of effects caused by artemisinins as well as new ACT partner drugs throughout pregnancy. For example, mefloquine given at any stage of pregnancy in Thailand has been associated with a fourfold increase in stillbirth (Nosten et al., 1999), whereas the same effect was not observed in pregnant women treated with mefloquine in Malawi (Steketee et al., 1996). This finding also requires further follow-up.
ANTIMALARIAL DRUG RESISTANCE
Although not every factor responsible for the emergence and spread of parasite resistance is fully known, what is clear is that antimalarial drug resistance can develop to any antimalarial drug, and that drug pressure is a key prerequisite. Other important contributors include drug elimination half-life, parasite biomass, and malaria transmission intensity (Talisuna et al., 2004).
How Resistance to Antimalarial Drugs Arises
The emergence of drug-resistant organisms can be considered in two discrete phases: the initial de novo event (a rare genetic occurrence) that first produces the resistant mutant, and the subsequent selection process that leads to its preferential transmission and spread. Resistance arises from spontaneous mutations or gene duplications, which are independent of drug pressure. Once formed, however, resistant mutants have a survival advantage in the presence of antimalarial drugs and, conversely, a survival disadvantage in the absence of at least certain antimalarial drugs. The resulting fitness cost may lead to a declining prevalence of resistance once drug pressure is removed (this pattern has been demonstrated for chloroquine resistance in Malawi [Kublin et al., 2003] although its operational significance is still uncertain).
Factors affecting the development of resistance include: the parasite mutation rate, the degree of resistance conferred by the genetic change, the fitness cost of the resistance mechanism, the proportion of all transmissible infection exposed to the drug, the drug concentration profile, the individual (e.g., dosing, duration, adherence) and community (e.g., quality, availability, distribution) patterns of drug use, and the immunity profile of the community (White, 1999).
The possible role of mass drug administration in accelerating the emergence of antimalarial drug resistance was first highlighted by Payne (Payne,
1988), who observed that chloroquine resistance in different sites had one common denominator: the long-term, local use of chloroquine for treatment. Later studies in coastal Kenya, Malawi, Mali and Bolivia found a positive correlation between patterns of drug use and in vitro parasite resistance or the prevalence of mutations linked to resistance (Diourte et al., 1999; Nzila et al., 2000). A recent study in Uganda observed that the prevalence of chloroquine resistance was higher in sites with high-frequency chloroquine use as reflected in detectable chloroquine metabolites in urine (Talisuna et al., 2002b). However, SP resistance was highest in high-transmission sites with relatively low SP use, suggesting that factors in addition to drug pressure influence the spread of SP drug resistance.
The role of drug elimination half-life in the development of parasite resistance has recently been reviewed, and modeled (Hastings et al., 2002). Drugs with long elimination phases are, in essence, “selective filters,” allowing infection by resistant parasites to flourish while the residual drug levels suppress infection by sensitive parasites (Watkins and Mosobo, 1993). Slowly eliminated drugs such as mefloquine (T 1/2=3 weeks) provide such a filter for months after drug administration. The resulting selection pressure can be enormous.
In Kenya, a potent selective pressure for SP resistance was found to operate even under conditions of supervised drug administration and optimal SP dosing (Watkins and Mosobo, 1993). Plasmodium falciparum infections appearing between days 15 and 52 after SP treatment were more likely to exhibit pyrimethamine resistance in vitro. The selective pressure of home-based use of SP (per the WHO strategy of home-based management of fevers) could accelerate the emergence of SP resistance to an even greater degree (Talisuna et al., 2004).
Finally, drug resistant mutant parasites are statistically more likely to emerge from infections involving large numbers of parasites. Such large parasite biomass infections are more common in nonimmune individuals, as demonstrated by the higher prevalence of chloroquine-resistant infections, and chloroquine treatment failures seen in African children compared to adults (Dorsey et al., 2000; Djimde et al., 2001; Talisuna et al., 2002a). Nonimmune patients infected with large numbers of parasites who receive inadequate treatment (either because of poor drug quality, adherence, vomiting of an oral treatment, etc.) are another potent source of resistance. This emphasizes the importance of correct prescribing and good adherence to prescribed drug regimens in slowing the emergence of resistance.
The Relationship between Resistance and Transmission Intensity
Recrudescence and onward transmission of a de novo resistant malaria parasite are essential for the propagation of resistance. Killing the transmis-
sible sexual stages (gametocytes) during the primary infection does not lessen resistance because these gametocytes derive from drug sensitive parasites. Gametocytes carrying the resistance genes will not reach transmission intensities until the resistant biomass has expanded to numbers close to those producing illness (>107 parasites) (Jeffery and Eyles, 1955). In order to decrease the spread of resistance, gametocyte production from the recrudescent resistant infection must be prevented.
One way to look at the relationship between resistance and transmission intensity is as follows. In low-transmission areas the majority of malaria is symptomatic, and selection therefore takes place at a time when relatively large numbers of parasites in a given individual encounter antimalarial drugs. In higher transmission areas, however, symptomatic disease usually occurs during the first years of life. Later on, malaria becomes less symptomatic due to imperfect immunity (sometimes called premunition), which holds the infection in check at levels below the symptom threshold.
Despite premunition, many residents of high-transmission areas still receive antimalarial treatments throughout their lives, often inappropriately. Fortunately, most “treatments” are largely unrelated to the peaks of parasitemia, thereby minimizing the selection of resistant strains. Immunity also reduces the emergence of resistance considerably (White and Pongtavornpinyo, 2003). Even if a resistant mutant does survive and multiply, the likelihood that it will produce sufficient gametocytes for transmission is reduced by the presence of other competing parasite genotypes (Dye and Williams, 1997).
Since significant symptoms generally are confined to young children in high-transmission areas, the fact that children comprise only 20 percent of the population, roughly speaking, and their blood volumes are much lower than adults’ (therefore containing fewer total parasites) are final curbs on the emergence of drug resistance in such areas. The net result overall is considerable reduction in the probability of de novo selection and subsequent transmission of a resistant parasite in high- versus low-transmission areas, as borne out by the historical emergence of chloroquine resistance and more rapid increase of antifol resistance in low transmission areas.3
Global Overview of Current Antimalarial Drug Resistance to P. falciparum
The historical emergence of drug-resistant P. falciparum is reviewed elsewhere in this report (see Chapter 5). The following is a brief summary of current levels of global drug resistance to chloroquine and SP based on in vivo assessment of therapeutic treatment failure (TTF). Data showing in vivo resistance to chloroquine and SP in Africa also are presented in maps (Figures 9-1, and 9-2).
In Africa, median chloroquine (CQ)-TTF among children younger than 5 years was above the critical value (TTF=25 percent) in all the countries in the eastern/great lakes (seven of seven), and central/southern (four of four) regions. However, in West Africa, only one of five countries had a median CQ-TTF above the critical value. The median CQ parasitologic failure (CQ-PF) varied from 38 to 91 percent in Southeast Asia, from 23 to 38
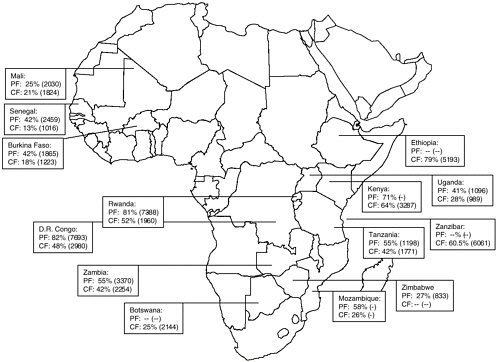
FIGURE 9-1 In vivo chloroquine resistance in selected countries, sub-Saharan Africa, 1996-2002 (median range).
PF = parasitologic failure; CF = clinical failure.
SOURCE: From Talisuna et al. (2004); Kazadi et al. (2003).

FIGURE 9-2 In vivo sulfadoxine-pyrimethamine resistance in selected countries, sub-Saharan Africa.
Dates of studies in upper right corner; PF = parasitologic failure; CF = clinical failure. Median (range) given. Superscript numbers following range = number of studies contributing to data; duration of follow-up in days.
SOURCE: From Talisuna et al. (2004); Kazadi et al. (2003); Plowe et al. (2004); CDC unpublished data.
percent in the Pacific region, from 46 to 77 percent in the Indian subcontinent, and from 12 to 69 percent in the Middle East. In South America, the lowest median CQ-PF was 33 percent, and the highest was 50 percent, while the corresponding lowest and highest estimates for CQ-TTF were 44 and 100 percent, respectively.
In Africa, the median SP-TTF varied from 0 to 35 percent and the median SP parasitologic failure (SP-PF) ranged from 0 to 76 percent. In Asia, the SP-TTF varied from 9 to 35 percent, and in South America it varied from 3 to 32 percent. The SP-PF was higher, and varied between 5 and 67 percent in Asia, and between 6 and 20 percent in South America.
In summary, these recent data indicate a high prevalence of CQ resistance (TTF>25 percent) in the eastern/great lakes and central/southern regions of Africa, where SP resistance is emerging. Such a situation could
quickly lead to multidrug resistance, particularly if SP is adopted widely as first-line treatment.
Resistance to Artemisinins
At present, although artemisinin-resistant strains of P. falciparum (Inselburg, 1985) and Plasmodium yoelii (Peters and Robinson, 1999) have been produced in the laboratory, there is no evidence for clinically relevant artemisinin resistance. Clinical and laboratory strains of P. falciparum also can vary in their artemisinin sensitivity in vitro (Le Bras and Durand, 2003) without clinical effects. Two P. falciparum-infected patients who failed artesunate therapy had parasites that were later shown to be sensitive to artemisinin in vitro (Luxemburger et al., 1998). In general, recrudescence after artemisinin monotherapy is due to host factors (such as drug metabolism) as opposed to failure due to parasite resistance.
In the absence of a functioning spleen, dead intraerythrocytic malaria parasites are not removed. The persistence of these parasites on blood smears—implying delayed clearance—may be misinterpreted as artemisinin resistance (Chotivanich et al., 2002). However, even when 7-day courses of artemisinin derivatives are given, cure rates generally do not exceed 90-95 percent. This failure to cure all patients with artemisinin does not reflect resistance per se but a curious phenomenon in which a subpopulation of very young parasites remain in an arrested state of development for over a week following treatment (Personal communication, D. Kyle, 1997). These parasites may later multiply and cause a recrudescence. However, the recrudescent parasites are no more resistant to artemisinin than the primary infection (White, 1998).
Multidrug resistant P. falciparum (with MDR amplification) are more resistant to artemisinin than non-MDR parasites but the differences do not influence therapeutic outcomes. They have, however, led to incorrect suppositions about the selection of resistance (Wongsrichanalai et al., 1999). For example, on the western border of Thailand, as mefloquine resistance worsened, in vitro susceptibility to dihydroartemisinin (the major metabolite of artesunate and artemether) also fell. However, full artemisinin efficacy along with mefloquine efficacy restored following the systematic use of mefloquine-artesunate in the region (Brockman et al., 2000).
Plasmodium vivax
Chloroquine Resistance
It was not until 1989—30 years after chloroquine resistance first emerged in P. falciparum—that a chloroquine-resistant strain of P. vivax
was first detected in Papua New Guinea (Rieckmann et al., 1989). Chloroquine-resistant P. vivax (CRPV) infections were subsequently reported from Indonesian New Guinea (Baird et al., 1996; Fryauff et al., 1998), Burma (Marlar-Than et al., 1995), India (Garg et al., 1995), and Guyana (Phillips et al., 1996). Sporadic CRPV infections have now surfaced in many geographic regions of the world but do not appear to be a growing problem in most areas (Mendis et al., 2001). One exception is eastern Indonesia, where the risk of therapeutic failure following chloroquine approached 80 percent 28 days after treatment (Baird et al., 1997). Fortunately, drug-sensitive parasites of P. vivax are at little disadvantage vis-à-vis drug resistant strains, largely because P. vivax gametocytes develop quickly in the blood of infected individuals, and they have less drug exposure than P. falciparum prior to transmission.
Primaquine Tolerance
Among the species of Plasmodium that infect humans, only P. vivax and P. ovale have latent liver hypnozoites that can trigger a clinical relapse of malaria after successful eradication of bloodstream parasites. Although primaquine, an 8-aminoquinoline drug, has been used for several decades to prevent relapse, some P. vivax and P. ovale infections acquired in Southeast Asia and the Southwest Pacific area are now tolerant to its effects. As a result, the new recommended adult dose of primaquine is 30 mg base drug daily for 14 days (420 mg total dose) as opposed to the previous 15 mg daily dose over the same time period. An alternative regimen for primaquine-tolerant infections is the weekly administration of 45 mg PQ for 8 weeks. This approach lessens primaquine’s likelihood of producing hemolysis in patients with mild G6PD deficiency while enhancing its efficacy against tolerant strains. In patients with severe G6PD deficiency, primaquine should be strictly avoided.
ASSESSING ANTIMALARIAL DRUG EFFICACY AND RESISTANCE
Four methods are currently available to study or measure antimalarial drug efficacy vis-à-vis drug resistance: in vivo, in vitro, animal model studies, and molecular markers (Bloland, 2001).
In Vivo Testing
Of all available methods, in vivo tests most closely predict the likelihood that parasites infecting a target human population will respond to a particular treatment regimen. The process of in vivo testing involves monitoring a malaria-infected subject following administration of known doses
of an antimalarial drug. In 1973, the World Health Organization defined criteria for the evaluation of chloroquine and amodiaquine and recommended a 28-day follow-up period. These protocols stipulated that patients remain in screened quarters for 28 days to avoid reinfection during the period of evaluation. The original WHO definitions of in vivo drug sensitivity (sensitive [S], and progressively worsening degrees of resistance [R1, R2 and R3]) are shown in Table 9-3.
The original protocols were generally used until the 1990s when shorter durations of follow-up (usually 14 days) were recommended—particularly in areas of high transmission—whereas some research groups adopted longer periods of follow-up (up to 63 days) when evaluating slowly eliminated antimalarial drugs. Shorter periods of follow-up in assessing antimalarial treatment often underestimate drug-resistant recrudescent infections, which, even in areas of high stable transmission, cause significant morbidity, especially anemia.
WHO’s modified protocol, with a shortened follow-up period (WHO, 1996), emphasized clinical over parasitologic outcomes, recognizing that clinical parameters are equally important indicators of treatment response. The most recent 2003 protocol was developed with the specific goal of unifying testing around the world under a single methodology and classification scheme. In this scheme, parasitologic failure was brought back into the classification scheme in recognition that even outwardly asymptomatic parasitemia has adverse health consequences (Price et al., 2001). In all areas, regardless of the intensity of malaria transmission, the evaluation of
TABLE 9-3 Original WHO Classification of in Vivo Antimalarial Drug Sensitivity
|
S (sensitive) |
Reduction to <25% of initial parasitemia on day 2 with smears negative for malaria from day 7 to end of follow-up (28 days, or longer for drugs with long half-lives, such as mefloquine) |
|
R1 response |
Initial clearance of parasitemia, a negative smear on day 7, followed by recrudescence 8 days or more after treatment |
|
R2 response |
Initial clearance or substantial reduction of parasitemia (<25% of the initial count on day 2) but with persistence or recrudescence of parasitemia days 4-7 |
|
R3 Response |
No significant reduction of parasitemia |
|
SOURCE: Bruce-Chwatt et al. (1985). |
|
antimalarials for uncomplicated malaria now emphasizes treatment efficacy in children under five years with clinically apparent malaria. The rationale for this requirement is that, even in populations with little acquired immunity (as in areas of low or highly seasonal malaria transmission), younger children typically have a less favorable therapeutic response to antimalarials than do older children and adults (ter Kuile et al., 1995).
The range of initial parasite densities appropriate for inclusion in vivo testing is between 1,000 and 100,000 asexual parasites/uL in areas of low to moderate transmission, and between 2,000 and 200,000 asexual parasites/uL in areas of high transmission, as supported by the currently accepted definition of hyperparasitemia (WHO, 2000) (Box 9-1).
In vivo methods tend to underestimate the number of true treatment failures and therefore underestimate the level of antimalarial drug resistance. The reasons for this are:
-
In areas of intermediate and high transmission, older children and adults have significant antiparasitic immunity. This leads to a high probability of self-cure giving a false impression of drug efficacy.
-
Short-term (<14 days) follow-up assessments miss treatment failures. In a recent review of more than 16,000 patients followed for 28 days in treatment arms of randomized trials conducted between 1990 and 2003, the widely used day 14 assessment missed between 63 and 100 percent of all treatment failures, and had no predictive value of the true failure rate; the 28 day assessment also missed one-third of treatment failures but did have predictive value for overall treatment rates (Stepniewska et al., in press).
In Vitro Testing
In vitro assays of antimalarial drug resistance are analogous to antibiotic susceptibility assays performed in hospital and commercial microbiology laboratories. In brief, malaria parasites obtained from venous blood samples are co-cultured in microtiter wells with known concentrations of antimalarial drugs. The drug effect is then quantified according a given agent’s ability to inhibit the growth of parasites or their maturation into schizonts (Rieckmann et al., 1978).
In vitro assays are useful because they allow multiple tests (including panels of new and experimental drugs) to be performed on single blood isolates. However, their results frequently differ from those of in vivo tests because the latter are influenced by multiple host factors, chiefly immunity. At present, in vitro tests are only used to test P. falciparum drug susceptibility. Their relative expense plus the technical demands of culturing fresh blood samples limit their wide-scale use under routine field conditions.
|
BOX 9-1 In vivo assessments of malaria drug efficacy are designed to estimate treatment success or failure rates among malaria patients who are representative of the larger population at risk in a given area. In these assessments, a relatively small number of patients (a minimum of 50 is recommended) with slide-confirmed malaria are recruited, treated with the drug or drug combination, and monitored over time. All treatments are observed to ensure adherence. Patients are monitored for recurrence or persistence of malaria parasitemia for 14 or 28 days (occasionally longer). Recurrence of parasitemia with or without accompanying clinical symptoms is noted. Patients’ responses to treatment are classified into early treatment failures (“ETF”—failure occurring within 3 days of initiation of treatment), late clinical failures (“LCF”—recurrence of parasitemia accompanied by fever between 4 and 14 or 28 days), late parasitologic failure (“LPF”—recurrence of parasitemia not accompanied by fever between 4 and 14 or 28 days), and adequate clinical and parasitologic response (“ACPR”—no recurrence of parasites for the duration of monitoring). Ideally, a system of sentinel sites (typically between four and eight sites) chosen to represent the epidemiologic, ecologic, and geographic variation within the country is used to characterize treatment efficacy within the country. Data from these sentinel sites are used to inform the development of malaria drug policy, and/or national malaria treatment guidelines. These data define when a specific malaria treatment can no longer be considered efficacious, and consequently should no longer be used within a national malaria treatment policy. For areas of high transmission intensity in sub-Saharan Africa, WHO recommends a cut-off of greater than 15% total clinical failure (ETF + LCF), and greater than 25% parasitologic failure (LPF) over 14 days (WHO, 2003). There are no clear recommendations, however, on how to reconcile conflicting data from different sentinel sites (i.e., should policy be changed when one of eight sites demonstrates high failure rates, when all eight do, or some time in between?). In any case, because changing and implementing a new treatment policy takes time, the process of policy revision needs to be initiated before these levels are reached. In many cases, programs have waited too long to initiate change, and resistance levels are much higher by the time new drugs are in use. |
Molecular Markers of Resistance
Recent advances in molecular biology—in particular, the use of polymerase chain reaction (PCR)—have allowed the characterization of several genetic markers of drug resistance in P. falciparum (Greenwood, 2002; Wongsrichanalai et al., 2002). At present, attention is focused on four drug resistance genes (Wernsdorfer and Noedl, 2003). The key genes determining resistance to the antifolate drugs pyrimethamine and sulfadoxine are the dihydrofolate reductase gene (pfdhfr) and the dihyropteroate syn-
thase gene (pfdhps). High-level pyrimethamine resistance results from the accumulation of mutations in the dhfr gene, principally at codons 108, 59, 51, and 164 (Plowe et al., 1997; Wang et al., 1997); quadruple mutants combining all four point mutations confer the most severe resistance (Wongsrichanalai et al., 2002). Point mutations in dhps have been associated with decreased susceptibility to sulfadoxine in vitro (Wang et al., 1995). Although the precise relation between mutations in the dhfr and dhps genes and clinical SP resistance is unclear, current data indicate that a sensitive dhfr allele is predictive of SP treatment success independent of the dhps allele (Sibley et al., 2001).
The principal determinant of chloroquine resistance is polymorphism in the P. falciparum chloroquine resistance transporter gene (pfcrt) which is located on chromosome 7 and encodes the digestive vacuole transmembrane protein PfCRT (Wellems et al., 1991; Djimde et al., 2001). Although many polymorphisms associated with chloroquine resistance have been identified, the substitution of threonine for lysine in codon 76 has now been shown to correlate unequivocally with in vitro resistance in P. falciparum isolates from Africa, South America, Asia, and Papua New Guinea (Fidock et al., 2000; Djimde et al., 2001). The same finding has been corroborated by an additional in vitro study of isolates of various geographic origins (Durand et al., 2001) as well as clinical studies conducted in Mali (Djimde et al., 2001), Cameroon (Basco and Ringwald, 2001), Sudan (Babiker et al., 2001), Mozambique (Mayor et al., 2001), Laos (Pillai et al., 2001), Thailand (Chen et al., 2001), and Brazil (Vieira et al., 2001). Paradoxically, although cumulative data indicate that the Thr76 mutation is necessary for expression of the resistant phenotype, Thr76 also has been found to lesser degrees in chloroquine-sensitive P. falciparum strains, suggesting that other polymorphisms or genes contribute to resistance (Wongsrichanalai et al., 2002). Conversely, because of the added contribution of host immunity, individuals from malaria-endemic areas harboring parasites with the Thr76 mutation also have had successful treatment outcomes following chloroquine (Djimde et al., 2001).
A fourth gene, known as the P. falciparum multi-drug resistance gene (pfmdr), encodes the transmemebrane protein P-glycoprotein homologue 1 (Pgh1), and modulates the resistance phenotype in some if not all PfCRT mutant parasites. (Wongsrichanalai et al., 2002). The aspartic acid to tyrosine point mutation in codon 86 has been positively associated with chloroquine resistance in Mali (Djimde et al., 2001), The Gambia (von Seidlein et al., 1997), Sudan (Babiker et al., 2001), Uganda (Flueck et al., 2000), Thailand (Price et al., 1999b), and Brazil (Zalis et al., 1998); however, the same mutation was not associated with chloroquine resistance in other studies conducted in Uganda (Dorsey et al., 2001), Laos (Pillai et al., 2001), Thailand (Chaiyaroj et al., 1999), and Brazil (Povoa et al., 1998).
The relationship between mefloquine sensitivity and pfmdr1 gene mutations and/or overexpression also was ambiguous until studies in Thailand identified amplification (increased copy number) of Pfmdr1 to be the main determinant of mefloquine resistance (Price et al., 1999b).
In summary, while molecular markers allow drug resistance testing of multiple samples in a relatively short time, a continuing difficulty of this approach—particularly for chloroquine and mefloquine—has been to identify the specific mutations conferring resistance. The problem becomes even more challenging when drug resistance stems from multiple mutations on one or more genes.
REFERENCES
Abdin MZ, Israr M, Rehman RU, Jain SK. 2003. Artemisinin, a novel antimalarial drug: Biochemical and molecular approaches for enhanced production. Planta Medica 69(4): 289-299.
Adjuik M, Agnamey P, Babiker A, Borrmann S, Brasseur P, Cisse M, Cobelens F, Diallo S, Faucher JF, Garner P, Gikunda S, Kremsner PG, Krishna S, Lell B, Loolpapit M, Matsiegui PB, Missinou MA, Mwanza J, Ntoumi F, Olliaro P, Osimbo P, Rezbach P, Some E, Taylor WR. 2002. Amodiaquine-artesunate versus amodiaquine for uncomplicated Plasmodium falciparum malaria in African children: A randomised, multicentre trial. Lancet 359(9315):1365-1372.
Alving AS, Johnson CF, Tarlov AR, Brewer GJ, Kellermeyer RW, Carson PE. 1960. Mitigation of the haemolytic effect of primaquine and enhancement of its action against exoerythrocytic forms of the Chesson strain of Plasmodium vivax by intermittent regimens of drug administration: A preliminary report. Bulletin of the World Health Organization 22:621-631.
Anonymous. 1982. Clinical studies on the treatment of malaria with qinghaosu and its derivatives. China cooperative research group on qinghaosu and its derivatives as antimalarials. Journal of Traditional Chinese Medicine 2(1):45-50.
Awad MI, Alkadru AM, Behrens RH, Baraka OZ, Eltayeb IB. 2003. Descriptive study on the efficacy and safety of artesunate suppository in combination with other antimalarials in the treatment of severe malaria in Sudan. American Journal of Tropical Medicine and Hygiene 68(2):153-158.
Babiker HA, Pringle SJ, Abdel-Muhsin A, Mackinnon M, Hunt P, Walliker D. 2001. High-level chloroquine resistance in Sudanese isolates of Plasmodium falciparum is associated with mutations in the chloroquine resistance transporter gene pfcrt and the multidrug resistance gene pfmdr1. Journal of Infectious Diseases 183(10):1535-1538.
Baird JK, Sustriayu Nalim MF, Basri H, Masbar S, Leksana B, Tjitra E, Dewi RM, Khairani M, Wignall FS. 1996. Survey of resistance to chloroquine by Plasmodium vivax in Indonesia. Transactions of the Royal Society of Tropical Medicine and Hygiene 90(4):409-411.
Baird JK, Wiady I, Fryauff DJ, Sutanihardja MA, Leksana B, Widjaya H, Kysdarmanto, Subianto B. 1997. In vivo resistance to chloroquine by Plasmodium vivax and Plasmodium falciparum at Nabire, Irian Jaya, Indonesia. American Journal of Tropical Medicine and Hygiene 56(6):627-631.
Bakshi R, Hermeling-Fritz I, Gathmann I, Alteri E. 2000. An integrated assessment of the clinical safety of artemether-lumefantrine: A new oral fixed-dose combination antimalarial drug. Transactions of the Royal Society of Tropical Medicine and Hygiene 94(4):419-424.
Barnes K, Folb P. 2003. Reducing Malaria’s Burden: Evidence of Effectiveness for Decision Makers (Technical Report). The Role of Artemisinin-Based Combination Therapy in Malaria Management. Washington, DC: Global Health Council.
Barnes KI, Mwenechanya J, Tembo M, McIlleron H, Folb PI, Ribeiro I, Little F, Gomes M, Molyneux ME. 2004. Efficacy of rectal artesunate compared with parenteral quinine in initial treatment of moderately severe malaria in African children and adults: A randomised study. Lancet 363(9421):1598-1605.
Basco LK, Ringwald P. 2001. Analysis of the key pfcrt point mutation and in vitro and in vivo response to chloroquine in Yaounde, Cameroon. Journal of Infectious Diseases 183(12):1828-1831.
Berman PA, Adams PA. 1997. Artemisinin enhances heme-catalysed oxidation of lipid membranes. Free Radical Biology and Medicine 22(7):1283-1288.
Bindschedler M, Lefevre G, Ezzet F, Schaeffer N, Meyer I, Thomsen MS. 2000. Cardiac effects of co-artemether (artemether/lumefantrine) and mefloquine given alone or in combination to healthy volunteers. European Journal of Clinical Pharmacology 56(5):375-381.
Bloland PB. 2001. Drug Resistance in Malaria. Geneva: World Health Organization.
Bray PG, Mungthin M, Ridley RG, Ward SA. 1998. Access to hematin: The basis of chloroquine resistance. Molecular Pharmacology 54(1):170-179.
Brewer TG, Grate SJ, Peggins JO, Weina PJ, Petras JM, Levine BS, Heiffer MH, Schuster BG. 1994. Fatal neurotoxicity of arteether and artemether. American Journal of Tropical Medicine and Hygiene 51(3):251-259.
Brockman A, Price RN, van Vugt M, Heppner DG, Walsh D, Sookto P, Wimonwattrawatee T, Looareesuwan S, White NJ, Nosten F. 2000. Plasmodium falciparum antimalarial drug susceptibility on the north-western border of Thailand during five years of extensive use of artesunate-mefloquine. Transactions of the Royal Society of Tropical Medicine and Hygiene 94(5):537-544.
Brossi A, Venugopalan B, Dominguez Gerpe L, Yeh HJ, Flippen-Anderson JL, Buchs P, Luo XD, Milhous W, Peters W. 1988. Arteether, a new antimalarial drug: Synthesis and antimalarial properties. Journal of Medicinal Chemistry 31(3):645-650.
Bruce-Chwatt, LJ. 1985. Essential Malariology. 2nd ed. New York: John Wiley and Sons.
Chaiyaroj SC, Buranakiti A, Angkasekwinai P, Looareesuwan S, Cowman AF. 1999. Analysis of mefloquine resistance and amplification of pfmdr1 in multidrug-resistant Plasmodium falciparum isolates from Thailand. American Journal of Tropical Medicine and Hygiene 61(5):780-783.
Chen N, Russell B, Staley J, Kotecka B, Nasveld P, Cheng Q. 2001. Sequence polymorphisms in pfcrt are strongly associated with chloroquine resistance in Plasmodium falciparum. Journal of Infectious Diseases 183(10):1543-1545.
Chotivanich K, Udomsangpetch R, Dondorp A, Williams T, Angus B, Simpson JA, Pukrittayakamee S, Looareesuwan S, Newbold CI, White NJ. 2000. The mechanisms of parasite clearance after antimalarial treatment of Plasmodium falciparum malaria. Journal of Infectious Diseases 182(2):629-633.
Chotivanich K, Udomsangpetch R, McGready R, Proux S, Newton P, Pukrittayakamee S, Looareesuwan S, White NJ. 2002. Central role of the spleen in malaria parasite clearance. Journal of Infectious Diseases 185(10):1538-1541.
Day NP, Pham TD, Phan TL, Dinh XS, Pham PL, Ly VC, Tran TH, Nguyen TH, Bethell DB, Nguyan HP, Tran TH, White NJ. 1996. Clearance kinetics of parasites and pigment-containing leukocytes in severe malaria. Blood 88(12):4694-4700.
Deen JL, von Seidlein L, Pinder M, Walraven GE, Greenwood BM. 2001. The safety of the combination artesunate and pyrimethamine-sulfadoxine given during pregnancy. Transactions of the Royal Society of Tropical Medicine and Hygiene 95(4):424-428.
Diourte Y, Djimde A, Doumbo OK, Sagara I, Coulibaly Y, Dicko A, Diallo M, Diakite M, Cortese JF, Plowe CV. 1999. Pyrimethamine-sulfadoxine efficacy and selection for mutations in Plasmodium falciparum dihydrofolate reductase and dihydropteroate synthase in Mali. American Journal of Tropical Medicine and Hygiene 60(3):475-478.
Djimde A, Doumbo OK, Cortese JF, Kayentao K, Doumbo S, Diourte Y, Dicko A, Su XZ, Nomura T, Fidock DA, Wellems TE, Plowe CV, Coulibaly D. 2001. A molecular marker for chloroquine-resistant falciparum malaria. New England Journal of Medicine 344(4): 257-263.
Dorsey G, Kamya MR, Ndeezi G, Babirye JN, Phares CR, Olson JE, Katabira ET, Rosenthal PJ. 2000. Predictors of chloroquine treatment failure in children and adults with falciparum malaria in Kampala, Uganda. American Journal of Tropical Medicine and Hygiene 62(6):686-692.
Dorsey G, Kamya MR, Singh A, Rosenthal PJ. 2001. Polymorphisms in the Plasmodium falciparum pfcrt and pfmdr-1 genes and clinical response to chloroquine in Kampala, Uganda. Journal of Infectious Diseases 183(9):1417-1420.
Dorsey G, Njama D, Kamya MR, Cattamanchi A, Kyabayinze D, Staedke SG, Gasasira A, Rosenthal PJ. 2002. Sulfadoxine/pyrimethamine alone or with amodiaquine or artesunate for treatment of uncomplicated malaria: a longitudinal randomised trial. Lancet 360(9350):2031-2038.
Durand R, Jafari S, Vauzelle J, Delabre JF, Jesic Z, Le Bras J. 2001. Analysis of pfcrt point mutations and chloroquine susceptibility in isolates of Plasmodium falciparum. Molecular and Biochemical Parasitology 114(1):95-102.
Dye C, Williams BG. 1997. Multigenic drug resistance among inbred malaria parasites. Proceedings of the Royal Society of London—Series B: Biological Sciences 264(1378):61-67.
Eckstein-Ludwig U, Webb RJ, Van Goethem ID, East JM, Lee AG, Kimura M, O’Neill PM, Bray PG, Ward SA, Krishna S. 2003. Artemisinins target the serca of Plasmodium falciparum. [See Comment]. Nature 424(6951):957-961.
Fidock DA, Nomura T, Talley AK, Cooper RA, Dzekunov SM, Ferdig MT, Ursos LM, Sidhu AB, Naude B, Deitsch KW, Su XZ, Wootton JC, Roepe PD, Wellems TE. 2000. Mutations in the P. falciparum digestive vacuole transmembrane protein pfcrt and evidence for their role in chloroquine resistance. Molecular Cell 6(4):861-871.
Flueck TP, Jelinek T, Kilian AH, Adagu IS, Kabagambe G, Sonnenburg F, Warhurst DC. 2000. Correlation of in vivo-resistance to chloroquine and allelic polymorphisms in Plasmodium falciparum isolates from Uganda. Tropical Medicine and International Health 5(3):174-178.
Fryauff DJ, Tuti S, Mardi A, Masbar S, Patipelohi R, Leksana B, Kain KC, Bangs MJ, Richie TL, Baird JK. 1998. Chloroquine-resistant Plasmodium vivax in transmigration settlements of west Kalimantan, Indonesia. American Journal of Tropical Medicine and Hygiene 59(4):513-518.
Garg M, Gopinathan N, Bodhe P, Kshirsagar NA. 1995. Vivax malaria resistant to chloroquine: Case reports from Bombay. Transactions of the Royal Society of Tropical Medicine and Hygiene 89(6):656-657.
Geary TG, Divo AA, Jensen JB. 1989. Stage specific actions of antimalarial drugs on Plasmodium falciparum in culture. American Journal of Tropical Medicine and Hygiene 40(3): 240-244.
Genovese RF, Newman DB, Petras JM, Brewer TG. 1998. Behavioral and neural toxicity of arteether in rats. Pharmacology, Biochemistry and Behavior 60(2):449-458.
Greenwood B. 2002. The molecular epidemiology of malaria. Tropical Medicine and International Health 7(12):1012-1021.
Hastings IM, Watkins WM, White NJ. 2002. The evolution of drug-resistant malaria: The role of drug elimination half-life. Philosophical Transactions of the Royal Society of London—Series B: Biological Sciences 357(1420):505-519.
Hien TT, Day NPJ, Phu NH, Mai NTH, Chau TTH, Loc PP, Sinh DX, Chuong LV, Vinh H, Waller D, Peto TEA, White NJ. 1996. A controlled trial of artemether or quinine in Vietnamese adults with severe falciparum malaria. New England Journal of Medicine 335(2):76-83.
Hien TT, Turner GD, Mai NT, Phu NH, Bethell D, Blakemore WF, Cavanagh JB, Dayan A, Medana I, Weller RO, Day NP, White NJ. 2003. Neuropathological assessment of artemether-treated severe malaria. Lancet 362(9380):295-296.
Imwong M, Pukrittakayamee S, Looareesuwan S, Pasvol G, Poirreiz J, White NJ, Snounou G. 2001. Association of genetic mutations in Plasmodium vivax dhfr with resistance to sulfadoxine-pyrimethamine: Geographical and clinical correlates. Antimicrobial Agents and Chemotherapy 45(11):3122-3127.
Inselburg J. 1985. Induction and isolation of artemisinin-resistant mutants of Plasmodium falciparum. American Journal of Tropical Medicine and Hygiene 34(3):417-418.
Jeffery GM, Eyles D. 1955. Infectivity to mosquitoes of Plasmodium falciparum as related to gametocyte density and duration of infection. American Journal of Tropical Medicine and Hygiene 4:781-789.
Karunajeewa HA, Kemiki A, Alpers MP, Lorry K, Batty KT, Ilett KF, Davis TM. 2003. Safety and therapeutic efficacy of artesunate suppositories for treatment of malaria in children in Papua New Guinea. Pediatric Infectious Disease Journal 22(3):251-256.
Kazadi WM, Vong S, Makina BN, Mantshumba JC, Kabuya W, Kebela BI, Ngimbi NP. 2003. Assessing the Efficacy of chloroquine and sulfadoxine-pyrimethamine for treatment of uncomplicated Plasmodium falciparum malaria in the Democratic Republic of Congo. Tropical Medicine and International Health 8(10):868-75.
Kissinger E, Hien TT, Hung NT, Nam ND, Tuyen NL, Dinh BV, Mann C, Phu NH, Loc PP, Simpson JA, White NJ, Farrar JJ. 2000. Clinical and neurophysiological study of the effects of multiple doses of artemisinin on brain-stem function in Vietnamese patients. American Journal of Tropical Medicine and Hygiene 63(1-2):48-55.
Klayman DL. 1985. Qinghaosu (artemisinin): An antimalarial drug from China. Science 228(4703):1049-1055.
Klayman DL, Lin AJ, Acton N, Scovill JP, Hoch JM, Milhous WK, Theoharides AD, Dobek AS. 1984. Isolation of artemisinin (qinghaosu) from Artemisia annua growing in the United States. Journal of Natural Products 47(4):715-717.
Korsinczky M, Chen N, Kotecka B, Saul A, Rieckmann K, Cheng Q. 2000. Mutations in Plasmodium falciparum cytochrome-b that are associated with atovaquone resistance are located at a putative drug-binding site. Antimicrobial Agents and Chemotherapy 44(8):2100-2108.
Krogstad DJ, Schlesinger PH. 1987. Acid-vesicle function, intracellular pathogens, and the action of chloroquine against Plasmodium falciparum. New England Journal of Medicine 317(9):542-549.
Kublin JG, Cortese JF, Njunju EM, Mukadam RA, Wirima JJ, Kazembe PN, Djimde AA, Kouriba B, Taylor TE, Plowe CV. 2003. Reemergence of chloroquine-sensitive Plasmodium falciparum malaria after cessation of chloroquine use in Malawi. Journal of Infectious Diseases 187(12):1870-1875.
Kumar N, Zheng H. 1990. Stage-specific gametocytocidal effect in vitro of the antimalaria drug qinghaosu on Plasmodium falciparum. Parasitology Research 76(3):214-218.
Le Bras J, Durand R. 2003. The mechanisms of resistance to antimalarial drugs in Plasmodium falciparum. Fundamental and Clinical Pharmacology 17(2):147-153.
Lefevre G, Looareesuwan S, Treeprasertsuk S, Krudsood S, Silachamroon U, Gathmann I, Mull R, Bakshi R. 2001. A clinical and pharmacokinetic trial of six doses of artemether-lumefantrine for multidrug-resistant Plasmodium falciparum malaria in Thailand. American Journal of Tropical Medicine and Hygiene 64(5):247-256.
Leonardi E, Gilvary G, White NJ, Nosten F. 2001. Severe allergic reactions to oral artesunate: A report of two cases. Transactions of the Royal Society of Tropical Medicine and Hygiene 95(2):182-183.
Li GQ, Guo XB, Yang F. 1990. Clinical Trials on Qinghaosu and Its Derivatives. China. Guangzhou Medicine Institute. Vol. 1.
Looareesuwan S, Wilairatana P, Chokejindachai W, Chalermrut K, Wernsdorfer W, Gemperli B, Gathmann I, Royce C. 1999. A randomized, double-blind, comparative trial of a new oral combination of artemether and benflumetol (Cgp 56697) with mefloquine in the treatment of acute Plasmodium falciparum malaria in Thailand. American Journal of Tropical Medicine and Hygiene 60(2):238-243.
Luo XD, Shen CC. 1987. The chemistry, pharmacology, and clinical applications of qinghaosu (artemisinin) and its derivatives. Medicinal Research Reviews 7(1):29-52.
Luxemburger C, Nosten F, Raimond SD, Chongsuphajaisiddhi T, White NJ. 1995. Oral artesunate in the treatment of uncomplicated hyperparasitemic falciparum malaria. American Journal of Tropical Medicine and Hygiene 53(5):522-525.
Luxemburger C, Brockman A, Silamut K, Nosten F, van Vugt M, Gimenez F, Chongsuphajaisiddhi T, White NJ. 1998. Two patients with falciparum malaria and poor in vivo responses to artesunate. Transactions of the Royal Society of Tropical Medicine and Hygiene 92(6):668-669.
Marlar-Than, Myat-Phone-Kyaw, Aye-Yu-Soe, Khaing-Khaing-Gyi, Ma-Sabai, Myint-Oo. 1995. Development of resistance to chloroquine by Plasmodium vivax in Myanmar. Transactions of the Royal Society of Tropical Medicine and Hygiene 89(3):307-308.
Martin SK, Oduola AM, Milhous WK. 1987. Reversal of chloroquine resistance in Plasmodium falciparum by verapamil. Science 235(4791):899-901.
Mayor AG, Gomez-Olive X, Aponte JJ, Casimiro S, Mabunda S, Dgedge M, Barreto A, Alonso PL. 2001. Prevalence of the k76t mutation in the putative Plasmodium falciparum chloroquine resistance transporter (pfcrt) gene and its relation to chloroquine resistance in Mozambique. Journal of Infectious Diseases 183(9):1413-1416.
McGready R, Cho T, Cho JJ, Simpson JA, Luxemburger C, Dubowitz L, Looareesuwan S, White NJ, Nosten F. 1998. Artemisinin derivatives in the treatment of falciparum malaria in pregnancy. Transactions of the Royal Society of Tropical Medicine and Hygiene 92(4):430-433.
McGready R, Brockman A, Cho T, Cho D, van Vugt M, Luxemburger C, Chongsuphajaisiddhi T, White NJ, Nosten F. 2000. Randomized comparison of mefloquine-artesunate versus quinine in the treatment of multidrug-resistant falciparum malaria in pregnancy. Transactions of the Royal Society of Tropical Medicine and Hygiene 94(6):689-693.
McGready R, Cho T, Keo NK, Thwai KL, Villegas L, Looareesuwan S, White NJ, Nosten F. 2001. Artemisinin antimalarials in pregnancy: A prospective treatment study of 539 episodes of multidrug-resistant Plasmodium falciparum. Clinical Infectious Diseases 33(12):2009-2016.
Mendis K, Sina BJ, Marchesini P, Carter R. 2001. The neglected burden of Plasmodium vivax malaria. American Journal of Tropical Medicine and Hygiene 64(1-2 Suppl):97-106.
Meshnick SR, Thomas A, Ranz A, Xu CM, Pan HZ. 1991. Artemisinin (Qinghaosu): The role of intracellular hemin in its mechanism of antimalarial action. Molecular and Biochemical Parasitology 49(2):181-189.
Miller LG, Panosian CB. 1997. Ataxia and slurred speech after artesunate treatment for falciparum malaria. New England Journal of Medicine 336(18):1328.
Myint HY, Tipmanee P, Nosten F, Day NPJ, Pukrittayakamee S, Looareesuwan S, White NJ. 2004. A systematic overview of published antimalarial drug trials. Transactions of the Royal Society of Tropical Medicine and Hygiene 98(2):73-81.
Nacher M. 2002. Malarial anaemia: A crossroad? Medical Hypotheses 59(3):363-365.
Newton PN, Angus BJ, Chierakul W, Dondorp A, Ruangveerayuth R, Silamut K, Teerapong P, Suputtamongkol Y, Looareesuwan S, White NJ. 2003. Randomized comparison of artesunate and quinine in the treatment of severe falciparum malaria. Clinical Infectious Diseases 37(1):7-16.
Nontprasert A, Nosten-Bertrand M, Pukrittayakamee S, Vanijanonta S, Angus BJ, White NJ. 1998. Assessment of the neurotoxicity of parenteral artemisinin derivatives in mice. American Journal of Tropical Medicine and Hygiene 59(4):519-522.
Nosten F, McGready R, Simpson JA, Thwai KL, Balkan S, Cho T, Hkirijaroen L, Looareesuwan S, White NJ. 1999. Effects of Plasmodium vivax malaria in pregnancy. Lancet 354(9178):546-549.
Nosten F, van Vugt M, Price R, Luxemburger C, Thway KL, Brockman A, McGready R, ter Kuile F, Looareesuwan S, White NJ. 2000. Effects of artesunate-mefloquine combination on incidence of Plasmodium falciparum malaria and mefloquine resistance in Western Thailand: A prospective study. Lancet 356(9226):297-302.
Nzila AM, Nduati E, Mberu EK, Hopkins Sibley C, Monks SA, Winstanley PA, Watkins WM. 2000. Molecular evidence of greater selective pressure for drug resistance exerted by the long-acting antifolate pyrimethamine/sulfadoxine compared with the shorter-acting chlorproguanil/dapsone on Kenyan Plasmodium falciparum. Journal of Infectious Diseases 181(6):2023-2028.
Oduola AM, Milhous WK, Salako LA, Walker O, Desjardins RE. 1987. Reduced in-vitro susceptibility to mefloquine in West African isolates of Plasmodium falciparum. Lancet 2(8571):1304-1305.
Oduola AMJ, Omitowoju GO, Gerena L, Kyle DE, Milhous WK, Sowunmi A, Salako LA. 1993. Reversal of mefloquine resistance with penfluridol in isolates of Plasmodium falciparum from South-west Nigeria. Transactions of the Royal Society of Tropical Medicine and Hygiene 87(1):81-83.
Omari AA, Preston C, Garner P. 2003. Artemether-lumefantrine for treating uncomplicated falciparum malaria. Cochrane Database of Systematic Reviews (2):CD003125.
Payne D. 1988. Did medicated salt hasten the spread of chloroquine resistance in Plasmodium falciparum? Parasitology Today 4(4):112-115.
Peters W. 1987. Chemotherapy and Drug Resistance in Malaria. 2nd ed. London: Academic Press.
Peters W, Robinson BL. 1999. The chemotherapy of rodent malaria. LVI: Studies on the development of resistance to natural and synthetic endoperoxides. Annals of Tropical Medicine and Parasitology 93(4):325-329.
Petras JM, Young GD, Bauman RA, Kyle DE, Gettayacamin M, Webster HK, Corcoran KD, Peggins JO, Vane MA, Brewer TG. 2000. Arteether-induced brain injury in Macaca mulatta. I. The precerebellar nuclei: The lateral reticular nuclei, paramedian reticular nuclei, and perihypoglossal nuclei. Anatomy and Embryology 201(5):383-397.
Phillips EJ, Keystone JS, Kain KC. 1996. Failure of combined chloroquine and high-dose primaquine therapy for Plasmodium vivax malaria acquired in Guyana, South America. Clinical Infectious Diseases 23(5):1171-1173.
Pillai DR, Labbe AC, Vanisaveth V, Hongvangthong B, Pomphida S, Inkathone S, Zhong K, Kain KC. 2001. Plasmodium falciparum malaria in Laos: Chloroquine treatment outcome and predictive value of molecular markers. Journal of Infectious Diseases 183(5):789-795.
Plowe CV, Cortese JF, Djimde A, Nwanyanwu OC, Watkins WM, Winstanley PA, Estrada-Franco JG, Mollinedo RE, Avila JC, Cespedes JL, Carter D, Doumbo OK. 1997. Mutations in Plasmodium falciparum dihydrofolate reductase and dihydropteroate synthase and epidemiologic patterns of pyrimethamine-sulfadoxine use and resistance. Journal of Infectious Diseases 176(6):1590-1596.
Plowe CV, Kublin JG, Dzinjalamala FK, Kamwendo DS, Mukadam RAG, Chimpeni P, Molyneux ME, Taylor TE. 2004. Sustained clinical efficacy of sulfadoxine-pyrimethamine for uncomplicated falciparum malaria in Malawi after 10 years as first line treatment: Five year prospective study. British Medical Journal 328(7439):545-548.
Povoa MM, Adagu IS, Oliveira SG, Machado RL, Miles MA, Warhurst DC. 1998. Pfmdr1 Asn1042asp and Asp1246tyr polymorphisms, thought to be associated with chloroquine resistance, are present in chloroquine-resistant and -sensitive Brazilian field isolates of Plasmodium falciparum. Experimental Parasitology 88(1):64-68.
Price RN, Nosten F, Luxemburger C, ter Kuile FO, Paiphun L, Chongsuphajaisiddhi T, White NJ. 1996. Effects of artemisinin derivatives on malaria transmissibility. Lancet 347(9016):1654-1658.
Price RN, Nosten F, Luxemburger C, van Vugt M, Phaipun L, Chongsuphajaisiddhi T, White NJ. 1997. Artesunate/mefloquine treatment of multi-drug resistant falciparum malaria. Transactions of the Royal Society of Tropical Medicine and Hygiene 91(5):574-577.
Price R, van Vugt M, Phaipun L, Luxemburger C, Simpson J, McGready R, ter Kuile F, Kham A, Chongsuphajaisiddhi T, White NJ, Nosten F. 1999a. Adverse effects in patients with acute falciparum malaria treated with artemisinin derivatives. American Journal of Tropical Medicine and Hygiene 60(4):547-555.
Price RN, Cassar C, Brockman A, Duraisingh M, van Vugt M, White NJ, Nosten F, Krishna S. 1999b. The pfmdr1 gene is associated with a multidrug-resistant phenotype in Plasmodium falciparum from the western border of Thailand. Antimicrobial Agents and Chemotherapy 43(12):2943-2949.
Price RN, Simpson JA, Nosten F, Luxemburger C, Hkirjaroen L, ter Kuile F, Chongsuphajaisiddhi T, White NJ. 2001. Factors contributing to anemia after uncomplicated falciparum malaria. American Journal of Tropical Medicine and Hygiene 65(5):614-622.
Pukrittayakamee S, Chantra A, Simpson JA, Vanijanonta S, Clemens R, Looareesuwan S, White NJ. 2000. Therapeutic responses to different antimalarial drugs in vivax malaria. Antimicrobial Agents and Chemotherapy 44(6):1680-1685.
Rieckmann KH, Campbell GH, Sax LJ, Mrema JE. 1978. Drug sensitivity of Plasmodium falciparum: An in-vitro microtechnique. Lancet 1(8054):22-23.
Rieckmann KH, Davis DR, Hutton DC. 1989. Plasmodium vivax resistance to chloroquine? Lancet 2(8673):1183-1184.
Sabchareon A, Attanath P, Chanthavanich P, Phanuaksook P, Prarinyanupharb V, Poonpanich Y, Mookmanee D, Teja-Isavadharm P, Heppner DG, Brewer TG, Chongsuphajaisiddhi T. 1998. Comparative clinical trial of artesunate suppositories and oral artesunate in combination with mefloquine in the treatment of children with acute falciparum malaria. American Journal of Tropical Medicine and Hygiene 58(1):11-16.
Scott MD, Meshnick SR, Williams RA, Chiu DT, Pan HC, Lubin BH, Kuypers FA. 1989. Qinghaosu-mediated oxidation in normal and abnormal erythrocytes. Journal of Laboratory and Clinical Medicine 114(4):401-406.
Sibley CH, Hyde JE, Sims PF, Plowe CV, Kublin JG, Mberu EK, Cowman AF, Winstanley PA, Watkins WM, Nzila AM. 2001. Pyrimethamine-sulfadoxine resistance in Plasmodium falciparum: What next? Trends in Parasitology 17(12):582-588.
Simon F, Le Bras J, Gaudebout C, Girard PM. 1988. Reduced sensitivity of Plasmodium falciparum to mefloquine in West Africa. Lancet 1(8583):467-468.
Sowunmi A, Oduola AM, Ogundahunsi OA, Falade CO, Gbotosho GO, Salako LA. 1997. Enhanced efficacy of chloroquine-chlorpheniramine combination in acute uncomplicated falciparum malaria in children. Transactions of the Royal Society of Tropical Medicine and Hygiene 91(1):63-67.
Srivastava IK, Rottenberg H, Vaidya AB. 1997. Atovaquone, a broad spectrum antiparasitic drug, collapses mitochondrial membrane potential in a malarial parasite. Journal of Biological Chemistry 272(7):3961-3966.
Steketee RW, Wirima JJ, Slutsker L, Khoromana CO, Breman JG, Heymann DL. 1996. Objectives and methodology in a study of malaria treatment and prevention in pregnancy in rural Malawi: The Mangochi Malaria Research Project. American Journal of Tropical Medicine and Hygiene 55(1 Suppl):8-16.
Stepniewska K, Taylor WRJ, Mayxay M, Smithuis F, Guthmann J-P, Barnes K, Myint H, Price R, Olliaro P, Pukrittayakamee S, Hien TT, Farrar J, Nosten F, Day NPJ, White NJ. In press. The in vivo assessment of antimalarial drug efficacy in falciparum malaria; the duration of follow-up. Antimicrobial Agents and Chemotherapy
Sutherland CJ, Alloueche A, Curtis J, Drakeley CJ, Ord R, Durasingh M, Greenwood BM, Pinder M, Warhurst D, Targett GAI. 2002. Gambian children successfully treated with chloroquine can harbor and transmit Plasmodium falciparum gametocytes carrying resistance genes. American Journal of Tropical Medicine and Hygiene 67:578-585.
Talisuna AO, Kyosiimire-Lugemwa J, Langi P, Mutabingwa TK, Watkins W, Van Marck E, Egwang T, D’Alessandro U. 2002a. Role of the pfcrt codon 76 mutation as a molecular marker for population-based surveillance of chloroquine (Cq)-resistant Plasmodium falciparum malaria in Ugandan sentinel sites with high Cq resistance. Transactions of the Royal Society of Tropical Medicine and Hygiene 96(5):551-556.
Talisuna AO, Langi P, Bakyaita N, Egwang T, Mutabingwa TK, Watkins W, Van Marck E, D’Alessandro U. 2002b. Intensity of malaria transmission, antimalarial-drug use and resistance in Uganda: What is the relationship between these three factors? Transactions of the Royal Society of Tropical Medicine and Hygiene 96(3):310-317.
Talisuna AO, Bloland P, D’Alessandro U. 2004. History, dynamics, and public health importance of malaria parasite resistance. Clinical Microbiology Reviews 17:235-254.
Targett G, Drakeley C, Jawara M, von Seidlein L, Coleman R, Deen J, Pinder M, Doherty T, Sutherland C, Walraven G, Milligan P. 2001. Artesunate reduces but does not prevent posttreatment transmission of Plasmodium falciparum to Anopheles gambiae. Journal of Infectious Diseases 183(8):1254-1259.
Taylor WRJ, White NJ. 2004. Antimalarial drug toxicity: A review. Drug Safety 27(1):25-61.
ter Kuile FO, Luxemburger C, Nosten F, Thai KL, Chongsuphajaisiddhi T, White NJ. 1995. Predictors of mefloquine treatment failure: A prospective study of 1590 patients with uncomplicated falciparum malaria. Transactions of the Royal Society of Tropical Medicine and Hygiene 89(6):660-664.
ter Kuile F, White NJ, Holloway P, Pasvol G, Krishna S. 1993. Plasmodium falciparum: In vitro studies of the pharmacodynamic properties of drugs used for the treatment of severe malaria. Experimental Parasitology 76(1):85-95.
Toovey S, Jamieson A. 2004. Audiotometric changes associated with the treatment of uncomplicated falciparum malaria with co-artemether. Transactions of the Royal Society of Tropical Medicine and Hygiene 98(5):261-267 [discussion 268-269].
Tran TH, Day NP, Nguyen HP, Nguyen TH, Tran TH, Pham PL, Dinh XS, Ly VC, Ha V, Waller D, Peto TE, White NJ. 1996. A controlled trial of artemether or quinine in Vietnamese adults with severe falciparum malaria. New England Journal of Medicine 335(2):76-83.
Van Vugt M, Brockman A, Gemperli B, Luxemburger C, Gathmann I, Royce C, Slight T, Looareesuwan S, White NJ, Nosten F. 1998. Randomized comparison of artemether-benflumetol and artesunate-mefloquine in treatment of multidrug-resistant falciparum malaria. Antimicrobial Agents and Chemotherapy 42(1):135-139.
Van Vugt M, Ezzet F, Nosten F, Gathmann I, Wilairatana P, Looareesuwan S, White NJ. 1999. No evidence of cardiotoxicity during antimalarial treatment with artemether-lumefantrine. American Journal of Tropical Medicine and Hygiene 61(6):964-967.
Van Vugt M, Angus BJ, Price RN, Mann C, Simpson JA, Poletto C, Htoo SE, Looareesuwan S , White NJ, Nosten F. 2000. A case-control auditory evaluation of patients treated with artemisinin derivatives for multidrug-resistant Plasmodium falciparum malaria. American Journal of Tropical Medicine and Hygiene 62(1):65-69.
Vieira PP, das Gracas Alecrim M, da Silva LH, Gonzalez-Jimenez I, Zalis MG. 2001. Analysis of the pfcrt K76t mutation in Plasmodium falciparum isolates from the Amazon region of Brazil. Journal of Infectious Diseases 183(12):1832-1833.
von Seidlein L, Duraisingh MT, Drakeley CJ, Bailey R, Greenwood BM, Pinder M. 1997. Polymorphism of the pfmdr1 gene and chloroquine resistance in Plasmodium falciparum in the Gambia. Transactions of the Royal Society of Tropical Medicine and Hygiene 91(4):450-453.
von Seidlein L, Milligan P, Pinder M, Bojang K, Anyalebechi C, Gosling R, Coleman R, Ude JI, Sadiq A, Duraisingh M, Warhurst D, Alloueche A, Targett G, McAdam K, Greenwood B, Walraven G, Olliaro P, Doherty T. 2000. Efficacy of artesunate plus pyrimethamine-sulphadoxine for uncomplicated malaria in Gambian children: A double-blind, randomised, controlled trial. Lancet 355(9201):352-357.
Wang P, Brooks DR, Sims PF, Hyde JE. 1995. A mutation-specific PCR system to detect sequence variation in the dihydropteroate synthetase gene of Plasmodium falciparum. Molecular and Biochemical Parasitology 71(1):115-125.
Wang P, Lee CS, Bayoumi R, Djimde A, Doumbo O, Swedberg G, Dao LD, Mshinda H, Tanner M, Watkins WM, Sims PF, Hyde JE. 1997. Resistance to antifolates in Plasmodium falciparum monitored by sequence analysis of dihydropteroate synthetase and dihydrofolate reductase alleles in a large number of field samples of diverse origins. Molecular and Biochemical Parasitology 89(2):161-177.
Warhurst DC. 2001. A molecular marker for chloroquine-resistant falciparum malaria. New England Journal of Medicine 344(4):299-302.
Watkins WM, Mosobo M. 1993. Treatment of plasmodium falciparum malaria with pyrimethamine-sulfadoxine: Selective pressure for resistance is a function of long elimination half-life. Transactions of the Royal Society of Tropical Medicine and Hygiene 87(1):75-78.
Wei N, Sadrzadeh SM. 1994. Enhancement of hemin-induced membrane damage by artemisinin. Biochemical Pharmacology 48(4):737-741.
Wellems TE, Walker-Jonah A, Panton LJ. 1991. Genetic mapping of the chloroquine-resistance locus on Plasmodium falciparum chromosome 7. Proceedings of the National Academy of Sciences of the United States of America 88(8):3382-3386.
Wernsdorfer WH, Noedl H. 2003. Molecular markers for drug resistance in malaria: Use in treatment, diagnosis and epidemiology. Current Opinion in Infectious Diseases 16(6):553-558.
White NJ. 1997. Assessment of the pharmacodynamic properties of antimalarial drugs in vivo. Antimicrobial Agents and Chemotherapy 41(7):1413-1422.
White NJ. 1998. Why is it that antimalarial drug treatments do not always work? Annals of Tropical Medicine and Parasitology 92(4):449-458.
White NJ. 1999. Delaying antimalarial drug resistance with combination chemotherapy. Parassitologia 41(1-3):301-308.
White NJ, Olliaro P. 1998. Artemisinin and derivatives in the treatment of uncomplicated malaria. Medecine Tropicale 58(3 Suppl):54-56.
White NJ, Pongtavornpinyo W. 2003. The de novo selection of drug-resistant malaria parasites. Proceedings of the Royal Society of London—Series B: Biological Sciences 270(1514):545-554.
WHO. 1996. Assessment of Therapeutic Efficacy of Antimalarial Drugs for Uncomplicated Falciparum Malaria in Areas With Intense Transmission. Geneva: World Health Organization.
WHO. 2000. Severe falciparum malaria. Transactions of the Royal Society of Tropical Medicine and Hygiene 94(Suppl 1):S1-S90.
WHO. 2001. Presentation at the meeting of the Antimalarial Drug Combination Therapy: Report of a WHO Technical Consultation. Geneva: World Health Organization.
WHO. 2003. Assessment and Monitoring of Antimalarial Drug Efficacy for the Treatment of Uncomplicated Falciparum Malaria. Geneva: World Health Organization. WHO/HTM/ RBM/2003.50.
WHO/RBM/UNDP/World Bank. 2003. Assessment of the Safety of Artemisinin Compounds in Pregnancy. Report of the two informal consultations convened by WHO in 2002. Geneva: World Health Organization.
Wongsrichanalai C, Wimonwattrawatee T, Sookto P, Laoboonchai A, Heppner DG, Kyle DE, Wernsdorfer WH. 1999. In vitro sensitivity of Plasmodium falciparum to artesunate in Thailand. Bulletin of the World Health Organization 77(5):392-398.
Wongsrichanalai C, Pickard AL, Wernsdorfer WH, Meshnick SR. 2002. Epidemiology of drug-resistant malaria. The Lancet Infectious Diseases 2(4):209-218.
Yayon A, Vande Waa JA, Yayon M, Geary TG, Jensen JB. 1983. Stage-dependent effects of chloroquine on Plasmodium falciparum in vitro. Journal of Protozoology 30(4):642-647.
York W, Macfie JWS. 1924. Observations on malaria made during treatment of general paralysis. Transactions of the Royal Society of Tropical Medicine and Hygiene 18(1&2):12-44.
Zalis MG, Pang L, Silveira MS, Milhous WK, Wirth DF. 1998. Characterization of Plasmodium falciparum isolated from the Amazon region of Brazil: Evidence for quinine resistance. American Journal of Tropical Medicine and Hygiene 58(5):630-637.
APPENDIX 9-A
Descriptions of Specific Antimalarial Drugs1
QUININE, first isolated from cinchona bark in 1820, remains a fundamental tool for treating malaria, especially severe disease. Quinine acts rapidly, targeting the bloodborne asexual stages of all malaria species. It is available in oral and injectable preparations and can be used in infants and pregnant women. Side effects—nausea, mood change, blurred vision, and ringing in the ears—are common but typically resolve after treatment ends.
Since P. falciparum parasites from most areas of the world respond well to quinine, short courses of the drug are often sufficient when paired with a second drug. In Southeast Asia, however, full course quinine treatment is necessary, usually given in combination with a second drug such as tetracycline.
CHLOROQUINE is a 4-aminoquinoline derivative of quinine first synthesized in 1934. It is safe in infants and pregnant women, and was the historical drug of choice for treatment of nonsevere or uncomplicated malaria and to prevent malaria in travelers. Chloroquine acts primarily against bloodborne asexual stages, although it also works against the bloodstream stage infective to mosquitoes. Because of widespread resistance to this drug, its usefulness is increasingly limited. Side effects are uncommon and generally mild.
AMODIAQUINE, which is closely related to chloroquine, fell out of favor because it caused adverse effects on bone marrow and liver when used for prophylaxis. Amodiaquine is currently being reevaluated as a co-formulation partner with artesunate. Concerns over toxicity remain.
ANTIFOL COMBINATION DRUGS include various combinations of dihydrofolate reductase inhibitors (proguanil, chlorproguanil, pyrimethamine, and trimethoprim) and sulfa drugs (dapsone, sulfalene, sulfamethoxazole, sulfadoxine, and others). The partner drugs in antifol combinations have similar mechanisms of action; consequently, they do not protect each other from resistance to the same degree as unrelated drugs. Current combinations include sulfadoxine-pyrimethamine (SP; Fansidar), sulfalene/pyrimethamine (Metakelfin), and sulfamethoxazole/trimethoprim (cotrimoxazole). Proguanil has also been used in combination with chloroquine for prophylaxis in areas of moderate chloroquine resistance, although it confers only minimal added benefit, especially with prolonged exposure
(Steffen et al., 1993). When used for prophylaxis, Fansidar can produce severe allergic reactions: in American travelers, Fansidar was linked to severe skin reactions (1 per 5,000 to 8,000 users) and mortality (1 per 11,000 to 25,000 users) (Miller et al., 1986). These adverse outcomes are not as frequent when a single dose of Fansidar is used for treatment. Concerns about sulfa drug use during pregnancy are outweighed by the known risks to mother and fetus of untreated malaria.
The latest antifol combination is chlorproguanil and dapsone, also known as Lapdap. This particular combination is inherently more effective than Fansidar (even in areas where resistance is present) and has a far shorter elimination time, which may decrease the likelihood of resistance (Watkins et al., 1997; Mutabingwa et al., 2001). On the other hand, its shorter half-life requires that Lapdap be given over 3 days rather than as SP’s one single dose.
TETRACYCLINE and derivatives such as doxycycline may be paired with other drugs for treatment or used as single agents for prophylaxis. In areas where quinine efficacy is diminished, tetracyclines are often added to quinine to improve cure rates. Tetracyclines are also used with shortened courses of quinine to decrease quinine-associated side effects. Tetracyclines are contraindicated in pregnant or breastfeeding women, or in children under age 8. Common side effects include nausea, vomiting, diarrhea, secondary yeast infections, and photosensitivity.
PRIMAQUINE, an 8-aminoquinoline, acts against malaria parasites in the liver, thereby reducing the likelihood of P. vivax or P. ovale relapse. Primaquine is also reasonably efficacious (74% efficacy against P. falciparum; 90 percent efficacy against P. vivax) when used for prophylaxis (Baird et al., 1995). Although it also has activity against blood-stage asexual parasites, drug concentrations that kill fully mature blood parasites are toxic. Primaquine is also a potent gametocidal drug, i.e., it kills the sexual stage of the malaria parasite infective to mosquitoes.
Primaquine can produce severe and potentially fatal hemolytic anemia in people with glucose-6-phosphate dehydrogenase (G6PD) enzyme deficiencies. The most severe Mediterranean B variant and related Asian variants of G6PD deficiency occur at high rates among several groups and regions: Kurdish Jews (62 percent), Saudia Arabia (13 percent), Burma (20 percent), and southern China (6 percent) and have now spread through migration and intermarriage. Primaquine should not be used in pregnancy.
TAFENOQUINE, a synthetic analog of primaquine, is currently being tested. It is highly effective against both liver and blood stages of malaria. Because of its long half-life (14 days versus 6 hours for primaquine),
tafenoquine may prove to be a valuable chemoprophylactic drug (Lell et al., 2000). As with primaquine, tafenoquine can produce acute hemolytic anemia in patients with G6PD deficiency.
MEFLOQUINE is a quinoline-methanol derivative of quinine that can be used for treatment or prevention in most areas with multidrug resistant malaria. Resistance to mefloquine occurs frequently in parts of Southeast Asia, however, and sporadic resistance has been reported in areas of Africa and South America. Mefloquine causes a relatively high incidence of neuropsychiatric side effects when used at treatment doses and to a lesser degree when used for prophylaxis. In one large study in Asia, mefloquine was associated with stillbirth when given in pregnancy (Nosten et al., 1999).
HALOFANTRINE is a phenanthrene-methanol compound with activity against the bloodborne stages of the malaria parasite. It is especially useful in areas where multidrug-resistant falciparum malaria is present. Cardiac conduction abnormalities (specifically, prolongation of the PR and QT intervals on a standard electrocardiogram) are halofantrine’s major drawback (Nosten et al., 1993). Taking halofantrine immediately following mefloquine or quinine therapy also increases the risk of cardiac complications. Halofantrine and mefloquine may exhibit clinical cross-resistance (Wongsrichanalai et al., 1992; ter Kuile et al., 1993).
CLINDAMYCIN is an antibiotic with weak antimalarial activity. It should only be used in combination with a fast-acting schizonticide, such as quinine, especially when treating patients with little or no immunity to malaria (Pukrittayakamee et al., 2000b; Parola et al., 2001). Clindamycin is considered safe for use in pregnant women and very young children (Pukrittayakamee et al., 2000a).
ARTEMISININ COMPOUNDS include the compounds artesunate, artemether, arteether, and dihydroartemisinin derived from the sesquiterpene lactone principle (artemisinin) of the plant Artemisia annua. In severe malaria, artemisinin compounds produce faster parasite clearance and resolution of fever than quinine. Artemisinins also reverse coma more quickly than quinine (Taylor et al., 1993; Salako et al., 1994). However, used alone for periods under 5 to 7 days, recrudescence rates are high. For nonsevere malaria, artemisinins are most successful when used in combination with a second drug (Nosten et al., 1994). The best documented combination is mefloquine plus 3 days of artesunate.
The safety of artemisinins in early pregnancy is of particular concern since the drugs have produced fetal resorption in experimental animals. Despite reassuring clinical data on over 600 carefully followed pregnancies
treated with artemisinins in the second and third trimesters of pregnancy, there are unresolved concerns about their effects in early human gestation.
A fixed-dose preparation of lumefantrine and artemether is commercially sold under the trade name of Coartem or Riamet. Lumefantrine (previously known as benflumetol) is an aryl-amino alcohol antimalarial compound. Although chemically related, lumefantrine does not appear to have the same cardiac effects as halofantrine (van Vugt et al., 1999).
Coartem is marketed in two packages: a six-dose (24-tablet) package intended for nonimmune patients and a four-dose (16-tablet) package for use by semi-immune patients. Until studies show conclusive efficacy of the four-dose regimen in semi-immune populations, all patients should receive the six-dose regimen (van Vugt et al., 2000). Until further safety data become available, lumefantrine is not recommended for treatment of pregnant women.
ATOVAQUONE PLUS PROGUANIL (MALARONE) is a fixed-dose combination containing 250 mg of atovaquone (a hydroxynaphthoquinone) and 100 mg of proguanil in a single adult-sized pill taken daily for prophylaxis. An adult treatment course of Malarone is 1,000 mg of atovaquone and 400 mg of proguanil daily for 3 days. Malarone has also been combined with artesunate for treatment of uncomplicated multidrug-resistant falciparum malaria (van Vugt et al., 2002). Malarone is also thought to be effective against bloodborne forms of P. vivax (Looareesuwan et al., 1996a).
PYRONARIDINE has been used in China for over 20 years. While it was reportedly 100 percent effective in a single trial in Cameroon, the drug was only 63 to 88 percent effective in Thailand (Ringwald et al., 1996; Looareesuwan et al., 1996b). Further testing is required before pyronaridine can be recommended for widespread use.
PIPERAQUINE is an orally active bisquinoline discovered in the early 1960s and developed for clinical use in China in 1973. In vitro testing in several laboratories has shown that piperaquine approximates chloroquine’s effects against sensitive parasites and is significantly more effective than chloroquine in treating resistant P. falciparum. In China, Vietnam and Cambodia, piperaquine is now available as a fixed combination with dihydroartemisinin (it has also been combined with trimethoprim and primaquine). Prospective clinical trial data are pending.
Appendix 9-A References
Baird JK, Fryauff DJ, Basri H, Bangs MJ, Subianto B, Wiady I, Purnomo, Leksana B, Masbar S, Richie TL, Jones TR, Tjitra E, Wignall FS, Hoffman SL. 1995. Primaquine for prophylaxis against malaria among nonimmune transmigrants in Irian Jaya, Indonesia. American Journal of Tropical Medicine and Hygiene 52(6):479-484.
Bloland PB, Williams H. 2002. Malaria Control During Mass Population Movements and Disasters. Committee on Population. National Research Council of the National Academies, Roundtable on the Demography of Forced Migration. Washington, DC: The National Academies Press.
Bloland PB, Ettling M, Meek S. 2000. Combination therapy for malaria in Africa: Hype or hope? Bulletin of the World Health Organization 78:1378-1388.
Doherty JF, Sadiq AD, Bayo L, Alloueche A, Olliaro P, Milligan P, von Seidlin L, Pinder M. 1999. A randomized safety and efficacy trial of artesunate plus sulfadoxine-pyrimethamine vs. sulfadoxine-pyrimethamine alone for the treatment of uncomplicated malaria in Gambian children. Transactions of the Royal Society of Tropical Medicine and Hygiene 93(5):543-546.
Greenberg AE, Ntumbanzondo M, Ntula N, Mawa L, Howell J, Davachi F. 1989. Hospital-based surveillance of malaria-related paediatric morbidity and mortality in Kinshasa, Zaire. Bulletin of the World Health Organization 67:189-196.
Greenwood BM. 1987. Asymptomatic malaria infections. Do they matter? Parasitology Today 3:206-214.
Kachur SP, Abdulla S, Barnes K, Mshinda H, Durrheim D, Kitua A, Bloland P. 2001. Letter to the editors. Tropical Medicine and International Health 6:324-325.
Kazadi WM, Vong S, Makina BN, Mantshumba JC, Kabuya W, Kebela BI, Ngimbi NP. 2003. Assessing the Efficacy of chloroquine and sulfadoxine-pyrimethamine for treatment of uncomplicated Plasmodium falciparum malaria in the Democratic Republic of Congo. Tropical Medicine and International Health 8(10):868-75.
Lell G, Faucher J-F, Missinou MN, Bormann S, Dangelmaier O, Horton J, Kremsner PG. 2000. Malaria chemoprophylaxis with tafenoquine: A randomized study. Lancet 255:2041-2045.
Looareesuwan S, Viravan C, Webster HK, Kyle DE, Hutchinson DB, Canfield CJ. 1996a. Clinical studies of atovaquone, alone or in combination with other antimalarial drugs for the treatment of acute uncomplicated malaria in Thailand. American Journal of Tropical Medicine and Hygiene 54(1):62-66.
Looareesuwan S, Olliaro P, Kyle D, Werndorfer W. 1996b. Pyronaridine. Lancet 347:1189-1190.
Miller KD, Lobl HO, Satrial RF, Kuritsky JN, Stern R, Campbell CC. 1986. Severe cutaneous reactions among American travelers using pyrimethamine-sulfadoxine (fansidar) for malaria prophylaxis. American Journal of Tropical Medicine and Hygiene 35:451-458.
Molyneux ME, Taylor TE, Wirima JJ, Borgstein J. 1989. Clinical features and prognostic indicators in paediatric cerebral malaria: A study of 131 comatose Malawian children. Quarterly Journal of Medicine 71:441-459.
Mutabingwa T, Nzila A, Mberu E, Nduati E, Winstanley P, Hills E, Watkins W. 2001. Chlorproguanil-dapsone for treatment of drug-resistant malaria in Tanzania. Lancet 358:1218-1223.
Nosten F, ter Kuile FO, Luxemburger D, Woodrow C, Kyle DE, Chongsuphajaisiddhi T, White NJ. 1993. Cardiac effects of antimalarial treatment with haolfantrine. Lancet 341 (8852):1054-1056.
Nosten F, Luxemburger C, ter Kuile FO, Woodrow C, Eh JP, White NJ. 1994. Treatment of multidrug-resistant Plasmodium falciparum malaria with 3-day artesunate-mefloquine combination. Journal of Infectious Diseases 170:971-977.
Nosten F, Vincenti M, Simpson J, Yei P, Kyaw Lay Thwai, De Vries A, Chongsuphajaisiddhi T, White NJ. 1999. The effects of mefloquine treatment in pregnancy. Clinical Infectious Diseases 28(4):808-815.
Nosten F, van Vugt M, Price R, Luxemburger C, Thway KI, Brockman A, McGready R, ter Kuile F, Loosareesuwan S, White NJ. 2000. Effects of artesunate-mefloquine combination on incidence of Plasmodium falciparum malaria in western Thailand: A prospective study. Lancet 356(9226):297-302.
Parola P, Ranque S, Badiaga S, Niang M, Blin O, Charbit JJ, Delmont J, Brousqui P. 2001. Controlled trial of 3-day quinine-clindamycin treatment versus 7-day quinine treatment for adult travelers with uncomplicated falciparum malaria imported from the tropics. Antimicrobial Agents and Chemotherapy 45:932-935.
Price RN, Nosten F, Luxemburger C, van Vugt M, Phaipun I, Chongasuphajaisiddi T, White NJ. 1997. Artesunate/mefloquine treatment of multi-drug resistant falciparum malaria. Transactions of the Royal Society of Tropical Medicine and Hygiene 91:574-577.
Pukrittyakamee S, Chantra A, Simpson JA, Vanijanonta S, Clemens R, Loosareesuwan S, White NJ. 2000a. Therapeutic responses to different antimalarial drugs in vivax malaria. Antimicrobial Agents and Chemotherapy 44:1680-1685.
Pukrittayakamee S, Chantra A, Vanijanonta S, Clemens R, Loosareesuwan S, White NJ. 2000b. Therapeutic responses to quinine and clindamycin in multidrug-resistant falciparum malaria. Antimicrobial Agents and Chemotherapy 44:2395-2398.
Radloff PD, Phillipps J, Nkeyi M, Hutchinson D, Kremsner PG. 1996. Atovaquone and proguanil for Plasmodium falciparum malaria. Lancet 347:1511-1513.
Ringwald P, Bickii J, Basco L. 1996. Randomised trial of pyronaridine versus chloroquine for acute uncomplicated falciparum malaria in Africa. Lancet 347:24-27.
Salako LA, Walker O, Sowunmi A, Omokhodion SJ, Adio R, Oduola AM. 1994. Artemether in moderately severe and cerebral malaria in Nigerian children. Transactions of the Royal Society of Tropical Medicine and Hygiene 88(Suppl 1):S13-S15.
Steffen R, Fuchs E, Schildknecht J, Naef U, Funk M, Schlagenhauf P, Phillips-Howard P, Nevill C, Stürchler D. 1993. Mefloquine compared with other malaria chemoprophylactic regimens in tourists visiting East Africa. Lancet 341:1299-1303.
Talisuna AO, Bloland P, D’Alessandro U. 2004. History, dynamics, and public health importance of malaria parasite resistance. Clinical Microbiology Reviews 17:235-254.
Taylor TE, Wills BA, Kazembe P, Chisale M, Wirima JJ, Ratsma EY, Molyneux ME. 1993. Rapid coma resolution with artemether in Malawian children with cerebral malaria. Lancet 341(8846):661-662.
Ter Kuile FO, Dolan G, Nosten F, Edstein MD, Luxemburger C, Phaipun L, Chongsuphajaisiddhi T, Webster HK, White NJ. 1993. Halofantrine versus mefloquine in treatment of multidrug-resistant falciparum malaria. Lancet 341(8852):1044-1049.
Van Hensbroek MB, Morris-Jones S, Meisner S, Jaffar S, Bayo L, Dackour R, Phillips C, Greenwood BM. 1995. Iron, but not folic acid, combined with effective antimalarial therapy promotes haematological recovery in African children after acute falciparum malaria. Transactions of the Royal Society of Tropical Medicine and Hygiene 89(6):672-676.
van Vugt M, Ezzet F, Nosten F, Gathmann I, Wilairatana P, Looareesuwan S, White NJ. 1999. No evidence of cardiotoxicity during antimalarial treatment with artemether-lumefantrine. American Journal of Tropical Medicine and Hygiene 61(6):964-967.
van Vugt M, Angus BJ, Price RN, Mann C, Simpson JA, Poletto C, Htoo SE, Looareesuwan S, White NJ, Nosten F. 2000. A case-control auditory evaluation of patients treated with artemisinin derivatives for multidrug-resistant Plasmodium falciparum malaria. American Journal of Tropical Medicine and Hygiene 62(1):65-69.
van Vugt M, Leonardi E, Phaipun L, Slight T, Thway KL, McGready R, Brockman A, Villegas L, Looareesuwan S, White NJ, Nosten F. 2002. Treatment of uncomplicated multidrug-resistant falciparum malaria with artesunate-atovaquone-proguanil. Clinical Infectious Diseases 35:1498-1504.
Von Seidlen L, Milligan P, Pinder M, Bojang K, Anyalebeschi C, Gosling R, Coleman R, Ude JL, Sadiq A, Duraisingh M, Warhurst D, Alloueche A, Targett G, McAdam K, Greenwood B, Walraven G, Olliaro P, Doherty T. 2000. Efficacy of artesunate plus pyrimethamine-sulphadoxine for uncomplicated malaria in Gambian children: A double-blind, randomized controlled trial. Lancet 355(9220):352-357.
Watkins WM, Mberu EK, Winstanley PA, Plowe CV. 1997. The efficacy of antifolate antimalarial combinations in Africa: A predictive model based on pharmacodynamic and pharmacokinetic analyses. Parasitology Today 13:459-464.
White N. 1999. Antimalarial drug resistance and mortality in falciparum malaria. Tropical Medicine and International Health 4:469-470.
WHO. 2003. Assessment and monitoring of antimalarial drug efficacy for the treatment of uncomplicated falciparum malaria Geneva: World Health Organization. WHO/HTM/ RBM/2003.50.
Wongsrichanalai C, Webster HK, Wimonwattrawatee T, Sookto P, Chuanak N, Thimasarn K, Wernsdorfer WH. 1992. Emergence of multidrug-resistant Plasmodium falciparum in Thailand: In vitro tracking. American Journal of Tropical Medicine and Hygiene 47(1):112-116.















































