
6
Action Agenda for the Pharmaceutical, Medical Device, and Health Information Technology Industries
CHAPTER SUMMARY
Pharmaceutical, medical device, and health information technology companies represent the chief drug product-related industry sectors of the medication-use system. If designed well, their products can improve the health and well-being of consumers, advance medical science, and enhance clinical practice. As with other components of the medication-use system, however, certain features of design processes and communication mechanisms warrant significant improvement to better serve the health needs of consumers and the practice needs of providers and, most important, prevent medication errors. This chapter provides an action agenda for the pharmaceutical, medical device, and health information technology industries that, in collaboration with appropriate government agencies, can begin to address key problems that affect the safety and quality of the medication-use system.
This chapter presents an action agenda first for the pharmaceutical industry, and then for the medical device and health information technology industries.
PHARMACEUTICAL INDUSTRY
As discussed in Chapter 2, improving the safety of medication use requires improving the quality of information generated by industry and
other researchers regarding drug products and their use in clinical practice. Also required are improvements in the way drug information is presented to providers and consumers through labeling and packaging since such materials have a direct effect on medication errors and adverse drug events (ADEs). This section reviews key problems involved in the generation and presentation of information that should be addressed by the pharmaceutical industry and the Food and Drug Administration (FDA).
Generation of Information
Current methods for generating information about medications are insufficient to meet the changing medical needs of the population, particularly given expected increases in the numbers of elderly people with multiple chronic conditions (IOM, 2000, 2001). While a comprehensive review of the drug research and development process and recommendations for its redesign are beyond the scope of this report, certain key aspects of information generation germane to medication safety merit discussion here.
Clinical Data
Determining that a medication error has occurred presumes that the correct dose of a drug for a given patient at a particular time is known, and that the indication for that drug is correct relative to alternative approaches to treatment. Unfortunately, this fundamental presumption is too often unwarranted.
The benefits of drugs can be categorized as improvement in longevity, improvement or stabilization of symptoms (improvement in quality of life), prevention of adverse events, or reduction in the costs of other medical interventions. To determine whether the benefits of a drug outweigh its risks, both the benefits and the risks must be measured in the population to whom the drug will be given for a relevant period of time (Yusuf et al., 1984; Prentice, 1989; Fleming and DeMets, 1996). Ideally, after these measurements have been made, individuals should be informed about both the benefits that can be expected and the potential risks. Since the benefits and risks are measured with different metrics, it is important to recognize that in the end, a subjective judgment regarding the balance of benefit and risk is necessary, since a ratio cannot be calculated (CERTS, 2003; Tsintis and La Mache, 2004; Edwards et al., 2005).
Over the past several decades, our understanding of therapeutic evaluation has advanced significantly. Nonetheless, the balance of benefit and risk of a drug compared with alternative treatments usually is not known. A variety of examples can be used to illustrate this point. Hormone replacement therapy, for instance, was once the most prevalent drug prescription
globally, with its most common indication for use being prevention of cardiovascular disease in postmenopausal women. Many years after the therapy was marketed, however, both the HERS (Heart and Estrogen/ progestin Replacement study) Trial and the Women’s Health Initiative demonstrated its association with an excess of vascular events (Hulley et al., 1998; WHI Steering Committee, 2004). As a second example, the COX-2 (cyclo-oxygenase-2) inhibitors were expected to be a safer alternative to nonsteroidal anti-inflammatory drugs (NSAIDs). Vioxx and Bextra (among others), however, were removed from the market after the FDA Advisory Committee meeting in February 2005. Some experts believed the true balance of benefit and risk was not known for any of the COX-2 inhibitors (Psaty and Furberg, 2005). Perhaps even more startling, it was pointed out at the hearing that the same could be said for the traditional NSAIDs, which had been considered safe enough to sell over the counter. As a final example, a variety of antihypertension drugs have been developed and marketed as superior to the older, generic drugs used for this indication. However, when the National Institutes of Health (NIH) funded a pragamatic clinical trial involving more than 40,000 patients, it was found that the newer drugs provided no greater protection against stroke, heart failure, or death than the generic drug chlorthalidone (ALLHAT, 2002). Given that these examples involve some of the most commonly used and intensively studied drugs, there is uncertainty that drugs receiving less attention are better characterized. Since the only way to be confident about the balance of benefit and risk is empirical measurement, this information is lacking for most prescriptions that are written, especially those for chronically administered drugs.
The above issues are magnified in certain populations that bear much of the risk of drug prescription and administration:
-
The majority of prescriptions written for children are off label,1 with no empirical demonstration of safety and efficacy (Roberts et al., 2003). The Best Pharmaceuticals Act for Children has stimulated a major increase in clinical trials in children, but the legacy of sparse evidence remains substantial, and few of these trials have provided definitive information about indications and doses for the drugs involved. Pediatric oncology has been at the forefront in terms of enrolling a significant number of children in trials and could possibly be used as a model for other drug categories.
-
Almost nothing is known about the balance of benefit and risk in the fastest-growing segment of the population—those over age 80. These patients have only recently been enrolled in clinical trials (Alexander and
-
Peterson, 2003). Given the major changes in organ physiology that occur in the elderly, people over age 80 have unique characteristics related to drug metabolism and pharmacodynamics.
-
Patients with renal dysfunction represent a large and growing population requiring more comprehensive studies. Over 10 percent of the population now has a creatinine clearance below 60 milligrams per deciliter (mg/ dl), indicating moderate or worse renal function (Reddan et al., 2003). The fact that many drugs are excreted by the kidneys raises obvious issues about dosing as a function of renal clearance. In addition, however, almost everyone with impaired renal function is either elderly or chronically ill, so that a simple mathematical calculation of clearance will not yield an accurate estimate of the balance of the benefit and risk of a drug at a particular dose.
-
Patients with multiple comorbidities are typically excluded from premarketing clinical trials, yet many of the major problems involving drug toxicity have occurred in those taking multiple medications because of multiple diseases (Gurwitz, 2004). Drug interactions and additive toxic effects are common, and while they can be anticipated based on studies in other populations, the cumulative effects of multiple drugs cannot be predicted accurately without empirical study.
-
Drugs for patients with psychiatric illnesses are particularly controversial. Most studies in these populations have been small and incapable of providing pragmatic, comparative information (March et al., 2005). Recent studies funded by the National Institute of Mental Health (NIMH) have fueled concern about the basic knowledge base for treatment of depression, manic-depressive illness, and schizophrenia.
The theory of clinical pharmacology has not been well supported by the academic community or the NIH. In particular, the characteristics of patients that determine the manner in which the pharmacokinetics and pharmacodynamics of drugs will be manifest are poorly understood and often overlooked (Fitzgerald, 2005). As a result of marketing considerations, the industry has tended to attempt to develop drugs that are given once a day and intravenous formulations that have fixed doses for ease of administration. Thus recommended doses are not specifically tailored to the needs of the individual patient.
The field of clinical pharmacology needs to be invigorated. Few training programs in this area exist today in the United States, prompting the Institute of Medicine (IOM) to initiate its own national course in drug development. With the anticipated availability of pharmacogenomic data, a cadre of experts will be needed to evaluate the modifiers of drug concentration and activity.
A large increase in the number of patients for whom clinical outcomes are measured is needed to elucidate the proper dosing of drugs in individu-
als (Califf and DeMets, 2002a,b). The case of anticoagulant drugs illustrates this need. These drugs are characterized by a complex balance of benefit (prevention of blood clots) and risk (bleeding) (Schünemann et al., 2004). Aspirin has been available for over 100 years and heparin for over 50 years. Yet the best dose of each for preventing arterial thrombosis remains controversial. Multiple new drugs, including direct antithrombins, low-molecular-weight heparins, P2Y12 inhibitors, and glycoprotein IIb/IIIa inhibitors, have been developed in the past two decades and have been demonstrated to provide a net balance of benefit on average in patients entered into clinical trials. Yet little is known about the appropriate dose of these agents in children, the very elderly, and patients with renal impairment. The adjusted dosing regimens for heparin and coumadin, each of which has been marketed for more than four decades, were delineated relatively recently after thousands of patients had been entered into clinical trials that included outcome measurement to determine the degree of anticoagulation with each agent that led to prevention of thrombosis without unacceptable bleeding.
Once a drug is on the market, the expansion to new indications continues throughout its life cycle. Most postmarket studies funded by industry are intended specifically to expand the market for a drug, and such studies are usually not undertaken unless the calculated probabilities indicate that the study will yield a positive financial return (Tunis et al., 2003). Direct comparisons of a drug with an alternative drug or other treatment rarely meet this financial test because there is too great a risk of finding that there is no difference or that the competing treatment is better.
An increasing number of reports over the past several decades have called for a marked increase in pragmatic clinical trials that answer questions relevant to clinical practice (Crowley et al., 2004). A new approach is needed that includes industry participation, but also independent oversight to stimulate more such trials. Lacking the results of such trials, neither prescriber nor patient can know what treatment plan is best.
A critical issue is where to draw the line between the premarketing development phase and the point at which the drug is allowed on the market. Scientifically, the gaining of knowledge about a drug should be a continuous process in which new information is used to refine understanding of the drug’s uses, benefits, and risks at particular doses in particular patients. In actuality, however, the development of scientific knowledge about drugs is quite discontinuous, and the process is dependent on clearing a series of hurdles with defined criteria. In particular, tremendous effort and expense go into the New Drug Application (NDA) required to obtain initial approval for marketing (see Chapter 2). Ideally, at the time of initial marketing, the balance of a drug’s benefit and risk would be known so that the label for its use could be clear. In reality, however, the costs of drug
development and the length of time required to develop this information, particularly for drugs used to treat chronic diseases, make this impractical (Wood, 1999). Accordingly, an increasing number of experts advocate greater use of provisional drug approval to allow access to new therapies, with the requirement for long-term studies in heterogeneous populations after the drug is on the market.
Key Finding
Providers require better-quality information about medications and their effects if they are to meet the needs of their patients. For example, to facilitate safety and quality in the medication-use system, there is a need for more comprehensive benefit/risk information, clinical outcome data, and effectiveness data. Open access to such data is important not only for developing clinical understanding, but also for populating clinical knowledge and decision-support systems.
Disclosure of Clinical Data
The current state of disclosure of the findings of clinical studies is inadequate to support safety and quality in medication use, although the situation is improving. Of the multitude of drug products on the market, mandatory registration of clinical trial data applies only to those used for serious and life-threatening conditions. Moreover, requirements do not include disclosure of the results of Phase III trials—a key tool for educating patients and health care providers about drug benefits and risks—while results for only selected Phase IV studies are voluntarily included. Registration of clinical trial data is done at http://www.ClinicalTrials.gov, a federal government website operated through the NIH.
Until recently, information about other drugs was scattered over several database systems operated by different entities (IOM, 2006). However, the International Consortium of Medical Journal Editors now requires registration of clinical trial data at ClinicalTrials.gov as a condition for journal publication. Until this new requirement and its associated public scrutiny, the overall quality of the information reported was low in terms of usefulness, comprehensiveness, and standardization. A recent report from ClinicalTrials.gov documents improvement in the quality of reporting, but considerable progress is still needed (Zarin et al., 2005).
Providers and others have historically relied on medical journals publications to obtain results of different types of clinical studies and compensate for the limitations of repositories. While medical journals will remain important to the dissemination of objective clinical trial and practice infor-
mation, a more flexible system is needed to ensure that the demands of the public for complete disclosure of drug benefits and risks are met.
The recently released IOM workshop report Clinical Trial Registration: Developing a National Registry to Improve Public Access and Reliability states that the best course of action to build the nation’s repository of information about therapeutics and improve the quality of that information may be a broad expansion of the ClinicalTrials.gov database (IOM, 2006). A single national registry populated with information generated through clinical studies of all drug products would be a critically important resource for all stakeholders in the medication-use system. Each stakeholder group (e.g., patients, providers, researchers, medical journal editors, pharmaceutical companies, health insurers, information technology vendors, and regulators) has different needs and uses for the information contained in such a registry (see Box 6-1) (IOM, 2006). For optimal functioning, the registry should serve several purposes:
-
List and track the status of ongoing clinical trials.
-
Provide information on patient recruitment.
-
Report results of clinical trials, including late Phase II, Phase III, and postmarketing studies; “head-to-head” comparisons of drugs; comparisons of drugs and alternative treatments; and effectiveness studies.
Full disclosure of the results of all clinical trials and postmarket studies in a national registry is particularly important to fill the current knowledge gaps that affect clinical practice, patient self-management, and medication safety. The distortion of information that results from the design of post-marketing studies has been described above. Well beyond this distortion, however, positive study results are much more likely to be published than negative results. This publication bias yields an incomplete picture of the drug characteristics that must be known for more accurate medication use and error prevention, and can therefore have a detrimental effect on patients. This has clearly been a major issue with COX-2 inhibitors and NSAIDs (see the discussion above). Thus all clinical trial results must be disseminated in a comprehensive, objective, and unbiased manner (IOM, 2006). Clear communication of risk information (not just benefits) is essential to preventing errors and potential adverse reactions.
The same holds true for other study results that should be incorporated into a national registry (i.e., postmarket, comparison, and effectiveness studies). Postmarket studies are especially important in relaying new safety information revealed as a drug is used in clinical practice. Comparative and effectiveness studies contribute further to understanding a drug’s characteristics and therapeutic value. Given the proposed national registry, patients, providers, and others would not have to search multiple database systems for these study results but could easily maneuver within one comprehensive
|
BOX 6-1 Diverse Expectations and Perceived Needs for a National Registry of Clinical Trial Data The public and various entities within the medical community have different expectations and perceived needs regarding a public registry of clinical trial data:
|
system to learn more about a particular medication. The registry would also facilitate more efficient use of clinical data for such purposes as cross-referencing patients’ response to a drug during clinical trials and their response in clinical practice, patients’ response to one drug and their response to another, and patients’ response to a drug and their response to other treatment options. Also, a drug’s overall effectiveness in terms of patient outcomes is becoming a valuable measure of therapeutic success.
Further discussion and recommendations concerning such a national
registry will be provided in the forthcoming report of the IOM Committee on the Assessment of the U.S. Drug Safety System.
Communication of Information
Drug information is communicated to providers and consumers through labeling and packaging, marketing practices, and advertisements. Poorly designed materials and inadequate representation of drug benefits and risks has led to errors across the medication-use continuum, such as inappropriate prescribing, confusion among products affecting dispensing and administration, and compromised ability to monitor a drug’s effects adequately. This section addresses these issues.
Recommendation 4: Enhancing the safety and quality of the medication-use process and reducing errors requires improved methods for labeling drug products and communicating medication information to providers and consumers. For such improvements to occur, materials should be designed according to designated standards to meet the needs of the end user. Industry, the Agency for Healthcare Research and Quality (AHRQ), the FDA, and others as appropriate (e.g., U.S. Pharmacopeia, Institute for Safe Medication Practices) should work together to undertake the following actions to address labeling, packaging, and the distribution of free samples:
-
The FDA should develop two guidance documents for industry: one for drug naming and another for labeling and packaging. The FDA and industry should collaborate to develop (1) a common drug nomenclature that standardizes abbreviations, acronyms, and terms to the extent possible, and (2) methods of applying failure modes and effects analysis to labeling and packaging.
-
Additional study of optimum designs for all drug labeling and information sheets to reflect human and cognitive factors should be undertaken. Methods for testing and measuring the effects of these materials on providers and consumers should also be established, including methods for field testing of the materials. The FDA, the National Library of Medicine (NLM), and industry should work with consumer and patient safety organizations to improve the nomenclature used in consumer materials.
-
The FDA, the pharmaceutical industry, and other stakeholders should collaborate to develop a strategy for expanding unit-of-use packaging for consumers to new therapeutic areas. Studies should be undertaken to evaluate different unit-of-use packaging
-
and design approaches that will best support various consumer groups in their medication self-management.
-
AHRQ should fund studies to evaluate the impact of free samples on overall patient safety, provider prescribing practices, and consumer behavior (e.g., adherence to the medication regimen), as well as alternative methods of distribution that can improve safety, quality, and effectiveness.
Naming, Labeling, and Packaging
Drug names that look or sound alike increase the risk of medication errors (Cohen, 2000). Confusion over the similarity of drug names for prescription, generic, and over-the-counter (OTC) products accounts for up to 25 percent of all errors reported to the U.S. Pharmacopeia (USP) (NCC MERP, 2001). Abbreviations, acronyms, certain dose designations, and other symbols used for labeling also have caused a number of errors (FDA, 2005b). Even the layout and presentation of drug information on the drug container or package label can be visually confusing, particularly when designed for the marketplace instead of clinical practice. From January 2000 to March 2004, close to 32,000 reports were submitted to USP’s MedMarx Reporting System that linked errors to look-alike or sound-alike drug names (Santell and Camp, 2004). The Joint Commission on Accreditation of Healthcare Organizations’ (JCAHO) National Patient Safety Goals reference several look-alike/sound-alike generic drug names that have contributed to 9 of 10 serious medication errors in the hospital setting (JCAHO, 2006). And labeling and packaging issues were cited as the cause of 33 percent of errors, including 30 percent of fatalities, reported to the USP–Institute for Safe Medication Practices (ISMP) Medication Error Reporting Program (MERP) database (USP, 1998). Box 6-2 outlines the major problems in drug naming, labeling, and packaging that contribute to medication errors. Addressing these problems requires understanding the processes and requirements involved in naming, labeling, and packaging drug products.
Drug naming is a complex process. Each drug has multiple names assigned by different organizations for different purposes (Berman, 2004). The chemical name is assigned by the International Union of Pure and Applied Chemistry and identifies molecular structure. It serves the needs of scientific researchers. The nonproprietary or generic name is assigned by the United States Adopted Name Council (USAN) using a series of guidelines to ensure uniformity and safety.2 These guidelines require that the
|
BOX 6-2 Examples of Major Naming, Labeling, and Packaging Problems
|
|
appropriate name stem from a standardized list and be incorporated into the generic name to give clinicians some indication of the chemical and/or therapeutic characteristics of the drug (USAN, 2005). The official title of a medication is determined by the USP Expert Committee on Nomenclature using the nonproprietary name plus dosage, formulation, and route of administration.3 The proprietary or brand name is created by pharmaceutical companies to facilitate brand recognition and promote brand loyalty (Berman, 2004). The brand name for a drug may be different among countries, and drugs marketed by more than one pharmaceutical company may have more than one brand name (Hoffman and Proulx, 2003). Nevertheless, all drugs are promoted and marketed in the United States to providers and consumers under their brand name, although many providers and payers prefer to use the nonproprietary name.
Mixups resulting in medication errors can occur with either generic or brand names. In cases where the generic names are similar, the brand name can be used to differentiate products. Brand names are almost always easier to pronounce, spell, and remember than generic names (Cohen, 2002). The reverse is also true: similar brand names can be differentiated by using or including the generic name. In very rare cases the generic and brand names are similar for a particular drug. Thus, using both the generic and brand name is one of the easiest means of decreasing the likelihood of medication errors due to name confusion (Cohen, 2000; Hoffman and Proulx, 2003; Berman, 2004). It is particularly important to use both names when a drug has been involved in a name mix-up that led to an adverse event.
Because generic names are assigned from a limited list of word stems, there are a limited number of ways to represent a drug, increasing the likelihood that a name similar to that of another drug will be selected. Analysis is usually based on peer review. In contrast, brand names are cleared and trademarked4 through the U.S. Patent and Trademark Office. In addition, brand names are analyzed by pharmaceutical companies themselves and the FDA (after submission of a regulatory approval application) using failure modes and effects analysis (FMEA). FMEA is a systematic approach used to identify and prevent product and process problems before they occur (IOM, 2004). With FMEA, a topic (e.g., drug name) is analyzed using a flow diagram of each step involved in the processes and subprocesses affecting the end user (e.g., using the drug in the clinical setting). A failure analysis is conducted to identify all possible points (i.e., modes) and causes of an error, and the severity and probability of each error. The final evaluation determines which modes to eliminate, control, or accept, and actions that can be taken to eliminate or reduce the error. If the company decides that the benefit/risk of the drug name is acceptable, it obtains a trademark for that name and includes the name in its application for regulatory approval.
From that point, the FDA’s Division of Medication Errors and Technical Support (DMETS) reviews brand names for prescription and certain OTC drugs to determine the potential for naming-related medication errors (FDA, 2005b).5 The FDA’s FMEA review includes several evaluations. First, FDA staff undertake a handwriting and verbal analysis (through internal testing) to determine the degree of confusion in visual appearance or pronunciation between the brand name and the names of other products on the market. Second, the FDA uses a computer software tool, the Phonetic Orthographic Computer Analysis (POCA) program, to identify names with similar spelling, letter strings, or syllables. Third, additional risk information (e.g., overlapping strengths, dosage forms, dosing recommendations, indications for use) and container labeling/packaging are evaluated (but not using FMEA) to identify areas of potential confusion and improvement (FDA, 2005a). When errors occur after approval, DMETS has limited ability to require manufacturers to make name or labeling changes.
Generally, FMEA is not used by either pharmaceutical companies or the FDA to evaluate external labeling and packaging. As a result, many of the problems listed in Box 6-2 (e.g., cluttered labeling, small font, serif
typeface, lack of background contrast, inadequate prominence of reminders and warnings, overemphasis on company logos and trade dress) continue to have a direct effect on the readability and comprehensibility of product labels, and hence on rates of medication errors (Cohen, 2000; Berman, 2004). An important example is the redesign of the labels for potassium chloride concentrate. The older bottle label was poorly designed and looked very similar to the label for dextrose. Many mix-ups between potassium chloride concentrate and dextrose occurred that resulted in fatalities (Cohen, 2000). The labeling was redesigned to eliminate clutter and emphasize the drug name, concentration, and warnings. Another example is lidocaine hydrochloride, used for cardiac arrhythmias, which is administered via a loading dose followed by a continuous intravenous infusion (Berman, 2004). The prefilled 100 mg syringes (most common loading dose) were frequently confused with the prefilled 1 or 2 g syringes (for injection into a bag of 5 percent dextrose) because of similarities in appearance and design.
When problems with labeling and packaging do occur, they are usually addressed on a case-by-case basis, if at all. An exception to this policy is related to the labeling for small vials and injection syringes and for high-alert medications, which are particularly susceptible to errors due to such problems (Cohen, 2000). In 1994, a USP–FDA advisory panel made a number of recommendations for improving labeling and safety for injectable medications (http:// www.nccmerp.org/council/council1997-09-16.html). Because USP functions as a standards organization for medication safety, most companies are complying with these recommendations and a few others cited in the Code of Federal Regulations (e.g., replacing “Federal Law Prohibits Dispensing without a Prescription” by “Rx Only”). In 1997, following the USP–FDA lead, the National Coordinating Council for Medication Error Reporting Programs developed a set of general recommendations for improving the labeling and packaging of all drug products. However, these are just recommendations and not formal requirements or standards, and compliance is inconsistent.
Once a product is on the market, adjustments to naming, labeling, and packaging are made only when providers and patient safety experts exert significant effort to get problems acknowledged and accepted by industry and FDA representatives. In many instances, however, known problems continue to be inadequately addressed over extended periods of time. For example, from 2000 until the present, ISMP, USP, and the Centers for Disease Control and Prevention (CDC) have notified the manufacturer and the FDA about repeated medication errors due to labeling confusion between tetanus toxoid and tuberculin vaccines (ISMP, 2005). Vaccine mixups can place hundreds, if not thousands, of patients at risk for serious ADEs. For about a year, the response by industry and the FDA to the error notification was that “providers should read the label.” The FDA did produce a safety video on the vaccine mix-up, but errors continued to be
reported. When the manufacturer sent the FDA a revised label for approval, it remained at the agency for 6 months without action being taken. Moreover, when the change in labeling was finally approved, the FDA would not discuss it and did not notify patient safety organizations or the public about it. This example illustrates the problems that can occur when human factors issues are not incorporated into labeling and packaging designs, and communications about problems are not transparent.
The FDA has held public meetings to address some of the problems identified above (e.g., naming, need for color coding). Progress is being made incrementally on certain naming issues. For example, the FDA has instituted a requirement that medications have bar codes to facilitate accurate drug dispensing and administration. And at the FDA’s request, the generic drug industry agreed to use a mix of upper- and lowercase letters to highlight the differences between similar generic names, such as vinBLAStine and vinCRIStine (FDA, 2005b). Still, an overall guidance document that formally and comprehensively advises companies on naming, labeling, and packaging for safety has not yet been produced. As a consequence, there is great inconsistency among products and companies as regards follow-through on the detailed aspects of labeling and packaging that can reduce medication errors. In contrast, the National Health Service in the United Kingdom recently released Guidance Note 25: Best Practices for Labeling and Packaging of Medicines, which expanded requirements to increase the clarity and safety of drug labeling on external packaging and blister pacs (MHPRA, 2003). This document addresses several of the issues (e.g., font size, color, design) discussed at FDA public meetings.
The proliferation of manufacturers, medications, formulations, and doses will likely continue, increasing providers’ difficulties in differentiating drug products (Berman, 2004). Thus, a formal action plan to address naming, labeling, and packaging problems is critical to improving the safety of medication use.
The committee believes strongly that industry and the FDA should take several specific actions to address the remaining key problems with drug naming, labeling, and packaging (see Box 6-3). This proposed action plan is founded on two overarching principles:
-
Product naming, labeling, and packaging should be designed for the end user—the provider in the clinical environment and/or the consumer.
-
Safety should always take precedence over commercial interests.
Unit-of-Use Packaging
Chapter 4 examines one possible way of improving consumers’ medication self-management—redesigning pharmacy containers and warning
|
BOX 6-3 Actions to Improve Drug Naming, Labeling, and Packaging
|
|
labels. Another method is the provision of medications to consumers in unit-of-use packaging. Unit-of-use packaging refers to drug products supplied in containers that provide enough medication for patients’ use during a specified time interval (Szeinbach et al., 2003). The unit can be dispensed directly to a patient without pharmacists’ repackaging or modification other than the application of a prescription label6 (USP, 1993).
The most common forms of unit-of-use packaging for solid medications (tablets or capsules) are (1) the blister pack—a sheet of 10 to 30 individually wrapped doses of a particular medication (more than one sheet may be dispensed to the consumer); (2) the calendar blister pack of individually wrapped doses, organized for administration according to a calendar, such as that used for oral contraceptives; and (3) the multidose packet or sachet, which contains doses of more than one medication (Ientile et al., 2004). Semisolids (creams) and liquids are typically packaged in unit-of-use plastic tubes or ampoules.
Unit-of-use packaging is employed broadly for both prescription and nonprescription drugs in Europe, Australia, Asia, and Latin America as an important part of product approval requirements (Ientile et al., 2004). There are several reasons for instituting unit-of-use packaging standards. First, such packaging promotes child safety by providing greater protection against death or serious injury from accidental poisoning (HCPC, 2003). Second, because medicines are often distributed in the manufacturer’s original packaging, errors that occur as a result of repackaging at the pharmacy can be minimized or prevented (HCPC, 2003). Third, several studies have shown that unit-of-use packaging is easier for consumers to use, facilitates more accurate self-administration, and improves adherence to treatment regimens (including complex regimens) and health outcomes (Becker et al., 1986; Wright et al., 1999; Huang et al., 2000; Simmons et al., 2000). Lastly, unit-of-use packaging is “tamper evident” (i.e., it is easy to detect product tampering), which can reassure consumers of product safety amidst growing concern about contamination or counterfeits (Allen, 2002).
In the United States, only certain medicines, such as oral contraceptives, azithromycin, prednisone, and many OTCs, are packaged in this manner (Schneider et al., 2006). Some vitamin and supplement combinations are available in multidose packets. Though the positive effect of unit-of-use packaging on many aspects of medication use are well documented in Europe and other regions, fewer than 20 percent of all prescription and OTC drugs in the United States are produced in blister packs (Erickson, 1998). Until recently, a few practical issues hindered more widespread adoption in the United States: (1) the cost to shift manufacturing from bulk distribution to unit-of-use packaging; (2) limited space and storage in community pharmacies; (3) rigidity of dispensing, making it more difficult to customize doses for patients; and (4) the lack of regulatory requirements (Allen, 2002). Changes taking place in the marketplace are now addressing many of these issues: (1) passage of the FDA’s final rule requiring unit-dose packaging and bar codes for all medicines distributed to hospitals (FR, 2004); (2) growth in the number of repackaging companies, resulting in competitive pricing for such services; (3) a shift among community pharmacies to just-in-time inventories; (4) advances in packaging machinery, making unit-of-use packaging more efficient and less costly; and (5) revisions to and adoption of international packaging standards by manufacturers (Allen, 2002; HCPC, 2003; FG, 2003). Some are even predicting that the higher costs of unit-of-use packaging could be offset by increased adherence and decreased waste (Valero, 2005). Current trends suggest that unit-of-use packaging will generate the highest worldwide growth prospects among all pharmaceutical packaging products (FG, 2003) and gain momentum in the United States, possibly achieving the same level of use as in European and other countries where it is standard (Szeinbach et al., 2003).
The potential to improve patient safety and prevent errors has generated interest among U.S. regulators, providers, consumers, industry representatives, and other stakeholders in expanding unit-of-use packaging to medications for chronic conditions (Schneider et al., 2006). The strategy of using calendar blister packs could help large numbers of patients (including seniors, children, and those challenged by cognitive, physical, or functional impairment) take their medication more reliably and safely and enhance their treatment outcomes. In a 2003 survey of state boards of pharmacy, two-thirds of respondents expressed their belief that unit-of-use packaging would improve efficiency, reduce errors in dispensing, improve patient compliance, and increase opportunities for patient counseling (Szeinbach et al., 2003). Schneider and colleagues (2006) believe that packaging prescription medicines in easy-to-remember forms should be an important component of health care redesign for quality and safety.
The committee believes that stakeholders should collaborate to develop a strategy for expansion of unit-of-use packaging to new therapeutic areas. Additional head-to-head studies should be undertaken to evaluate various approaches to unit-of-use packaging and determine optimum designs to support different consumer groups in their medication self-management.
Distribution of Free Samples
The prescription drug industry defines a drug sample as “… a package containing a limited quantity of pharmaceutical product sufficient to evaluate clinical response, distributed to authorized health care practitioners free of charge, for patient treatment” (Groves et al., 2003). Such distribution of samples to physicians during detailing visits is the number one promotional tool used by industry. According to IMS Health, the estimated retail value of free product samples distributed in 2003 was over $16 billion (IMS Health, 2004). The actual cost to the manufacturer is much less, however— about 20 to 30 percent of the retail price (Petersen, 2000).
Few studies have been conducted to evaluate the true impact of the distribution of drug samples (Groves et al., 2003). Those studies that have been carried out have found conflicting attitudes about the use of free samples (Chew et al., 2000). Some early studies emphasized the benefits of the practice, including allowing physicians to start patients on medications quickly, to evaluate early effectiveness or adverse effects, to adjust prescribed doses before a full prescription is filled, to offset the cost of drugs to indigent and underinsured patients, and to demonstrate appropriate use to patients (Rasmussen, 1988; Weary, 1988). One more recent study highlighted the benefits of a sample pack in helping consumers detect a drug dispensing error because of visual familiarity with the product (Dodds-Ashley et al., 2002). Visual familiarity can change, however, when a generic
product is dispensed, particularly since generics are manufactured by multiple different vendors, and have different colors (and sometimes shapes) to differentiate among competitors.
Historically, concerns about the safety of sample use were focused on the products themselves—on diversion of samples to the wholesale market for repackaging and retail sale, as well as outright counterfeiting (Rasmussen, 1988). Attempts by Congress to pass legislation banning or regulating the distribution of free samples were unsuccessful, and these concerns persist. In general over the past decade, there has been growing unease among the provider community and others about the use of samples and their direct effects on physician behavior and medication safety (see Box 6-4) (Chew et al., 2000).
Because sampling is reserved for newer, higher-priced, brand-name drugs, prescribing is skewed toward these drugs (versus generics, older drugs, or OTC medications). Overall health expenditures increase as a result of the cost of additional office visits to obtain more samples or the higher cost of the prescription (Taira et al., 2003).
In efforts to lower prescription drug costs to payers, several insurers, such as BlueCross BlueShield, have recently started to provide free samples of generics to health care providers to encourage use of these products (Davia, 2003; Sipkoff, 2003). The free generic samples are supplied on a trial basis for consumers who are currently using a brand name version or
|
BOX 6-4 Safety Issues Related to the Distribution of Free Samples A number of critical medication safety and quality-of-care issues related to the distribution of product samples can contribute to errors:
SOURCE: Chew et al., 2000; Groves et al., 2003; Taira et al., 2003. |
are about to start a new treatment. Other practices traditionally in the realm of brand name manufacturers, including aggressive advertising and voucher campaigns, are being used by payers to promote generics. Insurers mail thousands of vouchers to members, along with educational materials to help consumers understand the FDA review process for approving generics, as well as brand–generic comparative pricing information. Consumers bring the vouchers to their provider and receive the generic sample if clinically appropriate.
Comprehensive change in the delivery of free samples for prescription drugs is important to ensuring safety and quality in the medication-use system. Increasing numbers of health care providers are either banning drug samples altogether or experimenting with alternative means of dispensing them (Blumenthal, 2004; Simon et al., 2005; Brennan et al., 2006). Alternatives being evaluated include the use of coupons or vouchers to receive a sample dispensed by the pharmacy, policies restricting samples, and a smartcard system (Paterson and Anderson, 2002; Groves et al., 2003). Brennan and colleagues (2006) recently advocated a total ban on the direct provision of samples to physicians, and the institution of a voucher system for low-income patients or other arrangements that would distance a company and its products from physicians (Brennan et al., 2006). Some health care organizations have already instituted voucher systems. For example, the University of Wisconsin Hospital now uses a voucher program to replace free samples from drug companies (Charatan, 2001). Vouchers issued to patients at the hospital cover part of the cost of their prescription drugs. Participating manufacturers reimburse the hospital pharmacy for brand-name drugs, but the hospital pays for generic medications. The Everett Clinic in Washington State also has implemented a voucher program, which it believes has allowed greater assistance to the uninsured and financially impoverished (Charatan, 2001). Technology companies such as TrialCard Inc. (smart cards) and eMedRx (electronic prescribing) are developing systems to deliver pharmaceutical company coupons and vouchers electronically to physicians and/or pharmacies (Levy, 2002; Security Biometrics, Inc., 2004). Extensive studies are needed to evaluate the impact of free samples on physician and consumer behavior and patient safety and determine alternative methods of distribution.
MEDICAL DEVICE AND HEALTH INFORMATION TECHNOLOGY INDUSTRIES
Information technology systems and applications are valuable tools that can improve the safety and quality of care across the medication-use continuum. Some drug-related technologies are already in use, including
knowledge-based systems used for laboratory and pharmacy data, patient safety reporting systems, infusion pumps, and applications for computerized provider order entry (CPOE) and electronic prescribing (IOM, 2004). Bar code medication administration systems have been implemented in some institutions. A key feature of pharmacy database systems, infusion pumps, and bar code and decision-support applications is the alert function that warns clinicians of potential medication safety problems. In general, a fully developed set of drug alerts includes drug–dose defaults, drug–dose checking, allergy checking, drug interaction checking, drug–laboratory checking, drug–condition checking, and drug–diet (food) checking. Other rule-based alerts (e.g., a required laboratory test for the use of particular drug) and automated surveillance for ADEs and near misses also are important to improving safety and reducing errors. Yet most providers currently use these technologies as independent, stand-alone systems rather than as integrated components of comprehensive clinical information systems—the overarching goal in building the national health information infrastructure (IOM, 2004). Nurses rely on the medication administration record generated by infusion and bar code systems to administer medications; physicians rely on CPOE and, if linked, pharmacy database systems for prescribing; and pharmacists rely on their databases for preparation and dispensing of prescriptions. As a result, each component of the medication use-system remains compartmentalized, increasing safety risks.
The lack of common drug information standards and integration of pharmacy database, decision-support, infusion, and bar code systems can have particularly devastating effects on patient safety (Patterson et al., 2002; Han et al., 2005; Koppel et al., 2005). For example, Koppel and colleagues (2005) found that medication errors increased as a result of data fragmentation, failures of system integration, and poorly designed human– machine interfaces. Because all of the above systems produce medication administration records, they must be able to communicate with each other to produce a comprehensive view of the patient’s medication regimen. If they operate as stand-alone systems, no one has the full medication administration record, and clinicians have incomplete information.
Recommendation 5: Industry and government should collaborate to establish standards affecting drug-related health information technologies. Specifically:
-
The NLM should take the lead in developing a common drug nomenclature for use in all clinical information technology systems, based on standards for the national health information infrastructure.
-
AHRQ should take the lead in organizing mechanisms for safety alerts according to severity, frequency, and clinical importance to improve clinical value and acceptance.
-
AHRQ should take the lead in developing intelligent prompting mechanisms specific to a patient’s unique characteristics and needs; provider prescribing, ordering, and error patterns; and evidence-based best-practice guidelines.
-
AHRQ should take the lead in developing user interface designs based on the principles of cognitive and human factors and the context of the clinical environment.
-
AHRQ should support additional research to determine specifications for alert mechanisms and intelligent prompting, as well as optimum designs for user interfaces.
Data Standards
Unresolved problems with data standards inhibit the development and use of drug-related technologies, especially the alert functions described above. Data standards serve as the basis for representing and exchanging information electronically. Uniform data standards act as a common language, allowing communication and interoperability between different technologies. For example, a CPOE application on a handheld personal digital assistant (PDA) must be able to communicate with a pharmacy database system to process an electronic prescription. Although different types of data standards serve different functions, uniformity in the representation of similar data is required to optimize the usefulness and efficiency of technologies among systems and institutions.
Four problems are associated with data standards for drug information. First, there is no complete, standardized set of terms, concepts, and codes to represent drug information. Providers compensate for this lack of standards by piecing together different, incomplete datasets from multiple vendors, standards organizations, and internal sources. Second, there is no standardized method for presenting safety alerts, which should be ranked according to severity and/or clinical importance. Instead, providers are inundated with too many nonrelevant alerts, resulting in alert fatigue and high rates of alert overrides (Glassman et al., 2002; Hsieh et al., 2004). Third, systems lack intelligent or intuitive mechanisms for recognizing patient-specific data and relating those data to allowable overrides, such as those associated with a particular patient and drug allergy alert or duplicate therapy request (Abookire et al., 2000). Fourth, the bar codes stamped on drug packaging labels are designed differently by each vendor. Resolving these problems requires standardization on several levels: drug nomenclature, organization of alerts, intelligent prompting, and bar coding.
Drug Nomenclature
As the group overseeing the development of national data standards to support the technologies composing electronic health record systems, the National Committee on Vital and Health Statistics (NCVHS) should ensure that the appropriate organizations formulate a comprehensive set of standards for drug information (IOM, 2004). These standards should accomplish the following:
-
Representation of all attributes of a drug needed for electronic communication about prescriptions, medication administration, and monitoring
-
Representation of drug data specified according to the clinical needs of a specific patient population (e.g., pediatric, geriatric, pregnant women, those with renal or hepatic impairment)
RxNORM, developed by the NLM, standardizes certain components of the clinical drug nomenclature—active ingredient, strength, physical form, and dosage form. Work also is being carried out by the Veterans Health Administration (VHA) and the NLM to complete the national drug file reference terminology (NDF-RT), which will standardize many additional components (see Figure 6-1 and Box 6-5). NCVHS has designated RxNORM and NDF-RT as the core clinical drug nomenclature for electronic health records and the national health information network. However, these terminologies have not been widely adopted by most technology vendors or provider groups. Moreover, critical information needed for alert functions and for specific patient populations (e.g., dose limits, units of measure) have not been developed for NDF-RT. In the interim, proprietary standards for drug alerts developed by different pharmacy database vendors (e.g., First Data Bank, Multium) are being used in decision-support applications since they are the terminologies that cover the widest range of attributes listed in Table 6-1. To facilitate the transition to a standardized drug nomenclature, the NLM is planning to map the NDF-RT terminology to pharmacy database terminologies.
A similar effort is needed to address the lack of standardization among the drug terminologies used in medical devices (i.e., infusion pumps, patient-controlled analgesia [PCA]) and bar code medication administration systems. Infusion pumps with smart-pump technology contain datasets for drug libraries and error reduction software that facilitates programming of standardized concentrations, approved dosing units, general drug information, and dose limits (Vanderveen, 2005). The needs of specific populations (e.g., neonates, pediatric populations) can be addressed within one infusion system by programming the drug libraries to reflect the characteristics of the patient. However, the drug libraries used in smart-pump software are
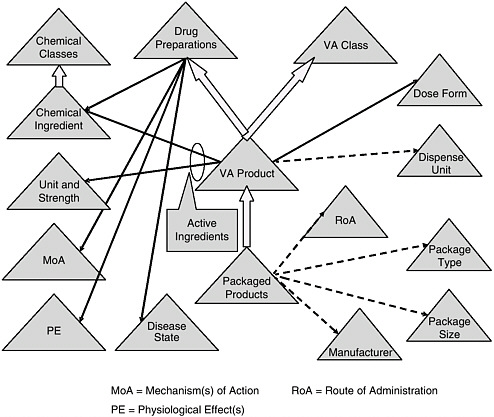
FIGURE 6-1 Veterans Health Administration’s national drug formulary reference terminology.
NOTE: See Box 6-5 for a discussion of this figure.
SOURCE: Brown, 2006
uploaded by each hospital using drug terminology from the pharmacy database system of choice. The device’s software program helps organize the data, but each hospital determines how the drug terminology will be used by its staff. Bar code medication administration systems are no different; they rely on the National Drug Code (NDC) for the bar code used on external package labels and on software programs that allow hospitals to upload drug terminology to generate drug alerts.
The lack of common data standards among provider organizations not only prevents systems from communicating with one another, but also compromises the comparability of data from one organization to another, as well as epidemiological analysis of medication errors and ADEs in all health care settings. To remedy this problem, the NCVHS, the NLM, and the VHA need to develop a strategy for completing the development of all attributes of a comprehensive drug nomenclature. Once this standardized
|
BOX 6-5 Overview of the Veterans Health Administration’s National Drug Formulary Reference Terminology (NDF RT) Figure 6-1 depicts NDF-RT, a drug terminology knowledge base derived from the U.S. Department of Veterans Affairs’ (VA) National Drug File. NDF-RT was developed over the past 5 years with input from a variety of government and other stakeholders. The figure outlines NDF-RT’s structure, and may be interpreted as follows. Each triangle represents a hierarchy of related concept definitions, with labels explained below. Thick arrows represent additional hierarchical connections. Solid black and dotted arrows represent semantic relationships. The triangles represent data contained in NDF-RT. The heart of NDF-RT is labeled VA Product, such as ASPIRIN 325MG ORAL TABLET. The VA Product is generally equivalent to the RxNORM Semantic Clinical Drug. Each VA Product is a “child” of two separate parents, a VA Class (e.g., ANALGESICS) and a Drug Preparation (e.g., ASPIRIN PREPARATIONS). Drug Preparations are described by relationships to their Mechanism(s) of Action (MoA triangle) (function at the cellular or subcellular level), Physiologic Effect(s) (PE triangle) (function at the organ, tissue, or body system level), and Disease State actions (diseases treated, caused, or prevented by the drug). Each VA Product also is characterized by its therapeutically active Chemical Ingredient(s), which in turn are characterized according to a structural class. In addition, each VA Product has a Dose Form (e.g., ORAL TABLET); a VA Dispense Units entry (e.g., TABLET); and a variety of other attributes, including links to RxNORM and commercial drug knowledge bases (not shown). For each active ingredient in a VA Product, there is an entry in the Unit Str. (Unit and Strength) hierarchy (325MG in the aspirin example). Each VA Product also encompasses some number of Packaged Products, which are identified by National Drug Code (NDC) numbers from the Food and Drug Administration (FDA). Each Packaged Product includes dispensing information, such as Route of Administration (RoA) (e.g., ORAL, INTRAMUSCULAR), Package Type (Pkg. Type) (e.g., BOX or BOTTLE), Package Size (Pkg. Size) (e.g., 500, 8 FL OZ), and Manufacturer (e.g., LILLY, SEARLE). NDF-RT’s multiaxial hierarchical structure is designed to provide a balance of rigor in terminology and compatibility with deployed systems, while simultaneously streamlining the maintenance required to keep pace with the thousands of changes to drug products that occur each month. Existing and planned extensions to NDF-RT (not shown) support a variety of clinical decision-support cases, such as dose adjustment based on individual pharmacogenomic characteristics. Currently, the VA is extending a commercial terminology management system to support the semiautomated integration of data from pharmacy personnel, the FDA’s Structured Product Label (SPL) project, and commercial drug knowledge sources. |
TABLE 6-1 Attributes Requiring Standardization for Drug Nomenclature
|
Basic Attributes |
Therapeutic Attributes |
Alert Attribute |
Payment Attributes |
Commercial Attributes |
|
|
|
|
|
drug nomenclature is completed, the NCVHS should ensure that all vendors incorporate it into their technology software.
Organization of Alerts
Standardizing the organization of alerts in pharmacy database systems, infusion pumps, and decision-support applications is required to reduce alert fatigue. The knowledge bases from which drug-related technologies derive their alerts are often highly inclusive, placing more emphasis on breadth of coverage than on clinical relevancy or severity of adverse events (Reichley et al., 2005). Unless alerts are ranked according to a severity– frequency scale and clinical importance, too many alerts tend to be delivered. The need to override excessive, inappropriate, nonspecified alerts can cause clinicians to miss critical safety alerts or to refuse the application altogether because of disruptions in workflow (van Bemmel and Musen, 1997). By the same token, many overrides are clinically appropriate and do not lead to ADEs (Hsieh et al., 2004).
Several studies have demonstrated improvements in clinician acceptance and reductions in inappropriate alerts through the ranking of alerts for all technologies (Kilbridge et al., 2001; DHA, 2002; Weingart et al., 2003; Shah et al., 2006). For example, a study of CPOE systems conducted by Shah and colleagues (2006) used a three-tiered alert structure:
-
Level 1—alerts of the highest severity (i.e., life-threatening or with the potential to cause permanent damage). Clinicians could not proceed with a prescription without either eliminating the contraindication or responding to specific information requested about the patient.
-
Level 2—alerts of strong severity (i.e., serious, capable of aggravating the patient’s condition). Clinicians could proceed if they provided a reason for an override.
-
Level 3—alerts of significant severity (i.e., important for the clinician to know). Clinicians could view clinical information in the alert, but the alert was noninterruptive.
Rather than using an all-inclusive knowledge base to determine the alerts, the researchers used a subset of only the most clinically relevant contraindications that pertained to the ambulatory care setting. Alerts of moderate to low severity were not included. The result was a significantly higher rate of acceptance among clinicians (67 percent) than that found in other studies (11 percent), although acceptance rates differed substantially by alert type (Weingart et al., 2003; Shah et al., 2006).
Alert fatigue and frequent overrides experienced with infusion pumps and bar code medication administration systems are no different than those
experienced with CPOE and pharmacy database systems. Alerts with infusion systems can be of particular concern since the highest percentage of medication errors is associated with intravenous medicines (Cohen, 2000; Billman, 2004). Smart-pump alerts may occur during programming or administration regardless of whether infusion rate limits are available for the drug (Malashock et al., 2004). The most common override occurs when the infusion rate is either above or below the maximum/minimum rate limit (Malashock et al., 2004). Other alerts can include informing the programmer of a duration change, a secondary stop, cancellation of drug selection, weight change–related dose recalculation, and same-drug infusion on multiple channels. Alerts can also occur for low battery power, venous occlusion, and similar conditions. The development of methods to rank the alert functions of infusion pumps could improve their functioning and safety.
While studies have clearly indicated that a tiered severity structure is important, additional work on how to differentiate, select, and integrate a separate tier for certain moderate-level alerts is required. A moderate-level warning may not be clinically important to one patient but may be for another, or may be important to a patient’s quality of life and adherence to medication therapy (Ahern and Kerr, 2003). A method for ranking the most frequent types of ADEs for each severity level also should be incorporated into the alert structure (Kilbridge et al., 2001; Miller et al., 2005). There have been many evidence-based studies identifying the frequency and severity of ADEs that can be used to determine these parameters (Classen et al., 1997; Forester et al., 2003, 2004; Gurwitz et al., 2005). Miller and colleagues (2005) suggest that the frequency rating might be based on percentage of total administrations, or 100 and 1,000 administrations. Table 6-2 provides a sample of the most common occurrences of alerts and the reasons for overriding them.
Standardization, however, does not imply rigidity. The alert configuration must remain flexible enough to reflect the inherent variability in clinical practice, such as the off-label use of a drug. In addition to severity and frequency, clinical importance is a third essential element of an alert structure. Alert ranking should be flexible enough to target the needs of specific patient populations (e.g., pediatric, geriatric) and medical disciplines (e.g., oncology, psychiatry) (Fortescue et al., 2003; Grasso et al., 2003). Most drug-related technologies allow for alert configurations according to patient age and weight, but are not designed to incorporate other individualized patient information. Hospital pharmacy database systems are the exception where laboratory test values can be linked to the patient’s medication management profile (IOM, 2004). As in the ambulatory care setting, alert rankings should reflect considerations specific to a patient’s condition or provider’s medical discipline (e.g., the alert ranking related to drug toxicity may be different for oncology than for nephrology). Such considerations do not
TABLE 6-2 Sample of Alert Types, Most Common Occurrences, and Most Common Reasons for Alert Override
|
Alert Type |
Most Common Occurrences of Highest Alerts |
Most Common Reasons for Alert Override |
|
Therapeutic duplication |
|
|
|
Drug–drug interaction |
|
|
|
Drug– laboratory |
Not documented |
|
|
Drug– disease |
|
|
|
Drug– pregnancy |
|
|
|
SOURCE: Shah et al., 2006 |
||
imply that known dangerous drug interactions or contraindications will change; instead, they reflect a recognition that in some cases, certain clinical preferences should take precedence. Thus, ranking of alerts according to all three dimensions—severity, frequency, and clinical importance—is necessary.
Intelligent Prompting
Technology that could use intelligent or intuitive mechanisms to prompt alerts would require the application of additional parameters beyond sever-
ity and frequency ranking and generalized patient data delineated only by age and weight. Such intelligent mechanisms would generate alerts specific to a patient’s unique characteristics and needs; physician prescribing, ordering, and error patterns; and evidence-based best-practice guidelines. For example, a patient might be allergic to one medication in a drug class but not others (Abookire et al., 2000). The software configuration should recognize the patient’s unique drug allergy without requiring that an alert be generated for every drug ordered in that class. As another example, a physician might have a preference for therapeutic duplication in transitioning a patient being prepared for discharge. The software configuration should accommodate the duration of the therapeutic duplication and the specific dosing transition of the two drugs without issuing repeated alerts requesting the same information. For this type of intelligent prompting to be possible, drug-related technologies must be linked not only to each other, but also to more comprehensive clinical information systems.
Incorporating rule-based physician monitoring features within prescribing systems is considered important for safety and learning. Anton and colleagues (2004) designed a more structured ranking of message severity according to seven categories, and system capabilities for monitoring based on storage functions and unique numbers for each prescription, provider, and patient. The system creates warnings using the incorporated rules and maintains a record of every occasion on which an alert is displayed. Each message can be linked to the user, the individual prescription key, and the outcome of the warning. Queries of the data were used to assess providers’ proficiency in preventing errors with the system and overall skill in using it (Anton et al., 2004).
In addition, evidence-based decision-support algorithms are necessary to ensure the adequacy of software configurations that incorporate specific protocols for real-time decision making and clinical action (Cole and Stewart, 1994; Sawa and Ohno-Machado, 2001; Fields and Peterman, 2005; Miller et al., 2005). Ideally, the algorithms should be developed according to three principles: (1) they should be system tested before full implementation; (2) they may have to be facility tailored based on process and workflow; and (3) they should be monitored and updated over time (Sawa and Ohno-Machado, 2001; Bates et al., 2003; Reichley et al., 2005). The algorithms, similar to any computer program, can never be finished or finalized as medicine is always changing; therefore, expiration labeling or update notices may be helpful to maintain currency.
One method for testing systems is to develop and test software configurations as well as train clinicians using simulation programs. The Anesthesia Patient Safety Foundation is the first medical community to adopt this technique successfully and apply it to anesthesia information management systems (AIMS) (Sawa and Ohno-Machado, 2001; Weinger and Slagle,
2002; Wachter et al., 2003; Pierce, 2006). Anesthesiologists administer anesthetics and observe the condition of their patients aided by multiple electronic monitors (e.g., electrocardiograph, pulse-oximeter, blood pressure monitors), which determine real-time decision making and actions. The AIMS records electronically all data generated by these technologies. The simulation programs allow clinicians to test themselves under various surgical situations. More recently, the Leapfrog Group commissioned the development of a simulation application both to test the decision-support algorithms of CPOE systems implemented in health care organizations and to train clinicians (Kilbridge et al., 2006). The methodology simulates different clinical scenarios using a wide variety of test patients and orders to evaluate how a hospital’s CPOE system responds to unsafe medication ordering and clinical situations.
Bar Coding
Another area requiring standardization is the bar codes used for drug labels and bar code medication administration systems. The ability of bar coding to affect medication error rates depends largely on the ability of hospitals to scan and interpret the data in the bar codes. A commonly used standard that scanners can easily read will have a greater impact on patient safety than a unique symbology that few scanners are programmed to read (FR, 2004).
A number of different stakeholders—drug manufacturers, distributors, repackagers/relabelers, manufacturers of bar code medication administration systems, and hospitals—use bar codes on drug products. As with the lack of a common drug nomenclature, there is no single, common bar code standard or symbology. Among hospitals, repackagers, and vendors of bar code medication administration systems, up to six different bar code standards are being used, each with its own special characteristics, features, and methods for encoding product information (see Figure 6-2). This situation creates several problems. First, the lack of a common standard drives costs up throughout the drug delivery system, particularly for hospitals that incur the expense of repackaging/relabeling drugs to the unit dose level and/or purchasing additional software or technology to read the different bar codes. Second, error rates associated with hospital relabeling are estimated at 17 percent nationwide, increasing the risk of ADEs (FR, 2004). Third, the multitude of standards inhibits integration of clinical systems. Designation of a single, common bar code standard could resolve these problems.
Efforts to standardize bar codes are linked to a rule establishing federal requirements for labeling of products down to the unit dose level. In the rule, the FDA requires all stakeholders using bar codes to choose one of two standards: (1) European Article Number/Uniform Code Council (EAN/

FIGURE 6-2 Bar code symbologies.
SOURCE: Combes, 2004.
UCC) or (2) Health Industry Business Communications Council (HIBCC). The NDC drug code must be incorporated into the bar code as it will serve as the unique product identifier. The different codes serve different purposes. The EAN/UCC standard was originally developed by the medical device industry, which uses Universal Product Numbers to meet the needs of retailers (HIBCC, 2001). Because no other standard was available at the time, drug manufacturers adopted the standard and integrated the NDC codes. However, many stakeholders consider the EAN/UCC standard to be inadequate for the specific applications and needs of the health care environment, especially those associated with patient safety (HIBCC, 2001). The HIBCC standard was designed to allow for more extensive and precise encoding and quicker tracking and tracing of specific drug products.
While narrowing choices down to two standards represents improvement, stakeholders believe that other aspects of the FDA rule need revision. In particular, the rule states that bar code symbology will be limited to a linear model that constrains the ability to encode a significant amount of information. This capability will be needed as health information technologies and clinical information systems advance. Therefore, these stakeholders are seeking to have the rule revised to allow for the use of three-dimensional models as well.
In addition, there are sizable cost implications when a hospital implements a bar code medication administration system. Thus, software programs will be required to be compatible with both the EAN/UCC and HIBCC standards, to accommodate various dimensional encoding models, and to be easily upgraded to meet demands for the encoding of additional
information. Further, radio frequency identification (RFID) technology may replace bar codes on external packaging altogether, particularly in light of the growing problems with counterfeit drug imports entering the U.S. market. However, RFID will not replace the need for standardized bar code systems for patient care.
User Interface
The ability of clinicians to use a medical device or decision-support system successfully depends on how well the technologies have been designed at the level of the human–machine interaction (i.e., user interface). From the user’s perspective, the interface is the system (Shortliffe et al., 2001). When interacting with technology, clinicians aim to carry out tasks in which information is assessed, manipulated, or created (van Bemmel and Musen, 1997). The quality and style of the interface directly affect this processing of information. Well-organized information that is presented in a logical and meaningful way results in a higher degree of usability, whereas the display of information in a cluttered, illogical, or confusing manner leads to decreases in user performance and satisfaction (van Bemmel and Musen, 1997). Most important, a poorly designed user interface can even contribute to medication errors for all drug-related technologies (Patterson et al., 2002; Ash et al., 2004; Koppel et al., 2005).
As noted earlier, several studies have confirmed that many medication errors resulting in patient harm involve intravenous infusion devices, with the most common cause of the errors being incorrect programming (Kaushal et al., 2001; Taxis and Barber, 2003; Tourville, 2003). Several problems with the interface design for these devices in terms of programming keys, display screens, and menu structure have contributed to these high rates of ADEs. In an effort to simplify programming and reduce pump size, a limited number of programming keys are provided on the pumps. Each key serves multiple functions, and clinical protocol is selected through scroll menus. However, menu structures are so complex that even skilled users could easily get confused (Nemeth, 2003). Device programming is often further complicated by small display screens that are difficult to read and follow. As a result, the state of the infusion pump is not always obvious during each step of the process. Even small data entry errors can result in numerous unforeseen medical complications that cause patient harm. Clinicians frequently must power down the pumps and start over to clear programming mistakes. Device manufacturers have been working to improve the user interface by incorporating the principles of human factors engineering into the pumps’ design structure. Standards for human factors design have been established by the Association for the Advancement of Medical Instrumentation (AAMI) and approved by the American National
Standards Institute (ANSI), and are a part of the FDA’s Good Manufacturing Practices (GMP) regulatory requirements (IOM, 2004). The standards do not go far enough to address user interface issues, however, and additional work is needed.
Medication errors also result from comparable problems in the user interface design for decision-support systems. A recent study of CPOE systems found that human–machine interface flaws facilitated 22 types of medication errors (Koppel et al., 2005).
A number of factors affect the ability of clinicians to interact effectively and efficiently with decision-support systems (whether CPOE, electronic health records, or pharmacy database). First, most of the commercial systems on the market were designed according to rigid machine rules that do not correspond appropriately to the clinician’s workflow and behavior (Koppel et al., 2005). The natural chain of clinical events is disrupted while clinicians are forced to accommodate the rigid data requirements of the technology (Han et al., 2005). Often, a second physician devoted solely to entering orders is needed when time-sensitive therapeutic interventions must be administered, such as in emergency or intensive care. Second, many interface designs are highly impractical or outdated. Information is presented in numerous lines of identical-looking text, without a windows-based structure or intuitive graphical navigation aids (Ash et al., 2004). Even when the information is there, it is difficult to find. Clinicians must click on multiple different screens to either retrieve all of a patient’s information or enter new clinical information. Information becomes fragmented, and clinicians lose their ability to develop a more comprehensive overview and conceptual understanding of the case (Ash et al., 2004). For example, in many inpatient CPOE systems, patient names are grouped alphabetically rather than by clinical staff or rooms. Thus similar names, combined with small fonts, hectic workstations, and interruptions, can easily be confused (Koppel et al., 2005). Equally troubling, a patient’s medication information is seldom synthesized on one screen; a clinician may need to access up to 20 screens to view all the medications included in the patient’s regimen. Although decision-support systems use standard computer monitors to display information, a significant amount of work is needed to develop optimal user interface designs that can make data capture and manipulation easier for clinicians and more accurate for patient safety.
Data presentation and the user interface affect the usability of bar code medication administration systems as well. Although there are no studies indicating that the design of such systems directly caused medication errors (Johnson et al., 2002), several studies have confirmed that negative unintended consequences resulting from the introduction of these systems may
create new paths to ADEs (Patterson et al., 2002; Patterson, 2003). The information display of the systems is much like that of other decision-support systems that rely on computer-based monitors and graphical interfaces. Specific issues with interface design vary depending on the vendor, but generally relate to the incompleteness of the medication information displayed and its effect on clinical coordination (Cipriano, 2003; Patterson, 2003). For example, the more inflexible systems require a long, confusing sequence of programming activities for a simple change to medication administration times. Moreover, important medication information is either not available or not displayed in a timely manner. Key problems identified include (1) pending and discontinued medication orders not displayed; (2) inability to document medications not displayed as administered when they had been administered; (3) automated removal of medications from bar code medication administration systems, resulting in confusion; (4) inability to view changes to medication orders without opening a patient record; (5) difficulty of undoing actions: (6) difficulty of revising database information once entered; and (7) poorly organized data screens, resulting in missed medications (Patterson et al., 2002; Rogers et al., 2005). The fragmentation of patient data also contributes to clinicians’ inability to obtain at a glance a comprehensive overview of patients’ medication information, as well as to degraded coordination between physicians and nurses—one of the more noted negative side effects of bar code medication administration systems (Patterson et al., 2002).
Efforts in the United Kingdom have started to address user interface issues through an agreement with Microsoft Corporation. Microsoft will develop a health-specific user interface for clinical systems used by the National Health Service to improve patient care and safety. Under the terms of the agreement, Microsoft will supply code based on the full shipping versions of its desktop software that can be used by independent vendors and supply customized versions of Office and Windows (NHS, 2004). However, use of common coding to link and present data is only one aspect of improving the user interface.
Addressing user interface issues will require greater attention to the cognitive and social factors influencing clinicians in their daily workflow and interaction with technologies (van Bemmel and Musen, 1997). Yet little emphasis has been placed on physicians’ ability to learn and use these systems or on the technologies’ effects on physicians’ reasoning. From the perspective of cognitive psychology, designers must develop a better understanding of how clinicians best comprehend information, as well as of the limits of human perception and memory. The context of the clinical environment, in which clinicians must perform multiple tasks simultaneously and manage numerous interruptions by beepers, telephones, and colleagues,
must be taken into account (Ash et al., 2004). Designers should understand that cognitive overload can result from overemphasizing complete information entry or retrieval.
A prime example of how comprehensive design strategies such as these have been successful in transforming health technology interfaces to improve patient safety is in the high-risk area of anesthesia (IOM, 2000; Hallinan, 2005; Pierce, 2006). Anesthesiology has reduced anesthesia mortality rates from two deaths per 10,000 administrations to one death per 300,000 administrations (JCAHO, 1998). This success was accomplished through a combination of the following:
-
Technical changes (new monitoring equipment, standardization of existing equipment)
-
Information-based strategies, including the development and adoption of guidelines and standards
-
Application of human factors to improve performance, such as the use of simulators for training
-
Formation of the Anesthesia Patient Safety Foundation to bring together stakeholders from different disciplines (physicians, nurses, manufacturers)
-
Having a leader who could serve as a champion for the cause (Leape et al., 1998; IOM, 2000)
No single one of these changes has been sufficient to have a clear-cut impact on mortality, yet, the application of human factors principles in conjunction with the other factors has been highly effective (Leape at al., 2002; Sawa and Ohno-Machado, 2002; Wachter at al., 2003). Measuring progress over time and regularly integrated lessons learned into clinical systems created a dynamic process for ongoing quality and safety improvement.
It can be challenging to capture the richness and complexity of clinical data in a manner that is concise and precise, but still comprehensive enough for medical care (Cimino et al., 2001). Screen layout and the visual salience of the information presented critically affect the way the information is interpreted by clinicians using decision-support systems (Kushniruk et al., 1996; Kaufman et al., 2003). Menus, graphics, and colors can all help differentiate data and make systems more attractive and simpler to learn and use. Interfaces must offer clear presentations, avoid unnecessary detail, and provide consistent interaction to be effective (Shortliffe et al., 2001). Designers also should recognize the inherent differences among clinician user groups, and seek to design multidimensional interfaces that can accommodate the information requirements of individual clinicians and comprehensive conceptual views of patient information.
REFERENCES
Abookire SA, Teich JM, Sandige H, Paterno MD, Martin MT, Kuperman GJ, Bates DW. 2000. Improving allergy alerting in a computerized physician order entry system. Proceedings of American Medical Informatics Association Symposium 2–6.
Ahern MD, Kerr SJ. 2003. General practitioners’ perceptions of the pharmaceutical decision-support tools in their prescribing software. Medical Journal of Australia 179(1):34–37.
Alexander KP, Peterson ED. 2003. Evidence-based care for all patients. American Journal of Medicine 114(4):333–335.
Allen D. 2002. The Next Chapter: Unit-of-Use Packaging. Pharmaceutical and Medical Packaging News. [Online]. Available: http://www.devicelink.com/pmpn/archive/02/11/004. html [accessed May 27, 2006].
ALLHAT (Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial). 2002. Major outcomes in high-risk hypertensive patients randomized to angiotensin-converting enzyme inhibitor or calcium channel blocker vs. diuretic: The Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT). Journal of the American Medical Association 288(23):2981–2997.
Anton C, Nightingale PG, Adu D, Lipkin G, Ferner RE. 2004. Improving prescribing using a rule-based prescribing system. Quality and Safety in Health Care 13(3):186–190.
Aronson JK. 2004. Medication errors resulting from the confusion of drug names. Drug Safety 3(3):167–172.
Ash JS, Berg M, Coiera E. 2004. Some unintended consequences of information technology in health care: The nature of patient care information system-related errors. Journal of the American Medical Informatics Association 11(2):104–112.
ASTM (American Society for Testing and Materials). 1988. Standard D4267-89. Philadelphia, PA: ASTM.
Bates DW, Kuperman GJ, Wang S, Gandhi T, Kittler A, Volk L, Spurr C, Khorasani R, Tanasijevic M, Middleton B. 2003. Ten commandments for effective clinical decision support: Making the practice of evidence-based medicine a reality. Journal of the American Medical Informatics Association 10(6):523–530.
Becker LA, Glanz K, Sobel E, Mossey J, Zinn SL, Knott KA. 1986. A randomized trial of special packaging of antihypertensive medications. Journal of Family Practice 22(4):357–361.
Berman A. 2004. Reducing medication errors through naming, labeling, and packaging. Journal of Medical Systems 28(1):9–29.
Billman G. 2004. A Medical Center’s Experience Using Smart Infusion Pumps to Manage Medication Administration. San Diego, CA: ALARIS Center for Medication Safety and Clinical Improvement.
Blumenthal D. 2004. Doctors and drug companies. New England Journal of Medicine 351(18): 1885–1890.
Brennan TA, Rothman DJ, Blank L, Blumenthal D, Chimonas SC, Cohen JJ, Goldman J, Kassirer JP, Kimball H, Naughton J, Smelser N. 2006. Health industry practices that create conflicts of interest: A policy proposal for academic medical centers. Journal of the American Medical Association 295(4):429–433.
Brown, SH. 2006. Diagram of NDF-RT Data Elements. Personal Communication.
Califf RM, DeMets DL. 2002a. Principles from clinical trials relevant to clinical practice: Part I. Circulation 106:1015–1021.
Califf RM, DeMets DL. 2002b. Principles from clinical trials relevant to clinical practice: Part II. Circulation 106:1172–1175.
CERTS (Centers for Education and Research on Therapeutics). 2003. Risk assessment of drugs, biologics, and therapeutic devices: Present and future issues. Pharmacoepidemiology and Drug Safety 12(8):653–662.
Charatan F. 2001. Hospital bans free drug samples. Western Journal of Medicine 174(4): 236–237.
Chew LD, O’Young TS, Hazlet TK, Bradley KA, Maynard C, Lessler D. 2000. A physician survey of the effect of drug sample availability on physicians’ behavior. Journal of General Internal Medicine 15(7):478–483.
Cimino JJ, Patel VL, Kushniruk AW. 2001. Studying the human-computer-terminology interface. Journal of the American Medical Informatics Association 8(2):163–173.
Cipriano PF. 2003. A nursing perspective on bedside scanning systems. Hospital Pharmacy 38(11):S14–S15.
Classen DC, Pestotnik SL, Evans RS, Lloyd JF, Burke JP. 1997. Adverse drug events in hospitalized patients. Excess length of stay, extra costs, and attributable mortality. Journal of the American Medical Association 277(4):301–306.
Cohen MR. 2000. Medication Errors: Causes, Prevention, and Risk Management. Sudbury, MA: Jones and Bartlett Publishers.
Cohen MR. 2002. Trade name, INNs, and medication errors. Archives of Internal Medicine 162(22):2636–2637.
Cole WG, Stewart JG. 1994. Human performance evaluation of a metaphor graphic display for respiratory data. Methods of Information in Medicine 33(4):390–396.
Combes JR. 2004. Understanding the Challenges of Implementation of Point Care Bar Code Systems. Presentation to the IOM Committee on Identifying and Preventing Medication Errors, March 19, 2004.
Crowley WF, Sherwoo, L, Salber P, Scheinberg D, Slavkin H, Tilson H, Reece EA, Catanese V, Johnson SB, Dobs A, Genel M, Korn A, Reame N, Bonow R, Grebb J, Rimoin D. 2004. Clinical research in the United States at a crossroads: Proposal for a novel public-private partnership to establish a national clinical research enterprise. Journal of the American Medical Association 291(9):1120–1126.
Davia J. 2003. Encourage Use of Generics to Rein in Cost of Medications. [Online]. Available: http://www.democratandchronicle.com/news/extra/fighting/health/story4.shtml [accessed March 12, 2006].
DHA (Australian Department of Health and Ageing). 2002. BMMS: Alerts Discussion Paper (Version 3.0 edition). Canbera, Australia: DHA.
Dodds-Ashley ES, Kirk K, Fowler VG. 2002. Patient detection of a drug dispensing error by use of physician-provided drug samples. Pharmacotherapy 22(12):1642–1643.
Edwards R, Faich G, Tilson H, and International Society of Pharmacovigilance. 2005. Points to consider: The roles of surveillance and epidemiology in advancing drug safety. Pharmacoepidemiology and Drug Safety 14(9):665–667.
Erickson G. 1998. Unit-of-Use Packaging: The Wave of the Future? Pharmaceutical and Medical Packaging News. [Online]. Available: http://www.devicelink.com/pmpn/archive/ 98/06/003.html [accessed May 27, 2006].
FDA (U.S. Food and Drug Administration). 2005a. Overview of the Office of Medication Errors and Technical Support. Submission to the IOM Committee on Identifying and Preventing Medication Errors. Rockville, MD: FDA.
FDA. 2005b, July–August 2005. Drug name confusion: Preventing medication errors. FDA Consumer Magazine.
FG (Freedonia Group). 2003. World Pharmaceutical Packaging: Forecasts to 2007 and 2012. [Online]. Available: http://www.gii.co.jp/sample/pdf/fd17351.pdf [accessed April 2, 2006].
Fields M, Peterman J. 2005. Intravenous medication safety system averts high-risk medication errors and provides actionable data. Nursing Administration Quarterly 29(1):78–87.
Fitzgerald GA. 2005. Opinion: Anticipating change in drug development: The emerging era of translational medicine and therapeutics. Drug Discovery 4(10):815–818.
Fleming TR, DeMets DL. 1996. Surrogate end points in clinical trials: Are we being misled? Annals of Internal Medicine 125:605–613.
Forester AJ, Murff HJ, Peterson JF, Gandhi TK, Bates DW. 2003. The incidence and severity of adverse events affecting patients after discharge from the hospital. Annals of Internal Medicine 138(3):161–167.
Forester AJ, Halil RB, Tierney MG. 2004. Pharmacist surveillance of adverse drug events. American Journal of Health System Pharmacists 61(14):1466–1472.
Fortescue EB, Kaushal R, Landrigan CP, McKenna KJ, Clapp MD, Federico F, Goldman DA, Bates DW. 2003. Prioritizing strategies for preventing medication errors and adverse drug events in pediatric inpatients. Pediatrics 111(4):722–729.
FR (Federal Register). 2004. Bar Code Label Requirements for Human Drug Products and Biological Products: Final Rule. Washington, DC: National Archives and Records Administration.
Glassman PA, Simon B, Belperio P, Lanto A. 2002. Improving recognition of drug interactions: Benefits and barriers to using automated drug alerts. Medical Care 40(12):1161– 1171.
Grasso BC, Rothschild JM, Genest R, Bates DW. 2003. What do we know about medication errors in psychiatry? Joint Commission Journal on Quality and Safety 29(8):391–400.
Groves KEM, Sketris I, Tett SE. 2003. Prescription drug samples: Does this marketing strategy counteract policies for quality use of medicines? Journal of Clinical Pharmacy and Therapeutics 28:259–271.
Gurwitz JH. 2004. Polypharmacy: A new paradigm for quality drug therapy in the elderly? Archives of Internal Medicine 164(18):1957–1959.
Gurwitz JH, Field TS, Judge J, Rochon P, Harrold LR, Cadoret C, Lee M, White K, LaPrino J, Mainard JF, DeFlorio M, Gavendo L, Auger J, Bates DW. 2005. The incidence of adverse drug events in two large academic long-term care facilities. The American Journal of Medicine 118(3):251–258.
Hallinan TJ. 2005. Once Seen as Risky, One Group of Doctors Changes Its Ways. [Online]. Available: http://webreprints.djreprints.com/1254400029287.html [accessed May 29, 2006].
Han YY, Carcillo JA, Venkataraman ST, Clark RSB, Watson RS, Nguyen TC, Bayir H, Orr RA. 2005. Unexpected increased mortality after implementation of a commercially sold computerized physician order entry system. Pediatrics 116(5):1506–1512.
HCPC (Healthcare Compliance Packaging Council). 2003. HCPC Response to Public Comments Requested in 68 FR 115, CPSC Petition PP 03-1. Falls Church, VA: HCPC.
HIBCC (Health Industry Business Communications Council). 2001. The Use of the Health Industry Bar Code for Product Labeling and Device Tracking. [Online]. Available: http:// www.hibcc.org/PUBS/WhitePapers/HIBCFeatures.pdf [accessed December 19, 2005].
Hoffman JM, Proulx SM. 2003. Medication errors caused by confusion of drug names. Drug Safety 26(7):445–452.
Hsieh TC, Kuperman GJ, Jaggi T, Hojnowski-Diaz P, Fiskio J, Williams DH, Bates DW, Gandhi TK. 2004. Characteristics and consequences of drug allergy alert overrides in a computerized physician order entry system. Journal of the American Medical Informatics Association 11(6):482–491.
Huang HY, Maguire MG, Miller ER, Appel LJ. 2000. Impact of pill organizers and blister packs on adherence to pill taking in two vitamin supplementation trials. American Journal of Epidemiology 152(8):780–787.
Hulley S, Grady D, Bush T, Furberg C, Herrington D, Riggs B, Vittinghoff E. 1998. Randomized trial of estrogen plus progestin for secondary prevention of coronary heart disease in postmenopausal women. Heart and Estrogen/progestin Replacement Study (HERS) Research Group. Journal of the American Medical Association 280(7):605–613.
Ientile C, Stokes J, Hendry M, Jensen A, Lewis G. 2004. Literature Review for the Effectiveness and Cost Effectiveness of Dose Administration Aids Project. Queensland, Australia: University of Queensland.
IMS Health. 2004. Total U.S. Promotional Spending by Type, 2003. Notes: Provided to IOM from PhRMA.
IOM (Institute of Medicine). 2000. To Err Is Human: Building a Safer Health System. Washington, DC: National Academy Press.
IOM. 2001. Crossing the Quality Chasm: A New Health System for the 21st Century. Washington, DC: National Academy Press.
IOM. 2004. Patient Safety: Achieving a New Standard for Care. Washington, DC: The National Academies Press.
IOM. 2006. Developing a Registry of Pharmacologic and Biologic Clinical Trials. Washington, DC: The National Academies Press.
ISMP (Institute for Safe Medication Practice). 2002. ISMP Medication Safety Alert July 10. Huntington Valley, PA: ISMP.
ISMP. 2005. Medication Safety Alert. December 15. Huntington Valley, PA: ISMP.
JCAHO (Joint Commission on Accreditation of Healthcare Organizations). 1998. Medication Use: A Systems Approach to Reducing Errors. Oakbrook Terrace, IL: JCAHO.
JCAHO. 2003. 2003 JCAHO National Patient Safety Goals: Practical Strategies and Helpful Solutions for Meeting These Goals (Special Report 2003). Oakbrook Terrace, IL: JCAHO.
JCAHO. 2006. National Patient Safety Goals: Look-Alike/Sound-Alike Drug List. [Online]. Available: http://www.jointcommission.org/PatientSafety/NationalPatientSafetyGoals [accessed June 13, 2006].
Johnson CL, Carlson RA, Tucker CL, Willette C. 2002. Using BCMA software to improve patient safety in Veterans Administration Medical Centers. Journal of Healthcare Information Management 16(1):46–51.
Kaufman DR, Patel VL, Hilliman C, Morin PC, Pevzner J, Weinstock RS, Goland R, Shea S, Starren J. 2003. Usability in the real world: Assessing medical information technologies in patients’ homes. Journal of Biomedical Informatics 36:45–60.
Kaushal R, Bates DW, Landrigan C, McKenna KJ, Clapp MD, Federico F, Goldmann DA. 2001. Medication errors and adverse drug events in pediatric inpatients. Journal of the American Medical Association 285(16):2114–2120.
Kilbridge P, Welebob E, Classen D. 2001. Overview of the Leapfrog Group Evaluation Tool for Computerized Physician Order Entry. Washington, DC: The Leapfrog Group.
Kilbridge PM, Welebob EM, Classen DC. 2006. Development of the Leapfrog methodology for evaluating hospital implemented inpatient computerized physician order entry systems. Quality and Safety in Health Care 15(2):81–84.
Koppel R, Metlay JP, Cohen A, Abaluck B, Localio AR, Kimmel SE, Strom BL. 2005. Role of computerized physician order entry systems in facilitating medication errors. Journal of the American Medical Association 293(10):1197–1203.
Kushniruk AW, Kaufman DR, Patel VL, Levesque Y, Lottin P. 1996. Assessment of a computerized patient record system: A cognitive approach to evaluating medical technology. MD Computing 13:406–415.
Leape LL, Kabcenell A, Berwick DM, Roessner J. 1998. Breakthrough Series Guide: Reducing Adverse Drug Events. Boston, MA: Institute for Healthcare Improvement.
Leape LL, Berwick DM, Bates DW. 2002. What practices will most improve safety? Evidence-based medicine meets patient safety. Journal of the American Medical Association 288(4): 501–507.
Levy S. 2002. TrialCard Puts Drug Samples in R.Ph.’s Hands. [Online]. Available: http:// www.drugtopics.com/drugtopics/article/articleDetail.jsp?id-116704 [accessed January 23, 2006].
Malashock CM, Shull SS, Gould DA. 2004. Effect of smart infusion pumps on medication errors related to infusion device programming. Hospital Pharmacy 39(5):460–469.
March JS, Silva SG, Compton S, Shapiro M, Califf R, Krishnan R. 2005. The case for practical clinical trials in psychiatry. American Journal of Psychiatry 162(5):836–846.
MHPRA (Medicines and Healthcare Products Regulatory Agency). 2003. Guidance Note 25: Best Practices for Labeling and Packaging of Medicines. London, United Kingdom: MHPRA.
Miller RA, Gardner RM, Johnson KB, Hripcsak G. 2005. Clinical decision support and electronic prescribing systems: A time for responsible thought and action. Journal of the American Medical Informatics Association 12(4):403–409.
NCC MERP (National Coordinating Council for Medication Error Reporting and Prevention). 2001. Recommendations to Reduce Medication Errors Associated with Verbal Medication Orders and Prescriptions. [Online]. Available: http://www.nccmerp.org/ council/council2001-02-20.html [accessed December 23, 2005].
NCVHS (National Committee on Vital and Health Statistics), 2003. Letter to Secretary of DHHS Tommy Thompson: PMRI Terminology Standards. [Online]. Available: http:// www.ncvhs.hhs.gov/reptrecs.htm [accessed May 23, 2006].
Nemeth C. 2003. Report on Infusion Pump Operation by Healthcare Professionals. Chicago, IL: Cognitive Technologies Laboratory.
NHS (National Health Service). 2004. Microsoft and Partners to Invest £40 Million in Development Resources to Improve Clinical Care in the NHS. [Online]. Available: http:// www.connectingforhealth.nhs.uk.news/0311041 [accessed March 15, 2006].
Paterson JM, Anderson GM. 2002. “Trial” prescriptions to reduce drug wastage: Results from Canadian programs and a community demonstration project. American Journal of Managed Care 8(2):151–158.
Patterson ES. 2003. Addressing human factors in bar code medication administration systems. Hospital Pharmacy 38(11):S16–S17.
Patterson ES, Cook RI, Render ML. 2002. Improving patient safety by identifying side effects from introducing bar coding in medication administration. Journal of the American Medical Informatics Association 9(5):540–553.
Petersen M. 2000, November 15. Growing opposition to free drug samples. New York Times. Business.
Pierce EC. 2006. 34th Rovenstine Lecture: Enhancing Patient Safety from the 1980’s through the Present. Pittsburgh, PA: Anesthesia Patient Safety Foundation.
Prentice RL. 1989. Surrogate end points in clinical trials: Definition and operational criteria. Statistics in Medicine 8:431–440.
Psaty BM, Furberg CD. 2005. COX-2 inhibitors—Lessons in drug safety. New England Journal of Medicine 352(11):1133–1135.
Rasmussen JE. 1988. Free drug samples. Archives of Determatology 124:135–137.
Reddan D, Szczech LA, O’Shea S, Califf RM. 2003. Anticoagulation in acute cardiac care in patients with chronic kidney disease. American Heart Journal 145(4):586–594.
Reichley RM, Seaton TL, Resetar E, Micek ST, Scott KL, Fraser VJ, Dunagan C, Bailey T.C. 2005. Implementing a commercial rule base as a medication order safety net. Journal of the American Medical Informatics Association 12(4):383–389.
Roberts R, Rodriguez W, Murphy D, Crescenzi T. 2003. Pediatric drug labeling: Improving the safety and efficacy of pediatric therapies. Journal of the American Medical Association 290:905–911.
Rogers ML, Patterson E, Chapman R, Render M. 2005. Usability Testing and the Relation of Clinical Information Systems to Patient Safety. [Online]. Available: http://www.ahrq.gov/ downloads/pub/advances/vol2/Rogers.pdf [accessed December 20, 2005].
Santell JO, Camp S. 2004. Similarity of drug names, labels, or packaging creates safety issues. U.S. Pharmacist 29(7).
Sawa T, Ohno-Machado L. 2001. Generation of dynamically configured check lists for intra-operative problems using a set covering algorithm. Proceedings of American Medical Informatics Association Symposium 593–597.
Schneider PJ, Murphy JE, Pedersen CA. 2006, In press. Adherence and treatment outcomes in elderly outpatients with hypertension using specially packaged medications. Journal of the American Pharmacists Association.
Schünemann HJ, Cook D, Grimshaw J, Liberati A, Heffner J, Tapson V, Guyatt G. 2004. Antithrombotic and thrombolytic therapy: From evidence to application. Chest 126(Suppl.): 688–696.
Security Biometrics, Inc. 2004. Security Biometrics, Inc. Subsidiary eMedRx, Forms Exclusive Strategic Alliance with Univec. [Online]. Available: http://findbiometrics.com/viewnews. php?id=831 [accessed January 23, 2006].
Shah NR, Seger AC, Seger DL, Fiskio JM, Kuperman GJ, Blumenfeld B, Recklet EG, Bates DW, Gandhi TK. 2006. Improving acceptance of computerized prescribing alerts in ambulatory care. Journal of the American Medical Informatics Association 13(1):5–11.
Shortliffe EH, Perreault LE, Wiederhold G, Fagan LM. 2001. Medical Informatics: Computer Applications in Health Care and Biomedicine. 2nd ed. New York: Springer.
Simmons D, Upjohn M, Gamble GD. 2000. Can medication packaging improve glycemic control abd blood pressure in Type 2 diabetes? Diabetes Care 23(2):153–156.
Simon SR, Majumdar SR, Prosser LA, Salem-Schatz S, Warner C, Kleinman K, Miroshnik I, Soumerai SB. 2005. Group versus individual academic detaining to improve the use of antihypertensive medications in primary care: A cluster-randomized controlled trial. American Journal of Medicine 118:521–528.
Sipkoff M. 2003. Getting Serious About Generics. [Online]. Available: http://www.managed caremag.com/archives/0301/0301.generics.html [accessed March 12, 2006].
Szeinbach SL, Baron M, Guschke T, Torkilson EA. 2003. Survey of state requirements for unit-of-use packaging. American Journal of Health-System Pharmacists 60(18):1863– 1866.
Taira DA, Iwane KA, Chung RS. 2003. Prescription drugs: Elderly enrollee reports of financial access, receipt of free samples, and discussion of generic equivalents related to type of coverage. American Journal of Managed Care 9(4):305–312.
Taxis K, Barber N. 2003. Ethnographic study of incidence and severity of intravenous drug errors. British Medical Journal 326(7391):684.
Tourville J. 2003. Automation and error reduction: How technology is helping Children’s Medical Center of Dallas reach zero-error tolerance. U.S. Pharmacist 28:80–86.
Tsintis P, La Mache E. 2004. CIOMS and ICH initiatives in pharmacovigilance and risk management: Overview and implications. Drug Safety 27(8):509–517.
Tunis SR, Stryer DB, Clancy CM. 2003. Practical clinical trials: Increasing the value of clinical research for decision making in clinical and health policy. Journal of the American Medical Association 290(12):1624–1632.
USAN (United States Adopted Name Council). 2005. Stem List. [Online]. Available: http:// www.ama-assn.org/ama1/pub/upload/mm/365/usanstmlist_10_19_05.doc [accessed December 23, 2005].
USP (U.S. Pharmacopeia). 1993. Unit-of-Use Packaging: Contemporary Issues. Rockville, MD: USP.
USP. 1998. USP Quality Review. No. 62. Rockville, MD: USP.
USP. 2005. CAPSLink Newsletter. Rockville, MD: USP.
Valero G. 2005. Patient Compliance via Unit-Dose Packaging. Pharmaceutical Business Strategies. [Online]. Available: http://www.pbsmag.com/ArticlePrinterFriendly.cfm?ID=176 [accessed May 27, 2006].
van Bemmel JH, Musen MA. 1997. Handbook of Medical Informatics. Heidelberg, Germany: Springer-Verlag.
Vanderveen T. 2005. Smart Pumps and Patient Controlled Analgesia Machines. Submission to the IOM Committee on Identifying and Preventing Medication Errors, July 2006.
Wachter SB, Agutter J, Syroid N, Drews F, Weinger MB, Westenskow D. 2003. The employment of an iterative design process to develop a pulmonary graphical display. Journal of the American Medical Informatics Association 10(4):363–372.
Weary PE. 1988. Free drug samples: Use and abuse. Archives of Determatology 124: 135–137.
Weingart SN, Toth M, Sands DZ, Aronson MD, Davis RB, Phillips RS. 2003. Physicians’ decisions to override computerized drug alerts in primary care. Archives of Internal Medicine 163(21):2625–2631.
Weinger MB, Slagle J. 2002. Human factors research in anesthesia patient safety. Journal of the American Medical Informatics Association 9(6):S58–S63.
WHI Steering Committee (Women’s Health Initiative). 2004. Effects of conjugated equine estrogen in postmenopausal women with hysterectomy: The Women’s Health Initiative randomized controlled trial. Journal of the American Medical Association 29:11701– 1712.
Wogalter MS, Vigilante WJ. 2003. Effects of label format on knowledge acquisition and perceived readability by younger and older adults. Ergonomics 46(4):327–344.
Wood AJ. 1999. The safety of new medicines: The importance of asking the right questions. Journal of the American Medical Association 281(18):1753–1754.
Wright JM, Htun Y, Leong MG, Forman P, Ballard RC. 1999. Evaluation of the use of calendar blister packaging on patient compliance with STD syndromic treatment regimens. Sexually Transmitted Disease 26(10):556–563.
Yusuf S, Collins R, Peto R. 1984. Why do we need some large, simple randomized trials? Statistics in Medicine 3:409–422.
Zarin DA, Tse T, Ide NC, 2005. Trial registration at ClinicalTrials.gov between May and October 2005. New England Journal of Medicine 353(26):2779–2787.
Zoeller J. 1999. Searle weighs Celebrex name change: New cox-2 has been confused with other similarly-named drugs. American Druggist 216(5):15.












































