
INTRODUCTION1
“We are launching a new initiative that will give us the capacity to respond faster and more effectively to bioterrorism or an infectious disease—a plan that will counter threats at home and strengthen public health abroad.”
—President Barack Obama, 2010 State of the Union Address
Safe and effective medical countermeasures, including vaccines, drugs, and diagnostics, are critical for responding to large-scale public health emergencies. Such situations, be they natural (e.g., pandemic influenza) or man-made (e.g., terrorism), have the potential to rapidly overwhelm public health and medical systems. America’s national security depends on having appropriately licensed chemical, biological, radiological, and nuclear medical countermeasures in its arsenal of defenses.
The Public Health Emergency Medical Countermeasures Enterprise (PHEMCE or countermeasures enterprise)2 encompasses diverse
partners from across federal, state, and local governments, industry, and academia. Despite its successes, certain structural, strategic, and technical elements of the countermeasures enterprise continue to impede research, development, and production of medical countermeasures. The National Institutes of Health (NIH) and the Department of Defense (DoD) support much of the basic research in the relevant health and disease areas. However, this research is not always aligned with the top priorities identified based on threat assessments, which limits the number of discoveries that are applicable for further development as medical countermeasures. Once potential candidates for advanced development are identified, they are often not yet at a stage of development where they can be handed off to the Biomedical Advance Research and Development Authority (BARDA)3 in the U.S. Department of Health and Human Services (HHS). Furthermore, because the commercial market is limited for most medical countermeasures, it can be difficult to engage private-sector pharmaceutical and biotechnology companies to participate in the development and manufacturing of these products.
To begin to address the efficiency and effectiveness issues of the PHEMCE, on December 1, 2009, HHS Secretary Kathleen Sebelius charged the “Office of the Assistant Secretary for Preparedness and Response [ASPR] to lead a review of its entire public health countermeasures enterprise, to be completed in the first quarter of next year.” Subsequently, in response to a request from the Assistant Secretary, the Institute of Medicine’s (IOM’s) Forum on Medical and Public Health Preparedness for Catastrophic Events and Forum on Drug Discovery, Development, and Translation jointly convened a workshop on February 22–24, 2010, titled The Public Health Emergency Medical Countermeasures Enterprise: Innovative Strategies to Enhance Products from Discovery Through Approval. The workshop was designed to examine federal policies and activities that affect medical countermeasure discovery, development, and approval, and to explore potential opportunities to enhance the countermeasures enterprise by
|
PHEMCE mission is to optimize national preparedness for public health emergencies, specifically by the creation, stockpiling, and use of medical countermeasures. |
|
3 |
BARDA’s mission is to provide countermeasures for chemical, biological, radiological, and nuclear threats, pandemic influenza, and emerging infectious diseases through product requirement setting, product development, stockpile acquisition/building, manufacturing infrastructure building, and product innovation. BARDA resides within ASPR, manages the PHEMCE, and has the procurement authority for Project BioShield acquisitions using the Special Reserve Fund (http://www.hhs.gov/aspr/barda/index.html). |
evaluating existing models or systems having similar goals of developing medical products with low commercial viability (Box 1).4
|
BOX 1 Workshop Objectives
|
About This Summary
This document highlights and summarizes the work presented at the workshop with the hope that this information will help federal officials to conduct a thorough review of the pipeline through approval spectrum of our national programs and to assist in the ultimate goal of improving the efficiency and effectiveness of the countermeasures enterprise. Whenever possible, unique ideas or concepts presented at the meetings are attributed in this report to the individual who first advanced those concepts. In situations where many attendees made similar points, the
|
4 |
Audio files, slides, and the meeting transcript are available for download via the Preparedness Forum’s website, http://www.iom.edu/preparednessforum. |
recurring themes are identified. The final section of the summary lists a number of suggestions for improving the medical countermeasures enterprise, including a number of suggestions focused on countermeasure regulation and licensure. They are compiled here as part of the factual summary of the workshop, and should not be construed as reflecting consensus or endorsement by the workshop, the Forums, or the National Academies. Investigating details about the feasibility and implementation of these ideas were beyond the scope of the workshop and this summary.
Charge to Workshop Participants
In her opening comments and charge to the workshop participants, the HHS Assistant Secretary for Preparedness and Response, Nicole Lurie, said that time and time again, it has been apparent that the United States does not necessarily have the countermeasures needed to respond to a public health emergency, regardless of whether it is natural or initiated by humans. Although signs of progress have been apparent in recent years, much more work is needed to protect the nation against the range of potential threats. Using the recent H1N1 influenza pandemic as an example, she also noted that even when countermeasures are available, low levels of public acceptance of the countermeasure can inhibit an effective response, and significant public education efforts may be required.
A primary goal of the end-to-end review of the public health countermeasures enterprise is to understand, in enough detail to be actionable, the challenges related to the current approach to develop countermeasures and the opportunities to improve them. Many of the challenges are already well known. Lurie urged workshop participants to be frank and forthcoming in offering creative solutions, calling for a very granular and specific focus on understanding the needs and developing strategies for systemic change. ASPR is seeking to understand how the incentive structures, policies, and procedures are, or are not, aligned with the needs of the pharmaceutical and biotechnology industries, the United States government, and the American people. The discussions at the workshop also helped inform the deliberations that were under way by the National Biodefense Science Board (NBSB),5 which was charged by
HHS to conduct a parallel examination of the related strategic management, leadership and accountability structure of the PHEMCE (NBSB, 2010a).
BACKGROUND
To aid their review, ASPR commissioned a set of briefings from PRTM Management Consultants that was presented at the workshop (Box 2). Two of these briefings, #1 and #3, were developed into white papers that serve as Appendixes D and E of this workshop summary.
|
BOX 2 Highlights of Commissioned White Papers Case Studies of the HHS Medical Countermeasure Programs: Briefing #1 Select case studies of Department of Health and Human Services (HHS) medical countermeasure programs were examined (anthrax, smallpox, hematopoietic acute radiation syndrome, viral hemorrhagic fevers, broad-spectrum antibiotics for bacterial threats) to evaluate: What were the successful elements of each program? What were the setbacks, real or perceived failures, of each program? What improvements could be made to improve future programs? Although there is not one event/characteristic that portends failure or guarantees success, there are shared risks identified in each case study, and some common factors that appear to increase the likelihood of success. Three factors that impact successful drug development are a failure in efficacy, a failure in safety (accounting for about two-thirds of failures), and failure in commercial considerations (e.g., cost to bring the product to market, perceived profitability of the product). Common factors of successful programs are strong leadership from the top, realistic expectations, experienced people, mature organizations, and adequate resources. |
|
guidance to the Secretary on other matters related to public health emergency preparedness and response” (http://www.hhs.gov/aspr/omsph/nbsb/). |
|
Optimizing the Medical Countermeasure Product Pipeline from the Science Base Through Advanced Development: Briefing #2 This briefing addresses how the product pipeline can be increased to improve the chances of producing approved products for the Public Health Emergency Medical Countermeasures Enterprise (PHEMCE), identifying challenges and looking to other programs for solutions. Although no single model or specific solution ensures success, observations from comparative research and development models suggest that better management structures, strategic decision making, better definition of requirements, target product profiles, and defined metrics of success may increase the PHEMCE pipeline of candidate products. Successful models have incorporated partnerships to optimize limited funding, market assurance, and pursuing products with multiuse potential. Synthesis of Business Models and Economic and Market Incentives for Vaccines and Therapeutics: Briefing #3 Increasing the level and mix of pharmaceutical and biotechnology company engagement can bring critical knowledge and experience to the PHEMCE. Based on interviews and the literature, three major deterrents to industry engagement in medical countermeasures development were identified: requirements (insufficient granularity/clarity about what the government wants, what companies are being asked to make, how it will be sold); return on investment (unpredictable, unsustainable market); and uncertainty in the regulatory pathway. This briefing explored multiple push and pull incentives for attracting industry participation that have been proposed or implemented in other contexts, but have not yet been applied to medical countermeasures development. No one push or pull incentive is sufficient to attract experienced companies to participate in medical countermeasures development. Similarly, there is no “silver bullet” combination of incentives. The right response depends on context. |
A report by the NBSB titled Optimizing Industrial Involvement with Medical Countermeasure Development was also presented as background for the discussions. John Grabenstein of Merck Vaccines, who is a member of the NBSB, said numerous chemical, biological, radiological, and nuclear countermeasures are still needed beyond those licensed medical countermeasures currently available in the Strategic National Stockpile (NBSB, 2010b). The Project BioShield Act provided for a procurement fund to foster the development of medical products that did not yet exist. Although subsequent legislation attempted to target resources for the advanced development of countermeasures, this funding has never been adequate. Although it is important to ensure the procurement resources remain available, Grabenstein explained, far
greater resources are currently required to ensure necessary countermeasures research and development.
In describing the findings from the NBSB report, Grabenstein said the U.S. government’s medical countermeasures enterprise has made several important advances in improving the environment for countermeasure development, including the creation of BARDA, the option for an Emergency Use Authorization (EUA),6 and the Animal Rule,7 as well as the new HHS and DoD commitment toward an “Integrated Portfolio,” and the PHEMCE holding stakeholder meetings and workshops. However, barriers hindering industry involvement in the development of countermeasures remain, including inadequate and inconsistent funding, opportunity costs (e.g., distractions from other company priorities), economics (e.g., financial margins and low volumes), uncertain regulatory pathways, finite human capital (a limited number of people having the necessary, specialized skill sets), the complexity of working with multiple federal agencies, inadequate federal government understanding of the commercial biopharmaceutical enterprise, and the use of an acquisition system that was originally created to procure complex mechanical equipment such as aircraft, vehicles, and ships (NBSB, 2010b). To begin to address these issues, the NBSB report offers a list of eight specific recommendation for the government, which are further detailed in the full report (Box 3).
|
6 |
Under Section 564 of the Federal Food, Drug, and Cosmetic Act, as amended by Project BioShield Act of 2004, the Commissioner of the Food and Drug Administration may authorize the use of an unapproved medical product, or an unapproved use of an approved medical product, during a declared emergency involving a heightened risk of attack on the public or U.S. military forces, or a significant potential to affect national security (http://www.fda.gov/RegulatoryInformation/Guidances/ucm125127.htm). |
|
7 |
The Animal Rule allows for the approval of drugs (21 C.F.R. 314.600) or biological products (21 C.F.R. 601.90) based on evidence of effectiveness from studies in animals under certain conditions when human efficacy studies are not ethical or feasible. |
|
BOX 3 Specific Recommendation of the National Biodefense Science Board to the U.S. Government
SOURCE: NBSB (2010b). |
The Public Health Perspective on Medical Countermeasure Development, Acquisition, and Use
A key challenge for the countermeasures enterprise is how to achieve the greatest health impact in the face of diminishing resources. Thomas Frieden, director, Centers for Disease Control and Prevention (CDC), said an effective response starts with several basic principles, as follows:
-
Define what is needed. Identify and characterize the threats; identify the at-risk groups and the specific needs of different at-risk subgroups (e.g., pediatric use); determine if new countermeasures are needed; and interface with the intelligence community. Defining what is needed involves a combination of pathogenesis, pathophysiology, the likelihood of use, and the likelihood of dispersal.
-
Decide what to make, and make it. Assess countermeasure availability; secure EUA as needed; develop stockpiling, distribution, and dispensing logistics; plan for countermeasure use and response; and secure licensure. This will require significant and consistent
-
investments, and a consistent way to work with industry productively, collaboratively, and perhaps most important, predictably.
-
Ensure that the countermeasures that are developed reach the people who need them most, using everyday systems that can be scaled up. This may require investing in the establishment or enhancement of more everyday systems (e.g., laboratory, epidemiological, vaccination, or healthcare systems). In this regard, Frieden cited the public health response to the recent H1N1 influenza pandemic. Over 100 million doses of vaccine were distributed with next-day delivery to more than 70,000 sites for vaccination. Despite several recalls, the distribution system for H1N1 vaccination worked extremely well because it used the infrastructure of the Vaccines for Children Program. In contrast, there were significant challenges with the distribution of antiviral medications because the public health system does not have an everyday route for dissemination.
-
Monitor the countermeasures and communicate with the public. Assess effectiveness, determine if supplies are sufficient to meet demand, determine how to increase demand to improve protection of the public, identify and interpret adverse effects, and look for changes in susceptibility of the pathogen to the countermeasure. Public acceptance of countermeasures depends on monitoring safety signals, analyzing risk, and communicating results frequently.
Going forward, Frieden said, better countermeasure delivery will require better intelligence about the presence, modification, and weaponization of different agents; storage and deployment logistics, evidence-based clinical recommendations and algorithms for use; and laboratory capacity that can adapt to the unexpected.
The FDA Perspective on the Countermeasures Enterprise: Moving Forward
In his keynote address to the workshop, Jesse Goodman, chief scientist and deputy commissioner for science and public health (acting) of the Food and Drug Administration (FDA) emphasized that the time is right for action on medical countermeasures and pandemic preparedness. The public health and national security needs are clear; there are multiple insights from the accomplishments and limitations of Project BioShield and from the experiences with 2009 H1N1 influenza; the public, policy makers, and the administration are interested; and there is bipartisan engagement and collaboration across agencies.
In that regard, Goodman highlighted the FDA’s February 24, 2010, announcement with the NIH of a new partnership to advance translation of innovations from basic science to products, including a focus on regulatory science. The agencies will establish a joint leadership council, and jointly issue a Request For Applications with the intent of awarding $6.75 million for research on novel technologies and approaches applicable to the development and regulatory review of medical products. Going forward, Goodman said, FDA is focusing on the following four key principles:
-
End-to-end partnering, including highly interactive and collaborative engagement and outcomes-oriented management. This means defining how products will be used up front, determining the pathways necessary to evaluate and regulate the product, and identifying scientific gaps. Making regulatory requirements clear is needed to reduce uncertainty. Oversight and review of progress at high levels is also necessary.
-
Increased attention to regulatory science, to expand agency capacity and knowledge and thereby enhance the quality and integrity of FDA decision making. Develop, assess, and provide tools, methods, models, standards, guidance, and pathways to evaluate product safety, efficacy, and quality (e.g., biomarkers; surrogate endpoints; adaptive and other flexible clinical trial designs; rapid scale-up of production; and rapid methods to assess purity, potency, quality, and contamination). Key elements include leadership and coordination within the agency, training and development of FDA staff, and targeted research within the agency.
-
More agile platform and multiuse technologies (e.g., vaccine, diagnostic, or monoclonal platforms) that can be rapidly adaptable to address new pathogens. (Goodman noted that platforms will not perform for all pathogens and diseases, and concrete experience with real products is needed to provide enhanced predictability of results and reduce regulatory requirements.)
-
Policies that meet public health needs. For example, although the EUA is a public health success, it can be cumbersome; the Animal Rule needs to reexamined in light of experience and scientific needs and realities; and consideration of accelerated approval approaches needs to be expanded. Are there other approaches or statuses short of full approval that should be considered?
Government Procurement of Science
Michael Kurilla, director of the Office of Biodefense Research Affairs at the National Institute of Allergy and Infectious Diseases (NIAID), explained that from the NIH perspective, there are three basic mechanisms for procuring science, with increasing focus and control. The first mechanism is geared toward increasing basic science knowledge and generating novel concepts, and the primary mechanism is grants. When the intent is vetting concepts (i.e., reduction to practice to demonstrate feasibility), the mechanism usually involved is a cooperative agreement, including small business grants and technology-transfer arrangements. When the goal is to take a product forward, from target identification to lead to candidate to human testing, the NIH relies on contracts with defined deliverables.
In addition to funding, NIH provides a number of services. Specialized services are available as needed and include, for example, sequencing, reagents, screening, animal model development, and containment. Gap-filling services are focused efforts to advance products, and involve traditional preclinical and clinical drug and vaccine development activities.
To facilitate the discussions, Kurilla offered a quick review of programmatic terminology (Table 1).
TABLE 1 National Institutes of Health Programmatic Terminology
|
Term |
Activities |
Management and Review |
|
Project |
Single effort focused on a specific candidate countermeasure against a specific agent |
Success in meeting milestones and time lines |
|
Portfolio |
Focused effort typically organized around a single threat agent with multiple countermeasures |
Adequacy of individual projects to cover the range of desired candidates, technical approaches, and developmental maturity |
|
Program |
Overall effort focused on multiple countermeasures against multiple threats |
Progress across total threat space with emphasis on desired approaches |
PARTNERS IN A SINGLE MISSION, DIVERSE CONCERNS AND CHALLENGES
Over the course of the workshop, participants highlighted some of the challenges, gaps, and barriers facing those involved in the countermeasures enterprise. While by no means a comprehensive review, these are some of the more pressing concerns that informed the subsequent
discussions on optimizing the countermeasures enterprise. A vast array of structural, strategic, technical, financial, and even cultural elements are involved in the research, development, production, and deployment of medical countermeasures for public health emergencies.
The Growing Threat of Bioweapons
D. A. Henderson, former director of the Office of Public Health Emergency Preparedness, and distinguished scholar at the Center for Biosecurity of University of Pittsburgh Medical Center (UPMC), stressed that countermeasures development needs to be approached with a real sense of urgency, noting that the situation with regard to anthrax is not much better than it was 8 years ago. Today, he said, we do not have that sense of real urgency we felt after 9/11, and yet there is an equal likelihood that an event could occur tomorrow.
Compared to nuclear and other weapons technology, bioterrorism is “relatively easy.” In a recent editorial, former senators Bob Graham and Jim Talent said they believed it is unlikely that the United States can ever prevent bioterrorism (Graham and Talent, 2009). Rather, the senators stressed that America’s best long-term strategy for biodefense is redefining its prevention efforts, striving to reach a level of preparedness that effectively removes bioweapons from the category of weapons of mass destruction (WMDs).
With this in mind, Graham, Talent, and Randy Larson, who served as executive director of the Commission on the Prevention of Weapons of Mass Destruction Proliferation and Terrorism, formed the Bipartisan WMD Terrorism Research Center, a 501(c)(3) organization operational as of March 1, 2010. The organization’s primary focus will be education, ensuring that those in leadership positions in the federal government understand the imminent threat that biological weapons present.
Larson urged workshop participants working with and within government on the countermeasures enterprise to request the Department of Homeland Security Office of Science and Technology’s population threat assessment briefing. The threat is real, Larson said, but people do not have a full understanding of that threat and therefore do not always apply themselves fully toward solutions.
Gaps and Barriers to International Collaboration
Maria Julia Marinissen of ASPR reminded participants that the threat of terrorism with chemical, biological, radiological, and nuclear agents,
and the spread of pandemics and other potential emerging infectious diseases, are global issues. The United States has experienced a steadily increasing demand for the supply of medical countermeasures to foreign countries. However, it is virtually impossible for a single country to fund research and development, acquisition, and stockpiling programs for medical countermeasures for all, or even most, threat agents. A global infrastructure for countermeasures is needed.
In its recommendations to the new administration, the IOM Committee on the U.S. Commitment to Global Health stated that “good health is a necessary condition for economic development and global prosperity” and concluded that this country can improve the lives of millions around the world, while reflecting America’s values and protecting and promoting the nation’s interests (IOM, 2008). However, the United States cannot become the world’s provider and pharmacy for medical countermeasures. A sustainable U.S. infrastructure depends on a larger marketplace for these products.
Over the past 2 years, Marinissen said, ASPR has been pursuing a strategy to work with international partners to build a sustainable global infrastructure for medical countermeasures. For developed countries, one effort under way uses the Global Health Security Initiative (GHSI).8 ASPR held GHSI medical countermeasure workshops in 2008 and 2009 to determine areas of interest for collaboration and to identify current gaps and barriers to international collaboration. GHSI will conduct an exercise to consider a single threat (anthrax) as a case study to identify gaps and concrete areas for collaboration. Major gaps and barriers to international collaboration identified included
-
Countries perceive threats differently. There is a need for improved surveillance of threats, increased information sharing, and joint development of assessment tools.
-
There is a need for information sharing to maximize resources and avoid duplication of efforts, for harmonized country regulatory requirements for market authorization and expedited clinical trial proc-
|
8 |
GHSI is a forum for high-level discussion concerning the coordination of public health emergency preparedness and response policies for CBRN threats and pandemic influenza. It was launched in 2001 by the ministers of health of Canada, France, Germany, Italy, Japan, Mexico, the United Kingdom, the United States, and the European Commission. The World Health Organization serves as an expert advisor. A ministerial-level summit is held every year to share information and coordinate efforts to improve global health security. See http://www.ghsi.org/. |
-
esses, and for innovative and modernized vaccine production processes.
-
Countries should collaborate on point-of-care diagnostic tools, stockpiles, emergency deployment plans, and harmonization of treatments and use policies.
With regard to the developing world, Marinissen said that ASPR is initiating international discussions on a framework and strategic plan to create regional, independent, and sustainable influenza vaccine production capacity in developing and emerging economy countries. Such capabilities could then be used as a platform for surge capacity for pandemic vaccine.
Gillian Woollett, chief scientist, Engel & Novitt, LLP, cautioned that the United States might not want biopharmaceutical companies selling their countermeasures all around the world, and questioned the ability to control the use of an effective countermeasure. This could make the situation worse, she said, if the United States spent large amounts of money to develop a countermeasure, and someone buys it in order to protect his or her own people or engineers a different or resistant threat.
Issues for Federal Agencies Engaged in Countermeasures Development
Systemic Concerns
Philip Russell, Major General, U.S. Army (ret.), former senior advisor in HHS’s Office of Public Health Preparedness and current member on the Board of Trustees of the Sabin Vaccine Institute, highlighted a number of systemic concerns impacting the effectiveness of the countermeasures enterprise. Reliance on an unwieldy and ineffective contracting process is a primary challenge across the board for all participants in the countermeasures enterprise. The Federal Acquisition Regulation (FAR) is unsuitable for product development in the pharmaceutical field, Russell said. The FAR restricts communication, and contractors generally lack experience and capability in the full range of skills needed to bring a medical product to licensure.
All product development paths must ultimately lead to the FDA, and the regulatory process can be cumbersome and fraught with uncertainty. Two key barriers were highlighted by workshop participants. The first, which will also be discussed later in this report, is the absence of a clear and consistently applied regulatory pathway for medical
countermeasures. The Animal Rule is also presenting itself as a barrier. The Animal Rule says FDA may grant approval when “the results of those animal studies establish that the drug is reasonably likely to produce clinical benefit in humans.” However, the guidance is significantly more restrictive than the Animal Rule itself and is being appropriately administered relative to the regulation of vaccines for biodefense, Mary Pendergast and Russell said.
As will be discussed later in the report, the lack of central leadership impacts the ability to bring together the numerous agencies involved in the countermeasures enterprise. (This topic was also a focus of a meeting hosted by the NBSB.) This viewpoint was shared by many workshop participants. However, others cautioned that although it is important to ensure that the DoD’s efforts are aligned and coordinated, it may also be important to maintain a level of independence due to complementary, but separate, missions.
Project Bioshield and BARDA Resources
The Project BioShield Act became law in July 2004 (Public Law 108-276) and provides for procurement of countermeasures. However, many of the products it seeks to acquire are not yet available for purchase. As BARDA Director Robin Robinson explained, the 2006 Pandemic and All Hazards Preparedness Act (Public Law 109-417) included a corrective measure, establishing BARDA and provisions for supporting advanced development of products. BARDA is responsible for advancing projects funded by the NIH and the DoD, by moving them across the high risk advanced development zone (the “valley of death”) to a point where acquisition and stockpiling can be achieved.
Although BARDA is off to an enormously good start, said Eric Rose, chief executive officer (CEO) and chair of Siga Technologies, it is a young organization on a steep learning curve. An economic analysis by Bradley Smith of the Center for Biosecurity, UPMC, and colleagues found that the advanced development mission of BARDA is underfunded by at least 10-fold and consequently its portfolio is very thin (Matheny, 2008).
As part of the Project BioShield Act, money for countermeasures procurement was set aside in escrow so that purchases could be made without needing to go back to Congress for an appropriation. Procurement authority for Project BioShield acquisitions using the Special Reserve Fund rests with BARDA. Chuck Ludlam, former counsel to Senator Joseph Lieberman and former principal lobbyist for the Biotechnology Industry Organization, and other participants
expressed concern that money from the Special Reserve Fund is being diverted to other initiatives. For example, in the 2009 Omnibus Appropriations Act, $412 million was transferred to other programs to support countermeasure advanced research and development and pandemic influenza preparedness and response. In FY2010 an additional $305 million has been proposed to be transferred to support countermeasure advanced research and development. This could have serious repercussions on the government’s ability to guarantee a suitable marketplace for future countermeasure procurement. This also leads to continued uncertainty among the private sector about long-term, stable funding for countermeasures research and development.
FDA Funding and Scientific Infrastructure
To highlight the challenges facing FDA today, Gail Cassell, workshop chair and vice president, Scientific Affairs, at Eli Lilly and Company reviewed the findings of the report FDA Science and Mission at Risk (FDA, 2007). The FDA Science Board Subcommittee on Science and Technology, chaired by Cassell, was charged by then-Commissioner Andrew von Eschenbach to review science and technology across the agency to answer the question of whether FDA is prepared to address emerging technologies in science. The subcommittee concluded that “science at the FDA is in a precarious position: the [a]gency suffers from serious scientific deficiencies and is not positioned to meet current or emerging regulatory responsibilities.” Major findings of the report are presented in Box 4. The Subcommittee found that the deficiencies had two main two sources:
-
The demands on the FDA have soared due to the extraordinary advance of scientific discoveries, the complexity of the new products and claims submitted to FDA for premarket review and approval, the emergence of challenging safety problems, and the globalization of the industries that FDA regulates.
-
The resources have not increased in proportion to the demands. The result is that the scientific demands on the agency far exceed its capacity to respond. This imbalance is imposing a significant risk to the integrity of the food, drug, and device regulatory system, and hence the safety of the public. This also raises the issues of the threats associated with a bioterror attack and the critical role of FDA in the development of medical countermeasures.
|
BOX 4 FDA Science and Mission at Risk: Major Findings
SOURCE: FDA (2007). |
Cassell noted that FDA had been given more than 100 unfunded mandates over the previous 15 years, while staffing did not increase concurrently to meet these new mandates. The agency is also responsible for conducting inspections at more than 300,000 sites in 100 countries. The FDA has a huge economic impact, regulating 25 cents of every dollar that Americans spend—over $1 trillion worth of products ranging from cosmetics to pet food. Yet in 2007, FDA had an appropriated budget of only $1.6 billion, which is about 1.5 cents per day per American.
While the agency has made progress in addressing each of the major deficiencies noted in the report, much more needs to be done because regulatory and information sciences are the very foundation of the FDA’s mission. They are critical to the agency’s role in development of medical countermeasures for biodefense. Although the world of drug discovery and development has undergone revolutionary change—shifting from cellular to molecular and gene-based approaches—FDA evaluation methods have remained largely unchanged over the past half century. Likewise, evaluation methods have not kept pace with major advances in medical devices and use of products in combination.
The Subcommittee noted that the impact of the deficiency is profound precisely because science is at the heart of everything FDA does. The world looks to FDA as a leader—to integrate emerging understandings of biology with medicine, technology, and computational mathematics in ways that will lead to successful disease therapies.
Today, not only can the agency not lead, it cannot even keep up with the advances in science. Due to constrained resources and lack of adequate staff, FDA is engaged in reactive regulatory priority setting or a firefighting regulatory posture instead of pursuing a culture of proactive regulatory science.
The Subcommittee identified the following eight emerging science and technologies that are the most challenging to the FDA: systems biology (including genomics and other “omics”), wireless healthcare devices, nanotechnology, medical imaging, robotics, cell- and tissue-based products, regenerative medicine, and combination products. Each of these emerging areas is developing at an exponential rate and each generates novel scientific, analytic, laboratory, and/or information requirements. These areas are also precisely those that have been identified as being critical to development of medical countermeasures. Furthermore, the FDA cannot fulfill its surveillance mission because of inadequate staff and IT resources to implement cutting-edge approaches to modeling, risk assessment, and data analysis. The status of regulatory and information sciences at FDA must consider our ability to successfully address the threats of bioterrorism. Other participants concurred, noting that there is no surge capacity at the FDA, or that in fact it is already operating at surge capacity.
Challenges Facing the Innovative Biopharmaceutical Industry
A focus of the workshop was to identify how to improve innovation in ways that respond to national priorities, including how to better engage the nation’s commercial drug, biologic, and device manufacturers in the countermeasures enterprise. Participants from industry described a variety of barriers and challenges to commercial involvement.
Risk and Uncertainty
Most new or in-development pharmaceutical products fail, said John Rex, vice president and medical director for infection at AstraZeneca. Philosophically, a company starts with that understanding, and designs programs to manage risk and to identify failures quickly and cheaply, without committing too many resources, until there is a reasonable level of confidence in the product. The pharmaceutical industry is very good at models and methods to help address the scientific, technical, formulation, and safety risks, for example. The risks that drive industry away occur when changes happen that cannot be readily anticipated—for example, when regulatory guidance is not clear.
Regulatory uncertainty at the FDA was a recurring theme during the discussions. For small companies in particular, this uncertainty can be compounded by a limited understanding of the regulatory pathway. The investment community also has a keen interest in the success or failure of industry pursuits. A participant from an investment bank, Stephen Brozak of WBB Securities, LLC, noted that “Wall Street hates uncertainty.” FDA adds uncertainty because financial analysts have no way to quantify how long the approval process will be for a product. The new draft guidance from the FDA caused significant uncertainty that hampered investors’ ability to predict regulatory, and consequently, revenue trends. If the FDA could establish a clear regulatory pathway in biodefense, it would allow the analysts some sort of metric to be able to say “if a company does this, that will happen.” That is likely to foster greater interest and investment in companies doing research in countermeasures, a point highlighted by multiple workshop participants.
Material threat determinations (the list of pathogens of concern) are public, but material threat assessments and population threat assessments are classified. So while the PHEMCE does provide some highly desirable predictability with regard to identification of the targets for discovery and development, companies have no information regarding the planning scenario for which they are trying to build a product. Although it is understandable that industry is not included in the PHEMCE, this leaves the countermeasures enterprise itself with a critical lack of business and capital markets expertise. The PHEMCE implementation plan itself provides limited guidance and is essentially a list of pathogens and agents that the government hopes to acquire. This kind of checkbox approach obscures product shortcomings and regulatory gaps. Participants also suggested that the implementation plan is somewhat counterproductive in that it defines the market, arbitrarily, as either above or below $100 million. Consequently this means that companies will not invest in developing products predicted to gross less than $100 million.
Not knowing how a product is going to be commercialized is also a risk that industry prefers to avoid. The manufacturing of biologics is complex, and there is an enormous difference in manufacturing, for example, 200,000 doses versus 40 million doses. Companies need guidance regarding volume so they can develop manufacturing plans.
The acquisition process (for initial stockpiles until product licensure), which is essentially guided by the Federal Acquisition Regulation, is perceived by the industry to be lengthy, opaque, unpredictable. In particular, the transition trigger from advanced
development to acquisition Request For Proposals (RFP) is unclear. After acquisition, there is a perceived improved communication compared to acquisition process, but some aspects remain unclear, particularly FDA coordination with BARDA. As a result, Goodman’s key principle of end-to-end partnering, including highly interactive and collaborative engagement and outcomes-oriented management, takes on an increasingly important role.
Financial and Resource Concerns
The most often mentioned financial barrier to engaging bio-pharmaceutical companies in the countermeasures enterprise is lack of market incentive. Wesley Yin, assistant professor in the Department of Economics at Boston University, said that firms simply are not going to be able to recoup the fixed costs of research and development of countermeasures. Unlike a standard low-prevalence disease, not only is demand low, but it is also uncertain. If there is demand, it usually comes in times of public health emergency. Plus, there is pressure, real or perceived, to sell these technologies at or just above marginal costs. In addition, if a company overcomes the revenue risks and pursues development of a product, it is at risk for product liability issues, which are also a financial and resource burden.
Lack of market incentive aside, Thomas Monath of Kleiner Perkins Caufield & Byers pointed out, participation in the countermeasures enterprise has a huge opportunity cost—taking away a company’s ability to focus on its commercial opportunity market. Woollett, of Engel & Novitt, added that the industry is actively making products that are saving lives, and asked whether those lives are any less important than a putative potential threat. Simply adding a capability is not an option unless we are prepared to take away from something else, she said.
Participants also highlighted that while companies are interested in countermeasures development, the long-term financing piece must be addressed to be able to make a more rational business case for devoting company resources to countermeasures. The ability to plan for the future can speed up everything tremendously (e.g., if the first step is successful, the company can move to the next step, and already be planning for the next clinical trial, without an interim funding step).
An issue for biotechnology companies is that they are generally small, unprofitable entities that are sustained by private capital and government grants and contracts. Many do not survive. But these companies are an integral part of the PHEMCE implementation plan. Rose of Siga Technologies said BARDA has been an excellent,
responsive development partner for the biotechnology industry. BARDA funding for direct project costs are reasonable, realistic, and flexible. He said, however, that the funding for indirect costs is only a fraction of actual costs, and has to be supplemented by in-house funding and private capital in order to keep these projects going for the 8 to 12 years that drug development generally takes. For small innovative companies, programs may start and stop, but funding cannot be discontinued and started again. Small organizations depend on that ongoing revenue to continue to employ staff.
Another enormous drain on resources for small biotechnology companies is the RFP process, said David Wurtman, vice president at NexBio. Although it can be quite constructive for companies to think through the entirety of a development-to-manufacturing plan, if the company is small, research may come to a halt as all hands focus on the RFP. Despite the efforts companies make to respond to an RFP, they often do not find out if they have been awarded a contract, which can present difficulties in planning for the future, especially if the company is small. From a human resources perspective, advanced development manufacturing is really an apprentice model. University training to grow a pool of talent is limited, if it even exists, said Phillip Gomez of PRTM. Therefore, it is important to provide opportunities for partnerships between academia and industry, where the advanced development manufacturing expertise rests. This will help grow the base of people with this expertise and help people learn from a variety of perspectives in the enterprise.
Intellectual Property and FDA Approval
The protection of intellectual property is at the core of the industry’s ability to earn a return on research investments and remain competitive. The ideal situation, according to Bruce Artim of Eli Lilly and Company, would be for an innovator to be awarded a patent by the Patent and Trademark Office (PTO) on the same day FDA approval is granted. However, in practice, FDA product approval review generally takes much longer than PTO patent application review, effectively reducing the patent protection period. A significant policy challenge is balancing two needs: (1) the need of the innovator drug company both to recoup costs and to profit and grow so it can continue to innovate, and (2) the need to bring less expensive generic versions of products to market. The 1984 Drug Price Competition Patent Term Restoration Act, commonly referred to as the Hatch-Waxman Law, is extremely complex, but at its core, it allows a generic drug manufacturer to refer to the pioneer drug
developer’s data when applying for FDA approval of the generic form.9 The pioneer also receives additional years of patent term to compensate for some of the time the drug was already on patent while still under the lengthy clinical development and FDA review periods. The pioneer also receives 5 years of data exclusivity. During that time, FDA cannot approve any generic drug applications for a comparable product. Basically, Artim said, this incentive system places more importance on patent term as an intellectual property tool than data protection. Therefore, companies invest resources where they believe they have strong patent protection. However, no correlation exists between the patent protection and the scientific or clinical value of the molecule.
Special Considerations for Antibiotics
Because small-molecule antibiotics have dual uses (both as standard medical care and as medical countermeasures), one might think they would be the countermeasure with the simplest development pathway. But this is not necessarily the case, said Rex of AstraZeneca.
A variety of considerations are specific to the development of antibiotics. First, Rex said, discovery and development are iterative. Simple “gateway indications” provide the entry point, and securing approval for the gateway indication (e.g., community-acquired pneumonia) opens the door to many other uses down the road, including countermeasures. But if a company cannot achieve approval for the basic clinical indication, nothing will follow. Second, bacterial resistance drives the need for novel antibiotics. The ideal comparative clinical trials—new drug versus the drug to which the organism is resistant, or a placebo-controlled superiority study—simply cannot be done for obvious ethical reasons. Rather, non-inferiority designs versus an active agent must be used. This approach has caused significant regulatory confusion. Non-inferiority trial design is more difficult to implement than superiority designs. Following approval, the new drug is subsequently perceived as only non-inferior rather than superior because its activity when other drugs would be resistant is not apparent. Finally, there is the paradox of antibiotic value. A new antibiotic may be deemed so important that it is not used, reserved only for situations when all else fails, which presents a problem for companies who plan to recover some of their development costs though sales. Pricing of the new antibiotic is also a challenge, especially when the new drug has only been shown to
be non-inferior to an existing generic drug, raising the question of why it should then cost more.
Research Infrastructure and Resources
Tools and Methodology
Infrastructure for rapid countermeasure development requires common data elements in both research and practice across the board, summarized Marietta Anthony from the Critical Path Institute. A unique scientific issue for countermeasures research is not having the disease to study in many cases, and the development of clinical disease/clinical injury models for trial simulation would be very useful to help speed the process. Innovation also needs to be brought to clinical trial design (e.g., adaptive clinical trial design). Biomarkers that are qualified for use by the FDA to reliably and accurately detect diseases in the field, or detect changes in the field, are also needed. Rapid point-of-care testing and resistance testing was cited as a need by state health departments.
Whether products are to be for engineered threats or natural pathogens, other research needs include vaccine adjuvants, cell culture manufacturing, expansion of biologics manufacturing capacity, and decontamination and remediation protocols after an attack or exposure.
Academia
In general, academic research, and to a large extent government research, are not intended to produce products. In academia, grant funding and publications are highly valued and are the currency for tenure or promotion, noted Brett Giroir, vice chancellor for research at Texas A&M. Product development, intellectual property, and commercialization, while not discouraged, are generally not fostered or rewarded.
The basic research funded by government and conducted in academia is, in general, not prioritized by national need. If research is successful in identifying a potential product, there are no transition partners lined up and no clear pathways for investigators to carry their discovery forward. As a result, it is likely that government and industry are aware of only a very small fraction of the innovations from academic laboratories that could eventually lead to products, Giroir said.
Liability
While believing that the 2006 Public Readiness and Emergency Preparedness (PREP) Act (Public Law 109-148)10 went a long way to address liability issues, a number of participants cautioned that there are gaps and holes that have not been filled. For example, it is unclear if PREP Act declarations preempt state tort law, which can result in continued liability concerns for end users. There is also liability in terms of what must be disclosed for informed consent in case of an emergency.
End Users: Challenges for Public Health and Providers
Although public health officials at the state and local levels are not directly involved in the research and development of countermeasures, they are responsible for ensuring the safety of the public by implementing whatever comes out of the countermeasures enterprise. Therefore, the needs of the public health as end users should inform the target product profiles. State and local public health and healthcare providers all play a critical role in the delivery of countermeasures. The need to integrate these individuals much earlier into the process of research and development of the countermeasures, perhaps through an advisory board to BARDA, was highlighted at the workshop as an opportunity.
Susan Cooper, commissioner of the Tennessee Department of Health, commented that although an abundance of product variations may seem like a benefit, it adds significant complexity to state implementation activities. For example, influenza vaccines come in single-dose syringes, multidose vials, and intranasal mists, each with its own labeled uses in different subpopulations. These different products are shipped as they became available, making distribution to different real-time providers a challenge. She also noted that the variety of forms of the vaccine confounds public health messaging. The Advisory Committee on Immunization Practices guidelines, for example, identified the priority groups to be vaccinated first as pregnant women, children with chronic diseases, healthcare workers, persons between the ages of 6
|
10 |
A “PREP Act declaration” by the HHS Secretary provides immunity from tort liability (except for willful misconduct) for claims of loss associated with the administration or use of medical countermeasures to threats that are deemed by the Secretary to constitute a public health emergency, to those involved in the development, manufacture, testing, distribution, administration, and use of such countermeasures (http://www.hhs.gov/disasters/discussion/planners/prepact/index.html). |
months and 24 years, and persons from ages 25 through 64 years who are at higher risk for novel H1N1 because of chronic health disorders or compromised immune systems. Unfortunately, following statewide media campaigns urging these groups to be vaccinated, the first product received in Tennessee was the intranasal mist—which cannot be given to pregnant women, children with chronic disease, and those over the age of 50, which would include many healthcare workers. So although states understand the challenges of developing countermeasures and acknowledge that choice is important, an abundance of choices can actually complicate implementation.
Although not a focus of this workshop, a recurring theme was the importance of investing in the public health infrastructure and delivery, noting that the most effective products have no value if you cannot get them to people who need them, or if people do not trust the product. State and local public health departments will require epidemiologic and laboratory capacity; robust emergency drill programs; management, logistics, and communication capacity; strong links with healthcare systems; and integration across a variety of non-health sectors (e.g., police, transportation, education). Participants specifically called out rapid point-of-care testing and resistance testing as current needs, noting that the ability to detect resistance in anything close to real time is very difficult.
EXAMPLES OF SUCCESSFUL COUNTERMEASURES DEVELOPMENT AND DEPLOYMENT
Features of Successful Government Countermeasures Efforts
Russell, identified earlier, presented several examples of products that were successfully developed by the government over the past 30 years, including the U.S. Army’s development of products for adenovirus, meningococcus, hepatitis A, nerve gas, and malaria, and recent HHS efforts on smallpox. Looking across these successful programs, Russell noted that several shared characteristics emerge, including good scientific direction and leadership, a strong pharmaceutical manufacturer as a partner, and the internal capability to move the candidate through pilot level (Box 5).
Captain Kenneth Cole, medical director of the Nuclear and Chemical and Biological Defense Programs at DoD, said a key element of the DoD approach to countermeasures research is an oversight mechanism that considers the entire portfolio. Within the DoD there is the Joint Science
and Technology Office, which handles basic research and early development; the Joint Program Executive Office for Chemical and Biological Defense, which handles advanced development and procurement; and the Joint Requirements Office, which is involved in planning, coordination, and oversight; defining desired capabilities; defining requirements needed to get there; setting key performance parameters; and determining how the product is going to be used. Within that program is a single authority that can make the decision as to when something translates from research and early development into advanced development. As part of the oversight process, a Medical Advisory Board looks at the full spectrum of research and advance development, and makes recommendations to this milestone decision authority on which candidates to advance.
This structure allows for oversight, accountability, prioritization, and translation of requirements all the way to the research level. Throughout the process, DoD leverages interagency as well as international partners (through 64 different bilateral, trilateral, and quadrilateral chem/biodefense relationships and treaties around the world).
|
BOX 5 Characteristics of Successful Government Product Development Programs
|
Transformational Medical Technologies Initiative
A new DoD initiative is the Transformational Medical Technologies Initiative (TMTI), specifically designed to look at the emerging and bioengineered threats the warfighter faces, the DoD’s Cole continued.
The goal is to move from the traditional “one bug–one drug” approach to the transformational “one drug–many bugs” approach, developing broad-spectrum countermeasures (e.g., that target common disease pathways or enhance the host’s immune response), as well as platform technologies to characterize unknown pathogens and rapidly develop medical countermeasures to newly identified threats. The TMTI approach integrates efforts within government, academia, the biotechnology industry, and small and large pharmaceutical corporations, providing seamless “end-to-end” product development.
Cole suggested that a national medical countermeasures strategy needs to be structured with an oversight and accountability mechanism that drives the requirements from research through advanced development and provides a focus to the program. In this regard, TMTI is an excellent model.
Transformational Medical Technologies Initiative: Demonstrated Capability
A rapid response biodefense capability is the overarching TMTI goal, including broad spectrum medical countermeasures and platform technologies. As a response to needs outlined in the 2006 Quadrennial Defense Review, since 2007 TMTI has seen two Investigational New Drug (IND) submissions submitted for hemorrhagic fever viruses and is preparing two broad spectrum antibiotic INDs, three broad spectrum antivirals, and is establishing efficacy for two innate immune activators. TMTI is also repurposing drugs already licensed for use as broad spectrum antibiotics. There are over 20 candidates in the pipeline. Most importantly, TMTI has demonstrated a capability to respond to emerging threats by producing an antiviral to the emerging H1N1 virus within seven days of receiving tissue samples and then proving efficacy in standard mice and ferret models. TMTI is so successful, the Department of Defense is transitioning it from an initiative to a program of record. The TMTI strategy is to move hemorrhagic fever viruses therapeutics through IND into the regulatory critical path, move intracellular bacterial pathogen therapeutics through IND submission into the regulatory critical path, advance animal models suitable for pivotal animal studies supporting licensure, develop pathogen gene lists and attributes for assessing pathways and target identification in order to recommend medical countermeasures, and finally to integrate all of these activities to identify candidate medical countermeasures against unknown pathogens. To date, hemorrhagic fever viruses candidates are moving onto clinical trials, intracellular bacterial pathogen candidates are moving into clinical trials,
animal models continue through development and validation, end-to-end pathogen evaluation and medical countermeasure development has been demonstrated, and discovery along with pre-clinical efficacy, safety, and toxicity studies continue on further candidates for pipeline replenishment.
Lessons from Pandemic 2009 H1N1 Influenza
Goodman of the FDA also cited the recent response to pandemic 2009 H1N1 influenza as an example of success. A great deal of investments were made and significant planning was done, including at FDA, and there were excellent public–private partnerships and interagency collaboration and communication. One important aspect, he said, was that FDA set up an Incident Command System (ICS) to address the urgent needs presented by 2009 H1N1, which was extremely valuable. FDA staff were able to collaborate with other federal agencies and rapidly respond in an ICS mode.
Daniel Jernigan, deputy influenza director for the CDC, described seven actions that serve as the strategy for diagnostic preparedness. They are as follows:
-
Developing new diagnostic tests and improved capabilities
-
Improving surge capacity
-
Implementing proficiency testing
-
Developing policy and regulatory preparedness to facilitate rapid responses
-
Improving access to viruses and reagents
-
Providing guidance for clinicians
-
Improving overall virologic surveillance
Another key feature of a successful program is adaptability, said Andrew Pavia of the University of Utah School of Medicine. Planning only works to a certain point. Problem solving and flexible approaches need to be integrated into countermeasure development and emergency planning. For example, because of a study that was funded by NIAID through the Collaborative Antiviral Study Group, there were data that had not yet been published on the appropriate dosing of Tamiflu® for children with influenza. Because of a flexible response by FDA and CDC, those unpublished data were able to be used in the EUA and providers were able to treat children and save lives.
Countermeasures Development in Industry
Focusing on Unmet Medical Need, Deriving Dual-Use Products
In response to the Graham-Talent Commission report on the Prevention of Weapons of Mass Destruction Proliferation and Terrorism described earlier, the Center for Arms Control and Non-Proliferation said in a statement published on January 26, 2010, “Direct targeting of effort and expenditure on natural disease threats would provide much greater public health benefit, and spinoffs from these programs would significantly strengthen resistance to bioterrorism” (Center for Arms Control and Non-Proliferation, 2010).
George Painter, CEO of Chimerix, said his company follows the strategy outlined by the Center for Arms Control and Non-Proliferation. The company is structured to focus on unmet medical need, and if there is an opportunity to expand from that unmet medical need into a countermeasure area, the company will pursue it. For example, Chimerix was actively developing an orally available broad-spectrum antiviral drug to treat double-stranded DNA viral infections in immunocompromised patients. Orthopoxviruses (which include Variola, the agent of smallpox) are also double-stranded DNA viruses, and the company bridged into biodefense on a grant from the NIAID. Similarly, the company has a robust hepatitis C drug development program. Hepatitis C virus and dengue virus are both flaviviruses, and the company will leverage what is learned as it takes the hepatitis C lead candidate into clinical development to help address dengue.
By managing the portfolio in this manner, the company has a clear regulatory pathway to approval and a definitive, definable market size that engenders interest on the part of private capital. The company can leverage that position if the opportunity arises to maintain a position in biodefense. Another advantage of pursuing only drugs with multiuse potential, Painter said, is that a clinical treatment protocol can be left open, and in the case of an emergency, one can broadly treat through that protocol.
Tyler Martin, chief medical officer of Dynavax, said that like Chimerix, Dynavax has focused its development efforts on commercial targets and then looks to see what other value can extracted from the research discoveries already made. Dynavax is a small biotechnology company of about 100 people, focused on exploiting the biology of Toll-like receptors for product development. “Portfolio management,” he said, “is something that a biotechnology company does every day, trying to create maximum value from a limited number of resources.” The way to
do that is with target product profiles. Martin described three dimensions of the profile that are malleable: Change the scope of the assignment (increase or decrease the target product profile), change the resources available for the assignment (money, people, etc.), or change the time line to complete the assignment.
Melinda Moree of BIO Ventures for Global Health noted that the United States is not necessarily focused on products for the developing world, but innovations for these regions may have a dual application for countermeasures development. For example, a heat-stable vaccine that does not require refrigeration and has a long shelf life would be more appropriate for a stockpile than a product with a shelf life of 18 months. David Gilbert of Providence Health & Services commented that efforts to develop new antibacterials for multidrug-resistant, gram-negative rods for the civilian population would benefit the countermeasures enterprise as well.
Countermeasures as Orphan Drugs
An orphan disease in the United States is one affecting fewer than 200,000 people. Marlene Haffner of Haffner Associates and former director of the Office of Orphan Products Development said that since passage of the Orphan Drug Act in 1983, 18 products have been designated as potential countermeasures for terrorism, 4 of which have been approved (for exposure to cyanide, cesium or thallium, and pediatric exposure to radioactive iodine).
Although the orphan drug program has been able to support development of products in the arena of counterterrorism, it has drawbacks. The Orphan Drug Act gives 7 years of exclusive marketing of that product for that indication, which means that another company cannot approach FDA with the same product for 7 years. This could be a concern if a goal of preparedness is to have redundancy. The other main incentive of the Act is tax credits for clinical trial expenditures. But because clinical trials may not be possible in some cases of countermeasures development, tax credits may not always serve as a compelling incentive.
PARTNERSHIPS AND ALTERNATIVE BUSINESS MODELS
Venture Philanthropy and Orphan Product Development Models
As discussed above, there are several examples of successful medical countermeasures development under the Orphan Drug Act. Nonprofit
disease research organizations and venture philanthropy groups are a force behind much of the progress in orphan product development. These organizations were primarily founded by patients because there was not enough research focus on their particular disease area (IOM, 2009). Their model is to derisk the research. Many of these groups are partnering successfully with industry, approaching biotechnology companies directly and offering funding for research in their area of interest. Margaret Anderson of FasterCures described two new rare disease-related activities that, if implemented effectively, may also serve as an opportunity for improved medical countermeasure development—the Cures Acceleration Network (CAN) and the Therapeutics for Rare and Neglected Diseases (TRND) program.
The Cures Acceleration Network Championed by Senator Arlen Specter and part of the recently passed Patient Protection and Affordable Care Act (Public Law 111-148) CAN seeks to cut the time between discovery and development of drugs and therapies through new grant-making mechanisms at the NIH; establishes CAN within the Office of the Director of NIH and authorizes grants to move discovery from the lab into the next generation of therapies; and integrates the FDA into the work that CAN will undertake, providing a vital link between NIH and the drug approval process.
Therapeutics for Rare and Neglected Diseases program at NIH TRND is a congressionally mandated effort to encourage and speed the development of new drugs for rare and neglected diseases. Specifically intended to stimulate research collaborations with academic scientists working on rare illnesses, TRND supports specific, preclinical research and product development, leveraging the in-house scientific capabilities needed to carry out much of the preclinical development work and contracting out other parts, as scientific opportunities dictate (http://rarediseases.info.nih.gov/TRND).
Pharmaceutical Shared-Risk Approaches
The private sector is also in the process of a significant reorganization, where it is establishing a large number of partnerships—public–private and private–private—that can leverage the expertise of multiple sectors, thus reducing costs, increasing probability of success, and sharing risk. For example, Eli Lilly has moved away from a “fully integrated pharmaceutical company” model, where everything is done (and correspondingly, funded) internally, to a “fully integrated pharmaceutical network” model, explained Paul Owens of Lilly
Research Laboratories. Lilly is looking to access global networks in a virtual fashion and thereby be more productive, optimizing the speed and cost of drug development. Companies can no longer afford to have the source of ideas, the source of capacity or capability, and the source of funding all under one roof, he said.
The Phenotypic Drug Discovery program is one example of the fully integrated pharmaceutical network approach. Lilly has opened up phenotypic screening models in five disease areas of interest (Alzheimer’s disease, osteoporosis, diabetes, cancer cell growth, and cancer antiangiogenesis) to any interested external drug discovery entities (e.g., academic institutions, small biotechnology companies, individual faculty). Investigators can submit a molecule into these phenotypic drug screening panels in a confidential manner. If there is a hit, a secondary, more target-based screening will be conducted. After the full set of five panels is completed, a report is sent to the investigator. The investigator retains the intellectual property, but Lilly has the first right of negotiation for a commercial opportunity on that particular hit for a defined period of time after the screen is completed. This program has attracted hundreds of samples from all over the world. A platform of this type could be applied to many different disease areas (http://www.PD2.lilly.com).
Another example is the TPG-Axon/NovaQuest collaboration with Lilly. TPG-Axon is a venture capital firm and NovaQuest is the investment arm of Quintiles, a global clinical research organization. Together, this three-way partnership will share the risk , and the potential return on investment, for the development of Lilly’s two lead candidates for the treatment of Alzheimer’s disease. Although Lilly is giving up some value of these assets, disseminating some of the risk allows the company to invest in a larger portfolio all the way back into discovery.
Planning for Failure
As described above, several industry participants described how failure is an inherent risk in biopharmaceutical development. The sign of a mature discovery group, said Rex of AstraZeneca, is the ability and willingness to stop pursuing drug targets or candidates. Scientists can become very attached to their targets, and can be unwilling to move on regardless of lack of progress. In addition, people are less willing to abandon the only targets they may have if they perceive their employment or the survival of their project is dependent on finding a candidate from a small pool of opportunities. To address this,
AstraZeneca has removed the process of hit-and-lead candidate identification from the responsibilities of individual biochemists. Instead, a small group is now charged with assessing leads for candidates that might be drugs. There is no emotional or historical attachment to a particular target or molecule, just a focus on identifying drug leads with good, drug-like properties.
Similar to the AstraZeneca approach, Eli Lilly accepts that failures will happen and looks to identify them early, commented Owens. Chorus is a small autonomous drug development group within Lilly focused on moving molecules from candidate selection through to proof of concept as quickly and inexpensively as possible. The goal is to eliminate as much risk as possible—pushing a molecule to a point where it works or not—and facilitating the decision of whether to invest in Phase III clinical trials or not.
Along the same lines, Lilly is now starting to develop a new lead-optimization program, a small, flexible group of scientists who are autonomous and independent of the larger Lilly Research Laboratories. They will take potential hits from the Phenotypic Drug Discovery program or another end-license opportunity, and pull together the required data elements to make decisions regarding candidate selection.
In addition, Owens noted that Lilly essentially suspends scale-up manufacturing and all major chemistry, manufacturing, control studies and formulation strategies until post-proof of concept, eliminating both time and cost.
Open Innovation Business Strategies
Open innovation is very important for the development of breakthrough medicines, said Teri Melese, director of Research Technologies and Alliances, University of California–San Francisco School of Medicine. More effective classification is needed from companies about what data and information can and cannot be shared (Melese et al., 2009). Melese cited several models of open innovation, which may serve as valuable models for the countermeasures enterprise (Box 6).
|
BOX 6 Models of Open Innovation Business Structures Novartis/Broad Institute Diabetes Genetics Initiative: The initiative will per- form whole-genome scans on DNA collected from type 2 diabetic patients worldwide to provide a comprehensive view of the DNA sequence variants associated with the disease. Genome data will be made publicly available. GlaxoSmithKline (GSK) Open Laboratory for Tropical Diseases: Up to 60 scientists from around the world will pursue their own projects as part of an integrated team and will be afforded access to the Open Lab based in Spain, as well as to the expertise, knowledge, and infrastructure of the company. GSK will also make its library of 13,500 malaria compounds publicly avail- able. Lilly, Merck, and Pfizer Asian Cancer Research Group, Inc.: This is an independent, not-for-profit company established to accelerate research and ultimately improve treatment for patients affected with the most commonly diagnosed cancers in Asia. In this precompetitive collaboration, the companies will combine their resources and expertise to advance knowledge of disease and disease processes. The Merck Gene Index: This is a collaborative effort begun in 1994 to re- lease vast human genome sequence information from Merck’s gene index into the GenBank and the public domain. |
Public–Private Partnerships
Public–private partnerships were of special interest to many participants representing all sectors of the countermeasures enterprise—small biotechs, large pharmaceutical companies, academia, and government. While industry involvement in medical countermeasure product research and development is important, it cannot be relied upon to solve all the problems. Large pharmaceutical companies have too many disincentives and will not enter this arena sufficiently to get the job done. The small biotechs, while likely to come up with some novel development candidates, lack the experience and capacity to take a development candidate through all the clinical testing and regulatory hurdles, and they lack manufacturing capacity. Public–private partnerships will also be valuable for improving access to the chemical libraries and chemistry expertise of the larger companies to develop antiviral and antibacterials in the preclinical phase and early discovery, something that likely cannot be accomplished alone through grant mechanisms. By leveraging the specific expertise of each of these sectors the entire countermeasures enterprise would be advanced, commented
Michael Goldblatt, president and CEO of Functional Genomics. In addition, the establishment of these partnerships would help entice further investment by industry. A variety of models were presented. Generally, industry supplies a list of priority areas of interest and problems they want to solve. For their role, the industry partner(s) supplies scientific advice and access to commercial resources (e.g., compound libraries, medicinal chemistry, manufacturing knowledge) to support the consortium. In return, the industry partner generally wants first rights to commercialize any resulting intellectual property.
One relevant, successful government-initiated public-private partnership is the National Space Biological Research Institute, which is a NASA-funded academic consortium to procure countermeasures for health-related issues associated with space flight. Industry members participate as partners, advisors, collaborators, and consultants, helping to accelerate product development (http://www.nsbri.org/).
Another example is the recently established Merck/Wellcome Trust Hilleman Laboratories, the result of a charity and a company investing equally to form a new entity. The nonprofit Hilleman Laboratories will leverage scientific expertise and platform technologies for discovery development of vaccines for the developing world.
Examples of product development partnerships that the Bill & Melinda Gates Foundation, the U.S. government, and others have funded to advance the development of vaccines, drugs, and diagnostics for poor populations around the world include the Malaria Vaccine Initiative, the Human Hookworm Vaccine Initiative, and the Global Alliance for TB Drugs. As a result of those investments, Rajeev Venkayya of the Bill & Melinda Gates Foundation said, 86 different products are in the pipeline at various stages of development (the majority in preclinical development and Phase I clinical trials). One aspect of these successful models that has been valuable, Venkayya said, is that the Foundation has offloaded the coordination of these activities. A benefit of product development partnerships, such as the Malaria Vaccine Initiative, is that there is a portfolio of products against one target. This facilitates learning across projects, and the ability to make go/no-go decisions based on an entire portfolio of products against one target.
Having looked at many models of success and failure, Russell, identified earlier, recommended public–private partnership as a critical aspect of product development success, and listed several key elements of such partnerships for countermeasures development (Box 7).
|
BOX 7 Elements of Public–Private Partnerships for Countermeasure Development
|
Gerald Parker, principal deputy assistant secretary in ASPR, suggested that in public–private partnerships involving product development, commercial scale-up, and manufacturing, the establishment of technical centers of excellence could provide dedicated capabilities and experienced technical personnel, particularly for these low-probability/high-consequence medical countermeasures that are needed for national security and emergency public health.
Another model would be a U.S. government support organization. The purpose would be to help specifically advance single-use targets where large pharmaceutical companies or small biotechs could not be relied upon to take on such targets due to minimal profit margins. This entity could focus on research and development for such products. Due to the intersection of the public and private sectors, leadership would be an important consideration, with an ideal person having background in industry to oversee discovery/development of important drug/vaccine products. This entity should have access to all the latest technology through licensing, if necessary, and would develop product candidates through phase 2. Further product development could then take place through contract research and development organizations and manufacturing could be contracted to commercial organizations. However, government manufacturing centers could also be used, if there is inadequate manufacturing capacity available for such products (such as vaccines).
A question was raised as to whether such synergy between the public and private sectors would raise concerns of conflict of interest, particularly partnership involving FDA and industry. Anthony Fauci, director of the National Institute of Allergy and Infectious Diseases, said the focus is on getting the FDA resources to be able to become
scientifically involved in aspects of development that can be used by any drug innovator (e.g., biomarkers, alternative clinical trial designs), so in that context there is no conflict. Goodman concurred, noting that working very iteratively and highly interactively with sponsors on trying to achieve an important public health outcome would not be a conflict of interest.
Independent Third-Party Facilitation of Collaboration
The Critical Path Institute was developed in response to the FDA’s Critical Path Initiative (FDA, 2004), with the intent of establishing a neutral ground where multiple companies could come together to work with the regulatory agencies (FDA, European Medicines Agency), with scientific experts from the NIH and academia, and with patient advocacy organizations.
Anthony of the Critical Path Institute said the real purpose of this model is to foster collaboration both among companies and between those who are regulated and the regulators. Anthony noted that typically, the contents of an individual company’s discussions with FDA are proprietary, and therefore any successes or failures remain confidential and other companies are subject to repeating those failures. This is an attempt to improve the process, where companies share data in the precompetitive area—not on products, but on tools and methods that can improve the process—with the goal of developing scientific consensus on tools and methods among those who are going to use them and those who will accept them (the regulators).
Anthony described three consortia that are models of collaboration: the Predictive Safety Testing Consortium, developing biomarkers for safety; the Patient-Reported Outcomes Consortium, developing, evaluating, and qualifying instruments for use in collecting information directly from the patient; and the Coalition Against Major Diseases, sharing information about basic mechanisms of disease.
Strategic Investor Model
Although every year $30–$65 billion in new investments from venture capital and industry support innovation and biotechnology, almost none of that money goes toward countermeasure development, commented Monath, of Kleiner Perkins Caufield & Byers (PhRMA, 2010). The government should act as a strategic investor, in partnership with private capital. In the case of countermeasures development, the
government would invest in technology development in a company, and the return on investment would be products. Thus the government would be rewarded for sharing the risk.
There is a precedent in government for this strategic investment approach. The Central Intelligence Agency established a venture capital company, In-Q-Tel, that makes investments in leading-edge commercial technologies that can help enable the agency’s mission and broadly serve national security interests (Box 8).
|
BOX 8 In-Q-Tel
|
ENGAGING INDUSTRY
Incentives: Push vs. Pull
Incentives used to engage the participation of commercial parties are generally thought of as either “push” or “pull” incentives, with push funding inputs, and pull funding or rewarding outputs. Push strategies should focus on cultivating partnerships and collaborations, James Guyton of PRTM said, while pull strategies should focus on increasing market sustainability. Guyton highlighted a number of push and pull incentives. Common incentives for industry are listed in Box 9.
A recent study by the London School of Economics supported the use of a combination of push and pull incentives (LSE, 2010). The study, described by Chantal Morel of the London School of Economics and Political Science, looked at incentives to spur research and development for antibiotics, specifically those for which there are high levels of pathogen resistance, and considered 24 types of incentive mechanisms.
As discussed above, pharmaceutical companies will assume the risk of failure in development if success can be expected to bring substantial market return. The industry is accustomed to raising capital to fund research when they choose to assume that risk. In that regard, some participants commented that industry does not need push incentives, but rather an assurance of a market (e.g., via a pull incentive). A report by BIO Ventures for Global Health supports the use of market-based incentives, in particular, advanced market commitments (BVGH, 2006). A workshop participant cautioned that paying incentives to industry up front (push incentives) raises the issue of the government “paying for failures.” Adopting a defense contractor model, where the government assumes all risk, is the most expensive and least productive way to proceed. Goal-line benefits (e.g., cash, patent benefits, liability protections) are a better approach.
Rex of AstraZeneca said, however, that push incentives are useful and appropriate for industry. Researchers, he said, must compete internally for company resources. If there is a tax incentive available for research on products for a particular threat, it is easier to make the business case internally that such research should be brought on board. Another participant noted that tax incentives are beneficial primarily for profitable companies, and therefore the majority of small companies that are innovating will not perceive tax credits as an incentive.
Offering a small-company perspective, Martin of Dynavax supported the concept of a prize-based funding strategy (retrospectively funding based on accomplishment of critical elements of a product profile), noting that such an approach creates a tremendous timing incentive. “If I can achieve the prize in one year by doing the critical experiment to show I have the appropriate attributes of the product profile, that is more interesting to me than achieving the same prize in 3 years,” Martin said.
|
BOX 9 Incentives That Could Be Applied in the Development of Medical Countermeasures Push incentives focus on cultivating partnerships between government and companies:
Pull incentives focus on market sustainability:
|
Regarding market exclusivity as an incentive, it was noted that medical countermeasures have very small, variable, or even nonexistent markets, and such exclusivity will not give rise to a strong private-sector reaction.
A question was also raised regarding how to encourage studies of appropriate countermeasure use in special populations, such as children. Although a pediatric incentive11 exists to encourage the testing of
products in children, in many situations involving countermeasures development, there will not be clinical trials at all, negating the current pediatric incentive.
Russell added that operationalizing many of these incentives is a real management challenge because they can be difficult to administer equitably and effectively.
Priority Review Voucher
The Priority Review Voucher was also discussed in some depth. As David Ridley of Duke University explained, this FDA program was enacted into law in September 2007 (Public Law 110-85) to establish an additional incentive for companies to invest in new drugs and vaccines. Under the program a company that develops a treatment for a neglected disease is awarded a Priority Review Voucher, which it can then (1) use when submitting an approval application for a different drug, or (2) sell to another company. Redeeming a voucher and receiving priority review would theoretically allow a company to bring a potential blockbuster drug to market sooner. Ridley noted that Novartis recently received the first voucher, but has not applied it yet. In support of the Priority Review Voucher, Smith of UPMC said there is a potential for significant benefit at minimal social cost.
A participant from a small company, however, expressed skepticism about the value of the Priority Review Voucher. As a small business enterprise, her company would be able to obtain a Priority Review Voucher, but most likely could not use it for the company for another product. Because no company has redeemed one yet, it is unclear what value it would hold for a large pharmaceutical company if the small company would try to sell it. Ridley acknowledged that until the vouchers begin to be used, there is great uncertainty associated with their value.
Incentives Not Needed?: Making a Strong Business Case
The government needs to try to speak the same language as industry when seeking collaboration. When approaching companies, the answer given is a function of the question being asked, said Douglas Pon, Pfizer’s assistant vice president, Licensing in Global Business Development.
Industry is consolidating, cutting back, and trying to become more efficient. Companies are assessing their portfolios, looking at medical need, technical feasibility, and regulatory hurdles. When industry looks at medical countermeasures in terms of the larger company portfolio, it sees low margin and low volume. Consequently, the U.S. government needs to establish a very good business case, with a solid product profile including, for example, the target population, mechanisms of action, and forecasted demand, Pon commented. This business case should also provide a gap analysis, defining what technologies are needed, what other intellectual property will be needed, and what would be required in terms of scale-up process development and manufacturing.
All of this could be done through a government-backed “incubator” or center of excellence, similar to how venture capital investors foster innovation, Pon said. Venture capitalists identify a need and identify the technology and intellectual property that are required, and they build a sound and strong scientific and operational management team. For any gaps, they form strategic partnerships. This is how the government may be able to engage the pharmaceutical industry. The government needs to approach senior management with a direct question and a defined plan to be able to expect a direct and sincere answer in response.
Pon said incentives may not be necessary if there is a true and urgent national need for these products. Whittle the long list of potential threats down to a priority few, define what is actually needed in terms of manufacturing, analytic support, and other capabilities, and come to the table with proposals ready for discussion. Follow the same model that companies use internally, Pon said, by approaching corporate management with a sound business case and request for resources.
An RFP-style process, Pon said, where government publishes a notice that there is an opportunity for collaboration, is not going to work. Government leadership needs to engage individual corporate leaders directly, with a plan in hand, and ask for help.
NEW PARADIGMS, STRATEGIES, AND TACTICS FOR ENHANCING THE COUNTERMEASURES DEVELOPMENT ENTERPRISE
In addition to the models and strategies discussed above that have already been applied to medical countermeasures development (e.g., central oversight/single decision authority, TMTI, Incident Command System; Unmet Medical Need/Dual-Use approach, countermeasures as orphan drugs) and the models that have been successful in other venues (e.g., shared-risk strategies, open innovation, public–private partnerships, consortia, government as a strategic investor), participants discussed a host of other potential paradigms, strategies, and tactics for consideration.
Outsourcing Program Management
Several participants suggested variations of a paradigm where the government would outsource the facilitation responsibilities associated with the coordination of countermeasures development activities to a single entity. Venkayya of the Gates Foundation said there are many capacities that small and medium-sized companies do not necessarily have, and it does not make sense for the U.S. government to build the capacity internally to manage the many industry partners and their needs. The government needs to set requirements and expectations, and be responsible for portfolio management, but not project management.
Robert House, president, DynPort Vaccine Company LLC, offered the concept of an integrator function that would be responsible for program management, contract management, and risk management (Figure 1). To manage risk, the integrator must be aware of both the technical challenges as well as the programmatic and logistical challenges. The integrator needs to be “agnostic,” that is, it must have no vested interest in seeing any particular technology taken forward. For any given need, the integrator would be responsible for pulling together all of the various components necessary: non-clinical, clinical, manufacturing, or other identified gap areas. The integrator would be staffed by technical experts and thus, for this approach to be successful, several participants said the government has to be willing to pay for a high-quality operation.
There can be regular engagement among the FDA, NIH, BARDA, and CDC, and this program coordination entity, as well as involvement of expert advisory boards and consultants. The government retains
portfolio management responsibility, stewardship responsibilities, and, ultimately, go/no-go decisions.
Advantages of such an approach are that multiple candidates could be continuously fed into this system, subcontractors could be chosen based on their expertise, and the project therefore need not be tied to any one company’s expertise. The system would allow technology innovators to concentrate on doing what they do best, which is discovery and early development of new technologies. Not all small companies want to be large companies, said House of DynPort Vaccine Company, and this approach would allow the small companies to continue to feed innovative product candidates into the countermeasures enterprise. This type of system would limit the need for BARDA to be deeply involved in managing the project and technology providers, so it could focus instead on overall portfolio success.
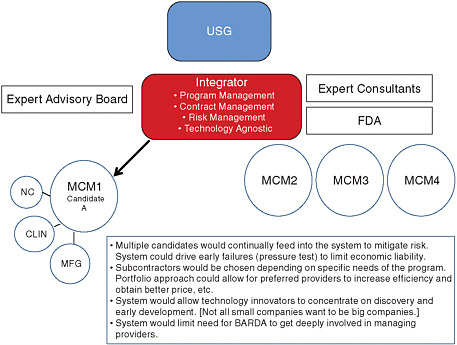
FIGURE 1 Model of medical countermeasures (MCM) development incorporating an integrator function. The U.S. government (USG) sets requirements and expectations and is responsible for portfolio management. The Integrator is responsible for project management.
NOTE: CLIN = clinical; MFG = manufacturing; NC = non-clinical.
SOURCE: Robert House. 2010. Presentation at IOM Workshop; The Public Health Emergency Medical Countermeasures Enterprise: Innovative strategies to enhance products from discovery through approval.
Government as a Strategic Partner
In addition to the “government as strategic investor” approach (à la In-Q-Tel) discussed above, Monath of Kleiner Perkins Caufield & Byers also suggested government approach industry as a strategic partner, looking to acquire technology, bring it in-house, and develop it. In this regard, government needs to provide small biotechnology companies with deals in terms that are familiar to them and the commercial space. For example, end-license the technology, provide upfront payments, and provide milestones when it reaches the appropriate point in its development. If there is procurement, and if doses are put into the Strategic National Stockpile, there is a royalty or an equivalent at the end. Monath said that biotechnology companies would be interested in seeing their technology used for that purpose by somebody else, in return for a deal in commercial terms.
Platform Technologies
There was much discussion of the development and use of platform technologies, as well as FDA approval of products developed on a new or established platform. Technology platforms can help address the unknown need, those threats and emerging pathogens that cannot be anticipated, meaning we cannot vaccinate against them.
The countermeasures enterprise should have a focus on multiuse products and multiuse platform technologies that span the entire public health product-development portfolio (i.e., existing and emerging public health problems and deliberately released public health problems), NIAID’s Fauci said. Focus should be on development of new platforms as opposed to going after a particular threat. As an example, Fauci cited a recent publication describing a broad-spectrum antiviral drug targeting enveloped viruses, noting that this could be effective against yet-to-be discovered enveloped viruses (Wolf et al., 2010).
Although the idea of platform technologies is attractive, the regulatory environment needs to be prepared to swiftly approve new platform technologies and to approve products developed on those platforms. As discussed previously, a clear regulatory pathway must be established for medical countermeasures. The first product through on any given platform is indeed a new product. The question then is to what degree second and subsequent products using the same platform can extrapolate data (perhaps similar to how the abbreviated new drug applications used for generic drugs can include data extrapolated from the pioneer product), commented Cassell, chair of the workshop.
One caution noted by Gomez of PRTM is that while it is fairly easy to know when a product has failed, it can be very difficult to tell when a platform has failed because there is always an improvement, always a new disease target.
Revised PHEMCE Implementation Plan
As discussed above, a challenge for industry is that the PHEMCE implementation plan itself provides limited guidance in that it is essentially a list of pathogens and agents of interest to the government. Rose proposed a use-based medical countermeasure acquisition matrix (Figure 2). The concept of operations is similar across all of the organisms listed. There is a need for pre-event prophylaxis, particularly for the military and first responders. Treatments are needed for people who develop symptomatic illness when an outbreak occurs. For those who are exposed but not yet ill, postexposure prophylaxis is needed to keep them from developing illness. Rose noted that the matrix is for healthy adults and a similar matrix for children or the elderly would show significantly more yellow and red.
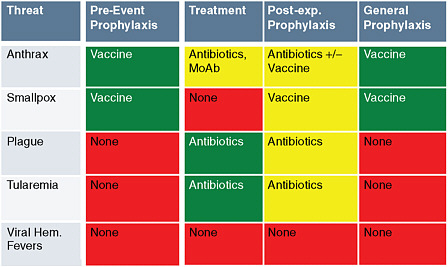
FIGURE 2 Use-based medical countermeasure acquisition matrix (Healthy Adults 18–64). Green: product is available; yellow: product is not yet approved; red: no products for development.
SOURCE: Eric Rose. 2010. Presentation at IOM Workshop; The Public Health Emergency Medical Countermeasures Enterprise: Innovative strategies to enhance products from discovery through approval.
EXISTING REGULATORY TOOLS AND APPROACHES THAT CAN BE APPLIED TO ADVANCE COUNTERMEASURES DEVELOPMENT
Boris Lushniak, assistant commissioner for Counterterrorism Policy at FDA, said the agency’s mission is very clear: safety, efficacy, and quality. Lushniak and Jeanne Novak, of CBR International Corp., both drew attention to policies and programs already in place that could be used to facilitate medical countermeasure development. However, throughout the workshop, it was repeatedly noted that there is a need for a clear regulatory pathway necessary to evaluate and regulate the biodefense products as well as a number of additional gaps and associated opportunities.
Opportunities for Accelerating Approval of Medical Countermeasures: Evolving the Regulatory Framework
Numerous individual suggestions were made about addressing the regulatory aspect of the medical countermeasures enterprise. They are compiled here as part of the factual summary of the workshop, and should not be construed as reflecting consensus or endorsement by the workshop, the Forums, or The National Academies. They are as follows:
-
Fund, support, and enable regulatory science. Develop, assess, and provide tools, methods, models, standards, guidance, and pathways to evaluate product safety, efficacy, and quality (e.g., biomarkers; surrogate endpoints; adaptive and other flexible clinical trial designs; rapid scale-up of production; rapid methods to assess purity, potency, quality, contamination).
-
Declare medical countermeasure to be a priority at the FDA, which would entail providing security clearances to senior FDA employees and briefing all medical officers across the entire agency about the real risks and the immediate need for products.
-
Create a designation indicating that a program is relevant to national security and establish priority review for medical countermeasure applications. For products so designated, FDA can then provide additional assistance, perhaps some form of priority review or acceleration of time lines consistent with the product being part of a national security countermeasures development program.
-
Consider creating an alternative approval mechanism for medical countermeasures with criteria more stringent than EUA and less stringent than normal licensure for commercial products.
-
Formalize the EUA process. The existing EUA mechanism works well as an emergency response process, but it is not a preparedness mechanism. A formal EUA approval process would use existing criteria for the EUA, with the exception of the actual declaration of an emergency, and would differ from the current process, in which submission of pre-EUA data is encouraged, but no response is required, and typically no response is generated.
-
Balance data needs according to risk/benefit. Because medical countermeasures will be used in situations of grave risk, the amount of data needed for approval may be less than that for a more mainstream product. Early HIV interventions were cited as a parallel situation.
-
Facilitate communication with product sponsors. In-person meetings and site visits between the FDA and with companies developing medical countermeasures could be done with increased frequency and flexibility, without having to formally request the existing FDA A, B, and C meeting categories.12
-
Send FDA agency staff into the field to assist companies as needed. This was done, for example, when Genentech was developing the first recombinant human growth hormone, a product for which there was urgent need. An FDA senior chemist spent 3 months at Genentech, working with its staff to prepare manufacturing for approval.
-
Abandon the Draft Guidance on the Animal Rule, which is significantly more restrictive than the Animal Rule itself. The Rule says FDA may grant approval when “the results of those animal
|
12 |
Through the 1997 FDA Modernization Act, the FDA established three main categories of meetings—A, B, or C—that describe the various types of formal industry meetings that occur (http://www.fda.gov/Drugs/DevelopmentApprovalProcess/FormsSubmissionRequirents/ElectronicSubmissions/DataStandardsManualmonographs/ucm071774.htm). |
-
studies establish that the drug is reasonably likely to produce clinical benefit in humans.”
-
Emphasize emergency preparedness throughout the FDA. Instill cultural change though an agency-wide focus on emergency preparedness/national security in the regulatory environment, and encourage use of many of the tools that already exist in the current regulatory laws and regulations, in a more efficient, focused way.
-
Provide regulatory and licensure guidance to funding agencies on development programs. FDA personnel can assist funding agency (HHS and DoD) reviews of medical countermeasures development programs. This would facilitate progress by providing a “unified” development method. Specialized Review and Development Teams could be formed at the FDA (center and division levels). Funding agencies would have access to the Specialized Review and Development Teams in order to seek expert FDA opinion about progress of a particular development program.
-
Reflect FDA data needs in NIH trial design and conduct. NIH funds many large clinical trials, which are then incorporated into an application for product approval. Many times the data are not adequate to support FDA decision making in terms of safety, efficacy, and labeling claims. If FDA is at the table with NIH when these trials are being planned, and when they are being executed, they can make sure the supporting data are available when needed for regulatory decisions.
The Way Forward: Themes from the Workshop
Additional suggestions regarding a wide variety of areas relevant to medical countermeasures development were presented by individuals during the workshop and are compiled here. Investigating details about the feasibility and implementation of these ideas were beyond the scope of the workshop. The additional suggestions are the following:
-
Institute a single, unified management structure for the federal countermeasures enterprise. The hallmark of a successful developmental program is strong leadership that can have a singular focus on developing products—a system that could direct basic and applied research and have the power to force alignment of all the
-
components. BARDA currently does not have this authority. However, consideration should be made for the complementary, but separate, missions of HHS and the DoD.
-
Facilitate improved end-to-end partnering between federal agencies (NIH, ASPR/BARDA, DoD, FDA), industry, and academia throughout research and development of medical countermeasures.
-
Declassify threat analysis data to allow transparency for potential product requirements, and thus to improve the understanding among the public, legislators, and industry about the scope of the threat.
-
Provide additional venues for classified briefings for all segments of the countermeasures enterprise, including private-sector stakeholders. Two examples are the Joint Civilian Orientation Conference sponsored by DoD and the FBI’s National Security Higher Education Advisory Board, where leaders in business and academia get security clearance, are briefed on issues, and contribute to discussions on national security matters. Something comparable could be established for the leadership from pharmaceutical and biotechnology companies.
-
Develop target product profiles. Define the product, including both the indication (i.e., disease/condition to be treated) and what an appropriate product would be to use in a public health emergency. Prospectively establish the metrics that define success for a product, then build an experimental plan that assesses whether a product has critical attributes.
-
Utilize grants and contracts to incentivize research and development in multi-use agents. BARDA, and other government grants and contracts, as well as milestone payments serve as important incentives for the development of multi-use agents, in particular for agents like antibiotics, which do not always have a very high profit margin.
-
Empower program managers. Provide increased authority to program managers to develop a vision of what a successful project or program encompasses, and then to go out and secure the resources
-
necessary to put that vision together. Small companies would benefit from project managers and/or FDA being able to reach out proactively at the beginning of a contract to discuss how the company could avoid potential hurdles and common mistakes related to complex areas such as the Animal Rule, GLP toxicology studies, etc.
-
Use the Federal Acquisition Regulation to create commercial markets for biodefense products. FAR § 12.102(f)(1) states that contracting officers “may treat any acquisition of supplies or services that, as determined by the head of the agency, are to be used to facilitate defense against or recovery from nuclear, biological, chemical, or radiological attack, as an acquisition of commercial items.” This establishes the authority to designate a countermeasure as a commercial item (not a cost-plus item) that can be compared to other drugs and vaccines already in the commercial markets.
-
Simplify the intellectual property system and improve alignment between FDA approval and when a patent is granted. Ascribe value to the data themselves. The data are what determine whether the product is of any value to the public. Consider a full 20-year patent term for government-chosen medical countermeasures and a data protection period of 20 years, both running from the day of FDA approval.
-
Establish incentives and public private partnerships to encourage greater investment by the private sector. Particular focus should also be made on how to incentive the development of countermeasures for children and other special populations.
-
Ensure integrity of Project Bioshield funds and increase funding for countermeasures research and development. It is important to guarantee that procurement resources remain available, but far greater resources are currently required to ensure necessary countermeasures research and development. Removing funds from Project Bioshield’s Reserve Fund for research and development could have serious repercussions on the government’s ability to guarantee a suitable marketplace for future countermeasure procurement. It also would contribute to uncertainty among the private sector about long-term, stable funding for countermeasures research and development.
-
Commit to multiyear federal funding of CBRN/pandemic countermeasures to ensure companies are able to maintain a continuous development program.
-
Consider a tax benefit for equipment purchased for use in the development or manufacturing of medical counter-measures. The Department of the Treasury could approve an accelerated depreciation schedule for equipment that is required for biodefense products exclusively (i.e., that has to be separate from the rest of the equipment in a research and development or manufacturing facility).
-
Make the federal government the defendant in all the tort claims resulting from countermeasures. The government is the defendant in the Vaccine Injury Compensation Act, but not in the existing PREP Act. Legislatively eliminate the cause of action and replace it with this administrative compensation system.
-
Make suspension of state liability in a federally declared emergency a condition for receipt of biodefense money for the states. The federal government cannot force states to pass legislation, but it can withhold preparedness funding if the state fails to comply with the conditions of the grant.
-
Institutionalize capacities. Planning is important, but adaptability and rapid responses are critical elements for success. Integrate problem solving and flexible approaches into countermeasure development and emergency planning. Institutionalize capacities throughout the countermeasures enterprise rather than reserve them for a crisis. The Illinois Department of Public Health, for example, sent 42 senior staff members to be trained in the National Incident Management System, providing them with useful background on an organizational approach to managing problems. These skills were of great value during the 2009 H1N1 pandemic.
-
Sponsor a contracting office training course, covering best practices to address countermeasure development, licensure, manufacturing, and procurement with biodefense, and updating officers on available options.
-
Establish more agile platforms and multiuse technologies (e.g., vaccine, diagnostic, or monoclonal platforms) that can be rapidly adaptable to address new pathogens.
-
Engage end users, specifically public health professionals and healthcare providers, in requirement setting. Incorporating end-user perspectives in the requirement setting for product development will help ensure that the final product that is stockpiled or distributed will be able to be administered effectively and efficiently to the target population. In this regard, the needs of special populations, including children, are part of planning, research, and development.
-
Improve public engagement. Robust and continued public engagement helps to ensure the legitimacy of the counter-measures enterprise, communicates the risk, communicates those plans currently in place, and helps produce the best possible result. Engage faith-based institutions. Use the educational system; develop a message that becomes part of a school curriculum. Work to dispel myths and misperceptions.
-
Create technical centers of excellence to help ensure the necessary expertise, including dedicated capabilities and core resources and manufacturing facilities.
-
Invest in career development strategies to ensure the necessary scientific and regulatory expertise. Define a curriculum at universities that could support the countermeasures enterprise. The government could sponsor educational opportunities to improve human capital capacity. For example, a fellowship for graduate degree candidates established by FDA and BARDA could enrich the countermeasures enterprise and bring new people in the system.
CONCLUSION
Deliberate acts of bioterrorism and emerging natural infections will continue to be a major public health concern. There is no one specific right incentive, or one specific model that is best suited to advanced development of countermeasures against chemical, biological, radiological, and nuclear agents. No single player is ideally positioned to meet the needs of the medical countermeasures enterprise.
There are at least two different ideological approaches to the development of medical countermeasures, as follows:
-
Consider it to be a critical national security issue, with an urgent need to develop countermeasures for an imminent threat.
-
Focus on addressing important unmet medical needs that are present today, and while these needs are somewhat different, some benefit can be derived for the national security mission in the end.
These approaches are not mutually exclusive, and the ideal countermeasures enterprise would have a multiuse focus on existing, emerging, and deliberately released threats. However even if cross-indications are feasible, a national threat may, or likely will, occur without notice. Therefore, without emphasis upfront on the national security implications at the federal level on development pathways, medical countermeasures are less likely to be available when needed. The use of these two approaches in combination could provide the basis of a robust development strategy and would allow federal systems to be in place to deal with imminent threats, in distinction from standard drug development processes.
Assistant Secretary Lurie reminded attendees that engaging industry in the countermeasures enterprise is essential. A dramatic transformation is needed in the way the federal government interacts with industry. Government and industry must come together as trusted partners, and this relationship must be sustained—not only in a time of a crisis. There is no one push or pull incentive that is sufficient to attract experienced companies to the enterprise. Similarly, there is no “silver bullet” combination of incentives that suits all situations.
Seeking private-sector participation in developing these products is not about saving the public money. It is about bringing innovation, human capital, entrepreneurship, and private-sector expertise to bear on important public health issues. Market incentives cost money. The U.S. government should be willing to pay for countermeasures, to the extent that there are national security and public health needs.
Throughout the workshop it was frequently asserted that partners in the countermeasures development enterprise should be able to expect the following:
-
A clear articulation of the threats
-
A very clear articulation of the target product profile (indication, formulation, manufacturing needs, predicted demand)
-
Clear time lines (what activities are on an urgency timeline vs. a long-term timeline)
-
Predictability of finance (e.g., if a company delivers according to specifications, there will be a purchaser on the other end, and that purchaser will be the U.S. government)
-
Predictability from the regulatory environment
Regardless of the approach taken, all participants in the medical countermeasures enterprise should remember the end goal: ensuring the health of the people. The medical countermeasures research and development enterprise should continue to develop and implement models that, while respecting industry and governmental needs and goals, foster collaborative relationships among government and industry to ensure maximum use of their respective strengths and capabilities for the protection of the nation’s health.
























































