
6
Advances in Technologies for Life Detection and Bioburden Reduction
At present, the most stringent bioburden reduction requirement is Viking post-sterilization (see Table 2.1), which imposes costs in resources and time. At the same time, new information from studies of microbial diversity suggests that NASA’s standard microbial monitoring procedures may underestimate the number of organisms that could compromise planetary protection efforts. This chapter examines new technologies that can aid in the detection and identification of living microorganisms and that may play a role in planetary protection, particularly in the analysis and reduction of bioburden1 in spacecraft assembly areas and on the spacecraft.
Ultimately, the use of advanced detection methods to identify the potential bioburden may lead to reduction techniques that are targeted at groups of organisms of specific concern for planetary protection. NASA is supporting research on some of these technologies (e.g., Dickinson et al., 2004a,b; Venkateswaran et al., 2001, 2003).2
EXAMPLES OF METHODS FOR ASSESSING TOTAL VIABLE CELL COUNT
Previously, a major obstacle to detecting and identifying microorganisms has been the limited capabilities of the available methodologies. Such microorganisms are of microscopic size and have relatively few morphological differences, and so detecting them and distinguishing individual taxa usually required the isolation and growth of microorganisms in the laboratory as pure cultures for metabolic and physiological investigations. However, as described in Chapter 5, most microorganisms are not amenable to laboratory culturing, and culturing is not an effective method for assessing the types of microorganisms in situ. Fewer than 1 percent of organisms in the environment are directly culturable using current techniques (Amman et al., 1995), and these organisms often represent minor members of natural populations in terms of both their numbers and their role in ecosystem processes (Ward et al., 1998). It is unlikely that the current NASA swab and culture approach is more successful at growing all spore formers present on spacecraft. Organisms that can be cultured in the laboratory are those that are most compatible with laboratory media, as opposed to organisms that have optimally adapted to a particular environment. The development of high-throughput culture conditions that mimic environmental conditions has reduced the bias of culturing microorganisms (Connon and Giovanonni, 2002; Kaeberlein et al., 2002), but this
laborious technology remains ineffective in assessing the total bioburden of spacecraft, since some microorganisms may still elude cultivation.
The current NASA approach (described in Chapter 2) of using the cultivation of spore-forming bacteria does not provide an accurate estimate of total bioburden. Moreover, it is possible that some of the organisms of potentially greatest concern for the forward contamination of Mars, such as psychrophiles (see Chapter 5), may not be well correlated with the counts of spore-forming bacteria on spacecraft. New rapid and less expensive methods that provide more accurate estimates of total viable bioburden, and that do not require growth of microorganisms in the laboratory, are described below.
Cytological Methods
Epifluorescent Microscopy
The use of epifluorescent microscopy3 and flow cytometry4 to directly count cells labeled with high-quantumyield fluorescent nucleic acids stains such as acridine orange, DAPI, and Sybr-Gold has increased estimates of microbial biomass in nature several-fold and has revised understanding of the importance of microorganisms in the natural environment. However, the detection of nucleic acids is not an indicator of viability. Fluorescent stains must be used in concert with other assays that enable the detection of essential metabolites or enzymes, if accurate estimates of total viable biomass are to be obtained.
Detection of Membrane Integrity
Membrane integrity is an indication of cell viability that can be detected using commercial fluorescence stains such as the BacLight Live/Dead kit.5 This kit uses two nucleic acid stains, SYTO 9 and propidium iodide, which differ in their ability to penetrate healthy cells. SYTO 9 can penetrate both live and dead cells, whereas propidium iodide can only penetrate cells with damaged membranes (dead or damaged cells). When those stains are used together, live cells fluoresce green and dead or damaged cells are red when viewed with an epifluorescent microscopic. Epifluorescent microscopy techniques will require either witness coupons (replicate coupons made of the same material as the spacecraft that can follow the spacecraft through the entire assembly and cleaning process and can be mounted under a microscope for viewing) or microscopic procedures in the clean-room and launch facilities to examine surfaces directly.6 Irrespective of how cells are stained, the sensitivity of all light microscopy techniques is constrained by the volume of material that can be viewed in a field. At a magnification of 1000×, the fluid volume represented in a viewing field will be ~0.05 µl. There is a 50 percent probability of finding a single cell in a field if the concentration of cells in the original sample is 2 × 103 cells per milliliter.
Flow Cytometry and Cell Sorting
Flow cytometers equipped with a fluorescently activated cell sorter (FACS) analyze particles in a laminar flow by their light-scattering and fluorescent properties.7 Flow cytometers are used to enumerate and characterize
selected microbial species in environmental samples by applying fluorescent labeled DNA or rRNA probes (see “Examples of Methods Relevant to Estimating Biodiversity” below; see also Amann et al., 1995) followed by selective sorting of labeled cells to further study their morphology and other characteristics.
Biomarker Methods
By focusing on organic and inorganic biomarkers, it is possible to avoid microscopy altogether. Such methods are highly sensitive and employ measurements of selected metabolic products as a proxy for biomass. These procedures are rapid and well suited for repeated use throughout the spacecraft assembly and launch process.
Limulus Amebocyte Lysate Assay
Most bacteria are either gram positive or gram negative. This classification relies on differences in the cell wall components that can be differentiated by stains that detect peptidoglycan in gram-positive cells and lipopolysacharide in gram-negative cells. Cell wall components can also serve as targets for certain biomarkers. For example, assays based on the limulus amebocyte lysate (LAL) from the horseshoe crab Limulus polyphpemus take advantage of a sensitive enzyme cascade that is triggered by the microbial cell wall components lipopolysaccharide and beta glucan.8
The LAL assay can detect 10–13 grams of lipopolysaccharide from Escherichia coli, which corresponds to one cell. However, the ability to detect a single cell is constrained by the upper limits of the sample size. Typically, each assay can test only 0.1 ml. To be confident at the 98 percent level of detecting a single organism in this small volume, the lower limit of cell concentration would have to exceed 40 cells per milliliter in the sample.9 The assay detects many gram-negative bacteria and fungi. Signals from gram-positive organisms are likely to represent contamination from soils, which can contain large amounts of gram-positive microbes. The technology is not useful for detection of archaean or eukaryan microorganisms and therefore is not effective in detecting total bioburden. A portable instrument that can produce results in near-real time is currently under development. The LAL assay provides no information about the diversity or viability of microorganisms in a sample, but the combination of speed and sensitivity renders it a potential tool for determining selective bioburdens on spacecraft surfaces. This technique may provide a good real-time indication of gram-negative bacterial life. In combination with molecular phylogenetic analyses (see below) LAL has the potential to detect and provide quantitative estimates of target organisms.
ATP Bioluminescence Assay
Unlike the LAL assay, the adenosine triphosphate (ATP) bioluminescence assay provides a very sensitive indicator of live cells (i.e., cells containing functional ATP). Assays for ATP are able to measure quantitatively the presence of microorganisms in an environmental sample within minutes rather than days. ATP is a stable molecule found in relatively high concentrations in metabolically active cells and it is the primary energy currency in all known living organisms. Commercial ATP-bioluminescence test kits measure the quantum output that occurs when ATP drives the luciferin-luciferase reaction in fireflies. The quantum output from the luciferin-luciferase
|
|
chromophores such as phycoerythrin and green fluorescent proteins. The fluorescence of the particles is then detected in several channels corresponding to the emission peak of the fluorophore and characterized and sorted according to this property. |
reaction is directly proportional to the amount of ATP within the cells, which can then be equated to viable cell concentration. Tests are available that can detect approximately 1000 cells. These kits are able to differentiate extracellular ATP from intracellular ATP, which in effect can provide a means to differentiate between live and dead cells. As with the LAL assay, ATP-bioluminescence assays are both rapid and adaptable to small instrument designs. Neither of these assays provides information on the diversity of microorganisms, but both are powerful tools for determining the total bioburden on selected spacecraft components.
A promising advance in detection of ATP is the use of a fusion protein containing ADK plus PolyP kinase (PPK)10 to amplify the amount of ATP in the sample. Standard bioluminescence can then measure the levels of ATP. Through this amplification mechanism, it is possible to detect ATP levels that correspond to a single cell. For planetary protection purposes, this would offer a rapid and sensitive method to detect a single cell, although the amplification assay is not linear in its correlation between luminescence and initial ATP concentrations. There is also a concomitant increase in the background noise relative to standard bioluminescence assays. This assay is still under development.
Both the LAL and ATP assays are examples of emerging technologies for rapid determinations of numbers of microbes without requiring their cultivation in the laboratory or observation with microscopy. Based on current technology, the limits of life detection can reach about 100 cells/ml,11 and future advances in LAL and ATP technology and sample handling may one day allow for the detection of one cell per milliliter. The application of these techniques and others to planetary protection will require the development of a standard certification process that can calibrate the relationship between biomarker levels and bioburden as measured in terms of numbers and kinds of organisms.
EXAMPLES OF METHODS FOR ESTIMATING BIODIVERSITY
Measurement of the number of microbial cells on a spacecraft provides a first estimate of total bioburden, but additional information is needed if planetary protection requirements are to be sensitive to organism type. For example, the ATP and membrane integrity assays will not detect spores, owing to their dormant state. More important, these estimates of biomass do not provide information about the kinds of organisms present on the spacecraft—important information to evaluate the potential for organisms to survive or propagate on Mars. Studies based on metagenomic analysis (sequence-based or function-based screening and analysis of genomes from a mixed assemblage of microorganisms) will one day provide a means to correlate physiological properties with phylogenetic assignments.12 Knowing which specific organisms are present on a spacecraft will allow scientists, engineers, and planetary protection officials to assess whether the bioburden poses a genuine threat to planetary protection. Such information on diversity is essential for making data-based decisions about the implementation of bioburden reduction measures that target particular kinds of organisms that might be present on spacecraft. As a first step to this goal, intensive surveys of clean-room and launch facilities could be conducted to identify resident types of microorganisms.
At present, the most effective means of detecting and identifying microbial diversity focuses on the small subunit ribosomal RNA (16S rRNA) gene to determine molecular phylogeny (Pace et al., 1986; Ward et al., 1992).
|
10 |
The ADK component plus AMP and ATP produces two molecules of ADP. The PPK in the presence of polyP (a linear polymer of many phosphate residues linked by high-energy phosphanhydride bonds) converts the ADP back to ATP. The excess AMP and polyP drive the ADK and PPK equilibrium toward ADP and ATP formation. |
|
11 |
See, for example, <las.perkinelmer.com/catalog/Product.aspx?ProductID=6016941>. |
|
12 |
Metagenomic technologies (genomic analysis of an assemblage of microorganisms) are constrained by the ability to prepare representative libraries from low-biomass samples and methods for annotating DNA sequences. In some cases, when very low numbers of cells are present, methods such as rolling circle amplification can be employed to amplify the signal; these methods are robust and are approaching the ultimate limit of single-cell whole genome amplification. The greater challenge may be functional annotation. Recent publications cite the use of metagenomic data such as environmental gene tags (EGTs: Tringe et al., 2005). The EGTs reflect the metabolic potential of an analyzed community, and their analysis involves the binning of sequences into functional metabolic groups from different environments containing a range of microbial communities. |
The ribosomal RNA gene sequences (16S and 18S) serve as a proxy for the occurrence of microorganisms from the three domains of life (Bacteria, Archaea, and Eukarya) in a sample because this gene is conserved in all known organisms and thus allows comparison of the relatedness between major phyla or between genera. Because of its slow rate of evolutionary change, it is not useful for differentiating between closely related bacterial strains. Diversity assessments made with molecular phylogenetic techniques rely on the amplification of homologous regions of 16S rRNA genes from an environmental sample using the polymerase chain reaction (see Box 6.1 and Figure 6.1.1), along with the analysis of the resultant sequences either by DNA sequencing or fingerprinting methods such as terminal restriction fragment length polymorphisms (T-RFLPs), denaturing gradient gel electrophoresis (DGGE), or microarrays as described below. The molecular phylogenetic approach can be quite useful for planetary protection because it is sensitive and has the potential of yielding quantitative information on individual groups of microorganisms.
Microorganism Identification by DNA Sequencing
Polymerase chain reaction (PCR) amplification of the 16S rRNA gene from a mixed population sample generates a pool of PCR fragments or amplicons of different base composition or sequence. The sequence of each unique PCR product serves as a molecular signature for a microbial species (Pace et al., 1986). By determining the sequence of each unique PCR product, it is therefore possible to detect members of the microbial community that are identical (or nearly so) to entries in DNA sequence databases without requiring their cultivation in the laboratory and to determine their phylogenetic position by using phylogenetic inference algorithms.13 DNA sequence analysis has become more rapid and cost-effective because of the use of fluorescent dyes to label products of DNA cycle-sequencing reactions and the development of high-throughput capillary instruments that can resolve the fragments of DNA sequencing reactions into ordered nucleotides. Future instruments have the potential to require even less preparation and to make use of nanotechnology, such as that in the Nanopore Project,14 which allows the analysis of single nucleic acid molecules using ion channels, an approach that could be useful for working with the low biomass that is found in spacecraft assembly rooms.
The molecular approach has been instrumental in revealing the diversity of and identifying novel microorganisms from a wide range of environments such as thermal springs (Reysenbach et al., 1994), the South Pole (Carpenter et al., 2000), solid ice (Priscu et al., 1998, 1999), and the ocean (DeLong et al., 1994; Béjà et al., 2001) that might otherwise escape detection.15 However, there are limitations and biases to the molecular approach. For example, not all cells are lysed easily during standard DNA extraction methods (Webster et al., 2003), PCR primers may not detect all organisms present (Baker and Cowan, 2004), and PCR reactions tend to preferentially amplify the genes of the most abundant template. With respect to analyzing sequence data, a sufficient number of nucleotides in an appropriate region of the gene must be analyzed in order to ensure that phylogenetic resolution may be achieved. Finally, it is important that a statistically appropriate number of sequences are screened from any given environment or sample to ensure that the diversity is appropriately surveyed (Altekruse et al., 2003; Hughes et al., 2001).
|
13 |
Phylogentic analyses use statistical procedures and models of mutation to estimate the evolutionary history of DNA sequences. However, different models and inference techniques can yield conflicting evolutionary reconstructions. For example, the deeper lineages of the Bacteria and the protists are controversial (Woese et al., 2000; Brochier and Philippe, 2002). Another potential artifact is caused by horizontal gene transfer among microorganisms. Because molecular trees reflect the evolutionary history at the sequence level, phylogenetic analyses of genes that have undergone horizontal transfer produce a phylogeny that does represent the history of the rest of the genome (Doolittle et al., 2003). |
|
14 |
Nanopore detectors are membranes containing a tiny pore called an ion channel, which are large enough to allow only a single strand of DNA to pass through. A voltage applied across the membrane generates an ionic current and pulls the negatively charged DNA molecules through the pore. When the DNA molecule blocks the opening of the nanopore, it causes a characteristic decrease in the current, allowing discrimination between individual DNA molecules. A computer trained by machine-learning techniques recognizes the signals generated by different DNA molecules. For further details, see <www.cbse.ucsc.edu/nanopore.html>. |
|
15 |
For example, for nearly a century, investigators considered pink filamentous tufts in Yellowstone thermal springs to represent dead cells or a precipitate. However, the molecular approach revealed them to be members of the Aquificales, a bacterial group important in thermal springs throughout Earth’s biosphere and of particular evolutionary interest because of its deep phylogenetic position in some molecular trees. |
|
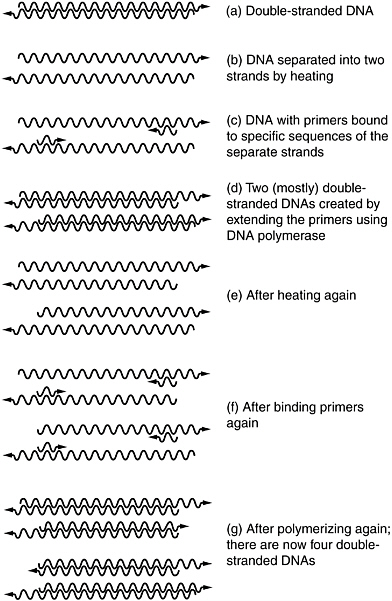
BOX 6.1 Before a cell can divide, it must make an identical copy of all of the genetic information encoded within its DNA. Each daughter cell must inherit all of the original genes after the process of DNA replication, which is mediated by the enzyme called DNA polymerase. The polymerase chain reaction (PCR) is a technique for amplifying (making many copies of) a defined portion of a DNA molecule by using the same biochemical reactions that cells use to copy their genes before they divide. With DNA polymerase as the catalyst, it is possible to convert one copy of a duplex DNA molecule into two copies identical to the first. Each time this process is repeated, the number of copies is doubled. In this way, two cycles generate 4 duplex DNA molecules, three cycles produce 8, four cycles produces 16 duplex molecules, and so on. This exponential increase can rapidly produce billions of copies of the original target genes, making the DNA relatively easy to detect in the laboratory. The process is called a chain reaction because the product of each reaction is more molecules that themselves serve as the substrates (templates) from which more reactions can occur. The process of PCR amplification is outlined below and shown in Figure 6.1.1.
SOURCE: Modified from NRC (2002), pp. 16-17. |
Microorganism Identification by Reverse-Transcriptase (RT)-PCR
RT-PCR employs the same amplification method as does PCR but targets RNA instead of DNA by reverse transcription (coding of an RNA molecule back to its DNA sequence). Because RNA is an essential component of active or metabolizing cells, it is indicative of live cells, whereas amplification of DNA may detect both active and inactive cells. However, this technology is expensive, and it still does not provide quantitative information about relative numbers of different kinds of organisms. It can also fail to identify minor members of the population.
Terminal Restriction Fragment Length Polymorphism (T-RFLP) Analysis
T-RFLP analysis is a rapid method that is easily applied to detect differences in community diversity among samples. The rRNA gene is amplified from genomic DNA and is labeled with a fluorescent dye. The products are then digested with restriction enzymes that recognize and cleave specific short DNA sequences. The fragments are then separated by gel electrophoresis using an automated sequencer. A charge-coupled device (CCD) camera detects the fluorescently labeled fragments; the data resulting from a single sample resemble a chromatograph in
that different phylotypes (sequence types) appear as separate peaks in the electropherogram (output from the sequencing instrument). This method suffers regarding reproducibility, and phylogenetically distinct peaks may be difficult to resolve, yielding no return of sequence data. However, if nucleic acid extraction and PCR amplification methods are standardized across samples, this method can be used to monitor the relative abundance of individual phylotypes, and large numbers of samples can be quickly analyzed. Additionally, individual peaks can be identified by using sequence data developed for the clean rooms.
Denaturing Gradient Gel Electrophoresis
Denaturing gradient gel electrophoresis (DGGE) is a highly sensitive and rapid technique often used to detect temporal and spatial shifts in microbial diversity. DNA fragments amplified from samples by PCR are separated on an acrylamide gel containing a gradient of denaturant according to base sequence differences. As the fragments migrate through a formamide/urea gradient gel, they melt at different points on the gel corresponding to their sequence.16 DGGE banding patterns provide a representation of the diversity of the sample, with each band representing a different phylotype. Differences in banding patterns among different samples reflect diversity differences that may result from temporal or spatial changes in community structure. DGGE methods are able to display subtle differences between complex microbial populations, but the assignment of any particular amplicon in a band to a phylotype still requires the determination of that amplicon’s DNA sequence. The use of DGGE is less labor intensive than traditional cloning, and it enables rapid estimation of diversity.
Real-Time Polymerase Chain Reaction
Real-time PCR is a method that allows for the enumeration of target gene copy number, a proxy for relative abundance of populations. PCR is performed with fluorescently labeled primers that enable the quantification of amplified PCR products using a CCD camera. The use of primers specific for particular groups of interest enables the comparison of relative population abundances. This method may be especially useful in evaluating the ability of microorganisms identified in clean rooms to survive and grow on Mars.
DNA Microarrays
Printed DNA microarrays consist of glass slides containing thousands of immobilized microscopic samples (probes), each containing oligonucleotide probes targeting the 16S rDNA (or other genes) of specific groups, genera, or species. Fluorescently labeled target DNA is loaded into the wells and any matching strands of DNA will bond to the microarray probes. Positive matches are detected using a fluorescent microscope, a CCD camera, a computer, and the appropriate image analysis software. A single printed slide can contain thousands of probes and can be reused more than 30 times, and the fluorescent dye reporter system can detect 10 attomoles (10–18 mole) of DNA but can only detect ribosomal RNAs that are complementary to probes on the array. Similar experimental strategies can be applied to “DNA chips” generated by lithographic processes that produce high densities of microscopic probes with defined oligonucleotide sequences on a matrix.17
Matrix-Assisted Laser Desorption Ionization–Time-of-Flight Mass Spectrometry
DNA and RNA methods are extremely effective at detecting organisms, are not cost prohibitive, and are becoming more rapid. However, there are situations in which these methods fail to resolve individual strains and populations. In the future, if particular organisms are determined to have the potential to colonize Mars, alternative
methods of detecting target organisms may be needed. Matrix-assisted laser desorption ionization–time-of-flight mass spectrometry (MALDI-TOFMS) is capable of rapidly detecting and identifying microbial species based on the protein profiles of individual species, and it is effective for both vegetative cells and spores. This method is well suited to the clean room and to identifying microbes on spacecraft because of its rapid throughput, high sensitivity, resolving power, and reproducibility. The drawbacks to this technology include cost and the need to create a database that includes protein profiles for all organisms of interest, a requirement that cannot be achieved without the use of pure cultures. Therefore, this method would not be useful until the microbial diversity of assembly rooms and spacecraft is thoroughly assessed.
None of these advanced detection methods is totally acceptable as a comprehensive measure of biomass and diversity, but neither are spore counts an acceptable proxy for biomass nor an indicator for which organisms may remain on spacecraft after cleaning and sterilization. Applying a combination of methods would offer significant improvements over current approaches to estimating the total bioburden and biodiversity of microorganisms on spacecraft and in the clean room. Moreover, many of the methods described above provide very rapid analysis of total bioburden, which could facilitate spacecraft development and assembly.
METHODS FOR REDUCING BIOBURDEN
Current Approaches and Their Limitations
The current approach to mission design is one in which spacecraft are constructed using conventional materials. This approach is in contrast to the approach taken for Viking missions, whereby materials and components had to be qualified to withstand dry-heat sterilization (see Chapter 2). Currently, spacecraft components and parts are not prequalified in terms of any bioburden reduction methods. Consequently, when a particular sterilization or disinfection procedure is selected, a major test is to determine its effect on spacecraft materials. Issues related to the compatibility of these materials are not typically considered in the hardware engineer’s initial model of design and should be determined subsequently. However, hardware can be designed for sterilization, albeit at greater cost, as long as the problem is approached at the onset of design. Incorporating planetary protection into future Mars mission programs at the earliest stages of design would give engineers a selection of effective bioburden reduction tools.18 The design phase of the Viking mission considered planetary protection requirements and supported investigations of heat sterilization to reduce total bioburden.
The scientific basis for planetary protection in the 1970s continues to guide the implementation of planetary protection protocols despite advances in technology and a dramatically expanded view of microbial diversity. The Viking 1 and 2 spacecraft were dry-heat sterilized at 111.7°C for 29.5 and 23.1 h, respectively (DeVincenzi et al., 1998), whereas spacecraft parts and systems were prequalified at 135°C. The sterilization cycle was based on bioburden determination and lethality correction at all temperatures, not just 111.7°C. That is, there are times when the temperatures increase but have not reached the maximum temperature and subsequently decrease after the maximum temperature has been reached. Document NPR 8020.12C (NASA, 2005) details these requirements.
The highest heat-resistance values for spores detected on spacecraft located in their final assembly and testing environments were used to design the sterilization cycle. Previous research for the Viking program showed that naturally occurring bacterial spores on spacecraft exhibit nonlogarithmic dry-heat inactivation (i.e., the survival curves on semilog graphs are polyphasic rather than linear). This situation reflects mixed populations of different spore formers that display different heat survival curves. These results would be expected, because naturally occurring spore populations are composed of organisms with a wide range of heat resistance. At one end of this spectrum were the least-resistant spore populations that were present in the highest numbers but were killed in the least amount of time. At the other end were the most-resistant spore populations that were present in relatively low numbers but survived for longer periods (Favero, 2004; Bond et al., 1970, 1971; Puleo et al., 1975).
Planetary protection has focused on heat-resistant, spore-forming bacteria, but it is just as important to consider other kinds of microbes. For example, the extremely radiation- and desiccation-resistant bacterium Deinococcus radiodurans may be capable of surviving space travel (Clark et al., 1999). Organisms representative of the community found on spacecraft and in clean rooms could be used in the future to validate estimates of bioburden on spacecraft. In addition, the potential for psychrophiles to grow on Mars is at least as likely as that for heat-resistant organisms or thermophiles. There are potential thermal environments on Mars, but the martian near-surface environments (see Chapter 4) are most likely compatible with psychrophiles and psychrotrophs (cold-loving and cold-tolerant organisms, respectively). However, little is known about the sensitivity of these specific organisms to heat treatment.19 It is possible that the high temperatures used to sterilize the Viking are not necessary, but that cannot be determined without knowing which organisms are present in clean rooms and on spacecraft, and their response to different heat treatments. It is possible that lower temperatures may be effective in reducing the viability of bioburden capable of growing on Mars and that more materials will thus be compatible with bioburden reduction.
The approach to spacecraft sterilization employed by the Viking program was to prequalify spacecraft hardware for dry-heat sterilization. Current bioburden reduction programs for spacecraft use no prequalification criteria for the selection of hardware, due to the added cost to the mission of conducting prequalification studies. Consequently, any bioburden reduction techniques beyond physical cleaning and environmental control should be evaluated to determine materials compatibility issues. When Viking-level sterilization is required, various sterilization procedures should be assessed for use to sterilize spacecraft or specific spacecraft components or systems.
Alternative Methods for Bioburden Reduction
The Viking spacecraft was sterilized by using dry heat. The components and parts of the spacecraft were screened and prequalified to withstand the dry-heat sterilization cycle—an effective but expensive approach. Recent spacecraft destined for Mars have faced a less stringent requirement than did the Viking spacecraft, and physical methods of cleaning (e.g., alcohol wipes) have been considered adequate to reduce the bioburden on the spacecraft to specified levels. However, if physical cleaning alone is not adequate to achieve certain bioburden reduction requirements, alternate methods need to be considered for use on spacecraft. A number of such methods are summarized below:
Heat Sterilization
Steam Sterilization. This procedure, one of the most effective sterilization methods, is used universally in industry, research, and hospitals. Its high moisture and heat content, however, is not compatible with spacecraft hardware.
Dry-Heat Sterilization. This effective sterilization method has been used to sterilize spacecraft. Its main disadvantage is that spacecraft must use hardware compatible with dry heat.
Radiation Sterilization
Electron-beam and gamma radiation are used for sterilization in the medical device industry. Both have the advantage of achieving sterilization at relatively low temperatures, and both penetrate packaging materials, have rapid cycles, leave no residuals (except for dead cells), and have a broad spectrum of microbiocidal activity (i.e., they can kill all types of microorganisms, including bacterial spores). Their disadvantages include the high cost of start-up, safety concerns, materials compatibility issues (including the possible production of ozone), and a requirement for specially trained staff. In the context of spacecraft sterilization, electron-beam sterilization would
be a more feasible approach than gamma sterilization, because gamma sterilizers approach the size of a small building, whereas electron-beam sterilizers can fit in a typical laboratory room.
Ambient-Temperature Sterilization
Ethylene Oxide (EO) Sterilization. This method was developed in the 1960s to sterilize instruments and medical devices that were heat labile and could not be steam sterilized. EO is a very effective sterilization system that can sterilize at ambient temperatures, is penetrating, and has a broad microbiocidal range of activity. However, EO is toxic, and there are significant safety concerns. It is used primarily by the medical device industry but much less in hospitals in recent years (Rutala, 1987; Favero and Bond, 2001). The Soviets proposed using EO and methyl bromide for terminal sterilization of their Mars 2 and 3 landers (Hall, 1972; Vashkov et al., 1971).
Newer Methods. A number of new ambient-temperature sterilization systems have been developed in the past decade. These include:
Hydrogen Peroxide Vapor. Hydrogen peroxide in the vapor phase is used commercially for the sterilization of glove boxes and pass-throughs, as well as for decontamination of laboratory incubators and laminar flow cabinets. Hydrogen peroxide is an oxidizing agent that accomplishes sterilization by oxidation of key cellular components. It is a bactericidal, virucidal, sporicidal, and fungicidal agent, even at low concentration and temperature.
Hydrogen Peroxide Gas Plasma. This process uses hydrogen peroxide in the vapor phase and low-temperature gas plasma for rapid inactivation of microorganisms and the removal of harmful residues. Gas plasmas are highly ionized gases composed of ions electrons, and neutral particles. A solution of hydrogen peroxide and water is vaporized and allowed to surround and interact with the devices to be sterilized. Applying a strong electrical field then creates plasma. The plasma converts hydrogen peroxide into water and oxygen, and there is thus no hydrogen peroxide residual left on sterilized loads and no hydrogen peroxide released into the environment nor exposure of technical staff. No aeration time is required, and the sterilized materials can be used immediately. This method is relatively rapid; for example, a load of surgical instruments can be sterilized in less than 1 h (Jacobs and Lin, 2001).
Ozone. Ozone is produced by passing dry air or oxygen between high-voltage electrodes that produce a coronal discharge, or by UV irradiation of air or oxygen. Because of its instability, the gas has to be produced at the point of use. Ozone is triatomic oxygen with one loosely bonded O atom; this readily attaches to other molecules and makes ozone a powerful oxidizing agent. It is a bactericidal, virucidal, sporicidal, and fungicidal agent and cycles between 4 to 5 h. Currently, a low-temperature sterilizer is sold in the United States and Canada. The sterilizer has been approved by the U.S. Food and Drug Administration for sale and is used to sterilize medical instruments and devices (Josyln, 2001).
Chlorine Dioxide. Chlorine dioxide in its gaseous form (CD) is approved for use as a sterilant by the U.S. Environmental Protection Agency. Its sporicidal effects are well documented and can be compared to those of vapor-phase hydrogen peroxide and hydrogen peroxide gas plasma. CD is not prepared by the vaporization of solution but is actually generated at the point of use. This is accomplished by using a method in which solid sodium chlorite contained in small plastic cartridges is contacted by a gas mixture of 2 percent chlorine and 98 percent nitrogen. The reaction products are CD and sodium chloride. CD is used for the sterilization of medical products and glove boxes and for the decontamination of small areas.
Liquid Chemical Disinfection
A number of liquid chemical germicides formulated as sterilants and high-level disinfectants are used for reprocessing of instruments and medical devices. They include formulations containing glutaraldehyde, orthophthalaldehyde, hydrogen peroxide, peracetic acid, and combinations of these. They are effective biocides and can
be used at low temperatures. In the context of spacecraft sterilization, the main disadvantages are materials incompatibility with oxidative formulations and the fact that they are in liquid form.
Encapsulated Bioburden
Levels of microbial contamination within solid materials, encapsulated components, occluded surfaces, lubricants, or surface finishes of Mars spacecraft have always been of concern. However, there is no published research investigating the actual bioburden contained within materials or its potential to survive if released (e.g., during a crash) because of the difficulty in assaying these regions (see Chapter 2). Furthermore, current estimates in use (see NPR 8020.12C; NASA, 2005, pp. 39-41) were developed for spacecraft materials and components 30 years ago and have not been updated.
Historically, mission engineers have worked hard to reduce accessible bioburden (e.g., on the surface of the spacecraft) to compensate for the poorly known encapsulated bioburden in the total bioburden estimate. Still, methods have not been developed for determining concentrations of bioburden encapsulated in spacecraft components, and little attention has been paid to improving understanding of this potential microbial reservoir. New methods should measure the true level of viable contamination in encapsulated and mated surfaces of contemporary and future spacecraft materials, and models should be developed for extrapolating bioburden levels to components that cannot be directly assayed.
SUMMARY
The use of swab cultures to test for spores (see Appendix C) has been a valuable approach for determining bioburdens on spacecraft, but it is not without limitations. Recent advances in microbial ecology argue that spores are an imperfect proxy for estimating microbial bioburden, and culturing methods do not provide a representative estimation of total bioburden on pre-launch spacecraft or in assembly areas. It is of great importance to know both the amounts of bioburden remaining on a spacecraft and the kinds of organisms that are present. As described in the section above titled “Biomarker Methods,” the ATP and LAL assays are promising emerging technologies that could offer rapid estimates of bioburden without the need for laboratory culturing. In addition, when the organisms present have been identified, cleaning and sterilization can be focused more specifically on organisms of importance, that is, those that have the potential to survive flight to Mars and then grow in the martian environment, rather than on spores per se. The committee recognizes that, given rapidly changing knowledge of potential special regions on Mars (see Chapter 4), no class of microorganisms can currently be identified with complete confidence as being that of greatest importance for preventing the forward contamination of Mars. This situation emphasizes the need to understand the full diversity of organisms on spacecraft and to adapt bioburden assessment and reduction methods to new planetary and biological knowledge. A number of rapid methods exist that will enable for more direct estimation and identification of viable cells, as well as many new methods of bioburden reduction and sterilization that could allow mission engineers a suite of materials with which to design spacecraft. The next step is to assess these methods with respect to NASA planetary protection guidelines, evaluate their efficacy in comparison to current spore and culture methods, determine the optimal methodological approach for updating planetary protection requirements, and develop a defined standard certification process that will allow engineers and planetary protection officers to implement these advances as early in the mission development process as possible.
Ultimately, the use of advanced detection methods to identify the potential bioburden may lead to bioburden reduction techniques that are targeted at groups of organisms of specific concern for planetary protection. NASA has been investigating some of these technologies, including LAL and ATP analysis (e.g., Dickinson et al., 2004a,b; Venkateswaran et al., 2001, 2003), and research on bioburden reduction methods has also been proceeding.20 However, additional research on these technologies is needed. At the very least, such research will provide the data needed to determine what future mix of methods is appropriate to minimize the forward contamination of Mars.
REFERENCES
Altekruse, S.F., F. Elvinger, Y. Wang, and K. Ye. 2003. A model to estimate the optimal sample size for microbiological surveys. Appl. Environ. Microbiol. 69: 6174-6178.
Amann, R.I., W. Ludwig, and K.-H. Schleifer. 1995. Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiol. Rev. 59(1): 143-169.
Baker, A. 2001. Space Hardware Microbial Contamination Workshops 1 and 2. A Report from Workshops at Moffett Field Calif. (December 1999) and Golden Colo. (June 2001). Contract No. A63616D(SXS). NASA Ames Research Center, Moffett Field, Calif.
Baker, G.C., and D.A. Cowan. 2004. 16S rDNA primers and the unbiased assessment of thermophile diversity. Biochem. Soc. Trans. 32: 218-221.
Béjà, O., E.N. Spudich, J.L. Spudich, M. Leclerc, and E.F. DeLong. 2001. Proteorhodopsin phototrophy in the ocean. Nature 411: 786-789.
Bond, W.W., M.S. Favero, N.J. Petersen, and J.H. Marshall. 1970. Dry-heat inactivation kinetics of naturally occurring spore populations. Appl. Microbiol. 20: 573-578.
Bond, W.W., M.S. Favero, N.J. Petersen, and J.H. Marshall. 1971. Relative frequency distribution of D125C values for spore isolates from the Mariner-Mars 1969 spacecraft. Appl. Microbiol. 21: 832-836.
Brochier, C., and H. Philippe. 2002. Phylogeny: A non-hyperthermophilic ancestor for bacteria. Nature 417: 244.
Carpenter, E.J., S. Lin, and D.G. Capone. 2000. Bacterial activity in South Pole snow. Appl. Environ. Microbiol. 66: 4514-4517.
Clark, B.C., A.L. Baker, A.F. Cheng, S.J. Clemett, D. McKay, H.Y. McSween, C.M. Pieters, P. Thomas, and M. Zolensky. 1999. Survival of life on asteroids, comets and other small bodies. Orig. Life Evol. Biosph. 29: 521-545.
Connon, S.A., and S.J. Giovannoni. 2002. High-throughput methods for culturing microorganisms in very-low-nutrient media yield diverse new marine isolates. Appl. Environ. Microbiol. 68: 3878.
DeLong, E.F., K.Y. Wu, B.B. Prezelin, and R.V. Jovine. 1994. High abundance of archaea in Antarctic marine picoplankton. Nature 371: 695-697.
DeVincenzi, D.L., M.S. Race, and H.P. Klein. 1998. Planetary protection, sample return missions and Mars exploration: History, status, and future needs. J. Geophys. Res. 103: 28577-28585.
Dickinson, D.N., M.T. La Duc, W.E. Haskins, I. Gornushkin, J.D. Winefordner, D.H. Powell, and K. Venkateswaran. 2004a. Species differentiation of a diverse suite of Bacillus spores using mass spectrometry based protein profiling. Appl. Environ. Microbiol. 70: 475-482.
Dickinson, D.N., M.T. La Duc, M. Satomi, J.D. Winefordner, D.H. Powell, and K. Venkateswaran. 2004b. MALDI-TOFMS compared with other polyphasic taxonomy approaches for the identification and classification of Bacillus pumilus spores. J. Microbiol. Methods 58(1): 1-12.
Doolittle, W.F., Y. Boucher, C.L. Nesbø, C.J. Douady, J.O. Andersson, and A.J. Roger. 2003. How big is the iceberg of which organellar genes in nuclear genomes are but the tip? Phil. Trans. R. Soc. Lond. B 358: 39-58.
Favero, M.S. 2004. Naturally occurring microorganisms and their resistance to physical and chemical agents. Pp. 1-14 in Disinfection, Sterilization and Antisepsis: Principles, Practices, Challenges, and New Research, W.A. Rutala, ed. Association for Professionals in Infection Control and Epidemiology, Inc., Washington, D.C.
Favero, M., and W. Bond. 2001. Chemical disinfection of medical surgical material. Pp. 881-917 in Disinfection, Sterilization and Preservation, 5th edition, S.S. Block, ed. Lippincott, Williams, and Wilkens, Philadelphia, Pa.
Hall, L.B. 1972. Memorandum on Soviet Planetary Quarantine Sterilization of Mars 1 and 2. NASA, Washington, D.C., June 1.
Hughes, J.B., J.J. Hellmann, T.H. Ricketts, and B.J.M. Bohannan. 2001. Counting the uncountable: Statistical approaches to estimating microbial diversity. Appl. Envir. Microbiol. 67: 4399-4406.
Jacobs, P., and S. Lin. 2001. Sterilization processes utilizing low-temperature plasma. Pp. 747-763 in Disinfection, Sterilization and Preservation, 5th edition, S.S. Block, ed. Lippincott, Williams, and Wilkens, Philadelphia, Pa.
Josyln, L.J. 2001. Gaseous chemical sterilization. Pp. 337-357 in Disinfection, Sterilization and Preservation, 5th edition, S.S. Block, ed. Lippincott, Williams, and Wilkens, Philadelphia, Pa.
Kaeberlein, T., K. Lewis, and S.S. Epstein. 2002. Isolating “uncultivable” microorganisms in pure culture in a simulated natural environment. Science 296: 1127-1129.
Kminek, G., and J.D. Rummel, eds. 2005. Planetary Protection Workshop on Sterilization Technologies. ESA WPP-243. ISSN 1022-6656, June.
Mullis, K.B., and F. Faloona. 1987. Specific analysis of DNA in vitro via a polymerase-catalysed chain reaction. Methods Enzymol. 155: 335-350.
NASA (National Aeronautics and Space Administration). 2005. Planetary Protection Provisions for Robotic Extraterrestrial Missions. NPR 8020.12C. NASA, Washington, D.C. Available at <planetaryprotection.nasa.gov>.
NRC (National Research Council). 2002. The Quarantine and Certification of Martian Samples. National Academy Press, Washington, D.C.
Pace, N.R., D.A. Stahl, D.J. Lane, and G.J. Olsen. 1986. The analysis of natural microbial populations by ribosomal RNA sequences. Pp. 1-55 in Current Microbial Ecology, K.C. Marshall, ed. Plenum Press, New York.
Priscu, J.C., C.H. Fritsen, E.E. Adams, S.J. Giovannoni, H.W. Paerl, C.P. McKay, P.T. Doran, and J.L. Pinckney. 1998. Perennial Antarctic lake ice: An oasis for life in a polar desert. Science 280: 2095-2098.
Priscu, J.C., E.E. Adams, W.B. Lyons, M.A. Voytek, D.W. Mogk, R.L. Brown, C.P. McKay, C.D. Takacs, K.A. Welch, C.F. Wolf, J.D. Kirstein, and R. Avci. 1999. Geomicrobiology of sub-glacial ice above Vostok Station. Science 286: 2141-2144.
Puleo, J.R., M.S. Favero, G.S. Oxborrow, and C.M. Herring. 1975. Methods for collecting naturally occurring airborne bacterial spores for determining their thermal resistance. Appl. Microbiol. 30: 786-790.
Reysenbach, A.L., G.S. Wickham, and N.R. Pace. 1994. Phylogenetic analysis of the hyperthermophilic pink filament community in Octopus Spring, Yellowstone National Park. Appl. Environ. Microbiol. 60: 2113-2119.
Rutala, W.A. 1987. Disinfection, sterilization and waste disposal. Pp. 257-282 in Prevention and Control of Nosocomial Infections, R.P. Wenzel, ed. Williams and Wilkins, Baltimore, Md.
Tringe, S.G., C. von Mering, A. Kobayashi, A.A. Salamov, K. Chen, H.W. Chang, M. Podar, J.M. Short, E.J. Mathur, J.C. Detter, P. Bork, P. Hugenholtz, and E.M. Rubin. 2005. Comparative metagenomics of microbial communities. Science 308: 554-557.
Vashkov, V.I., N.V. Rashkova, and G.V. Shcheglova. 1971. Planetary Quarantine Principles, Methods and Problems, L.B. Hall, ed. Gordon and Breach, New York.
Venkateswaran, K., M. Satomi, S. Chung, R. Kern, R. Koukol, C. Basic, and D. White. 2001. Molecular microbial diversity of spacecraft assembly facility. Syst. Appl. Microbiol. 24: 311-320.
Venkateswaran, K., N. Hattori, M.T. La Duc, and R. Kern. 2003. ATP as a biomarker of viable microorganisms in clean-room facilities. J. Microbiol. Methods 52: 367-377.
Ward, D.M., M.M. Bateson, R. Weller, and A.L. Ruff-Roberts. 1992. Ribosomal RNA analysis in microorganisms as they occur in nature. Adv. Microbiol. Ecol. 12: 219-286.
Ward, D.M., M.J. Ferris, S.C. Nold, and M.M. Bateson. 1998. A natural view of microbial biodiversity within hot spring cyanobacterial mat communities. Microbiol. Mol. Biol. Rev. 62: 1353-1370.
Webster, G., C.J. Newberry, J.C. Fry, and A.J. Weightman. 2003. Assessment of bacterial community structure in the deep sub-seafloor biosphere by 16S rDNA-based techniques: A cautionary tale. J. Microbiol. Methods 55: 155-164.
Woese, C.R., G.J. Olsen, M. Ibba, and D. Soll. 2000. Aminoacyl-tRNA synthetases, the genetic code, and the evolutionary process. Microbiol. Mol. Biol. Rev. 64: 202-236.














