
3
The Biomarker Evaluation Process
The previous chapter’s detailed exploration of biomarker evaluation efforts indicates a need for a unified, transparent process for the evaluation and adoption of biomarkers. Although the principal purpose for evaluation is to ensure that a biomarker is scientifically and clinically meaningful for specified purposes (Palou et al., 2009; Wagner, 2008; Wagner et al., 2007; Williams et al., 2006), evaluation also allows for informed decisions about which biomarkers to pursue and data to gather. This chapter begins to present the committee’s recommendations on the best ways to proceed (see Box 3-1 for the recommendations discussed in this chapter).
The committee’s biomarker evaluation framework was informed by the previously developed qualification frameworks discussed in Chapter 2; the committee determined there are three necessary components to biomarker evaluation: (1) analytical validation of relevant biomarker tests; (2) qualification, a description of the evidence relating to the biomarker in question—as measured using validated tests—to the intervention and disease outcome; and (3) utilization, the applicability of results from the analytical validation and the description of the evidence to the proposed use of the biomarker given the evidence assessment and proposed purpose and context of use. Thus, the committee’s framework has three distinct yet interrelated steps; they are not necessarily separated in time (i.e., some of the steps may occur concurrently) and conclusions in one step may require revisions or additional work in other steps (see Figure 3-1). Previous evaluation frameworks have not explicitly incorporated a process for reevaluating the three steps of the biomarker assessments based on new
data; the committee’s framework explicitly includes such a process, while allowing for timely, reliable, and effective decision making.
The evaluation framework is intended to be applicable across a wide range of biomarker uses, from exploratory uses for which less evidence is required to surrogate endpoint uses for which compelling evidence is required. The framework is meant for, but not limited to, use in research, clinical, product, and claim development in food, drug, and device industries as well as public health settings, and it is intended to function for
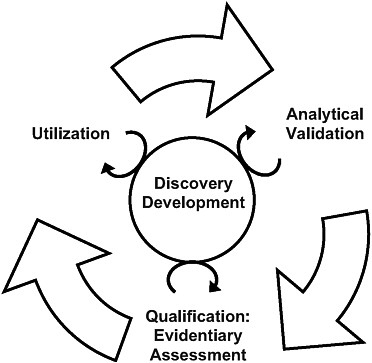
FIGURE 3-1 The steps of the evaluation framework are interdependent. While a validated test is required before qualification and utilization can be completed, biomarker uses inform test development, and the evidence suggests possible biomarker uses. In addition, the circle in the center signifies ongoing processes that should continually inform each step in the biomarker evaluation process.
panels of biomarkers in addition to single biomarkers and for both circulating and imaging biomarkers. While the report provides case studies of individual biomarkers, the committee concluded that sets of biomarkers need to be qualified using the same process. In some cases, individual biomarkers within the same set may need to be qualified individually.
This chapter explores the rationale behind the committee’s decision to separate evaluation into three interrelated steps before providing an in-depth examination of each step. This conceptual framework is meant to provide a clear, adaptable platform for statistically sound, evidence-based biomarker evaluation.
THE RATIONALE FOR AN INTERRELATED, THREE-STEP PROCESS
Recommendation 1:
The biomarker evaluation process should consist of the following three steps:
|
1a. |
Analytical validation: analyses of available evidence on the analytical performance of an assay; |
|
1b. |
Qualification: assessment of available evidence on associations between the biomarker and disease states, including data showing effects of interventions on both the biomarker and clinical outcomes; and |
|
1c. |
Utilization: contextual analysis based on the specific use proposed and the applicability of available evidence to this use. This includes a determination of whether the validation and qualification conducted provide sufficient support for the use proposed. |
The committee recognizes that including analytical validation in the evaluation framework and separating the evidentiary assessment from the utilization analysis is a departure from many previous attempts to develop biomarker evaluation systems, but found that these processes, although distinct, are interwoven in such a way that it is impossible to responsibly consider one without also considering the others. Although biomarker analytical validation and biomarker qualification will often be considered together (the statistical linkages of disease, biomarker, and drugs can depend on the analytical soundness of a biomarker assay) and have been used synonymously in the past (Biomarkers Definitions Working Group, 2001), differentiating these processes is important (Lee et al., 2006). A National Institutes of Health working group recommended the term “validation” be used for analytical methods (Biomarkers Definitions Working Group, 2001). The American Association of Pharmaceutical Scientists (AAPS), the Pharmaceutical Research and Manufacturers of America, and the Biomarkers Consortium, among other organizations, have worked to reinforce the distinction between analytical validation and qualification (Lee et al., 2005; Wagner, 2002). As discussed below, analytical validation is the process of assessing how well an assay quantitates a biomarker of interest; qualification is the evidentiary and statistical process linking a biomarker with biological processes and clinical endpoints (Biomarkers Definitions Working Group, 2001). The committee determined that qualification could be further separated into evidentiary assessment and utilization analysis, so that the different investigative and analytical processes required to evaluate evidence and contexts of use
are distinct. Details regarding methods for the gathering of evidence are discussed in the section on Recommendation 2.
It is important to emphasize the necessity of evaluating data relating to adverse events and unintended effects of biomarker use. In every step, the proposed use and its context are critical. For drug development and other medical uses, this entails a risk–benefit analysis, which weighs evidence supporting biomarker use against known inaccuracies and gaps in knowledge that present the possibility of error. For foods and supplements, this entails an analysis of the potential modifying matrix effects of the food or supplement that serves as the delivery vehicle and the dietary patterns associated with consumption of the nutrient or food substance.
The committee understands that a biomarker evaluation checklist of criteria to fulfill for given purposes would be more straightforward to use. But, given the complexities of biomarker utilization, the risks involved with their use, and the evolving nature of science and technology, a checklist-based approach was deemed to be infeasible. First, because any attempts to evaluate a biomarker must consider the context of and purpose for use of the biomarker, scientific and medical judgment plays a role in decision making. Because the purpose and context in each evaluation are unique, there are no precisely relevant past data to consult for guidance. Also, decisions made during the evaluation process are based on probabilistic rather than deterministic reasoning. Probabilistic reasoning emphasizes epidemiological and statistical relationships and acknowledges that the biology is not fully understood. Both statistical methods and decision analysis may be important tools for biomarker evaluations. Both of these were discussed in Chapter 2.
Despite these important caveats, a nuanced understanding of the strength of a biomarker is necessary to develop an evidence-based understanding of whether the biomarker is fit for its proposed purpose and context of use. The committee acknowledges that decisions resulting from the evaluation of a biomarker are dependent on the purposes for which the biomarker will be used. Although some have supported the idea of biomarker evaluations that can be viewed as general and definitive for any proposed purpose or context of use, the committee has determined that there has been no example of this so far and it does not expect to witness one in the future.
The committee recognizes that this approach will require some additional financial and human resources at the Food and Drug Administration (FDA), as was suggested in the Institute of Medicine (IOM) report The Future of Drug Safety and is discussed further in Chapter 5 (2007). However, the process fits well with the mechanisms that the FDA already uses to seek external advice (e.g., the scientific advisory committees). Also, this process would represent a modest investment compared to its
potentially broad benefits to society by ensuring a stronger evidence base underpinning FDA decisions. Benefit to the FDA itself and its commercial users may also be realized through more consistent and transparent expectations.
Analytical Validation
As previously defined, the term “biomarker” refers to a characteristic that is reliably and accurately measured and evaluated as an indicator of normal biological processes, pathogenic processes, or pharmacologic responses to a therapeutic intervention (Biomarkers Definitions Working Group, 2001). Thus, measurement itself must be an explicit component of any discussion of biomarker evaluation because it establishes the scientific basis and availability of experimental data that support or refute the context for qualification of a biomarker and its proposed application (Goodsaid and Frueh, 2007). The committee finds that analytical validation of a relevant biomarker test is a prerequisite for biomarker qualification.
Analytical validation is defined as an assessment of assays and their measurement performance characteristics, determining the range of conditions under which the assays will give reproducible and accurate data. Thus, analytical validation is an assessment of a biomarker test that includes the biomarker’s measurability and the test’s sensitivity for the biomarker, biomarker specificity, reliability, and lab-to-lab reproducibility. The terminology used in the recommendation, analytical performance, is not meant to describe how well a biomarker correlates with the clinical outcomes of interest. Instead, analytical validation of an assay includes the biomarker’s limit of detection, limit of quantitation, reference (normal) value cutoff concentration, and the total imprecision at the cutoff concentration. These specifications must be determined and met before data based on its use can be relevant in the qualification steps of biomarker evaluation. To ensure comparison across multiple laboratories and clinical settings, appropriate standards for ensuring quality and reproducibility need to be made available. Additionally, understanding the difference between individual assays is important to interpreting the findings of different studies monitored using different assays (Apple et al., 2007). For biomarkers used solely in laboratory testing, it would be beneficial to assess the ability to compare data from different assay platforms as much as possible and needed.
Though key guidelines and regulations have molded approaches to assay validation (Swanson, 2002), biomarker validation is distinct from pharmacokinetic validation and routine laboratory validation; however, an agreement for a uniform set of criteria for biomarker assay validation
has not been reached. Method validation requirements for assays that support pharmacokinetic studies have been the subject of intense interest, and the FDA has issued guidance for industry on bioanalytical method validation (CDER, 2001).1 This guidance, though, is directed at validation of assays looking at metabolism of conventional small-molecule drugs and is not directly related to the validation of assays for biomarkers for many other uses. Similarly, biomarker validation is, in many ways, different from routine laboratory validation. Laboratories that perform testing to support human diagnostics and health care are regulated by the Clinical Laboratory Improvement Amendments, or CLIA (Centers for Medicare & Medicaid Services), and accrediting organizations such as the College of American Pathologists (Swanson, 2002).
Because of the diverse purposes of biomarker research and the various locations in which these assays are performed (routine and novel biomarker assays are performed in both bioanalytical and clinical laboratories; novel biomarker assays are also performed in specialized laboratories), neither the FDA regulations nor the CLIA guidelines fully address all possible study objectives. Differences between biomarker assays and those of drug bioanalysis and diagnostics are described in detail in Table 1 of Lee et al. (2006), highlighting some of the unique validation challenges related to biomarker assays (Lee et al., 2006).
In the absence of uniform criteria for the validation of biomarker assays, analytical qualities and clinical performance of assays cannot be objectively evaluated (Apple et al., 2007). To address these challenges, the AAPS and Clinical Ligand Assay Society cosponsored a Biomarker Method Validation Workshop in October 2003 (Lee et al., 2005). It resulted in a validation approach for laboratory biomarker assays in support of drug development. This validation approach, though, was focused primarily on ligand-binding methods to gauge biomarkers measured ex vivo from body fluids and tissues (Lee et al., 2006).
Additionally, the International Federation of Clinical Chemistry and Laboratory Medicine Committee on Standardization of Markers of Cardiac Damage has recommended analytical and preanalytical quality specifications for a variety of assays, including those for natriuretic peptides and troponin assays (Apple et al., 2005, 2007; Writing Group Members et al., 2008). These guidelines were developed to guide both clinical and commercial laboratories that use the assays with the goal of establishing uniform criteria (Apple et al., 2007). The standardization and harmonization of biomarker assays is challenging due to the various analytical and biological factors that influence measurement (Swanson, 2002). By defini-
tion, biomarkers are dynamic and responsive to changes in the disease process, pharmacological intervention, and environment (Fraser, 2001; Ricos et al., 1999). For example, variability in biomarker level is affected by biology (e.g., gender, age, posture, diet), sample type (e.g., blood, urine), sample collection (e.g., transport and storage conditions, collection technique), and analytical factors (e.g., pipetting precision, antibody specificity) (Swanson, 2002). Sources of variability in biomarker measurements are summarized in Table 3-1. Though these background fluctuations affect
TABLE 3-1 Sources of Variability in Biomarker Measurements
|
Preanalytical Sources of Variability |
Analytical Sources of Variability |
|
|
Biological |
Sample Collection |
|
|
Sociodemographics (including age and gender) Posture Exercise Meals/fasting status Diet Diurnal biorhythm Seasonal biorhythm Concurrent diseases Concurrent medications Overall health/preexisting disease Gastrointestinal motility Anesthesia/surgical intervention Stress Pregnancy Menstrual cycle Dehydration Kidney function Body composition (obesity) |
Mislabeling Duration of tourniquet application Strength of collection vacuum Size of needle gauge Dead volume in catheters/collection tubes Anticoagulants Local effects of indwelling catheter Time and temperature prior to centrifugation Centrifugation speed, duration, temperature Evaporation/biomarker volatility Preservatives/biomarker instability Storage temperature Transport temperature Completeness of urine collection Hemolysis Effect of glass and plastic collection tubes Exposure to light Type of sample Time of clotting |
Purity of reference standards Lot-to-lot variation in reagents Antibody crossreactivity Loss during extraction Mislabeling of processing tubes Pre-assay incubation time and temperature Pre-assay amplifications Chemical interference by endogenous compounds Chemical interference by drugs Analyte or reagent instability in light Time between intermediate steps Fluctuations in instrument performance Correction for baseline/background levels Post-run calculation errors Matrix effects Reproducibility of sample |
|
SOURCE: Adapted from Swanson (2002). Copyright 2010, reprinted with permission from IOS Press. |
||
both the sensitivity and the specificity of biomarker measurements, and though it may not be possible to establish absolute accuracy, relative accuracy can be informative and the sources of variability can be understood and controlled, allowing for the delivery of high-quality assay results (Lee et al., 2006; Swanson, 2002).
Implementation of biomarker validation therefore requires both understanding and control of the various sources of variability in assay performance (Kristiansen, 2001). Results from biomarker assays are valid only if sample integrity is maintained from sample collection through analysis. It is important to devise standard protocols for sample collection, processing, and storage to achieve uniformity (Lee et al., 2005, 2006). The committee synthesized a variety of approaches to develop its key elements for biomarker validation. Table 3-2 lists important data for inclusion in package inserts and in peer-reviewed publications for biochemical biomarker assays in the preanalytic characteristics, calibration and standardization criteria, and analytic parameters. Other considerations may be needed for imaging and other types of biomarker assays; this is discussed further in Chapter 4.
Validation of biomarker tests should be done on a test-by-test basis and must then be deemed sufficient for the use proposed in the utilization step (ICH, 1994; Shah et al., 1992). Thus, the rigor of biomarker validation can be correlated with the intended use of the data (Lee et al., 2006). The committee finds that biomarker qualifications are often undermined by insufficiently validated tests, which may lack accuracy, sensitivity, and specificity. Additionally, use of tests after biomarker qualification and test validation depends on operator, reagent, and instrument variability, among other factors. In the case of clinical laboratory assays and reference ranges of common biomarkers, for example, absence of standardization can lead to interpretation mistakes (Rosner et al., 2007; Wu, 2010). The nature of health care is such that patients often use multiple laboratory facilities during the course of care (Wu, 2010). Diagnosis and management depend on the accuracy of testing across laboratories (Rosner et al., 2007). Therefore, proper standards and controls are necessary to ensure consistent delivery of high-quality biomarker data and the validation of biomarker tests prior to biomarker qualification. Box 3-2 introduces the case study exemplifying the issues found in analytical validation. Further detail can be found in Chapter 4.
Qualification
The second step of the committee’s evaluation framework is a factual description of the levels and types of available evidence. This objective analysis is a reproducible, systematic assembly and review of the evi-
TABLE 3-2 Information Needed for Package Inserts and Peer-Reviewed Publications Describing Biomarker Assays
|
Preanalytic |
Calibration/Standardization |
Analytic |
|
Sample handling Effect of storage time and temperature Influence of different anticoagulants (type and concentration) for plasma and whole blood measurements Influence of gel separator tubes Time and speed (relative centrifugal force) and temperature of sample centrifugation with the effects of various methods for tube filling, mixing, and centrifugation |
A low-level quality control (QC) sample with concentration close to reference value to monitor assay bias at cutoff A negative QC sample to monitor baseline drift Calibration frequency to be determined based on the imprecision and drift characteristics of the assay Calibration using defined biomarker calibrators to accommodate any subtle changes in assay calibration curve Defined limits for the zero calibrator’s reaction units |
For antibody assays, identification of antibody recognition epitopes For activity assays and immunoassays, identification of limiting substrates Linearity of signal Reactivity to various plasma biomarker forms (degree of equimolarity) Cross-reactivity with other related proteins in complex matrix (normal and disease) Identification of interferences from hemolysis, bilirubin, and lipemia, and potential interferences from heterophile antibodies, rheumatoid factors, and human antianimal antibodies and autoantibodies (neither of which are currently commercially available) Dilution response (i.e., linearity, recovery) over time and sites Assay limit of blank, limit of detection, and limit of quantitation Decision limits and precision at relevant concentrations Method comparison data, in particular if manufacturers offer both central laboratory and point-of-care assays Establishment of the decision limit of the distribution of healthy subject reference values |
|
BOX 3-2 Tumor Size and Analytical Validation (Recommendation 1a) Tumor size is a variously defined biomarker of efficacy of cancer therapeutics using tumor diameter, tumor volume, or tumor mass, as measured by a variety of platforms and techniques, including magnetic resonance (MR), computed tomography (CT), and positron emission tomography (PET). Different contrast agents and different protocols may be used, all of which affect the precision of measurement. Measurement precision is also affected by patient characteristics. Each protocol, which may also vary by tumor location, should undergo independent validation. There is a great deal of variability in the levels of evidence to support validation for different protocols; thus, analytical validation is complicated by multiple imaging platforms and other assay performance issues. The disparity in evidence impacts the interpretation and generalizability of these imaging endpoints. Assuming that at least one test is determined to be adequately validated, data collected for the qualification step have shown that tumor size may not always be linked to clinical benefit although tolerance for uncertainty of clinical benefit has been justified by the seriousness of cancer. For utilization, in 1992, the Food and Drug Administration started granting accelerated approval for drugs that are effective against serious diseases based on surrogate endpoints. Accelerated approvals for anticancer drugs or biologics have been granted on the basis of endpoints such as overall response rate, time to progression, or disease-free survival. Of those granted approval between 1992 and 2004, only about one-quarter have been converted to regular approval (i.e., demonstrating an effect on survival) (Lathia et al., 2009). All of them remain on the market. Concern exists that clinical benefit may be neglected in regulating this type of approval (Fleming, 2005). Tumor size is discussed in greater detail in the full case study found in Chapter 4. |
dence. Users of the evaluation framework will need to identify appropriate methods for gathering the evidence for this step. This is discussed with respect to the FDA in the section on recommendation 2. Fulfilling the qualification step requires: (1) evaluating the nature and strength of evidence about whether the biomarker is on a causal pathway in the disease pathogenesis, and (2) gathering available evidence showing that interventions targeting the biomarker in question impact the clinical endpoints of interest. If the biomarker–clinical endpoint relationship persists over multiple interventions, it is thought to be more generalizable.
It is important to note that although this is an objective, evidence-based assessment, the type of reasoning that may be used in this step is still probabilistic rather than deterministic. While deterministic reasoning ultimately means that every contributing factor to the biomarker– intervention–clinical endpoint link is defined and understood, probabi-
listic reasoning emphasizes epidemiological and statistical relationships, acknowledging that all contributing factors are generally not fully understood. Because this is almost always the case, clinical outcomes are fundamentally random in nature, requiring probabilistic reasoning to inform rational decision making. Thus, biomarker evidence allows for inferences, but rarely allows for certainties.
Evidence for a Link Between the Biomarker, the Disease Pathway, and the Clinical Endpoint: Hill’s Criteria
For the first part of the qualification step, evaluating the strength of evidence regarding the disease pathway can be done, in part, by using concepts described by Hill’s criteria (1965). Hill’s criteria were discussed in detail in Chapter 2; they evaluate characteristics such as strength of association, biological plausibility, and consistency, among others (see Box 2-2 and supporting text in the previous chapter) (Williams et al., 2006). Understanding the biology behind a biomarker is an important source of information on a biomarker’s relevance, specificity, and robustness (Koulman et al., 2009). However, biomarkers indicating differences between healthy and sick individuals may relate to consequences rather than the causes of the underlying disease pathology; as a result, these differences may not have predictive value (Koulman et al., 2009). As a result, these biomarkers need to undergo a rigorous multistep qualification process in order to become diagnostic tools (Koulman et al., 2009).
Given that biomarkers are “indicators”—in that they are not necessarily causal—and that an abnormal value or a gradient in level over time is not necessarily informative or predictive depending on the clinical situation, the committee instead used these criteria as a structure for assessing the prognostic value of the biomarker for the clinical outcomes of interest. Depending on the situation, not all of the criteria must be fulfilled; temporality and strength of association are generally necessary, however.
Different study designs have advantages and disadvantages. Prospective or cohort studies allow researchers to define study populations based on some relevant characteristic(s) in advance (e.g., level of a biomarker), then follow the development of health outcomes over time. This process is the most accurate and inclusive of possible outcomes, but it is slower than some other designs. On the other hand, cross-sectional studies define a population of interest and then collect data on both the characteristics of interest (e.g., level of a biomarker) and the health outcomes of interest simultaneously. Although these studies are faster, they have limitations. Cross-sectional designs do not allow for causal inferences to be made since biomarker–disease measurements occur simultaneously. Also, patients who died or experienced clinical outcomes that made them unavailable
for measurement would not be reflected in a cross-sectional population, leading to significant risk of incorrect conclusions. This is thought to be a reason that lower low-density lipoprotein (LDL) levels have been found to be associated with higher risk of death in patients after cardiac catheterization, even though lowering LDL cholesterol with statins has a large benefit in these same patients (Califf et al., 1992). Therefore it is important to consider the quality and strength of the data when conducting biomarker evaluation.
Evidence That Interventions Impacting the Biomarker Impact the Clinical Endpoint
For the second part of qualification—for surrogate endpoints, that is—prognostic value is a necessary but not sufficient criterion for the evaluation. Interventions targeting the biomarker in question should impact the clinical endpoints of interest. Although laboratory or preclinical data may indicate the effect of interventions on the biomarker and correspond to the effect on clinical outcome, robust, adequately controlled clinical study data using clinical endpoints (i.e., phase III data or equivalent studies) are necessary. Observational data in human populations and preliminary clinical data (e.g., phase I or II data) are considered, but are not sufficient to fully qualify a biomarker as a surrogate endpoint at this stage of evaluation. An informative evidence-based approach to qualification of a surrogate endpoint may be based on an overview analysis of multiple randomized trials, where the relationship of intervention’s effect on the biomarker is plotted against the intervention’s effect on the true clinical endpoint. Examples of this include an assessment of progression-free survival as a surrogate endpoint for overall survival for adjuvant treatment of colorectal cancer (Fleming, 2005; Sargent, 2004) and the assessment of blood pressure as a surrogate endpoint for cardiovascular risk (Staessen et al., 2003).
In biologic systems, a given intervention can exert multiple different, even contradictory, actions. There are challenges to determining what clinical trial data are necessary to document the value of interventions to target the specific clinical endpoint and predict benefit and harm. For example, postmenopausal hormone replacement therapy (HRT) was thought to protect women from cardiovascular disease based on both observational, epidemiologic data and the apparent beneficial effects of estrogen on lipoproteins and other cardiovascular disease biomarkers. However, several important inflammatory biomarkers (adhesion molecules) and prothrombotic biomarkers were not measured in the early studies. After several clinical trials, HRT was discovered to raise mortality from cardiovascular events and have other adverse unexpected effects.
For this reason, the committee recommends analyzing multiple mechanistic pathways leading to the same outcome when evaluating biomarkers.
Additionally, interventions may impact populations differently. The clinical consequences of an intervention may differ from a healthy population to a population with extensive comorbidities. A biomarker must not merely show a difference between healthy controls and individuals with a certain disease; the control group must also include subjects with other pathophysiologies in order to ensure the biomarker data show a distinctive difference between controls and individuals with the disease (Koulman et al., 2009). In the description of evidence about the biomarker, populations and conditions to which the assessment applies need to be articulated so they can be considered in the utilization step of the biomarker evaluation framework.
Box 3-3 introduces the case study exemplifying issues surrounding biomarker qualification. This example is discussed in Chapter 4.
|
BOX 3-3 CRP, Inflammatory Markers, and Qualification (Recommendation 1b) As the scientific understanding of atherosclerosis has evolved to include inflammation’s role in the disease process, researchers have sought inflammatory biomarkers. The one most extensively studied is C-reactive protein (CRP). In observational studies, CRP is an independent predictor of future vascular events, including myocardial infarction, ischemic stroke, peripheral vascular disease, and vascular death. In spite of CRP’s utility in cardiovascular risk prediction, its normal function and role in cardiovascular disease remains uncertain. The lack of understanding of CRP’s biological role in human physiology has elicited controversy over assertions of CRP’s causal role in cardiovascular disease. CRP satisfies the first step of the evaluation framework, analytic validity: CRP is easily measured by standardized high-sensitivity immunoassays and has negligible diurnal variation, does not depend on food intake, and has a long half-life. For qualification, CRP has prognostic value: a number of population cohorts have shown CRP to predict future vascular risk, though with some caveats (these are explained in Chapter 4). The evidence supporting CRP’s biologic association with cardiovascular disease is weak, and more research is needed to clarify determinants of CRP variation and utility in diverse populations. Although several interventions are known to lower CRP, it is unclear whether there is consistency of correlation between the effects of different interventions on CRP and clinical outcomes. Based on these findings, in the utilization step, CRP would not qualify for the context of use of surrogacy, but it may be used in risk prediction in certain populations. This matter is discussed in greater detail in the full case study found in Chapter 4. |
Utilization
The third step of the committee’s biomarker evaluation framework is a contextual analysis of the available evidence about a biomarker with regard to the proposed use of the biomarker. These evaluations should take place on a strictly designated fit-for-purpose basis, with consideration for the context of use, as knowledge and technology continually evolve. Defining the context of use requires explicit articulation of the populations and conditions for use to which the assessment applies. For surrogate endpoints, idealized statistical requirements are rarely or never achievable; subjective assessment is necessary to determine when surrogate endpoints can be used. This variability between evaluations can be minimized by consistently evaluating the critical and important factors, including risk assessment, as described by the committee.
The utilization step can be divided into several components. As discussed in Chapter 2 of this report, biomarkers have a multitude of uses in both clinical care and drug development, including for risk stratification, prevention, screening, diagnosis, prognosis, patient selection, and pharmacodynamics (see Table 2-1). In drug development, for example, there is a continuum of uses from early trials on one end to surrogate endpoints on the other end. A determination of the general category of use for which the biomarker is intended is necessary to inform the evaluation process as to whether analytical validation and qualification data are appropriate for that use, particularly when it may be involved in future regulatory or policy decisions. The list of uses is further expanded when biomarkers are discussed in the arenas of medical devices, biologics, and nutrients and foods. This determination can therefore be understood as a necessary first component in the utilization analysis step of biomarker evaluation.
The second component in the utilization analysis is consideration of factors related to defining the context for which a biomarker should be qualified. Generally, the earlier in the development of an intervention, the more flexibility there is in using a biomarker. The committee evaluated a multitude of factors, including prevalence of the disease, risks associated with the intervention, and concurrent and prior treatment, to develop its criteria. The exhaustive list of factors was synthesized into a concise list of Critical and Important Factors (see Table 3-3). The recommendations are meant to provide general guidelines that could be adapted for all uses of biomarkers that result in clinical, product, or claim development, or regulatory decisions, whether for drugs, biologics, or device development; for relationships between diet or nutrients and disease; or for public health monitoring and interventions. Thus, the criteria are broad in scope.
One of the principal considerations in biomarker evaluation is whether the biomarker is being used as a surrogate endpoint. If it is, the standards of evaluation are more rigidly defined. This scenario is discussed further
TABLE 3-3 Utilization: Critical and Important Factors for Consideration (Recommendation 1c)
|
|
Factors |
Rationale |
|
|
Critical |
1. |
Is the biomarker being used as a surrogate? |
If the biomarker is used as a surrogate, enhanced scrutiny would be necessary. |
|
|
2. |
What is the prevalence of the disease? What are the morbidities and mortalities associated with this disease? |
A highly prevalent or serious disease might have a lower threshold for use of biomarkers in clinical and regulatory decisions. |
|
|
3. |
What are the risks and benefits associated with the intervention? Has due attention been paid to both safety and efficacy? |
The benefits of the intervention must be weighed against the risks of biomarker failure to define a range of tolerable biomarker performance for each specific biomarker (Williams et al., 2006). |
|
|
4. |
What are the advantages and disadvantages associated with use of the biomarker when compared with the best available alternative? How does the biomarker benefit management and outcomes? |
The evaluation may proceed differently depending upon whether a variety of valid treatment options are available compared to if no treatments have yet been developed, for example. |
|
|
5. |
Is the biomarker for drugs, biologics, or device development; for relationships between diet or nutrients and disease; or for public health monitoring and interventions? |
While the highest level of scientific rigor is needed in biomarker evaluations for all uses, each category of use has different risks and regulatory frameworks, which carry implications for appropriate evidence thresholds and requirements for biomarker use. |
|
Important |
6. |
What is the biomarker’s purpose with respect to phase of development in clinical trials? |
For biomarkers that are likely to be used in a regulatory submission or as evidence supporting statements regulated by the FDA, consideration should be given to the need for additional data collection. |
|
|
7. |
Is the biomarker for primary or secondary disease prevention? |
Biomarkers used for these purposes carry especially high risk and should be evaluated with this consideration in mind. |
in the next section. For all biomarkers, including surrogate endpoints, the prevalence of the disease and the morbidities and mortalities associated with the disease are important contextual considerations. For example, in general, use of an intervention meant for primary prevention will have an extremely low tolerance for risk. Within this minimal tolerance, however, for risk reduction of a very common, serious chronic disease, more risk may be tolerated than for an intervention intended to prevent a less common or less serious disease. Likewise, an intervention meant to treat a rare but life-threatening disease may permit more tolerance of risk than an intervention meant to treat a more common but less serious disease. So, it may be easier to defend use of a surrogate endpoint for trials of rare and life-threatening diseases than for trials of primary prevention interventions for common but less serious or life-threatening diseases.
The safety and efficacy of biomarker use can be thought of in conjunction with the risks and benefits associated with the intervention targeting the biomarker. The benefits of the intervention must be weighed against the risk of biomarker failure to define a range of tolerable biomarker performance for each specific biomarker (Williams et al., 2006). Subjectivity can be minimized by thinking of biomarker utility as analogous to risk assessment, as discussed by Williams and colleagues (2006). In Williams’s proposed framework, the generation of knowledge links specific risk agents with uncertain, but possible, outcomes. Thus, a key factor is the perceived consequence that would result if the biomarker were to fail. Although quantitative information related to the degree and frequency of failure may be unavailable, the seriousness of this failure should be a factor in evaluation (Williams et al., 2006).
The extent to which a given surrogate endpoint can be a target of a therapeutic intervention depends on a patient’s or population’s specific constellation of risk factors, relative to the multiple components of risk found in the population as a whole. It is important to determine the mechanism dominating the clinical effect so that interventions most likely to affect that mechanism can be selected for particular patients or populations. For example, for patients with familial hypercholesterolemia, high LDL cholesterol (LDL-C) will likely be their most important cardiovascular risk factor; interventions that target LDL-C may be justified even when there is a lower level of supporting evidence (i.e., use of interventions approved on the basis of surrogate endpoint data). For the general population, on the other hand, where competing cardiovascular risks are from high LDL-C, hypertension, inflammation, smoking, and other dyslipidemias, the successful use of LDL-C as a surrogate endpoint for cardiovascular risk is less assured. Hence, better evidence is needed on the connection of LDL-C–lowering interventions and clinical outcomes
before use of those interventions can be recommended in the general population.
The committee finds that the hazards of making the wrong decision regarding a biomarker’s qualification is a critical factor in the decision-making process. Although the opportunity cost (i.e., the loss of the benefits of the next best alternative decision) differs depending on the stakeholders, the subjectivity of this consideration can be minimized. When a choice is made, the opportunity cost is the benefit that would have occurred had the second best option been chosen instead. To illustrate opportunity cost, someone choosing a breakfast food in the absence of health advice might choose a food high in calories and saturated fat or they might choose a bowl of healthy cereal with skim milk and an apple. The opportunity cost for one of these decisions over the other is the potential health benefit advantage or money saved that would have occurred should the other option have been chosen. In a related example, a box of cereal carries a claim recommending its healthy characteristics. In this case, an individual may choose the cereal with the healthy claim over a similar, cheaper cereal, or choose it over an unhealthy option. The opportunity cost would be the money that would have been saved by choosing the cheaper cereal, but the choice may also have prevented the individual from choosing an unhealthy breakfast. For the cereal manufacturer, the opportunity cost of not carrying the healthy claim would be the lost profits of more individuals choosing the manufacturer’s cereal, whereas the opportunity cost of carrying the healthy claim would be the money that could have been saved by not developing the claim, printing the new packaging, or carrying the legal liability of the claim.
As with considerations of competing risks, knowledge of the concurrent and prior treatments used in treating an individual patient or a patient population plays a role in contexts of use for which a biomarker may be qualified. The evaluation may proceed differently depending upon whether a variety of treatment options are available compared to if no treatments have yet been developed, for example. The committee believes it is important to value the costs of denial of an intervention to patients who would benefit.
The committee did not explicitly include analysis of a biomarker test’s or intervention’s cost effectiveness in the evaluation framework. Cost effectiveness is important for a subset of biomarker uses, particularly those involving changing the clinical practice of medicine. In such situations, evaluators may wish to include analysis of cost effectiveness of interventions in the utilization step. A great deal of research has been done on how to conduct such studies, although the committee cautions that definitive estimates of costs can be made only after clinical outcomes are measured.
|
BOX 3-4 Troponin and Utilization (Recommendation 1c) Use of troponin as a biomarker in acute settings is ubiquitous as a method to diagnose myocardial infarction (MI). MI causes cardiac muscle damage that results in a rise in troponin concentrations. Its use in chronic settings is more recent, and relies on developing high-sensitivity assays that still require validation. However, the criteria for such a validation are advanced compared to the current regulatory standards. Troponin can be elevated in patients who may suffer from a variety of chronic heart conditions, inflammatory conditions, side effects from drugs, or organ failures. These assays have not yet shown analytical validation. But, should one or several of the assays eventually show adequate sensitivity, specificity, and reproducibility, then the biomarker can be advanced to the qualification step. In qualification, it is apparent that clinical data from several different trials (Gupta and de Lemos, 2007; NACB Writing Group Members et al., 2007) show increased risk of mortality in individuals with elevated troponin levels. However, although there is evidence that prevention of MI reduces death rates, there is no evidence that using an intervention to decrease troponin levels rather than preventing the event in totality improves mortality risk. Finally, although use of troponin as a biomarker in phase I studies to indicate cardiac safety problems with tested drugs or to collect further information about the valuable applications of this biomarker is justified and valuable, use of troponin levels as a surrogate endpoint for interventions is not justified due to a dearth of evidence. This matter is discussed in greater detail in the full case study found in Chapter 4. |
Box 3-4 summarizes the case study for the biomarker troponin, which illustrates some of the judgments that can be made in the utilization step of biomarker evaluation. This case study is discussed in further detail in Chapter 4.
Evaluation of a Biomarker as a Surrogate Endpoint
In the case of chronic disease, where there are multiple pathogenetic pathways leading to development of clinical outcomes and multiple manifestations of disease, the probabilistic nature of predictions made using biomarker data means that no biomarker can give absolute certainty of an event’s future occurrence nor absolute certainty of the timing of the predicted event. Nonetheless, there are situations in which use of a biomarker as a surrogate endpoint in situations with regulatory impact may be supported, such as in situations where the need for interventions is urgent or where studies including clinical endpoints are not feasible because of technical or ethical reasons. Again, this is not meant to discourage use of biomarkers in product development; biomarkers play an important
role in research and decision making. Situations with regulatory impact are defined in the section on Recommendation 2. Finally, it is essential to remember that the information that an individual surrogate endpoint or clinical endpoint can give is inherently limited; as a result, it is important to emphasize the need to evaluate data relating to adverse events and unintended effects of biomarker use.
The committee does not intend to imply that selection of endpoints for clinical trials would be simple or risk free if investigators were simply to avoid surrogate endpoints. Clinical and surrogate endpoints have been defined in a way that may imply a clear distinction between the two, in that clinical endpoints typically reflect patient experience and surrogate endpoints do not. However, there is discussion surrounding this issue, which illustrates the scientific complexity of the distinction between clinical and surrogate endpoints. Some clinical endpoints have many similarities with biomarkers, and can be thought of as a step removed from patient experience, and therefore subject to similar potential failings as surrogate endpoints (i.e., pain scales). Some surrogate endpoints are highly robust (i.e., HIV-1 RNA for particular classes of viral-suppressing drugs). However, even these endpoints require an understanding of unrelated effects, the magnitude and duration of target effects, and optimization of use (such as timing related to initiation of viral-suppressing drugs). Clinical endpoints share many features of biomarkers, such as the need for analytical validation, but they differ from biomarkers in that clinical endpoints address how a patient feels, functions, or survives and also commonly utilize multiple diagnostic criteria. Nonetheless, the committee recognizes that selection of clinical endpoints is beyond the scope of this report. There are many important interests at stake in this discussion and some issues, such as the best way to choose endpoints for trials, may be context specific. In such settings, stakeholders such as industry, the public as represented by government and community representatives, and academic researchers, may benefit from convening to discuss these issues.
Utilization aims to establish whether the biomarker is being used as a surrogate endpoint; the prevalence, morbidity, and mortality of the disease; the risks and benefits associated with the intervention; and opportunity cost, among other factors. Box 3-5 introduces the case study on surrogate endpoint status of LDL and high-density lipoprotein (HDL) cholesterol.
APPLICATION OF THE EVALUATION FRAMEWORK
|
2b. |
Initial evaluation of analytical validation and qualification should be conducted separately from a particular context of use. |
|
2c. |
The expert panels should reevaluate analytical validation, qualification, and utilization on a continual and a case-by-case basis. |
Recommendation 2 provides further guidance on the application of the framework to uses of biomarkers that have regulatory impact. Specifically omitted from this recommendation are biomarker discovery activities and biomarkers for use in drug discovery, development, and other preclinical uses. This decision was made based on the sheer volume of
|
BOX 3-5 LDL and HDL Cholesterol and Surrogacy (Recommendation 1c) Low-density lipoprotein cholesterol (LDL-C) concentration is considered as a qualified surrogate endpoint for cardiovascular disease for both food-related disease claims and drugs. It is often viewed as the benchmark biomarker (Couzin, 2008; Rasnake et al., 2007). Thus, an examination of the evaluation of LDL-C not only highlights the strengths of the biomarker itself, but also the ways in which even qualified biomarkers face contextual caveats. The evidence supporting this biomarker rests almost entirely on the measurement of LDL-C even though it is only one part of the lipoprotein particle. Both apolipoprotein B and the quantity and the composition of LDL particles themselves have potential to be more indicative of cardiovascular disease risk than LDL-C for some populations (Berneis and Krauss, 2002; Rizzo and Berneis, 2007; Tardif et al., 2006), showing that even for qualified biomarkers, developing standard measures is an ongoing process. The strength of LDL-C as a surrogate endpoint is not absolute due to the heterogeneity of cardiovascular disease processes, the heterogeneity of LDL-lowering drug as well as food effects, and the heterogeneity of LDL particles themselves. Because cardiovascular disease is a multifactorial chronic disease, a single component of the disease (e.g., LDL-C) cannot fully account for all the variability that leads to a particular outcome (Libby and Theroux, 2005; Tardif et al., 2006). The C-reactive protein case study suggests that inflammation, for example, may also affect the cardiovascular disease pathway. Furthermore, sociodemographic factors have been shown to complicate these already complex disease dynamics; as a result, lowering LDL-C can never be assured to be a “perfect” indicator across all population groups or all interventions. Interventions to address a multifactorial disease introduce potentially unforeseen effects, particularly when the causal disease pathways, the mechanisms of action of the intervention, and the biochemical characteristics and function of the biomarker itself are not fully understood. High-density lipoprotein (HDL) does not qualify as a generic surrogate endpoint because these characteristics, particularly the latter, introduce high levels of variability. Furthermore, evidence is weak that elevation of HDL from therapeutics decreases cardiovascular disease risk. LDL and HDL are discussed in greater detail in the full case study found in Chapter 4. |
newly discovered biomarkers and the low probability that any given new biomarker will see application in either a regulatory submission for a new product or in a clinical setting. The committee sought ways to achieve a rigorous evaluation framework without stifling innovation. Experts qualified by experience and training are needed to conduct the evaluation reviews, focusing on utilization, as case-by-case analyses are the only way to ensure proper use of biomarkers given the state of the science. The committee has sought ways to support the FDA and other federal agencies in their important work to maximize public health and to regulate the food and drug industry. Maximizing public health means not only protecting the public from inconclusive or fraudulent claims about a food’s or drug’s benefits on health, but also allowing for rapid access to effective drugs and health-related information. Routes to evidence-based regulation need to be sought that will account for continued innovation and development of products and strategies to improve human health.
Situations having regulatory impact were defined by the committee as follows: circumstances where biomarker data will be submitted, is anticipated to be submitted, or may be requested for submission (as in the case of verification of certain claims on foods or supplements) to the FDA for a regulatory purpose. This definition allows for biomarker discovery and early product discovery activities without convening expert committees. The committee also considered situations in which generally accepted criteria for approval of drugs and other interventions cannot be followed due to insufficient numbers of patients, such as for development of interventions for rare diseases. In the case of rare diseases, product applications are submitted through the FDA’s Office of Orphan Products Development (OOPD). The committee suggests that OOPD will need to adapt use of Recommendation 2 to fit with its task.
Direct engagement by the FDA in the process of biomarker evaluation for regulatory decision making may be helpful. Because of the substantial expense, resources, and time that will be needed to qualify new biomarkers, particularly as surrogate endpoints, prospective and specific guidance on the potential or actual acceptance of biomarkers on the part of regulatory agencies for different purposes, and the agencies’ regulatory risk tolerance in qualifying biomarkers for each new use, would also be helpful. Ideally, this would lead to an agreement on the weight and specificity of data that would need to be submitted to qualify biomarkers for each purpose under proposed conditions for use. Such an agreement would help to justify the cost and risk of an elaborate biomarker research program in the same way that an end-of-phase II meeting does for phase III drug development. The IOM is currently conducting a study on “Accelerating Rare Diseases Research and Orphan Product Development”; the report will be released in late 2010 or early 2011 (IOM, 2010a).
This organized, large-scale approach to evaluation of a biomarker with regulatory impact requires the convening of an expert panel, similar to an FDA advisory committee, with (1) appropriate expertise, (2) a variety of stakeholders, and (3) attention to conflict of interest. Due to the complexity of data, the need for context-of-use analysis, and the need to deal with sometimes-contradictory evidence, expert input is essential to provide scientific judgment in areas of uncertainty. Experts qualified by experience and training are needed to conduct the evaluation reviews, as case-by-case analyses are the only way to ensure proper use of biomarkers. The same expert panel can discuss all steps in the evaluation framework provided that the panel contains all needed expertise. Panelists should encompass a range of backgrounds, as well as a full range of areas of expertise, including biologists, pharmacologists, clinicians, clinical trialists, and statisticians, as necessary, for decision making. The panelists must be knowledgeable about the biomarker evaluation process and represent a diversity of disciplines and perspectives.
Numerous entities, including the IOM in a 2009 report Conflict of Interest in Medical Research, Education, and Practice, have defined a need for attention to conflict of interest in order to protect the integrity of professional judgment. The expert panel for biomarker evaluation should be formulated with due attention to the 2009 IOM recommendations. The biomarker evaluation process inevitably requires judgments to be made by the expert panel; these judgments must be known to be made in good faith and without undue influence. A well-formulated conflict-of-interest policy does not prohibit individual and institutional relationships that might be questioned, but rather, manages these relationships as necessary and required by the policy (IOM, 2009).
As indicated in Figure 3-1, the steps in the recommended framework interact; they are not necessarily separated in time, and conclusions in one step may require revisions or additional work in other steps. For example, as the case study presented in Chapter 4 indicates, tumor size is a biomarker often used for determining efficacy of cancer drugs. Because of inconsistent definitions of tumor size and new findings about the prognostic value of tumor size for specific cancers, among other factors, tumor size is a biomarker that has been, and will continue to be, continually revisited.
Nonetheless, Recommendation 2b states that initial analytical validation and qualification of a biomarker can and should be conducted separately from a particular context of use. The committee understands that no decisions can be made about use of a biomarker without having its use in mind. The committee concluded that it was important to separate the parts of the evaluation framework that have the goal of being objective and those for which subjective judgment is necessary. Analytical valida-
tion and evidentiary qualification were viewed as objective tasks of gathering available evidence, and so they can be conducted separately from a particular context of use. It is also important to discuss briefly, how the committee envisions conduct of the data-gathering process. Data should be gathered from all available sources of evidence. When the evidence is to come entirely from the public domain, it can be gathered according to principles of systematic review (Cochrane Collaboration, 2009; IOM, 2010b). When data not generally publically accessible is made available, such as data owned by companies, for example, then gathering of such data would likely be subject to the same processes as data submission to the FDA for product review.
Evidence evolves even after a biomarker is evaluated; thus, it is imperative that biomarkers be reevaluated periodically so that both the scientific evidence and context-of-use analyses capture the current state of the science. By continual, the committee refers to the need for regular reevaluation on the basis of new scientific developments and data. For instance, continuing with the tumor-size case example, progression of gastrointestinal stromal tumors was found to occur within the original tumor boundaries. Although chemotherapeutic treatment of the tumors may result in decreased cell density and prolonged survival, tumor size (in terms of measurable diameters) was found to generally remain the same. These findings could be cause for reevaluation of the analytical validation step of the biomarker evaluation framework.
Ideally, research findings would dictate the necessity for reevaluation. Post-hoc review should be performed at regular intervals as new information is available to determine how new conclusions should modify the biomarker’s qualification and use. When new, potentially relevant evidence related to a biomarker is found, this evidence would be considered to determine the continued appropriate use of the biomarker across a variety of contexts. In practice, however, research efforts are often piecemeal and new findings may not readily be identified as cause for reevaluation of a biomarker. Additionally, the dynamic context of the regulatory environment may lead to reappraisal of the contexts for which a biomarker has been evaluated. For example, some regulatory environments may, despite attempts to minimize subjectivity, exhibit less caution when evaluating some contexts in which a given biomarker can be used. Thus, given the many demands and time constraints of the medical, scientific, and regulatory enterprises, the committee concludes that to incorporate and consider new research findings, biomarkers may be reevaluated within a reasonable time frame, such as every 4 years, for example. The committee does not intend such a time frame to dissuade more frequent reevaluation: Indeed, the rapidity of new knowledge available may dictate more immediate revisions in the contexts for which a biomarker may be used.
Rather, all biomarker evaluations should undergo reappraisal on at least such a time frame.
Each step needs to be reconsidered to the extent that research or context has changed since the previous evaluation. The reappraisal process need not consider the biomarker as though no previous evaluation had occurred. The monetary and opportunity costs of this kind of de novo evaluation would render such analyses prohibitive. Rather, the available data can be scrutinized in the context of what had been previously evaluated. By considering additional evidence, it is possible that the expert panel may alter its past findings by revoking recommendations for a previously accepted biomarker use, choosing not to recommend a biomarker for uses similar to those for which it was granted permission in the past, providing a more nuanced explanation as to how a biomarker should be used, or qualifying the biomarker for use in new contexts. Some of these scenarios are indicated in the case studies presented in Chapter 4. Nonetheless, it is essential that the utilization analysis be carried out by a panel of experts, as scientific and medical judgment is necessary to weigh the possible advantages and disadvantages of the proposed biomarker use.
SCIENTIFIC PROCESS HARMONIZATION
Recommendation 3:
The FDA should use the same degree of scientific rigor for evaluation of biomarkers across regulatory areas, whether they are proposed for use in the arenas of drugs, medical devices, biologics, or foods and dietary supplements. Congress may need to strengthen FDA authority to accomplish this goal.
Legislation and court decisions have created a regulatory environment in which different evidentiary and labeling requirements exist for drugs and biologics, devices, and foods and supplements. The committee has concluded that accurate and complete science is critical in all of these areas. While recognizing the differences between the different product categories, the committee emphasizes that none of these categories presents a situation so low in risk to consumers as to allow less rigorous scientific justification for claims. Box 3-6 summarizes the case study for a nutritionally relevant biomarker, blood levels of beta-carotene. This case study illustrates the need for collection of data for nutrition-related biomarkers.
To further illustrate the assertion that it is not safe to make assumptions about risks posed by products in a given category, consider the numbers of people exposed annually to several public health interventions that use food, compared to the numbers of people annually who
|
BOX 3-6 Blood Levels of β-Carotene Studies have consistently shown that diets rich in fruit and vegetables are associated with a reduced risk of chronic diseases such as heart disease and cancer (Block et al. 1992; Peto et al., 1981). Although fruits and vegetables offer many nutrients, years of epidemiological studies suggested that blood levels of β-carotene were associated with lower incidence of cardiovascular disease and cancer (Hennekens et al., 1984; Manson et al., 1993; Willett et al., 1984). β-carotene is a carotenoid and antioxidant known to be a precursor of vitamin A. To further corroborate the biomarker’s biological plausibility, β-carotene’s classification as an antioxidant provided a possible mechanism for a protective effect. Though there were no further animal studies or small-scale clinical trials performed, mounting pressures from multiple stakeholders, eager to prevent disease or improve the quality of life for persons at risk of chronic disease, quickly pushed the consideration of blood β-carotene levels as an effective chemopreventive biomarker and impelled large-scale intervention trials to test the possible benefits of increased intake of the nutrient itself were quickly initiated. Before results from the three large β-carotene trials (the Physicians’ Health Study) (Cook et al., 2000), the Beta Carotene and Retinal Efficacy Trial (CARET) (Omenn et al., 1996a, 1996b), and Alpha Tocopherol Beta Carotene Cancer Prevention Study (ATBC) (Albanes et al., 1996) had been confirmed, the belief in the “efficacy” of increased β-carotene intake became widespread based on the observational studies that demonstrated association, but not causality. This was based on the consistency, strength of association, dose–response gradient, and biological plausibility. Thus, the unfavorable and even deleterious results of the trials were surprising to physician, patient, research-scientist, and policy-maker proponents of β-carotene. These studies demonstrated that assumptions that β-carotene was a valid causal predictor of decreased lung cancer risk were in error and illustrate the public health value of proper preclinical research strategies and evaluative process before permitting claims. This matter is discussed in greater detail in the full case study found in Chapter 4. |
take a few common drugs. About 184 million people drank fluoridated water in the United States in 2006, about 62 percent of the entire population (CDC, 2006). Commercially available cereal flours and related products, milk and other dairy products, and fruit juices and drinks can be fortified with vitamin D. Milk and cereals are most frequently fortified (Calvo et al., 2004). Dietary intakes of vitamin D in the United States range from about 4.2 to 5.4 µg per person per day (depending on age and sex), most of which is from fortified foods (Moore et al., 2005). Additionally, about 27 percent of the U.S. adult population took
a supplement containing vitamin D in 2002 (Kaufman et al., 2002). In 2002, it was reported that about 5.2 percent of the U.S. adult population was taking statins (Freemantle, 2002; Kaufman et al., 2002). The most commonly used medication, acetaminophen, was taken by about 23 percent of the U.S. adult population in a given week (Kaufman et al., 2002). Just over 1 percent of U.S. adults were taking fluoxetine hydrochloride (Prozac) (Kaufman et al., 2002). These are among the most used over-the-counter and prescription medications in the United States. From these and other similar data, it can be concluded that exposure to some public health interventions is much more prevalent than exposure to the most common medications.
Further, many individuals are not aware that public health interventions involving food are not risk-free. Chapter 4 shows the risks of beta-carotene supplementation. The example above highlights a topic discussed more fully in Chapter 2: in order to make informed decisions, individuals need access to complete information (see Chapter 2 section titled “Biomarkers and Communication Strategies at the FDA”). Nonetheless, the ability to interpret this information depends on numeracy, and individuals making complex decisions may benefit from professional advice (see Chapter 2 section titled “Numeracy”). However, professional advice is generally not sought for dietary decisions, for example. Further discussion of issues related to the use of biomarker data and its impact on subsequent health-related decisions was discussed in Chapter 2 (see section titled “Cognitive Biases and Impacts of Evidence Gaps”).
Recommendation 3 is consistent with other recent efforts to improve the use of science at FDA and in European regulatory agencies. The renewed effort to strengthen the scientific base at FDA is discussed in Chapter 5 (see section titled “Tracking the Effects of Biomarker Use at the FDA”). Chapter 5 also goes into detail about the different requirements in different product areas. It discusses the use of regulatory authority and where better use may be needed. In order to implement this recommendation, the FDA will need to better implement some of its existing regulatory authority, and it may also need additional regulatory authority. Recommendation 3 is not meant to imply that an identical process be used across all of the centers. Instead, it means that rigorous, complete review of all available scientific evidence is necessary before regulatory decisions can be made. In the case of foods and supplements, for example, this may require Congress to enact legislation to allow the FDA to compel companies to gather and submit data relating to the safety and efficacy of proposed products and health claims, based on both the nutrients of interest alone and on the whole products within which they are contained.
Addressing Differences in Current Standards for Drugs, Biologics, Devices, Supplements, and Foods
Recommendation 4:
The FDA should take into account a nutrient’s or food’s source as well as any modifying effects of the food or supplement that serves as the delivery vehicle and the dietary patterns associated with consumption of the nutrient or food when reviewing health-related label claims and the safety of food and supplements. Congress may need to strengthen FDA authority to accomplish this goal.
Drugs, biologics, and devices are evaluated on the basis of the safety and efficacy of the entire product. The regulatory framework governing these products, foods, and supplements are explained in greater detail in Chapter 5. The committee concluded that for the utilization step of the biomarker evaluation framework, it is necessary to evaluate the biomarker’s proposed use in terms of the entire product in all situations. In addition, the committee concluded that it is important to evaluate efficacy as well as safety of proposed biomarker uses. Legislation may be required to implement this recommendation.
Currently, the safety of new food substances is evaluated for the individual substances within the context of intended conditions of use, and not on a product-specific basis as is done for drugs. Validity of claims made with respect to foods and supplements can be made on the basis of single ingredients in foods. There are some restrictions on the amount of fat, saturated fat, cholesterol, and sodium that foods bearing health claims can contain, and also on the need for a minimum amount of vitamin A, vitamin C, calcium, protein, fiber, or iron for foods bearing claims. Nonetheless, although review of proposed health claims takes into account the relationship of the specific substance that is the subject of the health claim to the health outcome of interest, it may not adequately consider the modifications of the substance’s effect on the disease outcome by other bioactive components in that food or the diet. For this reason, it is important to include an analysis of the connection between the biomarker and other factors associated with conditions that can affect its efficacy and safety in the qualification process.
In addition to the modifying effects of other material components of a food or supplement on the effect of a health claim based on a single ingredient, it is also important to consider the modifying effects of a health claim on the overall healthfulness of the diet. More research in this area is needed.
CONCLUSION
This approach to biomarker evaluation extends beyond reviewing the scientific literature to determine biomarker acceptance. The recommended comprehensive evaluation framework is a process by which consensus may be reached about the qualification of a biomarker and considers context-independent and context-dependent qualifications, as well as analytical validation. The committee finds it important to make analytical validation a necessary component to biomarker validation; without high-quality research data, biomarkers cannot be effectively used. Furthermore, it is important to know whether a biomarker has prognostic value and whether the science underlying its role in disease is well understood. Determining that a biomarker has prognostic value and a well-defined scientific basis, however, is distinct from knowledge that modifying the biomarker will bring about clinical benefit or harm. Utilization, the process of making assessments of whether a proposed biomarker is fit for the purpose for which it is being proposed, is the third essential component of the biomarker evaluation process. The committee concludes that these three steps therefore warrant separation to ensure each receives its full consideration. For decisions involving regulatory bodies, the committee recommends that an expert panel conduct the evaluation reviews. Biomarker evaluations need to be continually updated to reflect the current state of the science.
Importantly, the committee has recommended that the scientific information used to inform policy decisions regarding biomarkers should be equally rigorous across proposed uses and product categories. Finally, in the special case of foods and supplements, accommodations are needed to ensure that the entire food or supplement is taken into account when evaluating biomarkers for nutrition-related uses.
REFERENCES
Albanes, D., O. P. Heinonen, P. R. Taylor, J. Virtamo, B. K. Edwards, M. Rautalahti, A. M. Hartman, J. Palmgren, L. S. Freedman, J. Haapakoski, M. J. Barrett, P. Pietinen, N. Malila, E. Tala, K. Liippo, E. R. Salomaa, J. A. Tangrea, L. Teppo, F. B. Askin, E. Taskinen, Y. Erozan, P. Greenwald, and J. K. Huttunen. 1996. Alpha-tocopherol and beta-carotene supplements and lung cancer incidence in the alpha-tocopherol, beta-carotene cancer prevention study: Effects of base-line characteristics and study compliance. Journal of the National Cancer Institute 88(21):1560–1570.
Apple, F. S., M. Panteghini, J. Ravkilde, J. Mair, A. H. B. Wu, J. Tate, F. Pagani, R. H. Christenson, and A. S. Jaffe. 2005. Quality specifications for B-type natriuretic peptide assays. Clinical Chemistry 51(3):486–493.
Apple, F. S., A. H. B. Wu, A. S. Jaffe, M. Panteghini, R. H. Christenson, NACB Committee Members, C. P. Cannon, G. Francis, R. L. Jesse, D. A. Morrow, L. K. Newby, A. B. Storrow, W. H. W. Tang, IFCC Comittee on Standardization of Markers of Cardiac Damage Members, F. Pagani, J. Tate, J. Ordonez-Llanos, and J. Mair. 2007. National Academy of Clinical Biochemistry and IFCC Committee for Standardization of Markers of Cardiac Damage Laboratory Medicine Practice Guidelines: Analytical issues for biomarkers of heart failure. Circulation 116(5):e95–e98.
Berneis, K. K., and R. M. Krauss. 2002. Metabolic origins and clinical significance of LDL heterogeneity. Journal of Lipid Research 43(9):1363–1379.
Biomarkers Definitions Working Group. 2001. Biomarkers and surrogate endpoints: Preferred definitions and conceptual framework. Clinical Pharmacology and Therapeutics 69(3):89–95.
Block, G., B. Patterson, and A. Subar. 1992. Fruit, vegetables, and cancer prevention: A review of the epidemiological evidence. Nutrition and Cancer 18(1):1-29.
Califf, R. M., K. S. Pieper, W. R. Harlan 3rd, and K. L. Lee. 1992. Serum cholesterol in patients undergoing cardiac catheterization for suspected coronary artery disease: Diagnostic and prognostic implications. Annals of Epidemiology 2(1-2):137–145.
Calvo, M. S., S. J. Whiting, and C. N. Barton. 2004. Vitamin D fortification in the United States and Canada: Current status and data needs. The American Journal of Clinical Nutrition 80(Suppl):1710S–1716S.
CDC (Centers for Disease Control and Prevention). 2006. Water fluoridation statistics for 2006. http://www.cdc.gov/Fluoridation/statistics/2006stats.htm (accessed October 5, 2009).
CDER (Center for Drug Evaluation and Research). 2001. Guidance for Industry on Bioanalytical Method Validation: Availability. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM070107.pdf (accessed December 29, 2009).
Cochrane Collaboration. 2009. Cochrane handbook for systematic reviews of interventions version 5.0.2 [updated September 2009]. Higgins, J. P. T., and S. Green (editors). http://www.cochrane-handbook.org/ (accessed March 15, 2010).
Cook, N. R., I. M. Le, J. E. Manson, J. E. Buring, and C. H. Hennekens. 2000. Effects of beta-carotene supplementation on cancer incidence by baseline characteristics in the Physicians’ Health Study (United States). Cancer Causes and Control 11(7):617–626.
Couzin, J. 2008. Clinical trials and tribulations. Cholesterol veers off script. Science 322(5899): 220–223.
Fleming, T. R. 2005. Surrogate endpoints and the FDA’s accelerated approval process. Health Affairs 24(1): 67-78.
Fraser, C. G. 2001. Biological variation: From principles to practice. Washington, DC: AACC Press.
Freemantle, N. 2002. Medicalisation, limits to medicine, or never enough money to go around? Spending on preventive treatments that help a few is unaffordable. BMJ 324(7342):1241–1242.
Goodsaid, F., and F. Frueh. 2007. Biomarker qualification pilot process at the U.S. Food and Drug Administration. AAPS Journal 9(1):E105–E108.
Gupta, S., and J. de Lemos. 2007. Use and misuse of cardiac troponins in clinical practice. Progress in Cardiovascular Diseases 50(2):151–165.
Hennekens, C. H., M. J. Stampfer, and W. Willett. 1984. Micronutrients and cancer chemo-prevention. Cancer Detection and Prevention 7(3):147-158.
Hill, A. B. 1965. The environment and disease: Association or causation? Proceedings of the Royal Society of Medicine 58:295–300.
ICH (International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use). 1994. ICH Guidelines: Text on validation of analytical procedures, Q2A. Geneva, Switzerland: ICH.
IOM (Institute of Medicine). 2007. The future of drug safety. Washington, DC: The National Academies Press.
IOM. 2009. Conflict of interest in medical research, education, and practice. Washington, DC: The National Academies Press.
IOM. 2010a. Accelerating rare diseases research and orphan product development. http://www8.nationalacademies.org/cp/projectview.aspx?key=49099 (accessed March 15, 2010).
IOM. 2010b. Standards for systematic reviews of clinical effectiveness research. http://www8.nationalacademies.org/cp/projectview.aspx?key=49124 (accessed March 15, 2010).
Kaufman, D. W., J. P. Kelly, L. Rosenberg, T. E. Anderson, and A. A. Mitchell. 2002. Recent patterns of medication use in the ambulatory adult population of the United States: The Slone Survey. Journal of the American Medical Association 287(3):337–344.
Koulman, A., G. Lane, S. Harrison, and D. Volmer. 2009. From differentiating metabolites to biomarkers. Analytical and Bioanalytical Chemistry 394(3):663–670.
Kristiansen, J. 2001. Description of a generally applicable model for the evaluation of uncertainty of measurement in clinical chemistry. Clinical Chemistry and Laboratory Medicine 39(10):920–931.
Lathia, C. D., D. Amakye, W. Dai, C. Girman, S. Madani, J. Mayne, P. MacCarthy, P. Pertel, L. Seman, A. Stoch, P. Tarantino, C. Webster, S. Williams, and J. A. Wagner. 2009. The value, qualification, and regulatory use of surrogate end points in drug development. Clinical Pharmacology and Therapeutics 86(1):32–43.
Lee, J. W., R. S. Weiner, J. M. Salistad, R. R. Bowsher, D. W. Knuth, P. J. O’Brien, J. L. Fourcroy, R. Dixit, L. Pandite, R. G. Pietrusko, H. D. Soares, V. Quarmby, O. L. Vesterqvist, D. M. Potter, J. L. Witliff, H. A. Fritche, T. O’Leary, L. Perlee, S. Kadam, and J. A. Wagner. 2005. Method validation and measurement of biomarkers in nonclinical and clinical samples in drug development: A conference report. Pharmaceutical Research 22(4):2495–2499.
Lee, J. W., W. Devanarayan, Y. C. Barrett, R. Weiner, J. Allinson, S. Fountain, S. Keller, I. Weinryb, M. Green, L. Duan, J. A. Rogers, R. Millham, P. J. O’Brien, J. Salistad, M. Khan, C. Ray, and J. A. Wagner. 2006. Fit-for-purpose method development and validation for successful biomarker measurement. Pharmaceutical Research 23(2):312–328.
Libby, P., and P. Theroux. 2005. Pathophysiology of coronary artery disease. Circulation 111(25):3481–3488.
Manson, J. E., J. M. Gaziano, M. A. Jonas, and C. H. Hennekens. 1993. Antioxidants and cardiovascular disease: A review. Journal of the American College of Nutrition 12(4): 426–432.
Moore, C. E., M. M. Murphy, and M. F. Holick. 2005. Vitamin D intakes by children and adults in the United States differ among ethnic groups. The Journal of Nutrition 135(10): 2478–2485.
NACB Writing Group Members, A. H. B. Wu, A. S. Jaffe, F. S. Apple, R. L. Jesse, G. L. Francis, D. A. Morrow, L. K. Newby, J. Ravkilde, W. H. Wilson Tang, R. H. Christenson, NACB Committee Members, R. H. Christenson, F. S. Apple, C. P. Cannon, G. L. Francis, R. L. Jesse, D. A. Morrow, L. K. Newby, J. Ravkilde, A. B. Storrow, W. H. Wilson Tang, and A. H. B. Wu. 2007. National Academy of Clinical Biochemistry Laboratory Medicine practice guidelines: Use of cardiac troponin and B-type natriuretic peptide or N-terminal proB-type natriuretic peptide for etiologies other than acute coronary syndromes and heart failure. Clinical Chemistry 53(12):2086–2096.
Omenn, G. S., G. E. Goodman, M. D. Thornquist, J. Balmes, M. R. Cullen, A. Glass, J. P. Keogh, F. L. Meyskens Jr., B. Valanis, J. H. Williams Jr., S. Barnhart, M. G. Cherniack, C. A. Brodkin, and S. Hammar. 1996a. Risk factors for lung cancer and for intervention effects in CARET, the Beta-Carotene and Retinol Efficacy Trial. Journal of the National Cancer Institute 88(21):1550–1559.
Omenn, G. S., G. E. Goodman, M. D. Thornquist, J. Balmes, M. R. Cullen, A. Glass, J. P. Keogh, F. L. Meyskens Jr., B. Valanis, J. H. Williams Jr., S. Barnhart, and S. Hammar. 1996b. Effects of a combination of beta-carotene and vitamin A on lung cancer and cardiovascular disease. New England Journal of Medicine 334:1150–1155.
Palou, A., C. Pico, and J. Keijer. 2009. Integration of risk and benefit analysis—the window of benefit as a new tool? Critical Reviews in Food Science and Nutrition 49(7):670–680.
Peto, R., R. Doll, J. D. Buckley, and M. B. Sporn. 1981. Can dietary beta-carotene materially reduce human cancer rates? Nature 290(5803):201–208.
Rasnake, C. M., P. R. Trumbo, and T. M. Heinonen. 2007. Surrogate endpoints and emerging surrogate endpoints for risk reduction of cardiovascular disease. Nutrition Reviews 66(2):76–81.
Ricos, C., V. Alvarez, F. Cava, J. V. Garcia-Lario, A. Hernandez, C. V. Jimenez, J. Minchinela, C. Perich, and M. Simon. 1999. Current databases on biological variation: Pros, cons and progress. Scandinavian Journal of Clinical and Laboratory Investigation 59(7):491–500.
Rizzo, M., and K. Berneis. 2007. Erratum: Low-density lipoprotein size and cardiovascular risk assessment (Q J Med (2006) 99(1-14)). QJM 100(2):147.
Rosner, W., R. J. Auchus, R. Azziz, P. M. Sluss, and H. Raff. 2007. Utility, limitations, and pitfalls in measuring testosterone: An Endocrine Society position statement. The Journal of Clinical Endocrinology and Metabolism 92(2):405-413.
Sargent, D. 2004. Disease-free survival (DFS) vs. overall survival (OS) as a primary endpoint for adjuvant colon cancer studies. http://www.fda.gov/ohrms/dockets/ac/04/transcripts/4037T2.htm (accessed March 15, 2010).
Shah, V. P., K. K. Midha, S. Dighe, I. J. McGilveray, J. P. Skelly, A. Yacobi, T. Layloff, C. T. Viswanathan, C. Edgar Cook, R. D. McDowall, K. A. Pittman, S. Spector, K. S. Albert, S. Bolton, M. Dobrinska, W. Doub, M. Eichelbaum, J. W. A. Findlay, K. Gallicano, W. Garland, D. J. Hardy, J. D. Hulse, H. Thomas Karnes, R. Lange, W. D. Mason, G. McKay, E. Ormsby, J. Overpeck, H. D. Plattenberg, V. P. Sha, G. Shiu, D. Sitar, F. Sorgel, J. T. Stewart, and L. Yuh. 1992. Analytical methods validation: Bioavailability, bioequivalence and pharmacokinetic studies. Sponsored by the American Association of Pharmaceutical Chemists, U.S. Food and Drug Administration, Internationale Pharmaceutique, Health Protection Branch (Canada) and Association of Official Analytical Chemists. International Journal of Pharmaceutics 82(1-2):1–7.
Staessen, J. A., J. G. Wang, and L. Thijs. 2003. Cardiovascular prevention and blood pressure reduction: A quantitative overview updated until 1 March 2003. Journal of Hypertension 21(6):1055–1076.
Swanson, B. N. 2002. Delivery of high-quality biomarker assays. Disease Markers 18:47–56.
Tardif, J.-C., T. Heinonen, D. Orloff, and P. Libby. 2006. Vascular biomarkers and surrogates in cardiovascular disease. Circulation 113(25):2936–2942.
Wagner, J. A. 2002. Overview of biomarkers and surrogate endpoints in drug development. Disease Markers 18(2):41–46.
Wagner, J. A. 2008. Strategic approach to fit-for-purpose biomarkers in drug development. Annual Review of Pharmacology and Toxicology 48:631–651.
Wagner, J. A., S. A. Williams, and C. J. Webster. 2007. Biomarkers and surrogate end points for fit-for-purpose development and regulatory evaluation of new drugs. Clinical Pharmacology and Therapeutics 81(1):104–107.
Willett, W. C., B. F. Polk, B. A. Underwood, M. J. Stampfer, S. Pressel, B. Rosner, J. O. Taylor, K. Schneider, and C. G. Hames. 1984. Relation of serum vitamins A and E and carotenoids to the risk of cancer. New England Journal of Medicine 310(7):430–434.
Williams, S. A., D. E. Slavin, J. A. Wagner, and C. J. Webster. 2006. A cost-effectiveness approach to the qualification and acceptance of biomarkers. Nature Reviews Drug Discovery 5(11):897–902.
Writing Group Members, F. S. Apple, A. H. Wu, A. S. Jaffe, M. Panteghini, R. H. Christenson, NACB Committee Members, R. H. Christenson, F. S. Apple, C. P. Cannon, G. S. Frances, R. L. Jesse, D. A. Morrow, L. K. Newby, A. B. Storrow, W. H. Tang, A. H. Wu, IFCC Committee on Standardization of Markers of Cardiac Damage (C-SMCD) Members, F. S. Apple, C. P. Cannon, A. S. Jaffe, F. Pagani, J. Tate, J. Ordonez-Llanos, and J. Mair. 2008. National Academy of Clinical Biochemistry and IFCC Committee for Standardization of Markers of Cardiac Damage Laboratory Medicine practice guidelines: Analytical issues for biomarkers of heart failure. Clinical Biochemistry 41(4-5):222–226.
Wu, Alan H.B. 2010. Standardization of assays for clinically important enzymes that have biologic variation: what is all the fuss about? Clinical Chemistry and Laboratory Medicine 48(3)299–300.


































