
C
Geochemistry of Actinides During the Long-Term Storage and Disposal of Spent Nuclear Fuel*
Nikolay P. Laverov, Vasily I. Velichkin, B. I. Omelyanenko, and S. V. Yudintsev
Institute of Geology of Ore Deposits, Petrography, Mineralogy, and Geochemistry
Russian Academy of Sciences
The main source of high-level radioactive wastes (HLW) is the spent nuclear fuel (SNF) as a product of atomic power stations, as well as naval-propulsion and research reactors. Greater than 95 percent of total radioactivity of the materials involved in the human activity is concentrated in the SNF. The SNF contains 95–98 percent UO2 and several percent of various radioisotopes formed in nuclear reactions. The SNF removed from reactors preserves greater than 90 percent of its energy resource. It contains greater than 96 percent U and up to 1 percent Pu, which are suitable for reuse. They are separated by radiochemical treatment of SNF with the formation of a large volume of liquid radioactive wastes. A part of the liquid wastes, small by volume but high in concentration of highly radioactive isotopes, belongs to high-level wastes. In accordance with current technologies, the HLW would be transformed to solidified form, stored for some time, and disposed in underground repositories.
The storage, recycling, and disposal of liquid radioactive wastes pose significant economic and environmental problems, and the expediency of SNF recycling is variously assessed. SNF is considered disposable waste in the United States, Canada, Sweden, and Finland. It is treated as recyclable nuclear material in Great Britain, France, and Japan. A decision about the SNF treatment has not been reached yet in Argentina, Brazil, and Slovenia. Switzerland plans to recycle one-third of its SNF in Great Britain and France and dispose of two-thirds of SNF (Schneider et al., 1998). It is probable that all countries will use SNF for
separation of valuable components after its controlled storage, because the 239Pu, 235U, and 238U contents do not change in SNF while its radioactivity decreases. The fuel removed from reactors should be called irradiated and considered a potential energy resource, rather than waste.
In Russia some SNF types (from VVER-440, BN-600, and BN-350 reactors, some naval-propulsion reactors, and research reactors) are recycled at the RT-1 plant (Mayak in the Chelyabinsk region). Some other SNF types (from VVER-1000 reactors) are planned to be recycled at the RT-2 plant under construction (Zheleznogorsk near Krasnoyarsk). The SNF from RBMK, AMB, EGP-6, some transport facilities and research reactors, and faulty fuel is in storage, and its further treatment has not yet been considered. The primary portion of radioactivity (greater than 3 billion Ci) of SNF is accumulated in the RBMK reactors. This SNF has relatively low contents of fissionable species, and it will probably be recycled only in the far future. The problem of long-term safe storage and disposal of SNF in underground repositories is considered in this paper.
General geological recommendations for the long-term safe storage of SNF are deposition in the low-permeable rocks in seismically stable blocks with low velocities of vertical displacements and free of active volcanism and mineral deposits (Krauskopf, 1988a; Laverov et al., 2001). If these requirements have been met, the probability of the repository destruction, its exposure to erosion, transportation of radionuclides by magmas, and penetration of the repository by mining workings is minimized. The environmental pollution in this case can be related to radionuclide removal from the repository by groundwater in dissolved or colloidal forms. This mechanism is always taken into account in the assessment of the security level of HLW repositories.
SNF radioactivity decreases progressively during its storage (see Figure 1). For example, the radioactivity of SNF of a pressurized water reactor (PWR) (counted upon one ton of uranium) 10, 100, and 1000 years after its removal from the reactor will be approximately 400,000, 40,000, and 1700 Ci, respectively (Roddy et al., 1985). The strongest environmental impact would be expected if radionuclides escaped to groundwater at the earlier stages of SNF disposal, when the SNF contains short- and medium-lived radioisotopes with very high radioactivity. These isotopes decay in 500–1000 years. Current technology of long-term storage and disposal of HLW envisages HLW isolation from groundwater by engineered barriers for this or longer periods. The barriers comprise concrete tanks, corrosion-resistant containers, envelopes for nuclear fuel, and bentonite backfill. The SNF can interact with groundwater only after the engineered barriers lose their insulative properties. From this point on, actinides will present the main hazard for the biosphere. The geological medium will become the only barrier retaining the actinide migration. Below we analyze the conditions of safe SNF disposal provided by the insulative properties of the geological medium.
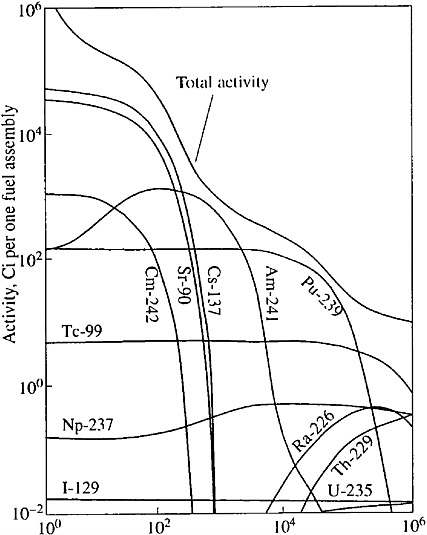
FIGURE 1 Variation of radioactivity of the SNF assembly of a PWR reactor with time (Brookins, 1984).
SPENT NUCLEAR FUEL PROPERTIES DEFINING THE CONDITIONS OF LONG-TERM STORAGE AND DISPOSAL
The principal constituent of the nuclear fuel is a heat-generating element (fuel rod) that is a long thin tube of corrosion-resistant zirconium alloy (or some other materials). The tube is filled with UO2 pellets, with the proportion of 235U isotopes several times higher than in natural minerals. Several fuel rods compose
the fuel assemblies; for example, the fuel assembly of the RBMK reactor consists of 18 fuel rods, and the assemblies of the VVER-1000 reactor consists of 317 fuel rods. The pellets are produced by pressure and have a density of 94–95 percent of the theoretical density of uraninite. The UO2 grains in pellets are smaller than a few microns. During irradiation the UO2 pellets acquire numerous fractures, and the intergranular spaces expand. This results in an increase of surface area of the pellets and consequently UO2 interaction with groundwater if the containers with SNF have failed.
The UO2 is a conserving matrix for all elements produced in a nuclear reaction. Some elements (Pu, Am, Cm, Np, Th, rare earth elements [REE], Nb, and Zr) are incorporated into the UO2 crystal structure. Its stability precludes release of these elements into groundwater. Some other elements (Tc, Se, I, Cs, Sn, and Sr) and their fission products are included as nonstructural admixtures. They are accumulated in the intergranular boundaries and microfractures in the UO2 matrix. These elements can be partly leached by groundwater even in the conditions of the UO2 stability. The leaching intensity will decrease with depletion of these elements in the areas in contact with groundwater.
In accordance with existing technologies for long-term storage and disposal, the SNF must be placed in metallic canisters and stored at depths of several hundred meters from the surface. Various schemes of SNF storage in underground workings and wells have been proposed (see Figures 2 and 3). According to these schemes the engineered barriers comprise bentonite backfill, canister, capsules, and fuel-rod coatings. These barriers must isolate the UO2 pellets from groundwater for 500–1000 years. The short- and medium-lived radioisotopes will decay during this period, and 98 percent of the residual radioactivity of SNF will be related to Pu and Am (see Figure 1). When the geological medium becomes the only barrier retaining the radionuclide release, the repository safety in most cases will be determined by the intensity of the Pu and Am escape from the SNF and the specific conditions of their migration in groundwater. The UO2 stability is very important for repository security, because Pu and Am occur as isomorphous impurities in this mineral. The analysis of this problem considers the data on U behavior in the geological medium and experimental results on dissolution of SNF and natural uraninite in groundwater.
URANIUM DEPOSITS AS NATURAL ANALOGUES OF SNF REPOSITORIES
The analysis of natural observations is crucial in the study of problems related to the long-term safe underground disposal of actinide-bearing wastes. It allows one to characterize the actinide behavior at various chemical conditions and obtain information about very slow processes with results notable only after thousands and millions of years.
Many researchers consider the U and Th deposits as natural analogues of the
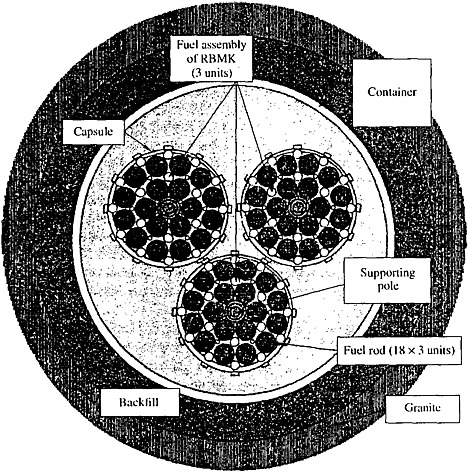
FIGURE 2 A scheme of the RBMK reactor SNF location in a well (Ivashkin et al., 2000).
HLW repositories (Brookins, 1984; Krauskopf, 1988b; Laverov et al., 1991, 1994). This is particularly appropriate for the SNF containing greater than 95 percent UO2, which presents the conserving matrix for the other radionuclides. The study of U deposits shows that uranium oxides can be highly stable at certain conditions in the geological medium. Ores of U deposits are generally composed of uranium oxides. The ore bodies are usually located in highly permeable zones composed of cataclasites, densely fractured rocks, or water-saturated sedimentary rocks. Therefore, a high portion of U minerals of the ore bodies may have contacted with groundwater, but many U ore bodies stored for hundreds of millions of years are almost unaltered, even in the zones with high water permeability.
It was found that the occurrence of reducing or nearly neutral groundwater is the principal factor facilitating the high stability of U minerals. The groundwa-
ter attains such properties during its continuous interaction with country rocks, when its salinity decreases and oxygen is spent for oxidation of ferrous iron (Kiryukhin et al., 1993; Krainov and Shvets, 1992; Ryzhenko et al., 1996). The oxygen-rich water normally penetrates to depths from several tens to a few hundreds of meters and reaches 1.0–1.5 km only in the Alpine regions. We suggest that the conditions of high stability of UO2 occur at depths below 500 m in most regions (except lofty mountains) (Laverov et al., 2001).
It is important that the reduced groundwater is always saturated in uranium derived from country rocks. Uranium in the rocks occurs as
-
disseminated U atoms and their clusters
-
accessory U minerals
-
isomorphous impurity in the U-bearing accessory minerals
-
U-rich segregations along grain boundaries
-
atoms sorbed on specific minerals (Omelyanenko et al., 1983)
The equilibrium uranium concentration in groundwater at the reducing hydrogeochemical conditions is very low, less than or equal to 10−8 mol/L (see Figure 4).
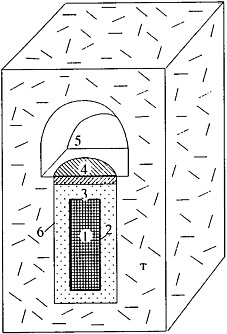
FIGURE 3 A scheme of the SNF location in an underground mining working. (1) SNF, (2) metallic canister, (3) bentonite backfill, (4) concrete seal, (5) horizontal working, (T) cell.

FIGURE 4 Eh-pH diagram for the U-C-O-H system and a part of the Fe-S-O-H system (Brookins, 1988). Activities of the dissolved particles: U = 10−6, −8, −10, C = 10−3, Fe = 10−6, and S = 10−3. The contour boundary of the field of Eh-pH values of groundwater is in equilibrium with unoxidized uranium ores.
At such conditions uranium is redistributed among various minerals and within single grains. The proportion of sorbed U increases at the expense of the other forms. These processes are most efficient in the cataclastic zones, where the interaction area and proportion of secondary minerals (the best sorbents for U) increase. The Fe(Ti)-bearing minerals and particularly their alteration products serve as sorbents for U in crystalline rocks. Similar processes resulting in the formation of the secondary dispersion halos occur around the uranium ore
bodies. These processes are very slow (but continuous) and their results become notable only over geologically long time intervals. The diffusion-driven redistribution cannot significantly affect the radionuclide release from SNF.
The Cigar Lake deposit in Northern Saskatchewan, Canada, is frequently referred to as a natural analogue of an SNF repository (Johnson and Shoesmith, 1988). The average U content in its ores is 7.9 wt percent (up to a few tens of percent in the richest blocks; Pagel et al., 1993). The deposit is dated at 1.1 Ga. Rich ores lie at a depth of 400 m, but no signs of their occurrence are seen on the surface. The ores are cut by water-permeable fractures, which may indicate that the ore contacted groundwater for a long time. The uraninite is only slightly altered by the reducing properties of groundwater. The U concentration in water from an ore body with 40 percent U is 10−8 mol/L and is almost equal to the background concentration (Cramer, 1986).
It is pertinent to note that the data on higher U concentrations in groundwater either correspond to the oxidizing conditions or characterize total contribution of dissolved and colloidal forms. This is also verified by experiments on uraninite dissolution under the reducing conditions, when U concentrations were determined both in the unfiltered liquids and in the solutions that passed through the filters with 2 nm pores. The results of natural observations convincingly indicate a high stability of uraninite at reducing, almost neutral hydrogeochemical conditions.
The question arises: If uraninite is irradiated in the fuel rod, does it behave in the geological medium like natural uraninite, because the intensity of an irradiation on the SNF surface is two to three orders of magnitude higher than in the uranium ore? Let us consider the data on the deposits of the Franceville uranium ore district in Gabon in West Africa to solve this problem. Oklo is the most famous deposit of the district. Some indications of natural nuclear reactions were discovered in ores of this deposit, that is, depletion of some ore bodies in 235U and occurrence of radioisotopes or their fission products resulting from nuclear reaction. The deposits of the Franceville ore district formed at about 2 Ga in sandstones with organic matter at depths of (3–3.5) × 10−3 km. It was calculated that the 235U content in ore at that time was 3.25 wt percent, which is close to that in fuel of modern power reactors. Processes similar to those in nuclear reactors occurred intermittently for about 500 Ma in some Oklo ore bodies in the presence of water serving as a neutron moderator (IAEA, 1978).
The study of these ore bodies located at different depths from the surface described the behavior of uraninite and associated products of nuclear reactions both in the reducing and oxidizing hydrogeochemical conditions; for example, the uraninite in an ore body at a depth of 250 m under reducing conditions is not affected by secondary processes (Pourcelot and Gauthier-Lafaye, 1998). Detailed studies of trace-element concentrations and isotopic compositions in such ores demonstrated that the elements incorporated in the uraninite crystal structure were preserved there until their complete decay.
Such behavior of Am, Pu, and Np is justified by the distribution of 209Bi and 207Pb, the final products of their fission, which were generally preserved in the uraninite, regardless of their dissimilar geochemical properties to uranium, and were only partially redeposited near the ore bodies (Brookins, 1984). The nonstructural elements, such as Rb, Cs, Sr, Mo, Cd, Xe, and I were almost completely eliminated from the ore bodies, while Ru and Sn were partially removed. It was suggested that diffusion was the main mechanism of migration of the nonstructural elements (Cowan, 1978).
Geochemical studies of natural reactor zones indicate that uraninite is highly stable under reducing conditions. It not only retains actinides but also strongly restrains migration of the nonstructural elements. Thus, the study of the Oklo ore bodies shows that the geological medium can provide the conditions for the long-term safe storage of SNF.
The interaction with oxygen-rich water facilitates the U removal and replacement of U oxides by the secondary uranyl-ion minerals. The upper parts of some steep ore bodies are strongly altered, while their lower levels are well preserved. The removal of uranium together with all mixtures from uraninite was detected in the Oklo ore body, which is located at a depth of 100 m and shows evidence of nuclear reactions (Pourcelot and Gauthier-Lafaye, 1998). The strongest decomposition of uraninite was found in the Bangombi zone located at a depth of 10 m from the surface directly beneath the lateritic weathering mantle. In this zone the uraninite was strongly modified, the organic matter in the host rocks was oxidized, and the reducing properties of these rocks declined. A part of the uranium removed from uraninite was redeposited as phosphates. The formation of secondary ore accumulations with hexavalent U is typical of the deposits located near the surface. These data, together with numerous examples of partial and complete transformation of uranium ores of many other deposits, indicate that the oxidizing conditions are inappropriate for the long-term underground storage of SNF.
EXPERIMENTAL STUDY OF SPENT NUCLEAR FUEL DISSOLUTION IN GROUNDWATER
Many experim|ents on interaction of natural and synthesized UO2, as well as SNF and its imitators, and groundwater were performed under oxidized conditions and are related to the U.S. project on long-term HLW storage in the Yucca Mountain repository in Nevada (LLNL, 1998). Seventy thousand tons of HLW (generally SNF) are supposed to be stored in this repository. The experimental results show high SNF solubility in aerobic conditions (U content reached 10−3 mol/L) and a high rate of release of all radionuclides from the spent fuel. Scientists attempted to follow all stages of uraninite transformation under the effect of oxygen-rich water.
During its interaction with water uraninite was oxidized from the surface
and along the microfractures and grain boundaries. The intergranular spaces, and consequently the interaction area, increased. The oxidized uraninite microparticles (UO2+x) were separated from the main mineral mass. Secondary phases of the hexavalent U, for example, haiweeite, schoepite, and soddyite, formed as suspension on the SNF surface during the longest runs. If the Yucca Mountain repository were located in the zone with conditions of continuous interaction with groundwater, uraninite would not serve as an immobilizing matrix; however the repository is located in a tuff sequence in the aeration zone above the groundwater level. The assessment of the repository safety is based on the suggestion that without approaching the SNF, water will vaporize for several hundred years because of elevated temperature. The conditions of probable interaction of SNF with water will be reached only after the temperature decreases below 90°C. It is suggested that water will approach the SNF intermittently and only in small amounts because of the dry climate and scarce atmospheric precipitation in the repository area. Thus, the safety of the Yucca Mountain repository is controlled by the amount of atmospheric precipitation instead of by the low solubility of UO2.
It was calculated that one SNF assembly (3140 kg) UO2 will interact with less than 20 liters of water per year. Even in such conditions about 0.1 percent of total 129I, 99Tc, and 137Cs amounts will escape in a year (LLNL, 1998; Chen et al., 1999). A larger 99Tc release (up to 0.8 percent per year) is also reported (Finn et al., 1998). The geochemical isolation of Np in such conditions is also problematic (Buck et al., 1998; Chen et al., 1999). Generally, because of the oxidizing conditions, some experts believe that the long-term safe storage of SNF in the Yucca Mountain repository is not possible (Ewing, 2002). It is emphasized that the water volume interacting with SNF can significantly increase as a result of climatic changes or some other reasons. It is pertinent to note that Yucca Mountain is the only repository with aerobic conditions projected for long-term storage and disposal of HLW.
Numerous experiments have demonstrated that uraninite solubility is very low (U concentration is less than or equal to 10−8 mol/L) under the reducing almost neutral conditions and independent of temperature or composition of groundwater (Johnson and Shoesmith, 1988; Redkin et al., 1989). The extent to which the data on solubility of the natural uraninite are applicable to SNF should be especially considered. The natural uraninite and UO2 of SNF are analogous in their crystal structure but differ in impurity elements, O/U ratio, and radioactivity. The natural uraninite grains are highly dense, and their boundaries are indistinguishable even under an electron microscope. SNF uraninite is broken by numerous microfractures; the boundaries among its grains in the SNF are wider and better seen than in natural uraninite aggregates.
Let us compare experimental results on solubility of natural uraninite and UO2 in SNF of various types. The deionized water and the water in equilibrium with granites, clays, salts, and other rocks were used as dissolvents. The experi-
ments showed that SNF is stable under the reducing conditions (Johnson and Shoesmith, 1988). Under similar conditions the solubility of natural uraninite, nuclear fuel imitators, and SNF are of the same order of magnitude. Insignificant differences in the solubility of individual uraninite samples are generally related to their oxidation degree: the higher the O/U ratio, the higher the solubility. The SNF solubility in water in equilibrium with atmospheric Ar and N strongly depends on the SNF-oxidation degree and is 10 times higher for the oxidized SNF than for unoxidized (Loida et al., 2001). The authors believe that SNF can be oxidized during its storage. The material of steel canisters and their corrosion products serve as inhibiting agents for SNF dissolution; for example, the solubility of the oxidized SNF decreased 20 times in equilibrium with iron powder. The radionuclide concentration in the solution in this experiment was comparable with that in runs performed under the reducing conditions. Thus, the experiments show that the reducing conditions maintain high SNF stability in the geological media.
THE EFFECT OF RADIOLYSIS ON SPENT NUCLEAR FUEL SOLUBILITY
The SNF radiation does not necessarily result in reducing conditions. The radiation can cause water dissociation on the SNF surface. Although the oxidizing (O2, H2O2) and reducing (H,) components are produced in equal molar quantities, the higher diffusion rate of hydrogen can lead to the formation of the local oxidizing conditions near the SNF surface, which facilitate the UO2 dissolution by the reaction
The possible effect of radiolysis on radionuclide release was emphasized by many researchers (see, for example, Brookins, 1984; Chapman and Savage, 1984; Chapman and McKinley, 1988; Krauskopf, 1988b). They noted, however, that radiolysis is not capable of significantly modifying the scale of radionuclide escape because of the large reducing capacity of the geological medium.
Rocks and backfill materials continuously supply ferrous iron to groundwater. The interaction of groundwater with a metallic canister and its corrosion products with hydrogen separation by the reaction
also counteract the formation of the oxidizing conditions.
The question of which processes will prevail on the SNF surface, oxidation due to radiolysis or reduction due to interaction with the ambient medium, is very important for the SNF disposal. This problem cannot be solved solely on a theoretical basis.
The experiments take into account that the SNF interaction with water will
begin only after the engineered barriers have failed, that is, in ≥500 years), the main mass of β sources has decayed, and the main process capable of changing the redox conditions is radiolysis caused by α radiation. The α-particle tracks in water are about 30–50 µm long, and radiolysis can occur only in a thin water film on the SNF surface. It is induced by no more than 1 percent of all α-particles (Neretnieks, 1997). It was found that the radiolysis does not affect the uraninite solubility in the reducing conditions; for example, the runs performed over a year at 70°C in equilibrium with hydrogen verified a high stability of SNF under the reducing conditions (Spahiu and Sellin, 2001). The uranium concentration in groundwater from granite was less than10−9 mol/L, and the other radionuclides had not been leached from SNF. The effect of radiolysis is best pronounced in experiments in an oxygen-free atmosphere of nitrogen and argon. The role of a radiolysis of groundwater in the formation of the oxidizing conditions on the SNF surface in an underground repository was proved to be insignificant (Forsyth and Werme, 1986); however the experiments with the intensity of α radiation being one to two orders of magnitude higher than that on the SNF surface showed that radiolysis-induced oxidizing conditions can exist locally (Johnson and Shoesmith, 1988).
New experimental data on the effect of radiolysis on the SNF solubility were obtained recently. Samples of pure uraninite and uraninite with 0.1 and 10 percent 238Pu isotope (a source of α particles) were used in these experiments (Rondinella et al., 1999, 2001). Specific activities at the surface of the samples doped with 238Pu were 2.71 × 106 and 2.71 × 108 Bq/cm2, which are one and three orders of magnitude higher than on the SNF surface 500 years after its removal from the reactor. The runs were performed with deionized water in equilibrium with a nitrogen atmosphere. The Eh values in the 60-day runs with uraninite with 0, 0.1, and 10 percent 238Pu were 180, 380, and 570 mV, respectively. Equilibrium concentrations of uranium were less than 10−8 mol/L in runs with pure UO2 and about 10−6 in runs with uraninite with 238Pu. It was found that the dissolved uranium was mostly redeposited on the vessel walls. The amount of the redeposited uranium increased proportionally with the increasing run duration and 238Pu concentration in uraninite. The experiments showed that the uranium release from uraninite doped with 238Pu is comparable to that from pure uraninite in the oxidizing conditions. Plutonium concentration in the solution was only 4 × 10−12 mol in runs with uraninite containing 0.1 percent 238Pu and four orders of magnitude higher in runs with uraninite containing 10 percent 238Pu.
As it follows from the experiments, active radiolysis can produce highly oxidative conditions when plutonium passes into the penta- and hexavalent states; however the canister material and products of its corrosion preclude the formation of such highly oxidative conditions even at very high SNF radioactivity. A very high rate of radionuclide leaching independent of the groundwater composition was observed in experiments on the groundwater interaction with SNF
powder in equilibrium with argon atmosphere (Loida et al., 2001). Radiolysis was the main process that induced the oxidizing conditions and the correspondingly high rate of SNF dissolution. Equilibrium element concentrations in water were 5.4 × 10−5 mol/L for U and 1.5 × 10−6 mol/L for Pu. After addition of iron powder to the system, the rate of SNF dissolution decreased by 20 times, equilibrium concentration of U in the solution decreased by two orders of magnitude, and that of Pu, Am, Eu, Np, and Sb by more than three orders of magnitude, which corresponds to the values observed under the reducing conditions. Note that in the 805-day runs iron was not oxidized completely, and the iron particles contacting the SNF particles were replaced by hematite (FeOOH). Thus, the reducing conditions were maintained by reactions of iron oxidation. The experimental results demonstrated that at the radiation level typical of SNF after 500 years of storage, radiolysis does not cause the transition of the tetravalent Pu to a more oxidized state.
Smith and Johnson (2000) estimated the effect of radiolysis on SNF stability from the experimental data on UO2 solubility, theoretical calculation of the amount of the oxidizing agents produced by SNF interaction with water, and the study of natural uraninite. Admitting that precise estimation was impossible, the authors concluded that the presence of reducing agents in host rocks and canister corrosion products strongly limits the radiolysis effect on the SNF solubility. The oxidizing species resulting from radiolysis will be spent primarily for oxidation of bivalent iron.
This conclusion is also justified by the absence of uraninite oxidation in many deposits. Uranium in the Oklo ores always occurred in the tetravalent state in spite of radiolysis. The organic matter and bivalent iron in host rocks and ores maintained the reducing conditions (Oversby, 2000). Local oxidizing conditions related to radiolysis were found near the uranium ore in some deposits (Cigar Lake, Canada). The low alteration degree of uraninite in these deposits shows that the oxidizing species were spent generally for oxidation of wall rock minerals with reducing components (Liu and Neretnieks, 1995).
Available data indicate that radiolysis cannot significantly affect the radionuclide migration from SNF if the host rocks and engineered barriers contain reducing species. The radiolysis effect needs further study, however. It is necessary to analyze the situation of high solubility of the uraninite matrix due to the local oxidizing conditions in the zone of SNF interaction with groundwater. In this case, the matrix cannot isolate actinides. Let us consider the capability of the geological medium to isolate actinides stored in repository from the biosphere.
MIGRATION AND ACCUMULATION OF ACTINIDES IN GEOLOGICAL MEDIUM
Crystalline rocks are the most probable medium for long-term SNF storage in Russia and most other countries. Safe physical isolation of SNF from interac-
tion with groundwater in crystalline rocks is impossible, because the rocks are intersected by fractures, and their amount can increase because of many factors. The most probable mechanism of biosphere pollution in this case is actinide migration from the repository with groundwater flows. The water-exchange intensity controls the rate and scale of the radionuclide migration from SNF. According to the general recommendations the repositories should be constructed in conditions of low water exchange. The interaction of groundwater with SNF in such conditions can be estimated in long-term experiments reaching steady concentrations of dissolved radionuclides (Wilson, 1990 a,b) and by results of the SNF study after its interaction with water (Finn et al., 1998b). It was found that the SNF transformation begins with uraninite oxidation accompanied by the widening of grain boundaries and detaching of tiny UO2+x particles from the grain surfaces. This process results in the formation of the secondary uranyl-ion phases on the SNF surface and areas loosened by oxidation, which inhibit further corrosion. Haiweeite, uranophane, and soddyite were identified there. The rate of uranium migration from SNF is 0.1–0.3 mg/m2 per day. The major part of the leached uranium is redeposited in the secondary uranyl-ion phases. The steady concentration of uranium in water is (4–8) × 10−6 mol/L, 98 percent of which occurs in colloidal form. Pu, Am, and Cm pass into water proportionally to U, while the mechanism of Np release is not yet understood. It was also found that the SNF type and degree of the fuel decay do not principally affect the rate of UO2 dissolution. On the contrary, the UO2 oxidation degree strongly influences the rate of its dissolution. For example, U3O8 dissolves two to three times faster than UO2. This is generally caused by the fact that the uraninite oxidation is accompanied by the formation of microfractures, thereby increasing the water penetration and interaction area.
During the dissolution of the uraninite matrix, actinides can pass into water in dissolved and colloidal forms, precipitate near SNF as isomorphous impurities in the secondary uranyl-ion minerals or as individual mineral phases, and accumulate in sorbed forms in host rocks. The actinide migration in dissolved form is inhibited by sorption of these elements by rocks. Thus, a more real mechanism of the actinide migration to the biosphere is due to their removal from the repository in colloidal form. Some brief information on radioactive colloids is given below.
Three mechanisms of formation of actinide-bearing colloids are distinguished. The first mechanism is related to actinide sorption by colloidal particles abundant in groundwater. Such particles form by groundwater interaction with minerals, mechanical and chemical weathering of rocks, detaching of the particles from the minerals covering the fracture walls, or due to the changes in the hydrogeochemical conditions that result in oversaturation of solutions with respect to some minerals. The colloidal particles are from 1 nm to 1 µm in size. Actinides can migrate only with smaller particles (less than 400 nm), because the larger particles rapidly precipitate. The colloidal particles normally consist of
clay minerals, chlorite, silica, iron hydroxides, zeolites, and organic matter. The amorphous phases of Si, Al, and Fe play a significant role among the colloidal particles. The content of the colloidal phases in groundwater varies significantly. The groundwater typical of granites has a constant temperature, flow velocity, ionic strength, pH, and Eh and normally contains less than 100 ng/ml colloidal particles. The disturbances in the hydrodynamic regime caused by drilling, mining, or water pumping can increase the amount of colloidal particles originated by detachment of mineral particles (including the coagulated colloids) from fracture walls. The concentration and composition of the colloidal particles near the repository can differ from the background values, which is a phenomenon related to the temperature gradient and introduction of extraneous materials into the geological medium.
The second mechanism is the formation of true radiocolloids. The radioisotopes released from the uraninite structure during its dissolution pass into saturated aqueous solution as suspended and colloidal particles, normally of amorphous hydroxides. The formation of true colloids can be favored by a decrease of actinide solubility in groundwater because of buffer reactions reducing the effect of radiolysis.
The third mechanism of the colloidal particle formation generally works in the disposed vitrified HLW. The products of their interaction with groundwater are represented by various crystalline and amorphous phases with radionuclides in isomorphous and sorbed forms. Separation of such phases from the glass surface and their migration as colloidal particles is the most probable mechanism of actinide release from the vitrified HLW. The colloidal particles from the uraninite matrix corrosion products form in similar manners during the long-term storage of SNF. The colloids can be composed of UO2+x particles separated from the SNF surface or of secondary uranyl-ion minerals containing actinides in isomorphous and sorbed forms.
It was found that the migration velocity of actinides dissolved in water is two to three orders of magnitude lower due to sorption than the velocity of the water flow. On the contrary, the velocity of the colloidal particle migration is close to the velocity of the water flow. In the steady-state hydrodynamic conditions the relation between the amount of colloidal particles that entered the groundwater and were removed from it is close to equilibrium. The amount of colloidal particles can strongly increase because of tectonic activity, water sampling, mining, or some other processes disturbing the hydrodynamic regime. The main mechanism restraining the migration of colloidal particles in geological medium is their adhesion and precipitation on the fracture walls as loose aggregates. Aging of the colloids favors the particle consolidation on the fracture walls. Note that the rocks contain only natural analogues of actinides (U, Th, REE) and do not contain actinides themselves, which can be derived only from SNF. As a result, the actinide concentration in groundwater decreases with increasing distance from SNF. This favors the transition of actinides from colloi-
dal particles to aqueous solution followed by their further sorption by rocks and other colloidal particles.
The low water permeability and water exchange, the absence of wide fractures, and the sealing of fractures by secondary minerals also counteract the migration of the actinide-bearing colloids. The bentonite backfill is an insuperable obstacle in the way of colloid migration. It was shown experimentally that the bentonite layer 2 mm thick stems the colloidal particles, and only dissolved actinides can migrate to some longer distances (Tsukamoto et al., 1995). That is because most of the modern projects of the HLW repositories envisage the use of a bentonite backfill.
When modeling the radionuclide migration in a colloidal form, it is difficult to constrain the time of occurrence of colloidal particles in groundwater and the distance of their transportation. Some suggestions should be made for calculations. For example, it was implied that for the Yucca Mountain repository, half of the colloidal particles precipitate from water to the fracture walls in 50 days (Ahn, 1997). The calculation showed that the actinide-bearing colloidal particles can migrate a distance of 50 m from the site of their formation in 30,000 years, but the reliability of this estimate and its acceptability for the other geological conditions are still uncertain.
Some reliable data indicate that actinides can migrate in colloidal form for significant distances and with high velocities (Kersting et al., 1999). The water samples collected from depth intervals of 701–784 and 1046–1183 m within the area of underground nuclear tests in Nevada contained actinide-bearing colloidal particles. The 240Pu/239Pu ratio testifies to the origin of these isotopes from radioactive glass produced by the nuclear explosion in 1968 at a depth of 1402 m and at a distance of 1.3 km from the sampling site. The major portion of actinides is confined to colloidal particles from 7 nm to 1 µm in size composed of illite, smectite, and zeolite. These minerals are typical products of secondary alteration of host rhyolites and tuffs. The dissolved actinides compose only 1 percent of the total actinide amount. Thus, the predominant actinide migration in colloidal form suggested from experiments was verified by natural observations.
PLUTONIUM MIGRATION IN GEOLOGICAL MEDIUM
Plutonium contents in uranium ores are negligible (Pu/U is approximately 10−12). It forms there when 238U atoms capture neutrons. This element is most dangerous among the SNF actinides (Korenkov, 1992). The 239Pu capability of maintaining a chain reaction requires conditions excluding critical-mass formation in the HLW repositories. It was demonstrated that the HLW forms used for storage and disposal preclude the formation of critical masses in individual packages and their assemblies (Allen, 1978). Data analysis shows that the probability of critical-mass formation due to selective leaching and redeposition of 239Pu in or near an SNF repository is very low (Kastenberg et al., 1996; Oversby, 1998).
This property of plutonium, together with its high toxicity, should be taken into account in projects of long-term storage of SNF.
If uraninite is dissolved and actinides pass from SNF to groundwater, the repository can be considered secure only if any one of the following conditions is met: (1) the concentration of Pu (and other actinides) in groundwater contacting with SNF is below the maximum concentration limit for drinking water or (2) the SNF actinides passed into groundwater are retained within the restricted volume of the geological medium, and only refined water suitable for domestic use reaches the zone of active water exchange. To estimate the possibility of realization of these conditions, we used the available information on the properties of synthesized actinides and their natural geochemical analogues, experimental results on SNF interaction with groundwater, data on actinide solubility in aqueous solutions and interaction of actinide-bearing solutions with rocks, experimental results obtained in underground laboratories on velocities of migration of elements that can and cannot be sorbed in fractured rocks, and information on the colloidal form of radioisotope migration with groundwater.
To predict the behavior of plutonium in the SNF under reducing conditions, we used data from the Oklo deposit. Plutonium, until its complete fission, was retained in this deposit in the uraninite matrix, regardless of its interaction with groundwater. The conservation of Pu and other actinides at the site of their formation was maintained by the reducing conditions and the high stability of uraninite under such conditions. This conclusion is also consistent with experimental data. The Pu concentration in water from granite or clay in equilibrium with SNF at 90°C under the reducing conditions is only 10−11 mol/L (Lelous et al., 1998). This value is close to the maximum concentration limit for water of domestic use. The highest Pu concentrations possible in groundwater under the oxidizing conditions can be estimated by experimental data on SNF dissolution in groundwater, which were obtained within the framework of the project for the HLW disposal in the Yucca Mountain repository (LLNL, 1998). The groundwater from tuffs with pH = 7.6 and Eh = 0.77 V was used as a dissolvent. It was found that, during the SNF dissolution, plutonium passed into water in dissolved and colloidal forms, precipitated on the vessel walls as solid phases, and was incorporated into the secondary uranium minerals. A high proportion of plutonium occurred in colloidal form.
The highest Pu concentration in dissolved form is approximately 2 × 10−8 mol/L at 25°C, which is close to the solubility of the amorphous Pu(OH)4 in groundwater and seawater (approximately 6 × 10−8 mol/L at 25°C) (Kulyako et al., 2001). The dissolved Pu concentration is significantly lower at 85°C (approximately 2 × 10−11 mol/L) due to extensive precipitation of solid phases. In experiments on the SNF interaction with groundwater in the noble gas atmosphere (any external oxidizing or reducing agents were absent), the redox conditions were generally controlled by radiolysis. The 545-day runs in sealed ampoules filled with Ar or mixtures of Ar and CO2 indicated a high SNF solubility
in groundwater of various compositions (Loida et al., 2001). For example, the element concentrations in groundwater from granites (pH = 7–8) were 5.4 × 10−5 mol/L for U and 1.5 × 10−6 mol/L for Pu. It was found using a 1.8 nm filter that 96 percent U and 99 percent Pu occurred in colloidal form. The element concentration in water in equilibrium with iron powder decreases by two orders of magnitude for U and by more than three orders of magnitude for Pu, Am, Np, and Eu. The authors emphasize that these concentrations are comparable with those obtained under the reducing conditions.
The above data indicate that the Pu concentration in groundwater interacting with SNF under oxidizing conditions can be as high as n × 10−8 mol/L for the dissolved form and up to 10−6 mol/L for totally dissolved and colloidal forms. These values are two to four orders of magnitude higher than the maximum concentration limit for drinking water. Therefore, the Pu solubility in groundwater is not low enough for SNF repository safety with respect to Pu in oxidizing conditions.
Geochemical analogues of plutonium under reducing conditions are thorium and uranium. All three elements occur in the tetravalent state under such conditions, and their dioxides (PuO2, ThO2, and UO2) are stable solid phases with very low solubility. Under the weakly and moderately oxidizing conditions, plutonium occurs in the tetravalent state and behaves like thorium, whereas uranium passes into the highly mobile hexavalent state. Plutonium can pass into the penta-and hexavalent states and behave like uranium only in highly oxidizing conditions (see Figure 5[a]), which are not possible in the SNF repositories. Unlike uranium and thorium, plutonium responds to the increase of the solution acidity by transition into the relatively mobile trivalent state in a moderately acidic medium. Acid water can form only in zones of sulfide ore oxidation or in active volcanic areas. In both cases the high acidity of the medium is caused by the formation of sulfuric acid solutions by oxidation of sulfide sulfur. The geological conditions in which the formation of such solutions is possible are unsuitable for SNF storage and disposal. The pH values of groundwater of crystalline rock massifs typically are 8 ± 1.5.
Thermodynamic modeling shows that plutonium under strongly reducing conditions (Eh is approximately −500 mV) at pH = 8 occurs generally in the trivalent state; tetravalent plutonium dominates at Eh from −100 to +150 mV; while penta- and hexavalent plutonium prevails under the strongly oxidizing conditions close to the boundary of water stability (Berry et al., 2002). Under the Eh and pH conditions typical of groundwater in crystalline rock massifs, plutonium occurs generally in the tetravalent state and behaves like thorium. The information on the thorium geochemistry in the geological medium is summarized below. These data are important for prediction of the behavior of plutonium.
Thorium belongs to hydrolyzates, shows a constant valence of four, and does not participate in redox reactions (see Figure 5[b]). Thorium can reach significant concentrations in acid solutions. The formation of the Th(OH)4 col-
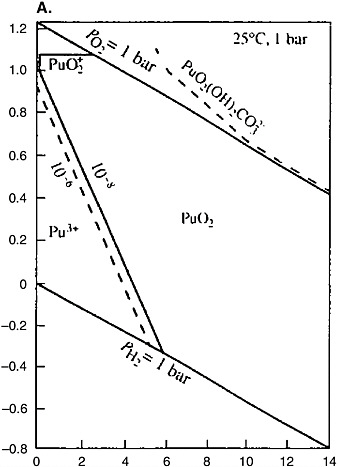
FIGURE 5 Eh-pH diagrams for the Pu-C-O-H (a) and Th-S-O-H (b) systems. Activities of the dissolved particles: Pu = 10−8, C = 10−3, Th = 10−6, −8, and S = 10−3 (Brookins, 1988).
loids followed by their precipitation begins at pH greater than 3.5. Aging of the sediment is accompanied by the irreversible reaction
Thorium can form complex ions (hydroxocomplexes, carbonate complexes, as well as complex compounds with organic acids). Thorium is readily sorbed by Fe-Ti minerals and products of their alteration. Thorium is two to four times more abundant in rocks than uranium. Its concentration in groundwater is one to
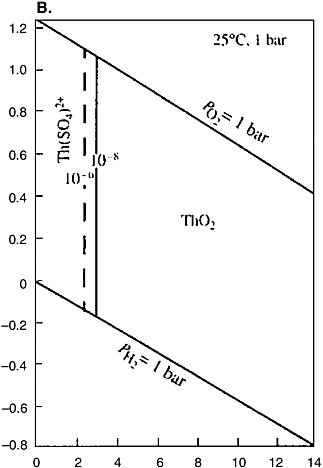
three orders of magnitude lower, which determines significantly lower natural mobility of Th compared with U. The Th concentrations in groundwater vary from n × 10−11 to n × 10−7 mol/L. Its highest concentrations are typical of acid sulfate water from the oxidation zone of REE deposits rich in Th. The Th concentration in groundwater below the zone of extensive hypergenesis never exceeds 10−9 mol/L, even in blocks with Th ore mineralization. Thorium in ground, river, and sea water generally occurs in suspended and colloidal forms. Minerals with high Th concentrations, for example, thorite, thorianite, monazite, and zirconolite, form by crystallization from magmas or alkaline and acidic high-
temperature hydrothermal solutions. These minerals are stable in natural conditions. Thorium-bearing minerals decompose only in chemical weathering in acidic conditions. Thorium can pass into aqueous solutions by incongruent leaching or complete dissolution. The study of weathered rocks indicates a very slow decomposition of these minerals (Eliseyeva and Omelyanenko, 1987). Thorium removed by acid leaching can be partly redeposited in a sorbed form below the kaolinite layer; this is related to the increase in pH values in groundwater with increasing depth.
The analysis of natural observations helped us to conclude that the interaction of the Th minerals with fracture water in the crystalline rock massifs below the hypergene zone cannot result in significant migration of thorium in a dissolved state from the disposal site. By analogy with thorium showing geochemical similarity to plutonium, we suggest that groundwater can have very low contents of dissolved plutonium whose migration will be retained by sorption and that plutonium can be transported for significant distances only as colloidal particles.
Like thorium, plutonium can be extensively sorbed by minerals. Many experiments simulated Pu sorption from aqueous solutions with rock powders. The partition coefficient values (Kd) significantly vary depending on the compositions of rocks and aqueous solutions, pH and Eh values, temperature, and some other factors. Kd values almost always exceed 100 cm3/g and in many cases are a few thousand or even a few hundred thousand cubic centimeters per gram. Kd values are 550 cm3/g for sand, 1200 cm3/g for loam, and 1800 cm3/g for organic soil (Ways of Migration, 1999). Kd values are higher under reducing conditions than under oxidizing conditions (Brookins, 1984). The effect of rock composition and CO2 concentration (Baston et al., 2000), as well as redox conditions (Berry et al., 2002), on the Pu sorption was also studied. The basalt, sandstone, and argillite powders (less than 0.25 mm fractions) mixed with water in the proportion 1:5 were used in experiments. Kd, values ranged from 1000 to a few hundred thousand cubic centimeters per gram. The experimental results cannot be accounted for only by the influence of the studied parameters. It is more probable that the samples contained various amounts of minerals with high sorptive capacity, for example, Fe, Ti, Mn oxides and hydroxides, and organic matter; for example, the oxides concentrate up to 60 percent of the total plutonium sorbed in tuff, although the oxide content in this rock is only 1 percent (Vaniman et al., 1996).
It is pertinent to note that when characterizing the potential sorptive capacities of various rocks and minerals, the experiments with powders do not reflect real conditions of the water-rock interaction and the actual insulative properties of geological media. Unlike the experimental conditions, only a part of crystalline rocks adjacent to the water-conducting fractures participates in sorption. Experiments in underground laboratories demonstrate that radionuclides can
move away from a fracture for less than 2 mm (up to several millimeters along microfractures and brecciation zones) (Neretnieks, 1993). The microfractures increase the water-rock interaction area and stimulate sorption.
Experiments on interaction of Pu-bearing solutions with plate-, cube-, or disk-shaped rock samples are also important in this context. It was found that plutonium was mostly concentrated on the sample surfaces and only partly penetrated inside along microfractures. The more prolonged the interaction, the more plutonium penetrated inside the sample along microfractures and pores (Zakharova et al., 1998). Desorption experiments indicate a stable immobilization of plutonium in microfractures.
Many experiments on Pu sorption and desorption on core samples from igneous rocks (mainly quartz monzonites) of the Canadian Shield were also performed (Vandergraaf and Abry, 1982). The groundwater from granites used in the experiments contained approximately 5 × 10−14 mol/L Pu with 19.5 percent 237Pu. In this case the partition coefficient was expressed in centimeters, because Kd values in runs with samples characterized the ratio of the element content per 1 cm2 of the sample area to its concentration in 1 cm3 of solution (g/cm2:g/cm3). The surface area of the samples in these experiments was 9.5 cm2; the solution volume 10 cm3; and the run duration up to 28 days. Kd of Pu ranged for various samples from 1.4 to 70.0 cm with an average value of 14 cm.
It was found that the mafic minerals sorb plutonium better than quartz and feldspars. Consequently, the mafic rocks can sorb more plutonium than the felsic rocks. Thus, the crystalline rocks present reliable barriers for migration of dissolved Pu. The velocity of the Pu plume propagation is hundreds and thousands of times lower than the groundwater flow velocity (Krauskopf, 1998a). The distance that the dissolved plutonium can migrate in groundwater strongly depends on the water flow velocity, which is highly dissimilar in crystalline rock massifs.
The experimental results obtained in underground laboratories show that the weakly sorbed elements migrate along some fractures with a velocity of a few tens of meters per year, whereas the migration of the easily sorbed elements is much slower than the water flow (Neretnieks, 1993). The easily sorbed elements, such as Cs, Sr, Eu, Nd, Th, and U, did not pass the distance of 5–10 m after six months and were immobilized near the injection site. The increasing duration of the experiments resulted in deeper penetration of the easily sorbed elements into the rock matrix along the microfractures and did not considerably change the migration distance. These results can be extended to plutonium, which also belongs to the most easily sorbed elements. The above data indicate that the geological medium can prevent the Pu transition to the biosphere in dissolved form.
The Pu migration in colloidal form seems to be more hazardous for the environment. PuO2 · H2O and Pu(OH)4 prevail among the true radiocolloids. Their role is insignificant, because Pu is extensively sorbed by minerals in rocks
and colloids in groundwater. The groundwater colloids are most important for Pu transportation. Based on the suggestions that half of colloidal particles in groundwater precipitate on the fracture walls in 50 days and that the Pu-bearing colloidal particles can migrate distances of less than 50 m from the site of their formation, Ahn (1997) calculated that the Pu concentration on the fracture walls near the source in 30,000 years will be 0.025 kg/m2. This is much lower than the critical concentration of Pu; however, the distance and scale of the Pu transportation in colloidal form are not known definitely.
Most researchers believe that Pu cannot be transported for significant distances either in dissolved or in colloidal forms; these suggestions are not consistent with the above-mentioned fact of Pu migration for 1.3 km (Kersting et al., 1999). Plutonium concentration in water in this case is 10−14 mol/L, and 99 percent of the total Pu amount is confined to colloidal particles from 7 nm to 7 µm in size, which are composed of illite, smectite, and zeolites. The repository safety with respect to Pu can be maintained generally by the factors preventing colloid migration, that is, low water permeability and water exchange, the absence of wide fractures, sealing of the fractures by secondary minerals, and the presence of bentonite backfill. Additional factors increasing the insulative properties of geological medium are the mafic composition of crystalline rocks and the distant location of the repository from the site of groundwater discharge (Laverov et al., 2001).
AMERICIUM MIGRATION IN GEOLOGICAL MEDIUM
Americium is the second element (after plutonium) among the actinides with risk to the environment (Korenkov, 1992). By the time of the container failure in 500–1000 years, Am and Pu will account for 98 percent of the total radioactivity of SNF. The 241Am isotope will play the major part among the actinides (see Figure 1). The general knowledge of the Am behavior in geological media can be deduced from the data on its geochemical analogues.
Am has similar geochemical properties to Nd and Eu, whose average abundances in the crustal rocks are n × 10−3 and n × 10−4 percent. The Nd and Eu in rocks and minerals compose from 10–25 to 0.5–2 percent of the total rare earth element amount. The highest contents of these elements are typical of the alkaline massifs. Nd and Eu occur in many minerals as isomorphous impurities. They are mobile in high-temperature alkaline solutions, which is evidenced by the close relation of most rare earth element deposits with high-temperature alkaline metasomatites. Rare earth elements in these deposits are accompanied by Nb, Ta, Th, Zr, and U. The rare earth elements are also mobile in acid media, where they migrate as sulfate and fluoride complexes. Rare earth element contents in uraninite are variable. The uraninite from the Oklo deposit contains 0.39 wt percent Nd2O3. The accessory minerals, such as monazite, orthite, and xenotime are the main concentrators of Nd and Eu. These minerals are stable to
interaction with groundwater, are preserved during mechanical weathering, and are accumulated in sands. These accessory minerals undergo dissolution with removal of poorly soluble elements (rare earth elements, Th, Nb, Ta, and Zr) only during chemical weathering by organic acids and accompanied by the formation of kaolinite layers. The solubility of these elements decreases with neutralization of the unconfined water, and they accumulate in the zone of rock disintegration (Burkov and Podporina, 1967). Sorption on secondary minerals is the principal mechanism of this accumulation.
The data on Nd and Eu concentrations in groundwater are scarce. The element concentrations in the CO2-rich water passed through a 100 nm filter ranged in (0.42–7.55) × 10−10 mol/L for Nd and (0.026–0.82) × 10−10 mol/L for Eu (Michard et al., 1987). The Nd and Eu concentrations in water from a uranium mine are 6.5 × 10−10 and 0.3 × 10−10 mol/L, respectively (Ivanov, 1997). The water samples from granite massifs of the French Pyrenees collected in deep wells and thermal springs (pH = 9.0, T = 75°C) and passed through a 450 nm filter contain 7.1 × 10−9 mol/L Nd and 3.5 × 10−11 mol/L Eu and decrease by one order of magnitude after passing the samples through a 10 nm filter (Michard et al., 1991). The authors concluded that the colloidal forms of these elements were dominant in the studied samples. Among the natural waters in the active volcanic areas worldwide, the highest Nd (7 × 10−7 mol/L) and Eu (2 × 10−8 mol/L) concentrations were found in the acid sulfate water (pH = 1.33, T = 43°C) passed through a 100 nm filter (Michard, 1989). The concentrations of these elements are four orders of magnitude lower in nearly neutral water. It was demonstrated that Nd and Eu concentrations in water are independent of the rock composition but strongly depend on the solution acidity and increase with decreasing pH. The hydrothermal rock alteration typically does not result in REE leaching (Sturchio et al., 1986); for example, a rhyolite alteration with strong redistribution of major elements does not change REE concentrations. Rare earth elements were leached from the rock-forming minerals during the rhyolite alteration and were sorbed in the secondary clay minerals and zeolites. Based on the above data, we can suggest that the Nd and Eu concentrations in groundwater do not exceed n × 10−9 mol/L and n × 10−10 mol/L, respectively, and are much lower in most cases. The Nd and Eu concentrations can exceed these values only in the specific conditions of intensive weathering of the REE ores or due to an ultra acidic composition of the solution. In most cases the main part of these elements in groundwater occurs in colloidal form. REE sorption on minerals is the leading factor retaining their migration in dissolved form. The elevated activity of the complex ions in groundwater stimulates the REE transition from the isomorphous to the easily sorbed forms and facilitates REE accumulation in the suspended particles with high sorptive properties. This phenomenon can be responsible for the correlation between the carbonate-ion and REE concentrations in groundwater, as well as the domination of the colloidal form. Similar behavior in
geological medium can be suggested for Am. Some additional facts supporting this conclusion are given below.
Americium can occur only in the trivalent state within the whole interval of hydrogeochemical conditions typical of the HLW repositories. The trivalent form is most stable even in the near-surface conditions (for example, in soils) (Ways of Migration, 1999). The tetravalent state is possible only in the strongly oxidizing alkaline conditions, which are unreal for the HLW repositories. Americium is rapidly hydrolyzed in water, and its equilibrium concentration in aqueous solution is determined by Am(OH)3 solubility. The latter can be represented by crystalline and more soluble amorphous phases. The Am(OH)3 solubility strongly depends on the composition and properties of groundwater. The ![]() and
and ![]() ions, as well as humic and fulvic acids, increase the Am(OH)3 solubility because of the formation of specific complexes. The highest mobility of americium is typical of the circulation zones of acid sulfate water, which usually forms during the oxidation of sulfide deposits or in active volcanic areas. Such conditions are unsuitable for the construction of HLW repositories. Am(OH)3 solubility is very low in the nearly neutral reducing conditions typical of the deep zones with low water exchange.
ions, as well as humic and fulvic acids, increase the Am(OH)3 solubility because of the formation of specific complexes. The highest mobility of americium is typical of the circulation zones of acid sulfate water, which usually forms during the oxidation of sulfide deposits or in active volcanic areas. Such conditions are unsuitable for the construction of HLW repositories. Am(OH)3 solubility is very low in the nearly neutral reducing conditions typical of the deep zones with low water exchange.
The hydroxocomplexes and carbonate complexes probably are the most abundant soluble forms under such conditions. According to Pokrovsky (2003), the percentage of the dissolved Am forms in seawater with pH = 8 are as follows: 43.2 for ![]() , 22.4 for Am(OH)2+, 15.0 for
, 22.4 for Am(OH)2+, 15.0 for ![]() , 12.0 for
, 12.0 for ![]() , 4.1 for Am3+, 1.5 for AmCl2+, 1.1 for
, 4.1 for Am3+, 1.5 for AmCl2+, 1.1 for ![]() , and 0.8 for
, and 0.8 for ![]() . Taking into account a great abundance of carbonates in rocks and CO2 dissolved in groundwater and the typically nearly neutral alkaline properties of groundwater, it is possible that the carbonate complexes will be the dominant dissolved species and stable solid Am(OH)(CO3) will occur together or instead of Am(OH)3 under natural conditions. The solubility of these compounds in groundwater is rather high. For example, the Am concentrations in dissolved and colloidal forms in groundwater from rhyolite tuffs (pH = 8.6) in equilibrium with Am(OH)(CO3), Am(OH)3(cryst), and Am(OH)3(amorph) are 1.7 × 10−8, 3.7 × 10−5, and 6.5 × 10−4 mol/L, respectively (LLNL, 1998). It is very difficult to determine the concentration of the dissolved Am alone because of its easy sorption on the tiny colloidal particles. Passing the solution through the filters decreased the Am concentrations in all experiments. The finer the filters, the more Am was retained on them. The experiments on Am(OH)3 solubility in groundwater from the Gorleben area in Germany showed a stable Am concentration of 10−6.3 mol/L (IAEA, 1992). The Am concentration in the aqueous solution decreased after it passed through filters with decreasing pore sizes. After a 1 nm filter, it decreased by almost four orders of magnitude and reached 10−10 mol/L. The specified values characterize the Am concentrations in water in equilibrium with highly soluble phases and are the highest possible concentrations. They considerably exceed the maximum Am concentration limit for drinking water (2 × 10−12 mol/L).
. Taking into account a great abundance of carbonates in rocks and CO2 dissolved in groundwater and the typically nearly neutral alkaline properties of groundwater, it is possible that the carbonate complexes will be the dominant dissolved species and stable solid Am(OH)(CO3) will occur together or instead of Am(OH)3 under natural conditions. The solubility of these compounds in groundwater is rather high. For example, the Am concentrations in dissolved and colloidal forms in groundwater from rhyolite tuffs (pH = 8.6) in equilibrium with Am(OH)(CO3), Am(OH)3(cryst), and Am(OH)3(amorph) are 1.7 × 10−8, 3.7 × 10−5, and 6.5 × 10−4 mol/L, respectively (LLNL, 1998). It is very difficult to determine the concentration of the dissolved Am alone because of its easy sorption on the tiny colloidal particles. Passing the solution through the filters decreased the Am concentrations in all experiments. The finer the filters, the more Am was retained on them. The experiments on Am(OH)3 solubility in groundwater from the Gorleben area in Germany showed a stable Am concentration of 10−6.3 mol/L (IAEA, 1992). The Am concentration in the aqueous solution decreased after it passed through filters with decreasing pore sizes. After a 1 nm filter, it decreased by almost four orders of magnitude and reached 10−10 mol/L. The specified values characterize the Am concentrations in water in equilibrium with highly soluble phases and are the highest possible concentrations. They considerably exceed the maximum Am concentration limit for drinking water (2 × 10−12 mol/L).
The data on Am concentrations in groundwater interacting with SNF is of great interest for current purposes. Let us emphasize that Am is incorporated in the UO2 crystal structure and its transition into the aqueous solution strongly depends on the uraninite solubility: The experimental data on SNF interaction with aqueous solutions indicate that Am is not leached if uraninite is stable and passes into solution congruently with U and other actinides during uraninite dissolution. In the Oklo deposit uraninite was stable because of the reducing conditions, and Am, like Pu, was retained at the site of the mineral formation until its complete fission.
The experimental data help us to suggest possible Am concentrations in groundwater under the oxidizing conditions induced by radiolysis. The Am concentration in the solution was (2.5–3.6) × 10−8 mol/L after the interaction of SNF with a high degree of fuel decay with groundwater from granites (pH = 7–8) after 545 days in closed ampoules filled with Ar or mixtures of Ar and CO2 (Loida et al., 2001). After ultrafine filtration, 99 percent of the total Am amount was retained on a 1.8 nm filter. The authors believe that microcolloidal particles smaller than 1.8 nm can also occur in the solution.
Many experiments on SNF interaction with groundwater in conditions open with respect to atmospheric oxygen were performed within the frames of the project for the HLW disposal in the Yucca Mountain repository (LLNL, 1998). SNF fragments 2–3 mm in size from reactors of various types and groundwater from rhyolite tuffs with pH = 7.6 were used in experiments. The runs were performed in closed and open ampoules at temperatures of 25°C and 85°C over several months. In these cases UO2 composing 98 percent of the SNF and acting as an immobilizing matrix for the other radionuclides could not maintain their secure isolation, because the groundwater had oxidizing properties and was significantly undersaturated with respect to U. The experiments showed that the Am concentration in water rapidly increased in the beginning of the experiment, reached a maximum, and then decreased to become constant at a certain level. The authors explain the concentration decrease by the formation of secondary uranyl-ion phases on the grain surfaces. These phases have lower solubility under oxidizing conditions as compared to uraninite.
The equilibrium Am concentration in the solutions passed through a 400 nm filter was approximately 1.5 × 10−10 mol/L. After ultrafine filtration through a 1.8 nm filter, the Am concentration decreased by five times, which indicates the prevalence of the colloidal form. The measured Am concentrations were notably lower than the concentrations expected in equilibrium with Am(OH)CO3 or Am(OH)3. The solid phases controlling the Am solubility were not found in the material retained by filters. The authors believe that the smaller-than-expected Am concentrations could be related to high concentrations of lanthanides in SNF, whose secondary phases can confine Am as an isomorphous impurity. The equilibrium Am concentration was two orders of magnitude lower at 85°C than at 25°C. This fact indicates a more rapid formation of solid phases controlling the Am solubility at higher temperature. The highest possible concentration of
Am dissolved in groundwater at the contact with SNF was estimated to be 10−9 mol/L. Note that a similar value was obtained in experiments on groundwater interaction with vitrified HLW.
Thus, the repository safety with respect to Am in the oxidizing conditions cannot be maintained only by low Am solubility in groundwater. The insulative capacity of the geological medium is also very important in this case. An easy sorption of Am on solid phases is a favorable factor in the natural environment. Many experimental results on Am sorption by various rocks and minerals, as well as mixtures of bentonite with sand, are now available. Depending on the run conditions, for example, composition, pH, Eh, solution temperature, Am concentration, and pounding degree of rocks and minerals, the partition coefficients (Kd) of Am vary from several tens to several thousands of cm3/g and reach several tens of thousands of cm3/g during water interaction with clay minerals and zeolitized tuffs (Baston et al., 1995). The Kd value for Am in interaction with sand is 2000 ml/g and is still higher in interaction with loam and soil (IAEA, 1992). In runs at 70°C, the Kd of Am was 7.5 × 104 for the mixture of bentonite (15 percent) and sand (85 percent) and from 0.46 × 104 to 7.5 × 104 cm3/g for tuff (Baston et al., 1995). The Kd of Am strongly increases if the rocks contain iron oxides and hydroxides, alteration products of titanium-bearing minerals, and chlorite. Depending on the rock composition and geochemical conditions, the retention factors (Rf) of Am range from a few hundred to a thousand and are comparable to those of Pu.
Summarizing the above data, we can conclude that the Am migration from the SNF repository to the biosphere in dissolved form in concentrations hazardous for the environment is impossible; its migration in colloidal form is much more probable. This conclusion is also verified by natural observations; for example, the study of Lake Trawsfynydd in northern Wales, which receives the liquid wastes from the Magnox reactor plant, demonstrated that almost all Am is confined to colloidal particles. Because of this property of Am, it is almost immobile in soils, regardless of the occurrence of organic matter (Ways of Migration, 1999). The scale of the Am release from SNF in colloidal form can be estimated only for a certain geological medium by experiments in an underground laboratory using tracers (for example, the tracers containing Nd as a geochemical analogue of Am). The secure isolation of Am, like Pu, within a repository can be maintained by the media retaining the colloid migration. The host rocks should not contain extended open fractures, while SNF should be isolated with bentonite backfill. All previous comments regarding Pu pertain equally to Am.
CONCLUSIONS
-
The spent nuclear fuel in an underground repository can interact with groundwater only after the engineered barriers fail, that is, in 500–1000 years. The main radioactivity in this case will be related to Pu and Am.
-
The SNF contains 95–98 percent UO2 with isomorphous mixtures of Pu and Am. Thus, the scale of the Pu and Am release depends on the uraninite stability. The latter compound is insoluble in hydrogeochemical conditions of reducing, nearly neutral to slightly alkaline groundwater saturated with respect to uraninite.
-
The groundwater attains such properties at certain depths due to its long-term interaction with the host rock masses under conditions of low water exchange. The repository safety of SNF is provided due to its placement in massifs of crystalline rocks below 500 m from a surface.
-
The formation of local oxidizing conditions in the SNF repositories induced by radiolysis or some other processes is not probable, because the host medium contains reducing agents. If the oxidizing conditions do appear, the Pu and Am migration in dissolved form in concentrations hazardous for the environment will be precluded by their rapid sorption on minerals.
-
The sorption of Pu and Am on colloidal particles followed by their migration to the discharge areas is the main real mechanism of environmental pollution. The distance and scale of Pu and Am migration in colloidal form can be estimated only for a certain geological medium by experiments in underground laboratories. The absence of wide fractures, low water permeability, and isolation with bentonite backfill are efficient factors retaining the actinide migration in colloidal form.
ACKNOWLEDGMENTS
The study was supported by the Russian Foundation for Basic Research, project numbers 02-OS-64007 and 02-05-64008.
REFERENCES
Ahn, J. 1997. Transport of weapons-grade plutonium and boron through fractured geologic media. Nuclear Technology 117:316–328.
Allen, E. J. 1978. Criticality Analysis of Thermal Underground Nuclear Waste Disposal. Report no. ORNL/TM-7405. Springfield, VA: Union Carbide Corp.
Baston, G. M., J. A. Berry, M. Brownsword, et al. 1995. Sorption of plutonium and americium on repository, backfill and geological materials relevant to the JNFL low-level radioactive waste repository at Rokkasho-Mura. Proceedings of the Symposium on Scientific Basis for Nuclear Waste Management XVIII 353(part 2):957–964.
Baston, G. M., J. A. Berry, M. Brownsword, et al. 2000. Effect of carbonate concentration on the sorption of plutonium onto geological materials. Proceedings of the Symposium on Scientific Basis for Nuclear Waste Management XXIII 608:293–298.
Berry, J. A., M. Brownsword, D. J. Ilett, et al. 2002. Effect of redox conditions on the sorption of plutonium on to geological materials. Proceedings of the Symposium on Scientific Basis for Nuclear Waste Management XXV 713:693–697.
Brookins, D. G. 1984. Geochemical Aspects of Radioactive Waste Disposal. New York: Springer.
Brookins, D. G. 1988. Eh-pH Diagrams for Geochemistry. Berlin: Springer.
Buck, E. C., R. J. Finch, P. A. Finch, and J. K. Bates. 1998. Retention of neptunium in uranyl alteration phases formed during spent fuel corrosion. Proceedings of the Symposium on Scientific Basis for Nuclear Waste Management XXI 506:87–94.
Burkov, V. V., and E. K. Podporina. 1967. Rare earths in weathering crust of granitoids. Doklady Akademy Nauk SSSR 177(3):691–694.
Chapman, N. A., and L. G. McKinley. 1988. The Geological Disposal of Nuclear Waste. Chichester: Wiley and Son.
Chapman, N. A., and D. Savage. 1984. Mineralogical aspects of the safe disposal of high-level radioactive waste. Fortschr. Miner. Bd. 62(H1):17–32.
Chen, Y., E. Siegmann, P. Mattie, et al. 1999. A mechanistic model of spent fuel dissolution, secondary mineral precipitation, and Np release. Symposium on Scientific Basis for Nuclear Waste Management XXII 556:471–478.
Cowan, G. A. 1978. Migration Paths for Oklo Reactor Products and Applications to the Problem of Geological Storage of Nuclear Wastes, Natural Fission Reactors. Pp. 693–698 in Proceedings of the Technical Committee Meeting. Paris: IAEA.
Cramer, J. J. 1986. A natural analog for a fuel waste disposal vault. Pp. 697–699 in Proceedings of the Canadian Nuclear Society International Conference on Radioactive Waste Management. Toronto: Canadian Nuclear Society.
Eliseyeva, O. P., and B. I. Omelyanenko. 1987. Uranium behavior in the process of formation of weathering profiles of the Alexeyevskoe kaolinite deposit, northern Kazakhstan. Geokhimiya 6:847–854.
Ewing, R. C. 2002. Yucca Mountain. Science 296:659–660.
Finn, P. A., R. Finch, E. Buck, and J. Bates. 1998a. Corrosion mechanisms of spent fuel under oxidizing conditions. Proceedings of the Symposium on Scientific Basis for Nuclear Waste Management XXI 506:123–131.
Finn, P.A., D. J. Wronkiewicz, R. J. Finch, J. C. Hoh, C. J. Mertz, J. W. Emery, E. C. Buck, J. A. Fortner, S. F. Wolf, L. A. Neimark, and J. K. Bates. 1998b. Yucca Mountain Project-Argonne National Laboratory, Annual Progress Report, F.Y. 1997. No. ANL-98/12. Argonne, Ill.: Argonne National Laboratory.
Forsyth, R., and L. O. Werme. 1986. The corrosion of spent UO2 fuel in synthetic groundwater. Proceedings of the Symposium on the Scientific Basis for Nuclear Waste Management IX 327–332.
IAEA (International Atomic Energy Agency). 1978. Natural Fission Reactors. Vienna: IAEA.
IAEA. 1992. Geochemistry of Long-Lived Transuranic Actinides and Fission Products. Final Report of a Coordinated Research Programme 1987–1991. Vienna: IAEA.
Ivanov, V. V. 1997. Ekologicheskaya geokhimiya elementov (Ecological Geochemistry of Elements). Book 6. Moscow: Ekologiya.
Ivashkin, N. V., G. V. Kovalenko, V. V. Leaonkov, et al. 2000. Numerical Modeling of Change in the State of Geological Environment and Migration of Radionuclides in the Period up to 10,000 Years in the Deep Disposal. Ohshchie Voprosy Radiatsionnoy Bezopasnosty 1:3–18.
Johnson, L. H., and D. W. Shoesmith. 1988. Spent fuel. Pp. 635–698 in Radioactive Waste Forms for the Future, W. Lutze and R. C. Ewing, eds. Amsterdam: North-Holland.
Kastenberg, W. E., R. E. Peterson, J. Ahn J. Burch, G. Casher, P. Chambré, E. Greenspan, D. R. Olander, J. Vujic, B. Bessinger, N. G. W. Cook, F. M. Doyle, B. Hilbert. 1996. Mechanisms for Autocatalytic Criticality of Fissile Materials in Geologic Repositories, Report Submitted to Los Alamos National Laboratory. UCB-NE-4214. Berkeley: University of California.
Kersting, A. B., D. W. Efurd, D. L. Finnegan, et al. 1999. Migration of plutonium in ground water at the Nevada test site. Nature (Lond.) 397:56–59.
Kiryukhin, V. A., A. I. Korotkov, and S. L. Shvartsev. 1993. Gidrogeokhimiya (Hydrogeochemistry). Moscow: Nedra.
Korenkov, A. P. 1992. To the problem of classification of solidified radioactive wastes. Atomic Energy 73(2):129–131.
Krainov, S. Z., and V. M. Shvets. 1992. Gidrogeokhimiya (Hydrogeochemistry). Moscow: Nedra.
Krauskopf, K. B. 1988a. Geology high-level nuclear waste disposal. Annual Review of Earth and Planetary Science 16:173–200.
Krauskopf, K. B. 1988b. Radioactive Waste Disposal and Geology. London: Chapman and Hall.
Kulyako, Yu. M., S. Perevalov, T. Trofimov, et al. 2001. Experimental studies of solubility of U(VI), Np(V), Pu(IV), and Am(III) hydroxides in simulated solutions of ground and sea water. P. 43 in Eighth International Conference on Chemistry and Migration Behavior of Actinides and Fission Products in the Geosphere. Migration ’01. Abstracts. Bregenz, Austria.
Laverov, N. P., A. V. Kantsel, B. I. Omelyanenko, et al. 1991. Principal problems of radioecology in connection with radioactive waste disposal. Atomic Energy 71(6):523–534.
Laverov, N. P., B. I. Omelyanenko, and V. I. Velichkin. 1994. Geological aspects of the problem of radioactive waste disposal. Geoekologiya 6:3–20.
Laverov, N. P., V. I. Velichkin, and B. I. Omelyanenko. 2001. Insulative properties of crystalline rock: on the problem of high level radioactive waste disposal. Geologia Rudn. Mestorozhdeny 43(1):6–23.
Lelous, K., S. Constantin, J. L. Paul, D. Sambuaaro, and E. Vernaz. 1998. Leaching of spent fuel and simulated fuel in the presence of environmental materials: integral experiments. Proceedings of the Symposium on Scientific Basis for Nuclear Waste Management XXI 506:175–188.
Liu, J., and L. Neretnieks. 1995. Some evidence of radiolysis in a uranium ore body: quantification and interpretation. Proceedings of the Symposium on Scientific Basis for Nuclear Waste Management XVIII 353(part 2):1179–1186.
LLNL (Lawrence Livermore National Laboratory). 1998. Waste Form Characteristics Report. UCRL-ID-132375. CD-ROM Version. Livermore, CA: LLNL.
Loida, A., B. Grambow, and H. Geckeis. 2001. Congruent and incongruent radionuclide release during matrix dissolution of partly oxidized high burnup spent fuel. Proceedings of the Symposium on Scientific Basis for Nuclear Waste Management XXIV 663:417–426.
Michard, A. 1989. Rare earth element systematics in hydrothermal fluids. Geochimica et Cosmochimica Acta 53(3):745–770.
Michard, A., C. Beaucaire, and G. Michard. 1987. Uranium and rare earth elements in CO2-rich waters from Vals-les-Bains (France). Geochimica et Cosmochimica Acta 51(4):901–909.
Michard, G., G. Negrel, G. Ouzounian, P. Toulhoat, et al. 1991. Major and trace elements regulation in natural granitic waters: application to deep radioactive waste disposal, high-level radioactive waste management. Proceedings, Second Annual International Conference, Las Vegas, Nevada 1:169–175.
Neretnieks, L. 1993. Solute transport in fractured rock—applications to radionuclide waste repositories. Pp. 39–123 in Flow and Contaminant Transport in Fractured Rock. San Diego: Academic Press.
Neretnieks, L. 1997. Modeling oxidative dissolution of spent fuel. Proceedings of the Symposium on Scientific Basis for Nuclear Waste Management XX 465:573–580.
Omelyanenko, B. I., P. S. Kozlova, O. P. Eliseyeva, and L. L. Simonova. 1983. Local distribution of uranium in rocks and minerals as an indicator of its geochemical history. Pp. 140–163 in Problemy Radiogeology (Problems of Radiogeology), pp. 140–163. Moscow: Nauka.
Oversby, V. M. 1998. Criticality in repository for spent fuel: lessons from Oklo. Proceedings of the Symposium on Scientific Basis for Nuclear Waste Management XXI 506:781–788.
Oversby, V. M. 2000. Using information from natural analogues in repository performance analysis: examples from Oklo . Proceedings of the Symposium on Scientific Basis for Nuclear Waste Management XXIII 608:545–550.
Pagel, M., F. Ruhlmann, and P. Bruneton. 1993. The Cigar Lake uranium deposit, Saskatchewan, Canada. Canadian Journal of Earth Sciences 30:651–652.
Pokrovsky, O. S. 2003. Forms of finding and coefficients of activity of Am(III) and Np(V) in marine water. Geokhimiya 3:337–341.
Pourcelot, L., and F. Gauthier-Lafaye. 1998. Weathering conditions and behavior of fission products in Oklo reactors. Proceedings of the Symposium on Scientific Basis for Nuclear Waste Management XXI 506:1071–1072.
Redkin, A. F., N. I. Savelyeva, E. I. Sergeyeva, B. L. Omelyanenko, I. P. Ivanov, I. L. Khodakovsky. 1989. Investigation of uraninite UO2 solubility under hydrothermal conditions. Sciences Géologiques Bulletin Strasbourg 42(4):329–334.
Roddy, J. W., H. C. Claiborne, R. C. Ashline, P. J. Johnson, and B. T. Rhyne. 1985. Physical and Decay Characteristics of Commercial LWR Spent Fuel. Report no. ORNL/TM-9591/V. 1. Oak Ridge, Tenn.: Oak Ridge National Laboratory.
Rondinella, V. V., J. Cobos, H. J. Matzke, T. Wiss, P. Carbol, and D. Solite. 2001. Leaching behavior and α-decay damage accumulation of UO2 containing short-lived actinides. Proceedings of the Symposium on Scientific Basis for Nuclear Waste Management XXIV 663:391–398.
Rondinella, V. V., H. J. Matzke, J. Cobos, and T. Wiss. 1999. α-radiolysis and α-radiation damage effects on UO2 dissolution under spent fuel storage conditions. Proceedings of the Symposium on Scientific Basis for Nuclear Waste Management XXII 556:447–454.
Ryzhenko, B. N., V. L. Barsukov, and S. N. Knyazeva. 1996. Chemical characteristics (composition, pH, Eh) of the rock-water system: I. Granitoids-water system. Geokhimiya 5:436–454.
Schneider, J. W., P. Zuidema, P. A. Smith, et al. 1998. Preliminary safety studies fuel, MOX and vitrified high-level waste. Proceedings of the Symposium on Scientific Basis for Nuclear Waste Management XXI 506:1077–1078.
Smith, P. A., and L. H. Johnson. 2000. Spent fuel dissolution: an examination of the impacts of alpha-radiolysis. Proceedings of the Symposium on Scientific Basis for Nuclear Waste Management XXII 608:29–39.
Spahiu, K., and P. Sellin. 2001. Spent fuel alteration/dissolution and the influence of near field hydrogen. Proceedings of the Symposium on Scientific Basis for Nuclear Waste Management XXVI 663: 765–772.
Sturchio, N. C., K. Muehlenbachst, and M. G. Seitz. 1986. Element redistribution during hydrothermal system: Yellowstone drill cores Y-7 and Y-8. Geochemica et Cosmochimica Acta 50(5): 1619–1631.
Tsukamoto, M., T. Ohe, T. Fujita, R. Hesbol, and Y. P. Yermansson. 1995. Diffusion of radionuclides in compacted bentonite: results from combined glass dissolution and migration tests. Proceedings of the Symposium on Scientific Basis for Nuclear Waste Management XVII 353(part 1):291–298.
Vandergraaf, T. T., and D. R. M. Abry. 1982. Radionuclide sorption on drill core material from the Canadian Shield. Nuclear Technology 57:399–412.
Vaniman, D., A. Furlando, S. Chipera, J. Thompson, and L. Triay. 1996. Microautoradiography in studies of Pu(V) sorption by trace and fracture minerals in tuff. Proceedings of the Symposium on Scientific Basis for Nuclear Waste Management XIX 412:639–646
1999. Ways of Migration of Man-Made Radionuclides in the Environment: Radioecology after Chernobyl (Puti migratsii iskusstvermykh radionuklidov v okruzhayushchei srede. Radioekologiya posle Chernobylya). Moscow: Mir.
Wilson, C. N. 1990a. Results from NNWSI Series 2 Spent Fuel Dissolution Tests. PNL-7170. Richland, Wa.: Pacific Northwest National Laboratory.
Wilson, C. N. 1990b. Results from NNWSI Series 3 Spent Fuel Dissolution Tests. PNL-7170. Richland, Wa.: Pacific Northwest National Laboratory.
Zakharova, E.V., A. A. Menyailo, K. A. Menyailo, E. P. Kaimin, et al. 1998. Assessment of process of transforming rocks enclosing radioactive waste storages on exposure to chemical and thermal factors. Pp. 149–158 in Issledovanie granitoidov Nizhnekanskoyo massiva dlya zakhororreniya RAO (The Study of Granitoids from Nizhnekansky Massif for Radioactive Waste Disposal). Zheleznogorsk: KNTS.































