
Appendix A
Contributed Manuscripts
A1
THE CASE FOR PATHOGEN-SPECIFIC THERAPY1
Arturo Casadevall2
Albert Einstein College of Medicine
At the beginning of the twenty-first century, the treatment of microbial diseases is increasingly complicated by drug resistance, the emergence of new pathogenic microbes, the relatively inefficacy of antimicrobial therapy in immunocompromised hosts, and the reemergence of older diseases, often with drug-resistant microbes. Some of these problems can be traced to the switch between pathogen-specific antibacterial therapy and the nonspecific antibacterial therapy that followed the transition from serum therapy to modern antimicrobial chemotherapy. The widespread availability of cheap, effective, nontoxic wide-spectrum antibacterial therapy for almost 75 years fostered a culture of therapeutic empiricism that neglected diagnostic technologies. Despite unquestioned lifesaving efficacy for individuals with microbial diseases, the use of broad-spectrum antimicrobials was associated with fungal superinfections and antibiotic-associated
colitis, helped to catalyze the emergence of resistance, and is now tentatively associated in the pathogenesis of certain chronic diseases, including atopy, asthma and – perhaps – certain forms of cancer. This article briefly reviews these trends and suggests that the current strategy of nonspecific therapy is fundamentally unsound because it damages the microflora and – consequently – the human symbiont. The essay argues for the development of immunotherapy and pathogen-specific therapies, especially with regard to bacterial and fungal diseases, and suggests possible routes to that future.
1.
The Problematic Status Quo
Current antimicrobial therapy is largely pathogen-specific for viral diseases and nonpathogen-specific for bacterial, fungal, and parasitic diseases (Casadevall, 1996). Although some of the latter diseases are sometimes treated with pathogen-specific drugs, such as the use of isoniazid for tuberculosis, the overwhelming majority of compounds targeting bacteria, fungi, and parasitic diseases have activity against multiple microbes. Furthermore, these compounds target both pathogenic and nonpathogenic microbes. This current antimicrobial paradigm is currently in use at a time of significant upheaval in the therapy of microbial diseases, which is the only field of medicine in which one can argue that therapeutic options have declined over time. For example, in the 1950s Jawetz noted that the then currently available antimicrobial drugs were satisfactory for the treatment of bacterial diseases (Jawetz, 1956). However, in recent years the field of infectious diseases has seen dramatic increases in antimicrobial resistance, an increasing prevalence of bacterial and fungal superinfections in treated individuals, a relatively low therapeutic efficacy of antimicrobial therapy in individuals with impaired immunity, the emergence of new infectious diseases, and the reemergence of older microbial diseases, often with highly resistant microbes such as XDR-Tb. Given this status quo, it behooves us to ask the questions: How did we get here? What are the consequences of the choices made then and now? Can we do better and how do we get there?
2.
How Did We Get Here?
Effective antimicrobial therapy can be dated to the introduction of serum therapy in the 1890s, which, for the first time, provided physicians with the ability to intervene and cause a favorable outcome for an infectious disease. Serum therapy was developed against numerous bacterial and viral diseases, including pneumococcal pneumonia, meningococcal meningitis, erysipelas, anthrax, and measles (for reviews, see refs Casadevall and Scharff, 1994; Casadevall and Scharff, 1995; Buchwald and Pirofski, 2003). The heyday of serum therapy was the 1930s, but the modality was rapidly abandoned because serum could not compete with small-molecule antimicrobial therapy, such as sulfonamides and
penicillin, with regard to price, stability, ease of use, and (low) toxicity. For some diseases such as meningococcal meningitis, small-molecule antimicrobial therapy was clearly more effective than serum therapy; however, for pneumococcal pneumonia the difference in efficacy was less clear. In addition to serum therapy, the few other therapies available (e.g., quinine for malaria, salvarsan for syphilis, optochin for pneumococcus, and phage therapy) were all pathogen specific. In a prior essay (Casadevall, 2006), I argued that the time of serum therapy and the subsequent era of therapy with small molecules constituted the two first ages of antimicrobial therapy. When viewed through the prism of microbial specificity, the greatest difference in the therapeutic approach between the first and second ages of antimicrobial therapy was a switch from pathogen-specific to nonspecific therapy with regard to antibacterial therapeutics. In this essay, I argue that this change was to have enormous implications, which are root causes for some of the problems we face today.
In evaluating the therapeutic paradigm for microbial diseases, it is worthwhile contrasting it with the therapy of cancer. Like therapy for infectious diseases, the treatment of tumors has relieved [sic] heavily on antibiotics made by microorganisms; adryamicin, actinomycin D, bleomycin etc. are all microbial products. Like antimicrobial antibiotics, these antimetabolite antibiotics are each nonspecific in the sense that they are cytotoxic to multiple tumors. However, unlike most antimicrobial antibiotics, these agents have tremendous toxicity for the host and, consequently, are never used empirically. Hence, oncology practice has placed great emphasis on diagnosis and in exploiting subtle pharmacological differences between these agents to enhance their therapeutic index.
In fairness to infectious diseases, it noteworthy that the temporal kinetics of microbial infections and tumorogenesis favored a more deliberate approach to diagnosis as tumors, which unlike microbes, seldom killed the host rapidly. Nevertheless, the analogy is relevant because it provides an inkling of how the practice of infectious diseases might have developed if early antimicrobials had more significant toxicity, as evidenced by the hesitant empiric use of amphotericin b and Ara-C for fungal and herpetic diseases, respectively, Consistent with this notion, the development of the relatively nontoxic antiherpetic drug acyclovir as a replacement for Ara-C was followed with significantly greater empiric use, especially in neonates and cases of encephalitis. Similarly, the introduction of low-toxicity azoles and echinochandins as replacements for the highly toxic amphotericin b has promoted the empirical use of antifungal therapy. Hence, the advantage of low toxicity has the perverse effect of promoting empirical and inappropriate use.
In comparing the ages of antimicrobial therapy, it is clear that the change in the specificity of therapeutic agents did not affect all types of antimicrobial therapy equally. Serum therapy for viral diseases was specific and current antiviral drugs remain largely pathogen-specific, with the caveat that some drugs like acyclovir have activity against multiple herperviruses [sic]. For mycobacterial
diseases, there was no effective therapy in the preantibiotic era and most drugs that were subsequently developed (isoniazid, ethambutol, and others) were used primarily for the therapy of tuberculosis. For fungal diseases, there was no effective therapy prior to the late 1950s when amphotericin B was introduced; a compound active against most fungal pathogens and antifungal therapy has always relied on nonpathogen-specific agents. For bacterial diseases, the change from serum to small-molecule therapeutics was a revolution, as therapeutic specificity was abandoned in favor of agents with increasingly greater spectrum of antimicrobial activity. However, what made the switch from pathogen-specific to non-pathogen-specific therapy so significant with regard to antibacterial therapy is that the human host is a symbiont, with microflora consisting mostly of desired commensal bacteria. By contrast, there are no known desirable commensal viruses and the known fungal flora is limited to a few fungal species where Candida spp predominate. Unlike bacteria, a beneficial function has not been demonstrated for the host-associated fungal microflora. Hence, the use of nonspecific bacterial therapy carried an inherent potentially detrimental effect in damaging the associated bacterial microflora, and thus the human symbiont.
3.
The Consequences of Nonspecific Antimicrobial Therapy
The nonspecificity of antibacterial, and to a lesser extent antifungal, therapies was to have profound consequences on the practice and outcome of infectious diseases that reverberate to current times. The availability of nonspecific antibacterial therapies with broad spectrum and low toxicity allowed physicians to rapidly treat many infectious diseases without a need for a microbial diagnosis. For individuals with bacterial diseases, such therapy was often lifesaving. However, the ability to effectively treat many diseases safely without making a diagnosis deemphasized diagnostic clinical microbiology and fostered a culture of empiricism. For example, the diagnosis of pneumococcal pneumonia with the identification of the offending serotype took approximately 6 – 8 h in the 1930s and used the mouse peritoneal infection assay followed by typing with rabbit type-specific serum. This methodology was developed to rapidly ascertain the presence and serotype of pneumococcus in sputum because the efficacy of serum therapy depended on matching the bacterial serotype with the specificity of the antiserum. Despite the problems in unequivocally diagnosing pneumonia from sputum, this approach was successful for selecting therapeutic sera and supported the use of serum therapy. However, the introduction of penicillin and later antimicrobial drugs made the test much less relevant and it was abandoned as a diagnostic tool. Currently, a definitive diagnosis of pneumococcal pneumonia is possible only when accompanied by bacteremia, information that requires 48 h. For fungal diseases, a full embrace of empiric therapy was checked by the toxicity of amphotericin b, but by the late 1990s, the availability of relatively nontoxic azole and echinocandin-type drugs had ushered greater empiric use. By contrast,
for conditions that required specific therapy, such as viral and mycobacterial diseases, the practice ethos supported continued emphasis on diagnostic identification of the causative microbe.
For bacterial and later fungal diseases, the availability of relatively nontoxic broad-spectrum therapy contributed to the emergence of resistance among both targeted and nontargeted microbes. Although specific therapy can also elicit resistance, as witnessed by the emergence of isoniazid-resistant Mycobacterium tuberculosis, only nonspecific therapy can elicit resistance among nontargeted microbes such as common inhabitants of the microflora. Furthermore, only non-specific therapy can damage the microflora to create alterations that foster the emergence of usually commensal microbes such as Candida and Enteroccocus spp, first as major pathogenic microbes and then as drug-resistant pathogenic microbes. Consequently, the discipline of infectious diseases may be the only specialty of medicine where previously effective therapeutic options have to be abandoned because of drug resistance creates [sic] obsolescence.
Another consequence of nonspecific antibacterial and antifungal therapy was damage to the human symbiont. There is rapidly accumulating evidence that the human microflora is established early in life through complex steps and that there are individual differences in microbial species composition, a fact that could reflect differences in the timing of acquisition or modulation by the host immune system. The microbial flora is essential for development of the immune system, helps digestion, provides numerous nutrients including vitamins, and protects the human host by niche-denial to more pathogenic microbes. There is conclusive evidence that damage to the microflora by nonspecific antibacterial therapy can translate into antibiotic-associated colitis and fungal diseases such as oral thrush and candidal vaginitis. However, there are ominous signs that nonspecific antimicrobial use might translate into certain chronic diseases such as atopy (Kusel et al., 2008), asthma (Kozyrskyj et al., 2007), and even some types of cancer (Velicer et al., 2004), possibly by altering the development of the immune system in childhood and/or affecting metabolites produced by the microflora. In this regard, it is noteworthy that there is a temporal association between widespread antimicrobial use and the increase in immunoreactive diseases such as allergies and asthma, although it is premature to conclude causality as there may be confounding variables (Wickens et al., 2008). Nevertheless, the available evidence does provide reason for concern.
In summary, the development of effective, nontoxic, nonspecific antibacterial and antifungal therapy has had great consequences, some positive and some negative. Positive consequences include a significantly enhanced capacity to treat bacterial and fungal diseases early and effectively, which has translated to reduced mortality. Furthermore, the ability to treat early, safely, and without knowledge of the causative microbe has created a permissive environment for the development of complex surgeries, aggressive chemotherapy for tumors, and organ transplantation, procedures that would have unacceptable mortality without
such drugs. However, the same approach has also created a culture of empiricism that promoted antibiotic use, which in turn selected for resistance in targeted and nontargeted microbes, promoted the phenomenon of superinfection and damaged the symbiont with consequences that are only now beginning to be understood. In this regard, empiricism was a practice largely dictated by clinical findings and historical probability that essentially rejected causality in favor of associations.
4.
Can We Do Better and How to Get There?
Of course we can do better. Even for the short historical time that effective antimicrobial therapy has been available it is clear that the effectiveness of therapy and diagnosis has fluctuated with time. In a previous essay (Casadevall, 2006), I argued that we are in the throes of a major paradigm shift that will usher in the third age of antimicrobial therapy. This age can be envisioned as an equilateral triangle with pathogen-specific therapy, greatly improved diagnostics, and immunotherapy at each apex. Nonspecific therapy will always have a role for the treatment of polymicrobial diseases and to insure proper coverage in individuals with fulminant disease but its use could be limited by the combination of rapid diagnostics and pathogen-specific drugs. Even for such polymicrobial diseases as abdominal sepsis originating from a ruptured viscus there is evidence that damage is caused by only a few microbial species and their identification would permit employment of pathogen-specific drugs. In this age, immunotherapy, whether with large molecules, such as antibodies or small-molecular-weight immuno-modulators, would have co-equal status with therapies designed to directly kill or inhibit the microbe. Although this author believes that third-age therapeutics will arrive in the twenty-first century, significant scientific, economic, and behavioral hurdles must be overcome for the realization of this vision.
On the scientific front, drug discovery would have to move from trying to identify common therapeutic pathways among phylogenetically distant bacteria to exploiting differences in physiology and virulence mechanisms and/or to augmenting host mechanisms that promote microbial clearance, which, interestingly, are nonspecific. This formidable task is made even more difficult by the economics of antimicrobial drug discovery. As for other diseases, the economics of drug development is a function of the prevalence of the disease, which dictates market size. However, in antimicrobial drug discovery this formula is further modified by the fact that the market size is directly proportional to the width of the drug antimicrobial spectrum. Given the cost of drug development, the economics are stacked against pathogen-specific drugs in favor of broad-spectrum drugs. One caveat in this analysis is that drug resistance can disproportionately shorten the useful life of broad-spectrum drugs and that the emergence of resistant microbes can in itself create new market opportunities. For example, the emergence and spread of methicillin-resistant Staphylococcus aureus (MRSA) creates a niche such that a new staphylococcal-specific drug active against methicillin- and
possibly vancomycin-resistant isolates would probably be developed clinically if available. The use of pathogen-specific drugs would necessitate advances in diagnostics to provide rapid and accurate information to support their use, and this would require new investments in research and laboratory assays. Finally, physicians would have to change their approach to patients with presumed infectious diseases, emphasizing the need for diagnosis to select appropriate therapy in an echo to the practices of physicians in the age of serum therapy.
Perhaps the hurdles are so high that pathogen-specific therapy is only in the far horizon. If that is the case, there are concrete actions that can be taken in the present to slow the spread of drug resistance and damage to the human microbial flora. For example, educational campaigns aimed at physicians and the general public can promote more prudent use of antimicrobial drugs. At a political level, policy makers should be made aware of the economic and regulatory hurdles that slow the development of rapid diagnostic tests and pathogen-specific drugs. However, perhaps things can change more rapidly that one can anticipate. Certainly, if future research was to associate disturbances in the microflora with such chronic diseases as asthma, atopy, and cancer, this would create tremendous medical and legal disincentives in the use of nonspecific microbial therapy. Another powerful force could be the categorization of such complications of broad-spectrum therapy as C. difficile colitis and candidiasis as medical errors, which would be followed by aversion of third-party payers for hospital and physician reimbursements. At the same time, economic incentives for the development of pathogen-specific therapy by industry could be created by linking the patent protection time of antimicrobial drugs to the width of the antimicrobial spectrum and inclusion of narrow-spectrum drugs as orphan drugs. For example, patent policy could be amended such that narrow-spectrum drugs with small markets enjoy much longer patent protection than broad-spectrum drugs. Although in 2009 a revolution in the antimicrobial therapeutic paradigm seems distant, it is worth noting that only a generation ago smoking was widely permitted and accepted in most public places. For smoking, it was the realization that second-hand smoke was dangerous that catalyzed the creation of smoke-free environments in most public places. Perhaps increased awareness of the consequences of long-term damage to the human flora will have a similar catalytic effect in promoting pathogen-specific antimicrobial therapies.
The re-introduction of pathogen-specific therapy for bacterial diseases, and its extension to fungal diseases, would require a concerted effort and collaboration between intellectual leaders in the field, industry, and government to find mechanisms that would promote and encourage the development of such drugs. There are indications of movement in this direction. A recent report by the Institute of Medicine recommended ‘development of strategies that will selectively target pathogenic organisms while avoiding targeting the host and beneficial or benign organisms’, which in other words is pathogen-specific therapy.3 Several therapies
|
3 |
Available from http://www.nap.edu/catalog/11471.html. |
narrow-spectrum are currently in development, for example, the renewed interest in phage therapy, monoclonal antibody therapies, and drugs aimed primarily at targeting highly resistant bacteria. However, the task of refocusing anti-bacterial and antifungal therapy to pathogen specificity is too great for any individual party and cooperation from industry, government, and the medical community will be needed to effect change. There is an acute need for an economic model that would allow the development and use of pathogen-specific drugs. Despite these hurdles, it is clear that pathogen-specific therapy makes sense and, given that the current nonspecific strategies are increasingly bankrupt, it behooves all parties to begin a dialogue on how to get there, and get there sooner than later.
Declaration of Interest
The author states no conflict of interest and has received no payment in preparation of this manuscript.
Bibliography
Buchwald UK, Pirofski L. Immune therapy for infectious diseases at the dawn of the 21st century: the past, present and future role of antibody therapy, therapeutic vaccination and biological response modifiers. Curr Pharm Des 2003;9(12):945-68
Casadevall A. Crisis in Infectious Diseases: Time for a new paradigm? Clin Infect Dis 1996;23:790-4
Casadevall A, Scharff MD. “Serum Therapy” revisited: Animal models of infection and the development of passive antibody therapy. Antimicrob Agents Chemother 1994;38:1695-702
Casadevall A, Scharff MD. Return to the past: the case for antibody-based therapies in infectious diseases. Clin Infect Dis 1995;21:150-61
Casadevall A. The third age of antimicrobial therapy. Clin Infect Dis 2006;42(10):1414-6
Jawetz E. Antimicrobial therapy. Ann Rev Microbiol 1956;10:85-114
Kozyrskyj AL, Ernst P, Becker AB. Increased risk of childhood asthma from antibiotic use in early life. Chest 2007;131(6):1753-9
Kusel MM, de KN, Holt PG, Sly PD. Antibiotic use in the first year of life and risk of atopic disease in early childhood. Clin Exp Allergy 2008;38(12):1921-8
Velicer CM, Heckbert SR, Lampe JW, et al. Antibiotic use in relation to the risk of breast cancer. JAMA 2004;291(7):827-35
Wickens K, Ingham T, Epton M, et al. The association of early life exposure to antibiotics and the development of asthma, eczema and atopy in a birth cohort: confounding or causality? Clin Exp Allergy 2008;38(8):1318-24
A2
WAVES OF RESISTANCE: STAPHYLOCOCCUS AUREUS IN THE ANTIBIOTIC ERA4
Henry F. Chambers5 and Frank R. DeLeo6
Abstract
Staphylococcus aureus is notorious for its ability to become resistant to antibiotics. Infections that are caused by antibiotic-resistant strains often occur in epidemic waves that are initiated by one or a few successful clones. Methicillin-resistant S. aureus (MRSA) features prominently in these epidemics. Historically associated with hospitals and other health care settings, MRSA has now emerged as a widespread cause of community infections. Community or community-associated MRSA (CA-MRSA) can spread rapidly among healthy individuals. Outbreaks of CA-MRSA infections have been reported worldwide, and CA-MRSA strains are now epidemic in the United States. Here, we review the molecular epidemiology of the epidemic waves of penicillin- and methicillin-resistant strains of S. aureus that have occurred since 1940, with a focus on the clinical and molecular epidemiology of CA-MRSA.
Staphylococcus aureus is naturally susceptible to virtually every antibiotic that has ever been developed. Resistance to antibiotics is often acquired by the horizontal transfer of genes from outside sources, although chromosomal mutation and antibiotic selection are also important. This exquisite susceptibility of S. aureus led to Alexander Fleming’s discovery of penicillin, which ushered in the ‘antibiotic era’. Penicillin was truly a miracle drug: uniformly fatal infections could now be cured. However, by the mid 1940s, only a few years after its introduction into clinical practice, penicillin resistance was encountered in hospitals, and within a decade it had become a notable problem in the community.
A fundamental biological property of S. aureus is its ability to asymptomatically colonize healthy individuals. Approximately 30% of humans are asymptomatic nasal carriers of S. aureus (Kluytmans and Verbaugh, 1997; Gorwitz et al., 2008) such that in these individuals S. aureus is part of the normal flora. S. aureus
|
4 |
Reprinted with permission from Nature Reviews Microbiology 7, 629-641 (September 2009). |
|
5 |
Division of Infectious Diseases, Department of Medicine, San Francisco General Hospital, University of California, San Francisco, California 94110, USA. |
|
6 |
Laboratory of Human Bacterial Pathogenesis, Rocky Mountain Laboratories, National Institute of Allergy and Infectious Diseases, National Institutes of Health, 903 South 4th Street, Hamilton, Montana 59840, USA. Correspondence to H.F.C. e-mail: hchambers@medsfgh.ucsf.edu. doi:10.1038/nrmicro2200. |
carriers are at higher risk of infection and they are presumed to be an important source of the S. aureus strains that spread among individuals. The primary mode of transmission of S. aureus is by direct contact, usually skin-to-skin contact with a colonized or infected individual, although contact with contaminated objects and surfaces might also have a role (Miller and Diep, 2008; Kazakova et al., 2005; Lowy, 1998; Muto et al., 2003). Various host factors can predispose individuals to infection, including the loss of the normal skin barrier, the presence of underlying diseases such as diabetes or AIDS and defects in neutrophil function.
Infections that are caused by antibiotic-resistant strains of S. aureus have reached epidemic proportions globally (Tiemersma, 2006). The overall burden of staphylococcal disease, particularly disease caused by methicillin-resistant S. aureus (MRSA) strains, is increasing in many countries in both health care and community settings (Kaplan et al., 2005; Hersh et al., 2008; Klevens et al., 2007; Hope et al., 2008; Laupland et al., 2008; European Antimicrobial resistance Surveillance System, 2008). In the United States, the emergence of community associated MRSA (CA-MRSA) strains accounts for much of this increase, as it is a major cause of skin and soft-tissue infections (Moran et al., 2006; Fridkin et al., 2005). The rapidity and extent of the spread of CA-MRSA strains has been remarkable. In addition to the United States, CA-MRSA strains have been reported in Canada, Asia, South America and Australia as well as throughout Europe, including in countries that historically have a low prevalence of MRSA, such as Norway, the Netherlands, Denmark and Finland (Laupland et al., 2008; Larsen et al., 2007; Larsen et al., 2008; Wannet et al., 2005; Deurenberg et al., 2009; Vandenesch et al., 2003; Stam-Bolink et al., 2007; Huang et al., 2007; Nimmo and Coombs, 2008; Kanerva et al., 2009; Park et al., 2009; Gardella et al., 2008; Francois et al., 2008; Fang et al., 2008; Conly and Johnston, 2003). Globally, CA-MRSA strains have shown considerable diversity in the number of different clones that have been identified.
In addition to their increasing prevalence and incidence, CA-MRSA strains seem to be particularly virulent. Overwhelming and tissue-destructive infections, such as necrotizing fasciitis and fulminant, necrotizing pneumonia (Francis et al., 2005; Gonzalez et al., 2005; Kallen et al., 2009), were rarely seen before the emergence of CA-MRSA strains. The factor (or factors) that is responsible for this hypervirulent behaviour is not known, but Panton–Valentine leukocidin (PVL), which has been epidemiologically associated with severe skin infections and pneumonia that are caused by methicillin-susceptible S. aureus (MSSA) strains (Lina et al., 1999), is a leading candidate.
Antibiotics arguably constitute the most concentrated selective pressure on S. aureus in its long coevolutionary history with mankind. The consequences of this selective pressure, in conjunction with horizontal and vertical gene transfer, are discussed in this Review. Given their crucial importance as therapeutic agents, we focus on resistance to penicillins and the structurally related β-lactam antibiotics.
Epidemic Waves of Resistance
The emergence of antibiotic resistance in S. aureus can be visualized as a series of waves (Figure A2-1). The first wave began in the mid 1940s as the proportion of infections caused by penicillin-resistant strains of S. aureus increased in hospitals (Kirby, 1944; Barber and Rozwadowska-Dowzenko, 1948). These strains produced a plasmid-encoded penicillinase, which hydrolyses the β-lactam ring of penicillin that is essential for its antimicrobial activity. Penicillin-resistant strains soon began to cause community infections, and by the early 1950s they had become pandemic (Roundtree and Freeman, 1956). These infections, both in hospitals and in the community, were frequently caused by an S. aureus clone known as phage type 80/81 (Roundtree and Freeman, 1956; Blair and Carr, 1960; Bynoe et al., 1956; Roundtree and Beard, 1958). Pandemic phage type 80/81 S. aureus infections largely disappeared after the introduction of methicillin (Jevons and Parker, 1964), but the prevalence of penicillinase-producing strains from other S. aureus lineages has remained high.
The introduction of methicillin marks the onset of the second wave of resistance (Figure A2-1). The first reports of a S. aureus strain that was resistant to methicillin were published in 1961 (Barber, 1961; Jevons, 1961). Although the specific gene responsible for methicillin resistance (mecA, which encodes the low-affinity penicillin-binding protein PbP2a (also known as PbP2′)) was not identified until over 20 years later, it was appreciated early on that the resistance mechanism involved was different from penicillinase-mediated resistance because drug inactivation did not occur. Unlike penicillinase-mediated resistance, which is narrow in its spectrum of activity, methicillin resistance is broad, conferring resistance to the entire β-lactam class of antibiotics, which include penicillins, cephalosporins and carbapenems. Among the earliest MRSA clinical isolates was the archetypal MRSA strain COL, a member of the ‘archaic’ clone of MRSA and perhaps the most studied MRSA strain, which was isolated from a patient in Colindale, UK, in 1960 (Jevons, 1961). COL is a member of the most successful MRSA lineage, which includes both hospital and community-associated strains.
Archaic MRSA strains circulated in hospitals throughout Europe until the 1970s (Crisostomo et al., 2001). There were also isolated reports of MRSA in hospitals in the United States (Barrett et al., 1968; Bran et al., 1972), but the rest of the world was largely unaffected, and these early MRSA strains never gained a foothold in the community. By the 1980s, for reasons that remain unclear, the archaic MRSA clone had largely disappeared from European hospitals, marking the end of the second and the beginning of the third wave of antibiotic resistance. Descendants of the archaic MRSA clone (for example, the Iberian and Rome clones (Mato et al., 2004) and other, highly successful MRSA lineages emerged (Enright et al., 2002; Robinson and Enright, 2003; Deurenberg and Stobberingh, 2008) (Table A2-1). Outbreaks of infections caused by MRSA strains were reported in hospitals in the United States in the late 1970s, and by the mid 1980s
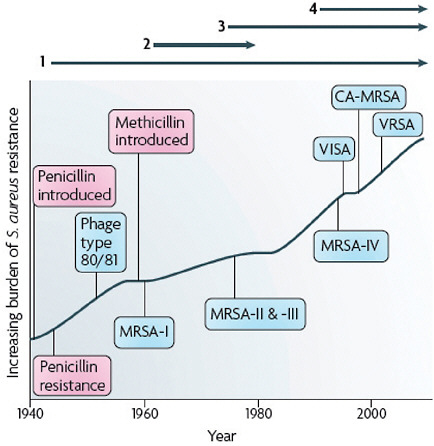
FIGURE A2-1 The four waves of antibiotic resistance in Staphylococcus aureus. Wave 1 (indicated above the graph), which continues today, began shortly after the introduction of penicillin into clinical practice in the 1940s. The first pandemic antibiotic-resistant strains, from the lineage known as phage type 80/81, were penicillin-resistant and produced Panton-Valentine leukocidin (PVL). Wave 2 began almost immediately following the introduction of methicillin into clinical practice with the isolation of the first MRSA strain (an archaic clone), which contained staphylococcal chromosome cassette mecl (SCCmecl) (indicated on the graph as MRSA-I); this wave extended into the 1970s in the form of the Iberian clone. Wave 3 began in the mid to late 1970s with the new emergence of MRSA strains that contained the new SCCmec allotypes, SCCmecll and SCCmeclll (MRSA-II and MRSA-III), marking the ongoing worldwide pandemic of MRSA in hospitals and health care facilities. The increase in vancomycin use for the treatment of MRSA infections eventually led to the emergence of vancomycin-intermediate S. auereus (VISA) strains. Wave 4, which began in the mid to late 1990s, marks the emergence of MRSA strains in the community. Community-associated MRSA (CA-MRSA) strains were susceptible to most antibiotics other than β-lactams, were unrelated to hospital strains and contained a new, smaller, more mobile SCCmec allotype, SCCmecIV (MRSA-IV) and various virulence factors, including PVL. Vancomycin-resistant S. aureus (VRSA) strains, ten or so of which have been isolated exclusively in health care settings, were first identified in 2002.
TABLE A2-1 Lineages of Common Nosocomial MRSA Strains
|
Clonal complex |
Sequence type |
Common name(s) |
Comment and SCCmec allotypes |
|
CC5 |
ST5 |
USA100, NewYork or Japan clone |
The most common health care-associated MRSA strain in the United States; SCCmecII |
|
|
ST5 |
EMRSA-3 |
SCCmecI |
|
|
ST5 |
USA800 or paediatric clone |
Prevalent in Argentina, Colombia and the United States; SCCmecIV |
|
|
ST5 |
HDE288 or paediatric clone (in Portugal) |
SCCmecVI |
|
CC8 |
ST250 |
Archaic |
The first MRSA clone to be identified, includes the COL strain; SCCmecl |
|
|
ST247 |
Iberian clone or EMRSA-5 |
A descendant of COL-type strains; SCCmecl |
|
|
ST239 |
Brazilian or Hungarian clone |
SCCmeclll |
|
|
ST239 |
EMRSA-1 |
An Eastern Australian epidemic clone of the 1980s; SCCmeclll |
|
|
ST239 |
AUS-2 and AUS-3 |
Common Australian multidrug-resistant clones of the early 2000s; SCCmeclll |
|
|
ST8 |
Irish-1 |
Common hospital-acquired isolate in the 1990s in Europe and the United States; SCCmecll |
|
|
ST8 |
USA500, EMRSA-2 or EMRSA-6 |
SCCmecIV |
|
CC22 |
ST22 |
EMRSA-15 |
An international clone that is prominent in Europe and Australia; SCCmecIV |
|
CC30 |
ST36 |
USA200 or EMRSA-16 |
The single most abundant cause of MRSA infections in UK hospitals and the second most common cause of MRSA infections in US hospitals in 2003; SCCmecll |
|
CC45 |
ST45 |
USA600 |
SCCmecll |
|
|
ST45 |
Berlin clone |
SCCmecIV |
|
CC, clonal complex; MRSA, methicillin-resistant Staphylococcus aureus; SCCmec, staphylococcal chromosome cassette mec, ST, sequence type. |
|||
these strains were endemic (Crossley et al., 1979; Peacock et al., 1980), leading to the worldwide pandemic of MRSA in hospitals that continues to the present time. Although global in its distribution and impact, MRSA was still confined mainly to hospitals and other institutional health care settings, such as long-term care facilities. The ever-increasing burden of MRSA infections in hospitals led to the increased use of vancomycin, the last remaining antibiotic to which MRSA strains were reliably susceptible. This intensive selective pressure resulted in the emergence of vancomycin-intermediate S. aureus (VISA) strains, which are not inhibited in vitro at vancomycin concentrations below 4–8 μg ml–1 (Hiramatsu et al., 1997), and vancomcyin-resistant S. aureus (VRSA) strains, which are inhibited only at concentrations of 16 μg ml–1 or more (Weigel et al., 2003).
The MRSA invasion of the community constitutes the fourth and most recent wave of antibiotic resistance (Figure A2-1). Some of the earliest cases of CA-MRSA infection occurred in indigenous populations in Western Australia in the early 1990s (O’Brien et al., 2004; Coombs et al., 2004; Udo et al., 1993). These MRSA strains were distinguishable from the contemporary clones or genotypes that were circulating in Australian hospitals by their pulsed-field gel electrophoresis patterns and their susceptibility to most antibiotics other than β-lactams, suggesting that they were either remote, ‘feral’ descendants of hospital strains or community strains that had acquired mecA by horizontal gene transfer. In the United States, the first well-documented cases of MRSA infection that were truly community associated occurred in otherwise healthy children from 1997 to 1999 (CDC, 1999). These children had no risk factors for developing MRSA and all died with overwhelming infection, suggesting that these CA-MRSA strains were especially virulent. Like their Australian counterparts, these CA-MRSA isolates were unrelated to hospital associated clones and were susceptible to most antibiotics. The CA-MRSA epidemic in the United States can be traced back to the early 1990s on the basis of retrospective data from 1993 to 1995, which show a dramatic increase in MRSA infections in Chicago among children who lacked risk factors for hospital-associated MRSA exposure (Herold et al., 1998). CA-MRSA has since been reported in numerous populations, including American Indians and Alaskan natives (Baggett et al., 2004), Pacific Islanders (CDC, 2004), athletes (Kazakova et al., 2005), jail and prison inmates (Aiello et al., 2006), men who have sex with men (Diep et al., 2008), contacts of patients with CA-MRSA infection (Johansson et al., 2007), military personnel (Aiello et al., 2006), adult emergency room patients (Moran et al., 2006) and children in day care centres (Adcock et al., 1998). CA-MRSA clones have also gained a foothold in hospitals and are increasingly being identified as a cause of hospital-onset and heath care-associated infections (Klevens et al., 2007; Laupland et al., 2008; Park et al., 2009; Liu et al., 2008; Seybold et al., 2006).
The epidemic wave of CA-MRSA in the United States and Canada (Gilbert et al., 2006; Mulvey et al., 2005) is actually two overlapping epidemics. The USA400 clone, which was isolated from the paediatric cases described above,
was most prevalent before 2001 (Lowy, 1998; CDC, 1999; Stemper et al., 2004) and remains a common cause of community-onset disease among indigenous populations in Alaska and the Pacific Northwest (David et al., 2008). A second epidemic clone, MRSA strain USA300, which is unrelated to USA400 and has largely displaced it in most other locations, emerged between 1999 and 2001 and now causes most of the CA-MRSA infections in the United States (Lowy, 1998; Kazakova et al., 2005; Pan et al., 2003; Pannaraj et al., 2006; Diep et al., 2004; Chavez-Bueno et al., 2005).
Outbreaks and epidemics of CA-MRSA now occur worldwide and have a similar epidemiology, although the specific clones that have emerged vary with geographical location. CA-MRSA strains are not merely escapees from health care facilities; their genotypes indicate that they are not closely related to endemic hospital clones and they are susceptible to numerous antibiotics to which hospital strains are routinely resistant. Two molecular markers that are not found in typical hospital MRSA strains are strongly associated with the emergence of CA-MRSA regardless of geographical origin: a specific cassette element encoding mecA and genes encoding PVL. These markers are discussed in detail below.
Molecular Epidemiology of S. aureus
S. aureus Clonal Complexes
Robust, sequence-based molecular methods for genotyping strains of S. aureus, and multilocus sequence typing (MLST) (Enright et al., 2000) in particular, have made it possible to study the evolutionary history of this pathogen (Box A2-1). MLST is carried out by sequence analysis of ~450 bp internal fragments of seven housekeeping genes (Figure A2-2). Isolates that have identical sequences at all seven loci are considered to be a clone and are assigned a unique sequence type (ST). STs that differ by single nucleotide polymorphisms (SNPs) at fewer than three loci are thought to be closely related and are grouped into clonal complexes (CCs). This grouping is accomplished by the eBURST algorithm, which uses MLST data to group closely related strains into a CC. It also predicts the probable founding clone, or ST, of each group and the recent evolutionary descent of all other strains in the CC from the founder (Feil et al., 2004; Turner et al., 2007). The analysis can be further refined to identify specific subclones by the addition of other methods, such as spa typing (Shopsin et al., 1999) or pulsed-field gel electrophoresis of genomic DNA (Box A2-1), or by the presence of other genetic markers (for example, toxin genes or specific plasmids).
Studies of MSSA strains, carriage isolates and hospital and community isolates causing disease that were collected worldwide between 1961 and 2004 show that 88% of the collected strains can be assigned to one of 11 clonal complexes (CC1, CC5, CC8, CC9, CC12, CC15, CC22, CC25, CC30, CC45 and CC51/121) (Enright et al., 2002, 2000; Feil et al., 2004, 2003; Tenover et al., 2008; Goering
|
BOX A2-1 Staphylococcus aureus Genotyping Multilocus sequence typing Multilocus sequence typing (MLST) is a sequence-based genotyping method based on single nucleotide varioations (each variant is termed an allele) of seven housekeeping genes in Staphylococcus aureus, providing a discriminatory allelic profile known as a sequence type (ST) (Enright et al., 2000) for each bacterial isolate. Because it indexes variations that accumulate slowly over time, MLST can be used to measure long periods of evolution among S. aureus lineages, and the results obtained are highly reproducible. S. aureus isolates that have identity at five or more of the seven housekeeping genes as determined by MLST are known as a clonal complex (CC) (Feil et al., 2004, 2003). Pulsed-field gel electrophoresis Pulsed-field gel electrophoresis (PFGE) has a more rapid clock speed than MLST and is suitable for the evaluation of more recent evolution among groups of strains. The method relies on the separation of Smal-digested S. aureus genomic DNA fragments in an agarose gel according to size. Related strains are clustered according to an 80% similarity coefficient (McDougal et al., 2003). The CDC has developed a national PFGE database for S. aureus, which uses the ‘USA’ designation; for example, USA300 refers to an ST8, Panton-Valentine leukocidin-positive community-associated MRSA strain (McDougal et al., 2003). spa typing spa typing (Shopsin et al., 1999) is based on the sequence analysis of variable-number tandem repeats in the gene that encodes protein A (spa). spa typing takes into account point mutations in the repeat region as well as the number of repeat variations. This method is suitable for the investigation of local or global S. aureus outbreaks. This sequence-based analysis of a single target locus is an inexpensive way of acquiring robust data that can be used to determine both epidemiological and phylogenetic relationships. |
et al., 2008; Hallin et al., 2007; Feng et al., 2008; Feil and Enright, 2004; Lindsay et al., 2006) (Figure A2-3a). For ten of these CCs, the percentage of isolates in each complex ranges from 2% to 9%; CC30 is an outlier, accounting for 21% of isolates.
The CCs for contemporary isolates are almost certainly the same as those of strains that were circulating before 1940. For example, the ST5 lineage (the founder of CC5) is estimated to have existed for over 2,000 years (Nubel et al., 2008). Gomes and colleagues (Gomes et al., 2006) genotyped 22 penicillin-susceptible and 67 penicillin-resistant MSSA blood culture isolates that were collected between 1957 and 1973 by the Statens Serum Institute in Copenhagen, which has collected and maintained every blood culture isolate from patients in Denmark from 1957 to the present. They found that 86% of the isolates fell into
seven CCs, the most common being CC8 and CC30, which together accounted for 46% of the isolates (Figure A2-3b). The distributions of penicillin-sensitive and penicillin-resistant isolates were similar. In this analysis, only a few isolates were tested and they all originated from a single country, which probably accounts for the absence of isolates from CC9, CC12, CC15 and CC22.
CC8 and CC30 have given rise to epidemics during each of the four waves of antibiotic resistance. The first well-characterized pandemic of antibiotic-resistant S. aureus that is attributable to a single clone was caused by phage type 80/81 strains, which belong to CC30 (Robinson et al., 2005). Phage type 80/81 strains were originally isolated in Australia in 1953 (Roundtree and Beard, 1958). They are penicillin resistant and have caused both hospital and community outbreaks on a global scale (Robinson et al., 2005). These strains are prevalent in collections that date back to 1927; they were thought to be highly transmissible and particularly virulent and were also among the first to be identified as penicillin resistant (Blair and Carr, 1960). Almost all of the phage type 80/81 isolates in a collection dating to the 1950s and 1960s encode PVL88, which is reminiscent of the association between PVL and resistance to methicillin in the contemporary epidemic CA-MRSA strains. For unknown reasons, phage type 80/81 strains virtually disappeared in the early 1960s, and this coincided with the first use of semisynthetic penicillins, which are resistant to penicillinase. Modern descendents of the CC30 lineage include the PVL-positive southwest Pacific (SWP) clone of CA-MRSA in Australia and the hospital-associated ST36 EMRSA-16 clone, a major cause of nosocomial infections and bacteraemia in both Australia and the United Kingdom (Robinson et al., 2005; Cox et al., 1995; Johnson et al., 2001).
MRSA CCs
The first MRSA clinical isolates, of which COL is an example, were ST250 and members of CC8. ST250 MRSA strains circulated in the United Kingdom and the rest of Europe before the 1970s but did not become established in the United States and had largely disappeared by the 1980s. However, other highly successful clones emerged, including the ST247 Iberian or EMRSA-5 clone, which is closely related to ST250. No fewer than nine other endemic nosocomial clones are descendants of the ST8 founder of this lineage. The CA-MRSA strain USA300 (which is PVL positive) that is prevalent in the United States is also ST8 (McDougal et al., 2003). MRSA strains have generally been found to be members of a subset of S. aureus CCs, including CC1, CC5, CC8, CC22, CC30 and CC45, although CA-MRSA strains have exhibited some diversity (discussed below). These CCs were widespread before the emergence of methicillin resistance (Crisostomo et al., 2001; Gomes et al., 2006), indicating that superior epidemicity preceded the acquisition of drug resistance and that the adaptations and innovations that make S. aureus clones successful can also favour their adaptation to antibiotic selective pressures.
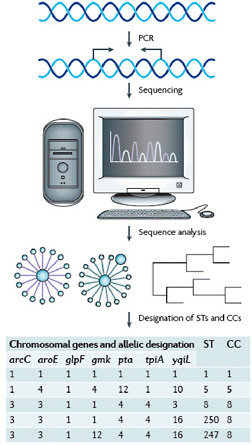
FIGURE A2-2 An example of a multilocus sequence typing scheme and the designation of clonal complexes. Multilocus sequence typing in Staphylococcus aureus involves PCR amplification and sequencing of approximately 450 nucleotides of seven chromosomal “housekeeping” genes that were selected for their presumed absence of selective pressure and their moderately stable nucleotide sequences (carbamate kinase (arc), shikimate dehydrogenase (aroE), glycerol kinase (glpF), guanylate kinase (gmk), phosphate acetyltransferase (pta), triose phosphate isomerase (tpiA) and acetyl-CoA acetyl-transferase (yqiL)). Each unique sequence within a gene locus is assigned a number. The numbers are concatenated left-to-right in the order shown to provide a seven-integer series of numbers, which is then assigned a sequence type (ST). Strains that are identical at all seven loci are classified as the same ST. Strains differing at one or two loci are related but, as they are not identical, they are assigned different STs. Closely related STs are grouped into a clonal complex (CC). In the example shown, ST1, ST5, and ST8 differ at most loci and so are not closely related; ST250 and ST247 differ from each other at one locus (gmk) and from ST8 at one (yqiL) and two loci (gmk, yqiL), respectively. Therefore, ST8, ST250 and ST247 are closely related and form CC8, so designated because the analysis of sequence identities and differences in a large collection of strains indicates that ST8 is the founder of this CC and the ancestor of both ST247 and ST250, and that ST247 is a descendant of ST250.
Staphylococcal Chromosome Cassette mec
The discovery by Hiramatsu and colleagues (Ito et al., 2001) that mecA is always found in a mobile cassette element was a great advance for our understanding of the biology of methicillin resistance and provided an additional tool for determining the evolutionary relationships among MRSA strains. Staphylococcal chromosome cassette mec (SCCmec) is integrated into orfX, an S. aureus gene of unknown function (Figure A2-4). To date, eight SCCmec allotypes, designated SCCmecI–SCCmecVIII (Deurenberg and Stobberingh, 2008; Ito et al., 2001; Ma et al., 2002; Oliveira et al., 2006; Higuchi et al., 2008; Zhang et al., 2009), have been described (Table A2-2), along with numerous subtypes, and more will probably be identified as sequence data become available for more MRSA strains (see the SCCmec website for additional descriptions and information). Similar elements are present in coagulase-negative staphylococci, which are commensal organisms that are part of the normal skin flora of humans and other mammals (Ruppe et al., 2009). Two gene complexes, mec and ccr (the recombination and excision locus encoding the gene or genes that mediate the integration and excision of the whole cassette into and out of orfX), are used to classify the SCCmec allotypes (Table A2-2). There are also other differences among the various SCCmec allotypes, particularly in terms of insertion sequences and antimicrobial resistance genes. However, as these are themselves mobile elements, they have not proved useful for the classification of the main allotypes, although they are useful for defining subtypes.
The class A mec gene complex is the prototype complex and is found in SCCmecII (Figure A2-4a), SCCmecIII and SCCmecVIII. It contains mecA, the complete mecR1 and mecI regulatory genes upstream of mecA, and the hypervariable region (HVR) and insertion sequence 431 (IS431) downstream of mecA. The class b mec gene complex is found in SCCmecI, SCCmecIV (Figure A2-4b) and SCCmecVI and is composed of mecA, a truncated mecR1 (resulting from the insertion of IS1272) upstream of mecA, and the HVR and IS431 downstream of mecA. There are two distinct class C mec gene complexes, both of which contain mecA, a truncated mecR1 (resulting from the insertion of IS431) upstream of mecA, and the HVR and IS431 downstream of mecA. In the class C1 mec gene complex, the IS431 elements upstream and downstream of mecA are in the same orientation, whereas in the class C2 mec gene complex, which is found in SCCmecV and SCCmecVII, the orientation of the IS431 upstream of mecA is reversed. C1 and C2 are regarded as different mec gene complexes, as they have probably evolved independently. The mecA, mecR1 and mecI sequences are highly conserved, with >99% nucleotide sequence identity.
The ccr gene complex consists of two adjacent genes, ccrA and ccrB, in SCCmecI–SCCmecIV, SCCmecVI and SCCmecVIII and one gene, ccrC, in SCCmecV and SCCmecVII. MRSA strains that were isolated before 1990, which were all nosocomial isolates, contained predominantly SCCmecI–SCCmecIII.
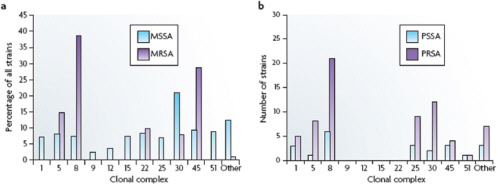
FIGURE A2-3 Distribution of antibiotic-susceptible and -resistant Staphylococcus aureus among clonal complexes. a| The distribution of methicillin-sensitive Staphylococcus aureus (MSSA) and methicillin-resistant S. aureus (MRSA) among the various clonal complexes. These data were collected from six continents between 1961 and 2004. b| The distribution of penicillin-susceptible S. aureus (PSSA) and penicillinresistant S. aureus (PRSA) among the various clonal conplexes. These data are from a single study of 89 isolates that were collected in Copenhagen from 1957 to 1973. See main text for details.
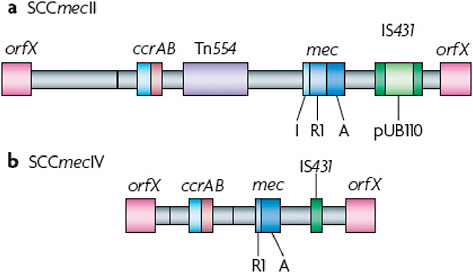
FIGURE A2-4 Comparison of the methicillin resistance cassettes that are typical of hospital- or community-acquired methicillin-resistant Staphylococcus aureus. Staphylococcal chromosome cassette mecII (SCCmecII) is most abundant in hospitals, whereas SCCmecIV is present in the most abundant community-acquired methicillin-resistant Staphylococcus aureus strains. The mecR1 gene (R1) in SCCmecIV is truncated, whereas the copy in SCCmecII is full-length. Transposon Tn554, which is present in SCCmecII but not in SCCmecIV, encodes resistance to macrolide-lincosomide-streptogramin B antibiotics and spectinmycin. pUB110 is an integrated plasmid that encodes a tobramycin resistance gene. SCCmecII therefore encodes resistance to multiple antibiotics, whereas SCCmecIV encodes resistance to methicillin alone. A, mecA; I mecl; IS431, insertion sequence 431.
CA-MRSA isolates most frequently contain variants of the SCCmecIV or SCCmecIV allotypes; less commonly, they contain SCCmecV (Francois et al., 2008; Okuma et al., 2002). SCCmecIV is also increasingly identified in contemporary hospital MRSA strains.
The three epidemic waves of MRSA correspond to evolutionary changes in SCCmec. The early MRSA strains (COL and other CC8 strains that circulated in the United Kingdom and Denmark in the early 1960s) all carried SCCmecI. They were replaced in the 1980s by new and arguably more successful lineages that eventually became established in hospitals throughout the world. These clones, which were predominantly CC5 and CC8, carried SCCmecII or SCCmecIII (for example, New York/Japan EMRSA, EMRSA-16 in Australia and the United Kingdom, the Brazilian clone and the Hungarian clone), or the type IA variant of the archaic SCCmecI (the Iberian clone). Why SCCmecII and SCCmecIII were more successful than SCCmecI is not known, but it could be that the recombinase genes, which are defective in SCCmecI but functional in SCCmecII and
TABLE A2-2 Comparison of Staphylococcal Chromosome Cassette mec Allotypes
|
Feature* |
SCCmec allotype |
|||||||
|
I |
II |
III |
IV |
V |
VI |
VII |
VIII |
|
|
Size (kb) |
34 |
53 |
67 |
21-24 |
28 |
24 |
41-49 |
32 |
|
mec complex |
B |
A |
A |
B |
C2 |
B |
C1 or C2 |
A |
|
ccr complex |
A1 and B1 |
A2 and B2 |
A3 and B3 |
A2 and B2 |
C |
A4 and B4 |
C2 and C8 |
A4 and B4 |
|
IS431(n) |
1 |
2 |
4 |
1 |
2 |
1 |
1 |
1 |
|
Tn554(n) |
0 |
1 |
2 |
0 |
0 |
0 |
0 |
1 |
|
pUB110 |
- |
+ |
- |
- |
- |
- |
- |
- |
|
pT181 |
- |
- |
+ |
- |
- |
- |
- |
- |
|
Pl258 |
- |
- |
+ |
- |
- |
- |
- |
- |
|
Other resistance genes |
None |
erm, spc, and tobra |
erm, tet, and Hg++ |
None |
None |
None |
None |
erm and spc |
|
*mec complex A has intact regulatory genes, mecR1 and mecl, upstream of mecA; mec complex B has regulatory gene deletions resulting from the insertion sequence 1272 (IS1272) insertion; mec complexes C1 and C2 have regulatory gene deletions resulting from the IS431 insertion; the ccr complex is the recombinase locus; pUB110, pT181 and pl258 are plasmids integrated at insertion sequences. erm, erythromycin resistance gene; Hg++, mercury resistance gene; IS431, insertion sequence 431; n, number of copies; spc, spectinomycin resistance gene, tet, tetracycline resistance gene; Tn554, transposon 554; tobra, tobramycin resistance gene. |
||||||||
SCCmecIII92, limited the potential for horizontal gene transfer of SCCmecI into new genomes.
SCCmecIV, which seems to have evolved from SCCmecI (although it has the ccrA and ccrB genes of SCCmecII (Lina et al., 2006)), gave rise to the most recent worldwide epidemic wave of CA-MRSA. Originally identified in the community-associated USA400 strain, MRSA strain MW2, the first occurrence of SCCmecIV in S. aureus might have been in the ST5 ‘paediatric’ clone that was circulating in hospitals in the late 1980s and the 1990s (Oliveira et al., 2001). The ultimate origins of mecA and SCCmec elements might never be known, but there is good evidence suggesting that coagulase negative staphylococci are the sources (Hanssen et al., 2004; Hanssen and Ericson Sollid, 2006; Wu et al., 1996).
The success of SCCmecIV is borne out by two observations. First, it is the most widely distributed SCCmec among S. aureus isolates. It has been found in nine distinct MRSA CCs or STs, whereas there are only two such lineages for SCCmecI, three for SCCmecII and two for SCCmecIII (Lina et al., 2006). Second, CA-MRSA strains containing SCCmecIV have faster growth rates than hospital MRSA strains carrying other SCCmec allotypes, and these growth rates are no different from MSSA isolates. In a rabbit bacteraemia model the fitness and virulence of USA300, which carries SCCmecIVA, were indistinguishable from those
of its isogenic MSSA variant (Diep et al., 2008). Thus, the SCCmecIV seems to confer little or no cost in fitness on the organism.
The Epidemiology of CA-MRSA
As mentioned above, the earliest reported cases of CA-MRSA infection in the United States were caused by a USA400 strain, MW2 (CDC, 1999). MW2 is closely related to the PVL-negative clone WA-1, which is an important CA-MRSA clone in Australia, and to the MSSA476 strain in the United Kingdom (Coombs et al., 2004). USA400 has been supplanted by USA300, which is currently by far the most frequent cause of CA-MRSA infections in the United States (Kennedy et al., 2008). The USA300 clone seems to be well adapted to the community, and there are reports of CA-MRSA infections caused by USA300 or its close relatives in Australia, Denmark and Colombia (Bartels et al., 2007; Gottlieb et al., 2008; Arias et al., 2008). USA300 strains can also cause health care-associated infections (Liu et al., 2008; Seybold et al., 2006; Maree et al., 2007; Gonzalez et al., 2006). Although there is evidence for the international spread of USA300 and USA400 (Wannet et al., 2005; Nimmo and Coombs, 2008; Tristan et al., 2007; Larsen et al., 2009), CA-MRSA strains that are not related to either USA300 or USA400 have been responsible for infections outside of the United States. ST80 is the predominant clone circulating in Europe, ST59 is the main clone in Taiwan and ST30 is the most frequent in Eastern Australia, demonstrating that CA-MRSA strains have evolved in separate geographical regions (Stam-Bolink et al., 2007; Huang et al., 2007; Nimmo and Coombs, 2008). There can also be considerable diversity in CA-MRSA strains from country to country. For example, in Australia 45 distinct clones of CA-MRSA have been identified; many of these are related to well-known MRSA lineages, but others seem to be new. The diversity of CA-MRSA isolates has also been noted by other studies (Wannet et al., 2005; Francois et al., 2008; Bartels et al., 2007; Tristan et al., 2007; Larsen et al., 2009). In the United Kingdom, most CA-MRSA infections are caused by EMRSA-15 (ST22) and EMRSA-16 (ST36), which are also important hospital-acquired clones (Rollason et al., 2008); ST80 is also present, but accounts for only a small proportion of isolates (Holmes et al., 2005). A CA-MRSA strain of swine origin that is transmissible to humans, ST398, has also been described (Huijsdens et al., 2006; Loeffler et al., 2009).
The epidemiology of CA-MRSA is similar regardless of the country of origin. Isolates tend not to be resistant to multiple drugs, SCCmecIV or SCCmecV is typically present, and infections of skin and soft tissue are the most common. The presence of PVL among CA-MRSA isolates is more variable. For example, in Australia and the United Kingdom most CA-MRSA clones do not produce PVL (Nimmo and Coombs, 2008; Rollason et al., 2008), and the prevalence of PVL among the more common CA-MRSA isolates from Denmark ranges from 17%
to 100% (Larsen et al., 2009). Conversely, isolates of clones that typically do not carry PVL genes (for example EMRSA-15 and EMRSA-16) have occasionally been found to be PVL-positive.
Nasal carriage of MRSA has increased in parallel with the emergence of MRSA as a community pathogen, which is not unexpected given that approximately 30% of individuals are asymptomatic nasal carriers of S. aureus. Between 2001 and 2004, carriage of MRSA strains in a US population-based study approximately doubled from 0.8% to 1.5% (Gorwitz et al., 2008), and the percentage of CA-MRSA genotypes increased from 7% to 24.2% (Tenover et al., 2008). Although the sites of carriage (for example, nares versus groin versus other sites) and the relationship between the carriage of CA-MRSA strains and disease are not entirely clear, CA-MRSA strains, especially USA300, seem to be more easily transmitted than other strains (Crum et al., 2006), which could account for the increasing carriage rates in the community. Thus, no individual or group can be considered not to be at risk for CA-MRSA infection.
The Virulence of CA-MRSA
CA-MRSA infections have been associated with fulminant and lethal infections and worse clinical outcomes than are seen with infections caused by health care-associated MRSA strains and community MSSA (Francis et al., 2005; Turner et al., 2007; Davis et al., 2007), giving rise to the impression that CA-MRSA strains, especially USA300, are more virulent than other strains. Much of our understanding of the unique virulence properties of CA-MRSA is based on studies of USA300 strains, the most extensively investigated clone. The USA300 core genome (the chromosome, excluding any mobile genetic elements) is similar to that of the early MRSA strain COL (Diep et al., 2006). However, studies in animal models indicate that USA300 is more virulent than COL (Voyich et al., 2005; Li et al., 2009). The expression of virulence factors by USA300 is high, and this and other closely related strains are more lethal than their more distant relatives and cause more extensive disease in animal models of infection (Li et al., 2009; Montgomery et al., 2008; Wang et al., 2007). The main difference between the COL and USA300 genomes is in their mobile genetic elements, which include prophages, plasmids, pathogenicity islands and transposons that have been acquired through horizontal gene transfer. These elements encode factors that can affect transmission, antibiotic resistance and virulence. Prophages ΦSA2 and ΦSA3, which are present in USA300 strains but not in COL, could contribute to the noted differences in virulence between these two lineages. Prophage ΦSA2 contains lukS–PV and lukF–PV, which encode PVL. Prophage ΦSA3 is present in strains other than CA-MRSA and encodes staphylokinase, staphylococcal complement inhibitor (SCIN) and S. aureus chemotaxis inhibitory protein (CHIPS), all of which are modulators of the innate immune system (Rooijakkers et al., 2006; van Wamel et al., 2006). In addition, USA300 contains
the pathogenicity island SaPI5, which is similar to the island that is present in COL. SaPI5 encodes two superantigens that are not present in COL, staphylococcal enterotoxin Q (SEQ) and staphylococcal enterotoxin K (SEK), which are also found in other MRSA and MSSA lineages. S. aureus produces many other molecules that promote host colonization, facilitate evasion of the innate immune system and alter immune responses (Wang et al., 2007; Deleo et al., 2009; Li et al., 2007) (see Supplementary information S1 [Table A2-S1]). Most of these molecules are not unique to CA-MRSA. The virulence factors that are found more commonly in CA-MRSA than in other strains, that are linked by epidemiology to CA-MRSA infections or that have been studied in animal models of CA-MRSA infection are discussed below.
PVL
PVL has been studied extensively since its discovery by Panton and Valentine 70 years ago (Wright, 1936). The role of PVL in the marked epidemicity and enhanced virulence of CA-MRSA is a subject of debate. PVL is composed of two subunits, LukS-PV and LukF-PV (Woodin, 1960), which are encoded by the horizontally acquired prophage ΦSA2 (Kaneko et al., 1998) and are secreted by the bacterium. These subunits bind to specific membrane receptors, which have yet to be identified, and associate to form pores in the membrane of host leukocytes (Meyer et al., 2009; Colin et al., 1994). At high concentrations (for example, 200 nM) PVL causes lytic cell death, but at sublytic concentrations (for example, 5 nM) it seems to partially activate neutrophils in a phenomenon known as priming, as they secrete potent mediators of inflammation, such as leukotriene b4 and interleukin 8, and also cause the release of neutrophil granule contents through exocytosis (Konig et al., 1995; Woodin and Wieneke, 1964; Genestier et al., 2005). In addition, PVL primes neutrophils for the enhanced production of reactive oxygen species on stimulation with the widely used neutrophil agonist fMLP (N-formyl-methionylleucyl-phenylalanine) (Colin and Monteil, 2003). Therefore, PVL could contribute to pathogenesis by causing an exaggerated inflammatory response and injury to the host. Several lines of evidence that are largely circumstantial indicate that PVL is associated with severe skin infections and severe necrotic haemorrhagic pneumonia (Lima et al., 1999; Gillet et al., 2002, 2007). Both USA300, which is now the leading cause of skin and soft tissue infections in the United States and a cause of extremely severe infections, and the penicillin-resistant phage type 80/81 strains that were associated with numerous outbreaks and severe disease in the 1950s produce PVL. The epidemiological association between PVL and the emergence of genetically unrelated CA-MRSA strains (that is, different and unrelated STs) that are geographically dispersed is striking.
There are other observations that call into question the presumption that PVL is driving the CA-MRSA epidemic. First, PVL is found infrequently in
other common, successful community strains. For example, the genes encoding PVL are present in only ~1–10% of MSSA clinical isolates (Goering et al., 2008; Kuehnert et al., 2006; Ellington et al., 2007). Second, although both USA300 and USA400 express PVL, USA300 has become the predominant CA-MRSA clone in the United States. This suggests that factors other than PVL are important for the recent emergence of CA-MRSA.
The experimental evidence does not provide a clear picture either. Voyich et al. (2006) found that USA300 and USA400 wild-type and isogenic PVL-deficient (∆pvl) strains caused virtually identical courses of infection in mouse abscess and sepsis models. Furthermore, there was no difference in neutrophil phagocytosis or lysis after uptake of the bacteria. However, because these experiments were carried out using culture supernatants, the results could reflect the action of multiple lytic factors. Similar results from a rat pneumonia model were reported by Montgomery and Daum (2009). Bubeck Wardenburg et al. (2007, 2008) also showed that USA300 and USA400 wild-type and isogenic ∆pvl strains were equally virulent in mouse abscess and pneumonia models. Diep et al. (2008) used two rabbit bacteraemia models to compare the haematogenous dissemination of wild-type and ∆pvl CA-MRSA strains to major organs: although PVL did not promote seeding of lungs, spleen or blood by USA300, there was a modest, transient contribution of PVL to colonization of the kidneys. In a series of experiments that used the same USA300 wild-type and mutant (∆pvl) strain pair as Voyich et al. (2006), Brown et al. (2008) found that the parent strain was more virulent than the ∆pvl mutant in mouse pneumonia and abscess models and that the disease caused by the wild-type strain was attenuated by immunization with recombinant LukF-PV or LukS-PV. In addition, Labandeira-Rey et al. (2007) found evidence to suggest that PVL might have a role in disease development in a mouse model of staphylococcal pneumonia: direct instillation of high doses of purified toxin provoked an inflammatory response in the lung and reduced survival. The authors used a laboratory strain of S. aureus that had been transduced with PVL-encoding bacteriophage to establish infection, and reported more severe disease in mice infected with this PVL-producing variant than in those infected with the PVL-negative parent. However, in addition to the presence of PVL, this transduced laboratory strain has substantial alterations in global gene expression that confounded the interpretation of the data. As PVL has no impact on protein or gene expression in USA300 or USA400 (Diep et al., 2008), it is possible that factors other than PVL accounted for the experimental results. Taken together, the data suggest that the contribution of PVL to CA-MRSA pathogenesis could be minor or perhaps dependent on an as-yet-unidentified bacterial factor or host susceptibility component.
α-Haemolysin
The pore-forming toxin α-haemolysin (also known as Hla or α-toxin) causes the destruction of a wide range of host cells, including epithelial cells, erythrocytes, fibroblasts and monocytes, and is lethal in animal models when injected in purified form (Bhakdi and Tranum-Jensen, 1991). α-haemolysin is ubiquitous among clinical isolates, although some strains lack an active α-toxin. Recent studies by Bubeck Wardenburg et al. (2007) showed that α-haemolysin is essential for USA300 and USA400 to cause lethal pneumonia in a mouse model of the disease. The amount of this toxin that is produced by these strains in vitro correlates with the severity of the resultant lung disease (Montgomery et al., 2008; Bubeck Wardenburg et al., 2007; Burlak et al., 2007).
α-Type Phenol-Soluble Modulins
α-type phenol-soluble modulins (PSMαs) are a newly discovered group of peptides in S. aureus that are similar to the PSMs of Staphylococcus epidermidis (Wang et al., 2007). High expression of PSMαs might contribute to the enhanced virulence of CA-MRSA; PSMs are produced at higher levels in vitro by prominent CA-MRSA strains, including USA300 and USA400, than by hospital-acquired MRSA strains (Wang et al., 2007). PSMα peptides recruit, activate and ultimately lyse human neutrophils, thereby promoting S. aureus pathogenesis, and greatly contribute to the virulence of USA300 and USA400 in mouse abscess and sepsis models. The study by Wang et al. (2007) was the first to identify molecules from CA-MRSA that could account at least in part for the enhanced virulence of USA300 and USA400.
Arginine Catabolic Mobile Element
The arginine catabolic mobile element (ACME) is a 30.9 kb segment of DNA that seems to be unique to USA300 (Diep et al., 2008). This element is adjacent to SCCmecIV and is mobilized by the recombinases that are encoded by SCCmec. It contains two potential virulence factors, a cluster of arginine catabolism (arc) genes that encode an arginine deiminase pathway and opp3, which encodes an oligopeptide permease (Coulter et al., 1998; Degnan et al., 1998). Deletion of ACME but not SCCmec has been shown to decrease the fitness of USA300 in a rabbit bacteraemia model (Diep et al., 2008). Therefore, ACME might contribute to the fitness and epidemic spread of USA300.
Although mobile genetic elements such as ACME are likely to play a part in the transmission of CA-MRSA, there are differences in virulence potential and human disease manifestation even among similar USA300 isolates. For example, Kennedy et al. (2008) used comparative whole-genome sequencing to determine whether USA300 arose by convergent evolution towards a
hypervirulent phenotype or from a recent common ancestor of high virulence potential. Ten USA300 isolates, including some from a wide range of clinical syndromes and from different geographical locations in the United States, were examined. The strains differed from the USA300 reference strain FPR3757 genome by only a few SNPs, ranging from 11 to 408 in number. Phylogenetic analysis indicated that 8 of the strains, differing on average by 32 SNPs from the reference strain and 50 SNPs from each other, clustered with the reference strain and had descended from a recent common ancestor. These nine closely related isolates constitute the epidemic USA300 clone. Eight of the nine strains were ACME positive and all nine contained the same SCCmecIVA subtype. The two other strains were outliers, both lacking ACME and carrying a different SCCmec subtype, type IVB. Unexpectedly, the virulence of the more closely related isolates was variable in animal infection models. Some of these isolates had caused dramatically different disease syndromes in humans (for example, necrotizing pneumonia versus abscesses were caused by isolates that differed by only 23 SNPs), which serves to highlight the importance of host factors in disease presentation and severity.
Treatment in the Era of CA-MRSA
CA-MRSA has had a marked impact on empirical therapy of suspected staphylococcal infection. Most β-lactam antibiotics, including all orally available agents, can no longer be assumed to be effective for a range of common staphylococcal infections, in particular for skin and soft-tissue infections. In regions where CA-MRSA is prevalent, antimicrobial therapy should be active against MRSA strains. However, there are few clinical data to support the use of agents other than vancomycin, daptomycin or linezolid. Despite a lack of rigorous clinical studies, the oral agents that are recommended for the treatment of CA-MRSA skin and soft-tissue infections include clindamycin, long-acting tetracyclines (doxycycline and minocycline) and trimethoprim–sulphamethoxazole, as well as rifampin and fusidic acid as adjunctive agents to be used in combination (Gorwitz et al., 2006; Barton et al., 2006; Nathwani et al., 2008).
Surgical incision and drainage is the treatment of choice for cutaneous abscesses; adjunctive antimicrobial therapy is of little or no benefit in most of these cases (Moran et al., 2006; Fridkin et al., 2005; Llera and Levy, 1985; Lee et al., 2004). Antibiotic therapy after drainage of CA-MRSA abscesses is not routinely recommended unless the patient has severe or extensive disease, has rapid progression in the presence of associated cellulitis, has symptoms of systemic illness, is very old or very young, has another illness or immune suppression (for example, type I diabetes, HIV infection or neoplastic disease), has an abscess in an area that is difficult to drain or has an abscess that is associated with septic phlebitis (Gorwitz et al., 2006).
Vancomycin is still the preferred drug for the treatment of serious MRSA infections. However, its effectiveness is limited by prolonged, persistent or recurrent bacteraemia during therapy (Khatib et al., 2009; Hawkins et al., 2007), high rates of microbiological and clinical failures (Dombrowski and Winston, 2008), nephrotoxicity (Lodise et al., 2008) and the increasing prevalence of non-susceptible strains (Steimkraus et al., 2007; Wang et al., 2006). Randomized clinical trials of alternative agents, such as linezolid and daptomycin, show that they are comparable or, more precisely, neither inferior nor superior to standard therapy (Arbeit et al., 2004; Shorr et al., 2005; Wunderink et al., 2003; Weigelt et al., 2005; Kaplan et al., 2003; Fowler et al., 2006). Resistance and drug toxicity will remain concerns regardless of the choice of agent.
One or more new compounds that are currently being developed are likely to become available for the treatment of MRSA infections in the near future (Lentino et al., 2008; Pan et al., 2008). Telavancin, dalbavancin and oritavancin are vancomycin derivatives that rapidly kill S. aureus in a concentration-dependent manner in vitro. Whether more rapid killing will translate into an improved efficacy over vancomycin for more serious infections, such as endocarditis or bacteraemia, remains to be determined. Carbapenems and cephalosporins that bind PBP2a, the penicillin-binding protein that mediates methicillin resistance, with much higher affinity than the currently available β-lactams have been developed (Koga et al., 2005). Two cephalosporins, ceftobiprole and ceftaroline, were shown to be clinically effective for the treatment of MRSA skin and soft-tissue infections (Parish and Scheinfeld, 2008; Anderson and Gums, 2008). One drawback with these and the other anti-MRSA β-lactams under development is that they are broad-spectrum antibiotics and are therefore not narrowly targeted treatments of MRSA infection. Further studies are needed to define their eventual role in the therapy of MRSA infections. Moreover, the vancomycin derivatives and anti-MRSA β-lactams, which can only be administered intravenously, do not address the need for orally administered agents. Orally bioavailable oxazolidinones that are active against MRSA are in the early stages of development (Shaw et al., 2008).
Several non-traditional approaches to the treatment and prevention of MRSA infections have been or are still being investigated. These include lysostaphin (Dajcs et al., 2001), antimicrobial peptides (Lawton et al., 2007) and other natural products (for example, tea tree oil) (Stapleton et al., 2007), as well as anti-staphylococcal vaccines (Bubeck Wardenburg and Schneewind, 2008). There are considerable challenges to be faced in the development of these agents, including prohibitively expensive costs, the potential for patient hypersensitivity (caused by the repeated administration of protein products), the short half-lives that are associated with systemic administration and the short-lived or only partially protective immunity that is gained from vaccines, as was the case with an anti-capsular vaccine that proved to be ineffective (Shinefield et al., 2002). These approaches are years away from being available in the clinic, if they make it at all. Prudent
use of the agents that are now available is essential to avoid further erosion of the antimicrobial armamentarium.
Concluding Remarks
S. aureus is an extraordinarily adaptable pathogen with a proven ability to develop resistance. The steady erosion of the effectiveness of β-lactam antibiotics since their first use only 60 years ago is particularly worrying. As we have described, there have been four waves of resistance over the past 60 years. Although the details vary, the basic themes of each successive wave of antibiotic resistance are similar. Often occurring as a consequence of horizontal gene transfer, resistance is initially encountered in hospitals and health care institutions, where the selective pressures for resistance are greatest. Resistant strains are temporarily contained in hospitals but eventually, through a series of modifications and adjustments, they find their way into or arise from within the community to emerge as fully fit and virulent pathogens. Our understanding of the forces that direct the evolution of virulent and drug-resistant organisms is not perfect, but the overuse and misuse of antibiotics is clearly a contributing factor. The discovery and development of new antimicrobials, although necessary, is unlikely to solve the problem of drug resistance for long. New technologies that lead to improved and more rapid diagnostics, a better understanding of the pathogenesis of staphylococcal disease and non-antimicrobial approaches to the prevention and treatment of infection will also be needed to forestall the coming of the post-antibiotic era.
References
Adcock, P. M., Pastor, P., Medley, F., Patterson, J. E. & Murphy, T. V. Methicillin-resistant Staphylococcus aureus in two child care centers. J. Infect. Dis. 178, 577–580 (1998).
Aiello, A. E., Lowy, F. D., Wright, L. N. & Larson, E. L. Meticillin-resistant Staphylococcus aureus among US prisoners and military personnel: review and recommendations for future studies. Lancet Infect. Dis. 6, 335–341 (2006).
Anderson, S. D. & Gums, J. G. Ceftobiprole: an extended-spectrum anti-methicillin-resistant Staphylococcus aureus cephalosporin. Ann. Pharmacother. 42, 806–816 (2008).
Arbeit, R. D., Maki, D., Tally, F. P., Campanaro, E. & Eisenstein, B. I. The safety and efficacy of daptomycin for the treatment of complicated skin and skin-structure infections. Clin. Infect. Dis. 38, 1673–1681 (2004).
Arias, C. A. et al. MRSA USA300 clone and VREF—a US–Colombian connection? N. Engl. J. Med. 359, 2177–2179 (2008).
Baggett, H. C. et al. Community-onset methicillin-resistant Staphylococcus aureus associated with antibiotic use and the cytotoxin Panton-Valentine leukocidin during a furunculosis outbreak in rural Alaska. J. Infect. Dis. 189, 1565–1573 (2004).
Barrett, F. F., McGehee, R. F. Jr & Finland, M. Methicillin-resistant Staphylococcus aureus at Boston City Hospital. Bacteriologic and epidemiologic observations. N. Engl J. Med. 279, 441–448 (1968).
Barber, M. Methicillin-resistant staphylococci. J. Clin. Pathol. 14, 385–393 (1961).
Barber, M. & Rozwadowska-Dowzenko, M. Infection by penicillin-resistant staphylococci. Lancet 1, 641–644 (1948).
Bartels, M. D., Boye, K., Rhod Larsen, A., Skov, R. & Westh, H. Rapid increase of genetically diverse methicillin-resistant Staphylococcus aureus, Copenhagen, Denmark. Emerg. Infect. Dis. 13, 1533–1540 (2007).
Barton, M. et al. Guidelines for the prevention and management of community-acquired methicillinresistant Staphylococcus aureus: a perspective for Canadian health care practitioners. Can. J. Infect. Dis. Med. Microbiol. 17 (Suppl. C), 4–24 (2006).
Bhakdi, S. & Tranum-Jensen, J. Alpha-toxin of Staphylococcus aureus. Microbiol. Rev. 55, 733–751 (1991).
Blair, J. E. & Carr, M. Distribution of phage groups of Staphylococcus aureus in the years 1927 through 1947. Science 132, 1247–1248 (1960).
Bran, J. L., Levison, M. E. & Kaye, D. Survey for methicillin-resistant staphylococci. Antimicrob. Agents Chemother. 1, 235–236 (1972).
Brown, E. L. et al. The Panton-Valentine leukocidin vaccine protects mice against lung and skin infections caused by Staphylococcus aureus USA300. Clin. Microbiol. Infect. 15, 156–164 (2008).
Bubeck Wardenburg, J., Bae, T., Otto, M., Deleo, F. R. & Schneewind, O. Poring over pores: α-hemolysin and Panton-Valentine leukocidin in Staphylococcus aureus pneumonia. Nature Med. 13, 1405–1406 (2007).
Bubeck Wardenburg, J. & Schneewind, O. Vaccine protection against Staphylococcus aureus pneumonia. J. Exp. Med. 205, 287–294 (2008).
Bubeck Wardenburg, J., Palazzolo-Ballance, A. M., Otto, M., Schneewind, O. & DeLeo, F. R. Panton-Valentine leukocidin is not a virulence determinant in murine models of community-associated methicillin-resistant Staphylococcus aureus disease. J. Infect. Dis. 198, 1166–1170 (2008).
Burlak, C. et al. Global analysis of community-associated methicillin-resistant Staphylococcus aureus exoproteins reveals molecules produced in vitro and during infection. Cell. Microbiol. 9, 1172–1190 (2007).
Bynoe, E. T., Elder, R. H. & Comtois, R. D. Phagetyping and antibiotic-resistance of staphylococci isolated in a general hospital. Can. J. Microbiol. 2, 346–358 (1956).
CDC. Community-associated methicillin-resistant Staphylococcus aureus infections in Pacific Islanders—Hawaii, 2001–2003. MMWR Morb. Mortal. Wkly Rep. 53, 767–770 (2004).
CDC. Four pediatric deaths from community-acquired methicillin-resistant Staphylococcus aureus— Minnesota and North Dakota, 1997–1999. MMWR Morb. Mortal. Wkly Rep. 48, 707–710 (1999).
Chavez-Bueno, S. et al. Inducible clindamycin resistance and molecular epidemiologic trends of pediatric community-acquired methicillin-resistant Staphylococcus aureus in Dallas, Texas. Antimicrob. Agents Chemother. 49, 2283–2288 (2005).
Colin, D. A. & Monteil, H. Control of the oxidative burst of human neutrophils by staphylococcal leukotoxins. Infect. Immun. 71, 3724–3729 (2003).
Colin, D. A., Mazurier, I., Sire, S. & Finck-Barbancon, V. Interaction of the two components of leukocidin from Staphylococcus aureus with human polymorphonuclear leukocyte membranes: sequential binding and subsequent activation. Infect. Immun. 62, 3184–3188 (1994).
Conly, J. M. & Johnston, B. L. The emergence of methicillin-resistant Staphylococcus aureus as a community-acquired pathogen in Canada. Can. J. Infect. Dis. 14, 249–251 (2003).
Coombs, G. W. et al. Genetic diversity among community methicillin-resistant Staphylococcus aureus strains causing outpatient infections in Australia. J. Clin. Microbiol. 42, 4735–4743 (2004).
Coulter, S. N. et al. Staphylococcus aureus genetic loci impacting growth and survival in multiple infection environments. Mol. Microbiol. 30, 393–404 (1998).
Cox, R. A., Conquest, C., Mallaghan, C. & Marples, R. R. A major outbreak of methicillin-resistant Staphylococcus aureus caused by a new phage-type (EMRSA-16). J. Hosp. Infect. 29, 87–106 (1995).
Crisostomo, M. I. et al. The evolution of methicillin resistance in Staphylococcus aureus: similarity of genetic backgrounds in historically early methicillin susceptible and -resistant isolates and contemporary epidemic clones. Proc. Natl Acad. Sci. USA 98, 9865–9870 (2001).
Crossley, K., Landesman, B. & Zaske, D. An outbreak of infections caused by strains of Staphylococcus aureus resistant to methicillin and aminoglycosides. II.Epidemiologic studies. J. Infect. Dis. 139, 280–287(1979).
Crum, N. F. et al. Fifteen-year study of the changing epidemiology of methicillin-resistant Staphylococcus aureus. Am. J. Med. 119, 943–951 (2006).
Dajcs, J. J. et al. Lysostaphin is effective in treating methicillin-resistant Staphylococcus aureus endophthalmitis in the rabbit. Curr. Eye Res. 22, 451–457 (2001).
David, M. Z., Rudolph, K. M., Hennessy, T. W., Boyle- Vavra, S. & Daum, R. S. Molecular epidemiology of methicillin-resistant Staphylococcus aureus, rural southwestern Alaska. Emerg. Infect. Dis. 14, 1693–1699 (2008).
Davis, S. L. et al. Epidemiology and outcomes of community-associated methicillin-resistant Staphylococcus aureus infection. J. Clin. Microbiol. 45, 1705–1711 (2007).
Degnan, B. A. et al. Inhibition of human peripheral blood mononuclear cell proliferation by Streptococcus pyogenes cell extract is associated with arginine deiminase activity. Infect. Immun. 66, 3050–3058 (1998).
Deleo, F. R., Diep, B. A. & Otto, M. Host defense and pathogenesis in Staphylococcus aureus infections. Infect. Dis. Clin. North Am. 23, 17–34 (2009). Review of the virulence factors found in S. aureus.
Deurenberg, R. H. & Stobberingh, E. E. The evolution of Staphylococcus aureus. Infect. Genet. Evol. 8, 747–763 (2008).
Deurenberg, R. H. et al. Cross-border dissemination of methicillin-resistant Staphylococcus aureus, Euregio Meuse-Rhin region. Emerg. Infect. Dis. 15, 727–734 (2009).
Diep, B. A. et al. The arginine catabolic mobile element and staphylococcal chromosomal cassette mec linkage: convergence of virulence and resistance in the USA300 clone of methicillin-resistant Staphylococcus aureus. J. Infect. Dis. 197, 1523–1530 (2008).
Diep, B. A., Sensabaugh, G. F., Somboona, N. S., Carleton, H. A. & Perdreau-Remington, F. Widespread skin and soft-tissue infections due to two methicillinresistant Staphylococcus aureus strains harboring the genes for Panton-Valentine leucocidin. J. Clin. Microbiol. 42, 2080–2084 (2004).
Diep, B. A. et al. Emergence of multidrug-resistant, community-associated, methicillin-resistant Staphylococcus aureus clone USA300 in men who have sex with men. Ann. Intern. Med. 148, 249–257 (2008).
Diep, B. A. et al. Complete genome sequence of USA300, an epidemic clone of community-acquired meticillin-resistant Staphylococcus aureus. Lancet 367, 731–739 (2006). Comparative genomics of USA300 and other MRSA strains.
Diep, B. A. et al. Contribution of Panton-Valentine leukocidin in community-associated methicillin-resistant Staphylococcus aureus pathogenesis. PLoS ONE 3, e3198 (2008).
Dombrowski, J. C. & Winston, L. G. Clinical failures of appropriately-treated methicillin-resistant Staphylococcus aureus infections. J. Infect. 57, 110–115 (2008).
Enright, M. C. et al. The evolutionary history of methicillin-resistant Staphylococcus aureus (MRSA). Proc. Natl Acad. Sci. USA 99, 7687–7692 (2002). Description of the MRSA clones and SCCmec allotypes present in a worldwide collection of mainly nosocomial isolates.
Enright, M. C., Day, N. P., Davies, C. E., Peacock, S. J. & Spratt, B. G. Multilocus sequence typing for characterization of methicillin-resistant and methicillin susceptible clones of Staphylococcus aureus. J. Clin. Microbiol. 38, 1008–1015 (2000). Description of the MLST method and how it can be applied to elucidate the population structure of S. aureus.
Ellington, M. J. et al. Is Panton-Valentine leucocidin associated with the pathogenesis of Staphylococcus aureus bacteraemia in the UK? J. Antimicrob. Chemother. 60, 402–405 (2007).
European Antimicrobial Resistance Surveillance System. Annual Report 2007. (EARSS, Bilthoven, 2008).
Fang, H., Hedin, G., Li, G. & Nord, C. E. Genetic diversity of community-associated methicillin-resistant Staphylococcus aureus in southern Stockholm, 2000–2005. Clin. Microbiol. Infect. 14, 370–376 (2008).
Feil, E. J. & Enright, M. C. Analyses of clonality and the evolution of bacterial pathogens. Curr. Opin. Microbiol. 7, 308–313 (2004).
Feil, E. J., Li, B. C., Aanensen, D. M., Hanage, W. P. & Spratt, B. G. eBURST: inferring patterns of evolutionary descent among clusters of related bacterial genotypes from multilocus sequence typing data. J. Bacteriol. 186, 1518–1530 (2004).
Feil, E. J. et al. How clonal is Staphylococcus aureus? J. Bacteriol. 185, 3307–3316 (2003).
Feng, Y. et al. Evolution and pathogenesis of Staphylococcus aureus: lessons learned from genotyping and comparative genomics. FEMS Microbiol. Rev. 32, 23–37 (2008).
Fowler, V. G. Jr et al. Daptomycin versus standard therapy for bacteremia and endocarditis caused by Staphylococcus aureus. N. Engl. J. Med. 355, 653–665 (2006).
Francis, J. S. et al. Severe community-onset pneumonia in healthy adults caused by methicillin-resistant Staphylococcus aureus carrying the Panton-Valentine leukocidin genes. Clin. Infect. Dis. 40, 100–107 (2005).
Francois, P. et al. Methicillin-resistant Staphylococcus aureus, Geneva, Switzerland, 1993–2005. Emerg. Infect. Dis. 14, 304–307 (2008).
Fridkin, S. K. et al. Methicillin-resistant Staphylococcus aureus disease in three communities. N. Engl. J. Med. 352, 1436–1444 (2005). First large study characterizing the outbreak of CA-MRSA that was caused by USA300 in the United States.
Gardella, N. et al. Community-associated methicillin-resistant Staphylococcus aureus, eastern Argentina. Diagn. Microbiol. Infect. Dis. 62, 343–347 (2008).
Genestier, A. L. et al. Staphylococcus aureus Panton- Valentine leukocidin directly targets mitochondria and induces Bax-independent apoptosis of human neutrophils. J. Clin. Invest. 115, 3117–3127 (2005).
Gilbert, M. et al. Outbreak in Alberta of community acquired (USA300) methicillin-resistant Staphylococcus aureus in people with a history of drug use, homelessness or incarceration. Can. Med. Assoc. J. 175, 149–154 (2006).
Gillet, Y. et al. Association between Staphylococcus aureus strains carrying gene for Panton-Valentine leukocidin and highly lethal necrotising pneumonia in young immunocompetent patients. Lancet 359, 753–759 (2002).
Gillet, Y. et al. Factors predicting mortality in necrotizing community-acquired pneumonia caused by Staphylococcus aureus containing Panton-Valentine leukocidin. Clin. Infect. Dis. 45, 315–321 (2007).
Goering, R. V. et al. Molecular epidemiology of methicillin-resistant and methicillin-susceptible Staphylococcus aureus isolates from global clinical trials. J. Clin. Microbiol. 46, 2842–2847 (2008).
Gomes, A. R., Westh, H. & de Lencastre, H. Origins and evolution of methicillin-resistant Staphylococcus aureus clonal lineages. Antimicrob. Agents Chemother. 50, 3237–3244 (2006). Analysis of penicillin-susceptible and penicillin resistant genotypes of S. aureus, carried out before the emergence of MRSA.
Gonzalez, B. E. et al. Community-associated strains of methicillin-resistant Staphylococcus aureus as the cause of healthcare-associated infection. Infect. Control Hosp. Epidemiol. 27, 1051–1056 (2006).
Gonzalez, B. E. et al. Pulmonary manifestations in children with invasive community-acquired Staphylococcus aureus infection. Clin. Infect. Dis. 41, 583–590 (2005).
Gorwitz, R. J. et al. Strategies for clinical management of MRSA in the community: summary of an expert’s meeting convened by the Centers for Disease Control and Prevention. CDC [online], http://www.cdc.gov/ncidod/dhqp/ar_mrsa_ca.html (2006).
Gottlieb, T., Su, W. Y., Merlino, J. & Cheong, E. Y. Recognition of USA300 isolates of community-acquired methicillin-resistant Staphylococcus aureus in Australia. Med. J. Aust. 189, 179–180 (2008).
Gorwitz, R. J. et al. Changes in the prevalence of nasal colonization with Staphylococcus aureus in the United States, 2001–2004. J. Infect. Dis. 197, 1226–1234 (2008).
Hallin, M. et al. Genetic relatedness between methicillin-susceptible and methicillin-resistant Staphylococcus aureus: results of a national survey. J. Antimicrob. Chemother. 59, 465–472 (2007).
Hanssen, A. M., Kjeldsen, G. & Sollid, J. U. Local variants of staphylococcal cassette chromosome mec in sporadic methicillin-resistant Staphylococcus aureus and methicillin-resistant coagulase-negative staphylococci: evidence of horizontal gene transfer? Antimicrob. Agents Chemother. 48, 285–296 (2004).
Hanssen, A. M. & Ericson Sollid, J. U. SCCmec in staphylococci: genes on the move. FEMS Immunol. Med. Microbiol. 46, 8–20 (2006).
Hawkins, C. et al. Persistent Staphylococcus aureus bacteremia: an analysis of risk factors and outcomes. Arch. Intern. Med. 167, 1861–1867 (2007).
Herold, B. C. et al. Community-acquired methicillinresistant Staphylococcus aureus in children with no identified predisposing risk. JAMA 279, 593–598 (1998). A report of CA-MRSA in children in Chicago, which stimulated an awareness of the scope of the epidemic.
Hersh, A. L., Chambers, H. F., Maselli, J. H. & Gonzales, R. National trends in ambulatory visits and antibiotic prescribing for skin and soft-tissue infections. Arch. Intern. Med. 168, 1585–1591 (2008).
Higuchi, W., Takano, T., Teng, L. J. & Yamamoto, T. Structure and specific detection of staphylococcal cassette chromosome mec type VII. Biochem. Biophys. Res. Commun. 377, 752–756 (2008).
Hiramatsu, K. et al. Dissemination in Japanese hospitals of strains of Staphylococcus aureus heterogeneously resistant to vancomycin. Lancet 350, 1670–1673 (1997).
Holmes, A. et al. Staphylococcus aureus isolates carrying Panton-Valentine leucocidin genes in England and Wales: frequency, characterization, and association with clinical disease. J. Clin. Microbiol. 43, 2384–2390 (2005).
Hope, R., Livermore, D. M., Brick, G., Lillie, M. & Reynolds, R. Non-susceptibility trends among staphylococci from bacteraemias in the UK and Ireland, 2001–2006. J. Antimicrobiol. Chemother. 62 (Suppl. 2), 65–74 (2008).
Huang, Y. C., Hwang, K. P., Chen, P. Y., Chen, C. J. & Lin, T. Y. Prevalence of methicillin-resistant Staphylococcus aureus nasal colonization among Taiwanese children in 2005 and 2006. J. Clin. Microbiol. 45, 3992–3995 (2007).
Huijsdens, X. W. et al. Community-acquired MRSA and pig-farming. Ann. Clin. Microbiol. Antimicrob. 5, 26 (2006).
Ito, T. et al. Structural comparison of three types of staphylococcal cassette chromosome mec integrated in the chromosome in methicillin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 45, 1323–1336 (2001). Comparison of the genetic structure and organization of SCCmecI, SCCmecII and SCCmecIII.
Jevons, M. P. & Parker, M. T. The evolution of new hospital strains of Staphylococcus aureus. J. Clin. Pathol. 17, 243–250 (1964).
Jevons, M. “Celbenin”-resistant staphylococci. BMJ 1, 124–125 (1961).
Johansson, P. J., Gustafsson, E. B. & Ringberg, H. High prevalence of MRSA in household contacts. Scand. J. Infect. Dis. 39, 764–768 (2007).
Johnson, A. P. et al. Dominance of EMRSA-15 and -16 among MRSA causing nosocomial bacteraemia in the UK: analysis of isolates from the European Antimicrobial Resistance Surveillance System (EARSS). J. Antimicrob. Chemother. 48, 143–144 (2001).
Kallen, A. J. et al. Staphylococcus aureus community acquired pneumonia during the 2006 to 2007 influenza season. Ann. Emerg. Med. 53, 358–365 (2009).
Kaneko, J., Kimura, T., Narita, S., Tomita, T. & Kamio, Y. Complete nucleotide sequence and molecular characterization of the temperate staphylococcal bacteriophage ΦPVL carrying Panton-Valentine leukocidin genes. Gene 215, 57–67 (1998).
Kanerva, M. et al. Community-associated methicillinresistant Staphylococcus aureus, isolated in Finland in 2004 to 2006. J. Clin. Microbiol. 7, 2655–2657 (2009).
Kaplan, S. L. et al. Linezolid versus vancomycin for treatment of resistant Gram-positive infections in children. Pediatr. Infect. Dis. J. 22, 677–686 (2003).
Kaplan, S. L. et al. Three-year surveillance of community-acquired Staphylococcus aureus infections in children. Clin. Infect. Dis. 40, 1785–1791 (2005).
Kazakova, S. V. et al. A clone of methicillin-resistant Staphylococcus aureus among professional football players. N. Engl. J. Med. 352, 468–475 (2005).
Kennedy, A. D. et al. Epidemic community-associated methicillin-resistant Staphylococcus aureus: recent clonal expansion and diversification. Proc. Natl Acad. Sci. USA 105, 1327–1332 (2008). Deep sequence analysis of closely related USA300 strains and a comparison of their virulence in a mouse model.
Khatib, R. et al. Persistent Staphylococcus aureus bacteremia: incidence and outcome trends over time. Scand. J. Infect. Dis. 41, 4–9 (2009).
Kirby, W. Extraction of a highly potent penicillin inactivator from penicillin resistant staphylococci. Science 99, 452–453 (1944).
Klevens, R. M. et al. Invasive methicillin-resistant Staphylococcus aureus infections in the United States. JAMA 298, 1763–1771 (2007).
Kluytmans, J., van Belkum, A. & Verbrugh, H. Nasal carriage of Staphylococcus aureus: epidemiology, underlying mechanisms, and associated risks. Clin. Microbiol. Rev. 10, 505–520 (1997). Review of S. aureus colonization of humans.
Koga, T. et al. In vitro and in vivo antibacterial activities of CS-023 (RO4908463), a novel parenteral carbapenem. Antimicrob. Agents Chemother. 49, 3239–3250 (2005).
Konig, B., Prevost, G., Piemont, Y. & Konig, W. Effects of Staphylococcus aureus leukocidins on inflammatory mediator release from human granulocytes. J. Infect. Dis. 171, 607–613 (1995).
Kuehnert, M. J. et al. Prevalence of Staphylococcus aureus nasal colonization in the United States, 2001–2002. J. Infect. Dis. 193, 172–179 (2006).
Labandeira-Rey, M. et al. Staphylococcus aureus Panton-Valentine leukocidin causes necrotizing pneumonia. Science 315, 1130–1133 (2007).
Larsen, A. R. et al. Emergence and characterization of community-associated methicillin-resistant Staphyloccocus aureus infections in Denmark, 1999 to 2006. J. Clin. Microbiol. 47, 73–78 (2009).
Larsen, A., Stegger, M., Goering, R., Sorum, M. & Skov, R. Emergence and dissemination of the methicillin resistant Staphylococcus aureus USA300 clone in Denmark (2000–2005). Euro. Surveill. 12, 22–24 (2007).
Larsen, A. R. et al. Epidemiology of European community-associated methicillin-resistant Staphylococcus aureus clonal complex 80 type IV strains isolated in Denmark from 1993 to 2004. J. Clin. Microbiol. 46, 62–68 (2008).
Laupland, K. B., Ross, T. & Gregson, D. B. Staphylococcus aureus bloodstream infections: risk factors, outcomes, and the influence of methicillin resistance in Calgary, Canada, 2000–2006. J. Infect. Dis. 198, 336–343 (2008).
Lawton, E. M., Ross, R. P., Hill, C. & Cotter, P. D. Two-peptide lantibiotics: a medical perspective. Mini Rev. Med. Chem. 7, 1236–1247 (2007).
Lee, M. C. et al. Management and outcome of children with skin and soft tissue abscesses caused by community-acquired methicillin-resistant Staphylococcus aureus. Pediatr. Infect. Dis. J. 23, 123–127 (2004).
Lentino, J. R., Narita, M. & Yu, V. L. New antimicrobial agents as therapy for resistant gram-positive cocci. Eur. J. Clin. Microbiol. Infect. Dis. 27, 3–15 (2008).
Li, M. et al. The antimicrobial peptide-sensing system aps of Staphylococcus aureus. Mol. Microbiol. 66, 1136–1147 (2007).
Li, M. et al. Evolution of virulence in epidemic community-associated MRSA. Proc. Natl Acad. Sci. USA 106, 5883–5888 (2009).
Lina, G. et al. Staphylococcal chromosome cassette evolution in Staphylococcus aureus inferred from ccr gene complex sequence typing analysis. Clin. Microbiol. Infect. 12, 1175–1184 (2006). Sequence typing of SCCmec allotypes to define possible origins and evolution.
Lina, G. et al. Involvement of Panton-Valentine leukocidin-producing Staphylococcus aureus in primary skin infections and pneumonia. Clin. Infect. Dis. 29, 1128–1132 (1999). Epidemiological study suggesting that PVL is an important virulence factor in severe pneumonia.
Lindsay, J. A. et al. Microarrays reveal that each of the ten dominant lineages of Staphylococcus aureus has a unique combination of surface-associated and regulatory genes. J. Bacteriol. 188, 669–676 (2006).
Liu, C. et al. A population-based study of the incidence and molecular epidemiology of methicillin-resistant Staphylococcus aureus disease in San Francisco, 2004–2005. Clin. Infect. Dis. 46, 1637–1646 (2008). A population-based study of the USA300 epidemic in San Francisco, a city with a high prevalence of CA-MRSA.
Llera, J. L. & Levy, R. C. Treatment of cutaneous abscess: a double-blind clinical study. Ann. Emerg. Med. 14, 15–19 (1985).
Lodise, T. P., Lomaestro, B., Graves, J. & Drusano, G. L. Larger vancomycin doses (at least four grams per day) are associated with an increased incidence of nephrotoxicity. Antimicrob. Agents Chemother. 52, 1330–1336 (2008).
Loeffler, A. et al. First isolation of MRSA ST398 from UK animals: a new challenge for infection control teams? J. Hosp. Infect. 72, 269–271 (2009).
Lowy, F. D. Staphylococcus aureus infections. N. Engl. J. Med. 339, 520–532 (1998).
Ma, X. X. et al. Novel type of staphylococcal cassette chromosome mec identified in community-acquired methicillin-resistant Staphylococcus aureus strains. Antimicrob. Agents Chemother. 46, 1147–1152 (2002). Genetic structure and organization of SCCmecIV.
Maree, C. L., Daum, R. S., Boyle-Vavra, S., Matayoshi, K. & Miller, L. G. Community-associated methicillin-resistant Staphylococcus aureus isolates causing healthcare-associated infections. Emerg. Infect. Dis. 13, 236–242 (2007).
Mato, R. et al. Clonal types and multidrug resistance patterns of methicillin-resistant Staphylococcus aureus (MRSA) recovered in Italy during the 1990s. Microb. Drug Resist. 10, 106–113 (2004).
McDougal, L. K. et al. Pulsed-field gel electrophoresis typing of oxacillin-resistant Staphylococcus aureus isolates from the United States: establishing a national database. J. Clin. Microbiol. 41, 5113–5120 (2003).
Meyer, F., Girardot, R., Piemont, Y., Prevost, G. & Colin, D. A. Analysis of the specificity of Panton-Valentine leucocidin and gamma-hemolysin F component binding. Infect. Immun. 77, 266–273 (2009).
Miller, L. G. & Diep, B. A. Clinical practice: colonization, fomites, and virulence: rethinking the pathogenesis of community-associated methicillin-resistant Staphylococcus aureus infection. Clin. Infect. Dis. 46, 752–760 (2008).
Montgomery, C. P. & Daum, R. S. Transcription of inflammatory genes in the lung after infection with community-associated methicillin-resistant Staphylococcus aureus: a role for Panton-Valentine leukocidin? Infect. Immun. 77, 2159–2167 (2009).
Montgomery, C. P. et al. Comparison of virulence in community-associated methicillin-resistant Staphylococcus aureus pulsotypes USA300 and USA400 in a rat model of pneumonia. J. Infect. Dis. 198, 561–570 (2008).
Moran, G. J. et al. Methicillin-resistant S. aureus infections among patients in the emergency department. N. Engl. J. Med. 355, 666–674 (2006).
Mulvey, M. R. et al. Community-associated methicillin-resistant Staphylococcus aureus, Canada. Emerg. Infect. Dis. 11, 844–850 (2005).
Muto, C. A. et al. SHEA guideline for preventing nosocomial transmission of multidrug-resistant strains of Staphylococcus aureus and Enterococcus. Infect. Control Hosp. Epidemiol. 24, 362–386 (2003).
Nathwani, D. et al. Guidelines for UK practice for the diagnosis and management of methicillin-resistant Staphylococcus aureus (MRSA) infections presenting in the community. J. Antimicrob. Chemother. 61, 976–994 (2008).
Nimmo, G. R. & Coombs, G. W. Community-associated methicillin-resistant Staphylococcus aureus (MRSA) in Australia. Int. J. Antimicrob. Agents 31, 401–410 (2008).
Nubel, U. et al. Frequent emergence and limited geographic dispersal of methicillin-resistant Staphylococcus aureus. Proc. Natl Acad. Sci. USA 105, 14130–14135 (2008). Evidence that MRSA infections are locally derived as opposed to internationally translocated, and that SCCmec has entered S. aureus strains on numerous occasions.
O’Brien, F. G. et al. Diversity among community isolates of methicillin-resistant Staphylococcus aureus in Australia. J. Clin. Microbiol. 42, 3185–3190 (2004).
Okuma, K. et al. Dissemination of new methicillin-resistant Staphylococcus aureus clones in the community. J. Clin. Microbiol. 40, 4289–4294 (2002).
Oliveira, D. C., Milheirico, C. & de Lencastre, H. Redefining a structural variant of staphylococcal cassette chromosome mec, SCCmec type VI. Antimicrob. Agents Chemother. 50, 3457–3459 (2006).
Oliveira, D. C., Tomasz, A. & de Lencastre, H. The evolution of pandemic clones of methicillin-resistant Staphylococcus aureus: identification of two ancestral genetic backgrounds and the associated mec elements. Microb. Drug Resist. 7, 349–361 (2001).
Pan, E. S. et al. Increasing prevalence of methicillin-resistant Staphylococcus aureus infection in California jails. Clin. Infect. Dis. 37, 1384–1388 (2003). The first description of USA300.
Pan, A., Lorenzotti, S. & Zoncada, A. Registered and investigational drugs for the treatment of methicillinresistant Staphylococcus aureus infection. Recent Pat. Antiinfect. Drug Discov. 3, 10–33 (2008).
Pannaraj, P. S., Hulten, K. G., Gonzalez, B. E., Mason, E. O. Jr & Kaplan, S. L. Infective pyomyositis and myositis in children in the era of community-acquired, methicillin-resistant Staphylococcus aureus infection. Clin. Infect. Dis. 43, 953–960 (2006).
Parish, D. & Scheinfeld, N. Ceftaroline fosamil, a cephalosporin derivative for the potential treatment of MRSA infection. Curr. Opin. Investig. Drugs 9, 201–209 (2008).
Park, S. H. et al. Emergence of community-associated methicillin-resistant Staphylococcus aureus strains as a cause of healthcare-associated bloodstream infections in Korea. Infect. Control Hosp. Epidemiol. 30, 146–155 (2009).
Peacock, J. E. Jr, Marsik, F. J. & Wenzel, R. P. Methicillin-resistant Staphylococcus aureus: introduction and spread within a hospital. Ann. Intern. Med. 93, 526–532 (1980).
Robinson, D. A. et al. Re-emergence of early pandemic Staphylococcus aureus as a community-acquired meticillin-resistant clone. Lancet 365, 1256–1258 (2005).
Robinson, D. A. & Enright, M. C. Evolutionary models of the emergence of methicillin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 47, 3926–3934 (2003).
Rollason, J. et al. Epidemiology of community-acquired meticillin-resistant Staphylococcus aureus obtained from the UK West Midlands region. J. Hosp. Infect. 70, 314–320 (2008).
Rooijakkers, S. H. et al. Early expression of SCIN and CHIPS drives instant immune evasion by Staphylococcus aureus. Cell. Microbiol. 8, 1282–1293 (2006).
Roundtree, P. & Freeman, V. Infections caused by a particular phage type of Staphylococcus aureus. Med. J. Aust. 42, 157–161 (1956).
Roundtree, P. & Beard, M. Further observations on infections with phage type 80 staphylococci in Australia. Med. J. Aust. 2, 789–795 (1958).
Ruppe, E. et al. Diversity of staphylococcal cassette chromosome mec structures in methicillin-resistant Staphylococcus epidermidis and Staphylococcus haemolyticus strains among outpatients from four countries. Antimicrob. Agents Chemother. 53, 442–449 (2009).
Seybold, U. et al. Emergence of community-associated methicillin-resistant Staphylococcus aureus USA300 genotype as a major cause of health care-associated blood stream infections. Clin. Infect. Dis. 42, 647–656 (2006).
Shaw, K. J. et al. In vitro activity of TR-700, the antibacterial moiety of the prodrug TR-701, against linezolid-resistant strains. Antimicrob. Agents Chemother. 52, 4442–4447 (2008).
Shinefield, H. et al. Use of a Staphylococcus aureus conjugate vaccine in patients receiving hemodialysis. N. Engl. J. Med. 346, 491–496 (2002).
Shopsin, B. et al. Evaluation of protein A gene polymorphic region DNA sequencing for typing of Staphylococcus aureus strains. J. Clin. Microbiol. 37, 3556–3563 (1999).
Shorr, A. F., Kunkel, M. J. & Kollef, M. Linezolid versus vancomycin for Staphylococcus aureus bacteraemia: pooled analysis of randomized studies. J. Antimicrobiol. Chemother. 56, 923–929 (2005).
Stam-Bolink, E. M., Mithoe, D., Baas, W. H., Arends, J. P. & Moller, A. V. Spread of a methicillin-resistant Staphylococcus aureus ST80 strain in the community of the northern Netherlands. Eur. J. Clin. Microbiol. Infect. Dis. 26, 723–727 (2007).
Stapleton, P. D., Shah, S., Ehlert, K., Hara, Y. & Taylor, P. W. The β-lactam-resistance modifier (-)-epicatechin gallate alters the architecture of the cell wall of Staphylococcus aureus. Microbiology 153, 2093–2103 (2007).
Steinkraus, G., White, R. & Friedrich, L. Vancomycin MIC creep in non-vancomycin-intermediate Staphylococcus aureus (VISA), vancomycin-susceptible clinical methicillin-resistant S. aureus (MRSA) blood isolates from 2001–2005. J. Antimicrob. Chemother. 60, 788–794 (2007).
Stemper, M. E., Shukla, S. K. & Reed, K. D. Emergence and spread of community-associated methicillin resistant Staphylococcus aureus in rural Wisconsin, 1989 to 1999. J. Clin. Microbiol. 42, 5673–5680 (2004).
Tenover, F. C. et al. Characterization of Staphylococcus aureus isolates from nasal cultures collected from individuals in the United States in 2001 to 2004. J. Clin. Microbiol. 46, 2837–2841 (2008).
Tiemersma, E. Emergence and resurgence of meticillin-resistant Staphylococcus aureus as a public-health threat. Lancet 368, 874–885 (2006).
Tristan, A. et al. Global distribution of Panton-Valentine leukocidin positive methicillin-resistant Staphylococcus aureus, 2006. Emerg. Infect. Dis. 13, 594–600 (2007).
Turner, K. M., Hanage, W. P., Fraser, C., Connor, T. R. & Spratt, B. G. Assessing the reliability of eBURST using simulated populations with known ancestry. BMC Microbiol. 7, 30 (2007).
Udo, E. E., Pearman, J. W. & Grubb, W. B. Genetic analysis of community isolates of methicillin-resistant Staphylococcus aureus in Western Australia. J. Hosp. Infect. 25, 97–108 (1993).
Vandenesch, F. et al. Community-acquired methicillin-resistant Staphylococcus aureus carrying Panton-Valentine leukocidin genes: worldwide emergence. Emerg. Infect. Dis. 9, 978–984 (2003).
van Wamel, W. J., Rooijakkers, S. H., Ruyken, M., van Kessel, K. P. & van Strijp, J. A. The innate immune modulators staphylococcal complement inhibitor and chemotaxis inhibitory protein of Staphylococcus aureus are located on β-hemolysin-converting bacteriophages. J. Bacteriol. 188, 1310–1315 (2006).
Voyich, J. M. et al. Is Panton-Valentine leukocidin the major virulence determinant in community-associated methicillin-resistant Staphylococcus aureus disease? J. Infect. Dis. 194, 1761–1770 (2006).
Voyich, J. M. et al. Insights into mechanisms used by Staphylococcus aureus to avoid destruction by human neutrophils. J. Immunol. 175, 3907–3919 (2005).
Wang, G., Hindler, J. F., Ward, K. W. & Bruckner, D. A. Increased vancomycin MICs for Staphylococcus aureus clinical isolates from a university hospital during a 5-year period. J. Clin. Microbiol. 44, 3883–3886 (2006).
Wang, R. et al. Identification of novel cytolytic peptides as key virulence determinants for community-associated MRSA. Nature Med. 13, 1510–1514 (2007).
Wannet, W. J. et al. Emergence of virulent methicillin-resistant Staphylococcus aureus strains carrying Panton-Valentine leucocidin genes in The Netherlands. J. Clin. Microbiol. 43, 3341–3345 (2005).
Weigel, L. M. et al. Genetic analysis of a high-level vancomycin-resistant isolate of Staphylococcus aureus. Science 302, 1569–1571 (2003).
Weigelt, J. et al. Linezolid versus vancomycin in treatment of complicated skin and soft tissue infections. Antimicrob. Agents Chemother. 49, 2260–2266 (2005).
Woodin, A. M. Purification of the two components of leucocidin from Staphylococcus aureus. Biochem. J. 75, 158–165 (1960).
Woodin, A. M. & Wieneke, A. A. The participation of calcium, adenosine triphosphate and adenosine triphosphatase in the extrusion of the granule proteins from the polymorphonuclear leucocyte. Biochem. J. 90, 498–509 (1964).
Wright, J. Staphylococcal leucocidin (Neisser-Wechsberg type) and antileucocidin. Lancet 227, 1002–1005 (1936).
Wu, S., Piscitelli, C., de Lencastre, H. & Tomasz, A. Tracking the evolutionary origin of the methicillin resistance gene: cloning and sequencing of a homologue of mecA from a methicillin susceptible strain of Staphylococcus sciuri. Microb. Drug Resist. 2, 435–441 (1996).
Wunderink, R. G., Cammarata, S. K., Oliphant, T. H. & Kollef, M. H. Continuation of a randomized, doubleblind, multicenter study of linezolid versus vancomycin in the treatment of patients with nosocomial pneumonia. Clin. Ther. 25, 980–992 (2003).
Zhang, K., McClure, J. A., Elsayed, S. & Conly, J. M. Novel staphylococcal cassette chromosome mec type, tentatively designated type VIII, harboring class A mec and type ccr gene complexes in a Canadian epidemic strain of methicillin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 53, 531–540 (2009).
Acknowledgments
F.R.D. is supported by the Intramural Research Program of the National Institute of Allergy and Infectious Disease (NIAID) and the National Institutes of Health (NIH) and H.F.C. is supported by the NIH, NIAID grant number R01 AI070289.
Supplementary Information
TABLE A2-S1 Virulence Factors of Staphylococcus aureus
|
Target cell, host factor or response |
Gene(s) |
Protein or molecule |
Putative function or effect on immune system |
|
Factors that interfere with bacterial killing |
|||
|
Antimicrobial peptides |
aur |
Zinc metalloproteinase aureolysin, Aur |
Degrades LL-37 |
|
|
dlt operon |
Dlt operon, DltABCD |
Promotes resistance to cationic antimicrobial peptides and group IIA phospholipase A2 |
|
Target cell, host factor or response |
Gene(s) |
Protein or molecule |
Putative function or effect on immune system |
|
|
icaA, icaD, icaB, icaC, icaR |
Polysaccharide intercellular adhesion, PIA |
Resistance to cationic antimicrobial peptides |
|
|
isdA, isdB |
Iron-regulated surface determinants of S. aureus, IsdA and IsdB |
Resistance to antimicrobial peptides, skin fatty acids, and neutrophil reactive oxygen species |
|
|
mprF |
Multiple peptide resistance factor, MprF |
Promotes resistance to cationic antimicrobial peptides |
|
|
sak |
Staphylokinase |
Inhibits host α-defensins |
|
Oxygen-mediating bacterial killing |
ahpC, ahpF |
Alkylhydroperoxide reductase subunits C and F, AhpC and AhpF |
Promotes resistance to ROS |
|
|
crtM, crtN |
Carotenoid pigment, staphyloxanthin (S. aureus golden pigment) |
Promotes resistance to reactive oxygen species |
|
|
isdA, isdB |
Iron-regulated surface determinants of S. aureus, IsdA and IsdB |
Resistance to neutrophil reactive oxygen species |
|
|
sodA, sodM |
Superoxide dismutase, SodA, SodM |
Promotes resistance to reactive oxygen species |
|
Hemolysins and anti-platelet factors |
|||
|
Erythrocytes |
hla, hly |
Alpha-hemolysin (α-hemolysin), Hla |
Causes cell lysis (also affect epithelial cells, fibroblasts, and monocytes) |
|
|
hld |
Delta-hemolysin, Hld |
Causes cell lysis |
|
|
hlgA, hlgB, hlgC |
Gamma-hemolysin subunits A, B, and C; HlgA, HlgB, HlgC; two-component leukocidin |
Causes cell lysis |
|
Platelets |
clfA |
Clumping factor A ClfA |
Causes platelet activation |
|
|
fnbA, fnbB |
Fibronectin-binding proteins A and B, FnbA and FnbB |
Causes platelet activation |
|
|
katA |
Catalase, KatA |
Detoxifies hydrogen peroxide |
|
|
sodA, sodM |
Superoxide dismutase, SodA, SodM |
Promotes resistance to reactive oxygen species |
|
Leucocidins and anti-phagocytic factors |
|||
|
Polymorphonuclear leukocytes |
cap5 or cap8 genes |
Capsular polysaccharide |
Inhibits phagocytosis |
|
|
clfA |
Clumping factor A, ClfA |
Inhibits phagocytosis |
|
Target cell, host factor or response |
Gene(s) |
Protein or molecule |
Putative function or effect on immune system |
|
|
eap |
Extracellular adherence protein, Eap |
Inhibits leukocyte adhesion |
|
|
hlgA, hlgB, hlgC |
Gamma-hemolysin subunits A, B, and C; HlgA, HlgB; HlgC; two-component leukocidin |
Causes cell lysis |
|
|
lukD, lukE |
Leukocidin D and E; LukD and LukE; two-component leukocidins |
Causes leukocyte lysis |
|
|
lukS-PV, lukF-PV |
Leukocidin S-PV and F-PV subunits; two-component leukocidin, PVL |
Causes leukocyte lysis |
|
|
psm |
Phenol-soluble modulin-like peptides, PSMs |
Cause leukocyte lysis |
|
|
sbi |
IgG-binding protein, Sbi |
Sequesters host IgG |
|
|
scn |
Staphylococcal inhibitor of complement, SCIN |
Inhibits complement |
|
|
ssl5 |
Staphylococcal superantigen-like 5, SSL5 |
Binds P-selectin glycoprotein ligand-1 and inhibits neutrophil rolling |
|
Chemotaxis |
chp |
Chemotaxis inhibitory protein of S. aureus, CHIPS |
Inhibits chemotaxis |
|
|
ecb |
Extracellular complement-binding protein, Ecb |
Inhibits C5a generation |
|
|
efb |
Extracellular fibrinogen-binding protein, Efb |
Inhibits C5a generation |
|
|
sbi |
IgG-binding protein, Sbi |
Sequesters host IgG |
|
|
scn |
Staphylococcal inhibitor of complement, SCIN |
Inhibits complement |
|
|
ssl7 |
Staphylococcal superantigen-like 7, SSL7 |
Binds to C5a and IgA |
|
Superantigens |
|||
|
T-cells |
sea, seb, secn, sed, see, seg, she, sei, sej, sek, sel, sep |
Staphylococcal enterotoxins; SEA, SEB, SECn, SED, SEE, SEG, SEH, SEI, SEJ, SEK, SEL, and SEP |
Activate T-cells (superantigen) |
|
|
tst |
Toxic shock syndrome toxin-1, TSST-1 |
Activate T-cells (superantigen) |
A3
SUBLETHAL ANTIBIOTIC TREATMENT LEADS TO MULTIDRUG RESISTANCE VIA RADICAL-INDUCED MUTAGENESIS7
Michael A. Kohanski,8,9,10,11,12 Mark A. DePristo,9,10,11,13 and James J. Collins8,9,10,11,12,14,*
Summary
Antibiotic resistance arises through mechanisms such as selection of naturally occurring resistant mutants and horizontal gene transfer. Recently, oxidative stress has been implicated as one of the mechanisms whereby bactericidal antibiotics kill bacteria. Here, we show that sublethal levels of bactericidal antibiotics induce mutagenesis, resulting in heterogeneous increases in the minimum inhibitory concentration for a range of antibiotics, irrespective of the drug target. This increase in mutagenesis correlates with an increase in ROS and is prevented by the ROS scavenger thiourea and by anaerobic conditions, indicating that sublethal concentrations of antibiotics induce mutagenesis by stimulating the production of ROS. We demonstrate that these effects can lead to mutant strains that are sensitive to the applied antibiotic but resistant to other antibiotics. This work establishes a radical-based molecular mechanism whereby sublethal levels of antibiotics can lead to multidrug resistance, which has important implications for the widespread use and misuse of antibiotics.
Introduction
There are a number of mechanisms whereby bacteria can develop antibiotic resistance (Dwyer et al., 2009; Hegreness et al., 2008; Livermore, 2003; McKenzie and Rosenberg, 2001), including horizontal transfer of resistance genes (Davies, 1994), drug-specific selection of naturally occurring resistant
|
7 |
Reprinted from Molecular Cell 37(3), Kohanski, M. A., M. A. DePristo, and J. J. Collins, Sublethal antibiotic treatment leads to multidrug resistance via radical-induced mutagenesis, pages 311-320, with permission from Elsevier. Copyright (2010). |
|
8 |
Howard Hughes Medical Institute. |
|
9 |
Department of Biomedical Engineering. |
|
10 |
Center for BioDynamics. |
|
11 |
Center for Advanced Biotechnology Boston University, Boston, MA 02215, USA. |
|
12 |
Boston University School of Medicine, Boston, MA 02118, USA. |
|
13 |
Department of Organismic and Evolutionary Biology, Harvard University, Cambridge, MA 02138, USA. |
|
14 |
Wyss Institute for Biologically Inspired Engineering, Harvard University, Boston, MA 02115, USA. |
|
* |
Correspondence: jcollins@bu.edu. |
variants within a population, and increased mutagenesis in hypermutator strains (Andersson, 2003; Chopra et al., 2003). Quinolones, which are DNA-damaging antibiotics, can stimulate the emergence of drug resistance via SOS-independent recombination (López et al., 2007) and through the induction of RecA-mediated processes, including homologous recombination (Drlica and Zhao, 1997) and SOS-regulated error-prone polymerases (Cirz et al., 2005). β-lactams can also induce the SOS response via RecA (Kohanski et al., 2007) and the DpiAB two-component system (Miller et al., 2004), and these drugs have been shown to induce DinB in an SOS-independent fashion, resulting in increased frameshift mutations (Pérez-Capilla et al., 2005).
Antibiotic treatment can also result in multidrug resistance (Cohen et al., 1989), which has been associated with mutations in multidrug efflux pumps, such as AcrAB (Ma et al., 1993). These drug efflux pumps can be regulated by a number of transcription factors, including the superoxide-responsive SoxRS system (Greenberg et al., 1990). In addition, there is evidence that low level antibiotic treatment can lead to mutations that cause resistance (Girgis et al., 2009); however, the mechanisms underlying this effect are not well understood.
Bactericidal antibiotics, including β-lactams, quinolones, and aminoglycosides, can stimulate bacteria to produce reactive oxygen species (ROS) (Dwyer et al., 2007; Kohanski et al., 2007, 2008), which are highly deleterious molecules that can interfere with the normal functions of oxygen-respiring organisms (Brumaghim et al., 2003; Fridovich, 1978; Imlay, 2006). Certain ROS, such as hydroxyl radicals, can directly damage DNA and lead to an accumulation of mutations (Demple and Harrison, 1994; Friedberg et al., 2006; Imlay et al., 1988). Oxidative DNA damage also activates the error-prone SOS response (Carlsson and Carpenter, 1980; Imlay and Linn, 1986, 1987) and error-correcting repair systems such as the “GO” repair system (Michaels and Miller, 1992; Miller, 1996). In this study, we hypothesized that ROS formation due to treatment with low levels of bactericidal antibiotics leads to an increase in mutation rates, which can result in the emergence of multidrug resistance. We thus consider a possible molecular mechanism whereby bactericidal antibiotics act as active, reactive mutagens.
Results
To test the above hypothesis, we examined mutation rates in E. coli strain MG1655 following treatment with low levels of the bactericidal antibiotics norfloxacin (quinolone), ampicillin (β-lactam), and kanamycin (aminoglycoside), respectively. Mutation rates were determined by plating aliquots of treated cultures onto rifampicin plates, counting rifampicin-resistant colonies, and using the MSS maximum likelihood method (Rosche and Foster, 2000) to estimate the number of mutation events per culture (see Experimental Procedures for additional details). The mutation rate for untreated wild-type E. coli was approximately 1.5 × 10−8 mutations/cell/generation.
Treatment with 1 μg/ml ampicillin, 3 μg/ml kanamycin, 15 ng/ml norfloxacin, or 50 ng/ml norfloxacin resulted in significant increases in the mutation rate relative to an untreated control (Figure A3-1). Treatment with 1 μg/ml kanamycin resulted in a modest increase in mutation rate (Figure A3-1). The largest increases in mutation rate were seen following treatment with ampicillin or 50 ng/ml norfloxacin (Figure A3-1A). These changes were on par with the increase in mutation rate observed following treatment with 1mM hydrogen peroxide (Figure A3-1A), a concentration of hydrogen peroxide known to induce hydroxyl radical formation via Fenton chemistry (Imlay et al., 1988). To determine if there is a correlation between these changes in mutation rate and ROS formation, we examined radical levels using the radical-sensitive dye 3′-(p-hydroxyphenyl) fluorescein (HPF) (Setsukinai et al., 2003) (see Experimental Procedures for more details). We found a significant correlation (R2 = 0.8455) between the fold change in mutation rate and peak HPF signal for the treatments described above (Figure A3-1B).
The strong correlation between ROS formation and fold change in mutation rate following treatment with bactericidal antibiotics suggests that ROS actively contribute to bactericidal drug-induced mutagenesis. To test if this is indeed the case, we added 100 mM thiourea to wild-type E. coli treated with antibiotics or hydrogen peroxide at the concentrations noted above (Figure A3-1C). Thiourea is a potent hydroxyl radical scavenger that mitigates the effects of hydroxyl radical damage in both prokaryotes and eukaryotes (Novogrodsky et al., 1982; Repine et al., 1981; Touati et al., 1995). We have previously shown that thiourea reduces hydroxyl radical formation and cell killing following treatment with bactericidal antibiotics (Kohanski et al., 2007).
The addition of thiourea significantly reduced the mutation rate to near untreated levels following the addition of 1 mM hydrogen peroxide, norfloxacin, or ampicillin (Figure A3-1). Interestingly, we were unable to detect any rifampicin-resistant colonies after plating up to 109 cells following treatment with both 3 μg/ml kanamycin and thiourea (Figure A3-1C). However, we were able to detect rifampicin-resistant colonies after scaling up the system to 1 L flasks and plating up to 1010 cells following treatment with both 3 μg/ml kanamycin and thiourea (data not shown). These results suggest a role for kanamycin-mediated interference with ribosome function and translation, in the absence of oxidative stress, on significantly lowering mutation rate.
To further demonstrate that antibiotic-mediated ROS formation has a mutagenic component, we examined mutation rates under anaerobic growth conditions (see Experimental Procedures for additional details) following treatment of wild-type E. coli with antibiotics or hydrogen peroxide as described above (Figure A3-1D). We observed mutation rates near untreated levels for all antibiotic treatments tested (Figure A3-1D). Treatment with 1 mM hydrogen peroxide, which results in direct addition of an oxidant, led to an increase in mutation rate relative to the no drug control under anaerobic growth conditions (Figure A3-1D), but
![FIGURE A3-1 Low levels of bactericidal antibiotics increase mutation rate due to reactive oxygen species formation. (A) Fold change in mutation rate (mean ±95% confidence interval [CI]) relative to an untreated control (no drug) for wild-type E. coli (MG1655) following an overnight treatment with 1 μg/ml ampicillin, 1 μg/ml kanamycin, 3 μg/ml kanamycin, 15 ng/ml norfloxacin, 50 ng/ml norfloxacin, or 1 mM hydrogen peroxide (H2O2). (B) Correlation between oxidative stress levels (HPF fluorescence) and fold change in mutation rate for wild-type E. coli for the treatments described in (A). (C and D) Fold change in mutation rate (mean ±95% CI) relative to an untreated control (no drug) for wild-type E. coli following an overnight treatment with 100 mM thiourea and no drug, 1 μg/ml ampicillin, 1 μg/ml kanamycin, 3 μg/ml kanamycin, 15 ng/ml norfloxacin, 50 ng/ml norfloxacin, or 1 mM hydrogen peroxide (H2O2) under aerobic growth conditions with 100 mM thiourea (C) or anaerobic growth conditions (D). See also Figure A3-S2.](/openbook/12925/xhtml/images/p2001c3c7g119001.jpg)
FIGURE A3-1 Low levels of bactericidal antibiotics increase mutation rate due to reactive oxygen species formation. (A) Fold change in mutation rate (mean ±95% confidence interval [CI]) relative to an untreated control (no drug) for wild-type E. coli (MG1655) following an overnight treatment with 1 μg/ml ampicillin, 1 μg/ml kanamycin, 3 μg/ml kanamycin, 15 ng/ml norfloxacin, 50 ng/ml norfloxacin, or 1 mM hydrogen peroxide (H2O2). (B) Correlation between oxidative stress levels (HPF fluorescence) and fold change in mutation rate for wild-type E. coli for the treatments described in (A). (C and D) Fold change in mutation rate (mean ±95% CI) relative to an untreated control (no drug) for wild-type E. coli following an overnight treatment with 100 mM thiourea and no drug, 1 μg/ml ampicillin, 1 μg/ml kanamycin, 3 μg/ml kanamycin, 15 ng/ml norfloxacin, 50 ng/ml norfloxacin, or 1 mM hydrogen peroxide (H2O2) under aerobic growth conditions with 100 mM thiourea (C) or anaerobic growth conditions (D). See also Figure A3-S2.
this increase was considerably smaller than that exhibited under aerobic growth conditions (Figure A3-1A).
Antibiotic-resistant strains can arise via drug-mediated selection of preexisting antibiotic-resistant variants that occur naturally within a population (Livermore, 2003). Antibiotic-induced oxidative stress may be an additional mechanism that allows for the accumulation of mutations that increase resistance to drugs, irrespective of the drug target of the applied antibiotic. To test this, we measured changes in the minimum inhibitory concentration (MIC) of wild-type E. coli over a period of 5 days of selective growth for the following antibiotics: norfloxacin, kanamycin, ampicillin, tetracycline, and chloramphenicol. During the growth period, the cultures were exposed to no drug, norfloxacin, ampicillin, or kanamycin (see Experimental Procedures for more details). In all cases, growth in the absence of antibiotics did not change the MIC for any of the drugs tested (data not shown).
Treatment with 25 ng/ml norfloxacin led to an increase in the MIC for norfloxacin and kanamycin (Figure A3-S1A). The observed increases in MIC following treatment with norfloxacin were concentration dependent (see Supplemental Information for more details). Treatment of wild-type E. coli with 3 μg/ml kanamycin led to an increase in the MIC for kanamycin and minimal increases in the MIC for norfloxacin and ampicillin, respectively (Figure A3-S1C). The MIC for tetracycline and chloramphenicol did not change (Figure A3-S1C), indicating that kanamycin treatment may not lead to mutants resistant to other classes of ribosome inhibitors.
Treatment of wild-type E. coli with 1 μg/ml ampicillin for 5 days led to an increase in the MIC to different levels for ampicillin, norfloxacin, kanamycin, tetracycline, and chloramphenicol (Figure A3-2A). These results show that treatment with a β-lactam can stimulate formation of mutants that are potentially resistant to a wide range of antibiotics. Cultures that had been grown for 5 days in the presence of low levels of ampicillin were shifted to a drug-free environment and grown without any ampicillin for 2 additional days. The MICs, which were increased after 5 days of ampicillin treatment (Figure A3-2A), remained elevated and did not change significantly following 2 days of growth in the absence of ampicillin (Figure A3-S1D). These findings demonstrate that the observed increases in MIC are stable and not due to a transient adaptation to growth in the presence of ampicillin.
To determine if the observed increases in MIC were related to antibiotic-mediated ROS formation, we measured the MIC for ampicillin, norfloxacin, kanamycin, tetracycline, and chloramphenicol, respectively, following treatment with no drug or 1 μg/ml ampicillin under anaerobic growth conditions. Untreated anaerobic growth had no effect on MIC relative to untreated aerobic growth (data not shown). Following treatment with 1 μg/ml ampicillin under anaerobic conditions, we observed almost no increase in MIC for ampicillin, kanamycin, tetracycline, or chloramphenicol (Figure A3-2B). The MIC for norfloxacin exhib-
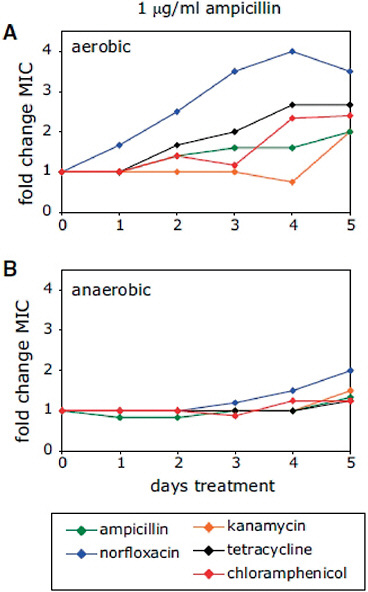
FIGURE A3-2 Low levels of bactericidal antibiotics can lead to broad-spectrum increases in MIC due to ROS-mediated mutagenesis. (A and B) Fold change in MIC relative to an aerobic no-drug control for ampicillin, norfloxacin, kanamycin, tetracycline, and chloramphenicol, following 5 days of growth in the presence of 1 μg/ml ampicillin under aerobic (A) or anaerobic (B) growth conditions. See also Figure A3-S1.
ited a small increase by day 5 (Figure A3-2B); however, this change in MIC was much smaller than the increase in MIC for norfloxacin following ampicillin treatment under aerobic growth conditions (Figure A3-2A). These results suggest that ROS formation due to treatment with low levels of bactericidal antibiotics can lead to mutagenesis and the emergence of bacteria resistant to a wide range of antibiotics.
Drug resistance may not always be uniform throughout a population. Some cells within a population may remain susceptible to the antibiotic, whereas other cells display varying degrees of drug resistance (de Lencastre et al., 1993), a phenomenon referred to as heteroresistance. Antibiotic-stimulated, ROS mediated mutagenesis could be a mechanism that stimulates the formation of a range of mutations that result in varying MICs within a population of cells. We sought to determine if the observed increases in population-level MIC for ampicillin following 5 days of treatment with 1 μg/ml ampicillin (Figure A3-2A) exhibited heterogeneity in MIC at the single-colony level.
We isolated individual colonies following ampicillin treatment and measured the MIC of each clone to ampicillin. We found that these isolates exhibited a range of resistance to ampicillin (>2.5–12.5 μg/ml), with some isolates remaining completely susceptible (≤2.5 μg/ml) to treatment with this drug (Figure A3-3A). We also found that the MICs for these isolates to norfloxacin ranged from <100 ng/ml (completely susceptible) to ≥1000 ng/ml (Figure A3-3B). Although levels of resistance from clinical isolates are typically quite high (with MICs in the range of 10–1000 μg/ml for norfloxacin [Becnel Boyd et al., 2009]), the upper ranges of the MICs for ampicillin or norfloxacin observed here (Figure A3-3) are near the peak serum concentrations for these drugs (Bryskier, 2005), indicating that these MICs might be near the limit for the amount of drug a human can tolerate. These data show that heterogeneous increases in MIC to ampicillin arise in E. coli following treatment with low levels of ampicillin, and treatment with one drug class can lead to heterogeneous increases in MIC against other classes of antibiotics.
Resistance to multiple antibiotics has been linked to mutations in drug-efflux systems such as the AcrAB multidrug (MDR) efflux pump (George and Levy, 1983; Ma et al., 1993) as well as mutations in transcription factors controlling these systems, such as MarA (Alekshun and Levy, 1997), Rob (Ariza et al., 1995), and SoxS (Greenberg et al., 1990). Our results suggest that ROS-mediated DNA damage induced by low levels of bactericidal antibiotics can result in mutations in a wider range of genes, potentially in some unrelated to the applied antibiotic and drug efflux systems. This implies that treatment with ampicillin, for example, may generate mutants that are not ampicillin resistant but are resistant to other antibiotics.
To determine if these types of resistant strains arise, we examined multidrug resistance following 5 days of treatment with 1 μg/ml ampicillin or no treatment. Mutants from ampicillin treated or untreated cultures were selected on plates
FIGURE A3-3 Ampicillin treatment of E. coli results in heterogeneous increases in MIC for ampicillin and norfloxacin. (A and B) Shown are the distributions of ampicillin (A) or norfloxacin (B) MICs for 44 ampicillin-treated isolates. The maximum growth-inhibitory concentration tested for norfloxacin was 1000 ng/ml, and the MICs for these isolates may be ≥1000 ng/ml.
containing norfloxacin, ampicillin, kanamycin, tetracycline, and chloramphenicol, respectively. From this primary selection, we determined cross-resistance to the other four antibiotics via replica plating (see Experimental Procedures for additional details). We found substantially more primary resistant colonies and higher rates of cross-resistance following ampicillin treatment as compared to no treatment (Table A3-1). Ampicillin-selected mutants displayed a range of cross-resistance to the other classes of antibiotics and showed a strong correlation (89% cross-resistance) with norfloxacin resistance (Table A3-1). We also found that ampicillin-treated cells selected originally on the basis of norfloxacin or kanamycin resistance were only 75% and 63% cross-resistant to ampicillin, respectively (Table A3-1). Interestingly, primary resistance selection with the static drugs tetracycline and chloramphenicol yielded isolates that were always
TABLE A3-1 Cross-Resistance Following Ampicillin Treatment and Primary Resistance Selection with Five Different Classes of Antibiotics
|
E. coli Control Strain |
Percent Cross-Resistant Following Ampicillin Treatment |
||||
|
|
Norfloxacin |
Ampicillin |
Kanamycin |
Tetracycline |
Chloramphenicol |
|
Primary Selection |
|
|
|
|
|
|
Norfloxacin |
100% (40/40) |
75% (30/40) |
25% (10/40) |
23% (9/40) |
23% (9/40) |
|
Ampicillin |
89% (77/87) |
100% (87/87) |
20% (17/87) |
54% (47/87) |
21% (18/87) |
|
Kanamycin |
20% (17/83) |
63% (52/83) |
100% (83/83) |
7% (6/83) |
0% (0/83) |
|
Tetracycline |
79% (63/80) |
100% (80/80) |
14% (11/80) |
100% (80/80) |
78% (62/80) |
|
Chloramphenicol |
87% (67/77) |
100% (77/77) |
35% (27/77) |
100% (77/77) |
100% (77/77) |
|
|
Percent Cross-Resistant Following No-Drug Treatment |
||||
|
|
Norfloxacin |
Ampicillin |
Kanamycin |
Tetracycline |
Chloramphenicol |
|
Primary Selection |
|
|
|
|
|
|
Norfloxacin |
100% (10/10) |
0% (0/10) |
0% (0/10) |
0% (0/10) |
0% (0/10) |
|
Ampicillin |
0% (0/10) |
100% (10/10) |
0% (0/10) |
0% (0/10) |
0% (0/10) |
|
Kanamycin |
0% (0/15) |
0% (0/15) |
100% (15/15) |
0% (0/15) |
0% (0/15) |
|
Tetracycline |
100% (1/1) |
0% (0/1) |
0% (0/1) |
100% (1/1) |
0% (0/1) |
|
Chloramphenicol |
100% (1/1) |
0% (0/1) |
0% (0/1) |
0% (0/1) |
100% (1/1) |
|
Wild-type E. coli were treated with 1 mg/ml ampicillin or no drug for 5 days. These ampicillin-treated or untreated cells were spread on plates containing norfloxacin, ampicillin, kanamycin, tetracycline, or chloramphenicol, and mutants resistant to the individual drugs were isolated. Resistance to the other four classes of antibiotics was determined by replica plating of the primary-selected strains onto plates containing the respective antibiotic. Shown is percent resistance (resistant colonies/total primary resistant colonies). Note: Double the volume of no-drug control cells were plated for primary resistance selection for E. coli as compared to the ampicillin-treated cells. |
|||||
(100%) cross-resistant to ampicillin (Table A3-1); this effect deserves further study. Also of note, ampicillin-treated, kanamycin-resistant strains were found to have very low cross-resistance to tetracycline (7%) and no cross-resistance with chloramphenicol (Table A3-1). This is consistent with previous work showing a lack of cross-resistance to tetracycline or chloramphenicol following selective treatment with aminoglycosides (Grassi, 1979). While the majority of these multidrug cross-resistant strains exhibit resistance against the treatment drug, ampicillin, our results demonstrate that treatment with ampicillin can also generate mutants that are not resistant to ampicillin yet are resistant to other classes of antibiotics.
We sought to determine if some of the ampicillin-treated, cross-resistant isolates had acquired mutations in specific antibiotic targets or in genes making up the common oxidative damage cell death pathway induced by bactericidal antibiotics (Kohanski et al., 2007, 2008), or if the observed cross-resistance (Table A3-1) was solely a function of altered drug efflux. We examined six norfloxacin-resistant isolates, six kanamycin-resistant isolates, and the untreated control strain. We sequenced the following genes where mutations could potentially lead to an increase in drug resistance: gyrA, gyrB, rpsL, ampC, icdA, arcA, cpxA, sdhB, iscR, tolC, marRA and its promoter region, and acrA and its promoter region. gyrA and gyrB code for the subunits of DNA Gyrase; the known target of quinolones, rpsL, encodes a component of the 30S subunit of the ribosome and has been associated with aminoglycoside resistance; ampC has been associated with ampicillin resistance; icdA, arcA, cpxA, sdhB, and iscR are genes involved in the common mechanism of cell death; and tolC, marRA and its promoter region, and acrA and its promoter region are involved in multidrug efflux.
We found that 3 of the 6 norfloxacin-resistant isolates contained point mutations in gyrA that resulted in a substitution of glycine for aspartic acid at amino acid 82 in one isolate and a substitution of tyrosine for aspartic acid at amino acid 87 in two other isolates (Figure A3-4A). We also found that 1 of these 6 norfloxacin-resistant isolates, which did not have a mutation in gyrA, had a point mutation resulting in the conversion of serine to phenylalanine at residue 464 of GyrB (Figure A3-4B). Interestingly, the point mutations we found in gyrA and gyrB are all in the quinolone resistance-determining regions of GyrA and GyrB, respectively, and these mutations have been observed in clinical isolates of Bacteroides fragilis (Oh et al., 2001), Salmonella enterica (Weill et al., 2006), and Pseudomonas aeruginosa (Mouneimné et al., 1999).
As noted above, mutations in rpsL have been associated with aminoglycoside resistance. We found that 2 of the 6 kanamycin-resistant isolates had point mutations in rpsL. These mutations led to a frameshift and truncated form of RpsL in both isolates (Figure A3-4D). It is possible that these mutations contribute to kanamycin resistance in these isolates.
Among the ampicillin-treated, drug-resistant mutants, we did not find any mutations in ampC (data not shown), a gene associated with ampicillin resistance.
FIGURE A3-4 Ampicillin treatment leads to the formation of norfloxacin-resistant isolates with mutations in gyrA, gyrB, or the acrAB promoter (PacrAB) and kanamycin-resistant isolates with mutations in rpsL or arcA. (A and B) Isolates with point mutation resulting in a D82G or D87Y substitution in GyrA (A) or a S464F substitution in GyrB (B). (C) T-to-A DNA base pair mutation in the AcrR/EnvR binding site of the -35 region of PacrAB. PacrAB is partially annotated to show the -10 and -35 regions (bold), the transcription start site (capitalized A), and the AcrR/EnvR binding site (underlined). (D) Isolates with insertion between base pair 92 and 93 (K58) and between base pair 78 and 79 (K62) resulting in truncation of RpsL. (E) Isolate with a single base pair insertion between base pair 211 and 212 resulting in a truncated ArcA protein missing the majority of the helix-turn-helix (HTH) DNA binding domain. See also Table A3-S1.
We also did not find any mutations in icdA, cpxA, sdhB, or iscR (data not shown). Interestingly, we did find a single insertion mutation in arcA in one of the drug-resistant isolates. ArcA is a two-component system transcription factor containing a sensor domain and a DNA-binding domain, and the mutation we found results in a truncated ArcA protein that is missing the DNA-binding element of the protein (Figure A3-4E). We have previously shown that two-component systems are important elements in the common mechanism of cell death, and a knockout of arcA is more tolerant to treatment with ampicillin and kanamycin compared to norfloxacin (Kohanski et al., 2008). This isolate is resistant to ampicillin and kanamycin, but not to norfloxacin. This result suggests that mutations leading to low-level antibiotic resistance can occur in genes that are involved in the common mechanism of cell death.
We did not find any mutations in tolC, marRA, the marRA promoter, or acrA (data not shown); however, we did find a T-to-A conversion in the promoter upstream of acrA (Figure A3-4C) in one of the norfloxacin-resistant isolates that also had a mutation in gyrA (Figure A3-4A). This promoter mutation occurs within the annotated -35 site of the promoter and the binding site for the repressor transcription factors AcrR and EnvR (Keseler et al., 2005; Miller et al., 2002). The observed mutations could reduce the ability of these repressors to bind to the acrAB promoter which would result in increased pump expression and drug resistance. These sequencing results demonstrate that ampicillin treatment can lead to the formation of norfloxacin-resistant strains with mutations in DNA Gyrase and/or mutations that can affect drug efflux pump activity, which likely contribute to the emergence of multidrug resistance.
To demonstrate that sublethal levels of bactericidal antibiotics can lead to an increase in multidrug cross-resistance in Gram-positive as well as Gram-negative bacteria, we also examined multidrug cross-resistance in Staphylococcus aureus following treatment with low levels of ampicillin (35 ng/ml) for 5 days. Previously, we demonstrated antibiotic-mediated ROS formation in S. aureus (Kohanski et al., 2007). In the present study, we found substantially more primary resistant S. aureus colonies and higher rates of cross-resistance following ampicillin treatment as compared to no treatment (Table A3-2). Interestingly, we were unable to enrich for tetracycline- or chloramphenicol-resistant S. aureus isolates following treatment with low-level ampicillin as compared with the no-drug treatment. This may be due to the lower level of ROS formation we have observed with S. aureus (Kohanski et al., 2007).
To demonstrate that these effects are not limited to lab strains, we considered a clinical isolate of E. coli from a patient with diarrhea (NCDC C771). We examined multidrug cross-resistance in the clinical isolate following treatment with 1 μg/ml ampicillin (see Experimental Procedures for more details). As with the wild-type strains, we found substantially more primary resistant colonies and higher rates of cross-resistance in the clinical isolates following ampicillin treatment as compared to no treatment. We also found that ampicillin-treated cells
TABLE A3-2 Cross-Resistance for S. aureus Following Ampicillin Treatment and Primary Resistance Selection with Five Different Classes of Antibiotics
|
S. aureus |
Percent Cross-Resistant Following Ampicillin Treatment |
||||
|
|
Norfloxacin |
Ampicillin |
Kanamycin |
Tetracycline |
Chloramphenicol |
|
Primary Selection |
|
|
|
|
|
|
Norfloxacin |
100% (59/59) |
64% (38/59) |
56% (33/59) |
36% (21/59) |
19% (11/59) |
|
Ampicillin |
41% (29/71) |
100% (71/71) |
18% (13/71) |
25% (13/71) |
14% (10/71) |
|
Kanamycin |
13% (9/68) |
60% (41/68) |
100% (68/68) |
18% (12/68) |
15% (10/68) |
|
Tetracycline |
0% (0/2) |
100% (2/2) |
0% (0/2) |
100% (2/2) |
0% (0/2) |
|
Chloramphenicol |
0/0 |
0/0 |
0/0 |
0/0 |
0/0 |
|
|
Percent Cross-Resistant Following No-Drug Treatment |
||||
|
|
Norfloxacin |
Ampicillin |
Kanamycin |
Tetracycline |
Chloramphenicol |
|
Primary Selection |
|
|
|
|
|
|
Norfloxacin |
100% (19/19) |
5% (1/19) |
26% (5/19) |
0% (0/19) |
5% (1/19) |
|
Ampicillin |
0% (0/13) |
100% (13/13) |
0% (0/13) |
8% (1/13) |
0% (0/13) |
|
Kanamycin |
2.6% (1/38) |
2.6% (1/38) |
100% (38/38) |
0% (0/38) |
8% (3/38) |
|
Tetracycline |
0/0 |
0/0 |
0/0 |
0/0 |
0/0 |
|
Chloramphenicol |
0/0 |
0/0 |
0/0 |
0/0 |
0/0 |
|
Wild-type S. aureus were treated with 35 ng/ml ampicillin or no drug for 5 days. These ampicillin-treated or untreated cells were spread on plates containing norfloxacin, ampicillin, kanamycin, tetracycline, or chloramphenicol, and mutants resistant to the individual drugs were isolated. Resistance to the other four classes of antibiotics was determined by replica plating of the primary selected strains onto plates containing the respective antibiotic. Shown is percent resistance (resistant colonies/total primary resistant colonies). |
|||||
selected originally on the basis of norfloxacin or kanamycin resistance were only 11.5% and 21.5% cross-resistant to ampicillin, respectively (Table A3-3). This further affirms that treatment with ampicillin can generate mutants that are not resistant to ampicillin yet are resistant to other classes of antibiotics.
Discussion
Here, we establish a radical-based molecular mechanism whereby sublethal levels of antibiotics can lead to multidrug resistance. This occurs via bactericidal antibiotic-mediated radical formation that results in the formation of mutations, some of which confer antibiotic resistance. Low-level resistance likely provides a first step toward clinically significant resistance (Goldstein, 2007), and the mechanism we propose and validate here establishes an antibiotic-stimulated mutagenic effect that likely works in conjunction with SOS-induced mutagenesis in the emergence of mutations that confer drug resistance.
TABLE A3-3 Cross-Resistance for E. coli Clinical Isolate NCDC C771 Following Ampicillin Treatment and Primary Resistance Selection with Four Different Classes of Antibiotics
|
E. coli Clinical Isolate |
Percent Cross-Resistant Following Ampicillin Treatment |
|||
|
|
Norfloxacin |
Ampicillin |
Kanamycin |
Chloramphenicol |
|
Primary Selection |
|
|
|
|
|
Norfloxacin |
100% (78/78) |
11.5% (9/78) |
1.3% (1/78) |
10.3% (8/78) |
|
Ampicillin |
13.2% (5/38) |
100% (38/38) |
2.6% (1/38) |
23.9% (9/38) |
|
Kanamycin |
15.2% (12/79) |
21.5% (17/79) |
100% (79/79) |
7.6% (6/79) |
|
Chloramphenicol |
41.4% (29/70) |
45.7% (32/70) |
22.9% (16/70) |
100% (70/70) |
|
|
Percent Cross-Resistant Following No-Drug Treatment |
|||
|
|
Norfloxacin |
Ampicillin |
Kanamycin |
Chloramphenicol |
|
Primary Selection |
|
|
|
|
|
Norfloxacin |
0/0 |
0/0 |
0/0 |
0/0 |
|
Ampicillin |
0/0 |
0/0 |
0/0 |
0/0 |
|
Kanamycin |
2.8% (1/36) |
11.1% (4/36) |
100% (36/36) |
2.8% (1/36) |
|
Chloramphenicol |
0/3 |
0/3 |
0/3 |
100% (3/3) |
|
E. coli strain NCDC C771 was treated with 1 μg/ml ampicillin or no drug for 5 days. These ampicillin-treated or untreated cells were spread on plates containing norfloxacin, ampicillin, kanamycin, or chloramphenicol, and mutants resistant to the individual drugs were isolated. Resistance to the other three classes of antibiotics was determined by replica plating of the primary-selected strains onto plates containing the respective antibiotic. Shown is percent resistance (resistant colonies/total primary resistant colonies). Tetracycline cross-resistance was not quantified for NCDC C771, as this strain is resistant to tetracycline (MIC > 35 μg/ml). |
||||
Clinical situations where bacteria are exposed to low levels of antibiotics can occur with incomplete treatment of an infection, noncompliance with antibiotic treatment (e.g., a missed pill), and reduced or limited drug accessibility to certain tissues (e.g., bone or cerebrospinal fluid [Bryskier, 2005]). It is possible that mutations arising via antibiotic-mediated oxidative stress could be maintained in the normal bacterial flora of the body and transferred to virulent bacteria via horizontal gene transfer, a mechanism that can be induced by DNA damage (Beaber et al., 2004). Novel therapeutics targeting ROS-forming systems or error-prone DNA damage repair systems may help reduce and contain the spread of new antibiotic-resistant bacteria.
Experimental Procedures
Strains, Media, and Antibiotics
All experiments were performed with wild-type E. coli strain MG1655 (ATCC 700926) in Luria-Bertani (LB) medium (Fisher Scientific; Waltham, MA). For all treatment conditions, we used 1 mM hydrogen peroxide (VWR;
West Chester, PA) and the following bactericidal antibiotics: norfloxacin (Sigma; St. Louis), ampicillin, and kanamycin (Fisher Scientific). Bactericidal antibiotics were used at concentrations of 15 ng/ml norfloxacin, 50 ng/ml norfloxacin, 1 μg/ml ampicillin, 1 μg/ml kanamycin, or 3 μg/ml kanamycin. Tetracycline (MP Biomedical; Solon, OH) and chloramphenicol (Fluka; St. Louis) were used for MIC assays, rifampicin (Sigma) for determination of antibiotic resistant rates, and thiourea (Fluka) for radical-quenching experiments. Anaerobic media was made by heating LB in 17 ml Bellco glass hungate tubes (FisherScientific) under anaerobic conditions in a Coy anaerobic chamber (Coy Laboratory Products Inc.; Grass Lake, MI) to drive out dissolved oxygen (Norris and Ribbons, 1969). Resazurin (10 mM) (Sigma), which turns clear in the absence of oxygen, was used as an indicator for anaerobic conditions. Multidrug resistance was also determined in wild-type S. aureus (ATCC 25923) and the E. coli clinical isolate NCDC C771 (ATCC 23985).
Determination of Mutation Rate
Mutation rates were examined following 24 hr of growth in the presence of a bactericidal antibiotic. Drug levels were chosen such that there was an observable effect on growth or survival within the first 6 hr after drug addition (Figure A3-S2), followed by “recovery” of the culture to near untreated colony density 24 hr after treatment. All treatment conditions exhibited recovery to near untreated colony density levels, with the exception of 50 ng/ml norfloxacin. This allowed us to compare mutation frequencies for cultures of similar densities following treatment with an antibiotic.
Mutation rates were determined using a rifampicin-based selection method (Giraud et al., 2001). Briefly, an overnight culture of E. coli was diluted 1:10,000 into 50 ml LB in a 250 ml baffled flask and grown for 3.5 hr at 37°C and 300 rpm. Cultures were grown at high shaking speeds and in baffled flasks to maximize aeration and ROS formation. The culture was diluted 1:3 into fresh LB containing no drug, an antibiotic, or hydrogen peroxide at the concentrations described above. For experiments with thiourea, thiourea in solid form was added to each diluted culture for a final concentration of 100 mM. Aliquots (1 ml; ten replicates) of these diluted cultures were grown in 14 ml tubes for 24 hr at 37°C and 300 rpm. Aliquots of each treatment were serially diluted and plated on LB-agar plates for colony forming unit per milliliter (cfu/ml) determination. Aliquots of each treatment were also plated on LB-agar plates containing 100 μg/ml rifampicin and grown for 48 hr at 37°C. Colonies were counted at 24 and 48 hr, and the colony count from the 48 hr time point was used to estimate mutation rates. For experiments in anaerobic conditions, cells were diluted 1:10,000 into 15 ml anaerobic LB in sealed hungate tubes to minimize exposure to oxygen. Antibiotic treatments, growth temperature, shaking speed, and sample collection were as described above for the aerobic growth conditions.
The colony counts from the ten replicates were then used in the MSS maximum-likelihood method (Rosche and Foster, 2000; Sarkar et al., 1992) to estimate the number of mutational events per culture. The MSS maximum likelihood method is a recursive algorithm based on the Lea-Coulson function for solving the Luria-Delbruck distribution for a given number of mutational events (Sarkar et al., 1992); its utility has been demonstrated in vitro (Rosche and Foster, 2000). The mutation rate was determined by dividing the number of mutational events per culture by the total number of bacteria plated on the rifampicin plates (Rosche and Foster, 2000). Fold change in mutation rate was determined for all treatments and conditions relative to an untreated MG1655 control. Three biological replicates were run for each treatment condition, and the averages are shown in Figure A3-1.
ROS Detection Using HPF
To detect ROS formation, we used the fluorescent reporter dye HPF (Invitrogen; Carlsbad, CA) and flow cytometry as previously described (Kohanski et al., 2007). Average fluorescence was determined at 0 (baseline), 1, 3, and 6 hr (normalized to a no-dye control) following antibiotic treatment at the concentrations described above, and peak fluorescence levels were used to determine the change in mean fluorescence relative to baseline (Figure A3-1B).
Determination of MIC
For wild-type E. coli, MICs for norfloxacin, ampicillin, kanamycin, tetracycline, and chloramphenicol were measured over 5 days of treatment with no drug, 25 ng/ml norfloxacin, 50 ng/ml norfloxacin, 1 μg/ml ampicillin, or 3 μg/ml kanamycin. Briefly, an overnight culture of E. coli was diluted 1:10,000 into 50 ml LB in a 250 ml baffled flask and grown for 3.5 hr at 37°C and 300 rpm. The culture was diluted 1:3 into fresh LB containing no drug or antibiotics at the above concentrations. Aliquots (1 ml) of these diluted cultures were grown in 14 ml tubes for 24 hr at 37°C and 300 rpm. Each day thereafter for 5 days, in order to avoid mutations arising due to evolution during stationary phase (GASP mutants) (Zinser and Kolter, 2004), cells were diluted 1:1000 into 1 ml LB in a 14 ml tube containing the respective antibiotic and grown for 24 hr at 37°C and 300 rpm.
MICs were also measured for anaerobically grown E. coli over 5 days of treatment with no drug or 1 μg/ml ampicillin. Briefly, an overnight culture of E. coli was diluted 1:1000 into 15 ml anaerobic LB in sealed hungate tubes containing no drug or 1 μg/ml ampicillin. These cultures were grown in the sealed hungate tubes for 24 hr at 37°C and 300 rpm. Each day thereafter for 5 days, cells were diluted 1:1000 into 15 ml anaerobic LB in a sealed hungate tube containing the respective antibiotic and grown for 24 hr at 37°C and 300 rpm.
To determine the MIC on each day, an aliquot of cells from each treatment condition was diluted 1:10,000 into LB and dispensed into 96-well plates (100 μl
total volume per well) containing various concentrations (ten replicates per drug concentration) of norfloxacin, ampicillin, kanamycin, tetracycline, or chloramphenicol. Plates were incubated at 37°C and 300 rpm for 24 hr, after which time the optical density at 600 nm (OD600) was measured using a SPECTRAFluor Plus (Tecan; Männedorf, Switzerland). The median OD600 was calculated for each drug concentration, and the MIC was determined as the concentration that inhibited 90% of growth based on OD600. Fold change in MIC was determined by dividing the treated MIC on each day by its respective MIC from day 0.
Determination of MIC Variability and Multidrug Resistance
Wild-type E. coli were grown for 5 days in the presence of 1 μg/ml ampicillin or no drug (untreated) as described above. These long-term-treated cultures were diluted 1:1000 into 25 ml LB in 250 ml flasks and grown for 3 hr at 37°C and 300 rpm. Aliquots (1 ml) were plated onto LB-agar plates containing 300 ng/ml norfloxacin, 7.5 μg/ml ampicillin, 15 μg/ml kanamycin, 8 μg/ml tetracycline, and 25 μg/ml chloramphenicol, respectively, and grown for 24 hr at 37°C. Approximately 100 ampicillin-treated colonies from each primary drug selection were purified by streaking them onto LB-agar plates containing the same selective antibiotic. Double the volume of untreated control cells were plated for primary resistance selection as compared to the ampicillin-treated cells, and these untreated colonies were also purified as described above. Plates were placed at 37°C for 24 hr; these strains were then transferred via replica plating onto LB-agar plates containing norfloxacin, ampicillin, kanamycin, tetracycline, or chloramphenicol. Cross-resistance for each primary antibiotic selection following the 5 day ampicillin treatment or the no-drug treatment was determined after 24 hr of growth at 37°C by counting the colonies that displayed growth on the various drug-containing replicated plates.
The MIC of 44 of the above isolates and the MG1655 control strain were determined for ampicillin and norfloxacin, respectively. Overnight cultures of each strain were diluted 1:10,000 into 100 μl LB plus varying concentration of antibiotic (four replicates per strain per drug concentration) in 96-well plates. Plates were incubated at 37°C and 300 rpm for 24 hr, after which time the OD600 was measured using a SPECTRAFluor Plus (Tecan). The median OD600 was calculated for each drug concentration, and the MIC was determined as the concentration that inhibited 90% of growth based on OD600.
Wild-type S. aureus were grown for 5 days in the presence of 35 ng/ml ampicillin or no drug (untreated) as described above. E. coli clinical isolate NCDC C771 was grown for 5 days in the presence of 1 mg/ml ampicillin or no drug (untreated) as described above. These long-term-treated cultures were diluted 1:1000 into 25 ml LB in 250 ml flasks and grown for 3 hr at 37°C and 300 rpm. For S. aureus, 1 ml aliquots were plated onto LB-agar plates containing 3 μg/ml
norfloxacin, 7.5 μg/ml ampicillin, 15 μg/ml kanamycin, 8 μg/ml tetracycline, and 25 μg/ml chloramphenicol, respectively, and grown for 24 hr at 37°C. For NCDC C771, 1 ml aliquots were plated onto LB-agar plates containing 400 ng/ml norfloxacin, 8.5 μg/ml ampicillin, 20 μg/ml kanamycin, and 15 μg/ml chloramphenicol. Tetracycline cross-resistance was not quantified for NCDC C771, as this strain is resistant to tetracycline (MIC >35 μg/ml). Approximately 100 ampicillin-treated colonies from each primary drug selection were purified by streaking them onto LB-agar plates containing the same selective antibiotic. An equal volume of untreated S. aureus or the E. coli clinical isolate cells were plated for primary resistance selection as compared to the ampicillin-treated cells, and these untreated colonies were also purified as described above. The remainder of the cell growth and cross-resistance determination was performed as described above for wild-type E. coli.
Sequencing of Ampicillin-Treated, Norfloxacin-Resistant, or Kanamycin-Resistant Mutants
Six ampicillin-treated, norfloxacin-resistant isolates and six ampicillin-treated, kanamycin-resistant isolates from the cross-resistance experiment described above, as well as the untreated MG1655 control strain, were grown to a cell density of approximately 109 cfu/ml. Genomic DNA was extracted from each sample using a QIAGEN genomic DNA extraction kit according to the manufacturer’s instructions. Primers from IDT (Coralville, IA) (Table A3-S1) were utilized to PCR amplify, using Phusion DNA Polymerase (Finnzyme; Espoo, Finland), the regions surrounding gyrA, gyrB, tolC, acrA, marRA, ampC, rpsL, icdA, iscR, sdhB, arcA, and cpxR. These samples were sequenced by Agencourt Bioscience Corporation (Beverly, MA) using primers from IDT (Table A3-S1). Sequences were analyzed using Clone Manager 7 (Scientific & Educational Software; Cary, NC) and Sequence Scanner v1.0 (Applied Biosystems; Foster City, CA).
Supplemental Information
Supplemental Information includes Supplemental Results, Supplemental References, two figures, and one table and can be found with this article online at doi:10.1016/j.molcel.2010.01.003.
Acknowledgments
We thank D. Dwyer, B. Davies, and K. Allison for helpful discussions during the course of this work. We thank Q. Beg for help running the anaerobic chamber. This work was supported by the National Institutes of Health through
the NIH Director’s Pioneer Award Program, grant number DP1 OD003644, and the Howard Hughes Medical Institute.
Received: June 15, 2009
Revised: October 18, 2009
Accepted: November 12, 2009
Published: February 11, 2010
References
Alekshun, M.N., and Levy, S.B. (1997). Regulation of chromosomally mediated multiple antibiotic resistance: the mar regulon. Antimicrob. Agents Chemother. 41, 2067–2075.
Andersson, D.I. (2003). Persistence of antibiotic resistant bacteria. Curr. Opin. Microbiol. 6, 452–456.
Ariza, R.R., Li, Z., Ringstad, N., and Demple, B. (1995). Activation of multiple antibiotic resistance and binding of stress-inducible promoters by Escherichia coli Rob protein. J. Bacteriol. 177, 1655–1661.
Beaber, J.W., Hochhut, B., and Waldor, M.K. (2004). SOS response promotes horizontal dissemination of antibiotic resistance genes. Nature 427, 72–74.
Becnel Boyd, L., Maynard, M.J., Morgan-Linnell, S.K., Horton, L.B., Sucgang, R., Hamill, R.J., Jimenez, J.R., Versalovic, J., Steffen, D., and Zechiedrich, L. (2009). Relationships among ciprofloxacin, gatifloxacin, levofloxacin, and norfloxacin MICs for fluoroquinolone-resistant Escherichia coli clinical isolates. Antimicrob. Agents Chemother. 53, 229–234.
Brumaghim, J.L., Li, Y., Henle, E., and Linn, S. (2003). Effects of hydrogen peroxide upon nicotinamide nucleotide metabolism in Escherichia coli: changes in enzyme levels and nicotinamide nucleotide pools and studies of the oxidation of NAD(P)H by Fe(III). J. Biol. Chem. 278, 42495–42504.
Bryskier, A. (2005). Antimicrobial Agents: Antibacterials and Antifungals (Washington, D.C.: ASM Press).
Carlsson, J., and Carpenter, V.S. (1980). The recA+ gene product is more important than catalase and superoxide dismutase in protecting Escherichia coli against hydrogen peroxide toxicity. J. Bacteriol. 142, 319–321.
Chopra, I., O’Neill, A.J., and Miller, K. (2003). The role of mutators in the emergence of antibiotic-resistant bacteria. Drug Resist. Updat. 6, 137–145.
Cirz, R.T., Chin, J.K., Andes, D.R., de Crécy-Lagard, V., Craig, W.A., and Romesberg, F.E. (2005). Inhibition of mutation and combating the evolution of antibiotic resistance. PLoS Biol. 3, e176.
Cohen, S.P., McMurry, L.M., Hooper, D.C., Wolfson, J.S., and Levy, S.B. (1989). Cross-resistance to fluoroquinolones in multiple-antibiotic-resistant (MAR) Escherichia coli selected by tetracycline or chloramphenicol: decreased drug accumulation associated with membrane changes in addition to OmpF reduction. Antimicrob. Agents Chemother. 33, 1318–1325.
Davies, J. (1994). Inactivation of antibiotics and the dissemination of resistance genes. Science 264, 375–382.
de Lencastre, H., Figueiredo, A.M., and Tomasz, A. (1993). Genetic control of population structure in heterogeneous strains of methicillin resistant Staphylococcus aureus. Eur. J. Clin. Microbiol. Infect. Dis. 12 (Suppl 1), S13–S18.
Demple, B., and Harrison, L. (1994). Repair of oxidative damage to DNA: enzymology and biology. Annu. Rev. Biochem. 63, 915–948.
Drlica, K., and Zhao, X. (1997). DNA gyrase, topoisomerase IV, and the 4-quinolones. Microbiol. Mol. Biol. Rev. 61, 377–392.
Dwyer, D.J., Kohanski, M.A., Hayete, B., and Collins, J.J. (2007). Gyrase inhibitors induce an oxidative damage cellular death pathway in Escherichia coli. Mol. Syst. Biol. 3, 91.
Dwyer, D.J., Kohanski, M.A., and Collins, J.J. (2009). Role of reactive oxygen species in antibiotic action and resistance. Curr. Opin. Microbiol. 12, 482–489.
Fridovich, I. (1978). The biology of oxygen radicals. Science 201, 875–880.
Friedberg, E.C., Walker, G.C., Siede, W., Wood, R.D., Schultz, R.A., and Ellenberger, T. (2006). DNA Repair and Mutagenesis, Second Edition (Washington, D.C.: ASM Press).
George, A.M., and Levy, S.B. (1983). Amplifiable resistance to tetracycline, chloramphenicol, and other antibiotics in Escherichia coli: involvement of a non-plasmid-determined efflux of tetracycline. J. Bacteriol. 155, 531–540.
Giraud, A., Matic, I., Tenaillon, O., Clara, A., Radman, M., Fons, M., and Taddei, F. (2001). Costs and benefits of high mutation rates: adaptive evolution of bacteria in the mouse gut. Science 291, 2606–2608.
Girgis, H.S., Hottes, A.K., and Tavazoie, S. (2009). Genetic architecture of intrinsic antibiotic susceptibility. PLoS ONE 4, e5629.
Goldstein, F. (2007). The potential clinical impact of low-level antibiotic resistance in Staphylococcus aureus. J. Antimicrob. Chemother. 59, 1–4.
Grassi, G.G. (1979). Drug-inactivating enzymes of bacteria grown in subminimal inhibitory concentrations of antibiotics. Rev. Infect. Dis. 1, 852–857.
Greenberg, J.T., Monach, P., Chou, J.H., Josephy, P.D., and Demple, B. (1990). Positive control of a global antioxidant defense regulon activated by superoxide-generating agents in Escherichia coli. Proc. Natl. Acad. Sci. USA 87, 6181–6185.
Hegreness, M., Shoresh, N., Damian, D., Hartl, D., and Kishony, R. (2008). Accelerated evolution of resistance in multidrug environments. Proc. Natl. Acad. Sci. USA 105, 13977–13981.
Imlay, J.A. (2006). Iron-sulphur clusters and the problem with oxygen. Mol. Microbiol. 59, 1073–1082.
Imlay, J.A., and Linn, S. (1986). Bimodal pattern of killing of DNA-repair-defective or anoxically grown Escherichia coli by hydrogen peroxide. J. Bacteriol. 166, 519–527.
Imlay, J.A., and Linn, S. (1987). Mutagenesis and stress responses induced in Escherichia coli by hydrogen peroxide. J. Bacteriol. 169, 2967–2976.
Imlay, J.A., Chin, S.M., and Linn, S. (1988). Toxic DNA damage by hydrogen peroxide through the Fenton reaction in vivo and in vitro. Science 240, 640–642.
Keseler, I.M., Collado-Vides, J., Gama-Castro, S., Ingraham, J., Paley, S., Paulsen, I.T., Peralta-Gil, M., and Karp, P.D. (2005). EcoCyc: a comprehensive database resource for Escherichia coli. Nucleic Acids Res. 33(Database issue), D334–D337.
Kohanski, M.A., Dwyer, D.J., Hayete, B., Lawrence, C.A., and Collins, J.J. (2007). A common mechanism of cellular death induced by bactericidal antibiotics. Cell 130, 797–810.
Kohanski, M.A., Dwyer, D.J., Wierzbowski, J., Cottarel, G., and Collins, J.J. (2008). Mistranslation of membrane proteins and two-component system activation trigger antibiotic-mediated cell death. Cell 135, 679–690.
Livermore, D.M. (2003). Bacterial resistance: origins, epidemiology, and impact. Clin. Infect. Dis. 36 (Suppl 1), S11–S23.
López, E., Elez, M., Matic, I., and Blázquez, J. (2007). Antibiotic-mediated recombination: ciprofloxacin stimulates SOS-independent recombination of divergent sequences in Escherichia coli. Mol. Microbiol. 64, 83–93.
Ma, D., Cook, D.N., Alberti, M., Pon, N.G., Nikaido, H., and Hearst, J.E. (1993). Molecular cloning and characterization of acrA and acrE genes of Escherichia coli. J. Bacteriol. 175, 6299–6313.
McKenzie, G.J., and Rosenberg, S.M. (2001). Adaptive mutations, mutator DNA polymerases and genetic change strategies of pathogens. Curr. Opin. Microbiol. 4, 586–594.
Michaels, M.L., and Miller, J.H. (1992). The GO system protects organisms from the mutagenic effect of the spontaneous lesion 8-hydroxyguanine (7,8-dihydro-8-oxoguanine). J. Bacteriol. 174, 6321–6325.
Miller, J.H. (1996). Spontaneous mutators in bacteria: insights into pathways of mutagenesis and repair. Annu. Rev. Microbiol. 50, 625–643.
Miller, K., O’Neill, A.J., and Chopra, I. (2002). Response of Escherichia coli hypermutators to selection pressure with antimicrobial agents from differentclasses. J. Antimicrob. Chemother. 49, 925–934.
Miller, C., Thomsen, L.E., Gaggero, C., Mosseri, R., Ingmer, H., and Cohen, S.N. (2004). SOS response induction by beta-lactams and bacterial defense against antibiotic lethality. Science 305, 1629–1631.
Mouneimné, H., Robert, J., Jarlier, V., and Cambau, E. (1999). Type II topoisomerase mutations in ciprofloxacin-resistant strains of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 43, 62–66.
Norris, J.R., and Ribbons, D.W. (1969). Methods in Microbiology (Orlando, FL: Academic Press Inc).
Novogrodsky, A., Ravid, A., Rubin, A.L., and Stenzel, K.H. (1982). Hydroxyl radical scavengers inhibit lymphocyte mitogenesis. Proc. Natl. Acad. Sci. USA 79, 1171–1174.
Oh, H., El Amin, N., Davies, T., Appelbaum, P.C., and Edlund, C. (2001). gyrA mutations associated with quinolone resistance in Bacteroides fragilis groupstrains. Antimicrob. Agents Chemother. 45, 1977–1981.
Pérez-Capilla, T., Baquero, M.R., Gómez-Gómez, J.M., Ionel, A., Martín, S., and Blázquez, J. (2005). SOS-independent induction of dinB transcription by beta-lactam-mediated inhibition of cell wall synthesis in Escherichia coli. J. Bacteriol. 187, 1515–1518.
Repine, J.E., Fox, R.B., and Berger, E.M. (1981). Hydrogen peroxide kills Staphylococcus aureus by reacting with staphylococcal iron to form hydroxylradical. J. Biol. Chem. 256, 7094–7096.
Rosche, W.A., and Foster, P.L. (2000). Determining mutation rates in bacterial populations. Methods 20, 4–17.
Sarkar, S., Ma, W.T., and Sandri, G.H. (1992). On fluctuation analysis: a new, simple and efficient method for computing the expected number of mutants. Genetica 85, 173–179.
Setsukinai, K., Urano, Y., Kakinuma, K., Majima, H.J., and Nagano, T. (2003). Development of novel fluorescence probes that can reliably detect reactive oxygen species and distinguish specific species. J. Biol. Chem. 278, 3170–3175.
Touati, D., Jacques, M., Tardat, B., Bouchard, L., and Despied, S. (1995). Lethal oxidative damage and mutagenesis are generated by iron in delta fur mutants of Escherichia coli: protective role of superoxide dismutase. J. Bacteriol. 177, 2305–2314.
Weill, F.X., Guesnier, F., Guibert, V., Timinouni, M., Demartin, M., Polomack, L., and Grimont, P.A. (2006). Multidrug resistance in Salmonella enterica serotype Typhimurium from humans in France (1993 to 2003). J. Clin. Microbiol. 44, 700–708.
Zinser, E.R., and Kolter, R. (2004). Escherichia coli evolution during stationary phase. Res. Microbiol. 155, 328–336.
Supplemental Information
Molecular Cell, Volume 37
Sublethal Antibiotic Treatment Leads to Multidrug Resistance via Radical-Induced Mutagenesis
Michael A. Kohanski, Mark A. DePristo and James J. Collins
Supplemental Results
Bactericidal Antibiotics Lead to Low-Level Increases in MIC for a Range of Antibiotics
Treatment with 25 ng/ml norfloxacin led to significant increases in the MIC for norfloxacin and kanamycin as well as modest, low-level increases in the MIC for ampicillin, tetracycline and chloramphenicol (Figure A3-S1A). This increase in MIC for norfloxacin was concentration dependent. Treatment with 50 ng/ml norfloxacin led to a 6-fold increase in the MIC for norfloxacin (Figure A3-S1B); however, we were unable to observe an increase in the MIC for ampicillin, kanamycin, tetracycline, or chloramphenicol following treatment
FIGURE A3-S1 Bactericidal antibiotics can lead to broad-spectrum increases in MIC. (A-C) Fold change in MIC relative to a no-drug control for ampicillin, norfloxacin, kanamycin, tetracycline and chloramphenicol, following 5 days of growth in the presence of (A) 25 ng/ml norfloxacin, (B) 50 ng/ml norfloxacin, or (C) 1 μg/ml kanamycin. (D) Ampicillin-mediated increases in MIC are stable. Fold change in MIC relative to a no-drug control for ampicillin, norfloxacin, kanamycin, tetracycline and chloramphenicol, following 5 days of growth in the presence of 1 μg/ml ampicillin and an additional 2 days of growth in the absence of drug.
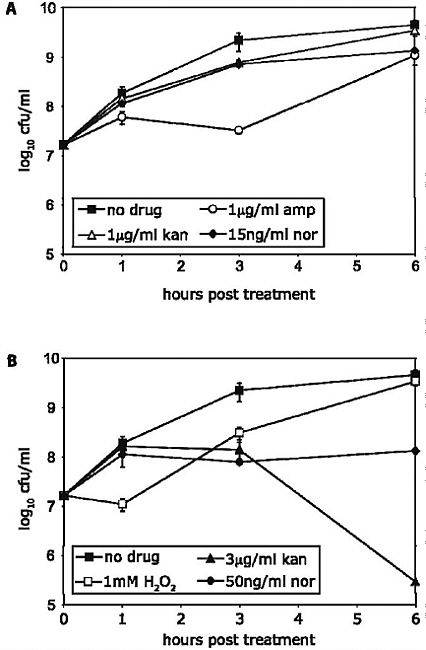
FIGURE A3-S2 Survival of E. coli following treatment with near-MIC levels of antibiotics. (A) Survival of MG1655 following treatment with no drug (filled squares), 1 μg/ml ampicillin (amp, open circles), 1 μg/ml kanamycin (kan, open triangles), and 15 ng/ml norfloxacin (nor, filled diamonds), respectively. (B) Survival of MG1655 following treatment with no drug (filled squares), 3μg/ml kanamycin (filled triangles), 1 mM hydrogen peroxide (H2O2, open squares), and 50 ng/ml norfloxacin (filled diamonds), respectively.
TABLE A3-S1 PCR Primers and Sequencing Primers
|
PCR Primers |
|
|
Primer Name |
Sequence (5′-3′) |
|
gyrA-F |
CCA GAC TTT GCA GCC TGG ACT T |
|
gyrA-R |
AAC TCA CCT TCC AGA TCC CAC CA |
|
gyrB-F |
TGA ACG CCT TAT CCG GCC TAC AA |
|
gyrB-R |
CTC TGA GCT TGA TGA TGA GCG TCG |
|
tolC-F |
TGA CTG CCG TTT GAG CAG TCA TGT G |
|
tolC-R |
TTA CGT TGC CTT ACG TTC AGA CGG |
|
marRA-F |
TAG CTA ACG GCA GCA ACA CCA C |
|
MarRA-R |
CAA TGT ATT TGG CTT GCG GTG GC |
|
acrAB-F |
TCG TAT GAG ATC CTG AGT TGG TGG TTC |
|
acrAB-R |
AAT GCC AGT AGA TTG CAC CGC |
|
acrAB-F2 |
ACT TAT TAC TAC GCG ATC GCC TGC T |
|
acrAB-R2 |
GCA GTG AAC CAG AAT AGC AAC GAC GA |
|
sdhB-F |
CTG CCA ACT TCC GTA CCG AAA G |
|
sdhB-R |
AGC TCT TGT CTA CGT AGT GGC TC |
|
icdA-F |
CTG GTA GAA CGT TGC GAG CT |
|
icdA-R |
GAC TAG TAG TAG AAC TAC CAC CTG ACC G |
|
iscR-F |
GTT ACC AAA GGT TCC GTC CAT CGT |
|
iscR-R |
CGT CTT ATC AGG CCT ACA GTG TAC AG |
|
cpxR-F |
CGA CAT GCT GCT CAA TCA TCA GC |
|
cpxR-R |
GCT TAA TGA ACT GAC TGC CAG CGT TGA |
|
arcA-F |
GAC TGC TCA ACT CTG CCG ATA G |
|
arcA-R |
TGC TGT TAA AAT GGT TAG GAT GAC AGC CGT |
|
ampC-F |
AGG CAA CGA CCA GAA ATG CAG CT |
|
ampC-R |
TAT GCA CCA CGC GAT GCA CGA T |
|
Sequencing Primers |
|
|
Primer Name |
Sequence (5′-3′) |
|
gyrA1 |
CAG GCA TTG GAT GTG AAT AAA GCG TAT AGG |
|
gyrA2 |
ATC ATT AAC GGT CGT CGC GGT ATT G |
|
gyrA3 |
TGC GTG ATG GTC TGT ACT ACC TGA |
|
gyrA4 |
TCC TCA CCG AGT TCA ACC GTC T |
|
gyrB1 |
TCA GTG CTG AAC ACG TTA TAG ACA TGT CGG |
|
gyrB2 |
GAC GGC AAA GAA GAC CAC TTA CAC T |
|
gyrB3 |
AAG CGC GCT TCG ATA GA TGC T |
|
gyrB4 |
GTT TGA TGT TCA CAC CAA TGC TGA GC |
|
tolC1 |
TAT GGC ACG TAA CGC CAA CCT |
|
tolC2 |
TAA CCT TGA TAA CGC GGT AGA GCA GC |
|
tolC3 |
GCT CAA GCG TGC CTG TAA CA |
|
marRA1 |
AGC TAG CCT TGC ATC GCA TTG A |
|
marRA2 |
CGG ACG AAG TGG CAA CAC TTG AGT AT |
|
marRA-M1 |
AGG TAT GAC GAT GTC CAG ACG CA |
|
marRA-M2 |
TGC GTC TGG ACA TCG TCA TAC CT |
|
acrA1 |
CAG CTG CTT TTG CAA TCT CGC |
|
Sequencing Primers |
|
|
Primer |
Name Sequence (5′-3′) |
|
acrA2 |
CTG CTC GGT ACT CAG TAC ATC AGT AAG C |
|
acrA3 |
TGC AGA AAG TGC GTC CTG GTG T |
|
acrA4 |
ATT ACC GCC ATC AAA GCG CAG |
|
acrA8 |
CTC CAT CAA TAA TCG ACG CCG TTC T |
|
acrA9 |
TGT AAG CCA GAT TGA TCC GCG CA |
|
acrA-M1 |
GTT CTG TAC CAA TGC GCC TTC CGT |
|
acrA-M2 |
ACG GAA GGC GCA TTG GTA CAG AAC |
|
icdA1 |
TAG CCT AAT AAC GCG CAT CTT TCA TGA CG |
|
icdA2 |
ATT CGC TTC CCG GAA CAT TGT GGT A |
|
icdA3 |
CTA CCC CAA AAC TAC CGA GGG GTT |
|
icdA4 |
CCA GTC TTT AAA CGC TCC TTC GGT |
|
icdA-R5 |
GGA GCG TTA CGC TCC CGT TAA TA |
|
icdA-M1 |
GGT ATC GAA TGG AAA GCA GAC TCT GC |
|
sdhB1 |
TCG ACT TCC CGG ATC GTG ATG ATG A |
|
sdhB2 |
TCC TTT GTT ACG CCT GAT GCG CT |
|
iscR1 |
TGG GTT GCG GAG TAG TCG AGA TAA |
|
iscR2 |
ATA TGG CGT TCA CGC CGC AT |
|
cpxR1 |
ACG ATG TTC GCT ATC CAG AAG CTC |
|
cpxR2 |
GCA GCG GTA ACT ATG CGC ATC ATT |
|
arcA1 |
GTG ACC CGT ATT ATC GAC TGG TAT GC |
|
arcA2 |
GTA CCC ACG ACC AAG CTA ATG ATG |
|
ampC1 |
TGG CTG CTA TCC TGA CAG TTG TCA |
|
ampC2 |
GTC TGT ATG CCA ACT CCA GTA TCG GT |
with 50 ng/ml norfloxacin for 5 days (Figure A3-S1B). Interestingly, selection of drug-resistant mutants following quinolone treatment is concentration dependent, with higher concentrations of quinolone selecting only quinolone-resistant strains and lower levels of quinolone selecting broadly for drugresistant mutants with mutations in a wide array of targets in E. coli (Drlica, 2003) and Mycobacterium tuberculosis (Zhou et al., 2000). It is possible that treatment with 50 ng/ml norfloxacin selects for naturally occurring quinolone-resistant mutants before the drug-induced mutagenesis has a chance to create mutants resistant to other drugs.
Supplemental References
Drlica, K. (2003). The mutant selection window and antimicrobial resistance. J Antimicrob Chemother 52, 11-17.
Zhou, J., Dong, Y., Zhao, X., Lee, S., Amin, A., Ramaswamy, S., Domagala, J., Musser, J.M., and Drlica, K. (2000). Selection of antibiotic-resistant bacterial mutants: allelic diversity among fluoroquinolone-resistant mutations. J Infect Dis 182, 517-525.
A4
ANTIBIOTIC-INDUCED RESISTANCE FLOW
Patrice Courvalin
Institut Pasteur, Unité des Agents Antibactériens, Paris, France
Introduction
The evolution of bacteria toward antibiotic resistance is unavoidable since it represents a particular aspect of the general evolution of bacteria. It results from two independent steps, emergence and dissemination; however, as we consider, the mechanism of the first one can largely influence the success of the second one. Resistance to antibiotics in bacteria is secondary to mutations in resident (housekeeping) structural or regulatory genes or to horizontal acquisition of foreign genetic information (Perichon and Courvalin, 2009). In this review, we consider the relationship between low levels of antibiotics and dissemination of resistance.
The emergence of resistance, an event that occurs by pure chance, can be a rare, even transient, event if it does not provide a selective advantage against a molecule present in the environment of the bacterium. Resistance potentially exists in nature, not only before the clinical use but also before the discovery, or even the design, of a new antibiotic. This is obvious for natural antibiotics, because the producing microorganisms must protect themselves against suicide by the products of their secondary metabolism, but it also holds true for semisynthetic (e.g., amikacin) or entirely synthetic (e.g., fluoroquinolone) antibiotics.
The bacterial genome is composed of the chromosome and of accessory genetic elements, self-transferable or mobilizable plasmids, integrative conjugative elements (ICEs), transposons, insertion sequences, and bacteriophages. The chromosome contains all the genetic information required for the life cycle of the bacterium, whereas, as their name indicates, accessory genetic elements carry genes that are dispensable, although under certain circumstances, they can provide major advantages for the survival of the host, such as antibiotic resistance. The chromosome is inherited vertically by the progeny of the cell and is not transferable horizontally, whereas accessory genetic elements can also be transmitted to other bacteria. As a result, resistance can thus be endogenous or exogenous. Endogenous resistance results from chromosomal mutations and is generally not infectious from bacteria to bacteria. In contrast, exogenous resistance is due to horizontal (lateral) transfer of DNA among bacteria, resulting in acquisition of mobile genetic elements.
The Classical View
Endogenous Resistance
The occurrence of chromosomal mutations is an efficient pathway to resistance. Mutations are considered rare because they occur at low frequency, generally between 10−7 and 10−10 and were considered errors that occurred during DNA replication. However, this limitation is easily overcome because, during infections in humans, bacterial populations are often very large. Mutations in chromosomal genes clearly represent the only mechanism of antibiotic resistance in genera such as Mycobacterium or strictly intracellular pathogens (such as Chlamydia, Rickettsia, Coxiella, Ehrlichia), which are not known to exchange DNA under natural conditions.
Exogenous Resistance
Dissemination of resistance has, in numerous instances, been shown to be closely associated with antibiotic use (Malhotra-Kumar et al., 2007), which stresses the importance of the prudent use of these molecules. In addition, resistance is, if at all, slowly reversible (Andersson, 2003). There are three levels of resistance dissemination, depending upon the vector: bacteria (clonal spread), replicons (plasmid epidemics), or genes (conjugative transposon [ICE] epidemics). These various levels of dissemination, which coexist in nature and thus account for the extraordinary rise in antibiotic resistance among bacteria, are not only infectious but also exponential because each is associated with DNA duplication. Clonal dissemination is associated with chromosome replication, plasmid conjugation with replicative transfer, and gene migration with replicative transposition.
It also turns out that conjugation has a very broad host range—plasmids and ICEs can transfer efficiently between phylogenetically remote bacterial genera—and that there are limited barriers to heterologous gene expression (Courvalin, 1994); that is, resistance genes can be expressed in very diverse hosts.
These observations led to the notion of a bacterial gene pool, in particular with respect to resistance, which means that genes are loosely bound to their hosts and can easily disseminate under natural conditions. This concept has many practical consequences, for example, in the case of the use of antibiotics as animal feed additives. Rather than discussing endlessly (and often in a biased fashion) whether the enterococci from animals and humans are similar (Phillips, 1999) (i.e., whether vancomycin-resistant enterococci from animals can stably colonize the human gut), the true question should rather be the following: Are the resistance genes (to glycopeptides, in this example) the same among bacteria of these two ecosystems? Along this line, studies published long ago that examined the biochemistry and genetics of aminoglycoside resistance, as well
as molecular study of the bacterial hosts (with the techniques available at that time), elucidated how the exclusive use of apramycin in animals could select gentamicin-resistant bacteria that were later found in humans (Chaslus-Dancla et al., 1986a, 1986b, 1991). This notion has since been largely documented, using more powerful techniques, for resistance to other drug classes (Stobberingh and van den Bogaard, 2000).
The Modern View
Endogenous Resistance
It was shown recently that bactericidal antibiotics kill bacteria by inducing the formation of highly deleterious hydroxyl radicals, reactive oxygen species, which can damage DNA (Kohanski et al., 2007). This oxidative stress leads to a significant increase in mutation rate either directly or indirectly by activation of the SOS DNA damage response pathway (Kohanski et al., 2010) as well as an increase in recombination (Figure A4-1). Thus, certain classes of antibiotics may behave as mutagens, in particular at low concentrations, and may select for resistance to other drug classes, whereas the mutant derivatives remain susceptible to the applied antibiotic (Kohanski et al., 2010).
The major human pathogen Streptococcus pneumoniae may represent a particular case of this mechanism. Evidence has been recently provided that the stress caused by low concentrations of certain antibiotics induces genetic transformability in pneumococci (Prudhomme et al., 2006). Transformation is a process inherent in this bacterial species that allows the transient uptake and integration of exogenous DNA in the recipient genome as well as the capability to kill noncompetent cells, a phenomenon referred to as fratricide. Low concentrations of bactericidal antibiotics, such as quinolones and aminoglycosides, induce full competence for genetic transformation, thereby increasing the rate of genetic exchange in S. pneumoniae and making chromosomal mutations horizontally transferable (Figure A4-2). Competence appears thus as a general stress response, playing a role similar to that of the SOS response in Escherichia coli that lacks in S. pneumoniae (Claverys and Havarstein, 2002). Fratricide is the killing of cells from the same species and can be considered a mechanism that is used by competent bacteria to acquire DNA from noncompetent pneumococci (Claverys et al., 2007). Considering the high incidence of asymptomatic carriage of and co-colonization by this human pathogen, inappropriate antibiotic use could accelerate the emergence of resistant clones, promote evolution toward virulence, and enrich in capsular types that are not included in the current vaccines. The latter observation represents an additional argument for not prescribing fluoroquinolones to children.
FIGURE A4-1 Antibiotic induced increase mutation rate in S. pneumoniae. Subinhibitory concentrations of bactericidal antibiotics promote production of reactive oxygen species (ROS) by bacteria via the stress response. This leads to DNA damage which i) increases recombination frequency and ii) induces a competence state resulting in transformation which both cause mutations.
Exogenous Resistance
Antibiotics can enhance gene transfer: they provide selective pressure for resistant bacteria to maintain and disseminate, but they can also induce the transfer of resistance genes. For example, it has been reported that (1) the use of subinhibitory concentrations of penicillins increased the conjugal transfer of plasmid DNA from Escherichia coli to Staphylococcus aureus and Listeria monocytogenes (Trieu-Cuot et al., 1993), (2) oxacillin increased the frequency of in vitro transfer of Tn916, an enterococcal ICE, from Enterococcus faecalis to Bacillus anthracis (Ivins et al., 1988), (3) the transfer frequency of conjugative transposons belonging to the Tn916/Tn1545 family (Figure A4-3), which contain a tetracycline resistance determinant, was increased 10- to 100-fold in vitro and in vivo in the presence of low concentrations of tetracycline (Doucet-Populaire et al., 1991), and (4) tetracycline also increased dramatically the transfer of a
FIGURE A4-2 Antibiotic promotes evolution of resistance in S. pneumoniae. The presence of an antibiotic generates a bacterial stress responsible for competence. The competence state induces transformation and fratricidy in which both can lead to antibiotic resistance and capsular switch.
Bacteroides conjugative transposon (Li et al., 1995). In the two latter cases, the antibiotic has a triple activity: as an antibacterial agent, as an inducer of resistance to itself, and as an inducer of the dissemination of resistance determinants. It thus appears that several antibiotics can behave like pheromones: they are synthesized by specific cells (such as the Actinobacteria), and they act on another cell, at low concentrations, on very specific targets to promote DNA exchange.
It was also shown that mitomycin C and ciprofloxaxin de-repressed the expression of genes necessary for transfer of an ICE in Vibrio cholerae (Beaber et al., 2004). This resulted in an unpredictable horizontal dissemination of the genetic element which confers resistance to chloramphenicol, trimethoprim, sulphonamides, and streptomycin.
Another example of increased resistance gene mobility by antibiotics is represented by the integrons (Mazel, 2006). These compact structures act as genetic systems for in vivo capture and expression of genes in the form of circular cassettes. These genes are the most tightly linked because they are not only adjacent but coexpressed from the same promoter. Integrons are thus typically responsible for coresistance: the stable association in the same cell of various resistance determinants, each conferring resistance to a drug class. Similarly to cross-resistance, which results in cross-selection, coresistance implies coselection: the use of any antibiotic that is substrate for a mechanism encoded by the integron will coselect for the other resistances. This genetic organization renders the consequences of
FIGURE A4-3 Transfer of an integrative conjugative element (ICE). ICEs are mobile genetic elements that carry one or several resistance genes. They excise by site-specific recombination between their flanking attachment sites, attR and attL, leading to the formation of an episomal ICE carrying an attI site and an empty attB site in the chromosome. They replicate during their transfer by conjugation and integrate in the chromosome of the recipient. Dissemination of resistance by ICEs is thus infectious and exponential.
the use of a single drug unpredictable. Because there is transcriptional attenuation along the operons in integrons (Collis and Hall, 1995), the use of an antibiotic selects not only for the neighboring resistance determinants but for a higher level of resistance to itself as well. This is achieved by the movement (excision, circularization, and re-integration) of the corresponding cassette that ends up downstream from the strong common promoter. Integrons, therefore, allow quantitative (self) and qualitative (nonself) alteration of resistance. Most interestingly, it has been shown recently that certain antibiotics such as mitomycin C, trimethroprim, the quinolones, and the β-lactams stimulate the intracellular mobility of the gene cassettes (Guerin et al., 2009).
Limitations to Dissemination
Genes from Gram-positive cocci can be transferred by conjugation (of plasmids or ICE) not only among these microorganisms but also to Gram-negative bacteria (Courvalin, 1994). The reverse is not true because of limitations in heterologous gene expression. This is due to the fact that the −35 and −10 sequences
and their spacing that constitute the promoters for expression of the genes, as well as the ribosome binding site, are conserved and are close to the consensus in Gram-positives; they are thus also functional in Gram-negatives. In contrast, these motifs are much more degenerate in Gram-negatives and cannot be accommodated by Gram-positives. Similarly, the promoters from Bacteroides fragilis and from E. coli are dissimilar, resulting in lack of gene expression from E. coli promoters (even strong promoters) in B. fragilis and the inactivity of B. fragilis promoters in other bacterial species (E. coli, Bacillus subtilis, and Clostridium perfringens) (Bayley et al., 2000; Smith et al., 1992). One can thus confidently predict that strains of B. fragilis will not, or will extremely inefficiently, act as intermediates in resistance gene transfer or represent a pool of origin of these genes for human pathogens.
Acquisition of resistance by bacteria corresponds to a gain of function and is, thus, generally associated with a biological cost. In other words, resistant derivatives have a lower degree of fitness than the parental strain lacking the resistance genes; that is, daughter cells are less competitive for growth in a given ecosystem and in the absence of antibiotic, than the mother cell. The proportion of resistant strains in a bacterial population depends on several factors, such as the concentration and type of antibiotic used, the biological cost of resistance to that antibiotic, and the ability of bacteria to compensate for the fitness cost of the resistance mechanism. Acquisition of antibiotic resistance is often associated with a biological cost because (1) bacteria acquire a new gene (or set of genes) responsible for new functions, (2) the resistance mutations occur in genes with essential functions, or (3) additional energy is required for replication and maintenance of plasmids that bear the resistance genes. The biological cost determines the stability and potential reversibility of resistance.
A compensatory evolution could occur to reduce the biological cost leading to stabilization of the resistant bacteria in a natural population. This process allows resistant strains to regain competitivity relative to their susceptible counterparts in an antibiotic-free environment (Hughes and Andersson, 2001).
However, it has been recently demonstrated that inducibility of resistance is a compensatory mechanism (Foucault et al., 2010). This accounts for the observation that the majority of horizontally acquired antibiotic resistance mechanisms is tightly regulated and that resistance evolves to become selectively neutral in the absence of antibiotics.
Conclusion
Regardless of the mechanism of action of a drug class, it must be realized that resistance already occurs in nature or will inevitably emerge. This is, perhaps, obvious for natural antibiotics, because the producing organisms must avoid self-destruction, but it also holds true for nonnatural drugs. It is thus clear that bacteria are able to resist every antibiotic, naturally or in an acquired
fashion, and selection of resistant bacteria can be regarded as the ultimate criterion for activity of an antibiotic. In addition, and by the mechanisms we have considered, resistance, either by mutation or after acquisition of foreign genetic information, can be drastically enhanced by low concentrations of antibiotics in the environment of the bacteria. Because dissemination of resistance is closely linked to the magnitude of the selective pressure, the only hope is to delay this dissemination. This leaves us with a single recommendation: antibiotics should be used cautiously.
References
Andersson, D. I. 2003. Persistence of antibiotic resistant bacteria. Curr. Opin. Microbiol. 6:452–6.
Bayley, D. P., E. R. Rocha, and C. J. Smith. 2000. Analysis of cepA and other Bacteroides fragilis genes reveals a unique promoter structure. FEMS Microbiol. Lett. 193:149–54.
Beaber, J. W., B. Hochhut, and M. K. Waldor. 2004. SOS response promotes horizontal dissemination of antibiotic resistance genes. Nature 427:72–4.
Chaslus-Dancla, E., G. Gerbaud, J. P. Lafont, J. L. Martel, and P. Courvalin. 1986a. Nucleic acid hybridization with a probe specific for 3-aminoglycoside acetyltransferase type IV: A survey of resistance to apramycin and gentamicin in animal strains of Escherichia coli. FEMS Microbiol. Lett. 34:265–8.
Chaslus-Dancla, E., J. L. Martel, C. Carlier, J. P. Lafont, and P. Courvalin. 1986b. Emergence of 3-aminoglycoside acetyltransferase IV in Escherichia coli and Salmonella typhimurium from animal in France. Antimicrob. Agents Chemother. 29:239–43.
Chaslus-Dancla, E., P. Pohl, M. Meurisse, M. Marin, and J. L. Lafont. 1991. High genetic homology between plasmids of human and animal origins conferring resistance to the aminoglycosides gentamicin and apramycin. Antimicrob. Agents Chemother. 35:590–3.
Claverys, J. P., and L. S. Havarstein. 2002. Extracellular-peptide control of competence for genetic transformation in Streptococcus pneumoniae. Front. Biosci. 7:1798–1814.
Claverys, J. P., B. Martin, and L. S. Havarstein. 2007. Competence induced fratricide in streptococci. Mol. Microbiol. 64:1423–33.
Collis, C. M., and R. M. Hall. 1995. Expression of antibiotic resistance genes in the integrated cassettes of integrons. Antimicrob. Agents Chemother. 39:155–62.
Courvalin, P. 1994. Transfer of antibiotic resistance genes between Gram-positive and Gram-negative bacteria. Antimicrob. Agents Chemother. 38:1447–51.
Doucet-Populaire, F., P. Trieu-Cuot, I. Dosbaa, A. Andremont, and P. Courvalin. 1991. Inducible transfer of conjugative transposon Tn1545 from Enterococcus faecalis to Listeria monocytogenes in the digestive tract of gnotobiotic mice.. Antimicrob. Agents Chemother. 35:185–7.35:185–7..
Foucault, M.-L., F. Depardieu, P. Courvalin, and C. Grillot-Courvalin. 2010 (in press). Inducible expression eliminates the fitness cost of vancomycin resistance in enterococci. Proc. Natl. Acad. Sci. USA.
Guerin, E., G. Cambray, N. Sanchez-Alberola, S. Campoy, I. Erill, S. Da Re, B. Gonzalez-Zorn, J. Barbé, M.-C. Ploy, and D. Mazel. 2009. The SOS response controls integron recombination Science 324:1034.
Hughes, D., and D. I. Andersson, eds. 2001. Antibiotic development and resistance. London, United Kingdom: Taylor and Francis.
Ivins, B. E., S. L. Welkos, G. B. Knudson, and D. J. Leblanc. 1988. Transposon Tn916 mutagenesis in Bacillus anthracis. Infect. Immun. 56:176–81.
Kohanski, M. A., D. J. Dwyer, B. Hayete, C. A. Lawrence, and J. J. Collins. 2007. A common mechanism of cellular death induced by bactericidal antibiotics. Cell 130:797–810.
Kohanski, M. A., M. A. DePristo, and J. J. Collins. 2010. Sublethal antibiotic treatment leads to multidrug resistance via radical-induced mutagenesis. Mol. Cell. 37:311–20.
Li, L. Y., N. B. Shoemaker, and A. A. Salyers. 1995. Location and characteristics of the transfer region of a Bacteroides conjugative transposon and regulation of transfer genes. J. Bacteriol. 177:4992–9.
Malhotra-Kumar, S., C. Lammens, S. Coenen, K. Van Herck, and H. Goossens. 2007. Effect of azithromycin and clarithromycin therapy on pharyngeal carriage of macrolide-resistant streptococci in healthy volunteers: A randomised, double-blind, placebo-controlled study. Lancet 369:482–90.
Mazel, D. 2006. Integrons: Agents of bacterial evolution. Nat. Rev. Microbiol. 8:608–20.
Perichon, B., and P. Courvalin. 2009. Antibiotic resistance. In Encyclopedia of microbiology, 3rd ed., edited by M. Schaechter. Oxford, United Kingdom: Elsevier. Pp. 193–204.
Phillips, I. 1999. The use of bacitracin as a growth promoter in animals produces no risk to human health. J. Antimicrob. Chemother. 44:725–8.
Prudhomme, M., L. Attaiech, G. Sanchez, B. Martin, and J. P. Claverys. 2006. Antibiotic stress induces genetic transformability in the human pathogen Streptococcus pneumoniae. Science 313:89–92.
Smith, C. J., M. B. Rogers, and M. L. McKee. 1992. Heterologous gene expression in Bacteroides fragilis. Plasmid 27:141–54.
Stobberingh, E. E., and A. E. van den Bogaard. 2000. Spread of antibiotic resistance from food animals to man. Acta Vet. Scand. 93(Suppl.):47–52.
Trieu-Cuot, P., E. Derlot, and P. Courvalin. 1993. Enhanced conjugative transfer of plasmid DNA from Escherichia coli to Staphylococcus aureus and Listeria monocytogenes. FEMS Microbiol. Lett. 109:19–24.109:19–24.
Vivan Miao and Julian Davies17
Abstract
The actinobacteria are arguably the richest source of small molecule diversity on the planet. These compounds have an incredible variety of chemical structures and biological activities (in nature and in the laboratory). Their potential for the development of therapeutic applications cannot be underestimated. It is suggested that an improved understanding of the biological roles of low molecular weight compounds in nature will lead to
|
15 |
Reprinted with kind permission from Springer Science + Business Media: Miao and Davies (2010). |
|
16 |
Keywords: Antibiotics · Bioactive molecules · Chemical diversity · Genomics · Molecular evolution · Natural products · Signaling · Therapeutics |
|
17 |
V. Miao and J. Davies (email) Department of Microbiology and Immunology, University of British Columbia, Vancouver, BC, Canada email: jed@interchange.ubc.ca. |
the discovery an inexhaustible supply of novel therapeutic agents in the next decade. To support this objective, a functional marriage of biochemistry, genomics, genetics, microbiology, and modern natural product chemistry will be essential.
The phylum actinobacteria, one of the largest groups in the domain Bacteria (Figure A5-1), largely consists of environmental bacteria and the denizens of many varied habitats: soils, the rhizosphere, marine and extreme arid environments. A number live in close association with higher organisms; for example, as components of different microbiomes they constitute more than a third of the healthy human microbiota. Members of the genus Frankia, on the other hand, can form symbiotic nodules in certain species of trees and shrubs, and fix atmospheric nitrogen to allow their hosts to survive in nutrient-limiting environments. Actinobacteria typically have elevated guanosine-cytosine contents (65-75% G + C) and their genome sizes range from the 2.5-Mb skin commensal Micrococcus luteus to the 9.7-Mb environmental strain Rhodococcus jostii. Since the discovery of antibiotics in the 1940s, the actinomycetes have received a great deal of attention, and Streptomyces species in particular have become renowned as the principal sources of therapeutic pharmaceuticals. There have been several good reviews on actinobacteria of late, notably that by Ventura et al. (2007) on evolutionary and genomic aspects, as well as occasional articles focusing on specific genera. Interest in the phylum in recent years is evidenced by the increasing number of citations; streptomycetes lead, of course, with the mycobacteria not far behind! However, other genera, including Rhodococcus, are beginning to excite more interest (Larkin et al., 2005, Kitagawa and Tamura, 2008) and who knows, Streptomyces may command less attention in the future.
Streptomycetes are demonstrably a rich source of compounds, but no more so than other members of the actinobacteria, also the Bacilli and bacterial genera such as the myxobacteria (Wenzel and Muller, 2009) and pseudomonads (Gross and Loper, 2009). Among the eukaryotes, fungal genomes are replete with biosynthetic gene clusters for encoding small molecule production. The ability to make bioactive small molecules is not exclusive to microbes. Plants are rich sources of a great variety of compounds that have been used as pharmaceuticals for millennia; this resource remains poorly understood and still largely untapped.
There is a global crisis in the treatment of infectious diseases; people are dying of infections that were previously treatable. Microbes are the source and the solution for the crisis, and for this reason it is imperative that the search for novel therapeutic agents be intensified. The constant moan of the pharmaceutical industry, that the natural reservoir of molecules with antibiotic activity is close to being exhausted and that they can no longer find useful bioactive compounds, is due in part to Waksman’s focus (see below) on the streptomycetes. It can also be explained by the inability to detect bioactive compounds when they are present only in low concentrations; the industry has found all the easily accessible
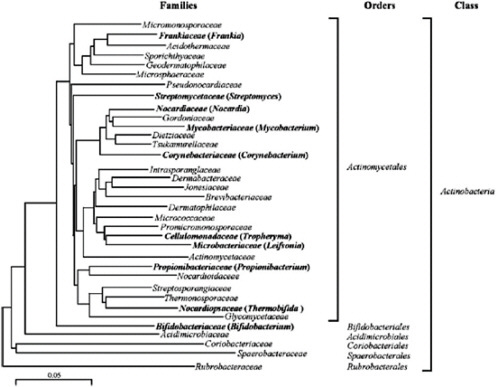
bioactives, the so-called “low hanging fruit” (Baltz, 2006). Presumably it was not considered essential to develop the technology to find compounds that were missed. In addition, actinobacteria as a whole have been ignored, even though they too possess the capacity to produce a huge number of bioactive small molecules; to date, only a small proportion have been examined for therapeutic purposes. We are now in the “genomic era” and in the case of streptomycetes, exciting new information coming from complete genome sequencing efforts reveals that most of these bacteria have the genetic capacity to produce many more structurally different bioactive compounds than suspected. As such, they represent an inexhaustible collection of hidden chemical and biochemical diversity. Moreover, creative techniques for generating some of these compounds are being developed and exploited (Baltz, 2008; Challis, 2008). We have no excuse for being short of compounds to screen (assuming that reliable screens are in place)! In this short article we present the case for a more extensive survey of the biology, properties and uses of natural molecules, especially those from other members of the actinobacteria. Now that we know the ubiquity and diversity of bioactive small molecules, the most important questions remain: “How do we find them?” and “What are their roles in nature?” When we have the answers to these questions, we will be far better equipped to harvest and exploit this vast chemical and biological wealth.
The “GOOD”
This characteristic refers mainly to the discovery and production of microbial small molecules with antibiotic activity that began with Waksman’s work on actinomycetes in the early 1940s. These seminal studies, together with the discovery of the fungal product penicillin by Fleming and co-workers and its characterization around the same time, were responsible for momentous and radical changes in medicine. A representative, but grossly incomplete, list is presented in Table A5-1; for more information see the review by Demain and Sanchez (2009). The availability of antimicrobial agents made possible, for the first time, the successful treatment of most types of infectious diseases. The discovery of antibiotics also presaged many other uses for microbial compounds in human and animal therapy and in agriculture. Recently these microbial sources have provided treatments for many non-infectious diseases including cancer and heart disease. Another role, often overlooked, is their use in prophylaxis and in immunosuppression prior to invasive surgery, which has been one of the most important factors in the development and success of organ transplantation. Hundreds of millions of dollars have been invested by pharmaceutical companies in saving the lives of millions of people—with profits of many billions of dollars! In addition, many actinobacterial strains have been developed for industrial applications such as bioremediation, the destruction of toxic xenobiotics, vitamins, fine chemical transformation and production and, more recently, for the development
TABLE A5-1 Some Beneficial Actinobacteria
|
Producing organism |
Compound |
Application |
|
Streptomyces aureofaciens |
Tetracycline |
Antibacterial |
|
Streptomyces griceus |
Streptomycin |
Antibacterial |
|
Streptomyces kanamycetius |
Kanamycin |
Antibacterial |
|
Streptomyces lactamdurans |
Cefotoxin |
Antibacterial |
|
Streptomyces mediterranei |
Rifamycin |
Antibacterial |
|
Streptomyces pristinaspiraelis |
Pristinamycin |
Antibacterial |
|
Streptomyces roseosporus |
Daptomycin |
Antibacterial |
|
Streptomyces spheroids |
Novobiocin |
Antibacterial |
|
Streptomyces venezuelae |
Chloramphenicol |
Antibacterial |
|
Amycolatopsis orientalis |
Vancomycin |
Antibacterial |
|
Micromonospora purpurea |
Gentamicin |
Antibacterial |
|
Saccharopolyspora erythraea |
Erythromycin |
Antibacterial |
|
Streptomyces avermitilis |
Ivermectin |
Antihelminthic |
|
Streptomyces clavuligerus |
Clavulanic acid |
β-Lactamase inhibitor |
|
Streptomyces hygroscopicus |
Bialophos |
Herbicide |
|
Streptomyces hygroscopicus |
Rapamycin |
Immunosuppressive |
|
Streptomyces noursei |
Nystatin |
Antifungal |
|
Streptomyces verticillus |
Bleomycin |
Anticancer |
of biofuel conversions. Novel uses of the extensive biosynthetic capacities of the Rhodococci are being discovered and one can predict their increasing importance as industrial microorganisms (Martinkova et al., 2009).
In spite of the numerous benefits accruing from these seemingly inexhaustible sources, the ecology and biology of actinobacteria and their roles in environmental communities are poorly understood and the functions of their myriad low-molecular-weight products in the environment are even less well studied. The development of these products as antibiotics led to the assumption that their primary (and only) function in nature was for use as molecular weaponry by their producers. The field was driven by the concept of antagonism: during the past half-century, their discovery and the proof of their biological activity relied solely on tests of their inhibition of the growth of other microbes under laboratory conditions. Given the number of bacterial genera and the inestimable number of compounds involved, this implies that the microbial world is nothing less than a constant theatre of war (Hibbing et al., 2009). There is very little sound evidence for this extreme concept and such an anthropocentric viewpoint needs to be discarded.
As with all biologically active compounds, the properties of bacterial products depend on the concentrations at which they are tested, immediately creating a dilemma: what exists and happens in nature is often quite distinct from what is found in a laboratory. Recent studies using sensitive promoter-reporter libraries or RNA microarrays have shown that at sub-inhibitory concentrations many microbial compounds modulate transcription patterns in a variety of bacterial and eukaryotic cells (Yim et al., 2006). Do the transcriptional effects provide the mechanistic basis for their wide range of biological activities? We believe so and have proposed that bioactive compounds act by binding to receptors in cells,
triggering cellular responses that are many and various; in other words they are cell-cell signaling agents (Davies et al., 2006; Fajardo and Martinez, 2008). In the past, studies of mechanisms of antibiotic activity in bacteria led to the identification of specific targets/receptor molecules and macromolecules. These include components of the cell wall, ribosomes, ribosomal RNA, DNA replication, RNA synthesis, as well as numerous enzymatic reactions, such as those involved in the synthesis of fatty acids. There is substantial genetic validation for these interactions and for the roles of single, specific targets in the cell.
Detailed studies with eukaryotic organisms are sparse, but recent results in studies of the different microbial populations that make up the human microbiome indicate that bioactive compounds play important roles in many aspects of human physiology that impact health and disease (Kaper and Sperandio, 2005). It can be predicted that studies of bacterial-mucosal and bacterial-tissue interactions and the role of bioactive small molecules in these processes will be pursued actively in the coming years. The native bacterial communities of humans and other organisms presumably use inter-cellular signaling mechanisms to modulate and control the activities of bacterial consortia and the essential interactions with their host. These interactions have significant implications on related issues such as the activity of probiotics and their roles in regulating immune responses. Similarly, the roles of small-molecule mediation in the operation of distributed metabolic networks in natural microbial communities is another topic that demands scrutiny in the future (Vallino, 2003). Will the next decade be the age of bioactive small molecules?
The evolutionary origins of the great diversity of bacterial products are poorly understood. How old are the actinomycetes? The photosynthetic cyanobacteria are associated with the appearance of oxygen in the earth’s atmosphere, and evidence of bacterial cells in fossilized stromatolites suggests that bacteria are as old as 2.7 Gya; the domain Bacteria includes the most ancient living organisms in the biosphere (Oren, 2004). Detection of hopanoids in ancient shales and also as cell membrane components that play a role in the structure of aerial hyphae in streptomycetes is another clue to the pathway of bacterial evolution (Taylor, 1984). Many actinomycetes, including streptomycetes and the Rhodococci, possess putative genes for gas vesicle production associated with the ability to survive in aqueous environments (as might be found under primordial conditions) (van Keulen et al., 2005).
Both the wide variety of amino acid derivatives found in meteorites and the seminal “primordial soup” experiments by visionaries such as Miller and Urey (Miller et al., 1976) provide chemical evidence for the presence of many types of non-protein amino acid derivatives in the prebiotic world. This leads to the probability that molecules similar to modern nonribosomal peptides are among the oldest bioactive molecules, as is borne out by their extant biological functions and their production by many types of microbes and plants. Their presence defined the evolutionary direction of the earliest forms of life. The evolution of
the biosynthetic pathways for nonribosomal peptides and other natural products such as the polyketides remains unclear, although tangible models for their being have been proposed (Nett et al., 2009; Ridley et al., 2008). The widespread use of similar classes of bioactive compounds in microbial and plant life, their coevolution and coexistence, are clearly of related interest.
From an historical point of view the first useful antibiotics to be discovered and used as such came not from actinomycetes but from members of the Bacilli! The peptide gramicidin was reported by Rene Dubos in 1938, and is still employed. (Check your local pharmacy if you don’t believe this.) There is every reason to believe that all bacteria have the capacity to make similar types of compounds; confirmation comes from the discovery of hybrid NRP-PK toxins produced by certain strains of E. coli (Putze et al., 2009). Recently, a novel non-ribosomal peptide derivative has been isolated from a strain of Staphylococcus (Magarvey, personal communication). This leads to the conclusion that the number of bioactive microbial compounds is, at a minimal estimate, equal to the number of microbial species; therefore, in terms of production of bioactives, all microbes are “good”. So much for suggestions that the supply is close to exhaustion!
We live in an occult universe of low-molecular-weight compounds. Suffice it to say, the antiquity of bioactive small molecules and their huge range of chemical space explains their ubiquity and enormous range of functions in cell biology. Paraphrasing the words of Douglas Adams, the author of The Hitchhikers Guide to the Galaxy: “Microbial chemical space is big. You just won’t believe how vastly, hugely, mind-bogglingly big it is!” As has been suggested on several occasions, the many roles of low-molecular-weight natural products justify their place as elements of the “central dogma” along with DNA, RNA, and protein (Schreiber, 2005). More focused efforts on their biology will reap many intellectual advances along with increasing medical and industrial applications.
However, a major stumbling block is the isolation and characterization of the organic compounds in this vast repertoire. Methods for the chemical identification of microbial products have improved significantly in recent years; however, it still requires an enormous effort to isolate, purify and determine the structures of natural products. Even with the most advanced instrumentation (nuclear magnetic resonance, mass spectroscopy, etc. and various combinations thereof), unraveling the structures of natural compounds remains a slow and highly specialized process. The throughput of current platforms does not in any way meet the needs for identifying thousands of diverse bioactive molecules with multiple biological roles. Until there is a revolutionary advancement in the structure determination process (akin to the effect of pyro-sequencing on genomics) studies of the world of small molecules will lag behind other fields. It is imperative that we decipher the language of small molecules in nature. This major undertaking would provide huge benefits, not the least, novel medicines and the identification of other bioactive molecules with applications in many areas of human and animal health and industry.
It is worth noting that an inventory of bioactive molecules will be only the prelude to developing the methodology required to systematically determine their functions, thereby effecting the metamorphosis of structural data into results and ultimately into molecular understanding. Precious little is known at present about the natural functions of these compounds.
One can identify antibiotics, siderophores, redox-active agents, transcription factors, transporters, and cell signals, etc. (Dietrich et al., 2008), but what are they actually doing in microbial population dynamics? The possibilities are many and the proposed functions must be confirmed under natural conditions (possibly using modern in situ imaging techniques). There is no doubt that working with well-characterized compounds will permit more sophisticated biochemical and genetic studies in target organisms, with the subsequent identification of unsuspected receptors and functions. If macromolecules such as the bacterial ribosome possess dozens of different receptor sites (Yassin and Mankin 2007) this will be a significant enterprise!
The “BAD”
We refer of course to parasitic strains that cause disease in other living organisms. From an evolutionary point of view any synergistic relationship can potentially lead to negative interactions; synergy with one partner or host can easily be translated into pathogenic interactions with another. The total number of known human and animal microbial pathogens is currently limited to a few thousand or so (including viruses); this is but a small percentage of the Bacteriaceae (Taylor et al., 2001). One can predict that the number may be much larger for plant pathogens.
We hear much about emerging pathogens in clinical studies: there are two broad classes: those organisms to which humans are newly exposed as a result of anthropogenic activities (reclaiming land, forest destruction, or social practices) and those dedicated pathogens that have recently acquired antibiotic resistance by mutation or horizontal gene transfer and thereby overcome/bypass the prevailing therapeutic options. Relatively few actinobacteria fall into the category of professional pathogens (that we know of) (Table A5-2).
Historically, M. tuberculosis is the most important pathogen and remains the most widely disseminated; there is evidence of human infection for 9,000 years (Hershkovitz et al., 2008). The total number of human deaths due to TB throughout history is not known, but it is estimated that M. tuberculosis caused at least 200 million deaths in the twentieth century (Kaufman and Van Helden, 2008). The first streptomycete-derived antibiotic and the most successful, streptomycin, was developed for the purpose of combating TB, the “White Plague”. At the present time, it is difficult to appreciate the incredible importance of the discovery of streptomycin; we have become nonchalant about the control of infectious diseases. Other critical drug discovery events in the 1940s built the reputation of
TABLE A5-2 Some Actinobacterial Pathogens (human, animal, and plant)
|
Mycobacterium avium |
Actinomyces bovis |
|
Mycobacterium avium complex |
Actinomyces israelii |
|
Mycobacterium bovis |
Clavibacter michiganensis |
|
Mycobacterium chelonae |
Corynebacterium diphtheria |
|
Mycobacterium fortuitum |
Leisonia xyli |
|
Mycobacterium leprae |
Nocardia asteroids complex |
|
Mycobacterium marinum |
Nocardia farcinia |
|
Mycobacterium tuberculosis |
Rhodococcus equi |
|
Mycobacterium ulcerans |
Streptomyces scabies |
|
Propionibacterium acnes |
Tropheryma whipplei |
|
Streptomyces somaliensis |
|
|
Streptomyces sudanensis |
|
the actinomycetes and established the bias towards this family of soil bacteria as producers of antibiotics. Although penicillin and several antibiotics from Bacillus spp. predated streptomycin in therapeutic use, they did not cure TB!
Other pathogenic mycobacteria are of significance, such as M. leprae (leprosy) and M. ulcerans (buruli ulcers). Among the actinomycetes, Rhodococcus equi has been recently identified as an equine infection that is an opportunistic pathogen for humans. Interestingly, the disease-causing actinomycetes evolved primarily by extensive genome reduction compared to their environmental precursors, rather than by the horizontal gene transfer of myriad pathogenicity islands associated with the Gram-negative pathogens such as E. coli, etc. M. tuberculosis is a notoriously difficult organism to work with due to its virulence, slow growth, and, until recently, lack of facile genetic manipulation; thus comparative studies of close relatives among the actinomycetes have provided important information on novel aspects of mycobacterial metabolism and mechanisms of virulence. For example, there is the important question of how M. tuberculosis survives in human macrophages. This has been revealed by comparative genomic analyses with the genome sequence of Rhodococcus jostii RHA1 that identified a gene cluster encoding a possible cholesterol degradation pathway (McLeod et al., 2006). The observation that this matched a closely related sequence in M. tuberculosis led to studies that have shown that the pathogen does indeed use cholesterol as a carbon source, providing critical information on its intracellular survival mechanisms and the possibility of novel targets for TB drug development (Van der Geize et al., 2007).
The “UGLY”
There are no ugly actinomycetes. However, for every silver lining there is a cloud, and this family is no different. It has been demonstrated that most of the common antibiotic resistance genes or their progenitors have their origins in environmental bacteria, and evidence suggests strongly that actinobacteria may be one of the main natural sources of clinically significant antibiotic resistance genes (Wright, 2007). On the other hand, the actinomycetes produce the
clavulanate-derived inhibitors of β-lactamases and also enzymes that degrade the acylhomoserine lactones, signal molecules that are responsible for the induction of virulence functions in a number of common pathogens. If we had been smart enough to recognize this fact earlier, it might have been possible to devise inhibitors of these resistance mechanisms and so defuse the pathogens prior to years of unrestricted antimicrobial therapy.
Afterthoughts
We have mentioned the “occult universe of small molecules” and will conclude with a few additional comments on this theme. The existence and the roles of low-molecular-weight organic compounds in biology have been all but ignored. Despite a century of studies of the chemistry, physiology, and critical roles of vitamins, neurotransmitters, pheromones, alkaloids, and other useful products of plants and animals, the chemical store of the microbial world remains a great mystery. Recent studies of the phenomenon of quorum sensing communication have taught microbiologists and chemists that small can be beautiful and meaningful (Atkinson and Williams, 2009; Winans and Bassler, 2008). However, quorum-sensing activities, like antibiotic effects, are still largely studied as laboratory phenomena that do not necessarily represent the environmental roles of organic compounds; it remains difficult to assess concentrations of the signaling compounds in the wild. What is needed is more science and much less anthropocentricity; the latter provides the substance of exciting movies but is bad science. (Admittedly, for convenience we have slipped into anthropomorphic mode by using the descriptors “good”, “bad” and “ugly”; this is almost as lamentable as saying that bacteria “decide”, or “make lifestyle choices”, phrases seen in many publications!)
Finally, the diversity and ubiquity of bioactive small molecules, their multitudinous sources, and their potential and critical roles in the functioning and interactions of all living things lead us to propose that there should be significant, targeted funding initiatives (and perhaps even institutes) devoted to their study: chemical (structural and synthetic), genetic, biological, physical, imaging, etc. Surely, the increasing interest in systems biology will benefit from a full understanding of small-molecule biology? We can do no better than to quote the proverb “from small beginnings come great things”.
Acknowledgments
We are grateful to Dorothy Davies for her patient editing assistance and Dr. Marco Ventura for permission to reproduce Figure A5-1. Funding has been provided by the National Science and Engineering Research Council, the Canadian Institutes for Health Research, Merck Research Laboratories, and the Tally Fund.
References
Atkinson S, Williams P (2009) Quorum sensing and social networking in the microbial world. J R Soc Interface 6:959–978
Baltz RH (2006) Marcel Faber roundtable: is our antibiotic pipeline unproductive because of starvation, constipation or lack of inspiration? J Ind Microbiol Biotechnol 33:507–513
Baltz RH (2008) Renaissance in antibacterial discovery from actinomycetes. Curr Opin Pharmacol 8:557–563
Challis GL (2008) Mining microbial genomes for new natural products and biosynthetic pathways. Microbiology 154:1555–1569
Davies J, Spiegelman GB, Yim G (2006) The world of subinhibitory antibiotic concentrations. Curr Opin Microbiol 9:445–453
Demain AL, Sanchez S (2009) Microbial drug discovery: 80 years of progress. J Antibiot (Tokyo) 62:5–16
Dietrich LEP, Teal TK, Price-Whelan A, Newman DK (2008) Redox-active antibiotics control gene expression and community behavior in divergent bacteria. Science 321:1203–1206
Fajardo A, Martinez JL (2008) Antibiotics as signals that trigger specific bacterial responses. Curr Opin Microbiol 11:161–167
Gross H, Loper JE (2009) Genomics of secondary metabolite production by Pseudomonas spp. Nat Prod Rep 26:1408–1446
Hershkovitz I, Donoghue HD, Minnikin DE, Besra GS, Lee OY, Gernaey AM, Galili E, Eshed V, Greenblatt CL, Lemma E, Bar-Gal GK, Spigelman M (2008) Detection and molecular characterization of 9000-year-old Mycobacterium tuberculosis from a neolithic settlement in the eastern Mediterranean. PLos One 3:e3426
Hibbing ME, Fuqua C, Parsek MR, Peterson SR (2009) Bacterial competition: surviving and thriving in the microbial jungle. Nat Rev Microbiol 8:15–25 Antonie van Leeuwenhoek
Kaper JB, Sperandio V (2005) Bacterial cell-to-cell signaling in the gastrointestinal tract. Infect Immun 73:3197–3209
Kaufman SHE, van Helden P (2008) Handbook of tuberculosis vol. 3: clinics, diagnostics, therapy and epidemiology. Wiley-VCH, Weinheim
Kitagawa W, Tamura T (2008) Three types of antibiotics produced from Rhodococcus erythropolis strains. Microbes Environ 23:163–171
Larkin MJ, Kulakov LA, Allen CC (2005) Biodegradation and Rhodococcus—masters of catabolic versatility. Curr Opin Biotechnol 16:282–290
Martinkova L, Uhnakova B, Patek M, Nesvera J, Kren V (2009) Biodegradation potential of the genus Rhodococcus. Environ Int 35:162–177
McLeod MP, Warren RL, Hsiao WW, Araki N, Myhre M, Fernandes C, Miyazawa D, Wong W, Lillquist AL, Wang D, Dosanjh M, Hara H, Petrescu A, Morin RD, Yang G, Stott JM, Schein JE, Shin H, Smailus D, Siddiqui AS, Marra MA, Jones SJM, Holt R, Brinkman FSL, Miyauchi K, Fukuda M, Davies JE, Mohn WW, Eltis LD (2006) The complete genome of Rhodococcus sp. RHA1 provides insights into a catabolic powerhouse. Proc Natl Acad Sci USA 103:15582–15587
Miller SL, Urey HC, Oro J (1976) Origin of organic compounds on the primitive earth and in meteorites. J Mol Evol 9:59–72
Nett M, Ikeda H, Moore BS (2009) Genomic basis for natural product biosynthetic diversity in the actinomycetes. Nat Prod Rep 26:1362–1384
Oren A (2004) Prokaryote diversity and taxonomy: current status and future challenges. Philos Trans R Soc B 359:623–638
Putze J, Hennequin C, Nougayre`de J-P, Zhang W, Homburg S, Karch H, Bringer M-A, Fayolle C, Carniel E, Rabsch W, Oelschlaeger TA, Oswald E, Forestier C, Hacker J, Dobrindt U (2009) Genetic structure and distribution of the colibactin genomic island among members of the family Enterobacteriaceae. Infect Immun 77:4696–4703
Ridley CP, Lee HY, Khosla C (2008) Evolution of polyketide synthases in bacteria. Proc Natl Acad Sci USA 105:4595–4600
Schreiber SL (2005) Small molecules: the missing link in the central dogma. Nat Chem Biol 1:64–66
Taylor RF (1984) Bacterial triterpenoids. Microbiol Mol Biol Rev 48:181–198
Taylor LH, Latham SM, Woolhouse MEJ (2001) Risk factors for human disease emergence. Philos Trans R Soc B 356:983–989
Vallino JJ (2003) Modeling microbial consortiums as distributed metabolic networks. Biol Bull 204:174–179
Van der Geize R, Yam K, Heuser T, Wilbrink MH, Hara H, Anderton MC, Sim S, Dijkhuizen L, Davies JE, Mohn WH, Eltis LE (2007) A gene cluster encoding cholesterol catabolism in a soil actinomycete provides insight into Mycobacterium tuberculosis survival in macrophages. Proc Natl Acad Sci USA 104:1947–1952
Van Keulen G, Hopwood DA, Dijkhuyizen L, Sawers RG (2005) Gas vesicles in actinomycetes: old buoys in novel habitats? Trends Microbiol 13:350–354
Ventura M, Canchaya C, Tauch A, Chandra G, Fitzgerald GF, Chater KF, van Sinderen D (2007) Genomics of actinobacteria: tracing the evolutionary history of an ancient phylum. Microbiol Mol Biol Rev 71:495–548
Wenzel SC, Muller R (2009) The impact of genomics on the exploitation of the myxobacterial secondary metabolome. Nat Prod Rep 26:1385–1407
Winans SC, Bassler BL (eds) (2008) Chemical communication among bacteria. ASM Press, Washington, DC
Wright GD (2007) The antibiotic resistome: the nexus of chemical and genetic diversity. Nat Rev Microbiol 5:175–186
Yassin A, Mankin AS (2007) Potential new antibiotic sites in the ribosome revealed by deleterious mutations in RNA of the large ribosomal subunit. J Biol Chem 282:24329–24342
Yim G, Wang HH, Davies J (2006) The truth about antibiotics. Int J Med Microbiol 296:163–170
A6
ANTIBIOTICS FOR EMERGING PATHOGENS18
Michael A. Fischbach19 and Christopher T. Walsh20,21
Antibiotic-resistant strains of pathogenic bacteria are increasingly prevalent in hospitals and the community. New antibiotics are needed to combat these bacterial pathogens, but progress in developing them has been slow. Historically, most antibiotics have come from a small set of molecular
|
18 |
Reprinted from Fischbach, M. A., and C. T. Walsh (2009). Antibiotics for emerging pathogens. Science 325(5944):1089-1093 with permission from AAAS. |
|
19 |
Department of Molecular Biology and Center for Computational and Integrative Biology, Massachusetts General Hospital, Boston, MA 02114, USA. E-mail: fischbach@fischbachgroup.org. |
|
20 |
Department of Biological Chemistry and Molecular Pharmacology, Harvard Medical School, Boston, MA 02115, USA. E-mail: christopher_walsh@hms.harvard.edu. |
|
21 |
Research in the authors’ laboratories is supported by the Department of Molecular Biology and the Center for Computational and Integrative Biology at Massachusetts General Hospital (M.A.F.) and by NIH grants GM20011 and GM49338 (C.T.W.). C.T.W. is a member of the board of directors of Achaogen (South San Francisco, CA). |
scaffolds whose functional lifetimes have been extended by generations of synthetic tailoring. The emergence of multidrug resistance among the latest generation of pathogens suggests that the discovery of new scaffolds should be a priority. Promising approaches to scaffold discovery are emerging; they include mining underexplored microbial niches for natural products, designing screens that avoid rediscovering old scaffolds, and repurposing libraries of synthetic molecules for use as antibiotics.
There is a perpetual need for new antibiotics: Whereas most drugs will be just as effective in the future as they are today, the inevitable rise of resistance will erode the utility of today’s antibiotics (Walsh, 2003). Two factors exacerbate this supply problem by creating unique disincentives for antibiotic development (Nathan et al., 2005). First, antibiotics are used in smaller quantities than other drugs. Prescriptions for chronic illnesses can last years or decades, whereas a standard course of antibiotics lasts only weeks; therefore, antibiotics yield lower revenues than most drugs. Second, whereas most newly approved drugs can be prescribed to all who would benefit, the use of a newly approved antibiotic may be restricted to the treatment of serious bacterial infections. The result is a quandary: Resistance is on the rise while antibiotic discovery and development are on the decline (Nathan, 2004; von Nussbaum et al., 2006).
The unfavorable economics of antibiotic development have had a chilling effect on industrial discovery programs, and policy-based efforts to reverse this decline deserve attention (Nathan, 2004). This perspective focuses on a different, yet no less formidable, challenge: finding new classes of antibiotics.
On the face of it, antibiotic discovery would seem to be straightforward. The goal is to kill an organism that is only distantly related to humans; unique, essential targets should be abundant, and novel antibiotics with low toxicity should be easy to find. Yet, the history of antibiotic development suggests otherwise. Since the early 1960s, only four new classes of antibiotics have been introduced, and none of these has made a major impact yet; the ~$30 billion global antibiotics market is still dominated by antibiotic classes discovered half a century ago. Since then, most “new” antibiotics have been chemically tailored derivatives of these well-worn scaffolds. In this review, we argue that the rise of resistant pathogens should redouble our focus on discovering not just new antibiotics, but new classes of antibiotics. We then highlight some promising approaches to scaffold discovery: mining under explored microbial niches for natural products, designing screens that avoid rediscovering old scaffolds, and repurposing libraries of synthetic molecules for use as antibiotics.
A New Generation of Resistant Pathogens
Three classes of antibiotic-resistant pathogens are emerging as major threats to public health (Figure A6-1). First, methicillin-resistant Staphylococcus aureus
FIGURE A6-1 Multidrug-resistant strains of these bacterial pathogens are on the rise.
SOURCE: Copyright Dennis Kunkel Microscopy, Inc.
(MRSA) is estimated to cause ~19,000 deaths per year in the United States (Klevens et al., 2007). Apart from their high mortality rate, MRSA infections lead to an estimated $3 billion to $4 billion of additional health care costs per year. Furthermore, the rising prevalence of MRSA increases the likelihood that vancomycin-resistant S. aureus (VRSA) (Weigel et al., 2003)—just as deadly as MRSA but more challenging to treat—will become a new scourge in hospitals.
Pathogens from the second class, multidrug-resistant (MDR) and pandrug-resistant (PDR) Gram-negative bacteria, are less prevalent than MRSA, but they pose the grave threat of infections that are truly untreatable (Falages et al., 2005). These strains of Acinetobacter baumannii, Escherichia coli, Klebsiella pneumoniae, and Pseudomonas aeruginosa are resistant to some (MDR) or all (PDR) of the antibiotic classes commonly used to treat Gram-negative bacteria: penicillins, cephalosporins, carbapenems, monobactams, quinolones, aminoglycosides, tetracyclines, and polymyxins (Falages et al., 2005). Prospects for finding new antibiotics for Gram-negative pathogens are especially poor: Their outer membrane blocks the entry of some antibiotics, and efflux pumps expel many of the remainder.
The third class comprises MDR and extensively drug-resistant (XDR) strains of Mycobacterium tuberculosis (MDR-TB and XDR-TB), which are a rising threat in the developing world (Dorman and Chaisson, 2007). MDR-TB treatment requires a 2-year course of antibiotics with serious side effects; XDR-TB is even more difficult to cure and often fatal (Kim et al., 2008). Cases of MDR-TB and XDR-TB have been reported in the United States and other developed countries.
In spite of the rise of resistant pathogens, the rate of new antibiotic approvals is dropping. Where will new antibiotics come from? In the past, this question has mostly been answered through synthetic tailoring of a small group of “scaffolds.”
Few Scaffolds, Many Generations of Tailoring
Members of each antibiotic class share a common core structure, or scaffold. For example, the cephalosporins share a β-lactam embedded in a fused 4,6-ring system (Figure A6-2). Most chemical scaffolds from which today’s antibiotics are derived were introduced between the mid-1930s and the early 1960s (Figure A6-3). Aside from the introduction of carbapenems in 1985, all antibiotics approved for clinical use between the early 1960s and 2000 were synthetic derivatives of existing scaffolds. Just four such scaffolds—cephalosporins, penicillins, quinolones, and macrolides—account for 73% of the antibacterial new chemical entities filed between 1981 and 2005 (Newman and Cragg, 2007).
During synthetic tailoring (Figure A6-2), the core of the antibiotic is left intact, preserving its activity, but the chemical groups at its periphery are modified to improve the drug’s properties. New generations are often designed to be active against pathogens that have become resistant to the previous generation. For example, second- (Neu and Fu, 1978) and third-generation (Dunn, 1982) cephalosporins like cefaclor and ceftazidime are more resistant to destruction by the resistance enzyme β-lactamase, and they can penetrate the Gram-negative outer membrane more effectively. When new β-lactamases emerged that can cleave third-generation cephalosporins, pharmaceutical companies developed fourth-generation molecules, like cefepime, that are less susceptible to cleavage by these enzymes (Garau et al., 1997). Cephalosporins and other semisynthetic antibiotics account for 64% of the new chemical entities filed between 1981 and 2005 (Newman and Cragg, 2007), suggesting that incremental synthetic tailoring of natural scaffolds has become the predominant mode of antibiotic discovery. The most useful scaffolds have therefore been those that are easy for medicinal chemists to tailor; this allows many derivatives to be synthesized and tested for improved properties.
Organic synthesis plays two other key roles in antibiotic discovery. First, scaffolds like the quinolones and oxazolidinones are derived entirely from chemical synthesis; these fully synthetic scaffolds account for an additional 25% of the
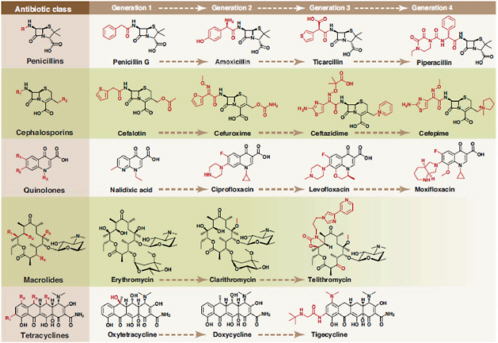
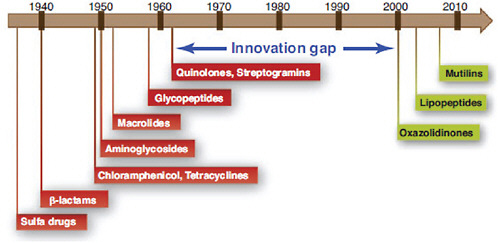
FIGURE A6-3 Between 1962 and 2000, no major classes of antibiotics were introduced.
antibiotic new chemical entities. Second, some natural scaffolds like carbapenems can now be produced entirely by organic synthesis, expanding the scope of accessible scaffold modifications.
The interplay between semisynthesis and total synthesis—and the ability of synthetic modifications to unlock the therapeutic potential of a scaffold—are exemplified by the tetracyclines. Resistance to this class of 30S-targeting antibiotics is mediated in part by a widely distributed gene encoding an efflux pump. Semisynthetic modifications to the tetracycline scaffold yielded the glycylcycline tigecycline (Figure A6-4) (Noskin, 2005). This third generation molecule (Figure A6-2) is no longer a substrate for the efflux pump, restoring its activity against tetracycline-resistant pathogens. A fully synthetic route to the tetracyclines (Charest et al., 2005) makes it possible to modify scaffold positions that are difficult to modify semisynthetically, further broadening the range of accessible derivatives.
Making incremental improvements to existing scaffolds is a good short-term strategy for refilling the antibiotic pipeline, but a presumably more sustainable way to combat resistance is to discover new scaffolds. Their utility will depend on three criteria: spectrum of activity against Gram-positive and Gram-negative pathogens, lack of cross-resistance to existing drugs, and amenability to generations of synthetic tailoring.
Next-Generation Scaffolds: Natural Products
More than two-thirds of clinically used antibiotics are natural products or their semisynthetic derivatives (Newman and Cragg, 2007). It is therefore troubling that natural product discovery efforts have waned in recent years (Li and
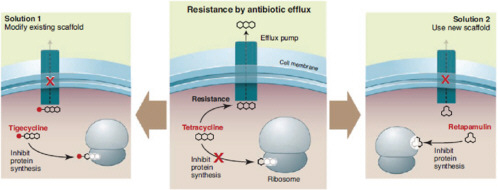
FIGURE A6-4 Surmounting resistance with scaffold alterations. Two ways of overcoming resistance are shown, using tetracycline (center) as an example. First, the tetracycline scaffold can be chemically modified, creating a tetracycline derivative like tigecycline that is no longer a substrate for the efflux pump (left). Second, a new scaffold like retapamulin, which is not a substrate for efflux and binds to a different site in the ribosome, can be used instead of tetracycline (right).
Vedesas, 2009); this decline is due in part to a rising rate of scaffold rediscovery (Baltz, 2006) and the accompanying difficulty in finding new antibiotics. Recent efforts to search new modalities—underexplored ecological niches, unmined bacterial taxa, and the genomes of even well-studied bacteria—have yielded novel molecules, whereas new screening strategies have begun to circumvent the time-consuming problem of rediscovery (Clardy et al., 2006).
New Places to Look
Most natural product antibiotics have come from soil actinomycetes, reflecting the historical bias of pharmaceutical screening programs toward these easily collected and cultured bacteria (Walsh, 2003). Searches of underexplored ecological niches and bacterial taxa have revealed new molecules. Marine niches are particularly promising; for example, a deep-sea sediment sample yielded an actinomycete that produces the abyssomicins (Bister et al., 2004), a new antifolate scaffold (Figure A6-5). Terrestrial and marine symbioses are also promising ecological niches; recent efforts to study bacterial symbionts of insects, ascidians, and fungi have yielded many new natural products (Donia et al., 2008; Partida-Martinez and Hertweck, 2005; Piel, 2009; Scott et al., 2008). Among underexplored bacterial taxa, myxobacteria are particularly prolific natural product producers, and their continued mining holds much promise for the discovery of new antibiotic scaffolds (Wenzel and Muller, 2009).
The genome sequences of a handful of actinomycetes and myxobacteria have revealed that these bacteria generally harbor >25 gene clusters encoding secondary metabolites. Given that only one to four natural products are known from a typical bacterium under various culture conditions, researchers may as yet have discovered only 10% of natural products from screened strains and just 1% of molecules from the global consortium of microbial producers (Watve et al., 2001). Taking this lesson to heart, several industrial and academic groups have carried out bioinformatics-based efforts to mine bacterial genomes for new natural products (Challis, 2008; McAlpine, 2009). Ecopia Biosciences (now Thallion Pharmaceuticals) has had particular success with their genome-scanning approach, including the discovery of ECO-0501, a new antibiotic scaffold (Banskota et al., 2006) (Figure A6-5). If the throughput of these genomics-based approaches to natural product discovery can be scaled up efficiently, their contribution to antibiotic discovery will be increasingly important.
Lastly, some promising candidate scaffolds for development may already be known. The founding members of the three most recently introduced antibiotic classes—mutilins, lipopeptides, and oxazolidinones—were each discovered at least 2 decades before they were introduced. Old patent literature seems a good place to start; on the basis of a 1985 patent from Eli Lilly, a group from Bayer recently isolated a series of acyldepsipeptide antibiotics that activate the bacterial chambered protease ClpP, leading to uncontrolled proteolysis and cell death
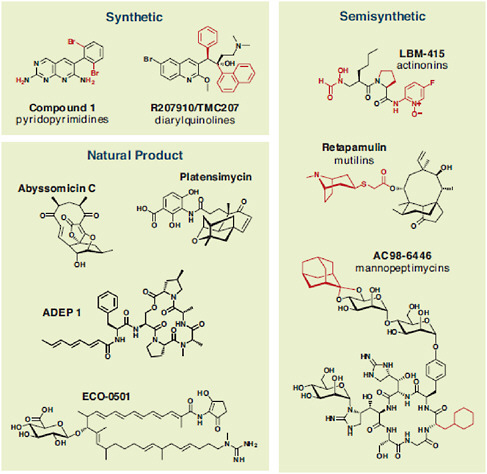
FIGURE A6-5 The chemical structures of new and underexplored antibiotic scaffolds mentioned throughout the text are organized by type into three categories: synthetic, semisynthetic, and natural product. For synthetic and semisynthetic scaffolds, core scaffolds are shown in black and variable positions are shown in red.
(Brotz-Oesterhelt et al., 2005) (Figure A6-5). Focusing development efforts on known but underexplored scaffolds can mitigate the risk of a costly and time-consuming de novo discovery program.
Combating Rediscovery
Out of 1000 randomly selected actinomycetes, about 10 will produce streptomycin, and 4 will produce tetracycline (Baltz, 2005). If extracts from these strains are screened against an indicator organism, most hits from the screen will be unhelpful rediscoveries. Two new screening strategies are beginning to
circumvent the problem of rediscovery. First, researchers at Cubist have developed a strain of E. coli that harbors resistance genes for the 15 most commonly rediscovered antibiotics (Gullo et al., 2006). Hits from their screening efforts are therefore preselected to be members of novel classes.
Second, a group at Merck has reported a bacterial antisense technology that allows them to knock down the expression of a given S. aureus gene, decreasing the amount of the encoded protein to the point that, in principle, it is present in growth-limiting quantities (Singh et al., 2007). Using this approach, they discovered platensimycin, the founding member of a new class of fatty acid biosynthesis inhibitors (Wang et al., 2006) (Figure A6-5), as well as several new protein synthesis inhibitory scaffolds.
Next-Generation Scaffolds: Synthetic Molecules
Fully synthetic molecules are a crucial component of the current antibiotic arsenal: The quinolones are highly effective broad-spectrum antibiotics, and the oxazolidinones are of increasing importance in the treatment of Gram-positive pathogens, including MRSA. However, recent efforts—based largely on high-throughput screens of novel targets identified by bacterial genomics—to discover and develop new synthetic scaffolds have not yet been successful (Payne et al., 2007).
Historically, synthetic scaffolds have originated outside of antibiotic discovery programs. The first drug in the sulfa class of antibiotics, Prontosil, was originally developed as a dye at Bayer, and the first quinolone was nalidixic acid, an intermediate in the synthesis of chloroquine. The oxazolidinones were discovered at DuPont as antibacterials but were originally developed to treat foliage diseases of plants.
Since the late 1990s, the rise of bacterial genomics held the promise of rejuvenating the discovery of synthetic antibiotics (Rosamond and Allsop, 2000). The genome sequences of pathogens like Haemophilus influenzae, S. aureus, Streptococcus pneumoniae, and E. coli made it possible to identify conserved enzymes that are essential for bacterial growth. These novel targets served as the basis for high-throughput screens of synthetic compound libraries, an approach that has been fruitful in other therapeutic areas. Genomics-based technologies have accelerated the process of identifying targets of existing drugs (Freiberg et al., 2005); however, they have not yet yielded new antibiotics (Payne et al., 2007; Mills, 2006).
Use External Libraries and a Whole-Cell Screen
The success of repurposing synthetic molecules from other development programs (Bogusli et al., 2009) and the failure of other approaches hold two important lessons for developing new synthetic antibiotics. First, look outside
antibacterial development programs for synthetic libraries to screen. Most pharmaceutical companies have invested considerable resources in synthesizing small molecule libraries for other therapeutic areas. Given the current level of uncertainty about which targets are relevant in an infected host (Brinster et al., 2009) and how antibiotics get into bacterial cells (Nikaido, 2003), libraries developed for other therapeutic areas may be just as likely to harbor hits as compound libraries developed for antibacterial screening.
Second, unbiased whole-cell screens have fewer pitfalls than other assays. The advantages of target-based screening—knowledge of the target and ease of optimization using a biochemical screen—are outweighed by the disadvantage of having to engineer cell permeability into a scaffold at a subsequent stage of the development process. Technologies like genome-wide expression profiling (Freiberg et al., 2005) and whole-genome resequencing of resistant mutants (Andries et al., 2004) have accelerated the bacterial infection, such as the hypoxia and oxidative stress that M. tuberculosis experiences in a host (Cho et al., 2007). A recent report has cast doubt on whether lipid synthesis is a viable target for Gram-positive pathogens; its authors argue that most models of infection fail to account for the fact that lipids in human serum can circumvent the inhibition of fatty acids synthesis (Brinster et al., 2009). Although future experiments will help resolve whether lipid synthesis inhibitors will be useful as drugs for Staphylococcus and Streptococcus, the mycolic acid pathway is already a well-validated target for M. tuberculosis. Any identified fatty acid synthesis inhibitors should therefore be tested against TB rather than being shelved for lack of efficacy against other Gram-positive pathogens.
A Recent Example of Success
A recent report from Pfizer demonstrates the utility of repurposing external compound libraries by screening them in a whole-cell antibacterial assay (Boguski et al., 2009). Miller and co-workers screened a one-million-compound library developed for eukaryotic protein kinase inhibition in an assay of E. coli killing, predicting that the low molecular weight ATP-mimetic molecules in the library might inhibit an essential bacterial enzyme and therefore exhibit antibacterial activity. They identified a set of pyridopyrimidines (Figure A6-5) that are subnanomolar inhibitors of the biotin carboxylase subunit of acetyl–coenzymeA (CoA) carboxylase (ACC), acting as competitive inhibitors of ATP binding. These molecules are selective for bacterial ACC over eukaryotic protein kinases and have potent activity against Gram-negative bacteria in vitro and in vivo. Similar efforts using other existing libraries could uncover new targets and scaffolds.
Is There Still a Role for Target-Based Antibiotic Discovery?
The failure of bacterial genomics to validate novel targets or yield new antibiotics has cast doubt on the utility of target-based discovery programs (Payne et
al., 2007; Mills, 2006). Nevertheless, retooled target-based strategies can play an important role in discovery. Examples include developing novel scaffolds for old targets and grouping new targets by inhibitor class.
A New Look at Old Targets
Most clinically used antibiotics inhibit enzymes from pathways that have been known for decades: peptidoglycan synthesis, ribosomal protein synthesis, folate synthesis, and nucleic acid synthesis and topoisomerization. Future generations of existing scaffolds should continue to have success in the clinic, and these classical targets will thus remain useful. However, a complementary and perhaps more promising strategy is to develop new scaffolds for these targets, thereby avoiding cross-resistance with existing drugs.
For example, the recently introduced mutilin retapamulin (Figures A6-4 and A6-5) targets the 50S subunit of the bacterial ribosome but is unaffected by resistance to other 50S-targeting classes like macrolides (Davidovich et al., 2007). Another target that deserves renewed focus is Lipid II; the success of glycopeptide antibiotics like vancomycin bodes well for other Lipid II–binding molecules like the mannopeptimycins (Figure A6-5) and lantibiotics (Breukink and de Kruijff, 2006).
Grouping Targets by Inhibitor Scaffold
To identify new targets, candidates are often grouped by a functional criterion, such as membership in a validated pathway or essentiality for growth in the laboratory. The attendant dangers of single-target bias (Payne et al., 2007) argue in favor of a strategy that begins with a wider funnel at its early stages.
A different way of grouping targets—by a common inhibitor scaffold rather than by pathway—may not only reveal new targets but also clues about how to inhibit them. For example, ATP binding enzymes are a group of targets that can be inhibited by ATP-mimetic scaffolds, and they deserve particular attention for two reasons.
First, bacterial genomes encode hundreds of ATP-binding proteins. They include well-validated targets like DNA gyrase, the target of the quinolones, and a host of new or underexplored targets: the chambered protease ClpP (Brotz-Oesterhelt et al., 2005), ATP synthase (Andries et al., 2004), aminoacyl-tRNA synthetases, and acyl-CoA carboxylase. The sensor kinase PhoQ is essential for the virulence of Salmonella (Bader et al., 2005), and several widely conserved essential genes encode proteins of unknown function that are predicted to bind ATP (Gerdes et al., 2003), suggesting that this class might include a particularly broad range of relevant targets. Insights from outside the antibiotic arena are also important for antibiotics; the observation that Zn-dependent hydrolases are efficiently inhibited by small molecules with Zn-chelating groups has led to the
development of inhibitors for a broad range of enzymes, including angiotensin-converting enzyme, histone deacetylases, and matrix metalloproteases. Indeed, semisynthetic derivatives of actinonin—a Zn-chelating natural product that inhibits the Zn-dependent bacterial enzyme peptide deformylase—have been considered as antibiotic candidates (Chen et al., 2000) (Figure A6-5).
Second, Miller and co-workers have demonstrated the feasibility of finding molecules from libraries of ATP-mimetic molecules that are selective for bacterial targets over human targets (Miller et al., 2009). Screening these libraries in whole-cell assays could simultaneously identify new targets and new lead compounds with scaffolds that can be optimized synthetically.
A More Inclusionary Approach?
In the heyday of antibiotic discovery, the pool of lead compounds was large enough for pharmaceutical companies to focus on broad-spectrum antibiotics for use as single-agent therapies and shelve compounds that failed these high therapeutic barriers. Today’s greater need for new antibiotics may encourage the development of lead molecules with characteristics that, until recently, have been seen as liabilities: narrow activity spectra and high intrinsic resistance rates.
The rule for antibacterial activity spectrum has been “broader is better.” However, the challenge of finding new broad-spectrum antibiotics and the rising threat from specific pathogens like MRSA have led to the development and approval of more agents with a narrower spectrum of activity, particularly those that kill Gram-positive but not Gram-negative bacteria. Extending this trend to near its logical limit, two groups recently reported Staphylococcus-selective antibiotics: One group used a repurposed series of eukaryotic cholesterol synthesis inhibitors to block the production of the gold pigment staphyloxanthin (Liu et al., 2008), from which the species name aureus is derived; the other group identified inhibitors of the tubulinlike protein FtsZ to block cell division (Haydon et al., 2008). It remains to be seen whether compounds with a spectrum this narrow find a therapeutic niche; one prerequisite for their use would be the availability of rapid diagnostics to identify the etiological agent of infection (Bootsma et al., 2006). Such genus-selective agents may have the benefit of sparing more of the endogenous microflora than conventional antibiotics, thereby avoiding complications like secondary Clostridium difficile infections.
Most bacterial infections are treated with a single antibiotic, ruling out the use of molecules with high intrinsic resistance rates. However, pairing these compounds into additive or synergistic combinations could rescue candidates formerly thought to be untenable for development. Although development of combination therapies carries the risk of unforeseen toxicity, precedents like amoxicillin-clavulanate and isoniazid-rifampicin-pyrazinamide-ethambutol all argue that antibacterial combination therapies can be quite successful, especially in suppressing the development of resistance. Whether natural or synthetic,
broad-spectrum or narrow, single agents or combinations, new scaffolds will be an essential component of a sustainable plan for combating resistance.
References
Andries, K. et al., Science 307, 223 (2005); published online 9 December 2004 (10.1126/science .1106753).
Bader, M. W. et al., Cell 122, 461 (2005).
Banskota, A. H. et al., J. Antibiot. (Tokyo) 59, 533 (2006).
Baltz, R. H., SIM News 55, 186 (2005).
Baltz, R. H., J. Ind. Microbiol. Biotechnol. 33, 507 (2006).
Bister, B. et al., Angew. Chem. Int. Ed. 43, 2574 (2004).
Boguski, M. S., K. D. Mandl, V. P. Sukhatme, Science 324, 1394 (2009).
Bootsma, M. C., O. Diekmann, M. J. Bonten, Proc. Natl. Acad. Sci. U.S.A. 103, 5620 (2006).
Breukink, E., B. de Kruijff, Nat. Rev. Drug Discovery 5, 321 (2006).
Brinster, S. et al., Nature 458, 83 (2009).
Brotz-Oesterhelt, H. et al., Nat. Med. 11, 1082 (2005).
Challis, G. L., J. Med. Chem. 51, 2618 (2008).
Charest, M. G., C. D. Lerner, J. D. Brubaker, D. R. Siegel, A. G. Myers, Science 308, 395 (2005).
Chen, D. Z. et al., Biochemistry 39, 1256 (2000).
Cho, S. H. et al., Antimicrob. Agents Chemother. 51, 1380 (2007).
Clardy, J., M. A. Fischbach, C. T. Walsh, Nat. Biotechnol. 24, 1541 (2006).
Davidovich, C. et al., Proc. Natl. Acad. Sci. U.S.A. 104, 4291 (2007).
Donia, M. S., J. Ravel, E. W. Schmidt, Nat. Chem. Biol. 4, 341 (2008).
Dorman, S. E., R. E. Chaisson, Nat. Med. 13, 295 (2007).
Dunn, G. L., J. Antimicrob. Chemother. 10 (suppl. C), 1 (1982).
Falagas, M. E. et al., BMC Infect. Dis. 5, 24 (2005).
Freiberg, C., H. P. Fischer, N. A. Brunner, Antimicrob. Agents Chemother 49, 749 (2005).
Garau, J., W. Wilson, M. Wood, J. Carlet, Clin. Microbiol. Infect. 3, S87 (1997).
Gerdes, S. Y. et al., J. Bacteriol. 185, 5673 (2003).
Gullo, V. P., J. McAlpine, K. S. Lam, D. Baker, F. Petersen, J. Ind. Microbiol. Biotechnol. 33, 523 (2006).
Haydon, D. J. et al., Science 321, 1673 (2008).
Kim, D. H. et al., Am. J. Respir. Crit. Care Med. 178, 1075 (2008).
Klevens, R. M. et al., JAMA 298, 1763 (2007).
Li, J. W., J. C. Vedesas, Science 325, 161 (2009).
Liu, C.-I. et al., Science 319, 1391 (2008); published online 14 February 2008 (10.1126/science .1153018).
McAlpine, J. B., J. Nat. Prod. 72, 566 (2009).
Miller, J. R. et al., Proc. Natl. Acad. Sci. U.S.A. 106, 1737 (2009).
Mills, S. D., Biochem. Pharmacol. 71, 1096 (2006).
Nathan, C., F. M. Goldberg, Nat. Rev. Drug Discovery 4, 887 (2005).
Nathan, C. Nature 431, 899 (2004).
Neu, H. C., K. P. Fu, Antimicrob. Agents Chemother. 13, 584 (1978).
Newman, D. J., G. M. Cragg, J. Nat. Prod. 70, 461 (2007).
Nikaido, H., Microbiol. Mol. Biol. Rev. 67, 593 (2003).
Noskin, G. A., Clin. Infect. Dis. 41 (suppl. 5), S303 (2005).
Partida-Martinez, L. P., C. Hertweck, Nature 437, 884 (2005).
Payne, D. J., M. N. Gwynn, D. J. Holmes, D. L. Pompliano, Nat. Rev. Drug Discovery 6, 29 (2007).
Piel, J., Nat. Prod. Rep. 26, 338 (2009).
Rosamond, J., A. Allsop, Science 287, 1973 (2000).
Scott, J. J. et al., Science 322, 63 (2008).
Singh, S. B., J. W. Phillips, J. Wang, Curr. Opin. Drug Discov. Dev. 10, 160 (2007).
von Nussbaum, F., M. Brands, B. Hinzen, S. Weigand, D. Habich, Angew. Chem. Int. Ed. 45, 5072 (2006).
Walsh, C. Antibiotics: Actions, Origins, Resistance [American Society for Microbiology (ASM) Press, Washington, DC, 2003].
Wang, J. et al., Nature 441, 358 (2006).
Watve, M. G., R. Tickoo, M. M. Jog, B. D. Bhole, Arch. Microbiol. 176, 386 (2001).
Weigel, L. M. et al., Science 302, 1569 (2003).
Wenzel, S. C., R. Muller, Curr. Opin. Drug Discov. Dev. 12, 220 (2009).
A7
AVERTING A POTENTIAL POST-ANTIBIOTIC ERA
Shelley Hearne22
The Pew Charitable Trusts
Introduction
Antibiotics save untold numbers of human lives every day. Modern medicine depends on our ability to treat and prevent infections. Yet a global crisis looms. Drug-resistant bacteria are spreading in our hospitals, our communities, and on our farms. Resistance is fueled by injudicious use of existing drugs and compounded by a failure to invest adequately in the development of new ones.
Dr. Thomas Frieden, director of the Centers for Disease Control and Prevention (CDC), has warned that we may be on the brink of “a post-antibiotic era” (Frieden, 2010). To prevent this warning from becoming an accurate prediction, we need to embrace what we already know from the science and heed the decades-long call to action by our leading health authorities and institutions, including the Institute of Medicine (IOM) and the World Health Organization (WHO). Several piecemeal legislative proposals exist that could address portions of the problem. But only a comprehensive policy framework designed to both preserve the efficacy of existing antibiotics and spur innovation of new drugs will provide a sustainable solution.
Antibiotic Resistance: An Inevitable and Growing Health Threat
Infections caused by bacteria can strike and kill anyone, including the young and the old, and the healthy and the chronically ill, but when antibiotics were discovered and developed beginning some 70 years ago, humanity turned a corner in
its ability to fight pathogens. Antibiotics quickly became the treatment of choice for staving off infections and saving lives.
But exposure to antibiotics inherently creates resistance among microorganisms (Levy, 2002; Wilkins, 1996). Their short generation time and the efficiency with which they develop and share resistance genes mean that no antibiotic remains effective forever (American Academy of Microbiology, 2009).
Resistance has increased rapidly among the major causes of bacterial illness in the United States, including Escherichia coli (Lewis et al., 2007), Salmonella (Winokur et. al., 2000), Campylobacter (Boucher et al., 2009), Enterococcus (McDonald, 2006), Streptococcus (Albrich et al., 2004), Staphylococcus (Klevens et al., 2007), and others (Rice, 2008).
One of the most widely known superbugs is methicillin-resistant Staphylococcus aureus (MRSA), which was once confined to already-vulnerable patients in hospitals and nursing homes. Now a community-acquired strain is also spreading among young, healthy individuals in everyday locations, among them schools, daycare centers, and locker rooms. Since 1998, the incidence of MRSA infections in children’s hospitals in the United States has increased 10-fold (see Figure A7-1) (Herigon et al., 2010). Researchers estimate that MRSA
FIGURE A7-1 Shifting balance. The number of hospital admissions with Staphylococcus aureus infections that remains sensitive (MSSA) to methicillin treatment has kept steady while that of resistant infections (MRSA) has been increasing.
SOURCE: Herigon et al. (2010). Reprinted with permission from Pediatrics, 125(6), pages e1294-e1300. Copyright © 2010 by the AAP.
alone causes almost 100,000 serious infections and 18,000 deaths every year in the United States (Klevens et al., 2007), and it costs $3 billion to $4 billion each year to treat (Fischbach and Walsh, 2009). Infections with resistant bacteria also result in longer and more costly hospital stays. Overall, antibiotic resistance was responsible for an estimated $16.6 billion to $26 billion per year in extra costs to the U.S. healthcare system (Roberts et al., 2009).
Other resistant infections are also on the rise and may pose even more serious challenges in the long term. The frequency of multidrug-resistant Acinetobacter baumannii infections, for example, is increasingly significant (Falagas et al., 2006; Munoz-Price and Weinstein, 2008) among U.S. military personnel returning from duty in Iraq and Afghanistan. Ordinarily, A. baumannii causes a variety of conditions, ranging from pneumonia to serious blood or wound infections, but in soldiers it also causes devastating prostheses infections and catheter-related sepsis (Crane et al., 2009). The bacterium is now spreading among patients in non-military U.S. hospitals and intensive care units (Perez et al., 2008). Some strains of A. baumannii are resistant to all known antibiotics, and estimates of death rates from resistant Acinetobacter infections range from 30 to 40 percent. A strain of the tuberculosis bacterium, extensively drug-resistant tuberculosis (TB), which has not yet become prevalent in the United States, is resistant to all currently available TB drugs, and is virtually untreatable (Shah et al., 2007). WHO warns that widespread multidrug resistance is making gonorrhea increasingly hard to treat (Tapsall, 2009).
A Dearth of Innovation Just When It Is Needed
Increasing the likelihood of a post-antibiotic future is an innovation slow-down: the pipeline of drugs to replace ineffective antibiotics has dwindled to a trickle (Boucher et al., 2009; Spellberg et al., 2004). Many major pharmaceutical companies have abandoned the antibiotics business in favor of medicines promising greater profits. Companies that remain engaged face both scientific and regulatory barriers that are compounded by limited return on investment.
Development of a new pharmaceutical costs hundreds of millions of dollars for basic and clinical research, including the investments related to drug candidates that fail. For antibiotics, revenue is limited by the fact the drugs tend to be short-course therapies that are completed in days, weeks, or at most months. Compared to revenues generated from sales of high blood pressure or cholesterol medications that patients take for many years or a lifetime, returns from antibiotics are low. Even an effective new treatment for MRSA, such as daptomycin (Cubicin®), is estimated to generate annual revenues of more than $500 million—which is not insubstantial—this is far below, for example, estimated revenues for a mid-market antipsychotic drug (Cubist Pharmaceuticals, 2010).
Another problem derives from a paradox of sorts. In an effort to preserve the effectiveness of a good new drug, clinicians will often use it only infrequently. In
this way, they aim to stave off the emergence of resistance to the new antibiotic, at least until the usefulness of older drugs is exhausted. This is an appropriate and prudent antibiotic-preserving practice, but it further diminishes the flow of at least near-term revenue that an antibiotic developer might expect from rolling out a new product.
An additional economic barrier to antibiotic development is the cost of regulatory approval, which has increased in recent years due to revisions and more stringent standards for clinical trials instituted by the Food and Drug Administration (FDA). Pharmaceutical companies argue that their inability to predict changes in FDA’s requirements prevents them from effectively planning for approval time and costs and further deters them from development of new antibiotics. The Pew Health Group has interviewed companies small and large, and the discussions suggest that this lack of clarity from the FDA about the standard of evidence required for approval discourages companies from pursuing innovative approaches to new antibiotics.
Growing Resistance from Injudicious Use
Overuse of antibiotics by doctors and their patients has long been a major threat to antibiotic efficacy. The medical community has known for decades that some practices, among them the repeated and inappropriate use of antibiotics in clinical settings, are a primary factor in the accelerated rise of antibiotic-resistant bacteria (Costelloe et al., 2010). Numerous international programs are successfully reducing antibiotics use and resistance through public and physician education and improved vaccination guidance (Anonymous, 2008; Goosens et al., 2008). In the United States, efforts to promote more judicious use of antibiotics in children with acute respiratory tract infections appear also to be having positive outcomes (Finkelstein et al., 2003; Grijaldi et al., 2009). That is a start, but countless people with viral infections (such as colds and influenza) incorrectly believe that an antibiotic will help, and they lobby their physicians for these treatments. Additionally, the medical community has underemphasized the strategy of preventing infectious disease as a way of reducing antibiotic use and thereby prolonging the drugs’ efficacy.
One critical but less appreciated and understood part of the microbial resistance dynamic is the long-term and unnecessary use of antibiotics in food animal production (Cohen and Tauxe, 1986; Sarmah et al., 2006). In the United States, it is common for growers of swine, poultry, and, to a lesser degree, cattle to administer low, sub-therapeutic doses of antibiotics to healthy food animals in their feed or water to encourage faster growth and as a prophylactic measure to hedge against overcrowding and other unsanitary and disease-friendly conditions. In addition, in contrast to Europe, most of the antibiotics used on industrial farms in the United States are obtained and administered without the consultation of a veterinarian.
Precise data about antimicrobial use in food animal production in the United
States is not publicly available, but analysts say existing data suggest U.S. consumption of antibiotics for these purposes greatly outpaces that of European countries (Aarestrup et al., 2010). Estimates indicate that the non-therapeutic use of antibiotics accounts for anywhere from 35 to 70 percent of all antibiotics sold in the United States (American Health Institute; Mellon et al., 2001)23. In terms of annual quantities, the mass of antibiotics used in animals amounts to between 100 and 1,000 times that used in humans (Feinmen, 1998; Levy, 1998; Witte, 1998). This makes the United States one of the biggest users of antibiotics in food animal production on a pound-per-pound basis in the world (see Figure A7-2) (Aarestrup, 2009; DANMAP, 2008).
Here in the United States, food animal producers use many antibiotics that are similar or even identical to those used in human medicine, among them penicillins, tetracyclines, macrolides, and sulfonamides (Chee-Sanford et al., 2009). This practice encourages the proliferation of resistance to the very drugs that doctors now rely on to save their patients’ lives (Ho et al., 2010). Administering human drugs to animals at sub-therapeutic dosages is like giving evolutionary intelligence away to pathogens so that they will be able to more quickly counter the drugs that might otherwise kill them off in a patient’s body.
Over the last four decades, researchers have demonstrated that feeding antibiotics to healthy food animals over a long period of time promotes the development of dangerous strains of drug-resistant bacteria that can spread to humans (Angulo et al., 2004; McDonald et al., 2001). Here are some of the seminal findings:
-
In 1969, a ground-breaking report from the United Kingdom concluded that the use of antimicrobials in food animal production, especially when used for growth promotion, was of great concern and should be limited and in some cases excluded from animal use altogether (Swann et al., 1969). Since then, a growing body of research has continually strengthened that conclusion.
-
In the 1970s, researchers gave chickens a diet that included tetracycline-supplemented feed. After 6 months, the scientists found that both the chickens and the farm workers were colonized with tetracycline-resistant bacteria (Levy et al., 1976).
-
Studies in the 1980s linked multidrug-resistant Salmonella infections in humans with exposure to cattle on dairy farms (O’Brien et al., 1982). Further studies and molecular subtyping revealed widespread emergence of resistance in Salmonella infections in humans in the United States, which researchers concluded were likely from food animals (GAO, 2004).
-
During the late 1990s and early 2000s, contemporaneous food and hospi-
-
tal surveillance linked the introduction of fluoroquinolones to U.S. broiler production with the emergence of resistant Campylobacter infections in humans (GAO, 2004). Although the use of fluoroquinolones in poultry was banned in the United States in 2005, this class of antibiotics is still approved for therapeutic use in cattle and swine and thus continues to exert a selective pressure for the emergence of fluoroquinolone-resistant bacteria that can be transferred to humans. Likewise, a broad collection of antibiotics beyond the fluoroquinolones continues to be approved for use in U.S. broiler production.
Recent data from North America also point to the public health benefits of reducing antibiotics in food production. In 2009, for instance, researchers in Canada found that removing third-generation cephalosporins from broiler hen production resulted in significant reductions in contamination of retail poultry products with ceftiofur-resistant Salmonella enterica and E. coli as well as commensurate reductions in third-generation cephalosporin-resistant Salmonella enterica infections in humans (Dutil et al., 2010).
Buying Time by Using Antibiotics More Judiciously
Some countries have been enacting laws and implementing agricultural practices that help protect the efficacy of antibiotics as well as the interests of food animal producers. In 2006, the European Union banned the use of antibiotics and related drugs for growth promotion purposes in livestock. Beginning in the late 1990s, Denmark became the leader in scaling back the routine use of antibiotics in industrial farming when it instituted a series of policy strategies for preventing antibiotic resistance in humans and animals. Since then, the country has experienced tremendous productivity growth in its swine production, little economic impact, and evidence of lower resistance rates in human and animal pathogens. Denmark is one of the world’s largest pork exporters, accounting for 17 percent of the global export market for pork as well as 22 percent of the world’s exports of bacon and ham (Hamann, 2006). Today, the Danish business interests and farmers are supportive of the actions to limit antimicrobial use in agriculture.
The Danish law ensured that antibiotics remained available to veterinarians for treating sick animals. To prevent misuse, the law stipulated that antibiotics for use in food animals must be accompanied by a prescription from a veterinarian acquired through a valid veterinarian-client-patient relationship and never used for growth promotion.
A key Danish provision prohibited veterinarians from selling antibiotics. In the United States, physicians have long been barred from selling pharmaceuticals to patients because this would constitute a conflict of interest. This restriction also serves as a critical means of avoiding overprescribing. In the United States, veterinarians generate much of their revenue in the sale of pharmaceuticals. This too
poses a conflict of interest and an incentive to use antimicrobials. Although this practice of serving simultaneously as both the animals’ doctor and the animals’ pharmacist should be eliminated, policy makers should address veterinarians’ potential loss of income with stricter requirements for actual animal veterinarian visits for prescribing and other incentives.
With pressure growing for the United States to adopt similar antibiotic restrictions, agribusiness opposition has also begun to mount. The American Veterinary Medical Association, for example, claims that the European antibiotic phase-out has caused increased animal deaths and economic hardship on livestock and poultry producers (American Veterinary Medical Association, accessed August 13, 2010).
Research by Danish scientists reveals that antibiotic consumption per kilogram of swine production on industrial farms dropped by more than half between 1992 and 2008, while production increased by 47 percent, from 18.4 million hogs in 1992 to 27.1 million in 2008 (Figure A7-3) (Aarestrup et al., 2010). At the same time, antibiotic-resistant bacteria in food animals have become less prevalent.
The WHO concurs that the antibiotics ban in Denmark has reduced the risks to human health without making a significant financial impact (WHO, 2002). Data from industry and from the Danish government reveal that livestock and poultry production has increased since the ban while antibiotic resistance on farms and in meat has declined (Hammerum et al., 2007; Letter from Dr. Jan Mousing, Chief Veterinary Officer of Denmark, to Congress, August 12, 2009). U.S. industry has expressed alarm over increased treatment of diarrhea and a rise in mortality in weaner pigs in the few years immediately after the ban. The WHO found that diarrhea in young pigs did increase following the ban, creating a short-term need to increase therapeutic antibiotic use. However, levels of diarrhea treatment began to decline after 7 months and were back to the pre-ban levels after 1 year. Weaner mortality has improved considerably in recent years (WHO, 2003).
The Danish Integrated Antimicrobial Resistance Monitoring and Research Programme (DANMAP) confirms that, in general, the numbers of antibiotic-resistant microbes in food animals rise and fall with changes in antibiotic usage (DANMAP, 2000 and 2008). For example, data reported by DANMAP in 2008 indicated that decreases in neomycin, spectinomycin, and macrolide use in pig farming correlated with declines in neomycin, spectinomycin, and erythromycin (a macrolide antibiotic) resistance in bacterial isolates from the pigs. Likewise, elimination of avoparcin (related to vancomycin) and virginiamycin (related to Synercid) as growth promoters resulted in significant reductions in bacteria resistant to these two critical antibiotics among poultry and swine. Similarly, a 24 percent increase in apramycin use in swine feed between 2006 and 2008 correlated with an increase in apramycin/gentamicin cross-resistance among S. typhimurium isolates from pigs (Jensen et al., 2006). And an increase in the use of tetracycline in pigs corresponded with an increase in tetracycline resistance
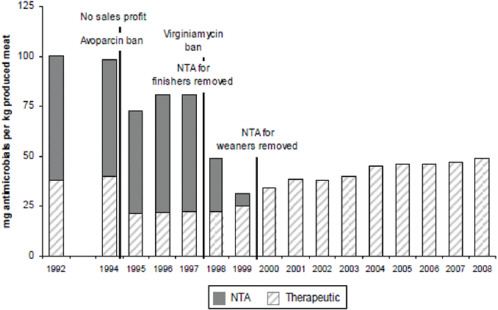
FIGURE A7-3 Danish laws limiting antimicrobial use in swine production resulted in a dramatic decline in non-therapeutic (NTA) use of these agents (dark gray) as well as an overall decline in antibiotic use per kilogram of meat produced.
SOURCE: Adapted from Aarestrup et al. (2010; 71(7): 726, Fig 2, p 730), with permission of the AVMA.
among S. typhimurium isolates from pigs and among human bacterial infections (DANMAP, 2000).
Surveillance data from the U.S. National Antibiotic Resistance Monitoring System reveals that resistance rates to some of the most commonly used antibiotics are high among bacteria from food animals. For example, retail meat and poultry surveys indicate that well above 50 percent of the Enterococcus isolates that contaminate these products are resistant to the streptogramins, tetracyclines, and lincosamides routinely used in poultry production. Likewise, aminoglycoside and tetracycline resistance is common among E. coli isolates from these same products. Reducing non-therapeutic antibiotic use will likely decrease resistance and increase the utility of these drugs for disease therapy.
Industry groups have argued that a ban on using human drugs as growth promoters on farms would lead to higher food prices for consumers. A study by the National Research Council indicates that a ban would have a negligible effect, increasing the price of meat by an estimated 0.013 to 0.06 dollars per pound. For consumers, this translates to $4.84 to $9.72 per person each year (National Research Council, 1999). In Denmark, consumers have not had price increases related to antimicrobial restrictions.
The Denmark example shows that it is possible to raise food animals profitably even while reducing the presence of antibiotic-resistant bacteria by eliminating unnecessary antibiotic use (Aarestrup et al., 2001). Pork producers initially opposed the ban but now acknowledge its successful implementation.
Owing to poor regulations and lack of oversight of drug use in food animal production, U.S. consumers do not know what their food is treated with or how often. Nor is there an adequate system in place to test meat and poultry for dangerous antibiotic-resistant bacteria. Government, industry, and professional leaders need to monitor and regulate the use of antibiotic drugs in poultry and livestock more carefully in order to limit the development of resistant bacteria in food animals and the unnecessary threat it poses to people. Any comprehensive antimicrobial preservation and discovery bill would have to make provisions for these functions.
The Way Forward
The rising tide of resistant infections demands a comprehensive policy response (Laxminarayan and Malini, 2007). The failure of the market to deliver effective new treatments must be addressed (Gilbert et al., 2010). But with the pipeline nearly empty, policy makers also must act to preserve the waning effectiveness of existing drugs. That requires a multipronged response to improve infection control and reduce injudicious use of antibiotics in both humans and animals (IOM, 1998). None of these elements can be effective in isolation.
Under the leadership of its co-chairs, the late Nobel Laureate Joshua Lederberg, and Margaret Hamburg, now Commissioner of the FDA, the IOM produced a consensus blueprint on how to best address the global crisis of reemerging microbial infections. That report, Microbial Threats to Health: Emergence,
Detection, and Response, included a series of commonsense policy strategies for addressing antibiotic resistance and the need for new antimicrobial drugs (IOM, 2003):
-
Limit antimicrobial use to medical situations in which their use will yield results. This means ending the practice of prescribing antibiotics to merely appease patients or because it has become the normal thing to do.
-
Discourage misuse, such as poor compliance by patients or low-dose regimens that only accelerate the rise of resistance bacteria. Specifically, the IOM urged the FDA to ban any classes of antibiotics used in human medicine from being used as growth promoters in the livestock and poultry.
-
Reduce the need for antibiotic treatment by reducing the rates of infection through better hygiene, vaccines, and other disease-prevention measures.
-
Develop and enact policies and incentives to spur innovation in new antibiotics and other tactics for treating infections.
These fundamental consensus points can provide the basic building blocks for comprehensive legislation that could preserve the medical value of antibiotics while fueling the next generation of therapies for microbial infection. Such a bill would recognize that antibiotics are a vital, shared public resource. Only a comprehensive policy framework designed to both preserve the efficacy of existing antibiotics and spur innovation of new drugs will provide a sustainable solution.
Currently, numerous specific legislative proposals exist that link to different components of the IOM framework:
-
The Strategies to Address Antimicrobial Resistance (STAAR) Act, backed by the Infectious Diseases Society of America (IDSA), would bolster existing surveillance, data collection, and research. It would strengthen the public health infrastructure essential to the long-term management of antibiotic-resistant diseases in such settings as hospitals, clinics, veterinarians’ offices, and animal production operations.
-
The Preservation of Antibiotics for Medical Treatment Act (PAMTA) of 2009 would phase out the routine use of seven classes of medically important antibiotics (penicillins, tetracyclines, macrolides, lincosamides, streptogramins, aminoglycosides, and sulfonamides) in healthy food animals unless manufacturers can prove reasonable certainty of no danger to public health from resistance. New drugs are required to meet the same standard. PAMTA critically shifts the burden of proof to the drug manufacturers to ensure antibiotics used in farm animal production have no human health impacts.
In addition to provisions like those in STAAR and PAMTA to curtail and manage antimicrobial resistance, a comprehensive antimicrobial preservations and discovery bill must include a set of powerful incentives to spur innovation by scientists and pharmaceutical companies to develop new antibiotics, better diagnostics for use at the “point of care,” and vaccines to prevent bacterial infections.
Public policy has long played a role in antibiotics innovation, beginning with the public-private partnership that led to the large-scale introduction of penicillin in 1944. Congress has taken a number of approaches to encourage pharmaceutical investment. The Orphan Drugs Act, passed in 1983, stimulates the development of drugs for rare but serious disorders, using a mix of pre-market, or “push,” incentives, such as research and development (R&D) tax credits and help with the cost of clinical trials, as well as post-market “pull” incentives, including longer periods of exclusivity during which the drug does not face generic competition. Similarly, the Best Pharmaceuticals for Children Act provides companies with extended exclusivity in exchange for conducting pediatric research on their products. And in 2006, Congress moved to create the Biomedical Advanced Research and Development Authority to facilitate the public- and private-sector R&D of antimicrobials and other emergency countermeasures to respond to potential bioterrorist, pandemic, or other urgent medical threats.
Any successful policy-driven effort to stimulate antibiotic development will have to recognize that new drug candidates may originate and move through a variety of pathways, including large pharmaceutical companies, small and mid-sized companies, or academic laboratories. Each enterprise will have distinct needs and may respond to different types of incentives. Therefore, the legislation will need to include a range of mechanisms. In addition to the specific measures mentioned above, possible innovation incentives include funding to defray cost of clinical development or approvals; grants or partnerships to facilitate transitional research; technical assistance, particularly for small companies that would benefit from help in navigating the federal agencies that facilitate drug development and approval; and advance market commitments, by which the sales volume of a product is guaranteed in advance.
It also is essential that the FDA provide clarity on the standards for approval of new antibiotics, which have been repeatedly revised in recent years. Approval standards must remain rigorous and scientifically appropriate in order to protect patients, but pharmaceutical companies must feel confident that they can embark on drug development with some predictable understanding of the scale and cost of the required trials.
Besides enacting antibiotics-preserving policy, better professional practices and behavior regarding antibiotic use is a must. In this regard, the FDA, the CDC, and professional health organizations, academia, agribusiness, and the pharmaceutical industry should increase their efforts to reduce the inappropriate use of antimicrobials in human and animal medicine. Important tactics here include
renewed efforts in outreach and improved education of healthcare professionals and the public about the dangers resulting from the misuse and overuse of antibiotics. These organizations also should encourage the development and routine use of rapid diagnostic tests to determine the specific viral or microbial causes of infections and ensure appropriate treatments are applied.
As Joshua Lederberg said repeatedly: “In the race against microbial genes, our best weapon is our wits, not natural selection on our genes” (Lederberg, 1997).
Scientists, physicians, and public health experts agree. The WHO, the American Academy of Pediatrics, the American Nursing Association, the American Society of Microbiology, IDSA, and the American Medical Association all echo the IOM recommendations and have repeatedly called for a policy response to the crisis of antimicrobial resistance. Yet, as a nation, we have failed to take action.
Donald Kennedy, president emeritus of Stanford University who served as commissioner of the FDA from 1977 to 1979, proposed eliminating the use of penicillin and tetracycline as growth promoters in food animals more than 30 years ago. Kennedy and Stanley Falkow, one of the nation’s leading microbiologists, later described the antibiotic debate as a “struggle between good science and strong politics” (Kennedy and Falkow, 2001). Agribusiness proponents of this application won that policy decision by pressuring Congress to shelve the FDA proposal to limit the practice. Kennedy and Falkow concluded that “science lost.”
With the impending threat to our crown jewel in medicine—antibiotics—we cannot afford to let science lose. Now, more than ever, we need to ensure that science effectively informs and drives our antibiotic policy strategies, not politics. In 1863, with the approval of President Abraham Lincoln, the U.S. Congress chartered the National Academy of Sciences for this very purpose: to advise the federal government on scientific and technological matters. As such, the nation needs the IOM to play a more prominent role in translating science for policy makers and advancing their existing recommendations. If the IOM does not lend a voice to its findings, not only will science lose again, but we may be sped along to Frieden’s dire predictions of a post-antibiotic era.
Acknowledgments
This manuscript was prepared with valuable assistance from Merry Buckley, Lance Price, and Pew Health Group staff members Allan Coukell, Laura Rogers, Gail Hansen, and Ivan Amato.
References
Aarestrup, F. M. 2009. Letter to Representative Pelosi and Powerpoint Presentation associated with visit on September 9, 2009, by a delegation of the Danish Technical University with four members of the House of Representatives. http://www.louise.house.gov/images/stories/attachments/2009.10.01.pamta.pdf (accessed August 13, 2010 ).
Aarestrup, F. M., A. M. Seyfarth, H. D. Emborg, K. Pedersen, R. S. Hendriksen, and F. Bager. 2001. Effect of abolishment of the use of antimicrobial agents for growth promotion on occurrence of antimicrobial resistance in fecal Enterococci from food animals in Denmark. Antimicrobial Agents and Chemotherapy 45(7):2054–9.
Aarestrup, F. A., V. F. Jensen, H.D. Emborg, E. Jacobsen, and H. C. Wegener. 2010. Changes in the use of antimicrobials and the effects on productivity in swine production in Denmark. American Journal of Veterinary Research 71(7):726–23.
Albrich, W. C., D. L. Monnet, S. Harbarth. 2004. Antibiotic selection pressure and resistance in Streptococcus pneumoniae and Streptococcus pyogenes. Emerging Infectious Diseases 10(3):514–17.
American Academy of Microbiology. 2009. Antibiotic resistance: An ecological perspective on an old problem. Washington, DC: American Academy of Microbiology.
American Veterinary Medical Association. Antimicrobial use and antimicrobial resistance FAQ. http://www.avma.org/public_health/antimicrobial_use.asp (accessed August 13, 2010).
Angulo, F. J., V. N. Nargund, and T. C. Chiller. 2004. Evidence of an association between use of anti-microbial agents in food animals and anti-microbial resistance among bacteria isolated from humans and the human health consequences of such resistance. Journal of Veterinary Medicine Series B 51(8–9):374–9.
Anonymous. 2008. Recent trends in antimicrobial resistance among Streptococcus pneumoniae and Staphylococcus aureaus isolates: The French experience. Eurosurveillance 13(46):1–6.
Boucher, H. W., G. H. Talbot, J. S. Bradley, J. E. Edwards, Jr., D. Gilbert, L. B. Rice, M. Scheld, B. Spellberg, and J. Bartlett. 2009. Bad bugs, no drugs: No ESKAPE! An update from the Infectious Diseases Society of America. Clinical Infectious Diseases 48(1):1–12.
Chee-Sanford, J. C., R. I. Mackie, S. Koike, I. G. Krapac, Y. F. Lin, A. C. Yannarell, S. Maxwell, and R. I. Aminov. 2009. Fate and transport of antibiotic residues and antibiotic resistance genes following land application of manure waste. Journal of Environmental Quality 38(3):1086–108.
Cohen, M. L., and R. V. Tauxe. 1986. Drug resistant Salmonella in the United States: An epidemiologic perspective. Science 234(4479):964–9.
Costelloe, C., C. Metcalfe, A. Lovering, D. Mant A. D. Hay. 2010. Effect of antibiotic prescribing in primary care on antimicrobial resistance in individual patients: Systematic review and meta-analysis. British Medical Journal 340:c2096.
Crane, D. P., K. Gromov, D. Li, K. Søballe, C. Wahnes, H. Büchner, M. J. Hilton, R. J. O’Keefe, C. K. Murray, and E. M. Schwarz. 2009. Efficacy of colistin-impregnated beads to prevent multi-drug-resistant A. baumannii implant-associated osteomyelitis. Journal of Orthopedic Research (August 2009):1108–15.
Cubist Pharmaceuticals. 2010. Cubist Pharmaceuticals 1Q10 total net revenues up 19% to $144.1 Million. Press Release. http://www.businesswire.com/portal/site/home/permalink/?ndmViewId=news_view&newsId=20100415006531&newsLang=en (accessed August 13, 2010).
DANMAP (Danish Integrated Antimicrobial Resistance Monitoring and Research Programme). 2000. Consumption of antimicrobial agents and occurrence of antimicrobial resistance in bacteria from food animals, food and humans in Denmark. http://www.danmap.org (accessed August 13, 2010).
DANMAP. 2008. Use of antimicrobial agents and occurrence of antimicrobial resistance in bacteria from food animals, foods and humans in Denmark. http//www.danmap.org (accessed August 13, 2010).
Dutil, L., R. Irwin, R. Finley, L. K. Ng, B. Avery, P. Boerlin, A.-M. Bourgault, L. Cole, D. Daignault, A. Desruisseau, W. Demczuk, L. Hoang, G. B. Horsman, J. Ismail, F. Jamieson, A. Maki, A. Pacagnella, and D. R. Pillai. 2010. Ceftiofur resistance in Salmonella enterica Serovar Heidelberg from chicken meat and humans, Canada. Emerging Infectious Diseases 16(1):48–54.
Falagas, M. E., I. A. Bliziotis, and I. I. Siempos. 2006. Attributable mortality of Acinetobacter baumannii infections in critically ill patients: A systematic review of matched cohort and case-control studies. Critical Care 10:R48, http://ccforum.com/content/10/2/R48. (accessed August 13, 2010).
Feinmen, S. 1998. Antibiotics in animal feed: Drug resistance revisited. American Society of Microbiology. News 24–30.
Finkelstein, J. A., C. Stille, J. Nordin, R. Davis, M. A. Raebel, D. Roblin, A. S. Go, D. Smith, C. C. Johnson, K. Kleinman, K. A. Chan, and R. Platt. 2003. Reduction in antibiotic use among U.S. children, 1996–2000. Pediatrics 112(3):620–7.
Fischbach, A., and C. T. Walsh. 2009. Antibiotics for emerging pathogens. Science 325(5944): 1089–93.
Frieden, T. 2010. Antibiotic resistance and the threat to public health. Testimony before the House Committee on Energy and Commerce, Subcommittee on Health, released April 28, 2010. http://energycommerce.house.gov/Press_111/20100428/Frieden%20Testimony%204.28.10.pdf (accessed August 13, 2010).
GAO (U.S. General Accounting Office). 2004. Report to Congressional Requesters No. 04-490: Antibiotic resistance: Federal agencies need to better focus efforts to address risk to humans from antibiotic use in animals. http://www.gao.gov/new.items/d04490.pdf (accessed August 13, 2010).
Gilbert, D. N., R. J. Guidos, H. W. Boucher, G. H. Talbot, B. Spellberg, J. E. Edwards, Jr., W. M. Scheld, J. S. Bradley and J. G. Bartlett. 2010. The 10 × 20 initiative: Pursuing a global commitment to develop 10 new antibacterial drugs by 2020. Clinical Infectious Diseases 50(8):1081–3.
Goossens, H., S. Coenen, M. Costers, S. De Corte, A. De Sutter, B. Gordts, L. Laurier, and M. J. Struelens. 2008. Achievements of the Belgian Antibiotic Policy Coordination Committee (BAP-COC). Eurosurveillance 13(46):1–4.
Grijaldi, C. G., J. P. Nuorti, and M. R. Griffin. 2009. Antibiotic prescription rates for acute respiratory tract infections in U.S. ambulatory settings. Journal of the American Medical Asssociation 302(7):758–66.
Hamann, K. 2006. An overview of Danish pork industry integration and structure. The Institute for Food Studies & Agroindustrial Development (IFAU), Denmark (presentation at the Banff Pork Seminar). http://www.cecmanitoba.ca/resource/hearings/22/21.pdf (accessed October 27, 2010).
Hammerum, A. M., O. E. Heuer, H. D. Emborg, L. Bagger-Skjøt, V. F. Jensen, A. M. Rogues, R. L. Skov, Y. Agersø, C. T. Brandt, A. M. Seyfarth, A. Muller, K. Hovgaard, J. Ajufo, F. Bager, F. M. Aarestrup, N. Frimodt-Møller, H. C. Wegener and D. L. Monnet. 2007. Danish integrated antimicrobial resistance monitoring and research program. Emerging Infectious Diseases 13(11):1632–9.
Herigon, J. C., A. L. Hersh, J. S. Gerber, T. E. Zaoutis, and J. Newland. 2010. Antibiotic management of Staphylococcus aureus infections in U.S. children’s hospitals, 1999–2008. Pediatrics 125(6):e1294–300.
Ho, P.-L., R. C. Wong, S. W. Lo, K.-H. Chow, S. S. Wong, and T.-L. Que. 2010. Genetic identity of aminoglycoside-resistance genes in Escherichia coli isolates from human and animal sources. Journal of Medical Microbiology 59(Pt 6):702–7.
IOM (Institute of Medicine). 1998. Antimicrobial resistance: Issues and options. Workshop report. Forum on Emerging Infections. Washington, DC: National Academy Press.
_____. 2003. Microbial threats to health: Emergence, detection, and response. Washington, DC: The National Academies Press.
Jensen, V. F., L. Jakobsen, H. D. Emborg, A. M. Seyfarth, and A. M. Hammerum. 2006. Correlation between apramycin and gentamicin use in pigs and an increasing reservoir of gentamicin-resistant Escherichia coli. Journal of Antimicrobials and Chemotherapy 58(1):101–7.
Kennedy, D., and S. Falkow. 2001. Antibiotics, animals, and people—again! Science 291(5503):397.
Klevens, R. M., M. A. Morrison, J. Nadle, S. Petit, K. Gershman, S. Ray, L. H. Harrison, R. Lynfield, G. Dumyati, J. M. Townes, A. S. Craig, E. R. Zell, G. E. Fosheim, L. K. McDougal, R. B. Carey, and S. K. Fridkin. 2007. Invasive methicillin-resistant Staphylococcus aureus infections in the United States. Journal of the American Medical Association 298(15):1763–71.
Laxminarayan, R., and A. Malini. 2007. Extending the cure: Policy responses to the growing threat of antibiotic resistance. Washington, DC: Resources for the Future.
Lederberg, J. 1997. Infectious disease as evolutionary paradigm. Emerging Infectious Diseases 3(4):417–23.
Levy, S. B. 1998. The challenge of antibiotic resistance. Scientific American 278(3):46–53.
Levy, S. B. 2002. The antibiotic paradox: How the misuse of antibiotics destroys their curative power. Cambridge, MA: Perseus.
Levy, S. B., G. B. Fitzgerald, and A. B. Macone. 1976. Changes in intestinal flora of farm personnel after introduction of a tetracycline-supplemented feed on a farm. New England Journal of Medicine 295(11):583–8.
Lewis, J. S., 2nd, M. Herrera, B. Wickes, J. E. Patterson, and J. H. Jorgensen. 2007. First report of the emergence of CTX-M-type extended-spectrum beta-lactamases (ESBLs) as the predominant ESBL isolated in a U.S. health care system. Antimicrobial Agents amd Chemotherapy 51(11):4015–21.
McDonald, L. C. 2006. Trends in antimicrobial resistance in health care-associated pathogens and effect on treatment. Clinical Infectious Diseases 42(S2):S65–71.
McDonald, L. C., S. Rossiter, C. Mackinson, Y. Y. Wang, S. Johnson, M. Sullivan, R. Sokolow, E. DeBess, L. Gilbert, J. A. Benson, B. Hill, and F. J. Angulo. 2001. Quinupristin-dalfopristin-resistant Enterococcus faecium on chicken and in human stool specimens. New England Journal of Medicine 345(16):1155–60.
Mellon, M., C. Benbrook, and K. L. Benbrook. 2001. Hogging it: Estimates of antimicrobial abuse in livestock. Cambridge, MA: Union of Concerned Scientists. www.ucsusa.org/assets/documents/food_and_agriculture/hog_chaps.pdf (accessed August 13, 2010).
Munoz-Price, L. S., and R. A. Weinstein. 2008. Acetinobacter infection. New England Journal of Medicine 258:1271–81.
National Research Council. 1999. The use of drugs in food animals: Benefits and risks. Board on Agriculture. Panel on Animal Health Food Safety and Public Health. Committee on Drug Use in Food Animals. Washington, DC: National Academy Press.
O’Brien, T., J. Holkins, E. Gilleece, A. Medeiros, R. Kent, B. Blackburn, M. Holmes, J. Reardon, J. Vergeront, W. Schell, E. Christenson, M. Bissett and E. Morse. 1982. Molecular epidemiology of antibiotic resistance in Salmonella from animals and human beings in the United States. New England Journal of Medicine 307(1):1–6.
Perez, A., A. Endiminia, and R. A. Bonomo. 2008. Why are we afraid of Baumannii acinetobacter? Expert Review of Anti-Infective Therapy 6(3):269–71.
Rice, L. B. 2008. Federal funding for the study of antimicrobial resistance in nosocomial pathogens: No ESKAPE. Journal of Infectious Diseases 197(8):1079–81.
Roberts, R. R., B. Hota, I. Ahmad, R. D. Scott, S. D. Foster, F. Abbasi, S. Schabowski, L. M. Kampe, G. G. Ciavarella, M. Supino, J. Naples, R. Cordell, S. B. Levya and R. A. Weinstein. 2009. Hospital and societal costs of antimicrobial-resistant infections in a Chicago teaching hospital: Implications for antibiotic stewardship. Clinical Infectious Diseases 49(8):1175–84.
Sarmah, A. K., M. T. Meyer, and A. B. A. Boxall. 2006. A global perspective on the use, sales, exposure pathways, occurrence, fate and effects of veterinary antibiotics (VAs) in the environment. Chemosphere 65(5):725–59.
Shah, N. S., A. Wright, G. H. Bai, L. Barrera, F. Boulahbal, N. Martin-Casabona, F. Drobniewski, C. Gilpin, M. Havelkova, R. Lepe, R. Lumb, B. Metchock, F. Portaels, M. F. Rodriguez, S. Rusch-Gerdes, A. Van Deun, V. Vincent, K. Laserson, C. Wells and J. P. Cegielski. 2007. Worldwide emergence of extensively drug-resistant tuberculosis. Emerging Infectious Diseases 13(3):380–7.
Spellberg, B., J. H. Powers, E. P. Brass, L. G. Miller and J. E. Edwards. 2004. Trends in antimicrobial drug development: Implications for the future. Clinical Infectious Diseases 38(9):1279–86.
Swann, M. M. (Chairman), K. L. Baxter, and H. I. Field. 1969. Joint Committee on the use of antibiotics in animal husbandry and veterinary medicine (“Swann Report”). London, United Kingdom: Her Majesty’s Stationery Office.
Tapsall, J. 2009. Multidrug-resistant Neisseria gonorrhoeae. Canadian Medical Association Journal 180(3):268–9.
WHO (World Health Organization). 2002. Impacts of antimicrobial growth promoter termination in Denmark: The WHO International Review Panel’s evaluation of the termination of the use of antimicrobial growth promoters in Denmark. Geneva, Switzerland: WHO.
_____. 2003. Impacts of antimicrobial growth promoter termination in Denmark: The WHO international review panel’s evaluation of the termination of the use of antimicrobial growth promoters in Denmark. http://whqlibdoc.who.int/hq/2003/WHO_CDS_CPE_ZFK_2003.1.pdf (accessed August 13, 2010).
Wilkins, A. S. 1996. Antibiotic resistance: Origins, evolution and spread. Ciba Foundation Symposium, 16–18 July 1996, London. Bioessays 18(10):847–8.
Winokur, P. L., A. Brueggemann, D. L. DeSalvo, L. Hoffmann, M. D. Apley, E. K. Uhlenhopp, M. A. Pfaller and G. V. Doern. 2000. Animal and human multidrug-resistant, cephalosporin-resistant Salmonella isolates expressing a plasmid-mediated CMY-2 AmpC β-lactamase. Antimicrobial Agents and Chemotherapy 44(10):2777–83.
Witte, W. 1998. BIOMEDICINE: Medical consequences of antibiotic use in agriculture. Science 279(5353):996–7.
A8
ANTIBIOTIC EFFECTIVENESS: NEW CHALLENGES IN NATURAL RESOURCE MANAGEMENT24
Markus Herrmann25 and Ramanan Laxminarayan26,27,28
Abstract
Problems of optimal natural resource extraction that were first addressed by economists in the contexts of fisheries and forests have reemerged in the context of a newly recognized resource: antibiotic effectiveness. This review
|
24 |
Reprinted with permission from Annual Reviews, posted online 20 April 2010. |
|
25 |
CRÉA-GREEN and CIRPÉE, Department of Economics, Université Laval, Québec, Québec G1V 0A6, Canada; email: markus.herrmann@ecn.ulaval.ca. |
|
26 |
Resources for the Future, Washington, DC 20036; email: ramanan@rff.org. |
|
27 |
Princeton Environmental Institute, Princeton University, Princeton, New Jersey 08544. |
|
28 |
Corresponding author. |
introduces economists to the growing literature on optimal use, innovation, and regulation of antibiotic effectiveness. Along the way, we draw links and parallels to similar problems in the management of other resources with which economists may be more familiar, and we address new questions that have arisen in the context of antibiotic effectiveness but that are also relevant to other resources.
1.
Antibiotic Effectiveness as a Natural Resource
Although humans may have known of antibiotics for centuries, the formal discovery of antibiotics occurred in 1929.29 Improvements in public health and medicine and a decline in infectious disease mortality preceded the widespread use of penicillin, but since the introduction of antibiotics in 1942, they have made possible further reductions in deaths and disability from infectious disease. Perhaps equally important, they have facilitated the vast expansion of other medical interventions, such as kidney and heart transplants, by allowing clinicians to prevent surgical site infections and to suppress the immunity of organ recipients.
Resistance to penicillin emerged soon after its introduction and was linked to patient deaths in the early 1950s (Abboud and Waisbren, 1959). Since then, bacteria have grown increasingly resistant to available antibiotics. In recent years, pan–drug resistance has emerged: Bacteria are resistant to nearly all antibiotics that were earlier active against them. The prevalence of high-level penicillin resistance in Streptococcus pneumoniae in the United States rose from 0.02% in 1987 to nearly 20% in 2004 (Laxminarayan et al., 2007). Over roughly the same period, the prevalence of methicillin-resistant Staphylococcus aureus (MRSA) in hospitals climbed from roughly 2% to more than 50% in many U.S. hospitals. Although the United States is among the heaviest users of antibiotics in the world on a per capita basis, the situation is even worse in some other countries, where infections spread more rapidly because of a lack of infection control in hospitals and inadequate water and sanitation in the community. For instance, in Vietnam, gram-negative organisms like Acinetobacter and Klebsiella are resistant to all antibiotics approved for human medicine; in addition, resistant organisms are commonly found in the environment (Duong et al., 2008).
Most antibiotics are derived from natural organisms like fungi, which use these compounds as weapons against bacteria. Resistance to antibiotics has always existed in bacteria, albeit at a very low frequency (perhaps one in a million or less), and predates the use of antibiotics as a treatment for infectious disease
(Levy, 1992). Human use of antibiotics has vastly tilted the balance of survival in favor of bacteria that evade antibiotics. In the changed environment with large quantities of antibiotic use, forces of natural selection favor resistant strains.30 However, carrying resistance genes is costly from an evolutionary standpoint and can be disadvantageous in an antibiotics-free environment. Some studies have demonstrated that resistant strains face an evolutionary disadvantage in an antibiotics-free environment. Biologists call this the fitness cost of resistance and have found it to be significant for some combinations of bacteria and antibiotics (Musher et al., 1977; Bennett and Linton, 1986; Bouma and Lenski, 1988), but not for others (Schrag et al., 1997; Björkman et al., 1998).
The problem of resistance is common in other efforts to control organisms that are harmful to humans and human enterprise. Resistance is observed in bacteria (to antibiotics), malarial parasites (to antimalarial drugs), viruses (to antivirals), and pests (to pesticides). In each case, application of control measures increases the likelihood that they will be less effective when used in the future. The effectiveness of the control agents can therefore be modeled as a natural resource in much the same way as are fish, trees, oil, or other resources. As with other resources, the optimal management of antibiotic effectiveness is determined by the biological dynamics of bacterial evolution of resistance, the spread of infection, and the demand for antibiotic treatment.
This review of the current literature on the economics of managing antibiotic effectiveness is organized as follows. In Section 2, we discuss the literature on the optimal use of antibiotics. Section 3 presents models in which antibiotic effectiveness is renewable. In Section 4, we address the impact of market structure on antibiotic use. Section 5 covers problems of managing antibiotic effectiveness as a global public good. In Section 6, we cover optimal investment in research and development (R&D) of new antibiotics. Section 7 discusses the economic costs of resistance. Section 8 concludes the paper and suggests future avenues for research.
2.
Optimal Use of Antibiotic Effectiveness
Brown & Layton (1996) discuss resistance as a dynamic externality.31 Laxminarayan & Brown (2001) were the first to use a dynamic disease framework to model antibiotics as a natural resource. In their formulation of an optimal con-
trol problem combining an economic, intertemporal objective with a deterministic compartment model of disease transmission derived from epidemiology, the relative proportion of individuals infected by antibiotic-susceptible bacteria to the overall infected population represents a measure for the treatment effectiveness of the antibiotic drug, the evolution of which depends on the use of antibiotics.
Antibiotic treatment implies social benefits and costs that are external to the person receiving treatment. Benefits include the treatment of sick patients, which also has the dynamic effect of reducing infections in the future. The cost of antibiotic use is not just the treatment cost that is borne by the patient or the insurance provider; it is also the shadow cost associated with the decline in its effectiveness. At the optimum, an antibiotic should be used when the full marginal benefits equal the full marginal costs.
Laxminarayan & Brown (2001) find that, depending on the relative production cost and the speed at which effectiveness declines, an initial phase may exist during which it is optimal to use only one antibiotic. For instance, when antibiotics have the same production cost but differ with respect to their level of effectiveness, the more effective drug should be used in the first phase because it procures at the margin a higher number of effective treatments and avoided future infections. This phase continues until the two antibiotics have equal effectiveness. It then becomes optimal to use them in a proportion that is inversely related to the speed at which their effectiveness declines.
The basic intuition underlying this conclusion parallels that of the optimal extraction of different ore qualities, when production costs depend on current extraction rates and remaining stocks (Weitzman 1976). As with resource pools with declining quality, there are three conditions under which simultaneous extraction from more than one resource pool is optimal. First, the marginal costs of extraction from the multiple pools may be identical, implying that it is optimal to engage in simultaneous extraction.
The other two reasons are unique to antibiotics. One is that the likelihood of resistance is a nonlinear function of antibiotic use. In most other resource problems, the stock of the resource decreases linearly with the use of the resource. With antibiotics, the marginal impact on effectiveness of antibiotic use is increasing. Therefore, simultaneously deploying two antibiotics reduces the likelihood that resistance to either of them will develop. The other reason is that, even if an infection is resistant to one antibiotic, it is treatable with a second antibiotic. So simultaneously treating the population with two antibiotics may lower resistance because effectiveness is regained when bacteria resistant to one antibiotic are treated with a different antibiotic.
Those considerations alter the standard prescription that resources should be used strictly in order of increasing marginal cost (Weitzman, 1976) and imply that when resistance arises as a consequence of antibiotic use, it may be shortsighted to use a single antibiotic on all patients just because that antibiotic appears to be the most cost-effective option (Laxminarayan and Weitzman, 2002). The
trade-off between economic costs and epidemiological advantage is described by Laxminarayan & Weitzman, who show that it may be optimal, from society’s point of view, to use different drugs on different but observationally identical patients and include on this menu of drugs some that may not be cost-effective from the individual patient’s perspective. The notion of treatment heterogeneity—the simultaneous use of different types of antibiotics—is consistent with the finding in Laxminarayan & Brown (2001) but addresses a different question: which antibiotics are optimal to include on a menu for simultaneous use at a population level. Other studies confirm the economic value of treatment heterogeneity (Bonhoeffer et al., 1997; Boni et al., 2008).
Thus far, we discuss models in which antibiotic effectiveness can be interpreted as a nonrenewable resource and in which more than one antibiotic is available to fight an infection. For antibiotics, the criterion of the most cost-effective treatment does not hold; instead, the overall social costs and benefits of using antibiotics must be considered. In the next section, we address models in which antibiotic effectiveness is renewable.
3.
Antibiotic Effectiveness as a Renewable Resource
Wilen & Msangi (2003) extend earlier models by assuming that the effectiveness of antibiotic efficacy represents a renewable resource. Such a modeling is appropriate when the drug-resistant bacterial strain incurs a positive fitness cost, in which case a low-enough treatment rate allows the population of bacteria to reach a sustainable equilibrium for the effectiveness of the drug. Whereas in the fishery case, multiple sustainable equilibria are attainable and depend on the regeneration rate of the remaining stock of fish in the sea, the regeneration of antibiotic effectiveness is independent of the stock of infection. In the model by Wilen & Msangi, the independence of the stock of infection occurs because the overall population is constant. When the economic objective is to minimize the discounted cost associated with infection, the authors show, the typical optimal solution combines an initially extreme treatment with subsequent intermediate controls. The extreme control corresponds to treating the overall infected population. This comes at the cost of decreasing the effectiveness of the drug but at the benefit of lowering, at least temporarily, the level of infection considerably below its steady state. The extreme control remains optimal as long as the marginal benefit of treating the infected population outweighs the marginal shadow cost of lowering antibiotic effectiveness. Once the two are in balance, an intermediate fraction of the infected population should get antibiotic treatment. This fraction eventually converges to the critical value at which the selection of the susceptible strain is exactly compensated by the selection of the resistant one.
In contrast, Rowthorn & Brown (2003) model two infections, each of which can be fought with a particular antibiotic only. At the time of treatment, the physician may be unaware of the specific bacterial strain that he or she is treating
and chooses the best possible treatment, knowing that a successful treatment may cure the patient but may also increase the likelihood of resistance in the future. The authors find that it makes sense to treat all patients with the antibiotic that is effective against the more prevalent strain, even if that antibiotic is relatively more expensive. Although one may not necessarily encounter the problem of two drugs used to treat two mutually exclusive diseases in a clinical setting, the model developed here offers a framework and provides a point of departure for more realistic variations of the problem.
Herrmann & Gaudet (2009) build on the model of renewable effectiveness by Wilen & Msangi (2003) and compare the optimal use of antibiotics with a market outcome in which drug producers have open access to a common pool of antibiotic effectiveness. As in the open-access fishery, economic rents are dissipated such that the price of the resource equals its average production cost and no producer accounts for the future evolution of the resource. The demand for the antibiotic plays a crucial role in the model dynamics: Demand shifts downward (upward) as the level of effectiveness decreases (increases). In equilibrium, this movement of the demand function makes the fraction of individuals buying the antibiotic adjust to the current level of antibiotic effectiveness and allows the latter to reach a sustainable level in the long run. This level may be higher or lower than the one that would be reached in the social optimum. Notably, when the average production cost is high, so is the price of the antibiotic, and a relatively small fraction of individuals buy the antibiotic over time. This comes, however, at the social cost of relatively high infection levels when out of steady state. To treat a higher fraction of the infected population, a lower-than-average price, which is lower than the average cost, may be optimal, implying that the production or the consumption of the antibiotic should be subsidized to make the market outcome coincide with the social optimum.
Finally, an important question of optimal use that has been discussed widely in the medical literature involves cycling. The optimality of treatment heterogeneity, discussed above, implies that cycling antibiotics may not be the best strategy, even though it has received much attention in the medical profession as a way to address the growing resistance of bacteria to antibiotics in hospitals (McGowan, 1986; Niederman, 1997; Bergstrom et al., 2000; John and Rice, 2000). Cycling hinges on the notion of the fitness cost of resistance: the evolutionary disadvantage placed on resistant strains in an antibiotics-free environment. If the fitness cost associated with bacterial resistance to antibiotics is high, the argument goes, then one can periodically remove an antibiotic from active use until it recovers its effectiveness. In contrast, if fitness cost is insignificant, then antibiotic effectiveness always declines, and it makes no sense to cycle antibiotics.
In this case, introducing economics can alter the conclusions reached by purely epidemiological models, as well as enrich their applicability to the real world, where economic costs play an important role. Cycling is suboptimal only
when antibiotic treatment costs are convex32 (Laxminarayan and Brown, 2001). This may not be the case in a hospital setting, where maintaining a drug on the hospital formulary entails a fixed cost, for shelf space, plus any cost associated with returning unused or expired products to the wholesaler. Furthermore, some drug companies offer volume discounts and even special prices if their products are put on the formulary and substitutes are excluded.33 Such factors introduce nonconvexities into the cost function and may make cycling of two antibiotics economically efficient. Switching from one antibiotic to another also entails its own costs, such as the administrative effort of taking one drug off the formulary and adding another one and the cost associated with educating physicians and nurse practitioners about a new drug. In the absence of these nonconvexities, there may be no economic rationale to cycle antibiotics.
4.
Market Structure and Antibiotic Use
The foregoing market equilibrium of open access represents a benchmark analysis for a generic industry selling an antibiotic once its patent has expired. Before that occurs, a single firm sells the antibiotic and thus controls at least to some extent the evolution of antibiotic effectiveness. More particularly, to what extent the evolution of antibiotic effectiveness can be controlled depends on whether the effectiveness of the antibiotic is linked to other antibiotics via a common resource pool of effectiveness.
Mechoulan (2007) and Herrmann (2009) consider the case in which the effectiveness of an antibiotic can be managed perfectly by a monopolist and the costs related to innovation can be considered sunk. Mechoulan (2007) shows that, although it may be socially optimal to eradicate a disease, a monopolist does not do so because the disease represents market size to the firm. With nonrenewable antibiotic effectiveness being added (in an ad hoc manner) into this model, the author shows that a reactivation of the patent after its initial expiration can be welfare improving. This occurs when the price charged by the monopolist is closer to the socially optimal price than to the price charged by the generic industry.
Herrmann (2009) characterizes the pricing policy and its impact on renewable antibiotic effectiveness and infection in a combined epidemiological-economic framework, as explained above. As the end of the patent approaches, the monopolist’s pricing policy bears greater resemblance to the myopic monopolist’s policy,
as less value is attached to the quality and market size of the antibiotic. This decreases the monopolist’s opportunity cost of selling the antibiotic, such that the amount of antibiotics sold increases, and decreases future levels of antibiotic efficiency and infection. This result depends crucially on the hypothesis that no profits are to be made in a generic industry—that is, there is open access to the pool of effectiveness. Whether a prolongation of the patent is socially desirable hinges on the relative values of antibiotic effectiveness and infection. Clearly, monopolistic pricing benefits the evolution of effectiveness more than the open-access outcome via lower treatment rates. However, this benefit comes at the cost of higher levels of infection because it represents a valuable asset to the monopolist. Thus, a prolongation of the patent is socially desirable only when infection is not an issue—when infection levels are relatively low compared with the level of antibiotic efficacy.
An important characteristic of an antibiotic is that it may have multiple end uses, implying multiple markets to which it can be sold. For instance, consider an antibiotic that can be used to treat humans and is also a growth-enhancing product for livestock. These two markets are likely to differ in size and in quantity responses to price changes. In such a context, Fischer & Laxminarayan (2004) show that a monopolist will extract the pool of antibiotic effectiveness faster than what is socially optimal, even if the demand in each market has a constant, but differing, price elasticity. Because there is only one resource pool with a unique shadow cost of extraction, it would be socially optimal to sell the antibiotic at identical prices in both markets. However, the monopolist discriminates between markets, lowering the drug’s price in the market characterized by the higher demand elasticity while increasing it in the other market compared with the social optimum. The combined effect of the price discrimination in both markets is such that antibiotic effectiveness is extracted at a higher rate over all periods.34
5.
Transboundary and Externality Problems
An early insight into the nature of antibiotic resistance was provided by Salant (2003), who likened resistance to a congestion problem. A key feature of the congestion problem is that enclosure of some resources but not others could lead to resource use that is suboptimal from a societal perspective (de Meza and Gould, 1992). For instance, efforts to reduce overgrazing and environmental degradation have focused on encouraging pastoralists to confine their animals to fenced enclosures, on the basis of the argument that overgrazing is more
likely to be avoided if pastoralists “own” the land. However, the effect of private enclosures on the remaining grazing lands that remain open access has often not been recognized. A possible regulatory response to the cross-resource spillover problem may be to impose a levy per animal to ensure against overgrazing. Alternatively, one could impose a quota restriction on the number of cattle allowed to graze on a common pasture.
Congestion spillovers across resources are also relevant in the case of antibiotic effectiveness. Patents permit enclosure of the effectiveness of new antibiotics but also confer monopoly rights. Other antibiotics have long been in use and are no longer under patent; they are in an open-access regime. Although patents may give a single firm the incentive to care about resistance to a drug, the patentee is likely to ignore the effect of her pricing decision on exacerbating resistance to antibiotics that may be in the generic domain, and she may overprice or under-use her antibiotic relative to the socially optimal level. Fischer & Laxminarayan (2009) analyze the optimality of price and quantity instruments in regulating resource use when there is uncertainty about congestion costs and show that taxes on antibiotics are preferable to quotas on antibiotic use, and strictly so when demand for antibiotic treatment is less than perfectly elastic.
The explanation arises from the fact that the tax still allows both markets—particularly, the enclosed market—to adjust to the cost shock, whereas the quota does not. This result differs from the well-known Weitzman result, in which the overall level of the pollution externality does not affect the marginal abatement cost curve and the relative slopes drive the preference for a tax or quota (Weitzman, 1974). Here, because the congestion externality for the open-access supply is defined by the difference between marginal and average costs, a shock shifts that market supply (average cost) curve in the same direction as the social marginal cost curve. Thus, whereas in the Weitzman case the tax fixes the price signal for producers, here the tax is not the price; rather, it influences the price, as do the cost shocks. A quota, in contrast, makes supply invariant to shocks, as in the Weitzman case. As a result, the relative trade-off is not between a too-rigid price and a too-rigid quantity but between a flexible, suboptimal price and a too-rigid quantity. Without the spillovers from partial enclosure, however, taxes are equally preferred to quotas.
Congestion spillovers across resources (antibiotics) are one challenge; spill-over of infection across one hospital, one health care institution, or one country is another. Smith et al. (2005) explore incentives for hospitals to invest in control of antibiotic-resistant bacteria when patients coming from other facilities are colonized (and therefore potentially infectious). In a result that is no surprise to economists, Smith et al. find that incentives to control drug resistance are greatest when there is only one hospital and decline as there are more hospitals. However, in a result that demonstrates the value added by disease models, these researchers find that investments in infection control initially increase in response to the growth in the influx rate of patients carrying resistant infections and then drop
to a minimum. This finding implies that efforts to manage antibiotic effectiveness in any single country or hospital has implications for incentives to manage elsewhere and that disease dynamics play a strong role in determining when such efforts are strategic substitutes across countries or institutions and when they are strategic complements.
Efforts to manage resistance across national borders would have to rely on international agreements and regulations (Walker et al., 2009) or on tax or subsidy instruments (Arrow et al., 2004). In the absence of such agreements and regulation, countries are unable to commit themselves to an optimal use of antibiotics, which would be in all countries’ interest. As a consequence, a country makes a too-intensive use of antibiotics as an input into its production at a macroeconomic level (Cornes et al., 2001). A supranational authority would have to consider both the externality benefits of antibiotic use, in terms of reducing infections, and the costs, in terms of resistance (Rudholm, 2002). Whether antibiotic consumption should be taxed or subsidized to reach the first-best outcome then depends on the relative magnitude of the externalities.
A relatively new class of antimalarial drugs, called artemisinins, requires a different way of thinking about optimal subsidies to manage resistance. When chloroquine, a once powerful antimalarial drug, became obsolete, the public health world was left with the challenge of using the last remaining effective drug class, artemisinins, in an effective manner. The World Health Organization (WHO, 2001) has recommended that artemisinin be used in combination with a partner drug that is unrelated in its mechanism of action and genetic bases of resistance, so that a single mutation cannot encode resistance to both components. Artemisinin combination treatments (ACTs), if used instead of monotherapies of either artemisinin or the partner drug, should slow down the emergence of anti-malarial resistance. However, the WHO guidelines are routinely flouted because monotherapies are much less expensive than ACTs. In response to this problem, an Institute of Medicine report (Arrow et al., 2004) recommended establishing an international fund to buy ACTs at producer cost and to resell them at a small fraction of that cost.
On economic efficiency grounds, there is a second-best case for subsidizing ACTs because the ideal policy of taxing monotherapies and other antimalarials according to the marginal external cost from the elevated risk of resistance evolution is infeasible, given the widespread use of these therapies in the informal sector. The efficiency argument is further strengthened by the positive externality to the extent that effective treatment of one individual reduces the risk of infection transmission to other individuals. Laxminarayan et al. (2007) show that it is possible to determine the optimal subsidy in a dynamic-disease-modeling framework. Bioeconomic analysis has been helpful for determining whether the social benefit from the subsidy, in terms of delayed resistance and saved lives, exceeds the social cost of resistance because of increased use of ACTs (Laxminarayan et al., 2006). Such analysis was also instrumental in turning an idea into the
Affordable Medicines Facility for malaria, a global financing system launched in early 2009.
6.
Antibiotic Innovation
In recent years, growing resistance levels have given rise to fears that antibiotic innovation cannot keep pace. Ellison & Hellerstein (1999) argue that a society that underevaluates antibiotic diversity’s contribution to addressing the problem of bacterial resistance also tends to value insufficiently the innovation of new antibiotics. They hypothesize that this effect is reinforced in the context of a competitive industry. Laxminarayan et al. (2007) argue that the private demand for new antibiotics may be considerably lower than what would be socially optimal, because private demand tends to be shortsighted. Consequently, if the current market supply of new antibiotics responds to the private demand, it may also be suboptimal.
As for any type of product innovation, a determining factor is patent protection. The patenting of new antibiotics has allowed firms to recover their previous spending on R&D.35 However, because of the particularly long regulatory control process (as exercised by the U.S. Food and Drug Administration) intended to ensure the safety of any new drug for human use, the nominal 20-year period of patent protection is considerably reduced, making the innovation of new antibiotics less profitable. A report by the Office of Technology Assessment (OTA, 1995) notes that the regulatory process shortens patent life by effectively seven to ten years. This clearly reduces the incentive for pharmaceutical firms to innovate.
A patent conveys an exclusive property right to sell a given antibiotic drug. Whether this incentive is sufficient for the patentee to incur the considerable R&D cost and, at the same time, to account for the intertemporal evolution of antibiotic effectiveness of its drug depends crucially on the corroboration of cross-resistance of the patented antibiotic with respect to other antibiotics. The OTA (1995) report advances the idea of increasing incentives for innovation by prolonging the duration of antibiotic length in exchange for restrictions on its use to fight a particular infection. Laxminarayan (2002) discusses the optimal breadth of a patent when there is a common pool of effectiveness related to antibiotic use in humans and livestock. The analysis shows that a narrower patent breadth is
associated with a more rapid exhaustion of antibiotic effectiveness by the patent-holding firm. The optimal patent breadth then brings into balance the deadweight loss, which results from the greater market power of a firm that holds a broader patent, and the social benefits of increasing a firm’s incentives to conserve antibiotic effectiveness. Broader patents may discourage marginal innovations, such as new drugs that are closely related to existing antibiotics, and instead encourage nonmarginal innovations of new classes of antibiotics and increase incentives to conserve their effectiveness.
Laxminarayan et al. (2007) suggest a sui generis right for antibiotic drugs whose patents have expired. In particular, the rights associated with drugs in the same pool of antibiotic effectiveness should be assigned to the same company or individual to permit better control of antibiotic effectiveness and to provide better incentives for further innovations. This type of right would be a surrogate for the physical territory and related property rights in the case of other natural resources.
A thorough modeling of the many aspects related to the innovation of antibiotics is still missing. Fischer & Laxminarayan (2005) provide a first sketch of how a single firm exploits successive pools of effectiveness and how this compares with the social optimum. Modeling antibiotic effectiveness as a nonrenewable resource and abstracting from issues related to cross-resistance, the authors address the sequential development and exploitation of a series of resource pools on behalf of a monopolist and compare the extraction path to the social optimum. The process of antibiotic innovation is captured by a fixed setup cost, which has to be incurred to access a subsequent resource pool. Whether the depletion of effectiveness, and thus the process of innovation, is faster under monopoly than in the social optimum depends principally on the demand schedule and the number of remaining resource pools. The opportunity cost of postponing the switch to the next resource pool depends on the current and future values of the monopolist’s and social planner’s optimization problem, as well as the speed at which an existing resource pool is extracted if no new pools are developed. In particular, the authors show that for a constant elasticity demand and zero extraction cost, the opportunity cost of waiting is higher for the monopolist than for the social planner when there are many resource pools left in the line, such that the monopolist extracts the resource relatively faster. The result is reversed when there are only a few resource pools left.
Cairns & Davis (2007) embed the former result in a more general setting, in which the setup cost may vary over time and in which the social surplus as well as the profits derived from the exploitation of the resource may be nonstationary. The formulation of the problem of when to invest, at a fixed cost, in a new resource pool leads to a reinterpretation of Hotelling’s r-percent rule. The rule applies not to the evolution of the scarcity rent of a resource unit when there are “lumpy” setup costs but rather to the rate of change in the net present value (also called forward value in finance) at the optimal investment date of the particular
resource project under consideration. When investment occurs at the optimal time, the current market value of the overall project rises at the rate of interest. When one is dealing with the sequential exploitation of multiple resource pools, it may be optimal not to extract the resource for a period of time, notably if the setup cost is high or the benefits derived from the use of the resource pool are low (because of the nonstationary nature of the problem). In particular, as long as the forward value of a yet-to-be-developed resource pool rises faster than the rate of interest, it is optimal to wait.
7.
Empirical Work
Two parameters drive optimal use of antibiotics: first, the relationship between antibiotic use and resistance and second, the magnitude of the biological fitness cost of resistance.
Antibiotic resistance is often positively correlated with antibiotic use. However, the direction of causality is unclear because, although antibiotic selection pressure contributes to resistance, higher resistance also necessitates greater antibiotic use. Moreover, the influence of antibiotic use on the level of infection complicates a direct estimation of the relationship. Comprehensive data sets are only now being assembled to estimate this function. Phelps (1989) calculates the resistance response for gentamicin and amikacin use and finds that a 1% increase in the use of each drug leads to a 0.15% increase and a 1.1% increase in resistance, respectively. Most other studies are unable to control for the effect of antibiotic resistance on use. For instance, Kaier & Frank (2008) document the use of fluoroquinolones and the incidence of hospital-acquired MRSA, finding a 0.55% increase in resistance in Geneva and a 0.32% increase in Belfast, but there is no evidence that fluoroquinolone use has any effect on MRSA. Considering not only antibiotic consumption but also a preventive measure, an alcohol-based hand sanitizer, on the incidence levels of MRSA and Clostridium difficile, Vernaz et al. (2008) can explain up to 57% and 17% of the variation in the level of infection, respectively. No particular policy related to antibiotic use was in place during their time series analysis. The consumption of antibiotics belonging to the fluoroquinolone and cephalosporin classes had a lagged effect of one month and four to five months, respectively, on MRSA; C. difficile was influenced only by broad-spectrum cephalosporins (with a one-month time lag).
The analysis by Aldeyab et al. (2008) also considers MRSA infections and generally confirms the aforementioned results. Their time series model explains up to 78% of the variation in hospital-acquired MRSA and accounts also for the influx of positively tested MRSA patients into the hospital and the number of patients tested for MRSA when in the hospital.
The second critical parameter is the fitness cost of resistance, which tells us how rapidly antibiotic effectiveness recovers when antibiotic selection pressure is removed. Studies in the medical literature have observed a decrease in
resistance after a period of sustained decrease in antibiotic use. In one Finnish study, a reduction in the overall use of macrolide antibiotics in the community was followed by a decrease in erythromycin-resistant Streptococcus pyogenes (Seppala et al. 1997). A similar study observed a decrease in penicillin-resistant S. pneumoniae following a decrease in use of antibiotics in children (Kristinsson, 1997). Sundqvist et al. (2007) find a significant but small estimated fitness cost of 1–2% of Escherichia coli resistant to trimethoprim. However, in vitro, no fitness cost is observed. This finding dampens the hope that a reduction in antibiotic consumption may reverse the rising trend of antibiotic resistance. Indeed, the authors find that a two year decrease of trimethoprim consumption to 15% of the former consumption level effectively halted the rise of resistance but led to only a slight reversal in resistance rates.
Another estimation challenge is the economic cost associated with bacterial resistance. In hospital settings, the challenge has been disentangling two effects: The longer the hospital stay, the greater is the likelihood of being infected with a resistant pathogen, and in turn, a hospital acquired infection with a resistant pathogen lengthens the hospital stay. In community settings, the challenge has been correctly estimating both the benefits and the costs of antibiotic use. Resistance-related costs alone are an insufficient reason to recommend that fewer antibiotics be used, because antibiotics bring benefits as well as costs. To date, there has been no reliable benefit-cost estimate of antibiotic use in either setting.
8.
Conclusions and Avenues for Further Research
The literature on the economics of antibiotic effectiveness as a natural resource has grown considerably in the past decade. Many of the problems faced in the management of antibiotic effectiveness bear resemblance to problems in both renewable and nonrenewable natural resources. For antibiotic effectiveness, as for optimal oil and mineral extraction, the effect of market structure on extraction rates, optimal investment in development of new resource pools, and the impact of taxation on antibiotic resistance are salient questions. And just as for fisheries or biodiversity conservation, problems in antibiotic effectiveness require understanding of biological and metapopulation dynamics, and the solutions involve management of open-access resources. However, those researching problems in antibiotic effectiveness face some unique challenges.
The first challenge is the realities of the health care system and pharmaceutical industry. Resource economists must learn the institutional details that will inform their models and assumptions. Second, the demand for antibiotics can be modified by reducing the need for antibiotics through better hospital infection control and vaccinations. Although antibiotic effectiveness is a scarce resource, its value depends on the number of individuals infected. Therefore, under some conditions it is optimal to use more antibiotics if that reduces the stock of infected individuals, although antibiotic effectiveness is somewhat decreased. Although
demand for energy or fish can also be modified, the dynamics of doing so are not tied closely to the problem of resource management, as is the case with antibiotics. This potential for a trade-off between smaller stocks of infected individuals in the present and a high level of antibiotic effectiveness in the future stems from discounting future infection costs. Additional insight should be gained by using other than utilitarian welfare functions. Third, qualitative conclusions reached by resource economists should be robust to assumptions about the size of the biological fitness cost of resistance. Although earlier papers have described a problem with either zero or high fitness costs, reality may not be so clear-cut. Therefore, deriving comparative statics with respect to the fitness cost of resistance would be valuable for policy.
Disclosure Statement
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
Literature Cited
Abboud FM, Waisbren BA. 1959. Correlation between in vitro studies and response to antibiotic therapy in staphylococcic bacteremia. AMA Arch. Intern. Med. 104(2):226–33
Aldeyab MA, Monnet DL, López-Lozano JM,Hughes CM, Scott MG, et al. 2008. Modelling the impact of antibiotic use and infection control practices on the incidence of hospital-acquired methicillinresistant Staphylococcus aureus: a time-series analysis. J. Antimicrob. Chemother. 62(3):593–600
Arrow KJ, Panosian CB, Gelband H, eds. 2004. Saving Lives, Buying Time: Economics of Malaria Drugs in an Age of Resistance. Washington, DC: Inst. Med.
Bassett EJ, Keith MS, Armelagos GJ. 1980. Tetracycline-labeled bone from ancient Sudanese Nubia. Science. 209:1532–34
Bennett PM, Linton AH. 1986. Do plasmids influence the survival of bacteria? J. Antimicrob. Chemother. 18(Suppl. C):123–26
Bergstrom CT, Lipsitch M, McGowan JE Jr. 2000. Nomenclature and methods for studies of antimicrobial switching (cycling). Presented at Conf. Antibiot. Resist. Glob. Policies Options, Cambridge, MA
Björkman J, Hughes D, Andersson DI. 1998. Virulence of antibiotic-resistant Salmonella typhimurium. Proc. Natl. Acad. Sci. USA 95(7):3949–53
Bonhoeffer S, Lipsitch M, Levin BR. 1997. Evaluating treatment protocols to prevent antibiotic resistance. Proc. Natl. Acad. Sci. USA 94(22):12106–11
Boni MF, Smith DL, Laxminarayan R. 2008. Benefits of using multiple first-line therapies against malaria. Proc. Natl. Acad. Sci. USA 105(37):14216–21
Bouma JE, Lenski RE. 1988. Evolution of a bacteria/plasmid association. Nature 335(6188):351–52
Brown G, Layton DF. 1996. Resistance economics: social cost and the evolution of antibiotic resistance. Environ. Dev. Econ. 1:349–55
Cairns RD, Davis GA. 2007. Strike when the force is with you: optimal stopping with application to resource equilibria. Am. J. Agric. Econ. 89(2):461–72
Comins HN. 1977. The management of pesticide resistance. J. Theor. Biol. 65(3):399–420
Comins HN. 1979. Analytic methods for management of pesticide resistance. J. Theor. Biol. 77(2):171–88
Cornes R, Van Long N, Shimomura K. 2001. Drugs and pests: intertemporal production externalities. Jpn. World Econ. 13(3):255–78
de Meza D, Gould JR. 1992. The social efficiency of private decisions to enforce property rights. J. Polit. Econ. 100(3):561–80
Duong HA, Pham NH, Nguyen HT, Hoang TT, Pham HV, et al. 2008. Occurrence, fate and antibiotic resistance of fluoroquinolone antibacterials in hospital wastewaters in Hanoi, Vietnam. Chemosphere 72(6):968–73
Ellison SF, Hellerstein JK. 1999. The economics of antibiotics: an exploratory study. In Measuring the Prices of Medical Treatments, ed. JE Triplett, 4:118–43.Washington, DC: Brookings Inst.
Fischer C, Laxminarayan R. 2004. Monopoly extraction of an exhaustible resource with two markets. Can. J. Econ. 37(1):178–88
Fischer C, Laxminarayan R. 2005. Sequential development and exploitation of an exhaustible resource: Do monopoly rights promote conservation? J. Environ. Econ. Manag. 49(3):500–15
Fischer C, Laxminarayan R. 2010. Managing partially protected resources under uncertainty. J. Environ. Econ. Manag. 59:129–41
Herrmann M. 2010. Monopoly pricing of an antibiotic subject to bacterial resistance. J. Health Econ. 29:137–150
Herrmann M, Gaudet G. 2009. The economic dynamics of antibiotic efficacy under open access. J. Environ. Econ. Manag. 57(3):334–50
Hueth D, Regev U. 1974. Optimal agricultural pest management with increasing pest resistance. Am. J. Agric. Econ. 56(3):543–53
John JF Jr, Rice LB. 2000. The microbial genetics of antibiotic cycling. Infect. Control Hosp. Epidemiol. 21(Suppl.):S22–31
Kaier K, Frank U. 2008. An econometric view of the dynamic relationship between antibiotic consumption, hand disinfection and methicillin-resistant Staphylococcus aureus. J. Antimicrob. Chemother. 63(3):630–31
Kingston W. 2000. Antibiotics, invention and innovation. Res. Policy 29(6):679–710
Kristinsson KG. 1997. Effect of antimicrobial use and other risk factors on antimicrobial resistance in pneumococci. Microb. Drug Resist. 3(2):117–23
Laxminarayan R. 2002. Antibiotic use in animal agriculture and the economics of resistance: How broad should the scope of antibiotics patents be? Am. J. Agric. Econ. 84:1287–92
Laxminarayan R, ed. 2003. Battling Resistance to Antibiotics and Pesticides: An Economic Approach. Washington, DC: RFF
Laxminarayan R, Brown GM. 2001. Economics of antibiotic resistance: a theory of optimal use. J. Environ. Econ. Manag. 42(2):183–206
Laxminarayan R, Malani A, Howard D, Smith DL. 2007. Extending the Cure: Policy Responses to the Growing Threat of Antibiotic Resistance. Washington, DC: RFF
Laxminarayan R, Over M, Smith DL. 2006. Will a global subsidy of new antimalarials delay the emergence of resistance and save lives? Health Aff. 25(2):325–36
Laxminarayan R, Weitzman ML. 2002. On the implications of endogenous resistance to medications. J. Health Econ. 21(4):709–18
Levy SB. 1992. The Antibiotic Paradox: How Miracle Drugs Are Destroying the Miracle. New York: Plenum
McGowan JE Jr. 1986. Minimizing antimicrobial resistance in hospital bacteria: Can switching or cycling drugs help? Infect. Control. 7(12):573–76
Mechoulan S. 2007. Market structure and communicable diseases. Can. J. Econ. 40:468–92
Musher DM, Baughn RE, Templeton GB, Minuth JN. 1977. Emergence of variant forms of Staphylococcus aureus after exposure to gentamicin and infectivity of the variants in experimental animals. J. Infect. Dis. 136(3):360–69
Niederman MS. 1997. Is “crop rotation” of antibiotics the solution to a “resistant” problem in the ICU? Am. J. Respir. Crit. Care Med. 156:1029–31
Office of Technology Assessment (OTA). 1995. Impacts of antibiotic-resistant bacteria. OTA-H-629, OTA, Washington, DC
Phelps CE. 1989. Bug/drug resistance: Sometimes less is more. Med. Care. 27(2):194–203
Rowthorn R, Brown G. 2003. Using antibiotics when resistance is renewable. In Laxminarayan 2003, pp. 42–62
Rudholm N. 2002. Economic implications of antibiotic resistance in a global economy. J. Health Econ. 21(6):1071–83
Salant S. 2003. Same infection, same time, same antibiotic? In Laxminarayan 2003, pp. 84–93
Schrag SJ, Perrot V, Levin BR. 1997. Adaptation to the fitness costs of antibiotic resistance in Escherichia coli. Proc. Biol Sci. 264(1386):1287–91
Seppala H, Klaukka T, Vuopio-Varkila J, Muotiala A, Helenius H, et al. 1997. The effect of changes in the consumption of macrolide antibiotics on erythromycin resistance in group A streptococci in Finland. Finnish Study Group for Antimicrobial Resistance. N. Engl. J. Med. 337(7):441–46
Smith DL, Levin SA, Laxminarayan R. 2005. Strategic interactions in multi-institutional epidemics of antibiotic resistance. Proc. Natl. Acad. Sci. USA 102(8):3153–58
Sundqvist M, Sjölund M, Runehagen A, Cars H, Abelson-Storby K, et al. 2007. A planned dramatic drop in trimethoprim consumption in a 180,000 population did not result in a related decrease in trimethoprim resistance in Escherichia coli. Int. J. Antimicrob. Agents 29:S32–33
Vernaz N, Sax H, Pittet D, Bonnabry P, Schrenzel J, Harbarth S. 2008. Temporal effects of antibiotic use and hand rub consumption on the incidence of MRSA and Clostridium difficile. J. Antimicrob. Chemother. 62(3):601–7
Walker B, Barrett S, Polasky S, Galaz V, Folke C, et al. 2009. Looming global-scale failures and missing institutions. Science 325(5946):1345–46
Weitzman M. 1976. The optimal development of resource pools. J. Econ. Theor. 12(3):351–64
Weitzman ML. 1974. Prices vs. quantities. Rev. Econ. Stud. 61(4):477–91
Wilen J, Msangi S. 2003. Dynamics of antibiotic use: ecological versus interventionist strategies to manage resistance to antibiotics. In Laxminarayan 2003, pp. 17–41
World Health Organ. (WHO). 2001. The use of antimalarial drugs. Report of a WHO informal consultation. WHO Geneva
A9
THE ROLE OF HEALTH CARE FACILITIES36
Ramanan Laxminarayan
Rapid improvements in medical technology have made possible lifesaving interventions that keep hospitalized patients alive for longer. However, the downside of these interventions is that patients tend to be sicker, spend longer periods of time in the hospital, and are more in need of intensive medical care than before, leading to an increased prevalence of many noso-
|
36 |
Reprinted from Laxminarayan and Malani (2007). Extending the cure: Policy responses to the growing threat of antibiotic resistance. Chapter 4: The role of health care facilities. © Resources for the Future 2007. All rights reserved. www.extendingthecure.com. |
comial infections.37 Also known as a hospital-acquired infection (HAI), a nosocomial infection is acquired in a hospital by a patient who was admitted for a reason other than that infection. Moreover, protracted illness and time on life support for these patients, many of whom are immuno-compromised, have increased reliance on antibiotics to help stave off infection, which in turn has resulted in increasing drug resistance among common, previously treatable HAIs.
According to the Centers for Disease Control and Prevention (CDC), HAIs account for an estimated 2 million infections and 90,000 deaths each year. Common HAIs include infections of surgical wounds, urinary tract infections, and lower respiratory tract infections. Infections acquired in health care institutions are among the top 10 causes of death in the United States: they are the primary cause of 1 percent of all deaths and are major contributors to an additional 2 percent of all deaths (Harrison and Lederberg, 1998). Many of the endemic bacteria causing these infections are resistant to one or more classes of antibiotics’ pose a major challenge to inpatient health, and significantly increase the costs of hospital stays. In fact, the United States has among the highest rates of drug-resistant hospital infections in the world, as described in Chapter 1. Vancomycin-resistant enterococci (VRE) and methicillin-resistant Staphylococcus aureus (MRSA) are among the most important HAIs because they now account for a large fraction of nosocomial infections, but they are not the only problematic pathogens: increasingly, resistant Gram-negative bacteria such as Pseudomonas aeruginosa and Klebsiella pneumoniae are causing serious infections in hospital patients. Hospitals and long-term care facilities like nursing homes and hospice care tend to use large quantities of antibiotics and are consequently significant reservoirs of resistant pathogens. The ability of these pathogens to persist may be due to multiple interacting factors, including excessive antibiotic use, poor hygiene by health care workers, high susceptibility of patients, establishment in long-term care facilities (as well as in prisons and in the community), and colonization of hospital staff or the hospital environment. Each of these factors contributes to the emergence and establishment of endemicity within a clinical setting. In addition to the impact of endemic antibiotic-resistant bacteria on their own patients, hospitals are significant reservoirs of resistant pathogens that can be transported to other facilities.
Strategies for lowering the resistance levels in hospitals fall into three categories.38 First is lowering antibiotic use by requiring preapproval for certain antibiotic prescriptions. Second is using creative antibiotic restriction strategies,
such as cycling and treatment heterogeneity. Third is better infection control, which is applicable not just to resistant pathogens, but to all HAIs. Studies suggest that the economic and health benefits of many common interventions to lower the prevalence of HAIs exceed the costs. In this chapter we explore the incentives for hospitals39 to invest in hospital infection control (HIC) and other measures to lower the prevalence of resistant bacteria in their facilities, as well as potential regulatory solutions to encourage greater reporting and improved infection control.
Economic Costs and Benefits
HAIs cost between $17 billion and $29 billion each year, and older studies have shown that a third of this burden can be lowered by adequate infection control programs (Haley et al., 1985). Numerous studies show that HAIs, especially resistant infections, cause longer hospital stays, greater risk of death, and much higher rates of hospitalization. There is also strong evidence that the overall economic benefits of infection control programs exceed costs by a wide margin and that “an effective infection control programme is one of the most cost-beneficial medical interventions available in modern public health” (Wenzel, 1995). However, there is considerable disagreement over who bears the principal economic burden of these infections, and this influences incentives for health care facilities to engage in better infection control. In this section, we review existing evidence on the economic benefits of hospital infection control and incentives for hospitals to engage in it.
Cost of Hospital-Acquired Infections
Numerous studies have documented the increased costs of nosocomial blood-stream infections, stretching back into the 1970s and 1980s. Pittet, Tarara et al. (1994) and Pittet and Wenzel (1995) found that during the 1980s, the incidence and risk of death from nosocomial bloodstream infections had risen markedly and that a patient with a nosocomial bloodstream infection was 35 percent more likely to die; for a patient who survived, extra costs attributable to the infection were approximately $40,000, and extra hospital costs, $6,000. Haley (1986) looked at all nosocomial infection costs and found that the average cost was about $1,800 per infection, with a maximum cost of about $42,000.
It is important to recognize the significant economic costs that nosocomial infections impose on both the hospital and the patient. The congressional Office of Technology Assessment has estimated the minimal hospital cost associated
with nosocomial infections caused by antibiotic-resistant bacteria to be $1.3 billion per year (in 1992) (OTA, 1995). This does not include the increased cost to patients, both monetarily and through the indirect and long-term morbidity and mortality consequences of resistant infections. In addition, most published studies have shown increased mortality risk on the order of 1.3 to 2 times, which may also have significant effects on indirect costs, such as long-term lost productivity. It is also important to understand that antibiotic resistance has an effect on many patients who do not become infected: they have to use stronger drugs, which may be more expensive, have more dangerous side effects, or be more toxic or possibly less effective than older or mainline drugs.
Those indirect costs aside, the cost of an antibiotic-resistant infection is still significant. According to Cosgrove, Qi et al. (2005), a nosocomial MRSA bacteremia significantly increases the length of hospital stays, the charges per patient, and hospital costs per case. They estimate that the excess cost of an MRSA bacteremia is $26,424 in patient charges and $14,655 in excess hospitals costs (total, $41,079 in excess charges) versus a control population. They also calculated costs for patients with methicillin-sensitive Staphylococcus infection (MSSA); these averaged $19,212 in excess patient charges and $10,655 in excess hospital costs (total, $29,867). McHugh and Riley (2004), similarly, estimated total per patient costs (as opposed to excess costs) of $9,699 for an MSSA infection versus $45,920 for an MRSA infection (an excess cost of $36,221).
Another important problem is surgical site infections, which are responsible for increased morbidity and mortality and cost hospitals more than $1.6 billion in extra charges each year (Martone and Nichols, 2001). Engemann, Carmeli et al. (2003) studied MRSA in surgical site infections in a large cohort at the Duke University Medical Center. MRSA in a surgical wound was found to result in more than a 12-fold increase in mortality versus non-infected patients and more than a 3-fold increase versus patients infected with MSSA. MRSA infections also cost patients about $40,000 more than an MSSA infection and about $84,000 more than an uninfected patient.
Vancomycin-resistant enterococci (VRE) are also associated with higher morbidity, mortality, and costs. Carmeli, Eliopoulos et al. (2002) found that a VRE infection led to longer hospital stays, a 2-fold increase in the rate of mortality, increased odds that a patient would require major surgery or be placed in the intensive care unit, and a 1.4-fold increase in hospital costs, which over the length of the study translated to excess costs of $2,974,478 (233 patients at an excess cost of $12,766 each). In addition, the authors found an increase in the likelihood that a patient would end up being discharged to a long-term care facility, meaning that the additional costs of a VRE infection are significantly understated in the study and that they continue for many patients. These estimates are lower than in another study (Stosoret al., 1998), which found that VRE bacteremia was associated with $27,190 in excess costs; yet another study (Song et al., 2003) found mean excess costs of VRE to be $81,208.
According to the Pennsylvania Health Care Cost Containment Council (PHC4) (PHC4, 2005), the average charge for Pennsylvania Medicare patients with HAIs was about $160,000, five times the $32,000 average charge for Medicare patients who did not contract infections. Among Medicaid patients, the average charge was approximately $391,000 for patients who contracted infections while hospitalized, compared with an average of $29,700 when infections did not occur. Private commercial insurers of businesses and labor unions that provide health insurance were billed for almost 23 percent, or 2,633, of the reported hospital-acquired infections, which added $604 million in extra hospital charges. The average charge for a hospital admission in which a commercially insured patient contracted an infection was almost $258,000, compared with an average of $28,000 for admissions when infections did not occur. The average charge for stays in which uninsured patients contracted infections reached almost $230,000, compared with $21,000 for an uninsured patient without an infection.
Benefits of Hospital Infection Control
There has been relatively little evaluation of the impact of programs to lower antibiotic use within hospitals, but greater attention has been paid to the benefits and costs of infection control programs. For example, a program of intensive surveillance and interventions targeted at reducing the risk of hospital-acquired ventilator-associated pneumonia at the University of Massachusetts Medical Center in 1997–1998 lowered the incidence of this pneumonia and resulted in a cost savings greater than $350,000 (Lai et al., 2003).
Similarly, a 1994 VRE outbreak at the University of Virginia Hospital prompted an active surveillance program and contact isolation of colonized patients. The costs of the program, including time spent collecting samples, additional length of hospital stays in isolation, and laboratory fees, were estimated at $253,099 during the two-year study, during which time only one primary VRE bacteremia occurred (Muto et al., 2002). At a control hospital, where no such program was in place, there were 29 cases of VRE bacteremia during the corresponding period, and these resulted in an estimated cost of $761,320, based on an estimate of excess costs of $27,190 per case of VRE (Stosor et al., 1998). Other per-case VRE cost estimates would value the program benefits at $357,448 (Carmeli et al., 2002) to $2,273,824 (Song et al., 2003), but even the lower end of these benefits far exceeded the costs of the program.
Two Charleston, S.C., hospitals implemented an active surveillance program and a contact isolation protocol as recommended by the Society for Health care Epidemiology of America (SHEA). Based on prior rates of nosocomial infections, the new programs and protocols prevented an estimated 13 MRSA bacteremias and 9 surgical site infections for a cost savings of about $596,960 for the prevented bacteremias ($45,920 per case, based on McHugh and Riley 2004) and $756,000 for the prevented surgical site infections ($84,000 in excess costs per
case, based on Engemann, Carmeli et al. 2003). The cost of implementing the program was $113,955, comprising $54,381 for surveillance and $59,574 for contact isolation (West et al., 2006).
Quality Control in U.S. Hospitals
This section provides an overview of how hospital quality, in general, and in particular with respect to infections, is currently measured and how hospitals are currently regulated or accredited.
Accreditation Process
Hospital accreditation organizations such as the Joint Commission on Accreditation of Health care Organizations (JCAHO) currently do not require standards for antibiotic use, resistance, or nosocomial infections.40 Hospitals are required to report only whether they follow a certain set of best practices for infection control, and not infection prevalence rates or resistance levels. JCAHO uses an onsite evaluation as the basis of accreditation. No long-term reporting is required for continued accreditation. Standards alone may not be able to solve the problem; a change in attitudes about hospital infections would come from a combination of education about the benefits of infection control and stronger incentives for hospitals to invest in control programs. Moreover, JCAHO clears more than 99 percent of all hospitals it inspects, which suggests that the current system is set up more to catch egregious violators of medical practices than to address pervasive problems like hospital-acquired infections and resistance (Gaul, 2005).41
Health Care Quality Organizations
The Leapfrog Consortium and other organizations that represent the interests of large purchasers of health care (such as automobile manufacturers) work with hospitals to encourage public reporting of health care quality and outcomes. They use a carrot-and-stick approach by rewarding hospitals that perform well and by leveraging consumer and health care purchaser choice to improve poor performers. Information on hospital infection control practices—including safety measures, hand washing, and vaccination of health care staff—is collected using self-reported surveys by hospitals. However, like JCAHO, Leapfrog may be better at separating good institutions from bad ones than at discerning finer indicators of performance, such as the prevalence of hospital acquired infection.
In general, hospital-acquired infections and resistance are not a focus for existing organizations like JCAHO and Leapfrog. Although drug resistance can be seen as a quality issue, the current system for determining hospital quality may not work well to improve reporting or compliance with better infection control practices.
HICPAC and SHEA Guidelines
Existing initiatives to improve hospital infection control—such as by CDC’s Health care Infection Control Practices Advisory Committee (HICPAC) (McKibbe et al., 2005) and SHEA (Muto et al., 2003)—provide guidance to hospitals to engage in greater infection control and thereby help prevent the spread of resistance. Both sets of guidelines are based on clinical evidence that the vast majority of MRSA and VRE infections are the result of transmission from patient to patient and not from de novo mutations, and thus they suggest that stringent infection control practices are probably the most important factor in limiting the spread of MRSA and VRE.
However, they differ in some important respects. In the context of MRSA and VRE, the SHEA guidelines call for active surveillance cultures to identify colonized patients, with barrier precautions for patients colonized or infected with MRSA and VRE. CDC guidelines, on the other hand, reject the need for active surveillance cultures on the grounds that they may impose unnecessary costs on hospitals. Nevertheless, the voluntary nature of these guidelines indicates that many hospitals are not likely to apply them unless they have a strong financial motivation for doing so.42
Reporting of Infections and Resistance in Hospitals
Since 1970, data on hospital infections and prevalence of MRSA and VRE (based on passive surveillance) have been voluntarily reported confidentially by hospitals participating in CDC’s National Nosocomial Infection Surveillance (NNIS) program. These hospitals provide general medical-surgical inpatient services to adults or children requiring acute care. With a few exceptions, most current understanding of the extent of HAIs and drug resistance comes from the NNIS surveillance. However, there are important problems with this system that restrict its usefulness in delivering a comprehensive, nationwide picture of hospital infections and resistance. First, the nearly 300 hospitals that participate in the program are self-selected and represent only about 2 percent of hospitals, mainly academic centers—raising strong concerns about selection bias. Second, reporting within hospitals can change significantly. For instance, hospitals do not necessarily report data from the same intensive care unit each year, making comparisons across years problematic. Third, NNIS data are generally not available
to researchers outside CDC because of confidentiality agreements signed with hospitals. This has restricted wider use of these data.
In recent years, under strong pressure from consumer advocates, some states have moved to require public reporting of hospital infections. In 2006, Consumers Union reported that six states (Illinois, Pennsylvania, Missouri, Florida, Virginia, and New York) had hospital infection disclosure laws, and 30 states had introduced similar legislation requiring hospitals to report their infection levels to state monitoring bodies (CU, 2006). By providing more transparency to consumers, better reporting of infection and resistance levels may give hospitals greater incentives to engage in infection control.
Incentives and Disincentives to Control Resistant HAIs
Hospital Incentives
Despite some awareness of the problem and new measures to tackle the growing threat, the overall trend of infections, both susceptible and resistant, appears to be upward, as seen in Chapter 1. Antibiotic restrictions and better infection control are the two main tools available to hospitals. Currently, antibiotic restrictions are the main strategy reported by hospitals. Cost containment had been the original reason for implementing these restrictions (to divert physicians from expensive antibiotics to cheaper generics), but these reasons have been reborn in the form of concerns about drug resistance.
Programs intended to control antibiotic-resistant infections associated with health care have been around for a long time; however, implementation of these programs has been highly variable across facilities. Moreover, the guidelines have mostly focused on contact precautions that require staff hand washing, staff cohorting,43 and use of protective equipment to prevent the spread of infection from patients identified as carrying an infection. Guidelines issued by SHEA in 2003, focused mainly on the spread of MRSA and VRE within the hospital setting, called existing measures insufficient and recommended active surveillance cultures to identify patients colonized but not infected with resistant pathogens (Muto et al., 2003).
Next we consider important reasons why hospitals may not invest heavily in infection control programs on their own.
Hospital Disincentives
The extent to which hospitals bear the cost of resistant HAIs is a subject of disagreement, as is the extent to which these costs are passed on to Medicare,
Medicaid, and private insurers. If reimbursement to the facility is tied to the number of days of hospitalization rather than by diagnosis-related group or episode of illness, the hospital may not bear any of the financial burden of extended hospital stays and may have few financial incentives for investing in HAIs even if the overall benefits of such investments exceed the costs.
A 1987 study that looked at charges associated with 9,423 nosocomial infections identified in 169,526 admissions, selected randomly from adult admissions to a random sample of U.S. hospitals, found that at least 95 percent of the cost savings obtained from preventing nosocomial infections represented financial gains to the hospital (Haley et al. 1987).
However, a series of recent reports from PHC4 find that Medicare and Medicaid bear the greatest burden of the additional cost of HAIs.44 Pennsylvania hospitals billed the federal Medicare program and Pennsylvania’s Medicaid program for 76 percent of the 11,668 hospital-acquired infections in 2004, with Medicare taking up much of the burden. The economic burden on government resources imposed by the additional hospital charges was estimated at $1 billion for Medicare and $372 million for Medicaid. Extrapolating from the figures in Pennsylvania to the entire country, PHC4 estimated that at least $20 billion was charged to Medicare to pay for HAIs during 2004. These figures indicate that hospitals may actually benefit by extending the length of stay and may have fewer incentives to control infection levels within the hospital.
Medicare is currently in the process of revising its rules on reimbursing for hospital-acquired infections, and these changes could have a significant impact on hospital incentives to invest in infection control. Some payers, such as Blue Cross–Blue Shield, have already made some payments contingent on lower rates of certain HAIs, and anecdotal evidence suggests that this has lowered the prevalence of those HAIs.
Impact of Lawsuits
Some hospitals have faced lawsuits from individual patients for HAIs, based on plaintiffs’ claims that defendants (hospitals) failed to adhere to the standard of care for infection control.45 A study from Philadelphia found that 72 percent of HAI malpractice cases in Philadelphia were either withdrawn or settled; when brought to trial, the plaintiff was more likely to prevail (Guinan et al., 2005). MRSA infections were the most common reason for lawsuits. Moreover, MRSA in class I surgical sites were more likely to result in a victory for plaintiffs because national data show lower rates of infection for these surgeries, with the
implication that these infections were preventable. The impact of lawsuits on infection control is unclear, but they may have made hospitals wary of reporting infection and resistance rates.
Short-Term Financial Considerations
Even if most of the costs of HAIs can be passed on to payers, hospitals and long-term care facilities may bear at least some of the burden associated with the high cost of treating resistant infections. However, even for these limited costs, short-term cost considerations may trump the long-term gains of lower levels of resistance and infection for facilities in financial trouble. Are financially troubled institutions more likely to cut back on infection control? Do hospitals and long-term care facilities really behave optimally, or do they tend to be myopic because they fail to recognize the effect of resistance management and infection control on future costs? Also, to what extent are hospitals prompted by the threat of lawsuits to do a better job of controlling nosocomial infections and resistant pathogens? Answering these questions is pivotal to making policy decisions on how best to incentivize hospitals to invest in stronger infection control programs.
Issues of Agency
Although the hospital as a whole may have an incentive to restrict the use of antibiotics and drug resistance, individual clinicians may not share the same incentives. Also, many physicians are not employees but consultants of hospitals and may therefore have a smaller incentive to care about costs imposed by resistance on hospitals. Conversely, the problem of resistance may be evident to infection control committees and clinicians, but they may not be able to convince senior management of the long-term financial benefits of lower levels of resistance. Management and operational structures of hospitals have implications both for investment in infection control and for implementation of control measures, but little is currently known about the influence of organizational culture and structure on infection and resistance levels.46
Incentives to Free-Ride
Hospital infection control is expensive and becomes more difficult and less effective when patients enter the hospital already carrying the resistant pathogens. Recent research on incentives for hospitals to control HAIs suggests that the large
spillovers of antibiotic-resistant bacteria among medical care facilities may be one factor that explains the lack of response (Smith et al., 2005). When institutions share patients, a person colonized in one facility may be responsible for introducing or increasing the prevalence of resistance in another facility.
Since any single hospital (especially in the current era of cost cutting and short-term financial pressures) may not see the benefits of its HIC program outside its own walls, hospitals may not benefit from decreasing the overall level of resistance in the catchment area when those patients are admitted later to other hospitals. Instead, hospitals may prefer to free-ride on the infection control investments of other hospitals. This results in an overall higher level of resistance.
Modeling shows that the selfishly “optimal” level of HIC that any hospital would undertake is lower the greater the number of hospitals that share a catchment area. In fact, it is in the interests of the hospital to spend less and free-ride on the efforts of other hospitals. When everyone free-rides, all hospitals will spend less on HIC, leading to epidemics that develop earlier and faster. A much better outcome can be achieved through regulation and the resulting coordination between facilities.
A good example comes from the Siouxland experience. An epidemic of VRE in the Siouxland region of Iowa, Nebraska, and South Dakota was first detected in late 1996. Within a short time, VRE had quickly spread to nearly half of the health care facilities in the region. In response, a VRE task force was constituted with representatives from acute care and long-term care facilities and public health departments in the region (Ostrowsky et al., 2001). Following a comprehensive two-year intervention (including aggressive culturing to identify VRE-colonized patients, isolation of patients, improved antibiotic use, sterile device measures, improved staff hand hygiene, and sharing of information among institutions), VRE was eliminated from all acute care facilities and significantly reduced in long-term care facilities in the region. This could not have happened without coordination. When hospitals are unwilling to coordinate on their own, regulation will ensure that no single hospital free-rides on the efforts of others. Regulations that require portability of patient records (which could show which patients are colonized) could help hospitals in identifying high-risk carriers of resistant pathogens.
The similarly successful experience of Dutch hospitals in lowering the prevalence of MRSA is described in Box A9-1.
Incentives to Report Infection Levels
Hospitals have a clear incentive to downplay infection levels in their facilities, since accurate reporting could decrease demand for their services. “Report cards” that provide patients with information on hospital quality, including nosocomial infection rates, may encourage hospitals to discriminate against sicker patients or those coming from long-term care facilities because they might be
|
BOX A9-1 The Dutch Experience with Controlling MRSA MRSA incidence rates in the Netherlands are among the lowest in the world—1.1 percent—in contrast to more than 25 percent in France, Spain, and Belgium and 43.5 percent in the United Kingdom (The National Institute for Public Health and the Environment (RIVM)) (see Figure 1.4). This extremely low rate is attributable to a decade-old national “search and destroy” policy to limit the spread of MRSA. The implementing guidelines are based on the premise that the best way to fight MRSA is to identify it as early as possible and to isolate infected or possibly infected patients. Patients and health care workers are categorized according to risk and screened regularly based on those risk assessments. For example, all patients treated in a foreign hospital are considered at high risk of being MRSA carriers and thus are isolated until cultures prove negative (Dutch Workingparty Infection Prevention, 2005). Most importantly, the policy requires the cooperation of all health care facilities and is enforced by the Dutch government. The policy has not been cheap to implement. Over the course of 10 years, the MRSA policy resulted in more than 2,265 lost hospitalization days (Vriens et al., 2002). Wards had to be closed 48 times, 29 health care workers had to temporarily discontinue working, and 78,000 additional cultures had to be performed. However, it is estimated that the 6 million euros realized as benefits of the campaign in terms of averted MRSA infections and increased vancomycin resistance in other bacteria (S. aureus and VRE) far outweighed the cost (2.8 million euros) of hospital infection control in the Netherlands during the period. A new strain of MRSA appeared in 1999 but was not immediately recognized as such because of the limited sensitivity of the tests at the time. Within weeks this new strain, still unrecognized, had spread to several health care facilities. By increasing the sensitivity of the tests and maintaining intensive screening of both patients and health care workers, by the end of 2003, the new strain of MRSA was under control (Vos et al., 2005). Controlling the spreading epidemic was possible only because of the national strategy: if any hospital had lapsed, MRSA would spread to all the other institutions fairly quickly. |
more likely to carry a resistant pathogen.47 To address this problem, Florida and some other states that publicly report outcome indicators by hospital risk adjust the data to account for the fact that some hospitals admit more patients who are sicker and require more resources than the average patient. An alternative strategy would be to monitor and subsidize inputs for hospital infection control rather than monitoring the outputs—that is, infection levels. Educational efforts to get
hospitals to recognize the long-term gains of infection control may also be part of the solution.
Hospital report cards also should be issued by an independent agency that is less susceptible to political pressure. These reports, if issued by government agencies, can be influenced or quashed by interference from the governor or state senators, who in turn are influenced by campaign contributions from wealthy doctors. Governmental policy can also influence the timing of the release of reports.
Some degree of enforcement is required, via periodic external surveillance cultures, withdrawal of approval for state Medicare reimbursement, or fines. Because reporting requirements can create perverse incentives—for example, hospitals that suspect high levels of resistance may cut back on surveillance expenditures—any reporting program needs to be designed to take these factors into consideration.
Recommendations
Hospitals are an important reservoir for resistant pathogens, and the problem of resistant infections is emblematic of broader problems with ensuring health care quality. The issue is not knowing how to address resistant infections in hospitals48—good examples exist, from both the United States and abroad, of how to maintain low levels of resistance in health care settings—but rather, understanding why some facilities have an incentive to invest in these programs while others do not.
Regulatory agencies play two important roles in the antibiotic resistance problem. One is to enable cooperative outcomes better than those attained if hospitals behave in their own self-interest. Regional coordination in infection control efforts may be one of several solutions to this dilemma (Kaye et al., 2006). Another is to make public the data on resistance and infection levels so that hospitals have an incentive to invest in addressing the problem. Here we propose ways to encourage reporting and control of resistant infections and improve surveillance, and we also recommend additional research.
Conclusions
-
Hospital reimbursement policies for HAIs could be linked to levels of infection and drug resistance. Tying Medicare and private insurance payments to a hospital to its levels of infection control may be one approach.
-
Subsidizing inputs for infection control and surveillance programs would
-
provide a greater incentive for hospitals to invest in them. Chapter 6, on health insurance and Medicare, describes such a program.
-
State requirements for reporting of hospital infections should adjust the data for risk so that hospitals that admit sicker patients are not penalized for having higher levels of antibiotic use and infection.
-
The national hospital infection and resistance surveillance system should be more comprehensive. Ideally, it would be separate from JCAHO and other accreditation groups and would take the approach used by several states: it would collect nationwide data not just on outcomes (infections and resistance) but also on inputs, such as antibiotic use, number of infection control nurses, and physical inputs for HIC. Given the incentive problems with reporting outcomes, independent monitoring and reporting of infections should be complemented with reports on infection control inputs.
-
Legal avenues for responding to resistance should be examined, perhaps involving a combination of workplace safety and labor laws (e.g., penalizing hospitals for a failure to protect nursing staff if they are at risk). Studies indicate that nurses are at-risk for infections caused by C. difficile and E. coli, however this risk is believed to be low (Sepkowitz, 1996a; 1996b).
-
Research needs to address the important policy-relevant questions. Little is known about the institutional characteristics (ownership structure,49 proximity to other hospitals and facilities) that predict resistance. We also know little about the costs of surveillance and infection control for a typical hospital and how these compare with other hospital expenses. Additional data will help determine the burden of infection control on hospital budgets and inform the design of taxes and subsidies for specific inputs for infection control.
-
A policy research program is needed to explore how to create incentives for hospitals to conduct surveillance and reporting, not just of infections but also of other important health care quality measures.
References
Bergstrom, C. T., M. Lo, et al. (2004). “Ecological Theory Suggests that Antimicrobial Cycling Will Not Reduce Antimicrobial Resistance in Hospitals.” Proceedings of the National Academy of Sciences of the United States of America 101(36): 13285-90.
Carmeli, Y., G. Eliopoulos, et al. (2002). “Health and Economic Outcomes of Vancomycin-Resistant Enterococci.” Archives of Internal Medicine 162(19): 2223-2228.
Cosgrove, S. E., Y. Qi, et al. (2005). “The Impact of Methicillin Resistance in Staphylococcus aureus Bacteremia on Patient Outcomes: Mortality, Length of Stay, and Hospital Charges.” Infection Control and Hospital Epidemiology 26: 166-174.
CU. (2006). http://www.consumersunion.org (accessed October 18, 2006). Consumers Union.
Dranove, D., D. P. Kessler, et al. (2002). “Is More Information Better? The Effects of ‘Report Cards’ on Health Care Providers.” Working paper bW8697. Cambridge, MA: NBER (National Bureau of Economic Research).
Engemann, J. J., Y. Carmeli, et al. (2003). “Adverse Clinical and Economic Outcomes Attributable to Methicillin Resistance among Patients with Staphylococcus aureus Surgical Site Infection.” Clinical Infectious Diseases 36: 592-598.
Gaul, G. M. (2005). “Accreditors Blamed for Overlooking Problems: Conflict of Interest Cited Between Health Facilities, Group That Assesses Conditions.” Washington Post. Washington, DC, July 25, A01.
Guinan, J. L., M. McGuckin, et al. (2005). “A Descriptive Review of Malpractice Claims for Health Care-Acquired Infections in Philadelphia.” American Journal of Infection Control 33(5): 310-2.
Haley, R. W. (1986). Managing Hospital Infection Control for Cost Effectiveness: A Strategy for Reducing Infectious Complications. Chicago: American Hospital Publishing, Inc.
Haley, R. W., D. H. Culver, et al. (1985). “The Efficacy of Infection Surveillance and Control Programs in Preventing Nosocomial Infections in US Hospitals.” American Journal of Epidemiology 121(2): 182-205.
Haley, R. W., J. W. White, et al. (1987). “The Financial Incentive for Hospitals to Prevent Nosocomial Infections under the Prospective Payment System. An Empirical Determination from a Nationally Representative Sample.” JAMA 257(12): 1611-4.
Harrison, P. F. and J. Lederberg (eds.). (1998). Antimicrobial Resistance: Issues and Options, Workshop Report. Forum on Emerging Infections. Washington, DC: Institute of Medicine.
Jevons, M. (1961). “Celbenin-Resistant Staphylococci.” BMJ 1: 124-5.
Kaye, K. S., J. J. Engemann, et al. (2006). “Favorable Impact of an Infection Control Network on Nosocomial Infection Rates in Community Hospitals.” Infection Control and Hospital Epidemiology 27(3): 228-32.
Lai, K. K., S. P. Baker, et al. (2003). “Impact of a Program of Intensive Surveillance and Interventions Targeting Ventilated Patients in the Reduction of Ventilator-Associated Pneumonia and Its Cost-Effectiveness.” Infection Control and Hospital Epidemiology 24(11): 859-863.
Laxminarayan, R., D. L. Smith, et al. (2005). “On the Importance of Incentives in Hospital Infection Control Spending.” Discovery Medicine 5(27): 303-308.
Martone, W. J. and R. L. Nichols. (2001). “Recognition, Prevention, Surveillance, and Management of Surgical Site Infections: Introduction to the Problem and Symposium Overview.” Clinical Infectious Diseases 33: S67-S68.
McHugh, C. G. and L. W. Riley. (2004). “Risk Factors and Costs Associated With Methicillin-Resistant Staphylococcus aureus Bloodstream Infections.” Infection Control and Hospital Epidemiology 25(5): 425-430.
McKibben, L., T. Horan, et al. (2005). “Guidance on Public Reporting of Health care-Associated Infections: Recommendations of the Health care Infection Control Practices Advisory Committee.” American Journal of Infection Control 33(4): 217-26.
Muto, C. A., E. T. Giannetta, et al. (2002). “Cost-Effectiveness of Perirectal Surveillance Cultures for Controlling Vancomycin-Resistant Enterococcus.” Infection Control and Hospital Epidemiology 23(8): 429-435.
Muto, C. A., J. A. Jernigan, et al. (2003). “SHEA Guideline for Preventing Nosocomial Transmission of Multidrug-Resistant Strains of Staphylococcus aureus and Enterococcus.” Infection Control and Hospital Epidemiology 24(5): 362-386.
Ostrowsky, B. E., W. E. Trick, et al. (2001). “Control of Vancomycin- Resistant Enterococcus in Health Care Facilities in a Region.” The New England Journal of Medicine 344(19): 1427-1433.
OTA. (1995). Impact of Antibiotic-Resistant Bacteria: A Report to the U.S. Congress. Washington, DC: Government Printing Office. Office of Technology Assessment.
PHC4. (2005). “Reducing Hospital-Acquired Infections: The Business Case.” Research Brief No. 8. http://www.phc4.org/reports/researchbriefs/111705/docs/researchbrief2005report_hospacq-infections_bizcase.pdf (accessed March 5, 2007). Pennsylvania Health Care Cost Containment Council.
PHC4. (2006). Hospital-acquired Infections in Pennsylvania 2005. http://www.phc4.org/reports/hai/05/default.htm (accessed December 11, 2006). Pennsylvania Health Care Cost Containment Council.
Pittet, D., D. Tarara, et al. (1994). “Nosocomial Bloodstream Infection in Critically Ill Patients. Excess Length of Stay, Extra Costs, and Attributable Mortality.” JAMA 271(20): 1598-1601.
Pittet, D. and R. P. Wenzel. (1995). “Nosocomial Bloodstream Infections. Secular Trends in Rates, Mortality, and Contribution to Total Hospital Deaths.” Archives of Internal Medicine 155(11): 1177-1184.
RIVM. (2005). “European Antimicrobial Resistance Surveillance System (EARSS).” http://www.rivm.nl/earss/ (accessed on January 18, 2005). The National Institute for Public Health and the Environment.
Sepkowitz, K. A. (1996a). “Occupationally Acquired Infections in Health Care Workers: Part I.” Annals of Internal Medicine 125(10): 826-834.
Sepkowitz, K. A. (1996b). “Occupationally Acquired Infections in Health Care Workers: Part II.” Annals of Internal Medicine 125(11): 917-928.
Smith, D. L., S. A. Levin, et al. (2005). “Strategic Interactions in Multi- Institutional Epidemics of Antibiotic Resistance.” Proceedings of the National Academy of Sciences of the United States of America 102(8): 3153-8.
Sohn, A. H., B. E. Ostrowsky, et al. (2001). “Evaluation of a Successful Vancomycin-Resistant Enterococcus Prevention Intervention in a Community of Health Care Facilities.” American Journal of Infection Control 29(1): 53-57.
Song, X., A. Srinivasan, et al. (2003). “Effect of Nosocomial Vancomycin-Resistant Enterococcal Bacteremia on Mortality, Length of Stay, and Costs.” Infection Control and Hospital Epidemiology 24(4): 251-256.
Stosor, V., L. R. Peterson, et al. (1998). “Enterococcus faecium Bacteremia: Does Vancomycin Resistance Make a Difference?” Archives of Internal Medicine 158(5): 522-527.
Vos, M. C., A. Ott, et al. (2005). “Successful Search-and-Destroy Policy for Methicillin-Resistant Staphylococcus aureus in The Netherlands.” Journal of Clinical Microbiology 43(4): 2034-2035.
Vriens, M., H. Blok, et al. (2002). “Costs Associated with a Strict Policy to Eradicate Methicillin-Resistant Staphylococcus aureus in a Dutch University Medical Center: A 10-Year Survey.” European Journal of Clinical Microbiology and Infectious Diseases 21(11): 782-786.
Wagenvoort, J. H. (2000). “Dutch Measures to Control MRSA and the Expanding European Union.” Euro Surveillance 5(3): 26-28.
Wenzel, R. P. (1995). “The Lowbury Lecture. The Economics of Nosocomial Infections.” The Journal of Hospital Infection 31(2): 79-87.
West, T. E., C. Guerry, et al. (2006). “Effect of Targeted Surveillance for Control of Methicillin-Resistant Staphylococcus aureus in a Community Hospital System.” Infection Control and Hospital Epidemiology 27(3): 233-238.
WIP. (2005). Policy for Methicillian-resistant Staphylococcus aureus. http://www.wip.nl (accessed May 31, 2006). Dutch Workingparty Infection Prevention.
A10
RESPONDING TO THE GLOBAL ANTIBIOTIC RESISTANCE CRISIS: THE APUA CHAPTER NETWORK
Stuart B. Levy50
Tufts University School of Medicine, Alliance for the Prudent Use of Antibiotics51
The Alliance for the Prudent Use of Antibiotics
The Alliance for the Prudent Use of Antibiotics (APUA) emerged in 1981 following a meeting on plasmids and drug resistance in the Dominican Republic (Levy et al., 1981). This was one of the first meetings to which scientists from the developing world, supported by the conference organizers, were invited to discuss the molecular basis for antibiotic resistance with representatives from industrialized countries. Many of the participants had sent novel resistant organisms to scientists in the industrialized world for study, but had never met face-to-face. The meeting forged new scientific relationships and collaborations. APUA, which began as a two-person part-time operation with about 30 members, has grown to several thousand members in over 100 countries with chapters in more than 60 countries on all continents (Figure A10-1).
The mission of APUA was then, and still is today, to “preserve the power of antibiotics.” This endeavor aims to control infectious diseases worldwide through appropriate access to these valuable therapeutics, as well as containment of antimicrobial resistance. The approach is to build local capacity through recruitment of individual APUA champions and building of APUA national chapters worldwide. The latter form strategic partnerships with other APUA chapters and individuals as well as public health organizations, such as the World Health Organization (WHO) and the Pan American Health Organization. The broad goals are to synthesize and disseminate the latest scientific information on antimicrobial resistance in each country and to conduct studies and activities to maintain and restore the efficacy of antibiotics.
More than half of the chapters are located in the developing world. Support for these chapters comes in the form of correspondence, meetings, lectures, and small grants. The small grants consist of several-thousand-dollar awards to undertake local projects (Figure A10-2). The findings from several of these projects have been published, one of which performed an inventory of antibiotics in home medicine cabinets in Russia (Stratchounski et al., 2003).
In Buenos Aires, Argentina, a training program on antibiotics and antibiotic

FIGURE A10-1 APUA global chapter network.
SOURCE: A. Sosa, Alliance for the Prudent Use of Antibiotics (personal communication, August 1, 2010).
resistance was initiated. Venezuela declared antibiotic resistance a public health issue and enacted a law that restricted the sale and dispensing of several antibiotic classes: macrolides, fluoroquinolones, third-generation cephalosporins, and rifampin (Figure A10-3). These activities are initiated locally, in large part through the members of the different country chapters. The rationale is to generate local interest and help the chapters devote attention to some local factor that affects antibiotic access and resistance in their own country.
In Chile, pharmacies are now required to obtain prescriptions before selling antibiotics. In just one year, that law translated into a dramatic drop in the sale of antibiotics (Figure A10-4). The lessons learned there can now be tested in other parts of South and Latin America to see if it is appropriate and feasible to limit the access to antibiotics without prescription.
Through journal articles, printed documents, and its website (www.apua.org), APUA gets its message out. One group targeted with information is journalists, for whom a training guide has been written (Figure A10-5). The first training was held in Addis Ababa, Ethiopa, in November 2007 with support from the U.S. Agency for International Development. Their initiatives and ability to

FIGURE A10-2 APUA Small Grants Program.
SOURCE: A. Sosa, Alliance for the Prudent Use of Antibiotics (personal communication, 2003).
reach diverse readers help deliver an understanding of the concept of antibiotic resistance to lay people.
In 2001, David Heymann invited me to chair a press conference accompanying the release of the WHO Global Strategy for Containment of Antimicrobial Resistance report. I had helped work on the document, but David was its champion, along with Rosamond Williams. Unfortunately, the press conference, scheduled in Washington, DC, on September 11, 2001, never happened because of the events which occurred that day.
Accompanying the report was an APUA-organized examination of 25 previous reports and recommendations from different organizations and countries throughout the world on the problem of antibiotic resistance (Levy and APUA, 2001). The document includes summary tables and text revealing a similarity in their conclusions and recommendations on the means to contain and reduce antibiotic resistance. The APUA report emphasizes that resistance is not a new problem but is an increasing threat and a public health priority. Moreover, each group argued that improper antibiotic use is the pivotal issue leading to resistance. Education of providers and patients on the problem and the factors affecting anti-

FIGURE A10-3 Venezuela declaration of public health threat by antimicrobial resistance (AMR).
SOURCES: Gaceta Oficial, Republica Bolivariana de Venezuela, 2002, and A. Sosa, Alliance for the Prudent Use of Antibiotics (personal communication, August 1, 2010).
biotic resistance is critical. The APUA document urged “action now,” citing the conclusions of the previous reports on the history and extent of the problem.
When APUA was established, we used the term “antibiotic” because bacteria were the major bearers of resistance in the form of transposons and plasmids. Today APUA looks at resistance as a general phenomenon in all microbes—bacteria, viruses, parasites, and fungi.
In 2005, APUA examined resistance globally, not just in bacteria but in viruses and parasites as well. The document, published as a supplement in Clinical Infectious Diseases, looked at resistance in HIV and other viruses, malaria, as well as many different bacteria (Levy and O’Brien, 2005). The situation was deemed epidemic, appropriately seen as “the shadow epidemic,” which clouded and interfered with effective therapy for all microbial diseases (Figure A10-6). APUA’s initial attention was, and continues to be, on developing countries, where resistance has its greatest impact.
We learned the following from work in these countries (A. Sosa, personal communication):
FIGURE A10-4 Effect of the need for a prescription on the sale of antibiotics in Chile.
SOURCE: Organizacion Panamericana de la Salud (2000). Printed with permissions from Dr. Luis Bavestrello, originally published in Spanish on Rev. méd. Chile v.130 n.11 Santiago Nov. 2002.
-
The government expenditure on health care is rarely 2 to 5 percent of the total budget.
-
There is serious concern about substandard drugs and counterfeits. We are examining this problem now through a situation analysis and needs assessment study under a Gates Foundation grant in Zambia and Uganda.
-
Sale and dispensing of antibiotics is often available without a prescription.
-
Child mortality from acute respiratory infections and diarrheal diseases of bacterial origin is high but is often overshadowed or even misdiagnosed as malaria, tuberculosis (TB), or AIDS. In fact, there are more deaths in children from bacteria than there are from the other microbial disease agents (Wardlaw et al., 2006).
-
There is often a “divorce” between the government and professional organizations.
-
Recognized key opinion leaders in these countries are often interested in

FIGURE A10-5 Training journalists.
SOURCE: http://www.tufts.edu/med/apua/Chapters/tripreport_new%20delhi.pdf.
-
leading efforts to institute change that will improve antibiotic access and prudent use. These individuals are key to forming APUA chapters.
-
The chapters often seek the involvement of WHO country offices, but usually in a cooperative partnership, not monetarily. The ongoing technical assistance is absolutely critical, as is continued communication with the chapters.
The short- and long-term goals are important because they aim to make sure that the chapter network and the chapters are able to do something useful, not only for themselves but also for other countries nearby. We have generated and supported regional meetings and activities.
We need to make resistance a public health issue that garners government ownership and financial support. Legislators and the media should be engaged to support and publicize efforts to educate the public. Activities should involve nongovernmental organizations, corporate entities, faith-based entities, and consumer protection agencies for advocacy mobilization. Local changes should draw a national picture of antimicrobial resistance priority issues, design a funding strategy to begin to sustain the efforts, and design a work plan with attainable

FIGURE A10-6 The APUA GAARD project reports a “shadow epidemic.” The 2005 Report of the Global Advisory on Antibiotic Resistance Data (GAARD), a project and publication of the Alliance for the Prudent Use of Antibiotics (www.apua.org).
SOURCE: www.apua.org.
goals. Efforts should seek cooperation with neighboring countries—something we are encouraging in Africa and in South America. A goal with very positive consequences is to publicize the findings and achievements, no matter how small. The critical issue in these countries is to get the message out and increase awareness with the public but also to stimulate interest and recognition of local leaders that something is happening on this issue. Important, as well, is getting WHO country offices to join APUA in this effort.
Antibiotics Are “Societal Drugs”
Country- and microbe-wide, there are key common concepts that can aid in the education about, and management of, resistance. Antibiotics and antimicrobials are unique therapeutic drugs. Unlike any other therapeutics in which only the individual is affected by therapy, antibiotics are truly “societal” drugs. Each individual use bears the potential for both narrow- and wide-ranging impacts
through the selection and propagation of resistant bacteria, the very target of the therapy.
This concept, which I first mentioned at the 25th anniversary of the Institute of Medicine in 1995, distinguishes these kinds of drugs from any other. Individual usage, by shedding of both the antibiotic and the bacteria and their resistance genes, affects the family unit, the community, and the larger society (Levy and Marshall, 2004).
There are a number of studies that illustrate this point. One by Bill Cunliffe’s group in England looked at the effect of antibiotic treatment of individuals for acne on other individuals living in the same home. Homes with acne-treated patients were compared with a cohort of similar age- and gender-matched homes without treatment (cotrimoxazole, erythromycin, tetracycline). As might be expected, the treated acne patients showed greatly increased carriage of resistant bacteria on their skin. Of note, after 7 to 10 days, the people sharing the household with an individual taking medication for acne began to carry high levels of resistant bacteria, mainly Staphylococcus, on their skin, though they were not taking the antibiotic. The control group remained unchanged during this experimental period. There was clearly a societal effect from the use of antibiotics for the acne patients (Cunliffe et al., 1996).
Antibiotics Have an Ecological Impact
By inference from studies among people (see above), antibiotics affect the environment, but their effect may not stay confined to one geographic location. When animals receive antibiotics—whether it be for therapy, prophylaxis, or growth promotion—that use affects farm workers and dwellers in that environment. For instance, the use of antibiotics on farms produces a broad spectrum of drug-resistant bacteria in both the animals and the farm dwellers (Levy et al., 1976). A more far-reaching effect comes through meats sold for food, fecal runoff into groundwater and streams, and manure being spread onto fruits and vegetables. Wildlife, such as birds and flies, have been shown to pick up resistant bacteria from feces and waste and move them to distant sites (Marshall et al., 1990). There is clearly a large environmental impact of antibiotic use.
The Amount of Resistance Reflects Selection Density
Another concept that is important when analyzing antibiotic use and resistance is the number of individuals getting the drug in that particular environment. If you give 100 grams of an antibiotic to one animal, that animal is the one that is going to be the “factory” producing antibiotic-resistant bacteria. If you take the same 100 grams and give it to 100 animals, you are going to have 100 times more animals producing resistant bacteria. I refer to this as “selection density” (Levy and Marshall, 2004). Thus, in order to understand the impact on nature, one has to
know how the antibiotic is being distributed. This information is somewhat more transparent in human medicine by the calculated defined daily dose per 1,000 individuals. This figure provides not merely the total quantity of antibiotic but also how much drug is used for how many people. Moreover, when total antibiotic use goes up or down, one should know how many individuals are involved in order to calculate the number of “producers” of resistant bacteria. Thus, the quantity of antibiotic used in one place does not produce a complete picture of the impact in terms of selection and propagation of resistant bacteria.
APUA supported a study in Nepal where a medical student, Judd Walson, looked at antibiotic resistance of fecal E. coli in three groups of people: those in Kathmandu, those 6 hours’ journey from Kathmandu, and those 3 days’ journey from Kathmandu (Walson et al., 2001). The total amount of antibiotics taken by individuals in the three different locations was the same. Drug sellers get to these distant areas. However, the amount of antibiotic resistance in the gut flora of the separate populations was dramatically different. The highest frequency was found in the group living in Kathmandu. This difference was linked to the presence of multiple health centers and a high density of people taking antibiotics there as compared to individuals in the two more distant areas. Although receiving the same amount of antibiotic, the more remote areas were much less densely populated and not subject to the selective force of antibiotics taken by others or to the spread of resistant bacteria.
Antibiotic “Life” After Treatment
Another factor potentially affecting resistance is the fate of the antibiotic after use. Antibiotics from hospitals, communities, and animals go into the environment. We see reports of antibiotics found in municipal waters downstream of a farm (Campagnolo et al., 2002). Resistant bacteria are isolated from vegetables (Levy, 1984). Also important, antibiotics are dispersed environmentally from our own use and leech into the wastewater from hospitals and homes.
One may ask, therefore: What is the biggest contributor to antibiotic resistance? Is it the treated individual getting the drug or the massive amounts of drug introduced into the environment? There are many more bacteria exposed to low-dose antibiotics released into the environment than confronted by drugs used therapeutically. This is particularly evident in veterinary medicine and agriculture.
Reservoirs of Antibiotic Resistance: The ROAR Project
The vast majority of bacteria sharing the environment do not cause disease; they are the commensal bacteria. Over the past decade, there has been an emphasis on the commensal flora as Reservoirs of Antibiotic Resistance (ROAR) (Marshall et al., 2009). Through a 5-year National Institutes of Health grant,
APUA, collaborating institutions, and individual scientists addressed this concept with sponsored research projects directed at this hypothesis: Do nonclinical strains bear resistance determinants found in clinical strains? The answer was a resounding “yes.”
An APUA project looked at what bacteria are affected by the dispersal of antibiotic. The ROAR project funded 12 different studies, all of which demonstrated the high levels of resistance carried by bacteria that were not causing disease but were in environments where antibiotics were available and spread was easy (www.apua.org).
We are currently engaged in a project sponsored by the National Biodefense Analysis and Countermeasures Center to examine reservoirs of antibiotic-resistant bacteria unassociated with disease in countries on different continents. Animal and environmental isolates are being sent from colleagues in individual APUA country chapter locations: India, South Korea, Turkey, South Africa, Georgia, Uganda, Vietnam, and Bangladesh. We are searching for trends and/or changes over time in antibiotic resistance and the genetic determinants found in one locale versus another. We may identify new resistance genes by analyzing isolates from soil, water, and healthy animals. The project aims to look at whether there are particular commensal and resistance determinants which are prominent in commensals of certain countries.
Antibiotics Warrant a Separate Drug Category
Should there be a separate drug class for antimicrobials? Antimicrobials are different. If they are placed in a separate drug category, then they would be regarded apart from other pharmaceuticals and dealt with as a unique class. For one thing, each individual use affects society. That should be enough; no other drug class can make that claim. Second, antibiotics have limited lifespans, because resistance that emerges in the microbes limits their long-term utility. We do not see that with other drugs.
Placing antimicrobials in a separate drug class:
-
recognizes that antibiotics are not like any other prescription drug;
-
emphasizes the consequences that the individual misuse has on society at large, which is not true of other drugs; and
-
allows special considerations for these drugs in terms of incentives that will allow industry to reenter the discovery field, which it has abandoned, and to develop new drugs.
The workshop explored wonderful new ideas for new antibiotics, but what is missing is who will fund the work and who will make the new drug?
There can be different incentives to enter or return to antimicrobial discovery, such as extended patent life, postmarketing surveillance to curb resistance,
tax reliefs, and preservation of antibiotic efficacy through combined efforts from producers and consumers. I am an optimist; I think we can find new drugs, but I also believe we have to learn how to use our current drugs more appropriately. The fewer individuals that are confronted by antibiotics, the less effect the drugs will have on wider society and the environment, as well as the other bacteria that are sharing that environment.
References
Campagnolo, E.R., K. R. Johnson, A. Karpati, C. S. Rubin, D. W. Kolpin, M. T. Meyer, J. E. Esteban, R. W. Currier, K. Smith, K. M. Thu, and M. McGeehin. 2002. Antimicrobial residues in animal waste and water resources proximal to large-scale swine and poultry feeding operations. The Science of the Total Environment 299(1–3):89–95.
Cunliffe, W. J., Y. W. Miller, E. A. Eady, R. W. Lacey, J. H. Cove, and D. N. Joanes. 1996. Sequential antibiotic therapy for acne promotes the carriage of resistant staphylococci on the skin of contacts. Journal of Antimicrobial Chemotherapy 38:829–37.
Levy, S. B. 1984. Antibiotic resistant bacteria in food of man and animals. In Antimicrobials and Agriculture, edited by M. Woodbine. London, United Kingdom: Butterworths, pp. 525-31.
Levy, S. B., and APUA (Alliance for Prudent Use of Antibiotics) (eds.). 2001. Antibiotic resistance: Synthesis of recommendations by expert policy groups. Geneva, Switzerland: World Health Organization.
Levy, S. B., and B. Marshall. 2004. Antibacterial resistance worldwide: Causes, challenges and responses. Nature Medicine 10:S122–9.
Levy, S. B., and T. O’Brien. 2005. Global antimicrobial resistance alerts and implications. Clinical Infectious Diseases 41(Suppl. 4):S219–88.
Levy, S. B., G. B. Fitzgerald, and A. B. Macone. 1976. Changes in intestinal flora of farm personnel after introduction of tetracycline-supplemented feed on a farm. New England Journal of Medicine 295:583–8.
Levy, S. B., R. C. Clowes, and E. L. Koenig (eds.). 1981. Molecular biology, pathogenicity and ecology of bacterial plasmids. New York: Plenum.
Marshall, B. M., D. Petrowski, and S. B. Levy. 1990. Inter and intraspecies spread of E. coli in a farm environment in the absence of antibiotic usage. Proceedings of the National Academy of Sciences USA 87:6609–13.
Marshall, B. M., D. J. Ochieng, and S. B. Levy. 2009. Commensals: Underappreciated reservoir of antibiotic resistance. Microbe 4:231–8.
Organizacion Panamericana de la Salud. 2000. Resistencia antimicrobiana en la Americas: Magnitud del problema y su contencion, edited by R. Salvatierra-Gonzalez and Y. Benguigui. Report OPS/HCP/HCT/163/2000, pp. 234–40.
Stratchounski, L. S., I. V. Andreeva, S. A. Ratchina, D. V. Galkin, N. A. Petrotchenkova, A. A. Demin, V. B. Kuzin, S. T. Kusnetsova, R. Y. Likhatcheva, S. V. Nedogoda, E. A. Ortenberg, A. S. Belikov, and I. A. Toropova. 2003. The inventory of antibiotics in Russian home medicine cabinets. Clinical Infectious Diseases 37:498–505.
Walson, J. L., B. Marshall, B. M. Pokhrel, K. K. Kafle, and S. B. Levy. 2001. Fecal carriage of antibiotic resistance in Nepal reflects proximity to Kathmandu. Journal of Infectious Diseases 184:1163–9.
Wardlaw, T. M., E. W. Johansson, M. Hodge. World Health Organization, and UNICEF. 2006. Pneumonia: The forgotten killer of children. Geneva, Switzerland: UNICEF/WHO.
A11
CHALLENGES AND OPPORTUNITIES IN ANTIBIOTIC DISCOVERY
Kim Lewis
Antimicrobial Discovery Center and the Department of Biology, Northeastern University52
The Nature of the Threat
It is a given that new antibiotics are needed to combat drug-resistant pathogens. However, this is only part of the need; we actually never had antibiotics capable of eradicating an infection. Currently used antibiotics have been developed against rapidly growing bacteria, most of them have no activity against stationary state organisms, and none are effective against dormant persister cells. The relative effectiveness of antibiotics in treating disease is largely a result of a cooperation with the immune system, which mops up after antibiotics eliminate the bulk of a growing population. But the deficiency of existing antibiotics against supposedly drug-susceptible pathogens is becoming increasingly apparent with the rise of immunocompromised patients (HIV infected, undergoing chemotherapy) and the wide use of indwelling devices (catheters, prostheses, heart valves), where the pathogen forms biofilms protecting cells from the components of the immune system. The ineffectiveness of the immune system leads to chronic diseases, which make up approximately half of all infectious disease cases in the developed world. The main culprit responsible for tolerance of pathogens to antibiotics are specialized survivors, persister cells (Lewis, 2007, 2010), which we examine in the following section.
Persisters
Persisters represent a small subpopulation of cells that spontaneously go into a dormant, nondividing state. When a population is treated with a bactericidal antibiotic, regular cells die, while persisters survive (Figure A11-1). In order to kill, antibiotics require active targets, which explains the tolerance of persisters. Taking samples and plating them for colony counts over time from a culture treated with antibiotic produces a biphasic pattern, with a distinct plateau of surviving persisters. By contrast, resistance mechanisms prevent antibiotics from binding to their targets (Figure A11-2).
Infectious disease is often untreatable, even when caused by a pathogen that is not resistant to antibiotics. This is the essential paradox of chronic infec-
|
52 |
Boston, MA. k.lewis@neu.edu, http://www.biology.neu.edu/faculty03/lewis03.html. |
FIGURE A11-1 Dose-dependent killing with a bactericidal antibiotic reveals a small subpopulation of tolerant cells, persisters.
tions. In most cases, chronic infections are accompanied by the formation of biofilms, which seems to point to the source of the problem (Costerton et al., 1999; Del Pozo and Patel, 2007). Biofilms have been linked to dental disease, endocarditis, cystitis, urinary tract infection, deep-seated infections, indwelling device and catheter infections, and the incurable disease of cystic fibrosis. In the case of indwelling devices, such as prostheses and heart valves, reoperation is the method of choice for treating the infection. Biofilms do not generally restrict penetration of antibiotics (Walters et al., 2003), but they do form a barrier for the larger components of the immune system (Jesaitis et al., 2003; Leid et al., 2002; Vuong et al., 2004). The presence of biofilm-specific resistance mechanisms was suggested to account for the recalcitrance of infectious diseases (Stewart and Costerton, 2001). However, the bulk of cells in the biofilm are actually highly
FIGURE A11-2 Resistance and tolerance. Bactericidal antibiotics kill cells by forcing the active target to produce corrupted products. Persister proteins act by blocking the target, so no corrupted product can be produced. By contrast, all resistance mechanisms prevent the antibiotic from binding to the target. MIC, minimal inhibitory concentration.
susceptible to killing by antibiotics; only a small fraction of persisters remains alive (Spoering and Lewis, 2001). Based on these findings, we proposed a simple model of a relapsing chronic infection: antibiotics kill the majority of cells, and the immune system eliminates both regular cells and persisters from the blood-stream (Lewis, 2001) (Figure A11-3). The only remaining live cells are then persisters in the biofilm. Once the level of antibiotic drops, persisters repopulate the biofilm, and the infection relapses. While this is a plausible model, it is not the only one. A simpler possibility is that antibiotics fail to effectively reach at least some cells in vivo, resulting in a relapsing infection.
Establishing potential causality between persisters and therapy failure is not trivial, because these cells form a small subpopulation with a temporary phenotype, which precludes introducing them into an animal model of infection. We reasoned that causality can be tested based on what we know about selection for high persister (hip) mutants in vitro. Periodic application of high doses of bactericidal antibiotics leads to the selection of strains that produce increased levels of persisters (Moyed and Bertrand, 1983; Wolfson et al., 1990). This is precisely what happens in the course of treating chronic infections: the patient is periodically exposed to high doses of antibiotics, which may select for hip mutants. But hip mutants would only gain advantage if the drugs effectively reach, and kill, the regular cells of the pathogen.
Patients with cystic fibrosis (CF) are treated for decades for an incurable P. aeruginosa infection to which they eventually succumb (Gibson et al., 2003).
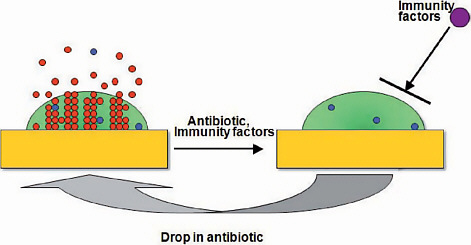
FIGURE A11-3 A model of a relapsing biofilm infection. Regular cells (red) and persisters (blue) form in the biofilm and are shed off into surrounding tissue and bloodstream. Antibiotics kill regular cells, and the immune system eliminates escaping persisters. The matrix protects persisters from the immune system, and when the concentration of the antibiotic drops, they repopulate the biofilm, causing a relapse.
The periodic application of high doses of antibiotics provides some relief by decreasing the pathogen burden, but it does not clear the infection. If hip strains of pathogens were selected in vivo, they would most likely be present in a CF patient. We took advantage of a set of longitudinal P. aeruginosa isolates from a single patient collected over the course of many years (Smith et al., 2006). Testing persister levels by monitoring survival after challenge with a high dose of ofloxacin showed a dramatic, 100-fold increase in surviving cells in the last 4 isolates (Mulcahy et al., 2010). Testing paired strains from additional patients showed that in most cases there was a considerable increase in persister levels in the late isolate from a patient. Interestingly, most of the hip isolates had no increase in minimal inhibitory concentration (MIC) compared to their clonal parent strain to ofloxacin, carbenicillin, and tobramycin, suggesting that classical acquired resistance plays little to no role in the recalcitrance of CF infection. These experiments directly link persisters to the clinical manifestation of the disease and suggest that persisters are responsible for the therapy failure of chronic CF infection. But why have the hip mutants with their striking survival phenotype evaded detection for such a long time?
The main focus of research in antimicrobials has been on drug resistance, and the basic starting experiment is to test a clinical isolate for its ability to grow in the presence of elevated levels of different antibiotics and to record any increases in the MIC. This is also the standard test employed by clinical microbiology laboratories. hip mutants are of course missed by this test, which explains why they had remained undetected in spite of a major effort aimed at understanding pathogen survival of antimicrobial chemotherapy. Given that hip mutants are the likely main culprit responsible for morbidity and mortality of the CF infection, it makes sense to test for their presence. Testing for persister levels is not that much more difficult as compared to an MIC test.
Is selection for hip mutants a general feature of chronic infections? We recently examined patients with chronic oral thrush caused by Candida albicans (LaFleur et al., 2010). These were cancer patients undergoing chemotherapy, and suppression of the immune system caused the fungal infection. In patients where the disease did not resolve, the C. albicans isolates were almost invariably hip mutants, as compared to patients where the disease cleared within 3 weeks of treatment with chlorhexidine. The eukaryotic C. albicans forms persisters (Al-Dhaheri and Douglas, 2008; Harrison et al., 2007; LaFleur et al., 2006) through mechanisms that are probably analogous, rather than homologous, to that of their bacterial counterparts. Given the similar lifestyles of the unrelated P. aeruginosa and C. albicans, we may expect that the survival advantage of a hip mutation is universal. Just as multidrug resistance has become the prevalent danger in acute infections, multidrug tolerance of persisters and hip mutants may be the main, but largely overlooked, culprit of chronic infectious disease.
Biofilms apparently serve as a protective habitat for persisters (Harrison et al., 2005a, 2005b, 2009; LaFleur et al., 2006; Spoering and Lewis, 2001),
allowing them to evade the immune response. However, a more general paradigm is that persisters will be critical for pathogens to survive antimicrobial chemotherapy whenever the immune response is limited. Such cases would include disseminating infections in immunocompromised patients undergoing cancer chemotherapy or infected with HIV. Persisters are also likely to play an important role in immunocompetent individuals in cases where the pathogen is located at sites poorly accessible by components of the immune system. These include the central nervous system, where pathogens cause debilitating meningitis and brain abscesses (Honda and Warren, 2009), and the gastrointestinal tract, where a hard-to-eradicate H. pylori causes gastroduodenal ulcers and gastric carcinoma (Peterson et al., 2000). Tuberculosis is perhaps the most prominent case of a chronic infection by a pathogen evading the immune system. The acute infection may resolve spontaneously or as a result of antimicrobial therapy, but the pathogen often remains in a “latent” form (Barry et al., 2009). It is estimated that 1 in every 3 people carry latent M. tuberculosis, and 10 percent of carriers develop an acute infection at some stage in their lives. Virtually nothing is known about this latent form that serves as the main reservoir of tuberculosis. One simple possibility is that persisters are equivalent to the latent form of the pathogen. The above analysis underscores the significance of drug tolerance as a barrier to effective antimicrobial chemotherapy. Given its significance—roughly half of all cases of infection (Figure A11-4)—the number of studies dedicated to tolerance is tiny compared to the number of publications on resistance. The difficulty in pinpointing the mechanism of biofilm recalcitrance and the formidable barriers to study persister cells account for the lack of parity between these two comparably significant fields. Hopefully a better balance will be achieved, and the following discussion summarizes recent advances in understanding the mechanism of tolerance.
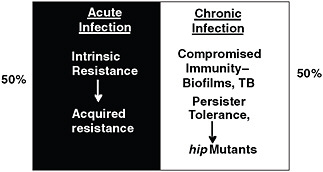
FIGURE A11-4 The two faces of recalcitrance. Drug resistance plays an important role in recalcitrance of acute infections, while drug tolerance is largely responsible for failures of chemotherapy in chronic infections. TB, tuberculosis.
Persisters were initially discovered in 1944, but the mechanism of their formation eluded us for a very long time. Only recently did the molecular mechanism of dormancy begin to emerge.
The most straightforward approach to finding an underlying mechanism of a complex function is by screening a library of transposon insertion mutants. This produces a set of candidate genes, and subsequent analysis leads to a pathway and a mechanism. This is indeed how the basic mechanisms of sporulation, flagellation, chemotaxis, virulence, and many other functions have been established. However, screening a Tn insertion library of E. coli for an ability to tolerate high doses of antibiotics produced no mutants completely lacking persisters (Hu and Coates, 2005; Spoering, 2006). With the development of the complete, ordered E. coli gene knockout library by the Mori group (Baba et al., 2006; the Keio collection), it seemed reasonable to revisit the screening approach. Indeed, there always remains a possibility that transposons missed a critical gene, or the library was not large enough. The use of the Keio collection largely resolves this uncertainty.
This advanced screen (Hansen et al., 2008), similar to previous efforts, did not produce a single mutant lacking persister, suggesting a high degree of redundancy. The screen did identify a number of interesting genes, with knockouts showing about a 10-fold decrease in persister formation. The majority of hits were in global regulators, DksA, DnaKJ, HupAB, and IhfAB. This is an independent indication of redundancy; a global regulator can affect expression of several persister genes simultaneously, resulting in a phenotype (Figure A11-5). The screen also produced two interesting candidate genes that may be more directly involved in persister formation: YgfA, which can inhibit nucleotide synthesis, and YigB, which may block metabolism by depleting the pool of flavine mononucleotide.
A similar screen of a P. aeruginosa mutant library was recently reported (De Groote et al., 2009). As in E. coli, no persisterless mutant was identified, pointing to the similar redundancy theme.
The main conclusion from the screens is that persister formation does not follow the main design theme of complex cellular functions—a single linear regulatory pathway controlling an execution mechanism. By contrast, persisters are apparently formed through a number of independent parallel mechanisms (Figure A11-5). There is a considerable adaptive advantage in this redundant design: no single compound will disable persister formation.
Screens for persister genes were useful in finding some possible candidate genes and pointing to redundancy of function. It seemed that a method better suited to uncover redundant elements would be transcriptome analysis. For this, persisters had to be isolated.
Persisters form a small and temporary population, making isolation challenging. The simplest approach is to lyse a population of growing cells with a β-lactam antibiotic and collect surviving persisters (Keren et al., 2004). This allowed enough E. coli cells to be isolated to perform a transcriptome analysis.
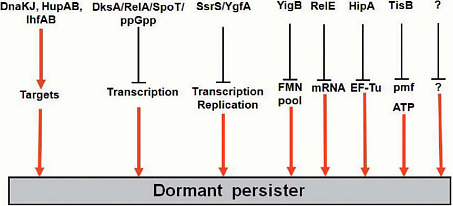
FIGURE A11-5 Candidate persister genes. Persisters are formed through parallel redundant pathways.
A more advanced method aimed at isolating native persisters was developed based on a guess that these are dormant cells with diminished protein synthesis (Shah et al., 2006). If the strain expressed degradable green fluorescent protein (GFP), then cells that stochastically enter into dormancy will become dim. In a population of E. coli expressing degradable GFP under the control of a ribosomal promoter that is only active in dividing cells, a small number of cells indeed appeared to be dim. The difference in fluorescence allowed for the sorting of the two subpopulations. The dim cells were tolerant of ofloxacin, confirming that they were persisters.
Transcriptomes obtained by both methods pointed to downregulation of biosynthesis genes and indicated increased expression of several toxin-antitoxin (TA) modules (RelBE, MazEF, DinJYafQ, YgiU). TA modules are found on plasmids where they constitute a maintenance mechanism (Gerdes et al., 1986a; Hayes, 2003). Typically, the toxin is a protein that inhibits an important cellular function, such as translation or replication, and it forms an inactive complex with the antitoxin. The toxin is stable, while the antitoxin is degradable. If a daughter cell does not receive a plasmid after segregation, the antitoxin level decreases due to proteolysis, leaving a toxin that either kills the cell or inhibits propagation. TA modules are also commonly found on bacterial chromosomes, but their role is largely unknown. In E. coli, MazF and an unrelated toxin RelE induce stasis by cleaving messenger RNA (mRNA), which of course inhibits translation, a condition that can be reversed by expression of corresponding antitoxins (Christensen and Gerdes, 2003; Pedersen et al., 2002). This property of toxins makes them excellent candidates for persister genes.
Ectopic expression of RelE (Keren et al., 2004) or MazF (Vazquez-Laslop et al., 2006) strongly increased tolerance to antibiotics. The first gene linked
to persisters, HipA (Moyed and Bertrand, 1983), is also a toxin, and its ectopic expression causes multidrug tolerance as well (Correia et al., 2006; Falla and Chopra, 1998; Korch and Hill, 2006; Vazquez-Laslop et al., 2006). Interestingly, a bioinformatics analysis indicates that HipA is a member of the Tor family of kinases, which have been extensively studied in eukaryotes (Schmelzle and Hall, 2000) but have not been previously identified in bacteria. HipA is indeed a kinase, it autophosphorylates on ser150, and site-directed mutagenesis replacing it or other conserved amino acids in the catalytic and Mg2+-binding sites abolishes its ability to stop cell growth and confer drug tolerance (Correia et al., 2006). The crystal structure of HipA in complex with its antitoxin HipB was recently resolved, and a pull-down experiment showed that the substrate of HipA is elongation factor EF-Tu (Schumacher et al., 2009). Phosphorylated EF-Tu is inactive, which leads to a block in translation and dormancy (Figure A11-6).
Deletion of potential candidates of persister genes noted above does not produce a discernible phenotype affecting persister production, possibly due to the high degree of redundancy of these elements. In E. coli, there are at least 15 TA
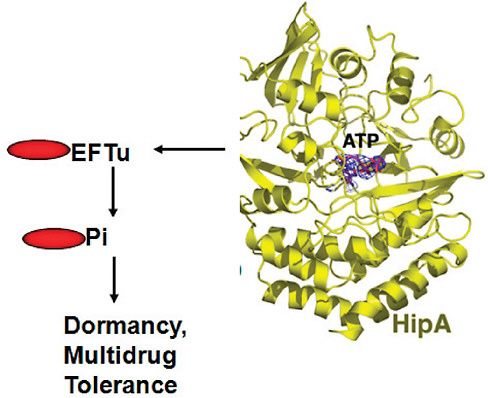
FIGURE A11-6 The HipA toxin causes dormancy in E. coli by phosphorylating elongation factor Tu, which inhibits protein synthesis. ATP, adenosine triphosphate.
modules (Alix and Blanc-Potard, 2009; Pandey and Gerdes, 2005; Pedersen and Gerdes, 1999) and more than 80 in M. tuberculosis (Ramage et al., 2009).
High redundancy of TA genes would explain the lack of a multidrug-tolerance phenotype in knockout mutants, and therefore it seemed useful to search for conditions where a particular toxin would be highly expressed in a wild-type strain and then examine a possible link to persister formation.
Several TA modules contain the Lex box and are induced by the SOS response. These are symER, hokE, yafN/yafO, and tisAB/istr1 (Courcelle et al., 2001; Fernandez De Henestrosa et al., 2000; Kawano et al., 2007; McKenzie et al., 2003; Motiejunaite et al., 2007; Pedersen and Gerdes, 1999; Singletary et al., 2009; Vogel et al., 2004). Fluoroquinolones induce the SOS response (Phillips et al., 1987), and we tested the ability of ciprofloxacin to induce persister formation (Dörr et al., 2009).
Examination of deletion strains showed that the level of persisters dropped dramatically, 10- to 100-fold, in a ∆tisAB mutant. This suggests that TisB was responsible for the formation of the majority of persisters under conditions of SOS induction. The level of persisters was unaffected in strains deleted in the other Lex box containing TA modules. Persister levels observed in time-dependent killing experiments with ampicillin or streptomycin that do not cause DNA damage were unchanged in the ∆tisAB strain. TisB only had a phenotype in the presence of a functional RecA protein, confirming the dependence on the SOS pathway.
Ectopic overexpression of tisB sharply increased the level of persisters. Drop in persisters in a deletion strain and increase upon overexpression gives reasonable confidence in the functionality of a persister gene. The dependence of TisB-induced persisters on a particular regulatory pathway, the SOS response, further strengthens the case for TisB as a specialized persister protein (Figure A11-7). Incidentally, a tisB mutant is not present in the otherwise fairly complete Keio knockout library, and the small open reading frame might have been easily missed by Tn mutagenesis as well, evading detection by the generalized screens for persister genes.
The role of TisB in persister formation is unexpected based on what we know about this type of proteins. TisB is a small, 29 amino acid hydrophobic peptide that binds to the membrane and disrupts the proton motive force (pmf), which leads to a drop in adenosine triphosphate (ATP) levels (Unoson and Wagner, 2008). Bacteria, plants, and animals all produce antimicrobial membrane-acting peptides (Garcia-Olmedo et al., 1998; Sahl and Bierbaum, 1998; Zasloff, 2002). Toxins of many TA loci found on plasmids belong to this type as well. If a daughter cell does not inherit a plasmid, the concentration of a labile antitoxin decreases, and the toxin—such as the membrane-acting hok—kills the cell (Gerdes et al., 1986b). High-level artificial overexpression of TisB also causes cell death (Unoson and Wagner, 2008). It is remarkable from this perspective that the membrane-acting TisB, under conditions of natural (mild) expression, has the exact opposite effect of protecting the cell from antibiotics.
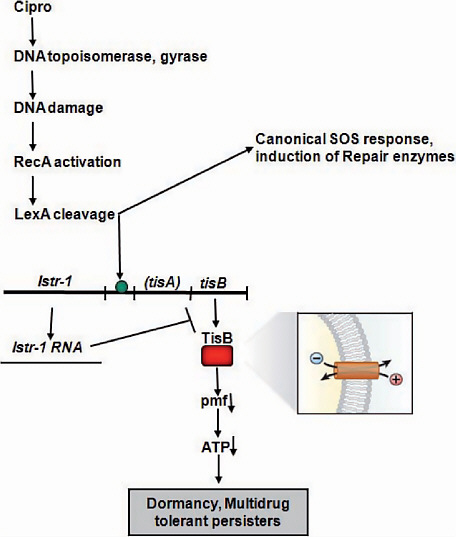
FIGURE A11-7 Persister induction by antibiotic. The common antibiotic ciprofloxacin causes DNA damage by converting its targets, DNA gyrase and topoisomerase, into endonucleases. This activates the canonical SOS response, leading to increased expression of DNA repair enzymes. At the same time, the LexA repressor that regulates expression of all SOS genes also controls transcription of the TisAB toxin-antitoxin module. The TisB toxin is an antimicrobial peptide, which binds to the membrane, causing an increase in pmf and ATP. This produces a systems shutdown, blocking antibiotic targets, which ensures multidrug tolerance.
Fluoroquinolones, such as ciprofloxacin, are widely used broad-spectrum antibiotics, and their ability to induce multidrug-tolerant cells is unexpected and a cause of considerable concern. Induction of persister formation by fluoroquinolones may contribute to the ineffectiveness of antibiotics in eradicating infections. Indeed, preexposure with a low dose of ciprofloxacin drastically increased tolerance to subsequent exposure with a high dose, and TisB persisters are multidrug tolerant.
The finding of the role of TisB in tolerance opens an intriguing possibility of a wider link between other stress responses and persister formation. Pathogens are exposed to many stress factors in the host environment apart from DNA-damaging agents—oxidants, high temperature, low pH, membrane-acting agents. It is possible that all stress responses induce the formation of surviving persisters.
Although resistance and tolerance are mechanistically distinct, there is sufficient reason to believe that tolerance may be a major cause for developing resistance. Indeed, the probability of resistance development is proportional to the size of the pathogen population, and a lingering chronic infection that cannot be eradicated due to tolerance will go on to produce resistant mutants and strains acquiring resistant determinants by transmission from other bacteria (Levin and Rozen, 2006). Combating tolerance then becomes a major component in preventing resistance.
The Discovery Challenge
Source Compounds
The discovery of penicillin was an isolated event, but development of screening for antimicrobial activity from soil actinomycetes by Salman Waxman produced the first, and also the only known, effective platform technology for antibiotic discovery (Schatz et al., 1944). Cultivable actinomycetes, however, are a limited resource; ~99 percent of microbes do not readily grow in the lab and are known as “uncultured” (Lewis et al., 2010). Overmining of actinomycetes by the early 1960s replaced the discovery of novel compounds with rediscovery of knowns.
In response to the dwindling returns in natural product antibiotic discovery, the industry focused on synthetics. Indeed, a number of antimicrobials are synthetic (metronidazole, trimethoprim, isoniazid, ethionamide, pyrazinamide, ethambutol), and there is one highly effective class of synthetic broad-spectrum antibiotics, the fluoroquinolones. Encouraged by these examples, and by dramatic advances in synthetic and combinatorial chemistry, high-throughput robotics, genomics, and proteomics, a new discovery platform emerged (Figure A11-8). Combinatorial chemistry provided a large number of test compounds, which were screened in high-throughput format against isolated essential target proteins determined by genomics. This platform, however, failed to produce a new
class of broad-spectrum antibiotics, leading to the closure of anti-infectives divisions in many of the Big Pharma companies. The main reason for failure is well understood: HTS hits were literally running into the penetration barrier of Gram-negative bacteria, which is made of transenvelope MDR pumps that extrude amphipathic compounds across the outer membrane barrier (Lomovskaya et al., 2008). Drugs have to be amphipathic in order to penetrate across the hydrophobic inner membrane, but this is precisely the feature that the outer membrane restricts and the MDRs recognize. There are few compounds that pass this seemingly impenetrable barrier rather effectively—the broad-spectrum aminoglycosides, tetracyclines, fluoroquinolones, some β-lactams, chloramphenicol, and azithromycin. Fluoroquinolones are the only synthetics on this list, and they were discovered 50 years ago.
But what about less challenging narrow spectrums, with good activity against at least Gram-positive species? Seventy high-throughput screens performed by GlaxoSmithKline, for example, against a large number of targets produced no viable leads (Payne et al., 2007). Glaxo scientists realized that penetration is a serious problem and therefore also performed in vivo screens against E. coli, but only obtained “nuisance” hits, such as membrane-acting compounds. One obvious conclusion from this negative experience is that the libraries do not carry good starting compounds. In part, this is due to the fact that libraries are constructed based on Lipinski rules (Lipinski, 2003), which are good for predicting druglike properties for compounds acting against mammalian cell targets but do not work well for bacteria because of peculiarities of permeation (O’Shea and Moser, 2008; Silver, 2008). Another important consideration is the probability of resistance development. Pathogen populations produce ~109 cells in an infected patient, which means that the probability of resistance development should be <109. This is readily achieved with most of the antibiotic classes currently in use, because they hit more than one target (fluoroquinolones attack DNA gyrase and topoisomerase, β-lactams inhibit several penicillin-binding proteins, and ribosomal inhibitors bind to rRNA which is coded by multiple genes) (Silver, 2007). This requirement severely limits the number of realistic targets for antimicrobial drug discovery.
The above analysis presents an extremely bleak picture: if we cannot even discover compounds acting against rapidly growing Gram-positive bacteria, what are the prospects of finding broad-spectrum antimicrobials acting against non-growing stationary cells and persisters?
Opportunities
There are many steps in the drug discovery pipeline, but if there are no viable leads, there is no pipeline. Indeed, at the last Interscience Conference on Antimicrobial Agents and Chemotherapy (ICAAC) meeting (2009), there was not a single broad-spectrum lead presented. This means that the number of realistic
broad-spectrum leads in the global antimicrobial drug discovery pipeline is zero. This is where the process needs to be restarted, and this is where allocation of resources will make a tangible impact.
A Fresh Look at Potential Sources of Compounds
Natural products There are two largely untapped and potentially enormous new sources of natural products—uncultured microorganisms, and silent operons coding for secondary metabolites.
A recent resurgence in cultivation efforts aimed at gaining access to uncultured microorganisms has been sparked by the vast diversity of uncultured bacterial groups revealed by environmental surveys of 16S rRNA (Aoi et al., 2009; Bollmann et al., 2007; Bruns et al., 2002; Connon and Giovannoni, 2002; Davis et al., 2005; Ferrari et al., 2005; Gavrish et al., 2008; Kaeberlein et al., 2002; Nichols et al., 2008; Rappe et al., 2002; Stevenson et al., 2004; Zengler et al., 2002). While some novel bacterial species were successfully cultured by varying media and growth conditions (Joseph et al., 2003), significant departures from conventional techniques were clearly in order, and indeed the new technologies substantially diverged from traditional cultivation methods by adopting single-cell and high-throughput strategies (Connon and Giovannoni, 2002; Nichols et al., 2008; Rappe et al., 2002; Zengler et al., 2002), better mimicking the natural milieu (Aoi et al., 2009; Bruns et al., 2002; Ferrari et al., 2005; Stevenson et al., 2004), increasing the length of incubation, and lowering the concentration of nutrients (Davis et al., 2005). High-throughput extinction culturing is based on the dilution of natural communities of bacteria to 1–10 cells per well in low-nutrient, filtered marine water. This strategy resulted in the cultivation of the first member of the ubiquitous, previously uncultured clade, SAR11 (Rappe et al., 2002). Our research group contributed to the effort by developing three cultivation methodologies (Gavrish et al., 2008; Kaeberlein et al., 2002; Nichols et al., 2008). All three strategies aim to provide microorganisms with their natural growth conditions by incubating them in simulated natural environments.
The diffusion chamber is designed to essentially “trick” cells into thinking they are growing in their natural environment by creating an incubation strategy that very closely mimics their natural habitat (Kaeberlein et al., 2002). The diffusion chamber consists of a stainless steel washer and 0.03-μm pore-size membranes (Figure A11-9). After gluing a membrane to one side of the washer, the inoculum (a mix of environmental cells and warm agar) is introduced, and the second membrane seals the chamber. Nutrients from the environment can diffuse into the chamber; therefore, it is not necessary to add them to the medium. Once inoculated and assembled, the chamber can be returned to the original location of sampling or in a simulated natural environment such as a block of sediment kept in an aquarium in a lab. Microcolonies grow in the chamber during such incubation.
FIGURE A11-9 A diffusion chamber for growing bacteria in situ. A sample from marine sediment is diluted, mixed with agar, and sandwiched between the two semipermeable membranes of the diffusion chamber, which is returned to the environment.
A recovery rate of 22 percent on average was observed in the diffusion chambers. In this study and follow-up research (Bollmann et al., 2007; Nichols et al., 2008), we isolated numerous species that did not grow in Petri dishes inoculated with environmental samples but were successfully grown in the diffusion chambers.
Reinoculation of material from both marine and soil environments from chamber to chamber produces “domesticated” variants that grow on regular media on a Petri dish and can be exploited for secondary metabolite production (Bollmann et al., 2007; Nichols et al., 2008).
The diffusion chamber typically produces a mixed culture, which requires considerable time to isolate, purify, and reinoculate individual colonies. In order to streamline this process into a high-throughput system, we developed a variant of the diffusion chamber for massively parallel microbial isolation. The Isolation Chip, or ichip for short (Nichols et al., 2010), consists of hundreds of miniature diffusion chambers that can be loaded with an average of one cell per chamber. The ichip enables microbial growth and isolation in a single step with hundreds of individual cultures incubating on a single chip.
Microorganisms that are particularly important for drug discovery, microscopic fungi and actinomycetes, grow by forming filaments capable of penetrating soft substrates. Because actinomycetes can pass through 0.2-μm pores, we reasoned this could be used to design a trap for the specific capture of these organisms (Gavrish et al., 2008). The trap is similar in design to the diffusion chamber, except the membranes have larger pores and the agar inside the trap is initially sterile when placed in the environment. Any growth observed afterward inside the trap is due to the movement of cells into the trap during incubation. The majority of organisms grown in the traps proved to be actinomycetes, some of
which represented rare and unusual species from the genera Dactylosporangium, Catellatospora, Catenulispora, Lentzea, and Streptacidiphilus.
We noticed that some organisms forming colonies in the diffusion chamber can grow on a Petri dish, but only in the presence of other species from the same environment (Kaeberlein et al., 2002; Nichols et al., 2008), suggesting that uncultured bacteria only commit to division in a familiar environment, which they recognize by the presence of growth factors released by their neighbors. In order to assess the commonality of the growth dependence of uncultured organisms on neighboring species and pick good models for study, we chose an environment where bacteria live in a tightly packed community (D’Onofrio et al., 2010). This is a biofilm that envelops sand particles of a tidal ocean beach (Figure A11-10). There were disproportionately more colonies appearing on densely inoculated plates compared with more dilute plates. This indicated that some of the cells that grew on the densely seeded plates were receiving growth factors from neighboring colonies. To test the possible growth dependence of microorganisms on neighboring species, pairs of colonies growing within a short distance of each other
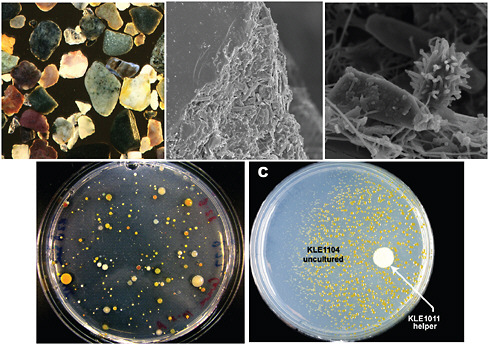
FIGURE A11-10 Understanding the mechanism of uncultivability. Marine sand particles are covered by a multispecies biofilm (upper panel). Cells from the biofilm form colonies on a densely seeded plate, and pairing them together reveals that some of them are uncultured bacteria (evenly spread on the plate) that will only grow in the presence of a helper species spotted on the same plate (lower panel).
were restreaked in close proximity to each other. Potential uncultured isolates were identified by their diminishing growth with increasing distance from the cultivable “helper” strain on the cross-streaked plates. Colonies of the culturable organism Micrococcus luteus KLE1011 (a marine sand sediment isolate 99.5 percent identical to Micrococcus luteus DSM 200030T according to 16S rRNA gene sequence) grew larger as their distance from other colonies increased (Figure A11-10). Approximately 100 randomly picked pairs of colonies were restreaked from the high-density plates, and 10 percent of these pairs showed this pattern of growth induction on cross-streaked plates.
In order to isolate growth factors, spent medium from the helper M. luteus KLE1011 was tested and shown to induce growth of the uncultured M. polysiphoniae KLE1104. An assay-guided fractionation led to isolation and structure determination of five different siderophores and each of them was able to induce growth of M. polysiphoniae KLE1104. This demonstrated that siderophores represent the growth factors responsible for the helping activity. The siderophores consisted of a central core with alternating N-hydroxycadaverine and succinic acid units and were of the desferrioxamine class (Challis, 2005). Close relatives of both known microorganisms and novel species were isolated by this approach. This study identified the first class of growth factors for uncultured bacteria and suggests that additional ones will come from analyzing organisms growing in co-culture.
Silent operons Whole-genome sequencing of several actinomycetes showed that there are many more potential biosynthetic pathways for production of secondary metabolites than there are known antibiotics made by these organisms (Ikeda et al., 2003). Ecopia has used fermentation in 40 different media to entice production of additional compounds and discovered a novel type of enediyne with anticancer activity (Zazopoulos et al., 2003). No novel antimicrobials emerged from this effort. However, in order to be effective, one needs to develop a high-throughput approach to induce production of such compounds. This is entirely doable.
Synthetics Are existing libraries, both commercially available and proprietary collections in Big Pharma, useless for antibiotic discovery? It does seem so, because they have obviously already been screened for actives, including non-biased screens for growth inhibition of whole cells, and produced no viable leads. But does it not seem strange that a screen of a collection of 600 dyes by Domagk produced the first viable antibiotics, while a screen of the total global library of ~107 compounds produced nothing at all? As the libraries grew, a number of innovations were introduced, aimed at improving the screening outcome—in vitro screening, targeted screens, Lipinski rules, and specificity validation. My feeling is that each time we tried to improve things, the result was to discard valuable compounds. I think that the existing libraries do harbor useful molecules; the question is how to identify them.
Good Compounds from Bad Libraries
Back to Domagk The first screen was also perfect: Domagk tested compounds against mice infected with Streptococci. The result was the discovery of prontosil, a sulfadrug that has no activity in vitro. The compound is cleaved in the intestine by gut bacteria, releasing the active sulfonamide moiety, which inhibits dehydrop-teroate synthase in the folate pathway. An in vitro test would have missed prontosil. There are obvious advantages to testing compounds in situ—the approach automatically eliminates the significant burden of toxic molecules, and it demonstrates efficacy, again automatically eliminating substances with problems of action in an animal, such as serum binding, instability, or poor tissue distribution. In addition, different types of compounds may be uniquely uncovered, such as those requiring activation in situ, and those hitting targets that are only important in an infection but not in vitro. Although this would theoretically be the perfect way to go, testing in 107 mice is not an option for a variety of reasons, including ethical considerations and the large amounts of required test compounds. We therefore considered a useful intermediate between in vitro and a mammal—an animal that, unlike mice, can be dispersed in microtiter wells. C. elegans can be infected with human pathogens by simply ingesting them, and we found that the worm can be cured by common antibiotics, such as tetracycline and vancomycin, and at concentrations typically achieved in human plasma (Moy et al., 2006). Worms infected with a pathogen such as E. feacalis die, stop moving, their shape changes from curved to straight, and they can be detected by typical eukaryotic vital dyes (Figure A11-11). Using these parameters, an automated approach was developed, and a large pilot screen of compound libraries uncovered hits, some of which had no activity in vitro (Moy et al., 2006, 2009). This approach shows that C. elegans points us in the right direction—back to Domagk, but with larger libraries.
Better Libraries and Rules of Penetration
Of course it would be great to have a better library, constructed based on “rules of penetration” and not on Lipinski rules. We have a small number of broad-spectrum compounds that are able to largely bypass the MDRs and get across the impermeable barrier of Gram-negative membranes: tetracycline, chloramphenicol, aminoglycosides, trimethoprim, β-lactams (these only need to traverse the outer membrane), fluoroquinolones, and metronidazole. This set is too small to enable us to discern rules for penetration. But testing a large number of unbiased compounds from a library for their ability to enter into the cytoplasm of Gram-negative bacteria should allow us to deduce general rules that favor penetration. Once these are available, this would drive the synthesis/combinatorial chemistry of new compound libraries specifically geared toward antimicrobial discovery.
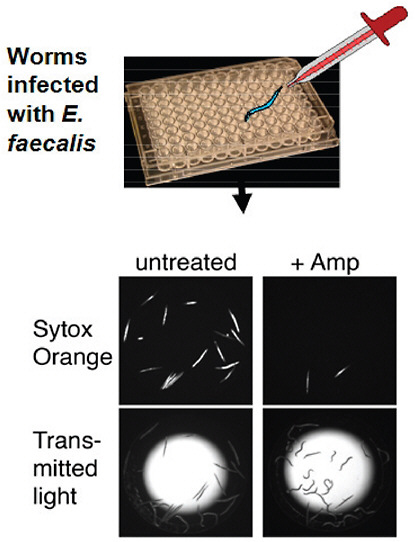
FIGURE A11-11 A high-throughput screen for antimicrobials in an animal model. C. elegans are infected with E. faecalis and cured with ampicillin. This provides for an assay of compounds that cure the worm in situ.
Prodrugs
It is useful to consider the theoretically perfect antibiotic from first principles and then decide whether it is realistic. Approaches we discussed so far did not address the daunting challenge of killing persister cells while showing broad-spectrum activity. It is useful to start with the end result: a highly reactive compound will kill all cells, including persisters. In order to spare the host, the compound must be delivered as a prodrug, and then a bacteria-specific enzyme will
activate it into a generally reactive molecule that will covalently bind to unrelated targets. Importantly, this mechanism creates an irreversible sink, largely resolving the issue of MDR efflux, so the antimicrobial is automatically broad spectrum. Is this realistic? Several existing antimicrobials closely match the properties of this idealized prodrug antibiotic. These are isoniazid, pyrazinamide, ethionamide, and metronidazole. The first three are anti-Mtb drugs, while metronidazole is a broad-spectrum compound acting against anaerobic bacteria. All four compounds convert into active antiseptic-type molecules inside the cell that covalently bind to their targets. It seems to be no accident that prodrug antibiotics make up the core of the anti-Mtb drug arsenal, because an ability to kill the pathogen is critical for treating the disease. Preferred targets have been identified for isoniazid and ethionamide (Vilcheze et al., 2005), suggesting a relatively limited reactivity of these compounds. The existence of preferred targets indicates that the prodrug products are not that reactive, and there is considerable room for developing better sterilizing antibiotics based on the same principle.
References
Al-Dhaheri, R. S., and L. J. Douglas. 2008. Absence of amphotericin B-tolerant persister cells in biofilms of some Candida species. Antimicrob. Agents Chemother. 52:1884–7.
Alix, E., and A. Blanc-Potard. 2009. Hydrophobic peptides: Novel regulators within bacterial membranes. Mol. Microbiol. 72:5–11.
Aoi, Y., T. Kinoshita, T. Hata, H. Ohta, H. Obokata, and S. Tsuneda. 2009. Hollow-fiber membrane chamber as a device for in situ environmental cultivation. Appl. Environ. Microbiol. 75:3826–33.
Baba, T., T. Ara, M. Hasegawa, Y. Takai, Y. Okumura, M. Baba, K. A. Datsenko, M. Tomita, B. L. Wanner, and H. Mori. 2006. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: The Keio collection. Mol. Syst. Biol. 2:2006.0008.
Barry, C. E., 3rd, H. I. Boshoff, V. Dartois, T. Dick, S. Ehrt, J. Flynn, D. Schnappinger, R. J. Wilkinson, and D. Young. 2009. The spectrum of latent tuberculosis: Rethinking the biology and intervention strategies. Nat. Rev. Microbiol. 7:845–55.
Bollmann, A., K. Lewis, and S. S. Epstein. 2007. Incubation of environmental samples in a diffusion chamber increases the diversity of recovered isolates. Appl. Environ. Microbiol. 73:6386–90.
Bruns, A., H. Cypionka, and J. Overmann. 2002. Cyclic AMP and acyl homoserine lactones increase the cultivation efficiency of heterotrophic bacteria from the central Baltic Sea. Appl. Environ. Microbiol. 68:3978–87.
Challis, G. L. 2005. A widely distributed bacterial pathway for siderophore biosynthesis independent of nonribosomal peptide synthetases. Chembiochem. 6:601–11.
Christensen, S. K., and K. Gerdes. 2003. RelE toxins from bacteria and Archaea cleave mRNAs on translating ribosomes, which are rescued by tmRNA. Mol. Microbiol. 48:1389–1400.
Connon, S. A., and S. J. Giovannoni. 2002. High-throughput methods for culturing microorganisms in very-low-nutrient media yield diverse new marine isolates. Appl. Environ. Microbiol. 68:3878–85.
Correia, F. F., A. D’Onofrio, T. Rejtar, L. Li, B. L. Karger, K. Makarova, E. V. Koonin, and K. Lewis. 2006. Kinase activity of overexpressed HipA is required for growth arrest and multidrug tolerance in Escherichia coli. J. Bacteriol. 188:8360–7.
Costerton, J. W., P. S. Stewart, and E. P. Greenberg. 1999. Bacterial biofilms: A common cause of persistent infections. Science 284:1318–22.
Courcelle, J., A. Khodursky, B. Peter, P. Brown and P. Hanawalt. 2001. Comparative gene expression profiles following UV exposure in wild type and SOS-deficient Escherichia coli. Genetics 158:41–64.
Davis, K. E., S. J. Joseph, and P. H. Janssen. 2005. Effects of growth medium, inoculum size, and incubation time on culturability and isolation of soil bacteria. Appl. Environ. Microbiol. 71:826–34.
De Groote, V. N., N. Verstraeten, M. Fauvart, C. I. Kint, A. M. Verbeeck, S. Beullens, P. Cornelis, and J. Michiels. 2009. Novel persistence genes in Pseudomonas aeruginosa identified by high-throughput screening. FEMS Microbiol. Lett. 297:73–9.
Del Pozo, J., and R. Patel. 2007. The challenge of treating biofilm-associated bacterial infections. Clin. Pharmacol. Ther. 82:204–9.
D’Onofrio, A., J. M. Crawford, E. J. Stewart, K. Witt, E. Gavrish, S. Epstein, J. Clardy, and K. Lewis. 2010. Siderophores from neighboring organisms promote the growth of uncultured bacteria. Chem. Biol. 17:254–64.
Dörr, T., K. Lewis, and M. Vulic. 2009. SOS response induces persistence to fluoroquinolones in Escherichia coli. PLoS Genet. 5:e1000760.
Falla, T. J., and I. Chopra. 1998. Joint tolerance to beta-lactam and fluoroquinolone antibiotics in Escherichia coli results from overexpression of hipA. Antimicrob. Agents Chemother. 42:3282–4.
Fernandez De Henestrosa, A. R., T. Ogi, S. Aoyagi, D. Chafin, J. J. Hayes, H. Ohmori, and R. Woodgate. 2000. Identification of additional genes belonging to the LexA regulon in Escherichia coli. Mol. Microbiol. 35:1560–72.
Ferrari, B. C., S. J. Binnerup, and M. Gillings. 2005. Microcolony cultivation on a soil substrate membrane system selects for previously uncultured soil bacteria. Appl. Environ. Microbiol. 71:8714–20.
Garcia-Olmedo, F., A. Molina, J. M. Alamillo, and P. Rodriguez-Palenzuela. 1998. Plant defense peptides. Biopolymers 47:479–91.
Gavrish, E., A. Bollmann, S. Epstein, and K. Lewis. 2008. A trap for in situ cultivation of filamentous actinobacteria. J. Microbiol. Methods 72:257–62.
Gerdes, K., P. B. Rasmussen, and S. Molin. 1986a. Unique type of plasmid maintenance function: Postsegregational killing of plasmid-free cells. Proc. Natl. Acad. Sci. USA 83:3116–20.
Gerdes, K., F. W. Bech, S. T. Jorgensen, A. Lobner-Olesen, P. B. Rasmussen, T. Atlung, L. Boe, O. Karlstrom, S. Molin, and K. von Meyenburg. 1986b. Mechanism of postsegregational killing by the hok gene product of the parB system of plasmid R1 and its homology with the relF gene product of the E. coli relB operon. Embo. J. 5:2023–9.
Gibson, R. L., J. L. Burns, and B. W. Ramsey. 2003. Pathophysiology and management of pulmonary infections in cystic fibrosis. Am. J. Respir. Crit. Care Med. 168:918–51.
Hansen, S., K. Lewis, and M. Vulić. 2008. The role of global regulators and nucleotide metabolism in antibiotic tolerance in Escherichia coli. Antimicrob. Agents Chemother. 52:2718–26.
Harrison, J. J., H. Ceri, N. J. Roper, E. A. Badry, K. M. Sproule, and R. J. Turner. 2005a. Persister cells mediate tolerance to metal oxyanions in Escherichia coli. Microbiology 151:3181–95.
Harrison, J. J., R. J. Turner, and H. Ceri. 2005b. Persister cells, the biofilm matrix and tolerance to metal cations in biofilm and planktonic Pseudomonas aeruginosa. Environ. Microbiol. 7:981–94.
Harrison, J. J., R. J. Turner, and H. Ceri. 2007. A subpopulation of Candida albicans and Candida tropicalis biofilm cells are highly tolerant to chelating agents. FEMS Microbiol. Lett. 272:172–81.
Harrison, J. J., W. D. Wade, S. Akierman, C. Vacchi-Suzzi, C. A. Stremick, R. J. Turner, and H. Ceri. 2009. The chromosomal toxin gene yafQ is a determinant of multidrug tolerance for Escherichia coli growing in a biofilm. Antimicrob. Agents Chemother. 53:2253–8.
Hayes, F. 2003. Toxins-antitoxins: Plasmid maintenance, programmed cell death, and cell cycle arrest. Science 301:1496–9.
Honda, H., and D. K. Warren. 2009. Central nervous system infections: Meningitis and brain abscess. Infect. Dis. Clin. North Am. 23:609–23.
Hu, Y., and A. R. Coates. 2005. Transposon mutagenesis identifies genes which control antimicrobial drug tolerance in stationary-phase Escherichia coli. FEMS Microbiol. Lett. 243:117–24.
Ikeda, H., J. Ishikawa, A. Hanamoto, M. Shinose, H. Kikuchi, T. Shiba, Y. Sakaki, M. Hattori, and S. Omura. 2003. Complete genome sequence and comparative analysis of the industrial microorganism Streptomyces avermitilis. Nat. Biotechnol. 21:526–31.
Jesaitis, A. J., M. J. Franklin, D. Berglund, M. Sasaki, C. I. Lord, J. B. Bleazard, J. E. Duffy, H. Beyenal, and Z. Lewandowski. 2003. Compromised host defense on Pseudomonas aeruginosa biofilms: Characterization of neutrophil and biofilm interactions. J. Immunol. 171:4329–39.
Joseph, S. J., P. Hugenholtz, P. Sangwan, C. A. Osborne, and P. H. Janssen. 2003. Laboratory cultivation of widespread and previously uncultured soil bacteria. Appl. Environ. Microbiol. 69:7210–5.
Kaeberlein, T., K. Lewis, and S. S. Epstein. 2002. Isolating “uncultivable” microorganisms in pure culture in a simulated natural environment. Science 296:1127–9.
Kawano, M., L. Aravind, and G. Storz. 2007. An antisense RNA controls synthesis of an SOS-induced toxin evolved from an antitoxin. Mol. Microbiol. 64:738–54.
Keren, I., D. Shah, A. Spoering, N. Kaldalu, and K. Lewis. 2004. Specialized persister cells and the mechanism of multidrug tolerance in Escherichia coli. J. Bacteriol. 186:8172–80.
Korch, S. B., and T. M. Hill. 2006. Ectopic overexpression of wild-type and mutant hipA genes in Escherichia coli: Effects on macromolecular synthesis and persister formation. J. Bacteriol. 188:3826–36.
LaFleur, M. D., C. A. Kumamoto, and K. Lewis. 2006. Candida albicans biofilms produce antifungal-tolerant persister cells. Antimicrob. Agents Chemother. 50:3839–46.
LaFleur, M. D., Q. Qi, and K. Lewis. 2010. Patients with long-term oral carriage harbor high-persister mutants of Candida albicans. Antimicrob. Agents Chemother. 54:39–44.
Leid, J. G., M. E. Shirtliff, J. W. Costerton, and A. P. Stoodley. 2002. Human leukocytes adhere to, penetrate, and respond to Staphylococcus aureus biofilms. Infect. Immun. 70:6339–45.
Levin, B. R., and D. E. Rozen. 2006. Non-inherited antibiotic resistance. Nat. Rev. Microbiol. 4:556–62.
Lewis, K. 2001. Riddle of biofilm resistance. Antimicrob. Agents Chemother. 45:999–1007.
_____. 2007. Persister cells, dormancy and infectious disease. Nat. Rev. Microbiol. 5:48–56.
_____. 2010 (in press). Persister cells. Annu. Rev. Microbiol.
Lewis, K., S. Epstein, A. D’Onofrio, and L. L. Ling. 2010 (in press). Uncultured microorganisms as a source of secondary metabolites. J. Antibiotics. (Epub ahead of print)
Lipinski, C. A. 2003. The practice of medicinal chemistry. Amsterdam, The Netherlands: Academic Press, 341 pp.
Lomovskaya, O., H. I. Zgurskaya, K. A. Bostian, and K. Lewis. 2008. Multidrug efflux pumps: Structure, mechanism, and inhibition. In Bacterial resistance to antimicrobials, edited by R. G. Wax, K. Lewis, A. A. Salyers, and H. Taber. Boca Raton, FL: CRC Press, pp. 45–70.
McKenzie, M. D., P. L. Lee, and S. M. Rosenberg. 2003. The dinB operon and spontaneous mutation in Escherichia coli. J. Bacteriol. 185:3972–7.
Motiejunaite, R., J. Armalyte, A. Markuckas, and E. Suziedeliene. 2007. Escherichia coli dinJ-yafQ genes act as a toxin-antitoxin module. FEMS Microbiol. Lett. 268:112–9.
Moy, T. I., A. R. Ball, Z. Anklesaria, G. Casadei, K. Lewis, and F. M. Ausubel. 2006. Identification of novel antimicrobials using a live-animal infection model. Proc. Natl. Acad. Sci. USA 103:10414–9.
Moy, T. I., A. L. Conery, J. Larkins-Ford, G. Wu, R. Mazitschek, G. Casadei, K. Lewis, A. E. Carpenter, and F. M. Ausubel. 2009. High-throughput screen for novel antimicrobials using a whole animal infection model. ACS Chem. Biol. 4:527–33.
Moyed, H. S., and K. P. Bertrand. 1983. hipA, a newly recognized gene of Escherichia coli K-12 that affects frequency of persistence after inhibition of murein synthesis. J. Bacteriol. 155:768–75.
Mulcahy, L. R., J. L. Burns, S. Lory, and K. Lewis. 2010 (in press). Emergence of Pseudomonas aeruginosa strains producing high levels of persister cells in patients with cystic fibrosis. J. Bacteriol.
Nichols, D., K. Lewis, J. Orjala, S. Mo, R. Ortenberg, P. O’Connor, C. Zhao, P. Vouros, T. Kaeberlein, and S. S. Epstein. 2008. Short peptide induces an “uncultivable” microorganism to grow in vitro. Appl. Environ. Microbiol. 74:4889–97.
Nichols, D., N. Cahoon, E. M. Trakhtenberg, L. Pham, A. Mehta, A. Belanger, T. Kanigan, K. Lewis, and S. S. Epstein. 2010. Use of ichip for high-throughput in situ cultivation of “uncultivable” microbial species. Appl. Environ. Microbiol. 76:2445–50.
O’Shea, R., and H. E. Moser. 2008. Physicochemical properties of antibacterial compounds: Implications for drug discovery. J. Med. Chem. 51:2871–8.
Pandey, D. P., and K. Gerdes. 2005. Toxin-antitoxin loci are highly abundant in free-living but lost from host-associated prokaryotes. Nucleic Acids Res. 33:966–76.
Payne, D. J., M. N. Gwynn, D. J. Holmes, and D. L. Pompliano. 2007. Drugs for bad bugs: Confronting the challenges of antibacterial discovery. Nat. Rev. Drug Discov. 6:29–40.
Pedersen, K., and K. Gerdes. 1999. Multiple hok genes on the chromosome of Escherichia coli. Mol. Microbiol. 32:1090–102.
Pedersen, K., S. K. Christensen, and K. Gerdes. 2002. Rapid induction and reversal of a bacteriostatic condition by controlled expression of toxins and antitoxins. Mol. Microbiol. 45:501–10.
Peterson, W. L., A. M. Fendrick, D. R. Cave, D. A. Peura, S. M. Garabedian-Ruffalo, and L. Laine. 2000. Helicobacter pylori-related disease: Guidelines for testing and treatment. Arch. Internal Med. 160:1285–91.
Phillips, I., E. Culebras, F. Moreno, and F. Baquero. 1987. Induction of the SOS response by new 4-quinolones. J. Antimicrob. Chemother. 20:631–8.
Ramage, H. R., L. E. Connolly, and J. S. Cox. 2009. Comprehensive functional analysis of Mycobacterium tuberculosis toxin-antitoxin systems: Implications for pathogenesis, stress responses, and evolution. PLoS Genet. 5:e1000767.
Rappe, M. S., S. A. Connon, K. L. Vergin, and S. J. Giovannoni. 2002. Cultivation of the ubiquitous SAR11 marine bacterioplankton clade. Nature 418:630–3.
Sahl, H. G., and G. Bierbaum. 1998. Lantibiotics: Biosynthesis and biological activities of uniquely modified peptides from gram-positive bacteria. Annu. Rev. Microbiol. 52:41-79.
Schatz, A., E. Bugie, and S. A. Waxman. 1944. Streptomycin, a substance exhibiting antibiotic activity against gram positive and gram negative bacteria. Proc. Soc. Exper. Biol. Med. 55:66.
Schmelzle, T., and M. N. Hall. 2000. TOR, a central controller of cell growth. Cell 103:253–62.
Schumacher, M. A., K. M. Piro, W. Xu, S. Hansen, K. Lewis, and R. G. Brennan. 2009. Molecular mechanisms of HipA-mediated multidrug tolerance and its neutralization by HipB. Science 323:396–401.
Shah, D., Z. Zhang, A. Khodursky, N. Kaldalu, K. Kurg, and K. Lewis. 2006. Persisters: A distinct physiological state of E. coli. BMC Microbiol. 6:53–61.
Silver, L. L. 2007. Multi-targeting by monotherapeutic antibacterials. Nat. Rev. Drug Discov. 6:41–55.
Silver, L. L. 2008. Are natural products still the best source for antibacterial discovery? The bacterial entry factor. Expert Opin. Drug Discov. 3:487–99.
Singletary, L. A., J. L. Gibson, E. J. Tanner, G. J. McKenzie, P. L. Lee, C. Gonzalez, and S. M. Rosenberg. 2009. An SOS-regulated type 2 toxin-antitoxin system. J. Bacteriol. 191:7456–65.
Smith, E. E., D. G. Buckley, Z. Wu, C. Saenphimmachak, L. R. Hoffman, D. A. D’Argenio, S. I. Miller, B. W. Ramsey, D. P. Speert, S. M. Moskowitz, J. L. Burns, R. Kaul, and M. V. Olson. 2006. Genetic adaptation by Pseudomonas aeruginosa to the airways of cystic fibrosis patients. Proc. Natl. Acad. Sci. USA 103:8487–92.
Spoering, A. 2006. GlpD and PlsB participate in persister cell formation in Escherichia coli. J. Bacteriol. 188:5136–44.
Spoering, A. L., and K. Lewis. 2001. Biofilms and planktonic cells of Pseudomonas aeruginosa have similar resistance to killing by antimicrobials. J. Bacteriol. 183:6746–51.
Stevenson, B. S., S. A. Eichorst, J. T. Wertz, T. M. Schmidt, and J. A. Breznak. 2004. New strategies for cultivation and detection of previously uncultured microbes. Appl. Environ. Microbiol. 70:4748–55.
Stewart, P. S., and J. W. Costerton. 2001. Antibiotic resistance of bacteria in biofilms. Lancet 358:135–8.
Unoson, C., and E. Wagner. 2008. A small SOS-induced toxin is targeted against the inner membrane in Escherichia coli. Mol. Microbiol. 70:258–70.
Vazquez-Laslop, N., H. Lee, and A. A. Neyfakh. 2006. Increased persistence in Escherichia coli caused by controlled expression of toxins or other unrelated proteins. J. Bacteriol. 188:3494–7.
Vilcheze, C., T. R. Weisbrod, B. Chen, L. Kremer, M. H. Hazbon, F. Wang, D. Alland, J. C. Sacchettini, and W. R. Jacobs, Jr. 2005. Altered NADH/NAD+ ratio mediates coresistance to isoniazid and ethionamide in mycobacteria. Antimicrob. Agents Chemother. 49:708–20.
Vogel, J., Argaman,L., Wagner, E.G., and Altuvia, S. 2004. The small RNA Istr inhibits synthesis of an SOS-induced toxic peptide. Curr. Biol. 14:2271–6.
Vuong, C., J. M. Voyich, E. R. Fischer, K. R. Braughton, A. R. Whitney, F. R. DeLeo, and M. Otto. 2004. Polysaccharide intercellular adhesin (PIA) protects Staphylococcus epidermidis against major components of the human innate immune system. Cell Microbiol. 6:269–75.
Walters, M. C., 3rd, F. Roe, A. Bugnicourt, M. J. Franklin, and P. S. Stewart. 2003. Contributions of antibiotic penetration, oxygen limitation, and low metabolic activity to tolerance of Pseudomonas aeruginosa biofilms to ciprofloxacin and tobramycin. Antimicrob. Agents Chemother. 47:317–323.
Wolfson, J. S., D. C. Hooper, G. L. McHugh, M. A. Bozza, and M. N. Swartz. 1990. Mutants of Escherichia coli K-12 exhibiting reduced killing by both quinolone and beta-lactam antimicrobial agents. Antimicrob. Agents Chemother. 34:1938–43.
Zasloff, M. 2002. Antimicrobial peptides of multicellular organisms. Nature 415:389–95.
Zazopoulos, E., K. Huang, A. Staffa, W. Liu, B. O. Bachmann, K. Nonaka, J. Ahlert, J. S. Thorson, B. Shen, and C. M. Farnet. 2003. A genomics-guided approach for discovering and expressing cryptic metabolic pathways. Nat. Biotechnol. 21:187–190.
Zengler, K., G. Toledo, M. Rappe, J. Elkins, E. J. Mathur, J. M. Short, and M. Keller. 2002. Cultivating the uncultured. Proc. Natl. Acad. Sci. USA 99:15681–6.
A12
POPULATION MOBILITY, GLOBALIZATION, AND ANTIMICROBIAL RESISTANCE
Douglas W. MacPherson53,54,55 and Brian D. Gushulak53
From the primordial soup
Out of the dim and gloom we came
We are animals
By any other name.
—“Primitive,” lyrics written by Ivan Corraliza and Roisin Marie Murphy
Human beings are a significant part of Earth’s biomass, with complex interactions and dependencies on our environment, including botantical and all other animal life. For the large part, our coexistence with microbial life is either a beneficial or a neutral experience. Our exposure to both the microbial flora that lives in and on us and the environmental microorganisms that surround us are important to our own survival … most times.
Increasingly, emerging and reemerging infectious diseases are seen as a threat to global and public health (IOM, 2001). While there are many reasons for this emergence of threat, human behavior and both the perception and the reality of risk are major contributors to this issue. One component of emerging threat conversion to an infectious disease risk is microbial resistance to drug therapy. While innate resistance to disease is a characteristic of all living organisms, the selective pressures of antimicrobial use by human beings is a major contributor to the relatively new phenomenon of induced antimicrobial drug resistance that leads to increased morbidity and mortality due to infectious diseases. The ability of microbes to attain resistance to antimicrobials was recognized soon after their introduction in clinical medicine.
There may be a danger, though, in underdosage. It is not difficult to make microbes resistant to penicillin in the laboratory by exposing them to concentrations not sufficient to kill them, and the same thing has occasionally happened in the body.
—Alexander Fleming’s Nobel Lecture, December 11, 1945 (Fleming, 1945)
|
53 |
Migration Health Consultants Inc., Cheltenham, Ontario, Canada/Singapore. |
|
54 |
Department of Pathology and Molecular Medicine, Faculty of Health Sciences, McMaster University, Hamilton, Ontario, Canada. |
|
55 |
Author contact information: Dr. Douglas W. MacPherson, 64 Maple Avenue, Dundas, Ontario, Canada L9H 4W4; e-mail: douglaswmacpherson@migrationhealth.com. Disclaimer: the views presented in this paper represent those of the authors only and should not be ascribed to any government, organization, institution, or membership society to which the authors are currently or in the past have been associated with. |
Microbiologists began to understand the nature and genetics of single- and multidrug resistance as it appeared in Japan and Europe in the 1950s and 1960s. By the 1970s, clinicians began to appreciate the importance of acquired and transferred resistance in both hospital- and community-acquired infections (Davies, 2007).
Resistance of microbes to drugs used for the treatment and prevention of infections is becoming a clinical and public health crisis (U.S. Congress, 2010), with cascading adverse consequences for other domains. Drug resistance affects domestic and international economics, trade, commerce, environment, security, and animal health. As therapeutic agents, with few exceptions, antimicrobials have the characteristic that, with use over time, they become less and less effective due to the selective pressures exerted by the drugs. These pressures for induced microbial resistance are not limited solely to the target organisms but act broadly through the exposed microbial environment. Because acquired resistance is often both permanent and transferable, the obsolescence associated with the use of antimicrobials over time does not contain itself only to the person or place of use (Kumarasamy et al., 2010).
From the global to the local setting, there are many identifiable vulnerable and susceptible populations for whom drug-resistant infections are and will increasingly become a significant risk. For the majority of their lives, most people coexist with their own normal and environmental microbial flora, but in those with access to health care few avoid the real or perceived need for antimicrobial therapy at some point. As more antibiotics are used, the implications of drug resistance correspondingly increase.
Resistance-associated diminished effectiveness of antimicrobial therapy is seen in three main areas:
-
First, there will be a progressive inability to treat common acute infections involving all body sites, including the ear and throat, meninges and brain, lung, bowels, urinary tract, and skin. This negative impact will be seen in both community- and institutional-based clinical practices.
-
Second, the use of antimicrobials for the prevention of infection will be compromised. Preventing wound infections in invasive surgery (e.g., bowel, cardiac surgery), protecting implanted prosthetic materials (e.g, joints, cardiac valves), or preventing invasive infections in immune-suppressed patients (e.g., cancer, AIDS, other congenital syndromes) will entail increased risks of clinical failure and death due to infection.
-
Last, the population-based approach and the use of antimicrobials to limit and contain diseases of public health significance, such as tuberculosis, leprosy, malaria, sexually transmitted infections, outbreaks of bacterial meningitis, certain other respiratory diseases including influenza, and others, will either be less effective or fail completely.
The Centers for Disease Control and Prevention (CDC) has observed that each year nearly 2 million patients acquire an infection while in hospital in the United States. Of those who do become infected in a hospital, about 90,000 will die as a result their infection. In addition, 70 percent of the bacteria that cause hospital-acquired infections are now resistant to at least one of the drugs most commonly used to treat them. Persons infected with drug-resistant organisms are more likely to have longer hospital stays and to require treatment with second- or third-choice drugs that may be less effective, more toxic with a greater incidence of adverse side effects, and/or be more expensive (CDC, 2001).
Hospital environments are associated with risks related to nosocomial infections, drug resistance, and death. However, the implications of resistance are not limited to healthcare institutions. Other determinants of human health—such as socioeconomic factors, behavior, genetics, and biology—are also important. In addition, in certain populations, primary infection with drug-resistant organisms contributes to the burden of morbidity and mortality. Examples include children with falciparum malaria or persons co-infected with human immunodeficiency virus (HIV) and tuberculosis.
Several concurrent trends noted by the World Health Organization (WHO, 2002) have accelerated the emergence and global spread of antimicrobial drug resistance:
-
Demographic changes (United Nations, 1999) have resulted in a growing proportion of vulnerable populations, especially elderly people, needing hospital-based interventions who are thus at risk of exposure to highly resistant pathogens found in these environments (see Figure A12-1).
-
The urbanization of human populations (United Nations Population Fund, 2007; United Nations Department of Economic and Social Affairs, 2006), with its associated overcrowding and poor sanitation, greatly facilitates the spread of such diseases as typhoid, tuberculosis, respiratory infections, and pneumonia (see Figures A12-2 and A12-3).
-
Pollution, environmental degradation, and changing weather patterns can affect the incidence and distribution of infectious diseases, especially those such as malaria, which are spread by insects and other vectors (IOM, 2008).
-
The AIDS epidemic has greatly enlarged the population of immune-compromised patients at risk of numerous infections, many of which were previously rare.
-
The resurgence of specific drug-resistant infectious diseases, such as malaria and tuberculosis, is now in total responsible for many millions of infections each year. Much of this burden is carried by low-income countries that lack social investments in infrastructure, training and education, and other resources to adequately contain and control emerging drug resistance.
FIGURE A12-1 Age pyramids for more and less developed regions, 1998 and 2050.
SOURCE: United Nations Population Division, 1999.
-
The enormous growth of global trade and travel has increased the speed and facility with which both infectious diseases and resistant microorganisms can spread between continents.
The last issue, linking population mobility, globalization, and antimicrobial resistance, was recently extensively reviewed (MacPherson et al., 2009). The movement of drug-resistant organisms, infections, and disease and the potential secondary transmission postarrival, due to the movement of human beings acting as physical vectors from areas of high to low prevalence, are documented for virtually every class of microbe (viruses, bacteria, mycobacteria, fungi, and parasites) and therapeutic agent. Addressing the challenges posed by human mobility acting as a vector for the movement and introduction of drug-resistant organisms are the numbers of those on the move. Identifying cost-effective interventions that can be targeted to the “at-risk” mobile populations can present both clinical and public health management and mitigation burdens. Given the large “denominator” of domestic and international movements that occur annually (approximately 2 billion arrivals per year), identifying the target is a challenge (see Table A12-1; sources of information include the Office of the United Nations High Commissioner of Refugees [UNHCR, 2008, 2010], the World Tourism Organization
FIGURE A12-2 Global population projection as percent urban, 2007, 2015, and 2030.
SOURCE: United Nations Population Fund (2007) and United Nations Division of Economic and Social Affairs (2006).
[UNWTO, 2009], the United Nations [United Nations Department of Economic and Social Affairs, Population Division, 2009], the British Council [Böhm et al., 2004], the International Labour Office [ILO, 2004], the International Labour Organization [2009], the U.S. Department of State [2008], and the International Air Transport Association [IATA, 2010]).
This “needle-in-the-haystack” issue exists in part due to the nature of the processes designed around facilitating the movement of goods and persons (the trade and commerce principles of laissez-faire and free pratique). At the same time, the health-associated activities of surveillance, notification, or reporting of events of “international public health significance” take place only after the event has occurred (WHO, 2008a). Trying to balance competing interests, as in this example with commerce and trade against public health when there is potentially a lack of appreciation and commitment to a common goal, is a significant barrier to the containment and prevention of emerging antimicrobial resistance globally.
This paper focuses on addressing the management of the human component contributing to antimicrobial resistance related to the following seven factors:
-
prescribers’ education, training, and invigilation in antimicrobial stewardship for good patient care and reduction of risk in emerging drug resistance;
TABLE A12-1 Mobile Populations by Category and Estimates of Domestic and International Arrivals
|
Category of migrant |
Population estimates (year) |
|
Regular immigrants between 2005 and 2010 |
Annual flow of 2.7 million per year with a stock of ~214 million international migrants in 2010 |
|
International students |
2.1 million (stock in 2003) (Böhm et al., 2004) |
|
Migrant workers |
~100 million (stock in 2009) |
|
Refugees |
15.2 million (stock beginning of 2009) (UNHCR, 2010) |
|
Asylum seekers or refugee claimants |
838,000 (stock beginning of 2009) (UNHCR, 2010) |
|
Temporary: recreational or business travel |
924 million per year (2008) (UNWTO, 2009) |
|
Trafficked (across international borders) |
Estimated 800,000 per year (2006) (U.S. Department of State, 2008) |
|
Internally displaced |
51 million (stock in 2007) includes those displaced by natural disasters and conflict (UNHCR, 2008) |
|
Domestic arrivals by air |
Estimated 900 million (2007) (IATA, 2010) |
|
SOURCES: Table adapted from MacPherson et al. (2009) and WHO (2010a). |
|
-
infection control training, certification, and practice;
-
laboratory methodology, proficiency testing, and quality management;
-
active and passive surveillance systems, rapid analysis, and reporting;
-
public health process and regulatory tools related to health outcomes;
-
pharmaceutical security systems for standard and quality medicines; and
-
animal and plant health-sector engagement.
These factors are addressed in detail throughout this paper.
Prescribers’ Education, Training, and Invigilation in Antimicrobial Stewardship for Good Patient Care and Reduction of Risk in Emerging Drug Resistance
Just say no to drugs.
—Nancy Reagan’s slogan in support of the Ronald Reagan Presidential Foundation to reduce recreational drug use by the youth of America (1980s)
In highly regulated medical and healthcare service environments where antimicrobials are controlled agents, the prescribing behavior of physicians has been a focus for change. The focus has largely been to reduce the use of antimicrobials for viral syndromes but also to promote the most effective drug choice, dosage, and duration of therapy for selected bacterial infection syndromes. Directly
modifying physician behavior is a complex undertaking, and it requires intensive interventions and ongoing programs for compliance and sustainability to reach the desired outcomes (Aagaard et al., 2010; Christian-Kopp et al., 2010; Frich et al., 2010; Heritage et al., 2010; Kennedy et al., 2010; Visscher et al., 2009). The challenge of addressing the physician role in antimicrobial prescribing and the physician’s contribution to drug resistance is diffuse and international, appearing in both inpatient and outpatient settings and many diverse national environments (Di Pentima and Chan, 2010; Esmaily et al., 2010; Hulscher et al., 2010; Huttner et al., 2010).
Part of addressing the challenge of excessive antimicrobial use, including drug selection, dose, interval, duration, and the simultaneous use of multiple or fixed-combination agents, is in identifying contributing factors other than physician prescribing practices. One of those factors is the pressure to treat. The office or clinic time required to write a prescription is often considerably less than that involved in clinical reasoning, medical judgment, or explaining and justifying why antimicrobials are not indicated for the clinical condition. Physician perception of demand and patient expectations for antimicrobial treatment are difficult to reconcile when they are in apparent or real conflict with not using these agents.
The use of laboratory technology to guide clinical decision making may be useful for physicians and reassuring for patients or their guardians in deciding to use or not use antimicrobials (Burkhardt et al., 2010; Cals et al., 2010; Neumark et al., 2010; Schuetz et al., 2010). In situations of clinical uncertainty or in which a “second opinion” is provided by bedside technology or diagnostic algorithms to support the physician’s recommendation, if it can also be obtained in a timely manner, this second opinion can be useful in reducing unnecessary antimicrobial use.
A third factor to be considered in promoting good antimicrobial stewardship is the administrative framework that directs and rationalizes antimicrobial use. One type is the use of evidence-based guidelines or best practices. These provide a reference point for clinical assessment and prescribing for both physicians and patients (Tenke et al., 2008). Equally, they provide a broader audit purpose for clinical practice monitoring and feedback (Coco et al., 2010). Clinical algorithms and defined “order sets” are available in many areas of medical care, but they are potentially underutilized in the management of the patient who is suspected or confirmed to have a clinically significant infection. Being able to effectively audit antimicrobial use and relevant outcomes is both an imperative and a challenge (Berrington, 2010).
Another form of administrative control on antimicrobial use practices are regulatory and pseudoregulatory barriers to access: licensing for availability, formularies for controlled accessibility or affordability through preapprovals, or limiting use. Examples of these include those used by the Centers for Medicare and Medicaid Services (http://www.cms.gov/) or other insurance coverage providers.
The above three factors relevant to antimicrobial stewardship (managed physician prescribing behavior, reducing pressure to treat, and use of administrative frameworks to rationalize antimicrobial use) are operative and interactive in most advanced economic regions with highly developed healthcare systems. However, there are large geographic and socioeconomic population bases where such controls may not exist, and where access to antimicrobial agents has few to no barriers for the consumer. Not infrequently, the lack of control mechanisms and easy access to antibiotics occurs where antimicrobial resistance is a recognized threat (National Intelligence Council, 2000). A taskforce to address a collaborative, international response to the global emergence of antimicrobial resistance was recently announced by U.S. President Obama and Swedish Prime Minister Fredrik Reinfeldt (U.S. and European experts, 2009). The challenge to this process will be to fruitfully engage the “gray” side of the factors driving toward antimicrobial resistance: unregulated or under-regulated environments of antimicrobial drug use, counterfeit and substandard drug marketing, and disparate and desperate use of treatment agents without clinical or diagnostic capabilities to direct care.
It is overly simplistic to believe that, as individuals or as collectives within society, we could “say no to drugs” that have contributed to both public health disease control efforts and beneficial patient-related healthcare services clinical outcomes. But the fact is that, as a therapeutic class, antimicrobials are becoming less effective, more expensive, and associated with increasingly important adverse clinical outcomes. Our ability to effectively treat many infectious disease syndromes and to prevent infections of both clinical and public health significance is being compromised by drug resistance. Antimicrobial stewardship is one component of delaying the slide into the post-antimicrobial era when medical treatment of infectious diseases may no longer be possible.
Infection Control Training, Certification, and Practice
This royal throne of kings, this scepter’d isle,
This earth of majesty, this seat of Mars,
This other Eden, demi-paradise,
This fortress built by Nature for herself
Against infection and the hand of war,
—John of Gaunt in King Richard II, ACT II, Scene 1, by William Shakespeare
On the international dimension, the ability to cloister ourselves from distant events has become increasingly difficult. Borders and physical barriers are porous to infectious diseases. Isolation and quarantine as infection control processes are often overcome by other considerations within globalization, including international trade and economics, civil security, environmental concerns, and mass transportation of goods, conveyances, and people (Gushalak et al., 2009; MacPherson and Gushalak, 2006).
Nonetheless, even before the acceptance of germs as the cause of transmissible disease and the advent of effective antimicrobial therapies, there was interest in the control of disease through personal and environmental hygienic practices. The emerging proponents of science in the early 19th century may have decried the somewhat moralistic tone to cleanliness in the prevention of pestilence, but subsequently the science of epidemiology did support the works of Semmelweis, Pasteur, and Lister in promoting hand washing and disinfection for the prevention of disease transmission. Countless thousands of lives, in particular the lives of postpartum women who were spared puerperal fever, were saved by institutionalizing the practice of hand washing between medical-surgical procedures and patient contact.
As antimicrobial resistance and nosocomial infections have reemerged as prevalent causes of institutional healthcare morbidity and mortality, the principles of infection prevention and control and patient safety have also risen in importance (International Federation of Infection Control, http://www.theific.org/default.asp; WHO, 2010b). While hand washing (WHO, 2008a, 2010c) has been heavily promoted recently to reduce the risk of both institutional infections, such as methicillin-resistant Staphylococcus aureus (MRSA) and Clostridium difficile toxin-associated diarrhea syndromes, and community-based diseases, including diarrhea, respiratory illnesses, and seasonal and pandemic influenza, the general principles of hygiene and social distancing remain core to infection prevention and control.
Ensuring quality of training, professional certification and regulation, and recognition of infection prevention and control as essential components of the global environment of transmissible infectious disease risk is an emerging consideration. The process of promoting and regulating infection control practice includes linking high-prevalence to low-prevalence regions, overcoming various barriers to implementation (such as social willingness and cultural beliefs), and balancing social and economic investments on return, language and communications, and behavioral change.
Laboratory Methodology, Proficiency Testing, and Quality Management
To promote the quality improvement of laboratories and related services for the public good and benefit of health professionals.
—Quality Management Program, Laboratory Services
The ability to peer into the swirling miasmas to detect the microscopic causes of transmissible human and animal disease freed humanity from ignorance, fear, and irrational responses to disease outbreaks. When ignorance, fear, and irrationality were the only responses available to outbreaks of the flux (cholera), the pox (smallpox), the Black Death (plague), and other diseases, inevitability and a fatalistic approach to public health and illness were all that could be expected.
The advent of the simple microscope and broth and agar culture techniques have been more recently supplemented by electron microscopy, immune diagnostic testing (serology and cell-mediated immunity), and direct antigen and genetic detection technologies that have vastly increased the sensitivity and specificity of the detection of infectious agents and the surveillance and diagnosis of disease.
There is a disproportionate distribution of laboratory technology and skilled technologists, technicians, and laboratory-trained medical professionals between higher-prevalence environments (economically disadvantaged regions of the world and rural environments) and lower-prevalence regions (economically advanced countries, major urban centers), with the possible exception of antimicrobial intensive-use settings, such as hospitals, in particular intensive care units, burn and trauma units, and academic health care settings. The latter represents significant local environments of selective pressures of drug use contributing to the nosocomial emergence of multidrug-resistant organisms (e.g., MRSA, vancomycin-resistant enterococci, and extended-spectrum β-lactamase-resistant microorganisms).
Whether laboratory-based or applied at the “bedside,” microbial detection and antimicrobial sensitivity and resistance testing technologies require rigorous performance and proficiency standards (National Foundation for Infectious Diseases, 2010). Unfortunately, these standards are not always attainable outside of the inspected and regulated laboratory environment (CDC, 1996).
International networks of collaborating laboratories have been created to address the concerns of shared technology and proficient performance. For example, the goal of the WHO laboratory network is to strengthen the performance of laboratories in support of immunization programs, detection, and reporting (WHO, 2010d). Similarly, the WHO has a coordinating role in laboratory biosafety and biosecurity (WHO, 2010e). In the words of their mandate,
[r]esponsible laboratory practices, including protection, control and accountability for valuable biological materials will help prevent their unauthorized access, loss, theft, misuse, diversion or intentional release, and contribute to preserving scientifically important work for future generations. The focus of WHO remains exclusively on the public health aspects of preparedness and response to global health events. However, a significant number of Member States are currently unequipped and unprepared to effectively address these complex issues. The vulnerabilities of these least-capable states reflect the vulnerabilities of the global community at large. No disease will remain eradicated or effectively contained unless and until Member States at every level of capacity participate in essential global health practices. The continuing appearance of highly virulent emerging and re-emerging infectious agents highlights the need for coordinated preparedness in support of global public health.
In distinction to rapid-onset and short-duration outbreaks of public health significance, like seasonal and pandemic influenza or severe acute respiratory
syndrome, slowly evolving health emergencies, such as widespread antimicrobial resistance affecting public health control and clinical management, also require robust and internationally connected laboratory practices. The nature of antimicrobial resistance will certainly be of longer duration and have potentially greater impact on morbidity and mortality worldwide. As a consequence, international leadership and coordination for laboratory safety, security, containment, detection, and reporting for the purposes of control are also essential.
Shared technology, sustainable capacity, and reference support structures will need to be components of international standards for laboratory performance in antimicrobial drug resistance testing, monitoring, and notification.
Active and Passive Surveillance Systems, Rapid Analysis, and Reporting
Hear now this, O foolish people, and without understanding; which have eyes, and see not; which have ears, and hear not.
—Jeremiah 5:21, Old Testament, King James version, The Bible
The first century of mass-produced, distributed, and dispensed antimicrobials is nearing an end. In this era of antimicrobials, countless deaths due to infectious disease have been averted or lives improved by limiting the consequences of infection. Yet, it has been known from the beginning that antimicrobial use begets antimicrobial resistance.
Pharmaceutical industry and academic science have countered emerging resistance with chemical modifications of existing drugs or increasingly rarely with new antimicrobial frameworks that lead to new classes of drugs. The microbial response to these innovations, virtually without exception and occurring rapidly, has been resistance.
The reproductive capacities and evolutionary biology of microbial populations continue to outstrip the abilities of infectious disease physicians. It has come down to our will and ingenuity versus their genetic plasticity.
In this context, it is essential that integrated and standardized surveillance systems exist that can detect, confirm, and create analytic reports on emerging risks and patterns of drug resistance. Regional to global networks, such as ProMed Mail (http://www.promedmail.org/pls/apex/f?p=2400:1000), Eurosurveillance (http://www.eurosurveillance.org/), GeoSentinel (http://www.istm.org/geosentinel/main.html), the Global Public Health Intelligence Network (http://www.phac-aspc.gc.ca/media/nr-rp/2004/2004_gphin-rmispbk-eng.php), and others, are proving to be useful in the notification role. Standardizing the approach to both active and passive components of surveillance, definitions, and detection technologies—as well as “intelligent systems” for shifting through the burden of events that enter a notification process for analysis—are needed areas of investment. As with all investments, there needs to be a bottom line. Knowing what is
happening in emerging drug resistance must translate into action for mitigation, prevention, and control for containment to be effective.
Public Health Process and Regulatory Tools Related to Health Outcomes
Insanity: doing the same thing over and over again and expecting different results.
—Albert Einstein
Doing the same thing over and over again and getting a good result in clinical and public health is desirable, but good outcomes are no longer predictable when the programs are largely dependent on the availability of effective antimicrobial therapy. This failure in predictability is largely due to emerging and spreading drug resistance.
The International Health Regulations (WHO, 2008b) and the International Sanitary and Phytosanitary Measures (World Trade Organization, 2010) are both well-known international tools “to protect human, animal or plant life or health.” Both of these tools try to achieve a balance in the benefits of mitigation versus the hazards of the interventions. To quote from the text of the Measures, while “[d]esiring to improve the human health, animal health and phytosanitary situation in all Members,” the Member State interventions are “subject to the requirement that these measures are not applied in a manner which would constitute a means of arbitrary or unjustifiable discrimination between Members where the same conditions prevail or a disguised restriction on international trade.”
Balancing health interests against trade and economics is a centuries-old challenge, particularly when the concern is “against” rather than “with.” The paradigm shift here, along the lines of Einstein’s quote, would be to seek a means to achieve both desired outcomes.
There are examples of standards in product production that are relevant to sustainability in both health and economics, such as the good manufacturing practices and quality systems for medical devices (FDA, 2010a) and pharmaceuticals (Health Canada, 2002). Implementing these concepts in practical and accountable programmatic means across diverse geographic and cultural environments is proving to be more challenging than expected (Beyer et al., 2010; Blossom et al., 2009).
Pharmaceutical Security Systems for Standard and Quality Medicines
Counterfeiting is greatest in regions where regulatory and enforcement systems for medicines are weakest.
—World Health Organization (2010f)
Counterfeit medicines are found everywhere in the world. They range from random mixtures of harmful toxic substances to inactive, ineffective preparations. Some contain a declared, active ingredient and look so similar to the genuine product that they can deceive health professionals as well as patients. But in every case, the source of a counterfeit medicine is unknown and its content unreliable. Counterfeit medicines are always illegal. They can result in treatment failure or even death. Eliminating them is a considerable public health challenge (WHO, 2010f).
Yet, not all medicines that are adulterated, altered, or substandard have been created with the intent to deceive and defraud. Errors in manufacturing, compounding, reformulation, labeling, storage, and transportation can also result in medicines that will not perform either on analysis or clinically as expected.
The importance of this issue relevant to antimicrobial drug effectiveness, patient safety, and emergence of resistance appeared in a United States Pharmacopeia drug quality report. Analyzing drugs obtained in U.S. Agency for International Development-associated countries indicated that antibiotics, antimalarials, antituberculous drugs, and antiretroviral agents for treatment of HIV/AIDS were found to be commonly substandard in content or completely counterfeit (Primo-Carpenter and McGinnis, 2006). Even in high-income nations, counterfeit (FDA, 2010b) or substandard drugs may enter the marketplace either directly from local illegal producers or through international portals, such as importation (Yankus, 2006) or Internet pharmacy access (FDA, 2006).
Although it is self-evident that substandard or counterfeit drugs would not produce clinical benefit, one of the consequences of sublethal antimicrobial drug dosing is microbial resistance (Kohanski et al., 2010). In 2006, the International Medical Products Anti-Counterfeiting Taskforce (IMPACT) was created. The aim of IMPACT is to protect people from buying and taking counterfeit medicines by preventing the manufacture and distribution of counterfeit medicines by focusing on five main areas: legislative and regulatory infrastructure, regulatory implementation, enforcement, technology, and communication. When human behavior for expedient and inexpensive access to medications is met through illegal providers whose goal is to increase their profits in environments of inadequate regulation and enforcement, substandard drugs will contribute to the emergence and spread of antimicrobial resistance.
Animal and Plant Health-Sector Engagement
Without the use of growth promoting antibiotics, the USA would require an additional 452 million chickens, 23 million more cattle and 12 million more pigs to reach the levels of production attained by the current practices.
—Animal Health Institute (1998)
Feeding the world is an increasing challenge. Global population growth and urbanization are two factors contributing to the demand for both total calories and protein to nourish the world’s people. To achieve adequate nutrition there is a shift from personal and local production to intensive, industrial agrifood techniques. The latter approaches are being employed with greater frequency, and food is increasingly becoming a globally traded and transported commodity.
There are estimates that, of all antimicrobial tonnage produced, 50 to 85 percent is used for animal and plant management for disease prevention, growth, or yield promotion, with a smaller amount used for actual infectious disease treatment. In some national agricultural jurisdictions, the use of antimicrobials as animal growth promoters, particularly those added at subtherapeutic levels in animal feed, are commonly used to specifically increase animal protein yields. Statements extolling the benefits of growth promoters are hotly debated. The counterclaims are that the increased yields are misrepresented, that the practice has both direct and indirect costs that exceed the claimed benefits, and that the practice of subtherapeutic antimicrobial use as growth promoters is contributing to emerging drug resistance (Delsol et al., 2003; Pasqualotto and Denning, 2007; Sharma et al., 2009).
The concern in human clinical and public health is that agrifood industrial practices contribute to the environmental burden of antimicrobial-resistant organisms that enter into human health and public health domains, and that the use of the same or similar drugs in animals renders them inaccessible for effective use in humans (Lipsitch and Samore, 2002; Roe and Pillai, 2003).
The WHO has also stated:
Veterinary prescription of antimicrobials also contributes to the problem of resistance. In North America and Europe, an estimated 50% in tonnage of all antimicrobial production is used in food-producing animals and poultry. The largest quantities are used as regular supplements for prophylaxis or growth promotion, thus exposing a large number of animals, irrespective of their health status, to frequently subtherapeutic concentrations of antimicrobials. Such widespread use of antimicrobials for disease control and growth promotion in animals has been paralleled by an increase in resistance in those bacteria (such as Salmonella and Campylobacter) that can spread from animals, often through food, to cause infections in humans (WHO, 2002).
While science and industry turn their resources to understanding the microbial factors in the development of drug resistance or the design and development of new drugs or classes of antimicrobial agents, the issue will remain that drug-resistant infections are innately a human problem.
Summary
The treatment and control of infectious diseases through the use of antimicrobials is a unique therapeutic challenge when the intervention itself is prone to obsolescence through drug resistance that rapidly globalizes. Motivation within the private commercial sector to invest in the high cost of research, development, and regulatory approval to bring to market a time-limited product has waned over the last several decades. Recently, newly available antimicrobial agents have been based on modifications of drugs in existing frameworks, and not all of those reaching the marketplace have proven effective or safe for human use (Liu, 2010).
Supporting new approaches to antimicrobial discovery and protecting new drug frameworks against obsolescence is a social imperative to protect, promote, and preserve human and animal health. In this context, the privatization of social investments may need to shift to an expectation of a social return on investment, where social forces—not market forces—determine the availability and accessibility to valuable antimicrobial resources.
In a similar vein, the seven factors outlined above must be global social commitments to protect and preserve the current and future availability of effective antimicrobial agents for clinical and public health purposes. Standalone approaches to any one of these factors will not be as effective as an integrated program that includes all seven factors that are implemented from the local to international levels.
References
Aagaard, E. M., R. Gonzales, C. A. Camargo, Jr., R. Auten, S. K. Levin, J. Maselli, and J. Metlay. 2010. Physician champions are key to improving antibiotic prescribing quality. Joint Commission Journal on Quality and Patient Safety 36:109–16.
Animal Health Institute. 1998. Antibiotic resistance back in the news. AHI Quarterly, 19:1–4.
Berrington, A. 2010. Antimicrobial prescribing in hospitals: Be careful what you measure. Journal of Antimicrobial Chemotherapy 65:163–8.
Beyer, T., M. Matz, D. Brinz, O. Rädler, B. Wolf, J. Norwig, K. Baumann, S. Alban, and U. Holzgrabe. 2010. Composition of OSCS-contaminated heparin occurring in 2008 in batches on the German market. European Journal of Pharmaceutical Sciences PMID:20399266 [Epub ahead of print].
Blossom, D., J. Noble-Wang, J. Su, S. Pur, R. Chemaly, A. Shams, B. Jensen, N. Pascoe, J. Gullion, E. Casey, M. Hayden, M. Arduino, D. S. Budnitz, I. Raad, G. Trenholme, and A. Srinivasan; Serratia in Prefilled Syringes Investigation Team Group. 2009. Multistate outbreak of Serratia marcescens bloodstream infections caused by contamination of prefilled heparin and isotonic sodium chloride solution syringes. Archives of Internal Medicine 169:1705–11.
Böhm, A., M. Follari, A. Hewett, S. Jones, N. Kemp, D. Meares, D. Pearce, and K. Van Cauteret. 2004. Vision 2020: Forecasting international student mobility, a UK perspective. http://www.britishcouncil.org/eumd_-_vision_2020.pdf (accessed April 25, 2010).
Burkhardt, O., S. Ewig, U. Haagen, S. Giersdorf, O. Hartmann, K. Wegscheider, E. Hummers-Pradier, and T. Welte. 2010. A simple procalcitonin-guided strategy results in safe reductions of antibiotic use in patients with symptoms of acute respiratory tract infections in primary care. European Respiratory Journal [Epub ahead of print].
Cals, J. W., M. J. Schot, S. A. de Jong, G. J. Dinant, and R. M. Hopstaken. 2010. Point-of-care C-reactive protein testing and antibiotic prescribing for respiratory tract infections: A randomized controlled trial. Annals of Family Medicine 8:124–33.
CDC (Centers for Disease Control and Prevention). 1996. Clinical laboratory performance on proficiency testing samples—United States, 1994. Morbidity and Mortality Weekly Report 45:193–6.
_____. 2001. Campaign to prevent antimicrobial resistance in the healthcare settings. http://www.cdc.gov/drugresistance/healthcare/problem.htm (accessed May 2, 2010).
Christian-Kopp, S., M. Sinha, D. I. Rosenberg, A. W. Eisenhart, and F. W. McDonald. 2010. Antibiotic dosing for acute otitis media in children: A weighty issue. Pediatric Emergency Care 26:19–25.
Coco, A., L. Vernacchio, M. Horst, and A. Anderson. 2010. Management of acute otitis media after publication of the 2004 AAP and AAFP clinical practice guideline. Pediatrics 125:214–20 [Epub January 25, 2010].
Davies, J. 2007. Microbes have the last word. A drastic re-evaluation of antimicrobial treatment is needed to overcome the threat of antibiotic-resistant bacteria. EMBO Reports 8(7):616–21.
Delsol, A. A., M. Anjum, M. J. Woodward, J. Sunderland, and J. M. Roe. 2003. The effect of chlortetracycline treatment and its subsequent withdrawal on multi-resistant Salmonella enterica serovar Typhimurium DT104 and commensal Escherichia coli in the pig. Journal of Applied Microbiology 95:1226–34.
Di Pentima, M. C., and S. Chan. 2010. Impact of antimicrobial stewardhip program on vancomy-cin use in a pediatric teaching hospital. Pediatric Infectious Disease Journal [Epub ahead of print].
Esmaily, H. M., I. Silver, S. Shiva, A. Gargani, N. Maleki-Dizaji, A. Al-Maniri, and R. Wahlstrom. 2010. Can rational prescribing be improved by an outcome-based educational approach? A randomized trial completed in Iran. Journal of Continuing Education in the Health Professions 30:11–8.
FDA (U.S. Food and Drug Administration). 2006. FDA warns consumers not to buy or use prescription drugs from various Canadian websites that apparently sell counterfeit products. Press release. P06-123. Released August 30, 2006. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/2006/ucm108722.htm (accessed March 5, 2009).
_____. 2010a. Center for Devices and Radiological Health. Good manufacturing practices (GMP)/quality systems (QS) regulation. http://www.fda.gov/cdrh/devadvice/32.html (accessed March 5, 2009).
_____. 2010b. Counterfeit drugs. http://www.fda.gov/oc/initiatives/counterfeit/ (accessed March 5, 2009).
Fleming, A. 1945. Penicillin. Nobel Lecture, December 11, 1945. http://nobelprize.org/nobel_prizes/medicine/laureates/1945/fleming-lecture.pdf (accessed May 29, 2010).
Frich, J. C., S. Høye, M. Lindbaek, and J. Straand. 2010. General practitioners and tutors’ experiences with peer group academic detailing: a qualitative study. BMC Family Practice 11:12.
Gushulak, B. D., J. Weekers, and D. W. MacPherson. 2009. Migrants in a globalized world—health threats, risks and challenges: An evidence based framework. Emerging Health Threats Journal 2:e10.
Health Canada. 2002. Health Products and Food Branch Inspectorate. Good manufacturing practice guidelines. 2002 Edition. Version 2. http://www.hc-sc.gc.ca/dhp-mps/alt_formats/hpfb-dgpsa/pdf/compli-conform/2002v2_e.pdf (accessed March 5, 2009).
Heritage, J., M. N. Elliott, T. Stivers, A. Richardson, and R. Mangione-Smith. 2010. Reducing inappropriate antibiotics prescribing: The role of online commentary on physical examination findings. Patient Education and Counseling PMID:20223616 [Epub ahead of print].
Hulscher, M. E., R. P. Grol, and J. W. van der Meer. 2010. Antibiotic prescribing in hospitals: A social and behavioural scientific approach. Lancet Infectious Diseases 10:167–75.
Huttner, B., H. Goossens, T. Verheij, S. Harbarth, and the CHAMP consortium. 2010. Characteristics and outcomes of public campaigns aimed at improving the use of antibiotics in outpatients in high-income countries. Lancet Infectious Diseases 10:17–31.
IATA (International Air Transport Association). 2010. Fact sheet: IATA—International Air Transport Association. http://www.iata.org/pressroom/facts_figures/fact_sheets/iata.htm (accessed April 25, 2010).
ILO (International Labour Office). 2004. Towards a fair deal for migrant workers in the global economy. Report VI. International Labour Conference, 92nd Session. Geneva, Switzerland. http://www.ilo.org/wcmsp5/groups/public/dgreports/dcomm/documents/meetingdocument/kd00096.pdf (accessed April 25, 2010).
International Labour Organization. 2009. Facing the global jobs crisis: Migrant workers, a population at risk. http://www.ilo.org/global/About_the_ILO/Media_and_public_information/Feature_stories/lang-en/WCMS_112537/index.htm (accessed May 30, 2010).
IOM (Institute of Medicine). 2001. Emerging infectious diseases from the global to the local perspective: Workshop summary. Forum on Microbial Threats. Washington, DC: National Academy Press. http://www.iom.edu/Reports/2003/Emerging-Infectious-Diseases-from-the-Global-to-the-Local-Perspective-Workshop-Summary.aspx (accessed May 2, 2010).
_____. 2008. Global climate change and extreme weather events: Understanding the contributions to infectious disease emergence. Workshop summary. Washington, DC: The National Academies Press. http://www.nap.edu/catalog.php?record_id=12435#toc (accessed April 25, 2010).
Kennedy, K. M., L. G. Glynn, and B. Dineen. 2010. A survey of the management of urinary tract infection in children in primary care and comparison with the NICE guidelines. BMC Family Practice 11:6.
Kohanski, M. A., M. A. DePristo, and J. J. Collins. 2010. Sublethal antibiotic treatment leads to multidrug resistance via radical-induced mutagenesis. Molecular Cell 37:311–20.
Kumarasamy, K. K., M. A. Toleman, T. R. Walsh, J. Bagaria, F. Butt, R. Balakrishnan, U. Chaudhary, M. Doumith, C. G. Giske, S. Irfan, P. Krishnan, A. V. Kumar, S. Maharjan, S. Mushtaq, T. Noorie, D. L. Paterson, A. Pearson, C. Perry, R. Pike, B. Rao, U. Ray, J. B. Sarma, M. Sharma, E. Sheridan, M. A. Thirunarayan, J. Turton, S. Upadhyay, M. Warner, W. Welfare, D. M. Livermore, and N. Woodford. 2010. Emergence of a new antibiotic resistance mechanism in India, Pakistan, and the UK: a molecular, biological, and epidemiological study. Lancet Infectious Diseases. Early Online Publication, 11 August 2010.
Lipsitch, M., and M. H. Samore. 2002. Antimicrobial use and antimicrobial resistance: A population perspective. Emerging Infectious Diseases 8:347–54.
Liu, H. H. 2010. Safety profile of the fluoroquinolones: Focus on levofloxacin. Drug Safety 33:353–69.
MacPherson, D. W., and B. D. Gushulak. 2006. The basic principles of migration health. Population mobility across prevalence gaps and disparity. Emerging Themes in Epidemiology 3:3.
MacPherson, D. W., B. D. Gushulak, W. B. Baine, S. Bala, P. O. Gubbins, P. Holtom, and M. Segarra-Newnham. 2009. Population mobility, globalization, and antimicrobial drug resistance. Emerging Infectious Diseases 15(11). http://www.cdc.gov/EID/content/15/11/1727.htm (accessed April 25, 2010).
National Foundation for Infectious Diseases. 2010. 2010 Annual Conference on Antimicrobial Resistance. http://www.nfid.org/conferences/resistance10/ (accessed May 24, 2010).
National Intelligence Council. 2000. The global infectious disease threat and its implications for the United States. http://www.dni.gov/nic/special_globalinfectious.html# (accessed May 16, 2010).
Neumark, T., L. Brudin, and S. Mölstad. 2010. Use of rapid diagnostic tests and choice of antibiotics in respiratory tract infections in primary healthcare—a 6-y follow-up study. Scandinavian Journal of Infectious Diseases 42:90–6.
Pasqualotto, A. C., and D. W. Denning. 2007. Generic substitution of itraconazole resulting in sub-therapeutic levels and resistance. International Journal of Antimicrobial Agents 30:93–4.
Primo-Carpenter, J., and M. McGinnis. 2006. Matrix of drug quality reports in USAID-assisted countries: The United States Pharmacopeial Convention Inc. http://www.uspdqi.org/pubs/other/GHC-DrugQualityMatrix.pdf (accessed March 5, 2009).
Roe, M. T., and S. D. Pillai. 2003. Monitoring and identifying antibiotic resistance mechanisms in bacteria. Poultry Science 82:622–6.
Schuetz, P., M. Batschwaroff, F. Dusemund, W. Albrich, U. Bürgi, M. Maurer, M. Brutsche, A. R. Huber, and B. Müller. 2010. Effectiveness of a procalcitonin algorithm to guide antibiotic therapy in respiratory tract infections outside of study conditions: A post-study survey. European Journal of Clinical Microbiology and Infectious Diseases 29:269–77.
Sharma, R., F. J. Larney, J. Chen, L. J. Yanke, M. Morrison, E. Topp, T. A. McAllister, and Z. Yu. 2009. Selected antimicrobial resistance during composting of manure from cattle administered sub-therapeutic antimicrobials. Journal of Environmental Quality 38:567–75.
Tenke, P., B. Kovacs, T. E. Bjerklund Johansen, T. Matsumoto, P. A. Tambyah, and K. G. Naber. 2008. European and Asian guidelines on management and prevention of catheter-associated urinary tract infections. International Journal of Antimicrobial Agents 31(Suppl. 1):S68–78 [Epub November 14, 2007].
UNHCR (Office of the United Nations High Commissioner for Refugees). 2008. 2007 global trends: Refugees, asylum-seekers, returnees, internally displaced and stateless persons. http://www.unhcr.org/statistics/STATISTICS/4852366f2.pdf (accessed April 25, 2010).
_____. 2010. Refugee figures. http://www.unhcr.org/pages/49c3646c1d.html (accessed May 30, 2010).
United Nations. 1999. International year of older persons: Demographics of older persons. http://www.un.org/NewLinks/older/99/older.htm (accessed April 25, 2010).
United Nations Department of Economic and Social Affairs. 2006. World urbanization prospects: The 2005 revision. New York: Population Division, Department of Economic and Social Affairs, United Nations.
United Nations Department of Economic and Social Affairs, Population Division. 2009. International migration 2009. New York: United Nations. http://www.un.org/esa/population/publications/2009Migration_Chart/ittmig_wallchart09.pdf (accessed May 30, 2010).
United Nations Population Division. 1999. World population prospects: The 1998 revision. New York: United Nations.
United Nations Population Fund. 2007. State of world population 2007. Unleashing the potential of urban growth. New York: United Nations Population Fund.
UNWTO (United Nations World Tourism Oranization). 2009. International tourism challenged by deteriorating global economy. UNWTA World Tourism Barometer 7(1):1. http://unwto.org/facts/eng/pdf/barometer/UNWTO_Barom09_1_en_excerpt.pdf (accessed April 25, 2010).
U.S. and European experts applaud new transatlantic task force on antibiotic resistance threat. 2009. e! science news. http://esciencenews.com/articles/2009/11/06/us.and.european.experts.applaud.new.transatlantic.task.force.antibiotic.resistance.threat (accessed May 16, 2010).
U.S. Congress, House of Representatives, Committee on Energy and Commerce. Subcommittee Hearing on Antibiotic Resistance. April 28, 2010.
U.S. Department of State. 2008. Trafficking in persons. Washington, DC: U.S. Department of State. http://www.state.gov/g/tip/rls/tiprpt/2008/ (accessed April 25, 2010).
Visscher, K. L., C. M. Hutnik, and M. Thomas. 2009. Evidence-based treatment of acute infective conjunctivitis: Breaking the cycle of antibiotic prescribing. Canadian Family Physician 55:1071–5.
WHO (World Health Organization). 2002. Antimicrobial resistance. http://www.who.int/mediacentre/factsheets/fs194/en/ (accessed April 25, 2010).
_____. 2008a. Global hand washing day. http://www.who.int/gpsc/events/2008/15_10_08/en/ (accessed May 16, 2010).
_____. 2008b. International health regulations (2005), 2nd edition. Geneva, Switzerland: WHO. http://whqlibdoc.who.int/publications/2008/9789241580410_eng.pdf (accessed April 25, 2010).
_____. 2010a. Health of migrants: The way forward—Report of a global consultation. http://www.who.int/hac/events/consultation_report_health_migrants_colour_web.pdf (accessed May 29, 2010).
_____. 2010b. Infection control. http://www.who.int/topics/infection_control/en/ (accessed May 16, 2010).
_____. 2010c. Hand washing techniques. http://www.who.int/surgery/publications/HandWashing-Techniques.pdf (accessed May 16, 2010).
_____. 2010d. Laboratory network. http://www.who.int/immunization_monitoring/laboratory/en/ (accessed May 24, 2010).
_____. 2010e. Biosafety and laboratory biosecurity. http://www.who.int/ihr/biosafety/en/ (accessed May 25, 2010).
_____. 2010f. Medicines: Counterfeit medicines. http://www.who.int/mediacentre/factsheets/fs275/en/ (accessed May 26, 2010).
World Trade Organization. 2010. Sanitary and phytosanitary measures. http://www.wto.org/english/tratop_e/sps_e/spsagr_e.htm (accessed May 26, 2010).
Yankus, W. 2006. Counterfeit drugs: Coming to a pharmacy near you. New York: American Council on Science and Health. http://www.acsh.org/publications/pubID.1384/pub_detail.asp (accessed March 5, 2009).
A13
POPULATION MOBILITY, GLOBALIZATION, AND ANTIMICROBIAL DRUG RESISTANCE56
Douglas W. MacPherson, Brian D. Gushulak, William B. Baine, Shukal Bala, Paul O. Gubbins, Paul Holtom, and Marisel Segarra-Newnham57
Population mobility is a main factor in globalization of public health threats and risks, specifically distribution of antimicrobial drug–resistant organisms. Drug resistance is a major risk in healthcare settings and is emerging as a problem in community-acquired infections. Traditional health policy approaches have focused on diseases of global public health significance such as tuberculosis, yellow fever, and cholera; however, new diseases and resistant organisms challenge existing approaches. Clinical implications and health policy challenges associated with movement of persons across barriers permeable to products, pathogens, and toxins (e.g., geopolitical
borders, patient care environments) are complex. Outcomes are complicated by high numbers of persons who move across disparate and diverse settings of disease threat and risk. Existing policies and processes lack design and capacity to prevent or mitigate adverse health outcomes. We propose an approach to global public health risk management that integrates population factors with effective and timely application of policies and processes.
Human mobility is causing an increase in antimicrobial drug–resistant organisms and drug-resistant infectious diseases. International population movement is an integral component of the globalization process. Current population movement dynamics rapidly and effectively link regions of marked health disparity, and these linkages can be associated with risk for importation of drug-resistant infectious diseases.
During the past century, developments in public health sanitation (Centers for Disease Control and Prevention, 2004), infrastructure engineering (Thompson et al., 2003), vaccines (World Health Organization, 2009), and antimicrobial drugs have contributed substantially to the control of infectious diseases, markedly decreasing associated illness and death. These developments have largely occurred in economically advanced regions and have produced complacency and a belief that the public health threats posed by infectious diseases have been conquered. However, by the early 1990s, infectious diseases were again being identified as substantial domestic and international public health threats in and to western nations (Institute of Medicine, 1992).
Although many infections of clinical relevance are effectively managed with the use of vaccines, antimicrobial drugs, or newer therapies, challenges to the control of infectious diseases remain. These challenges occur in industrialized and in developing countries and result at least in part from the failure of antimicrobial drugs to meet expectations for management and control of disease in clinical and public health contexts. Declining antimicrobial drug effectiveness has current and future consequences that affect all elements of the health sector, e.g., research and development, public health policy, service delivery, and payment programs. The emergence of antimicrobial drug resistance adversely affects patient care and threatens effective management of public health infectious diseases globally (World Health Organization, 2007).
Antimicrobial drug failure may occur for many reasons, e.g., reduced adherence to drug therapy, suboptimal dosing, diagnostic and laboratory error, ineffective infection control, counterfeit or altered drugs, and resistance (innate or acquired). Although much attention is focused on resistance patterns of eubacteria (Tenover, 2006), resistance is being found for virtually all microbial agents including mycobacteria (Andini and Nash, 2006; Gagneux et al., 2006), viruses (Kuritzkes, 2006; Monto et al., 2006), parasites (Schunk et al., 2006; Xiao et al., 2006), and fungi (Katiyar et al., 2006; Mentel et al., 2006). Antimicrobial drug resistance phenotype is commonly described in terms of the resistance character-
istics of the microorganism. These characteristics are either constitutionally based intrinsic characteristics of the organism or resistance factors acquired through induced genetic expression or gene transfer between organisms.
Human activities strongly affect acquired resistance. Emergence of drug resistance in environments that enable sharing of drug-resistance genes between organisms has been documented. Human activities that contribute to ecological niche pressures, such as antimicrobial drug use (MacDougall et al., 2005) and manufacturing or biological waste disposal into the environment (Chee-Sanford et al., 2009; Nagulapally et al., 2009), can support the development of resistance.
Against this background of diverse antimicrobial drug resistance, interregional migration and the processes associated with international population mobility can affect the spread and distribution of resistant organisms. These mechanisms of spread become increasingly common when people move among locations with disparate delivery of health services, public health systems, and regulatory frameworks for therapeutic drugs, particularly antimicrobial agents. We describe the role of population mobility in the dispersal of drug-resistant organisms and the emerging need for global standards, programs, and policies in the management of drug resistance, especially for mobile populations.
Population Mobility and Association with Infectious Diseases and Microbial Resistance
Each year, ≈2 billion persons move across large geographic distances; approximately half cross international boundaries (Table A13-1). The International Air Transport Association reported that their members carried 1.6 billion passengers in 2007, among which 699 million flew internationally (International Air Transportation Association, 2009). The United Nations World Tourism Organization estimated 924 million international tourist arrivals in 2008 (United Nations World Tourism Organization, 2009). International movements for permanent resettlement by immigrants, refugees, asylum seekers, or refugee claimants, and temporary movement by migrant workers and others augment the total international movements each year. The International Labour Organization stated that in 2004, an estimated 175 million persons (3% of the world’s population) lived permanently outside their country of birth and that there were 81 million migrant workers (excluding refugees) globally (International Labour Organization, 2004).
Despite the magnitude of mobile populations, translating international movement statistics into imported disease risk is challenging for several reasons. Domestic surveillance systems generally report disease events and only occasionally refer to infection in the context of place of acquisition. Patients’ travel or migration history may not be routinely gathered as part of the reporting requirements. Nevertheless, considerable information supports the belief that international population mobility plays a role in introducing antimicrobial drug–resistant disease, as follows.
TABLE A13-1 Global Estimates of Annual Migrant Populations
|
Administrative category |
Population estimates and year |
Reference |
|
Refugees |
16 million in 2007 |
(United Nations High Commission for Refugees, 2008) |
|
Asylum seekers or refugee claimants |
650,000 in 2007 |
(United Nations High Commission for Refugees, 2008) |
|
Internally displaced persons |
51 million in 2007, includes those displaced by natural disasters and conflict |
(United Nations High Commission for Refugees, 2008) |
|
Temporary (recreational or business travel) movement |
924 million in 2008 |
(United Nations World Tourism Organization, 2009) |
|
Regular immigrants |
Annual flow of 2.4 million, reported in 2005 (from a stock of 200 million immigrants worldwide) |
(United Nations, 2006) |
|
International students |
2.1 million in 2003 |
(Böhm et al., 2004) |
|
Migrant workers |
81–86 million in 2005 |
(International Labour Organization, 2004) |
|
Trafficked (across international borders) persons |
Estimated 800,000 in 2006 |
(US Department of State, 2008) |
|
Domestic arrivals, by air |
Estimated 900 million in 2007 |
(International Air Transportation Association, 2009) |
Human Travel to Disease-Nonendemic or Low Disease-Endemicity Regions
Mobile population importation of drug-resistant infections and diseases is most evident where the expected frequency of the infection or disease is low or absent. For diseases in nonendemic areas, it can be fairly assumed that humans imported the disease. Many examples of imported multidrug-resistant (MDR) infectious diseases are associated with migrant populations, e.g., MDR Plasmodium falciparum malaria in immigrants, tourists, and returned foreign-born travelers (Chan et al., 2006; Klein and Bosman, 2005; Skarbinski et al., 2006). Tuberculosis in regions of low disease endemicity, such as western Europe and North America, is also related to the influx of persons from tuberculosis-hyperendemic areas (MacPherson and Gushalak, 2006). Tuberculosis in foreign-born persons can shift the local disease epidemiology from endemic to imported
and includes the risk for MDR TB (Centers for Disease Control and Prevention, 2006; Public Health Agency of Canada, 2009; Falzon and Desenclos, 2006, World Health Organization, 2009) and extensively drug-resistant (XDR) TB (World Health Organization, 2006; Committee on Homeland Security, 2007).
Geographic Tracking of Human-to-Human Transmitted Diseases and Drug Resistance over Time
The emergence of high-level resistance to penicillin G by Streptococcus pneumoniae, first described in South Africa in 1977, followed by resistance to multiple drugs is an example of international tracking of human-to-human disease and this organism over almost 4 decades. Modern molecular microbiologic techniques are now being used to confirm its global spread (Reinert et al., 2005).
Similar studies have been conducted on the international spread of drug-resistant gonorrhea (Sutrisna et al., 2006; Centers for Disease Control and Prevention, 2004). Neisseria gonorrhoeae resistant to penicillin, tetracycline, and multiple other drugs, detected in Southeast Asia during the 1960s and 1970s, has been an emerging public health issue in the United States (Maurer and Sneider, 1969; Researchers report HIV and STD statistics from Vietnam, 2000). The reported emergence of quinolone-resistant gonorrhea in the United States (Centers for Disease Control and Prevention, 1994) followed a similar pattern of reactive public health response to the contribution of human mobility to international and then intranational spread. Successive treatment guidelines emphasize the importance of population mobility and the dispersal of resistant organisms in this illness (reference 41 in online Technical Appendix, available from www.cdc.gov/EID/content/15/11/1727-Techapp.pdf). The convergence of a resistant threat with decreased access to effective alternative therapy (cefixime shortage) during 2002–2003 complicated management and control (reference 42 in online Technical Appendix). Increasingly, development of clinical management guidelines for diagnosing and treating illness caused by many resistant organisms will refer to international differences in drug-resistance patterns (reference 43 in online Technical Appendix).
Since multidrug–or methicillin–resistant Staphylococcus aureus (MRSA) was first reported in the United States in 1968, its prevalence in North American healthcare institutions has grown, contributing to increased (number and duration) hospital stays and an associated increased number and severity of cases and more deaths (references 44,45 in online Technical Appendix). Recent descriptions of primary community-associated MRSA infections causing death have raised concerns about the control and management of this organism in not only North America but other locales worldwide as well (references 46,47 in online Technical Appendix). Clinical and laboratory testing can link distant disease exposures to local isolation of resistant strains (references 48–50 in online Technical Appendix). A worrying development of antimicrobial drug resistance in S.
aureus has been the emergence and geographic extension of reduced susceptibility to vancomycin, which at one time was the reliable backup therapy for MRSA infections (references 51–53 in online Technical Appendix). Although MRSA is not uniquely a human pathogen, the nature of its clinical distribution and ability to be carried in asymptomatic persons supports its association with human-to-human transmission over large distances.
Humans as Asymptomatic Carriers or Mobile Vectors of Antimicrobial Drug-Resistant Organisms
As with MRSA, humans can asymptomatically carry and transmit other cutaneous, enteric, or respiratory microbial flora from zones of high to low prevalence. Some of these organisms may have innate drug resistance or may reflect acquired resistance patterns that are not typical of locally acquired disease. Typhoid disease, Shigella, and Campylobacter infections are a few of many other enteric infections for which humans are documented carriers (references 54–56 in online Technical Appendix).
Recently, the potential for drug-resistant influenza viruses with emergent and pandemic potential has captured considerable global health attention (references 57–59 in online Technical Appendix). The local appearance of novel influenza strains with rapid global distribution raises questions about the role of human mobility in the spread and distribution of drug-resistant viruses (reference 60 in online Technical Appendix). Although local antiviral drug pressure is associated with rapid appearance of resistance, drug-resistant strains of influenza have also been associated with importation (reference 61 in online Technical Appendix).
The role of international tourists, travelers, or migrants colonized with antimicrobial drug–resistant organisms, in terms of transmission potential when they arrive in areas of a low disease prevalence, is difficult to detect and largely unexplored (reference 62 in online Technical Appendix). The reality of this risk is illustrated when persons obtain healthcare services outside their normal place of residence. Wounded military personnel and a group often referred to as medical tourists are at increased risk of acquiring nosocomial infections caused by drug-resistant organisms and of subsequently importing their infections when they repatriate to their country of residency.
Additionally, the role of international facilities that provide dental, surgical, medical, diagnostic, and therapeutic services to international travelers is expanding (reference 63 in online Technical Appendix). Health services in other countries may be provided in regulatory and standardization environments that differ from those at the patients’ place of origin. The estimated risk for hospital-acquired infections in developing countries is 2–20× greater than that in industrialized countries (reference 64 in online Technical Appendix). Antimicrobial drug–resistance patterns may also differ, as may health services, infection control practices, and public health requirements for surveillance and reporting of anti-
microbial drug resistance. The extension and transfer of nosocomial infections between regions and within the community has been well documented at the national level (references 65–67 in online Technical Appendix). As more high-risk and vulnerable populations travel internationally, either requiring or planning medical or surgical care abroad, or as migrants enter countries seeking healthcare services not available in their own countries, the international consequences of imported drug-resistant infections will be seen more frequently.
In some scenarios, linking the emergence of antimicrobial drug resistance and international mobility can be challenging. Given the global prevalence of many common organisms, their role in causing infections in high-risk populations (e.g., the elderly and patients with concurrent conditions such as diabetes, renal failure, malignancy, or immune compromise or patients who have had abdominal surgery) or certain institutional environments (e.g., intensive care units, burn units, long-term care facilities) may create similar local pressures potentially leading to multifocal emergence of drug resistance. Regardless of whether simultaneous multifocal emergence of resistance is a factor, unaffected areas will be linked to affected areas through mobilization of persons from zones of high to low prevalence. Microbial identification and typing systems, antibiograms, and new technologies for identifying genetic clones and “fingerprints” of microbes are better at defining the origin and patterns of spread of MDR organisms.
Local monitoring of susceptibility patterns combined with knowledge of emerging drug resistance, regionally or internationally, is already recognized as a component of some resistant infections such as MDR TB and XDR TB. Growing population mobility makes local monitoring an increasingly important component of routine surveillance for antimicrobial resistance.
Roles of International Policies, Processes, and Globalization in the Control of Imported Antimicrobial Drug-Resistant Diseases
Since development of the first international maritime sanitation regulations in 1832, coordinated international responses have been required to manage common threats. Such undertakings have always had to balance the benefits of mitigation with the negative effects of disease control interventions on international trade and commerce (reference 68 in online Technical Appendix). The modern version of these regulations, the International Health Regulations, focuses on a limited number of diseases and outbreaks of international public health significance for surveillance and reporting but only peripherally addresses population mobility and drug-resistance patterns (reference 69 in online Technical Appendix).
The association of international movements of conveyances, goods, and people with introductions of disease and vectors has been long recognized (references 70–71 in online Technical Appendix). Human travel, trade, and commerce have frequently been implicated in the redistribution of diseases. Examples include yellow fever in the 18th and 19th centuries, anopheline mosquito malaria vectors
in the 1930s, and, more recently, Aedes albopictus and dengue, the extension of West Nile Virus infection into North America, and the spread of chikungunya infections in Europe (references 72–76 in online Technical Appendix).
No specific antimicrobial therapies are available for yellow fever, dengue, West Nile, and chikungunya viruses, among others. Expanding human population mobility will affect and influence the spread, introduction, and endemicity of resistant and untreatable microbes because infections are unequally and rather unpredictably distributed around the world.
Proposed Approach to Global Public Health Risk Management
As recently demonstrated by influenza A pandemic (H1N1) 2009 virus, the volume, rapidity, and complexity of international movements exceed current international disease control practices (reference 77 in online Technical Appendix). Effective responses require engagement of local capacities, standardization of practices, multisectorial partnerships, and rigorous health intelligence with threat and risk assessment. The spread and introduction of resistant infections may not be preventable; but planning, recognition, and coordinated response can mitigate the consequences. Specifically, to control antimicrobial drug resistance and international movement of disease risk associated with human mobility, greater international collaboration and standardization are needed in the following areas:
-
Prescriber education, training, and invigilation in terms of antimicrobial drug stewardship for good patient care and reduction of risk for emerging drug resistance.
-
Infection control training, certification, and practice.
-
Laboratory methods, proficiency testing, and quality management.
-
Active and passive surveillance systems, including routine gathering of travel and migration history, rapid analysis, and reporting.
-
Engagement of process and regulatory tools unrelated to public health but related to health outcomes, e.g., good manufacturing practices and quality systems for medical devices and pharmaceuticals (references 78,79 in online Technical Appendix).
-
Pharmaceutical security systems for standard and quality medicines. (The importance of this issue relevant to drug effectiveness, patient safety, and emergence of resistance appeared in a United States Pharmacopeia drug quality report from countries associated with the US Agency for International Development; the report indicated that antibiotic drugs, antimalarial drugs, antituberculous drugs, and antiretroviral agents for treatment of HIV/AIDS were found to be commonly substandard or counterfeit [reference 80 in online Technical Appendix]. Even in industrialized countries, counterfeit drugs may enter the marketplace either directly from local illegal producers
-
or through international portals such as importation or Internet pharmacy access [references 81–83 in online Technical Appendix.])
-
Animal and plant health sector engagement. (Not only do subtherapeutic, subquality antiinfective therapies and low-level environmental antimicrobial drugs affect illness and death at the human level, but they also have the potential for emergence of drug resistance at the microbial level [references 84–88 in online Technical Appendix.])
Although all the above-listed efforts are essential, none will be sufficient without integrating the role played by humans and their international movement into modeling the complex relationship with antimicrobial drug resistance and microorganisms (reference 89 in online Technical Appendix). Enhanced global surveillance and population mapping demarcating differential zones of disease prevalence and major health disparities will support targeted interventions such as routine drug sensitivity analyses for infections originating in certain situations.
Acknowledging the dynamic role of population mobility in emerging risks to public health is a first step in formulating an effective response, but other components will be needed if this risk is to be successfully mitigated (reference 90 in online Technical Appendix). Components of this response will include the following:
-
Accurate and robust assessment of threat to risk management based on modern population characteristics that include mobility, travel, and migration history.
-
Mitigation of risk through nonhealth partnerships in other sectors, including economics and trade, education, agriculture, and security, all of which will affect the determinants of health, regional disease outcomes, and critical decision making for effective intervention and control.
-
Augmenting local knowledge and timely communications related to populations expressing emerging disease threats and risks and linking early detection through diagnostic and confirmatory epidemiologic tools and medical technology.
Conclusions
Although the association of human movement with antimicrobial drug resistance is not new, the extent of risk to public health caused by population mobility and drug-resistant infections is increasing. A shift in the existing paradigm of pathogen-focused policies and programs would contribute to a healthier future for everyone. The shift should address population mobility as a part of an integrated approach to decrease globalization of infectious disease threats and risks.
Dr MacPherson is a clinician, laboratorian, researcher, and advisor to multiple governments and agencies on population health issues. His primary interest is advocating for “people first” in all aspects of medicine.
References
Andini N, Nash KA. Intrinsic macrolide resistance in Mycobacterium tuberculosis complex is inducible. Antimicrob Agents Chemother. 2006;50:2560–2. DOI: 10.1128/AAC.00264-06
Böhm A, Folari M, Hewett A, Jones S, Kemp N, Meares D, et al. Vision 2020—forecasting international student mobility, a UK perspective, 2004 [cited 2009 Jun 8]. Available from http://www.britishcouncil.org/eumd_-_vision_2020.pdf
Centers for Disease Control and Prevention. 150th anniversary of John Snow and the pump handle. MMWR Morb Mortal Wkly Rep. 2004;53:783.
____. Trends in tuberculosis—United States, 2005. MMWR Morb Mortal Wkly Rep. 2006;55:305–8 [cited 2009 Jun 8]. Available from http://www.cdc.gov/mmwr/preview/mmwrhtml/mm5511a3.htm
____. Decreased susceptibility of Neisseria gonorrhoeae to fluoroquinolones—Ohio and Hawaii, 1992–1994. MMWR Morb Mortal Wkly Rep. 1994;43:325–7.
____. Increases in fluoroquinolone- resistant Neisseria gonorrhoeae among men who have sex with men—United States, 2003, and revised recommendations for gonorrhea treatment, 2004. MMWR Morb Mortal Wkly Rep. 2004;53:335–8.
Chan CW, Lynch E, Spathis R, Hombhanje FW, Kaneko A, Garruto RM, et al. Flashback to the 1960s: utility of archived sera to explore the origin and evolution of Plasmodium falciparum chloroquine resistance in the Pacific. Acta Trop. 2006;99:15–22. DOI: 10.1016/j.actatropica.2006.05.011
Chee-Sanford JC, Mackie RI, Koike S, Krapac IG, Lin YF, Yannarell AC, et al. Fate and transport of antibiotic residues and antibiotic resistance genes following land application of manure waste. J Environ Qual. 2009;38:1086–108. DOI: 10.2134/jeq2008.0128
Committee on Homeland Security. The 2007 XDR TB incident—a breakdown at the intersection of Homeland Security and Public Health. September 2007 [cited 2009 Jun 8]. Available from http://homeland.house.gov/SiteDocuments/tbreport.pdf
Falzon D, Desenclos JC. World TB Day: European countries report over 400,000 tuberculosis cases in 2004. Euro Surveill. 2006;11 [cited 2009 Jun 8]. Available from http://www.eurosurveillance.org/ViewArticle.aspx?ArticleId=2928
Gagneux S, Burgos MV, DeRiemer K, Encisco A, Munoz S, Hopewell PC, et al. Impact of bacterial genetics on the transmission of isonizid-resistant Mycobacterium tuberculosis. PloS Pathology. 2006; 2:e61. Epub 2006 Jun 16.
Institute of Medicine. Emerging infections: microbial threats to health in the United States. In: Joshua Lederberg, Robert E. Shope, and Stanley C. Oaks, Jr., editors. Washington: The National Academies Press; 1992.
International Air Transportation Association. Fact sheet: IATA–International Air Transport Association [cited 2009 Jun 8]. Available from http://www.iata.org/pressroom/facts_fi gures/fact_sheets/iata.htm
International Labour Organization. Towards a fair deal for migrant workers in the global economy. International Labour Conference, 92nd Session, 2004. Report VI [cited 2009 Jun 8]. Available from http://www.ilo.org/wcmsp5/groups/public/---dgreports/---dcomm/documents/meeting-document/kd00096.pdf
Katiyar S, Pfaller M, Edlind T. Candida albicans and Candida glabrata clinical isolates exhibiting reduced echinocandin susceptibility. Antimicrob Agents Chemother. 2006;50:2892–4. DOI: 10.1128/AAC.00349-06
Klein S, Bosman A. Completeness of malaria notification in the Netherlands 1995–2003 assessed by capture-recapture method. Euro Surveill. 2005;10:244–6.
Kuritzkes DR. Report from the XV International HIV Drug Resistance Workshop. AIDS Clin Care. 2006;18:83–4.
MacDougall C, Powell JP, Johnson CK, Edmond MB, Polk RE. Hospital and community fluoroquinolone use and resistance in Staphylococcus aureus and Escherichia coli in 17 US hospitals. Clin Infect Dis. 2005;41:435–40. DOI: 10.1086/432056
MacPherson DW, Gushulak BD. Balancing prevention and screening among international migrants with tuberculosis: population mobility as the major epidemiological influence in low-incidence nations. Public Health 2006;120:712–23. DOI: 10.1016/j.puhe.2006.05.002
Maurer LH, Sneider TJ. Gonococcal urethritis in males in Vietnam: three penicillin regimens and one tetracycline regimen. JAMA. 1969;207:946–8. DOI: 10.1001/jama.207.5.946
Mentel M, Spirek M, Jorck-Ramberg D, Piskur J. Transfer of genetic material between pathogenic and food-borne yeasts. Appl Environ Microbiol. 2006;72:5122–5. DOI: 10.1128/AEM.00293-06
Monto AS, McKimm-Breschkin JL, Macken C, Hampson AW, Hay A, Klimov A, et al. Detection of influenza viruses resistant to neuraminidase inhibitors in global surveillance during the first 3 years of their use. Antimicrob Agents Chemother. 2006;50:2395–402. DOI: 10.1128/AAC.01339-05
Nagulapally SR, Ahmad A, Henry A, Marchin GL, Zurek L, Bhandari A. Occurrence of ciprofloxacin-, trimethoprim–sulfamethoxazole-, and vancomycin-resistant bacteria in a municipal wastewater treatment plant. Water Environ Res. 2009;81:82–90. DOI: 10.2175/106143008X304596
Public Health Agency of Canada. Drug-resistant tuberculosis among the foreign-born in Canada [cited 2009 Jun 8]. Available from http://www.phac-aspc.gc.ca/publicat/ccdr-rmtc/05pdf/cdr3104.pdf
Reinert RR, Jacobs MR, Appelbaum PC, Bajaksouzian S, Cordeiro S, van der Linden M, et al. Relationship between the original multiply resistant South African isolates of Streptococcus pneumoniae from 1977 to 1978 and contemporary international resistant clones. J Clin Microbiol. 2005;43:6035–41. DOI: 10.1128/JCM.43.12.6035-6041.2005
Researchers report HIV and STD statistics from Vietnam. AIDS Wkly. 2000;20:21–2.
Schunk M, Kumma WP, Miranda IB, Osman ME, Roewer S, Alano A, et al. High prevalence of drug-resistance mutations in Plasmodium falciparum and Plasmodium vivax in southern Ethiopia. Malar J. 2006;5:54. DOI: 10.1186/1475-2875-5-54
Skarbinski J, Eliades MJ, Causer LM, Barber AM, Mali S, Nguyen-Dinh P, et al. Malaria surveillance—United States, 2004. MMWR Surveill Summ. 2006;55(SS04):23–37.
Sutrisna A, Soebjakto O, Wignall FS, Kaul S, Limnios EA, Ray S, et al. Increasing resistance to ciprofloxacin and other antibiotics in Neisseria gonorrhoeae from East Java and Papua, Indonesia, in 2004—implications for treatment. Int J STD AIDS. 2006;17:810–2. DOI: 10.1258/095646206779307595
Tenover FC. Mechanisms of antimicrobial resistance in bacteria. Am J Infect Control. 2006;34(Suppl 1):S3–10; discussion S64–73.
Thompson T, Sobsey M, Bartram J. Providing clean water, keeping water clean: an integrated approach. Int J Environ Health Res. 2003;13(Suppl 1):S89–94. DOI: 10.1080/0960312031000102840
United Nations High Commission for Refugees. 2007 global trends: refugees, asylum-seekers, returnees, internally displaced and stateless persons. Statistics, 17 June 2008 [cited 2009 Jun 8]. Available from http://www.unhcr.org/statistics/STATISTICS/4852366f2.pdf
United Nations World Tourism Organization. UNWTO world tourism barometer [cited 2009 Jun 8]. Available from http://unwto.org/facts/eng/pdf/barometer/UNWTO_Barom09_1_en_excerpt.pdf
United Nations. International migration 2006. New York: The Nations [cited 2009 Jun 8]. Available from http://www.un.org/esa/population/publications/2006Migration_Chart/2006IttMig_chart.htm
US Department of State. Trafficking in persons report, June 4, 2008 [cited 2009 Jun 8]. Available from http://www.state.gov/documents/organization/105501.pdf
World Health Organization. Immunization service delivery and accelerated disease control. New vaccines and technologies [cited 2009 Jun 8]. Available from http://www.who.int/immunization_delivery/new_vaccines/en
World Health Organization. World health report 2007: a safer future. Global public health security in the 21st century [cited 2009 Jun 8]. Available from http://www.who.int/whr/2007/whr07_en.pdf
World Health Organization. Anti-tuberculosis drug resistance in the world. Report No. 3 [cited 2009 Jun 8]. Available from http://www.who.int/tb/publications/who_htm_tb_2004_343/en
World Health Organization. Addressing the threat of tuberculosis caused by extensively drug-resistant Mycobacterium tuberculosis. Wkly Epidemiol Rec. 2006;81:386–90 [cited 2009 Jun 8]. Available from http://www.who.int/wer/2006/wer8141.pdf
Xiao JC, Xie LF, Fang SL, Gao MY, Zhu Y, Song LY, et al. Symbiosis of Mycoplasma hominis in Trichomonas vaginalis may link metronidazole resistance in vitro. Parasitol Res. 2006;100:123–30. DOI: 10.1007/s00436-006-0215-y
A14
THE BACTERIAL CHALLENGE: A TIME TO REACT, EXECUTIVE SUMMARY
European Centre for Disease Prevention and Control and European Medicines Agency Joint Working Group58,59
Main Findings
There is a gap between the burden of infections due to multidrug-resistant bacteria and the development of new antibiotics to tackle the problem.
-
Resistance to antibiotics is high among Gram-positive and Gram-negative bacteria that cause serious infections in humans and reaches 25% or more in several EU Member States.
-
Resistance is increasing in the EU among certain Gram-negative bacteria such as recently observed for Escherichia coli.
-
Each year, about 25000 patients die in the EU from an infection with the selected multidrug-resistant bacteria.
|
58 |
Reprinted with permission from European Centre for Disease Prevention (ECDC) and Control (ECDC) and European Medicines Agency. 2009. ECDC/EMEA JOINT TECHNICAL REPORT, The bacterial challenge: time to react: A call to narrow the gap between multidrug-resistant bacteria in the EU and the development of new antibacterial agents. http://www.ema.europa.eu/pdfs/human/antimicrobial_resistance/EMEA-576176-2009.pdf (accessed July 5, 2010). |
|
59 |
Dominique L. Monnet is Senior Expert & Programme Coordinator, Antimicrobial Resistance and Healthcare-Associated Infections, Scientific Advice Unit, European Centre for Disease Prevention and Control (ECDC), and his remarks to the workshop were largely based upon the findings and conclusions of this report. |
-
Infections due to these selected multidrug-resistant bacteria in the EU result in extra healthcare costs and productivity losses of at least EUR 1.5 billion each year.
-
Fifteen systemically administered antibacterial agents with a new mechanism of action or directed against a new bacterial target were identified as being under development with a potential to meet the challenge of multidrug resistance. Most of these were in early phases of development and were primarily developed against bacteria for which treatment options are already available.
-
There is a particular lack of new agents with new targets or mechanisms of action against multidrug-resistant Gram-negative bacteria. Two such agents with new or possibly new targets and documented activity were identified, both in early phases of development.
-
A European and global strategy to address this gap is urgently needed.
In 2007, the European Centre for Disease Prevention and Control (ECDC), the European Medicines Agency (EMEA) and the international network Action on Antibiotic Resistance (ReAct) entered into a discussion on the need to document the gap between the frequency of multidrug-resistant bacterial infections in the EU and the development of new antibiotics. As a result, an ECDC/EMEA Joint Working Group was established in 2008 to give an account of facts and figures that would allow reasonable predictions of the extent of the gap in the coming years.
The following antibiotic-resistant bacteria were selected because they frequently are responsible for bloodstream infections and because the associated antibiotic resistance trait is, in most cases, a marker for multiple resistance to antibiotics:
-
Staphylococcus aureus, methicillin resistance (MRSA);
-
S. aureus, vancomycin intermediate resistance and vancomycin resistance (VISA/VRSA);
-
Enterococcus spp. (e.g. Enterococcus faecium), vancomycin resistance (VRE);
-
Streptococcus pneumoniae, penicillin resistance (PRSP);
-
Enterobacteriaceae (e.g. Escherichia coli, Klebsiella pneumoniae), third-generation cephalosporin resistance;
-
Enterobacteriaceae (e.g. K. pneumoniae), carbapenem resistance; and
-
Non-fermentative Gram-negative bacteria (e.g. Pseudomonas aeruginosa), carbapenem resistance.
Trends and Burden of Infections Due to Multidrug-Resistant Bacteria in the EU
Data on these selected antibiotic-resistant bacteria in invasive infections (mainly bloodstream infections) were available from the European Antimicrobial
Resistance Surveillance System (EARSS) for EU Member States, Iceland and Norway for each year during the period 2002–2007.
The trends in the proportion of antibiotic-resistant isolates among blood isolates of the selected bacteria frequently responsible for bloodstream infections in Europe are shown in Figure A14-1.
In 2007, the average proportion of Staphylococcus aureus blood isolates that showed resistance to methicillin (% MRSA) was the highest proportion of antibiotic-resistant isolates among the selected bacteria frequently responsible for bloodstream infections in the European Union. However, this proportion has been decreasing in recent years (Figure A14-1). This is due to decreasing MRSA trends in several Member States, likely due to action plans at national level as documented for France, Slovenia and United Kingdom. The average proportion of MRSA has reached a level close to that of the selected antibiotic-resistant Gram-negative bacteria.
The proportion of S. aureus blood isolates that showed intermediate resistance to vancomycin (VISA) was very low (less than 0.1%) in EU Member States, Iceland and Norway. No vancomycin-resistant S. aureus isolates were reported to EARSS in 2007 (data not presented on Figure A14-1).
In contrast, the average proportion of Escherichia coli—the most common Gram-negative bacteria responsible for infections in humans—blood isolates showing resistance to third-generation cephalosporins has been rising steadily.
At the same time, there is no sign of decreasing resistance to third-generation cephalosporins in Klebsiella pneumoniae or to carbapenems in Pseudomonas aeruginosa (Figure A14-1).
In 2007, the proportion of K. pneumoniae blood isolates from EU Member States, Iceland and Norway that showed resistance to carbapenems was, in general, very low (median = 0%) with the exception of Greece, where it reached 42% (data not presented on Figure A14-1).
The human and economic burden of antibiotic-resistant bacteria could only be estimated for the following five antibiotic-resistant bacteria: MRSA, vancomycin-resistant Enterococcus faecium, third-generation cephalosporin-resistant E. coli and K. pneumoniae and carbapenem-resistant P. aeruginosa.
The study confirmed that MRSA was the most common, single, multidrug-resistant bacterium in the European Union. However, the sum of cases of common, antibiotic-resistant Gram-positive bacteria (mostly MRSA and vancomycin-resistant Enterococcus faecium) was comparable to that of common, antibiotic-resistant Gram-negative bacteria (third-generation cephalosporin-resistant E. coli and K. pneumoniae, and carbapenem-resistant P. aeruginosa).
Overall, it was estimated that in 2007 approximately 25000 patients died from an infection due to any of the selected five antibiotic-resistant bacteria in the European Union, Iceland and Norway. In addition, infections due to any of the selected antibiotic-resistant bacteria resulted in approximately 2.5 million extra hospital days and extra in-hospital costs of more than EUR 900 million.
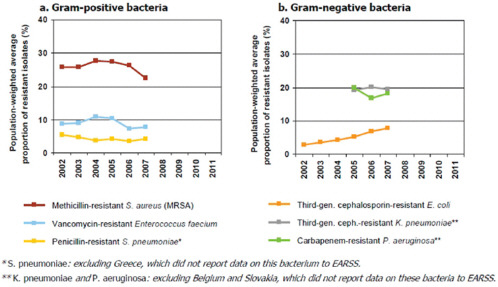
Subsequently, an estimate was made of loss of productivity due to these infections. Based on 2007 data, outpatient care costs were estimated at about EUR 10 million and productivity losses due to absence from work of infected patients were estimated at more than EUR 150 million, each year. Productivity losses due to patients who died from their infection were estimated at about EUR 450 million each year. Overall, societal costs of infections due to the selected antibiotic-resistant bacteria were estimated at about EUR 1.5 billion each year.
There are many reasons (e.g. limited range of included bacteria, outpatient infections not being considered, average cost of hospital care which does not take into account special patient care such as intensive care) to support a conclusion that these figures correspond to an underestimate of the human and economic burden of infections due to antibiotic-resistant bacteria.
Research and Development Pipeline of Antibacterial Agents
In order to assess the state of the antibacterial drug development pipeline, two commercial databases (Adis Insight R&D and Pharmaprojects) were queried for antibacterial agents in clinical development worldwide. It was decided not to perform an in-depth exploration of agents that had not yet reached clinical trials due to the high attrition rate during preclinical testing and the scarcity of data available for review.
Whenever possible, agents identified by the search were assessed for their antibacterial activity against the selected bacteria based on actual data available in the databases or in the literature. In the absence of actual in vitro data, reviewers also took into account reasonable assumptions of the activity of some agents based on the properties of similar agents (i.e. of the same class or with a common mechanism of action) in order to construct a ‘best-case scenario’.
Additionally, for each agent, reviewers were requested to indicate whether it was of a new class or belonged to an existing class of antibiotics and to indicate whether it:
-
acted on the same target and in the same way as that of at least one previously licensed antibacterial agent;
-
acted through a known mechanism of action on a new target; or
-
acted through a new mechanism of action.
The main results from this analysis were as follows:
-
Of 167 agents identified by the searches, there were 90 antibacterial agents with in vitro activity in a best-case scenario (based on actual data or assumed based on class properties of mechanism of action) against at least one organism in the panel of bacteria selected for their public health importance.
-
Of these 90 agents, 24 were new presentations of licensed antibacterial agents and 66 were new active substances.
-
Of the 66 new active agents, only 27 were assessed as having either a new target or a new mechanism of action, thus potentially offering a benefit over existing antibiotics.
-
Of these 27 agents, there were 15 that could be systemically administered.
-
Of the 15 agents with systemic administration, eight were judged to have activity against at least one of the selected Gram-negative bacteria.
-
Of the eight with activity against Gram-negative bacteria, four had activity based on actual data and four had assumed activity based on known class properties or mechanisms of action.
-
Of the four with activity against Gram-negative bacteria based on actual data, two acted on new or possibly new targets and none via new mechanisms of action.
Figure A14-2 shows the information on these 15 antibacterial agents. Notably, only five of these agents had progressed to clinical trials to confirm clinical efficacy (Phase 3 or later of clinical development).
The burden of bacterial resistance in the EU is already substantial and is likely to increase. Based on current data, it is expected that particular problems will arise in the coming years due to resistance among Gram-negative bacteria.
At the same time, there are very few antibacterial agents with new mechanisms of action under development to meet the challenge of multidrug resistance. There is a particular lack of new agents to treat infections due to multidrug-resistant Gram-negative bacteria.
This report has identified a gap between the burden of infections due to multidrug-resistant bacteria and the development of new antibacterial agents to tackle the problem. A European and global strategy to address the gap is urgently needed. Measures that spur drug development need to be put in place.
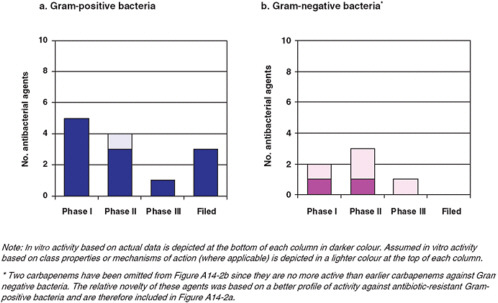
FIGURE A14-2 New systemic antibacterial agents with a new target or new mechanism of action and in vitro activity based on actual data (dark color bars) or assumed in vitro activity based on classproperties or mechanisms of action (light color bars) against the selected bacteria (best-case scenario), by phase of development (n = 15).
A15
THE EFFECTS OF ANTIBIOTIC AND PESTICIDE RESISTANCE ON PUBLIC HEALTH
David Pimentel
Department of Entomology, College of Agriculture and Life Sciences, Cornell University60
Introduction
Antibiotic and pesticide resistance caused by rapid genetic evolution in microbes and insects not only contributes to global disease outbreaks but also diminishes people’s ability to successfully control serious human illnesses. Over many decades microbe types, especially bacteria, have evolved resistance to antibiotics (CDC, 2007; Levy and Marshall, 2004; Whitney et al., 2000). Evolution also has occurred in various insect species, like flies and mosquitoes, with the result that they resist many types of insecticides (Pimentel, 2005).
In this report, the benefits and costs of antibiotics and pesticides (insecticides) are evaluated, and their impacts on human health due to antibiotic resistance by microbes and insecticide resistance by insects are assessed.
Antibiotics
For many years antibiotics have played a major role in treating microbial diseases in humans in the United States. Unfortunately antibiotic-resistant infections have been increasing in the United States due to microbe resistance and are now estimated to cause about 18,600 deaths each year (Messer, 2009). Now the number of deaths from antibiotic-resistant diseases in humans exceeds the annual number of deaths from HIV/AIDS (Messer, 2009). The cost of these antibiotic-resistant infections in humans in the U.S. healthcare system is in excess of $20 billion each year (Biomerieux, 2009).
The evolution of drug-resistant microbes can be surprisingly rapid. For example, in 1975, only 2.4 percent of the U.S. Staphylococcus aureus strains were resistant to penicillin, but today the percentage has grown to 95 percent (Goroncy-Bermes et al., 2001; Panililio et al., 1992; Silver Colloids, 2010). The rapid increase in drug resistance in disease organisms is caused by the widespread use and general overuse of the 150 to 300 antibiotics prescribed and used worldwide (ASM, 1995; Goldman, 2004). The United States is the largest producer of antibiotics, producing more than 11 million kilograms (kg) annually (Davies, 2008).
The rapid increase in antibiotic resistant S. aureus was confirmed in children recently in a 10-year study. The investigation reported that the number of methicillin-resistant Staphylococcus aureus infections in children during the study increased from 2 cases per 1,000 admissions in 1999 to 21 cases per 1,000 admissions in 2008 (CDC, 2007; Herigon et al., 2010).
The number of food infections in humans, many associated with livestock wastes, total 76 million per year (Jay, 2010). The hospitalizations total 300,000 per year and 5,000 deaths per year (Jay, 2010). Chicken, hog, and cattle wastes have polluted 35,000 miles of rivers in 22 states (Raettig, 2007).
Investigators suggest the greatest cause for the escalation of antibiotic resistance in microbes is the widespread use of antibiotics in livestock production (Union of Concerned Scientists, 2001). Specifically, from 70 to 87 percent of the antibiotics used in the United States go toward treating livestock prophylatically to increase animal production yields (Benbrook and Cattell, 2009; Environmental Health News, 2009). The total use of antibiotics in U.S. livestock is 11.4 million kg per year: 5 million kg in chickens, 4.5 million kg in hogs, and 1.8 million kg in cattle (Union of Concerned Scientists, 2001). On average, the use of antibiotics in livestock production increases the weight gain in livestock from 2 to 5 percent (Lam, 2010; University of Michigan, 2009). The total economic benefits are estimated to be between $1.2 billion and $2.5 billion per year (Goforth and Goforth, 2000). This is a relatively small increase in livestock production, at a high cost to human health problems of $20 billion per year.
The U.S. livestock population outweighs the human population by more than 5-fold (Pimentel et al., 2009). There are 100 million cattle, 60 million hogs, and 9 billion chickens plus other livestock fed forage, grains, and antibiotics (Pew Charitable Trusts, 2008).
Treating U.S. livestock with enormous quantities of antibiotics is not the only means of spreading antibiotic resistance in the environment. Many antibiotics end up in our sewer systems and some microbes evolve antibiotic resistance from this exposure and selection (Davies, 2008). Seagulls, geese, and ducks also have close contact with humans and have the capacity to transport resistant microbes long distances.
Microbes, especially bacteria, are extremely abundant in our soils. For example, in rich soil there may be more than 20,000 species of bacteria that weigh a total of 3,000 kg per hectare (Pimentel et al., 2006). With this abundance and diversity of species of microbes, the opportunity to evolve resistance to antibiotics is enormous (Allen et al., 2010).
Under current laws, the Food and Drug Administration is empowered to prevent agricultural antibiotics from use (FDA, 2007). Considering the costs to human public health, is the widespread use of antibiotics in livestock production prudent? There is a critical need to reduce or eliminate the routine treatment of livestock with antibiotics and to further withdraw the general use of antibiotics from livestock feed. There are several alternatives in animal husbandry to achieving similar weight
gain while achieving reduced infections in livestock. These alternatives include the use of straw bedding, reducing the density of livestock per unit space, more frequent cleaning, and better ventilation of the animal facilities (Jay, 2010).
Dr. Jørgen Schlundt of the World Health Organization, in his presentation before the Institute of Medicine’s workshop on Antibiotic Resistance: Implications for Global Health and Novel Intervention Strategies on April 6, 2010, in Washington, DC, reported that Denmark, Sweden, and the European Union have banned the routine use of antibiotics in livestock and this policy has been highly successful in terms of both human public health and livestock production (Kaufman, 2003; Schlundt et al., 2004).
About 70 percent of the 90,000 American deaths from bacterial infections acquired in hospitals are the result of infective bacteria that are resistant to at least one powerful antibiotic (Kennedy, 2010). The American Medical Association, the American Academy of Pediatrics, the American Pharmacists Association, the Infectious Diseases Society of America, the American Public Health Association, and the National Association of County City Health Officials are all urging Congress to phase out nontherapeutic antibiotic use in livestock (Kennedy, 2010). Antibiotics that are vital to humans should be restricted for human use.
Pesticides
About 3 billion kg of pesticides are applied throughout the world and about 500,000 kg are applied for pest control in the United States (Pimentel, 2009). Nicotine and arsenic were in widespread use in the early 1900s, but they were replaced by the newer agricultural insecticides, like dichlorodiphenyltrichloroethane (DDT) and parathion, starting in 1945 (Pimentel, 1997). The evolution of resistance to DDT and other insecticides was not detected until about 1950, when tests in houseflies confirmed the DDT resistance (Pimentel et al., 1950), and it was not detected in malaria-carrying mosquitoes until the late 1970s (Chapin and Wasserstrom, 1981). Initially, when DDT was used for mosquito control, it was applied to the inside of homes and was highly effective in countries like India that suffer from widespread malaria. The mosquitoes, after taking a blood meal, would fly to the nearest treated wall and digest some of the blood. While on the treated wall, mosquitoes picked up sufficient amounts of DDT to kill them.
There was initially no problem with the evolution of resistance in the mosquito population with the treatments inside homes, because less than one mosquito in one million was exposed to DDT. However, when the widespread use of DDT started in agriculture in India and other nations, DDT resistance was noted in the mosquito populations. Then widespread use of DDT in India and other nations resulted in the selection of resistant genotypes of Anopheline mosquitoes, and malaria incidence in humans increased (Knipling, 1952).
A highly desirable approach, in hindsight, would have been to restrict the use of DDT to inside homes and not to use it for agricultural use. This would
have been helpful in the control of malaria-carrying mosquitoes and in reducing pollution in the environment by DDT. This approach would have controlled mosquitoes and malaria without the problem of resistance subsequently developing in mosquito populations in the 1950s and later.
Today most malaria-carrying mosquito populations are highly resistant to most insecticides, and malaria has become one of the major diseases of the world (HealthMad, 2006). Malaria is responsible for about one million deaths each year. Resistance has also developed in the malarial protozoan organism to the potent drug artemisinin (Seppa, 2009). When artemisinin is used in combination with insecticides, resistance to artemisinin can be slowed.
In the United States today, about 1,000 agricultural pests are resistant to pesticides (Brown et al., 1999): specifically about 550 species of insects are resistant to insecticides, 330 species of plant pathogens are resistant to fungicides, and 220 species of weeds are resistant to herbicides. Roundup or glyphosate has been widely used with genetically engineered corn and soybeans and now with these crops and others there is growing resistance to this herbicide in agriculture. This pesticide resistance that has developed is costing the United States about $1.5 billion each year (Pimentel, 2005).
Worldwide, the widespread use of the 3 billion kg of pesticides is causing 26 million human poisonings plus 220,000 deaths (Richter, 2002). Roberts et al. (2007) reported about 300,000 nonfatal pesticide poisonings in the United States in farm workers and others. Despite the heavy use of pesticides, an estimated 40 percent of all potential food production is lost to pests worldwide and significant food production is lost to pests in the United States that have evolved pesticide resistance.
Pests Evolving Resistance to Biological Control Agents
The European rabbit was introduced into Australia in about 1859, and soon thereafter increased rapidly, destroying most of the forage grasses needed by sheep and other livestock (Bomford and Hart, 2002). Because of the increasing number of rabbits in Australia, wool production from sheep was reduced to only 32 million kg each year (Levin and Pimentel, 1981). In an effort to use biological controls to limit the numbers of rabbits, various natural enemies of the rabbit were introduced from Europe into Australia. But these were a failure, and the rabbits continued to increase.
Eventually some scientists in California investigating both the European rabbit and the South American rabbit found that biting insects transmitted a non-pathogenic virus in the South American rabbit to the European rabbit population (Kerr and McFadden, 2002). The South American rabbit virus was found to be highly pathogenic to the European rabbit. In 1950, the South American virus was introduced into the European rabbit in Australia (Kerr and McFadden, 2002). Immediately there was a 99.8 percent infection rate in the European rabbit and the rabbit population was reduced by 95 percent (Levin and Pimentel, 1981).
Slowly the European rabbit population in Australia started to increase again. Investigators found that the European rabbit population in Australia had evolved some resistance to the South American rabbit myxoma virus infections (Begon et al., 2006). In addition, attenuated strains of the virus started showing up in the rabbit population (Begon et al., 2006). The attenuated strains of the virus had an evolutionary advantage because biting insects were involved in the transmission. Thus, the longer the European rabbit infected with the virus lived, the greater the chance for the virus to be transmitted by the insects to noninfected European rabbits.
The rabbit and virus populations have stabilized, with the rabbit population existing at about 40 percent of the level of its original high numbers (Levin and Pimentel, 1981). Also, with the lower density of European rabbits, some of the natural predators are able to help keep the rabbit population at a lower level (Levin and Pimentel, 1981).
Conclusion
Evolved resistance to antibiotics, pesticides, and other chemicals by microbes, insects, and other organisms continues to increase and threaten the health of humans. The number of human deaths has increased over time, and the costs of health care have exploded due to antibiotic and pesticide resistance. Threats to U.S. food security and the natural environment have also increased due to increased resistance to antibiotics and pesticides.
References
Allen, H. K., J. Donato, H. H. Wang, K. A. Cloud-Hansen, J. Davies, and J. Handelsman. 2010. Call of the wild: Antibiotic resistance genes in natural environments. Nature Reviews Microbiology 8:251–9.
ASM (American Society for Microbiology). 1995. Report of the ASM task force on antibiotic resistance: Workshop Entitled “Antibiotic resistance: current status and future directions”: Report. Washington, DC: American Society for Microbiology.
Begon, M., C. R. Townsend, and J. L. Harber. 2006. Ecology: From individuals to ecosystems. New York: Wiley and Sons.
Benbrook, C., and M. Cattell. 2009. The science of organics. http://www.organic-center.org/report-fiels/Soc_Nut_Ed_Final.pdf (accessed January 10, 2010).
Biomeriux. 2009. Antibiotic-resistant infections cost the U.S. healthcare system in excess of $20 billion annually. http://www.biomerieux-usa.com/servlet/srt/bio/usa/dynPage?open=USA_NWS_NWS&doc=USA_NWS_NWS_G_PRS_RLS_73&crptprm=ZmlsdGVyPQ== (accessed June 29, 2010).
Bomford, M., and Q. Hart. 2002. Non-indigenous vertebrates in Australia. In Biological invasions: Economic and environmental costs of alien plant, animal, and microbe species, edited by D. Pimentel. Boca Raton, FL: CRC Press. Pp. 26–44.
Brown, L. R., M. Renner, B. Halweil, L. Starke, J. N. Abramovitz, and Worldwatch Institute. 1999. Vital signs 1999: The environmental trends that are shaping our future. New York: W.W. Norton & Co.
CDC (Centers for Disease Control and Prevention). 2007. MRSA: Methicillin-resistant Staphylococcus aureus in health care settings. Atlanta, GA: CDC.
Chapin, G., and R. Wasserstrom. 1981. Agricultural production and malaria resurgence in Central America and India. Nature 293(5829):181–5.
Davies, J. 2008. Antibiotic resistance and the future of antibiotics. In Microbial evolution and coadaptation: A tribute to the life and scientific legacies of Joshua Lederberg. Washington, DC: The National Academies Press. Pp. 160–72. http://www.nap.edu/catalog/125886.html (accessed February 22, 2010).
Environmental Health News. 2009. Crops absorb livestock antibiotics. http//www.environmental-healthnews.org/ehs/newsanntiboltics-in-crops (accessed December 18, 2009).
FDA (Food and Drug Administration). 2007. Antibiotics for Medical Treatment Act of 2007. S. 549, H.R. 962.
Goforth, R. L., and C. R. Goforth. 2000. Appropriate regulation of antibiotics in livestock feed. Boston College Environmental Affairs Law Review 28(1):39–77.
Goldman, E. 2004. Antibiotic abuse in animal agriculture: Exacerbating drug resistance in humans. Human and Ecological Risk Assessment: An International Journal 10(1):121–34.
Goroncy-Bermes, P., Schouten, M. A., and A. Voss. 2001. In vitro activity of a nonmedicated handwash product, chlorhexidine, and an alcohol-based hand disinfectant against multiply resistant gram-positive microorganisms. Infection Control & Hospital Epidemiology 22(4):194–6.
HealthMad. 2006. Major killer diseases of the world. http://healthmad.com/healthcare-industry/major-killer-diseases-of-the-world/ (accessed June 29, 2010).
Herigon, J. C., A. D. Hersh, J. S. Gerber, T. E. Zaoutis, and A. G. Newland. 2010. Antibiotic management of Staphylococcus aureus infections in US children’s hospitals, 1999-2008. Pediatrics 125(6):1294–300.
Jay, S. J. 2010. Alternatives to antibiotic use in food animal production. Presentation from Capital Hill briefing, March 2, 2010. Dr. Stephen J. Jay, Professor, Indiana School of Medicine, Indiana University. http://www.ikecoalition.org/Antibiotics%20in%20Animal%20Feed.pdf (accessed June 29, 2010).
Kaufman, M. 2003. WHO urges end to use of antibiotics for animal growth. The Washington Post, August 13, A01. http://www.washingtonpost.com/ac2/wp-dyn?pagename=article&node=&contentId=A51996-2003Aug12¬Found=true (accessed July 5, 2010).
Kennedy, D. 2010. Cows on drugs. The New York Times Sunday Opinion, April 18, 11.
Kerr, P., and G. McFadden. 2002. Immune response to myxoma virus. Viral Immunology 15(2):229–46.
Knipling, E. F. 1952. Present status of mosquito resistance to insecticides. The American Society of Tropical Medicine and Hygiene 1(3):389–94.
Lam, M. 2010. Beef, chicken, and fish. http://www.driam.com.articles/2002-BeefChickenFish.asp (accessed February 1, 2010).
Levin, S., and D. Pimentel. 1981. Selection of intermediate rates of increase in parasite-host systems. American Naturalist 117:308–15.
Levy, S. B., and B. Marshall. 2004. Antibacterial resistance worldwide: Causes, challenges and responses. Nature Medicine 10(12):S122–9.
Messer, T. 2009. Humans are at grave risks of antibiotic-resistant diseases. http://www.articles-base.com/alternative-medicine-articles/humans-are-at-grave-risk-due-to-anntibioticresistant-diseases-930037.html (accessed August 19, 2010).
Panililio, A. L., D. H. Culver, R. P. Gaynes, S. Banerjess, T. S. Henderson, J. S. Tolcon, and W. J. Martone. 1992. Methicillin-resistant Staphylococcus aureus in U.S. hospitals, 1975–1991. Infection Control Hospitals Epidemiology 13(10):582–6.
Pew Charitable Trusts. 2008. Testimony of Jay P. Graham Consultant, The Pew Commission on Industrial Farm Animal Production to the Committee on Senate Health, Education, Labor and Pensions. http://www.pewtrusts.org/news_room_detail.aspx?id=40810 (accessed July 5, 2010).
Pimentel, D., ed. 1997. Techniques for reducing pesticide use: Environmental and economic benefits. Chichester, UK: John Wiley and Sons. 444 pp.
Pimentel, D. 2005. Environmental and economic costs of the application of pesticides primarily in the United States. Environment, Development and Sustainability 7:229–52.
Pimentel, D. 2009. Biofuel food disasters and cellulosic ethanol problems. Special Issue, Agrofuels: Ecological, social, and energy ramifications, edited by M. A. Altieri. Bulletin of Science Technology and Society 29(3):205–12.
Pimentel, D., H. H. Schwardt, and L. B. Norton. 1950. House fly control in dairy barns. Journal of Economic Entomology 43:510–15.
Pimentel, D., T. Petrova, M. Riley, J. Jacquet, V. Ng, J. Honigman, and E. Valero. 2006. Conservation of biological diversity in agricultural, forestry, and marine systems. In Focus on ecology research, edited by A. R. Burk. New York: Nova Science Publishers. Pp. 151–73.
Pimentel, D., S. Williamson, C. E. Alexander, O.Gonzalez-Pagan, C. Kontak, and S. E. Mulkey. 2009. Reducing energy inputs in the U.S. food system. Human Ecology 36(4):459–71.
Raettig, K. A. 2007. Improvements needed in permitting CAFOS under the Clean Water Act. Prepared for the National Commission on Industrial Farm Animal Production. http://www.sec.nv.gov/cafo/tab_dd.pdf (accessed July 5, 2010).
Richter, E. D. 2002. Acute human pesticide poisonings. In Encyclopedia of pest management, edited by D. Pimentel. New York: Dekker. Pp. 3–6.
Roberts, E. M., P. B. English, J. K. Grether, G. C. Windham, L. Somberg, and C. Wolff. 2007. Maternal residence near agricultural pesticide application and autism spectrum disorders among children in California Central Valley. Environmental Health Perspectives 115(10):1482–9.
Schlundt, J., H. Toyofuku, J. Jensen, and S. A. Herbst. 2004. Emerging food-borne zoonoses. Review Science Technology Office International Epizootics 23(2):513–33.
Seppa, N. 2009. Malaria resists toughest medicine. American Society of Tropical Medicine and Hygiene Meeting, Washington, DC, November 18–22.
Silver Colloids. 2010. Antibiotic resistance a growing problem. http://www.silver-colloids.com/Pubs/antibiotic-resistance.html (accessed June 29, 2010).
Union of Concerned Scientists. 2001. 70 percent of all antibiotics given to healthy livestock. Press Release. Washington, DC: Union of Concerned Scientists. January 8.
University of Michigan. 2009. Resistant microbes, antibiotic abuse, and the threat to public health. http://www.fathom.com/course/21701753/session4.html (accessed June 29, 2010).
Whitney, C. G., M. M. Farley, J. Hadler, L. H. Harrison, C. Lexau, A. Reingold, L. Lefkowitz, P. R. Cieslak, M. Cetron, E. R. Zell, J. H. Jorgensen, A. Schuchat, and Active Bacterial Core Surveillance Program of the Emerging Infections Program Network. 2000. Increasing prevalence of multidrug-resistant Streptococcus pneumoniae in the United States. New England Journal of Medicine 343, Iss. 26; Part 1: 1917–24.
A16
CLINICAL ISSUES AND OUTCOMES ASSOCIATED WITH RISING ANTIMICROBIAL RESISTANCE
Louis B. Rice61
Louis Stokes Cleveland VA Medical Center and Case Western Reserve University
Measurable Clinical Impact of Antimicrobial Resistance
It is important, when discussing antimicrobial resistance and the problems that it generates, to be specific about the type of resistance under discussion. For example, resistance to β-lactam antibiotics in Streptococcus pneumoniae (Arason et al., 2006) is almost entirely a community problem and has its roots in the overuse of antimicrobial agents in the outpatient setting. As a result, it can be anticipated that efforts to reduce community antibiotic prescribing could impact levels of resistance in S. pneumoniae, as has been shown in Iceland (Arason et al., 2006). Resistances in Salmonella, Campylobacter, and to a significant extent Escherichia coli, on the other hand, are credibly attributed to the agricultural use of antibiotics (Angulo et al., 2004). The major resistance problems in the modern hospital occur in Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumanni, Pseudomonas aeruginosa and Enterobacter species (sometimes referred to as the ESKAPE pathogens; Rice, 2008). Resistance in these organisms cannot credibly be attributed to either the community use of antibiotics or to the use of antibiotics in animals, but rests squarely with physicians who prescribe antibiotics in the hospital setting.
These differences allow for substantial obfuscation when resistance issues are discussed. Physician groups frequently point to agricultural use of antibiotics as a major problem, conveniently ignoring our own culpability. When the agriculture industry is criticized, they correctly point to the fact that much resistance is not related to the use of antibiotics in animals. The reality is that resistance in S. pneumoniae is reason enough to reduce unnecessary prescription in the community setting. Resistance in Salmonella, Campylobacter and especially E. coli is reason enough to ban non-therapeutic use of antibiotics in animals. The growing problems in the ESKAPE pathogens are reason enough to focus efforts to limit antimicrobial selective pressure in the hospital.
For the remainder of this article, I will focus on resistance in the ESKAPE pathogens. These pathogens account for more than 40 percent of infections in intensive care units in the United States, according to recent data published by
|
61 |
Medical Service 111(W), 10701 East Blvd., Cleveland, OH 44106, Tel: 216-791-3800 x4800, Fax: 216-231-3289, e-mail: louis.rice@va.gov. |
the National Health Care Safety Network (Hidron et al., 2008). They account for a far higher percentage of infections caused by organisms that are resistant to two or more antibiotics, although precise numbers in that regard are unavailable. As impressive as these numbers are, it is fair to ask whether the occurrence of resistance truly exerts an impact in the clinical outcome of infections. The literature on the clinical impact of resistance in infections is complex and not always comparable (Cosgrove, 2006). Studies can vary in the outcome they assess (morbidity, mortality, cost). They can vary in the perspective they choose to adopt (hospital, payer, patient, society). They can use different control groups (patients infected with susceptible strains, uninfected patients, colonized patients) and they can be affected by a number of different confounders (length of stay, severity of illness, comorbid conditions). Despite this, there are reasonable data to support a real impact of resistance on important and measurable outcomes for infections caused by most of the ESKAPE pathogens (Cosgrove, 2006).
More Difficult-to-Measure Impacts of Antimicrobial Resistance
Beyond common and measurable outcomes, however, antimicrobial resistance changes our practice in ways that are more difficult to measure:
-
Resistance invalidates well-done studies.
-
Resistance makes it difficult to follow established guidelines.
-
Resistance forces the use of less effective antibiotics.
-
Resistance forces the use of agents that may be toxic or that we know little about.
Resistance Invalidates Well-Done Studies
Since the 1960s, the principles of therapy for patients who develop a fever while neutropenic from cancer chemotherapy are well-established. Febrile patients who are neutropenic are to be started on antibiotics immediately, before the results of bacterial cultures are available. This practice stems largely from a study in which 50 percent of untreated patients who ultimately grew P. aeruginosa from their blood were dead by day 3 after fever (or about when the culture results become available) (Whitecar et al., 1970). In an effort to reduce morbidity and mortality from infections further, investigators have performed a number of studies examining the efficacy of prophylactic use of fluoroquinolones in neutropenic patients. In 2005, a meta-analysis of several such studies (Gafter-Gvili et al., 2005) concluded that the use of fluoroquinolone prophylaxis was beneficial. That same year, Bucaneve and colleagues (Bucaneve et al., 2005) published a study confirming the usefulness of levofloxacin for preventing febrile episodes in neutropenic patient, but also reported the disturbing finding that 4/4 E. coli isolated from patients receiving levofloxacin prophylaxis were resistant to fluoroquinolones. In 2008, in a study from Italy, where community rates of fluoroquinolone resistance in are quite high, Cattaneo
et al. (2008) reported that fluoroquinolone-resistant E. coli was the most frequent isolate (20.1 percent of cases) and that the rate of fluoroquinolone resistance in E. coli over all was 86.7 percent, and 96.5 percent in those on prophylaxis. In such a setting, fluoroquinolone prophylaxis, shown to be effective a decade ago, can no longer be considered a viable therapeutic option.
Resistance Makes It Difficult to Follow Established Guidelines
The Infectious Diseases Society of America guidelines for the treatment of febrile neutropenia (Hughes et al., 2002) indicate that ciprofloxacin is adequate for low-risk patients and generally recommend anti-pseudomonal penicillins, cephalosporins, or carbapenems for empirical treatment of high risk patients. In Southern Europe, rates of cephalosporin resistance in Klebsiella pneumoniae approach 50 percent.62 These strains are generally also resistant to β-lactam-β-lactamase inhibitor combinations. In Greece, rates of resistance to carbapenems are also approaching 50 percent in K. pneumoniae. Although combinations with aminoglycosides are recommended, the rates of resistance to these agents are also quite high in the same countries. In such an environment, one could hardly argue with addition of peptide antibiotics, such as colistin or polymixin B, to empirical therapy, despite the absence of these agents from accepted guidelines.
Resistance Forces Use of Less Effective Antibiotics
The emergence and spread of methicillin-resistant Staphylococcus aureus (MRSA) has led to widespread use of vancomycin both empirically and for directed therapy of staphylococcal infections. When compared to β-lactams for the treatment of methicillin-susceptible S. aureus (MSSA), however, vancomycin administration has been found to be associated with longer periods of bacteremia and inferior results (Kim et al., 2008). It appears likely that vancomycin is even less effective against MRSA than against MSSA. In a recent study of staphylococcal bacteremia and endocarditis, vancomycin cure rates ranged from 30–45 percent and were especially poor for patients with left sided MRSA endocarditis (Fowler et al., 2006). Moreover, there is increasing concern that the “creep” of vancomycin minimal inhibitory concentrations (MICs) (Steinkraus et al., 2007) will have a significant impact of the therapeutic utility of the agent, especially for strains that have MICs of 2 μg/ml or greater.
Resistance Forces the Use of Agents That May Be Toxic or About Which We Know Little
The introduction of extended-spectrum cephalosporins, anti-pseudomonal carbapenems, aztreonam, and ciprofloxacin led to the virtual obsolescence of the
peptide antibiotics polymixin B and colistin (polymixin E). After all, little was known about the pharmacokinetics of these agents and they were perceived as both nephro- and neurotoxic. The emergence of multi-resistant K. pneumoniae, P. aeruginosa, and A. baumanni in the past decade has led to a dramatic increase in use of these agents. Fortunately, they do not appear to be as toxic as was previously thought (Markou et al., 2003), but we still have little idea about appropriate dosing or whether they are truly effective agents. The National Institutes of Health is currently funding a study to optimize dosing of colistin in human patients.
Strategies to Mitigate the Impact of Resistance in the Future
Fortunately, as a clinical phenomenon, antimicrobial resistance is relatively simple. When resistance problems arise, they are always caused by some combination of infection control lapses and antimicrobial selective pressure. It therefore stands to reason that tightening up our infection control practices and reducing antimicrobial selective pressure by minimizing antimicrobial overuse are important components of any successful strategy.
Infection Control
It is important to recognize that different strategies may have different effects, depending on the resistant organism involved. For example, the strict infection control measures put into place to control MRSA in the 1960s were remarkably successful in Scandinavian countries and the Netherlands (Rosdahl and Knudsen, 1991). To this day MRSA infections remain very rare in their hospitals.63 Some question, however, whether such measures can work in a decentralized health care system like the one in the United States, or whether rates at present are too high for such a strategy to be feasible or effective. The recently introduced plan in the U.S. Veterans Affairs Health System—in which universal screening for nasal colonization with MRSA, and subsequent isolation of those found to be positive—will be helpful in discerning the role of universal screening, although interpretation of the results may be confounded by the concomitant introduction of central line “bundle” practices (Pronovost et al., 2006), which by virtue of reducing central line-associated bacteremias almost certainly reduce rates of MRSA in many hospitals.
Infection control measures may not be as effective when the resistance at issue involves Gram-negative bacteria. Data published in several studies from Anthony Harris’s group at the University of Maryland (Harris et al., 2007a, 2007b; Johnson et al., 2009) indicate that patient-to-patient transmission of extended-spectrum β-lactamase (ESBL)-producing E. coli or imipenem-resistant P. aeruginosa in the intensive care unit may be responsible for as few as 11 percent of new coloniza-
tions. Acquisition of ESBL-producing K. pneumoniae was attributable to patient-to-patient transmission in 52 percent of cases. These data are consistent with earlier reports on ESBL-producing (Peña et al., 1998), where in many cases infection control interventions had little effect compared to adjustments in the antibiotic practices designed to minimize use of extended-spectrum cephalosporins.
Antibiotic Stewardship
It is clear, then, that if we are to truly minimize antimicrobial selective pressure, and therefore keep resistance to a minimum, we must use antibiotics judiciously. To address this need, antibiotic stewardship programs are being instituted at many medical centers. The impact of these programs on resistance is difficult to discern, as most are relatively new and most studies describing their impact analyze outcomes unrelated to resistance, such as antimicrobial costs. With the many questions that remain about the specific relationships between antimicrobial use and individual resistance phenotypes, it is difficult to predict which strategies will be most effective. In my opinion, strategies that emphasize “narrowing down” coverage will be unsuccessful, since in most cases the “narrow-spectrum” drug selected (ampicillin, for example) is not narrow spectrum at all. The only convincing narrowing down is stopping. I also believe that strategies designed to restrict use of antibiotics at a time of critical illness (“up-front restriction”) risk placing patients in danger and alienating staff by coming between a doctor and her patient. Therefore, the most rational strategy is to allow freedom to prescribe any antibiotics at the time of acute illness, but to force decisions at day 2 to discontinue antibiotics if either the cultures are negative or if controlled trials demonstrate that the infection can be safely treated with a 2- or 3-day course of therapy. In order to facilitate such programs, the National Institute of Allergy and Infectious Diseases has issued a Broad Agency Announcement to fund studies designed to test antibiotic usage strategies that will minimize resistance, among them shortened length of therapy studies (BAA NIAID-DMID-NIHAI2009058).
New Antibiotics
The traditional solution for the problem of resistance is the development of new antibiotics with activity against resistant bacteria. This strategy worked very successfully as recently as the last decade, with the introduction of several drugs effective for the treatment of MRSA (quinupristin-dalfopristin, linezolid, daptomycin, tigecycline, televancin). Despite only being a few years past this undeniable success, the possibilities for a new antibiotic solution to the problem of multi-resistant Gram-negative bacteria seem much more remote. The reasons for this skepticism are several, and include the exodus of several large pharmaceutical companies from antibiotics as a therapeutic area, confusion regarding the Food and Drug Administration requirements for acceptable clinical trials
to license new antibiotics, and the relatively poor financial return (compared to drugs for chronic disease and “lifestyle” drugs) of these agents (Boucher et al., 2009). Perhaps the most daunting obstacle, though, is the fact that the multi-resistant Gram-negative bacteria use a variety of synergistic mechanisms (multidrug efflux, porin reductions, biofilms) that are non-specific in nature, and just as likely to mediate resistance to a new antimicrobial as an old one. Nevertheless, with the extraordinary molecular techniques available to today’s scientists, it seems likely that, with enough investment, a brilliant new idea will emerge to lead us into a new era of antimicrobial therapy.
Conclusions
The emergence of antimicrobial resistance is inevitably associated with our use of antimicrobial agents. The major advantage in doubling time and genetic exchange enjoyed by human pathogenic bacteria virtually guarantees some level of resistance. Our goal must therefore be to keep resistance to a manageable level, one that does not significantly impact our ability to practice solid, evidence-based medicine. Clearly, what we are doing at present is not achieving that goal. If we are to have any chance at preserving our ability to treat bacterial infections (and as a consequence perform transplants, give aggressive cancer chemotherapy, or use implantable devices) we need to develop a coherent strategy for appropriate infection control measures, prudent antimicrobial use, and aggressive investment in the discovery of novel antimicrobials. It is not overstating the case to say that our way of practicing medicine is at stake.
References
Angulo, F. J., N. L. Baker, S. J. Olsen, A. Anderson, and T. J. Barrett. 2004. Antimicrobial use in agriculture: controlling the transfer of antimicrobial resistance to humans. Seminars in Pediatric Infectious Diseases 15(2):78–85.
Arason, V. A., J. A. Sigurdsson, H. Erlendsdottir, S. Gudmundsson, and K. G. Kristinsson. 2006. The role of antimicrobial use in the epidemiology of resistant pneumococci: A 10-year follow up. Microbial Drug Resistance 12(3):169–76.
Boucher, H. W., G. H. Talbot, J. S. Bradley, J. E. Edwards, D. Gilbert, L. B. Rice, M. Scheld, B. Spellberg, and J. Bartlett. 2009. Bad bugs, no drugs: no ESKAPE! An update from the Infectious Diseases Society of America. Clinical Infectious Diseases 48(1):1–12.
Bucaneve, G., A. Micozzi, F. Menichetti, P. Martino, M. S. Dionisi, G. Martinelli, B. Allione, D. D’Antonio, M. Buelli, A. M. Nosari, D. Cilloni, E. Zuffa, R. Cantaffa, G. Specchia, S. Amadori, F. Fabbiano, G. L. Deliliers, F. Lauria, R. Foa, and A. Del Favero. 2005. Levofloxacin to prevent bacterial infection in patients with cancer and neutropenia. New England Journal of Medicine 353(10):977–87.
Cattaneo, C., G. Quaresmini, S. Casari, M. A. Capucci, M. Micheletti, E. Borlenghi, L. Signorini, A. Re, G. Carosi, and G. Rossi. 2008. Recent changes in bacterial epidemiology and the emergence of fluoroquinolone-resistant Escherichia coli among patients with haematological malignancies: Results of a prospective study on 823 patients at a single institution. Journal of Antimicrobial Chemotherapy 61(3):721–8.
Cosgrove, S. E. 2006. The relationship between antimicrobial resistance and patient outcomes: Mortality, length of hospital stay, and health care costs. Clinical Infectious Diseases 42(Suppl. 2):S82–9.
Fowler, V. G., Jr., H. W. Boucher, G. R. Corey, E. Abrutyn, A. W. Karchmer, M. E. Rupp, D. P. Levine, H. F. Chambers, F. P. Tally, G. A. Vigliani, C. H. Cabell, A. S. Link, I. DeMeyer, S. G. Filler, M. Zervos, P. Cook, J. Parsonnet, J. M. Bernstein, C. S. Price, G. N. Forrest, G. Fatkenheuer, M. Gareca, S. J. Rehm, H. R. Brodt, A. Tice, and S. E. Cosgrove. 2006. Daptomycin versus standard therapy for bacteremia and endocarditis caused by Staphylococcus aureus. New England Journal of Medicine 355(7):653–65.
Gafter-Gvili, A., A. Fraser, M. Paul, and L. Leibovici. 2005. Meta-analysis: Antibiotic prophylaxis reduces mortality in neutropenic patients. Annals of Internal Medicine 142(12 Pt. 1):979–95.
Harris, A. D., M. Kotetishvili, S. Shurland, J. A. Johnson, J. G. Morris, L. L. Nemoy, and J. K. Johnson. 2007a. How important is patient-to-patient transmission in extended-spectrum beta-lactamase Escherichia coli acquisition. American Journal of Infection Control 35(2):97–101.
Harris, A. D., E. N. Perencevich, J. K. Johnson, D. L. Paterson, J. G. Morris, S. M. Strauss, and J. A. Johnson. 2007b. Patient-to-patient transmission is important in extended-spectrum beta-lactamase-producing Klebsiella pneumoniae acquisition. Clinical Infectious Diseases 45(10):1347–50.
Hidron, A. I., J. R. Edwards, J. Patel, T. C. Horan, D. M. Sievert, D. A. Pollock, and S. K. Fridkin. 2008. NHSN annual update: Antimicrobial-resistant pathogens associated with healthcare-associated infections: Annual summary of data reported to the National Healthcare Safety Network at the Centers for Disease Control and Prevention, 2006-2007. Infection Control and Hospital Epidemiology 29(11):996–1011.
Hughes, W. T., D. Armstrong, G. P. Bodey, E. J. Bow, A. E. Brown, T. Calandra, R. Feld, P. A. Pizzo, K. V. Rolston, J. L. Shenep, and L. S. Young. 2002. 2002 guidelines for the use of antimicrobial agents in neutropenic patients with cancer. Clinical Infectious Diseases 34(6):730–51.
Johnson, J. K., G. Smith, M. S. Lee, R. A. Venezia, O. C. Stine, J. P. Nataro, W. Hsiao, and A. D. Harris. 2009. The role of patient-to-patient transmission in the acquisition of imipenem-resistant Pseudomonas aeruginosa colonization in the intensive care unit. Journal of Infectious Diseases 200(6):900–5.
Kim, S. H., K. H. Kim, H. B. Kim, N. J. Kim, E. C. Kim, M. D. Oh, and K. W. Choe. 2008. Outcome of vancomycin treatment in patients with methicillin-susceptible Staphylococcus aureus bacteremia. Antimicrobial Agents and Chemotherapy 52(1):192–7.
Markou, N., H. Apostolakos, C. Koumoudiou, M. Athanasiou, A. Koutsoukou, I. Alamanos, and L. Gregorakos. 2003. Intravenous colistin in the treatment of sepsis from multiresistant Gram-negative bacilli in critically ill patients. Critical Care 7(5):R78–83.
Peña, C., M. Pujol, C. Ardanuy, A. Ricart, R. Pallares, J. Linares, J. Ariza, and F. Gudiol. 1998. Epidemiology and successful control of a large outbreak due to Klebsiella pneumoniae producing extended-spectrum ß-lactamases. Antimicrobial Agents and Chemotherapy 42(1):53–8.
Pronovost, P., D. Needham, S. Berenholtz, D. Sinopoli, H. Chu, S. Cosgrove, B. Sexton, R. Hyzy, R. Welsh, G. Roth, J. Bander, J. Kepros, and C. Goeschel. 2006. An intervention to decrease catheter-related bloodstream infections in the ICU. New England Journal of Medicine 355(26):2725–32.
Rice, L. B. 2008. Federal funding for the study of antimicrobial resistance in nosocomial pathogens: No ESKAPE. Journal of Infectious Diseases 197(8):1079–81.
Rosdahl, V. T., and A. M. Knudsen. 1991. The decline of methicillin resistance among Danish Staphylococcus aureus strains. Infection Control and Hospital Epidemiology 12(2):83–8.
Steinkraus, G., R. White, and L. Friedrich. 2007. Vancomycin MIC creep in non-vancomycin-intermediate Staphylococcus aureus (VISA), vancomycin-susceptible clinical methicillin-resistant S. aureus (MRSA) blood isolates from 2001-05. Journal of Antimicrobial Chemotherapy 60(4):788–94.
Whitecar, J. P., Jr., M. Luna, and G. P. Bodey. 1970. Pseudomonas bacteremia in patients with malignant diseases. American Journal of the Medical Sciences 60(4):216–23.
A17
WORLD HEALTH ORGANIZATION ACTIVITIES FOR CONTROL OF ANTIMICROBIAL RESISTANCE DUE TO USE OF ANTIMICROBIALS IN ANIMALS INTENDED FOR FOOD
Jørgen Schlundt64 and Awa Aidara-Kane
World Health Organization65
Introduction
The widespread use of antimicrobials for therapeutic purposes but also for growth promotion in livestock production has intensified the risk for the emergence and spread of resistant microorganisms. This raises particular concern since the same classes of antimicrobials are used in humans and animals. Food is generally considered to be the most important vector for the spread of antimicrobial resistance between humans and animals.
Concerned with the extensive use of antibiotics in food animal production, accelerating the development of resistant bacteria in animals and the transmission to humans via the food chain, the World Health Assembly (WHA) in 1998 adopted a resolution on antimicrobial resistance (WHO, 1998). The WHA is the top governing body of the World Health Organization (WHO), consisting of representatives of all 193 WHO Member States. This resolution urged Member States to encourage the reduced and rational use of antimicrobials in food animal production and resulted in the further development of “WHO global principles for the containment of antimicrobial resistance in animals intended for food” (WHO, 2000).
The Publication of the Global Principles has been followed by about 10 WHO expert consultations (some held jointly with the Food and Agriculture Organization of the United Nations [FAO] and the World Organisation for Animal Health66 [OIE]) to (a) assess the public health risk associated with the use of antimicrobials in animal husbandry (including aquaculture) and (b) propose high-level management options to address the identified risks. This consultative process, involving many of the leading scientists in this area, has demonstrated unequivocally that antimicrobial use in food animals can select for antimicrobial resistance in bacteria in the animal (especially in the gut). Subsequently these resistant bacteria or their genetic determinants can be transferred to humans via the food chain. The consultative process has resulted in three major outcomes:
|
64 |
Presently: Deputy Director, National Food Institute, Technical University of Denmark, Morkhoj Bygade 19, 2860 Soborg, Denmark, phone: +4535887703, e-mail: jors@food.dtu.dk. |
|
65 |
Department of Food Safety and Zoonoses, 20 Avenue Appia, CH-1211 Geneva 27, Switzerland, Phone: +41 22 791 24 03, e-mail: aidarakanea@who.int. |
|
66 |
Known as the Office International des Epizooties at that time. |
-
the development of a WHO list of Critically Important Antimicrobials (CIA),
-
the establishment of an Ad Hoc Intergovernmental Codex Task Force on Antimicrobial Resistance, and
-
the establishment of a WHO Advisory Group on Integrated Surveillance of Antimicrobial Resistance (AGISAR).
The Public Heath Impact of the Use of Antimicrobials in Food-Producing Animals
The First Alert
Realizing that modern livestock production throughout the world relied heavily on the use of antimicrobial substances, not only for treatment of infections but also for the promotion of growth and the prevention of diseases, WHO had already called for action in 1990. This was the first alert pointing at an increase of antimicrobial resistance in zoonotic and foodborne pathogens and the first call for action and intersectoral collaboration and harmonization of methods for monitoring and surveillance across sectors. An international, multidisciplinary working group was established by WHO to elaborate guidelines for uniform, regular, and systematic monitoring and surveillance of antimicrobial resistance susceptibility testing worldwide. The result was the Guidelines for Surveillance and Control of Antimicrobial Resistance (WHO, 1990). The guidelines stated that the
occurrence of antimicrobial resistant pathogenic bacteria in clinical materials, as well as in the normal flora of humans, in food and the environment urgently require elucidation to solve the problem encountered in treating the numerous infections due to such bacteria. In particular, systemic infections due to pathogenic bacteria such as Salmonella, which are common to animals and man and/or are transmissible from animals to man, is proving to be difficult to treat both in man and animals.
Following this, a WHO working group on antimicrobial resistance held its first meeting at the Central Veterinary Laboratory in Weybridge in 1991 to elaborate a small pilot project on “surveillance and assessment of antimicrobial resistance in microorganisms derived from animals, public and environmental health, and clinical medicine” by applying the common methodology outlined in the above-mentioned WHO guidelines (WHO, 1992).
Berlin 1997: Assessing the Medical Impact
In 1997 WHO convened an expert meeting on “The Medical Impact of the Use of Antimicrobials in Food Animals” in Berlin (WHO, 1997). The objectives
were to achieve international consensus on priority medical problems arising from the use of antimicrobials in the livestock production.
The meeting acknowledged that antimicrobial use can select resistant forms of bacteria in the ecosystem, and resistant bacteria and resistance genes can be exchanged among human, animal, and other ecosystems. The following adverse consequences of selecting resistant bacteria in animals were identified:
-
the transfer of resistant pathogens to humans via direct contact with animals or through the consumption of contaminated food or water,
-
the transfer of resistance genes to human bacteria,
-
increased incidence of human infections caused by resistant pathogens, and
-
potential therapeutic failures.
The meeting underlined the importance of monitoring antimicrobial resistance from farm to table and the importance of prudent use of antimicrobials as a risk-management tool at primary production level for the containment of antimicrobial resistance.
The following are some of the recommendations:
-
The use of any antimicrobial growth promoters should be terminated if they are used as human therapeutics or known to select for cross-resistance.
-
No antimicrobial should be administered to a food animal unless it has been evaluated and authorized by competent national authorities.
-
National authorities should maintain records of export/import figures of antimicrobials to quantify use.
-
National monitoring programs for antimicrobial resistance should allow for relating data obtained from animals, food, and humans.
-
WHO/FAO should convene an expert consultation to develop a code of practice for prudent use of antimicrobials in food animal production.
In summary, the meeting in 1997 concluded that the use of antimicrobials in food animals is a public health issue on which prudent use guidelines should be implemented, and that monitoring of both antimicrobial resistance as well as antimicrobial usage is warranted.
An Important Milestone in 2000: The WHO Global Principles for Containment of Antimicrobial Resistance in Animals Intended for Food
Based on a WHO consultation (with the participation of the FAO and the OIE) in 2000, the WHO Global Principles for the Containment of Antimicrobial Resistance in Animals Intended for Food (Global Principles) were developed
(WHO, 2000). These principles provide a framework of recommendations to reduce the overuse and misuse of antimicrobials in food animals for the protection of human health.
The development of the Global Principles represented a logical continuation of WHO’s activities on health implications of nonhuman use of antimicrobials. They endorsed and strengthened earlier WHO recommendations, such as the need to terminate the use of antimicrobial growth promoters and the need to establish surveillance systems on antimicrobial consumption and resistance.
It is especially noteworthy that already at this stage the process chosen for the development of these principles took into account the need for a broad partnership among all stakeholders. From the start, WHO consulted with a wide spectrum of interested groups. Collaboration between these and the other international organizations has been considered vital to identify complementary activities, to avoid duplication, and to coordinate efforts toward successful development and implementation of the Global Principles. Likewise, the participants at the WHO meeting on the Global Principles included experts from human and veterinary medicine, communicable disease surveillance, food safety, registration of medical and veterinary pharmaceuticals, marketing and sales of veterinary antimicrobials, and food animal production. In addition to the FAO and the OIE, many other governmental and nongovernmental international organizations and associations participated, including COMISA (World Federation of the Animal Pharmaceutical Industry).
The WHO consultation was characterized by the genuine desire among all participants to develop a set of recommendations that can be used by WHO Member States in their endeavors to minimize the public and human health risks from misuse of antimicrobials in animals intended for food.
The Global Principles are an important component of the general WHO Global Strategy for the Containment of Antimicrobial Resistance (WHO, 2001). This strategy aims to identify the key factors associated with emerging antimicrobial resistance related to human disease and to develop an effective implementing strategy that will reduce resistance development in general (i.e., related to both animal and human use of antimicrobials).
The Global Principles included general recommendations and recommendations directed to different agents, institutions, and stakeholders; excerpts of some of the more important are briefly mentioned here.
In General
-
National governments should adopt a national strategy for the containment of antimicrobial resistance.
-
Relevant authorities should develop strategies that reduce the actual and potential risk to public health from antimicrobial-resistant bacteria and resistance genes.
Pre- and Postapproval
-
Decisions concerning the licensing of veterinary antimicrobial substances should consider the impact on human health of antimicrobial resistance.
-
No antimicrobial should be administered to animals unless it has been evaluated and authorized for such use by relevant authorities.
-
A risk-based evaluation of the potential human health effects of all uses of antimicrobial drugs in food-producing animals should be conducted. Those antimicrobials judged to be essential for human medicine should be restricted and their use in food animals should be justified.
-
Relevant authorities should ensure that all antimicrobials for disease control in animals are classified as prescription-only medicines.
Distribution, Sales, and Marketing
-
Special attention should be paid to the distribution and sale of counterfeit, subpotent, and misbranded veterinary antimicrobials.
-
If sufficient evidence exists that profit from the sale of antimicrobials negatively affects prescribing practices, appropriate countermeasures should be taken to ensure prudent use.
-
The advertising and promotion of prescription-only antimicrobial products should be directed only to veterinary professionals.
Antimicrobial Growth Promoters
-
Use of antimicrobial growth promoters that belong to classes of antimicrobial agents used (or submitted for approval) in humans and animals should be terminated or rapidly phased out in the absence of risk-based evaluations.
-
Risk-based evaluations of all antimicrobial growth promoters should be continued.
Surveillance of Antimicrobial Resistance and Antimicrobial Usage
-
Data generated from the surveillance of antimicrobial resistance and antimicrobial usage should play a key role in the development of national policies for the containment of antimicrobial resistance.
-
Programs to monitor antimicrobial resistance in animal pathogens, zoonotic agents, and indicator bacteria should be implemented on bacteria from animals, food of animal origin, and humans.
-
Relevant authorities should establish systems to determine and publicize the amounts of antimicrobials given to food animals and compare such data to data on antimicrobial resistance.
Guidelines on Prudent Use
-
Policies should provide advice on optimal therapeutic effect and on the control of antimicrobial resistance in animal and zoonotic bacteria.
-
Guidelines on the prudent use of antimicrobials in animals should be developed with multidisciplinary involvement and revised at regular intervals.
-
Veterinarians should prescribe antimicrobials only for animals under their direct care. Veterinarians are expected to have examined clinically affected animals and developed a treatment protocol prior to prescribing medication.
-
Antimicrobials should be prescribed only when indicated.
-
It is the responsibility of the producers to ensure that production systems promote animal health and welfare. Antimicrobial usage should always be a part of, not a replacement for, an integrated animal health program. Veterinarians together with producers should be jointly responsible for the health of animals on the farm.
Prophylactic Use of Antimicrobials
-
Use of antimicrobials for prevention of disease can only be justified where it can be shown that a particular disease is present or likely to occur.
-
Prophylactic use of antimicrobials in control programs should be regularly assessed for effectiveness and whether use can be reduced or stopped.
Education and Training
-
Education strategies emphasizing the importance and benefits of prudent use principles must be developed and implemented to provide relevant information on antimicrobial resistance for producers and stakeholders.
-
The public should be informed of the human health aspects of antimicrobial use in food animals.
While these WHO Global Principles have had a significant impact in a number of countries, for instance in relation to the phasing out of antimicrobial growth promoters, a major challenge is still to translate the Global Principles into national rules and regulations. This will only occur if we succeed in engaging in an open, transparent, and collaborative effort at national as well as international levels, bringing together all stakeholders in the complex process of reducing health risks from the misuse and overuse of antimicrobials in animals intended for food.
Tripartite WHO/FAO/OIE Work on Antimicrobial Resistance
Recognizing the importance of WHO’s long-standing preventive work related to antimicrobial resistance, and considering that antimicrobial usage and
resistance is a problem that requires a multidisciplinary approach, the Executive Committee of the Codex Alimentarius Commission (Codex) at its 48th meeting (2001) suggested that WHO and FAO should convene a multidisciplinary expert consultation in cooperation with OIE to advise the Commission on possible directions to be taken for Codex work in this area. In response, the three organizations decided to have two separate consultations to be consistent with the risk analysis framework recommended by Codex: a first workshop on risk assessment and a second workshop on risk management options.
First Workshop, December 2003, Geneva, Switzerland: Scientific Assessment of the Risk
This expert workshop was convened by FAO, WHO, and OIE to undertake a scientific assessment of the human health risk associated with use of antimicrobials in animals intended for food, taking into account all available information.
During the workshop the main scientific findings on the relation between use of antimicrobials in animals and antimicrobial resistance in human bacteria were presented and discussed, followed by conclusions, recommendations, and definition of data gaps (WHO, 2004a). The main findings and conclusions of the workshop are described in brief.
Antimicrobial resistance emerges in primary food production in response to antimicrobial selective pressure. An association between use of antimicrobial agents in food animals and antimicrobial resistance among bacteria isolated from humans is most evident for Salmonella and Campylobacter, and to a lesser degree for enterococci and Escherichia coli. Several lines of evidence demonstrate an association between use of antimicrobial agents in food animals and antimicrobial resistance among bacteria isolated from humans, including (1) outbreak investigations, (2) epidemiological investigations, (3) field studies, (4) case reports, (5) ecological and temporal associations, and (6) molecular subtyping.
Although previous WHO consultations identified only limited data about treatment failures in humans due to antimicrobial resistance, examinations of previous and more recent studies at the time of the Geneva consultation provided accumulating evidence of this and other adverse human health consequences due to resistant organisms. These consequences can be divided into two categories: (1) infections that would otherwise not have occurred and (2) increased frequency of treatment failures and increased severity of infections.
1. Infections that would otherwise not have occurred Use of antimicrobial agents in humans and animals disturbs the microbiota of the intestinal tract, placing such individuals at increased risk of certain infections. Individuals taking an antimicrobial agent, for any reason, are therefore at increased risk of becoming infected with pathogens resistant to the antimicrobial agent. This increased risk
can be expressed in the form of an “attributable fraction,” which is defined as the proportion of Salmonella infections that would not have occurred if the Salmonella were not resistant.
2. Increased frequency of treatment failures and increased severity of infection Increased frequency of treatment failures and increased severity of infection may be manifested by prolonged duration of illness, increased frequency of bloodstream infections, increased hospitalization, or increased mortality. Prolonged duration of illness has been demonstrated in four case-control studies of fluoroquinolone-resistant Campylobacter. The association between an increased frequency of antimicrobial-resistant Salmonella and an increased frequency of hospitalization has been demonstrated in several studies; in addition, greater case-fatality rates have been found for outbreak with resistant Salmonella than for outbreaks caused by susceptible infections.
Second Workshop, March 2004, Oslo, Norway: Risk-Management Options
Based on the outcome of the first workshop in Geneva, as well as other relevant input (e.g., reports of previous WHO and OIE workshops), the second workshop, in Oslo, considered the broad range of possible risk-management options for antimicrobial resistance from nonhuman use of antimicrobials. In particular, it focused on potential directions of future Codex, FAO, OIE, and WHO work in this area in order to prevent and minimize antimicrobial resistance at the global level. To ensure that the conclusions of the second workshop reflected the perspectives of affected parties, the major stakeholder groups (e.g., pharmaceutical industry, farmers,67 food processors, consumers, regulatory agencies, and veterinarians) participated in the meeting (WHO, 2004b).
Among the important conclusions were the following:
-
Through stringent implementation of good agricultural practices, including good animal husbandry and good veterinary practices, it is possible to reduce the need for antimicrobials.
-
The need for rapid implementation by governments and all stakeholders of the WHO Global Principles for the Containment of Antimicrobial Resistance in Animals Intended for Food and the OIE Guidelines is emphasized.
-
The concept of “thresholds of resistance” should be pursued as a tool for risk management. If these thresholds are exceeded, this should trigger a range of risk-management actions.
-
The concept of “critically important” classes of antimicrobials for people should be developed by WHO with a view to enabling specific resistance-preventive actions for such antimicrobials related to nonhuman use.
-
There is need for capacity building, networking, and coordination to facilitate implementation of surveillance programs in various countries, particularly in developing countries. FAO, WHO, and OIE should take a leading role in this.
-
A Codex/OIE task force should be established to develop risk-management options for antimicrobial resistance related to nonhuman use of antimicrobials.
One of the conclusions of the consultative workshop was that a list of antimicrobials that are “critically important” for human medicine needed to be defined and identified by an expert group appointed by WHO. The classification should be reviewed on a regular basis. Antimicrobials that are critically important in veterinary medicine should be identified and listed by OIE to complement the identification of such antimicrobials used in human medicine. The overlap of critical lists for human and veterinary medicine can provide further information so that an appropriate balance may be struck between animal health needs and public health considerations.
The outcome of the consultative process was discussed in detail at the Codex Alimentarius Commission (CAC) meeting in June 2004 in Geneva resulting in the establishment of an Ad Hoc Intergovernmental Codex Task Force on Antimicrobial Resistance in 2006.
The WHO List of Critically Important Antimicrobials for Human Health
There are many serious infections in people (including enteric infections) for which few or no alternate antimicrobials can be used if antimicrobial resistance develops. These antimicrobial classes can be classified under various names such as “critically important,” “essential,” “reserve,” or “last resort.” Antimicrobial classes could be classified as critically important when the drug is in a class that is the only available therapy or one of a limited number of drugs available to treat serious human disease or enteric pathogens that cause foodborne disease.
WHO initiated its work in this area through the organization of an expert consultation in Canberra in 2005 with the overall scope to develop a list of critically important antimicrobial agents for human medicine (WHO, 2005). The resulting list has subsequently been reexamined and updated during two expert meetings, both held in Copenhagen, in 2007 (WHO, 2007a) and in 2009 (WHO, 2009). These reports present the updated (second and third editions) of the WHO list of critically important antimicrobials for human medicine.
To develop the WHO list of critically important antimicrobials, all antimicrobials used to treat bacterial infections in people were classified into three categories of importance: critically important, highly important, and important antimicrobial agents. Each antimicrobial agent (or class) was assigned to one of the three categories on the basis of two criteria: (1) sole therapy or one of few alternatives to treat serious human disease and (2) antimicrobial used to treat diseases caused by organisms that may be transmitted via nonhuman sources or diseases causes by organisms that may acquire resistance genes from nonhuman sources. Critically important antimicrobials are those that meet both criteria 1 and 2. Highly important antimicrobials are those that meet criteria 1 or 2. Important antimicrobials are those that do not meet either criteria.
It is important to appreciate that if resistance develops to one chemical group of antimicrobials then generally all the other antimicrobials in that group are also affected due to cross-resistance. The WHO classification should be considered a core list of the most “critical” antimicrobials agents globally. However, considerations, such as cost and availability of antimicrobials in various geographic areas as well as local resistance rates, could cause the list of critically important agents to be altered for regional use (e.g., an antimicrobial agent ranked highly important may become critically important in a particular region).
The WHO classification was mainly conceived to guide decisions in risk-management strategies of antimicrobial use. The list is updated regularly as new information becomes available, including data on resistance patterns, new and emerging diseases, and the development of new drugs. The history of the development of antimicrobial resistance shows that resistance may appear after a long period of usage. As an example, vancomycin resistance in Enterococcus was first detected after the drug had been in use for over 40 years. Conversely, however, it can also develop and disseminate rapidly like penicillinase production in S. aureus. Even if resistance has not developed to date in particular groups of bacteria, it could still develop in the future.
The WHO list should be used to support more comprehensive assessments of risk. Such comprehensive assessments should include information on the potential development of resistance in pathogens in animals (release assessment) and the potential spread of resistant organisms or their genes from animals to humans (exposure assessment); and integrating these data into a comprehensive assessment of risk and strategies to manage that risk.
Prioritization of agents within the critically important category has been undertaken in order to allow allocation of resources on the agents for which management of the risks from antimicrobial resistance are needed most urgently. This prioritization resulted in the designation of the classes for which comprehensive risk-management strategies are needed most urgently: quinolones, third- and fourth-generation cephalosporins, and macrolides. However, the prioritization of these three classes of drugs should not minimize the importance of other drugs categorized as critically important on the list.
Management Options for Critically Important Antimicrobials for Human Medicine
The development of the list of critically important antimicrobials is part of a more comprehensive approach to the public health issue of antimicrobial resistance in both animals and humans. There is some urgency to the development of such risk-management strategies, particularly for quinolones and third- and fourth-generation cephalosporins. In addition to management options for all antimicrobials, specific options include the following:
-
Do not use these drugs at all.
-
Use only in individual animals based on culture results and lack of alternatives.
-
Use only in individual animals.
-
Use in groups of animals after assessment demonstrates acceptable level of safety.
These options are listed in the order that will minimize selective pressure and are therefore least likely to contribute to the development and spread of resistant bacteria in animals treated with these agents.
Contingency plans could be developed to control or eradicate Salmonella and other zoonotic pathogenic bacteria resistant to two or more “critically important” antimicrobials when they appear in food production animals or in the food supply. Options include the following:
-
Recall associated foods.
-
Restrict movement of infected or colonized animals.
-
Use processing that guarantees removal of all resistant bacteria.
-
Destroy food items.
-
Destroy groups of animals infected or colonized.
These options are listed in the reverse order that will minimize the spread and persistence of these multiresistant bacteria and thus safeguard public and animal health.
The Ad Hoc Intergovernmental Codex Task Force on Antimicrobial Resistance
As mentioned above, an important outcome of the Geneva 2003 and Oslo 2004 workshops was the establishment of a Codex ad hoc Intergovernmental Task Force on Antimicrobial Resistance. The decision was taken at the 29th session of the CAC, Geneva, Switzerland, July 3–7, 2006.
The objective of the task force is to develop science-based guidance to help countries assess the risks to human health associated with the presence in food and feed and the transmission through food and feed of antimicrobial-resistant microorganisms and to develop risk-management advice based on that assessment to reduce such risk. Three sessions of the Codex ad hoc Intergovernmental Task Force on Antimicrobial Resistance were held, in 2007, 2008, and 2009, respectively. During these meetings the task force has made significant progress in advancing the Draft Guidelines for Risk Analysis of Foodborne Antimicrobial Resistance. The fourth and last session of the task force is scheduled to be held in October 2010 in the Republic of Korea and the finalization of its work will be the adoption of Guidelines for the Risk Analysis of Foodborne Antimicrobial Resistance at the next meeting of the CAC in Geneva, July 2011.
The WHO Advisory Group on Integrated Surveillance of Antimicrobial Resistance
The WHO-AGISAR (http://www.who.int/foodborne_disease/resistance/agisar/en/index.html) was established in December 2008 to support WHO’s effort to minimize the public health impact of antimicrobial resistance associated with the use of antimicrobials in food animals. In particular, the AGISAR will assist WHO on matters related to the integrated surveillance of antimicrobial resistance and the containment of food-related antimicrobial resistance. The terms of reference of WHO-AGISAR are as follows:
-
Develop harmonized schemes (including appropriate sampling) for monitoring antimicrobial resistance in zoonotic and enteric bacteria.
-
Support WHO capacity-building activities in Member States for antimicrobial resistance monitoring (AMR training modules for Global Food-borne Infections Network training courses and workshops; http://www.who.int/gfn/training/en/index.html).
-
Promote information sharing on AMR.
-
Provide expert advice to WHO on containment of antimicrobial resistance with a particular focus on human critically important antimicrobials.
-
Support and advise WHO on the selection of sentinel sites and the design of pilot projects for conducting integrated surveillance of antimicrobial resistance.
-
Support WHO capacity-building activities in Member States for antimicrobial usage monitoring.
WHO-AGISAR comprises over 20 internationally renowned experts in a broad range of disciplines relevant to antimicrobial resistance, appointed following a web-published call for advisers, and a transparent selection process. WHO-AGISAR
holds regular telephone conferences and annual face-to-face meetings. The first meeting of AGISAR was held in Copenhagen, Denmark, in 2009, and the second in Guelph, Canada, June 5–7, 2010. The four AGISAR subcommittees (antimicrobial usage monitoring, antimicrobial resistance monitoring, capacity building, and data management) are in the process of developing practical tools, guidelines, and protocols to support WHO Member States in their efforts to implement a national program for integrated surveillance of antimicrobial resistance.
Recent Developments in Food Safety: Focus on Human Health Risk and Disease Burden
It is clear that the problems related to antimicrobial resistance from animal use of antimicrobials relates directly to food safety, since the primary vehicle for human exposure to resistant bacteria from animals is food. A growing number of farmers and food producers globally are adopting modern food production technologies, including improvements in the efficient treatment of animals for food production. The transfer of technology has also included the other types of use of antimicrobials in food animals, including the use of antimicrobials for growth promotion and for so-called prophylactic, indiscriminate treatment of flocks and herds. In many cases agribusinesses and drug companies recover research and development costs through high levels of market share and profits in global food production markets. Unfortunately, the considerations of negative (health) effects from food safety problems related to such production systems do not seem to spread as efficiently as the systems themselves. Therefore, it is important for both consumers and producers that food safety issues are considered on a global level and not only at a national—or even regional—level. Fortunately, a number of important new developments in global food safety considerations have taken place over the last decades, some of which are briefly mentioned here.
Concurrently with the major developments in WHO’s work on antimicrobial resistance over the last 20 years, a major shift in the international food safety paradigm has taken place. This change provides an important background for the potential way forward in preventing the human health problems from animal use of antimicrobials.
The change seems to have occurred in three separate waves representing an increasingly focused framework toward disease prevention: (1) focus on hygiene, (2) focus on hazard, and (3) focus on risk. A food safety hazard is defined by WHO/FAO as a “biological, chemical, or physical agent in or property of food that may have an adverse health effect,” whereas food safety risk is defined as “a function of the probability of an adverse effect and the magnitude of that effect, consequential to a hazard in food.” While a resistance gene—or a resistant microorganism—is thus clearly a hazard, the risk is defined as the probability of a health effect caused by this hazard.
Wave 1 (1980s), Focus on Hygiene, recognized the need to have good hygiene in all parts of food production, preventing external contamination and keeping all food-related surfaces clean.
Wave 2 (1990s), Focus on Hazard, recognized the need to focus on the element present in the food with a potential to cause disease. If this element is not removed or reduced to a level that will not cause disease, any hygienic provisions will not prevent disease. The Hazard Analysis Critical Control System was originally defined by the U.S. National Aeronautics and Space Administration in order to ensure safe food for astronauts. Much later it was taken up by WHO, and, since 1997, international guidelines have existed defining how hazards control can be managed through simple means.
Wave 3 (New Millennium), Focus on Risk, recognized that the real driver for preventative efforts should be the actual human health risk recognized as human disease cases linked to specific food items. In simple terms, if most disease caused by Salmonella comes from inherent contamination of raw chicken reaching the consumer’s kitchen, good hygiene alone will not remove it, nor will hazard control in the production line. This leaves two major focus areas for lowering the risk of salmonellosis from chicken: (a) lowering the Salmonella prevalence in chicken at the farm and/or (b) ensuring good cooking practice and preventing cross-contamination in the kitchen.
It is important to realize that we do have hazards in food that cause no human health risk. Both certain chemicals and some bacteria in food may represent hazards but at a given (low) concentration do not represent a human health risk. On the other hand, chemical substances (or pathogenic bacteria) in levels clearly causing human disease risk need to be dealt with, irrespective of how low this level might be. (For some pathogens, very low concentrations will give high probability for disease.)
Responding to the third wave, the process of food safety risk analysis was defined by WHO and FAO in the late 1990s (WHO, 1995). Risk analysis is a process consisting of three components: risk assessment, risk management, and risk communication. Risk assessment is a scientifically based process estimating risk using quantitative or qualitative expressions and including indications of the attendant uncertainties. Risk management is the process of weighing policy alternatives and selecting appropriate prevention and control options, based on risk assessment and other relevant factors. Risk communication is the interactive exchange of information and opinions throughout the risk-analysis process. Within the framework of risk analysis the basic preconditions are (a) a functional separation of risk assessment and risk management, (b) a clear understanding of the need for transparent interaction between assessment and management, and (c) a general focus on the
importance of (two-way) risk communication throughout all steps in this process (see Figure A17-1). Within the risk-analysis framework, we should be able to define targets for risk reduction, realizing that the ultimate aim of food safety efforts should be to lower the significant risk to human health we know our present food production system presents.
Estimating the Real Risk: Foodborne Disease Burden
While a focus on risk clearly relates to a probability of disease, and although such probability can be predicted or modeled using existing data on exposure and effect, the final measurement of the levels of disease in a population caused by food does not necessarily correspond exactly to the models. We need specific data relating disease occurrence to food—data that are presently missing for most, if not all, countries.
Therefore, the full extent of the burden and cost of unsafe food is currently unknown. Growing international trade, migration, and travel accelerate the spread of dangerous pathogens, including in some cases antimicrobial resistance, through food, thus increasing our universal vulnerability. However, if we want to direct our resources and efforts toward the problems causing the highest burden, we obviously need comparable data on this burden of foodborne diseases.
WHO therefore hosted the “WHO Consultation to Develop a Strategy to Estimate the Global Burden of Foodborne Disease” in September 2006 (WHO,
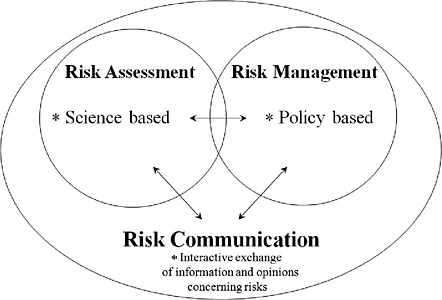
FIGURE A17-1 The WHO/FAO food safety risk analysis framework.
SOURCE: Schlundt (2010).
2007b). The consultation provided the strategic framework for the assessment of burden of foodborne diseases, including a roadmap for assembling existing information on the disease burden and a time frame outlining the individual strategic activities. The initiative will investigate disease burden from all major food causes using summary health metrics that combine morbidity, mortality, and disability in the form of the standard WHO metric: the disability-adjusted life year. The initiative will assemble, appraise, and report on the currently existing burden of foodborne disease estimates; conduct epidemiological reviews for mortality, morbidity, and disability; and provide models for each of the major foodborne diseases, including cause and source attribution models indicating links between specific foods and the relevant fraction of disease (attribution). The importance of antimicrobial resistance in foodborne pathogens is significant and will affect disease burden relative both to infections that would otherwise not have occurred and to increased frequency of treatment failures and increased severity of infection.
Realizing the International Dimension of Food Safety and the Interdependence of Food Safety Systems
The Beijing Declaration on Food Safety was adopted at the conclusion of the WHO High-Level International Forum on Food Safety in Beijing, November 2007 (WHO, 2007c). This declaration demonstrated the high-level political commitment of WHO Member States to resolving food safety problems through positive international collaboration rather than inefficient bilateral measures. It represents a consensus statement by the international community recognizing food safety as an individual human right and an essential public health function.
Following this push from Member States, WHO placed food safety on the agenda of its top governing body, the WHA, at its meeting in May 2010. Considering that the last time food safety was discussed at the WHA was in 2000, this represents a major recognition of the interest of Member States in improvements in this area. The outcome resolution (WHO, 2010) agreed by the WHA recognized that global trade in food is increasing, contributing to the risk of spread of pathogens across national borders, thereby necessitating more efficient global sharing of food safety information. It specifically acknowledged the need for closer collaboration between the health sector and other sectors, and increased action on food safety across the full length of the food production chain. It noted the need for updated and comprehensive internationally agreed-upon standards and agreements for risk assessments and scientific advice to support measures and interventions to improve the safety of food and recognized the importance of international agreement on global management of food safety. It thus requested that WHO continue to provide global leadership in providing tools for scientific estimations on foodborne risks and foodborne disease burden from all causes, provide support to Member States in building relevant capacity to improve cross-sectoral collaboration along the whole
food production chain, and promote research to support evidence-based strategies for the control and prevention of foodborne and zoonotic diseases.
The important developments in international food safety, now recognized by WHO Member States, will hopefully continue over the next decades, enabling safer food production methodology and thereby safer food. However, an important prerequisite for future improved efficiency in food safety relates to setting realistic targets for risk reduction and monitoring success or failure. In the past, food safety efforts have in most cases not been linked directly to foodborne disease risk. Compliance with existing regulation has often been the only measure of success. The lack of clearly communicated targets for disease reduction is still a major drawback of existing food safety systems, although some countries are now initiating major risk-based, target-driven efforts to improve food safety (e.g., national plans to lower the prevalence of Salmonella in food) or targeted—and monitored—efforts to reduce antimicrobial resistance levels through the phasing out of the use of antimicrobial growth promoters.
Conclusion: The Way Forward
WHO will work closely with partners at international, regional, and national levels to ensure the implementation of the WHO Global Principles for the containment of antimicrobial resistance in animals intended for food, in particular the ban of antimicrobial growth promoters, the rational prescription and use of veterinary drugs, and the restriction of use in animals of human critically important antimicrobials in animal husbandry, in particular quinolones and third- and fourth-generation cephalosporins.
WHO will work with FAO, OIE, as well as stakeholders, including industry, to achieve real reduction in the use of certain classes of antimicrobials in animals and the phasing out of the use of antimicrobial growth promoters in animals intended for food.
WHO will enhance the capacity of Member States, and in particular developing countries, through training courses and sentinel studies. Such capacity building will aim at supporting the implementation of:
-
surveillance of antimicrobial use and resistance,
-
intervention strategies to contain antimicrobial resistance, and
-
risk-assessment approaches to support selection of risk-management options.
The WHO-AGISAR will provide guidance to WHO on a framework for the development of an international network to promote and enhance collaboration on harmonization and data sharing.
The Ad Hoc Intergovernmental Codex Task Force on Antimicrobial Resistance will hold its last session in October 2010. Following the final approval at the CAC, June 2011, WHO will endeavor to provide to its Member States
support and guidance for implementation of the guidelines on risk analysis of antimicrobial resistance.
It is noteworthy that the theme for the next World Health Day (April 7, 2011) is antimicrobial resistance, in recognition of the importance of this global public health problem. This subject will of course cover all issues related to antimicrobial resistance, including resistance caused by both human and animal use of antimicrobials.
Member States should endeavor to put in place relevant policies to reduce problems related to the occurrence of antimicrobial resistance in pathogens. When covering the animal use area, the importance of international interaction is significant, recognizing that resistance caused by use practices in one country travels very quickly to other countries through food exports. The sharing of data and experiences between countries in this area is therefore extremely important and should be based on the setting up of surveillance systems according to existing WHO and OIE international standards. The documentation of the effects of specific national interventions to prevent antimicrobial resistance is increasingly being used across borders. Member States should support WHO efforts in sharing such experience internationally.
References
Schlundt, J. 2010. The contribution of antimicrobial use in food animal production to the emergence of antimicrobial resistance in humans. Presentation given at the April 6-7, 2010 public workshop on “Antimicrobial Resistance: Implications for Global Health & Novel Intervention Strategies” of the Institute of Medicine’s Forum on Microbial Threats, Washington, DC.
WHO (World Health Organization). 1990. Guidelines for surveillance and control of antimicrobial resistance, edited by B. Morgan, WHO/Zoonoses/90.167. Geneva, Switzerland: WHO. http://whqlibdoc.who.int/hq/1990/WHO_Zoonoses_90.167.pdf (accessed October 8, 2010).
____. 1992. Report of the WHO working group on antimicrobial resistance, Weybridge, UK, December 4, 1991. Geneva, Switzerland: WHO. http://whqlibdoc.who.int/hq/1992/VPH_92.105.pdf (accessed October 8, 2010).
____. 1995. Application of risk analysis to food standards issues. Report of the Joint FAO/WHO Expert Consultation, March 13–17. Geneva, Switzerland: WHO. http://www.who.int/foodsafety/publications/micro/march1995/en/index.html (accessed October 8, 2010).
____. 1997. WHO meeting on the medical impact of the use of antimicrobials in food animals, Berlin, October 13–17. WHO/EMC/ZOO/97.4. Geneva, Switzerland: WHO. http://www.who.int/foodborne_disease/resistance/amr_Oct97/en/ (accessed October 8, 2010).
____. 1998. Emerging and other communicable diseases: Antimicrobial resistance. Resolution WHA 51.17 adopted at the 51st World Health Assembly, Geneva, Switzerland, May. Geneva: WHO. http://www.paho.org/English/AD/DPC/CD/amr-eer-wha51-17-eng.pdf (accessed October 8, 2010).
____. 2000. WHO consultation on global principles for the containment of antimicrobial resistance in animals intended for food, Geneva, Switzerland, June 5–9. Geneva: WHO. http://www.who.int/foodborne_disease/resistance/amr_jun00/en (accessed October 8, 2010).
____. 2001. WHO global strategy for the containment of antimicrobial resistance. WHO/CDS/CSR/DRS/2001.2. Geneva, Switzerland: WHO. http://whqlibdoc.who.int/hq/2001/WHO_CDS_CSR_DRS_2001.2.pdf (accessed October 8, 2010).
____. 2004a. 1st joint FAO/OIE/WHO expert workshop on non-human antimicrobial usage and antimicrobial resistance: Scientific assessment, Geneva, Switzerland, December 1–5, 2003. Geneva, Switzerland: WHO. www.who.int/foodsafety/publications/micro/nov2003/en (accessed October 8, 2010).
____. 2004b. 2nd joint FAO/OIE/WHO expert workshop on non-human antimicrobial usage and antimicrobial resistance: Management options, Oslo, March 15–18. Geneva, Switzerland: WHO. www.who.int/foodsafety/publications/micro/mar04/en (accessed October 8, 2010).
____. 2005. Critically important antibacterial agents for human medicine for risk management strategies of non-human use: Report of a WHO working group consultation, February 15–18, Canberra, Australia. Geneva, Switzerland: WHO. www.who.int/entity/foodborne_disease/resistance/amr_feb2005.pdf (accessed October 8, 2010).
____. 2007a. Critically important antibacterial agents for human medicine: Categorization for the development of risk management strategies to contain antimicrobial resistance due to non-human use. Report of the second WHO Expert Meeting, Copenhagen, May 29–31. Geneva, Switzerland: WHO. www.who.int/foodborne_disease/resistance/cia/en (accessed October 8, 2010).
____. 2007b. WHO consultation to develop a strategy to estimate the global burden of foodborne diseases. Geneva, Switzerland, September 25–27, 2006. Geneva, Switzerland: WHO. http://www.who.int/foodsafety/publications/foodborne_disease/burden_ sept06/en/index.html (accessed October 8, 2010).
____. 2007c. The Beijing Declaration of Food Safety. Outcome of a high-level international forum on food safety “Enhancing Food Safety in a Global Community.” Beijing, November 26–27. Geneva, Switzerland: WHO. http://www.who.int/foodsafety/fs_management/meetings/forum07/en/index.html (accessed October 8, 2010).
____. 2009. Report of the 1st meeting of the WHO advisory group on Integrated Surveillance of Antimicrobial Resistance (AGISAR). Part 1: Third edition of the WHO list of critically important antimicrobials. Part 2: Strategic framework for WHO activities on integrated surveillance of antimicrobial resistance. Copenhagen, June 15–19. Geneva, Switzerland: WHO. www.who.int/foodborne_disease/resistance/agisar_June09/en/ (accessed October 8, 2010).
____. 2010. Advancing food safety initiatives. Resolution WHA 63.3 adopted at the 63rd World Health Assembly, Geneva, Switzerland, May. Geneva, Switzerland: WHO. http://apps.who.int/gb/ebwha/pdf_files/WHA63/A63_R3-en.pdf (accessed October 8, 2010).
A18
THE ANTIBACTERIAL PIPELINE: WHY IS IT DRYING UP, AND WHAT MUST BE DONE ABOUT IT?
University of California, Los Angeles
Introduction
It is difficult to define the limits of the enormous impact that antibacterial agents have had on the practice of medicine and on the health of the public in
the United States and throughout the world. The most fundamental impact of the introduction of antibacterial agents was a massive, immediate decline in death from infections of all types. For example, the overall mortality rate from infectious diseases in the United States fell from ~280 per 100,000 in 1936 (the year before sulfonamides were available in the United States), to ~200 per 100,000 in 1945 (the year before penicillin became widely available), to ~60 per 100,000 by the early 1950s (~15 years into the antibiotic era; Figure A18-1) (Armstrong et al., 1999).
Individual bacterial infections were massively affected by the sudden availability of antibacterial agents (Table A18-1). For example, within 1 year of availability, sulfonamides resulted in a 4-fold decline in mortality from cellulitis (Hoyne et al., 1939; Madsen, 1973). Penicillin led to a further 10-fold decline in mortality from cellulitis (Madsen, 1973). Indeed, within a period of 5 years of general availability, penicillin had reduced the mortality of cellulitis from ~11 percent in the pre-antibiotic era to ~0.3 percent, a 97 percent relative reduction in death (Spellberg et al., 2009); for every 9 patients with cellulitis treated with antibacterial agents, 1 life is saved. By comparison, the mortality from acute myocardial infarction in the placebo arm of a modern, international, randomized placebo-controlled study was 12 percent, and the reduction in mortality from aspirin or streptokinase was only 3 percent (the number needed to treat to save a life is 33) (ISIS-2 Collaborative Group, 1988). Who could have possibly imagined
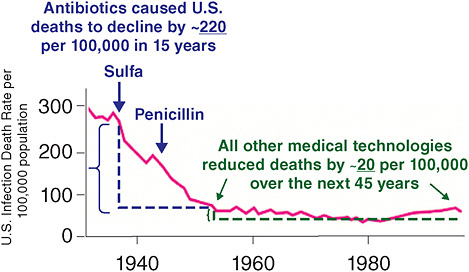
FIGURE A18-1 Change in deaths from infection in the United States following the introduction of antibiotics.
SOURCE: Adapted from Armstrong et al. (1999), Journal of the American Medical Association 281(1):61–6, with permission. Copyright © 1999 American Medical Association. All rights reserved.
TABLE A18-1 Antibiotic-Mediated Mortality Reductions for Specific Infections
|
Disease |
Pre-Antibiotic Mortality Rate |
Antibiotic Mortality Rate |
Change in Mortality |
|
Community pneumonia (Spellberg et al., 2008a) |
~35% |
~10% |
−25 |
|
Nosocomial pneumonia (Spellberg and Talbot, 2010) |
~60% |
~30% |
−30 |
|
Bacterial endocarditis (Christie, 1949; Guest and Harrison, 1948; Is endocarditis lenta always fatal, 1935; Kerr, 1955) |
~100% |
~25% |
−75 |
|
Bacterial meningitis (Chemotherapy of meningitis, 1938; Waring and Weinstein, 1948) |
>80% |
<20% |
−60 |
|
Skin infections (Madsen, 1973; Spellberg et al., 2009) |
~11% |
<0.5% |
−10 |
|
By comparison, treatment of myocardial infarction (i.e., heart attack) with aspirin or streptokinase (ISIS-2 Collaborative Group, 1988) |
−3 |
||
that, before antibiotics, the mortality of an infection as mundane as a simple skin infection (cellulitis) approached the mortality of “heart attacks” (myocardial infarction), or that antibiotics are far more effective at preventing death from cellulitis than aspirin or streptokinase for preventing death from myocardial infarction?
The mortality of community-acquired pneumonia was similarly affected (Armstrong et al., 1999; Kassowitz and Muscato, 1952). In the pre-antibiotic era, patients who were young (≤30 years) or had “good” baseline clinical status had a ~10 percent mortality rate from community-acquired pneumonia (Spellberg et al., 2008c). Those who were 30 to 59 years or had “fair” baseline status had a ~30 percent mortality rate, and those who were ≥60 years old or had a “poor” baseline status had a ~60 percent mortality rate. Antibacterial agents massively reduced these mortality rates to ~1 percent, ~5 percent, and ~15 percent, respectively. In absolute terms, these reductions in mortality are among the largest caused by any intervention in medicine. One of the few examples exceeding the mortality benefit of antibacterial agents for community-acquired pneumonia is the efficacy of antibacterial agents for treatment of bacterial endocarditis, which was 100 percent fatal in the pre-antibiotic era and experienced massive reductions in mor-
tality (to rates ≤30 percent fatal) with the availability of penicillin (Table A18-1) (Christie, 1949; Gorlin et al., 1950; Guest and Harrison, 1948; Is endocarditis lenta always fatal?, 1935; Kerr, 1955; McCartney, 1992). Similar huge mortality benefits are seen with antibacterial therapy for nosocomial pneumonia and bacterial meningitis (Table A18-1) (Chemotherapy of meningitis, 1938; Kassowitz and Muscato, 1952; Spellberg and Talbot, 2010; Trachsler et al., 1937; Waring and Weinstein, 1948).
Indeed, so massive were the mortality benefits of antibacterial agents that all subsequent medical advances since the early 1950s—including the advent of critical care medicine—have resulted in only minor further reductions in death from infections. As mentioned, from the late 1930s through the early 1950s (spanning the pre-antibiotic, sulfonamide, and penicillin eras), the mortality rate from infections in the United States fell by ~75 percent (5 percent per year), or by a remarkable ~220 per 100,000 population on an absolute basis (Armstrong et al., 1999). Over the ensuing 45 years, despite all intervening advances in medical care, mortality rates from infections declined only by an additional 20 per 100,000 (Armstrong et al., 1999). The federal government recognized this plateau effect in mortality reduction and understood that it was due to the remarkable power of antibacterial agents (National Center for Health Statistics, 1968; Schmeck, 1971; Stewart, 1968). The antibacterial-mediated decline in death from infections was so profound and rapid, health policy leaders believed that it could not be further improved upon by other medical technologies. This belief was central to the explicit decision made by the U.S. Surgeon General in the 1960s to shift federal healthcare priorities from acute illnesses to chronic illnesses, such as cancer, where it was hoped more dramatic gains could be made (Schmeck, 1971).
Beyond saving the lives of infected patients, the enormous efficacy of anti-bacterial agents enabled the conduct of complicated and deeply invasive surgery, aggressive myeloablative chemotherapy for treatment of cancer, fundamental elements of critical care (such as central venous catheter placement and mechanical ventilation), care for premature neonates, and solid and liquid organ transplantation. None of these medical advances would be feasible without effective antibacterial agents to deal with the infections, which result as a side effect of the advances themselves.
Dr. Lewis Thomas, one of the most prominent physicians of the 20th century—winner of the Lasker Award and a National Book Award and member of the National Academy of Sciences—described the experience of attending medical school in the pre-antibiotic era (Thomas, 1983). He wrote that, as a backlash against 19th-century medical chicanery, a central focus of Osler-inspired medical education during the first third of the 20th century was to emphasize to physicians that they could not alter the course of their patients’ illnesses. Medicine was a nontherapeutic, noninterventional field; the primary focus was on making an accurate diagnosis so an accurate prognosis could be provided to the patient or their family. Of his internship, Dr. Thomas wrote:
For most of the infectious diseases on the wards of Boston City Hospital in 1937, there was nothing that could be done beyond bed rest and good nursing care. Then came the explosive news of sulfanilamide, and the start of the real revolution in medicine. I remember the astonishment when the first cases of pneumococcal and streptococcal septicemia were treated in Boston in 1937. The phenomenon was almost beyond belief. Here were moribund patients, who would surely have died without treatment, improving … within a matter of hours … and feeling entirely well within the next day … we became convinced, overnight, that nothing lay beyond reach for the future. Medicine was off and running. (Thomas, 1983)
In short, the power of antibacterial therapy resulted in nothing less than a total revolution in the practice of medicine, fundamentally transforming the profession from a diagnostic, noninterventional field to a therapeutic, interventional profession.
The Dying of Antibacterial Research and Development
Fast forward seven decades from Dr. Thomas’s internship—from the introduction of antibacterial agents—and we find ourselves in the midst of a drying up antibacterial well. The first warning to the medical community about the decline in the development of antibacterial agents was published by Shlaes and Moellering in 2002 (Shlaes and Moellering, 2002). By that time, it was already clear that a combination of economic and regulatory forces were responsible for the effect (Projan, 2003; Shlaes, 2002, 2003). Reports from media sources and pharmaceutical insiders confirmed that most pharmaceutical companies had totally divested themselves of antibacterial research and development (R&D) programs, and that those companies with remaining programs had greatly diminished them (Wenzel, 2004).
In 2004, the first objective, quantitative analysis of new antibacterial approvals by the Food and Drug Administration (FDA), was published by members of the Infectious Diseases Society of America (IDSA) (Spellberg et al., 2004). That report described a 56 percent reduction in approvals of new systemic antibacterial agents between the 5-year periods of 1983–1987 and 1998–2002. At that time, a search of the publicly reported drug pipelines of the 15 largest pharmaceutical companies and 10 largest biotechnology companies identified only 6 antibacterial agents in advanced clinical development, which was fewer than the number of drugs in development for bladder hyperactivity, and was only one more than the number of drugs in development for erectile dysfunction. Such data became a cornerstone of the IDSA’s Bad Bugs, No Drugs white paper, which was used as a policy instrument with which to inform Congress and the media about this brewing public health crisis. After 4 years of active discussions and efforts to advance this issue, an update on antibacterial approvals was published (Spellberg et al., 2008b). In the interim, the situation had worsened. From 2003 to 2007, only
5 new antibacterial agents were approved by the FDA. The situation has further worsened since 2007 (see Figure A18-2) (Spellberg et al., 2008b). Only 1 new antibacterial agent has been approved between 2008 and the present, reflecting a 94 percent decline in approvals since 1983–1987.
It is critical that we understand the causes of the decline in antibacterial R&D so effective responses can be enacted. As mentioned previously, these causes can be generally grouped into two major categories: economic and regulatory.
Economic Forces Obstructing Antibacterial R&D
The predominant economic force obstructing antibacterial R&D, and the one most amenable to intervention, is the poor return on investment of antibacterial agents relative to other classes of drugs (Projan, 2003; Spellberg, 2009; Spellberg et al., 2008b). The financial return on investment for drugs which are taken chronically is far higher than for antibacterial agents, which are typically taken for 1 to 2 weeks and then are stopped because they result in cure of their target disease. Pharmaceutical insiders have extensively written about the Net Present Value calculation which has doomed antibacterial development in pharmaceutical R&D budgets (Projan, 2003; Shlaes, 2003). Given the remarkable increase (1,300 percent over 30 years; DiMasi, 1992; DiMasi et al., 1991, 2003) in costs of successfully developing a new drug, it is not surprising that companies have chosen to focus their R&D expenditures on developing drugs with higher return-on-investment potential than antibacterial agents.
When analyzing the poor return on investment for antibacterial agents, it is apparent that its impact is along the entire R&D cascade; there is no single,
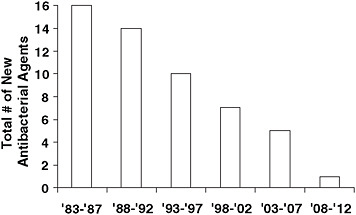
FIGURE A18-2 Number of new systemic antibacterial agents approved by the FDA per 5-year period. New molecular entities are considered. Data are accurate through June 2010.
rate-limiting block to overcome (Figure A18-3). Historically, the vast majority of antibacterial lead compound discovery and preclinical development has been done within industry. With the exit of large pharmaceutical companies from this activity, and the inability of academic scientists to get basic science research grants funded to conduct this translational/directed scientific activity, there has been a marked decline in lead compound discovery and preclinical development efforts. The bulk of the current activity is likely occurring in relatively small biotechnology companies and start-up/translational ventures growing out of academic laboratories. Such companies are relatively poorly capitalized, and venture and investment funds are drying up for antibacterial R&D due to the fundamental economic and regulatory forces that make it decreasingly likely that these drugs can be successfully developed to regulatory approval. Furthermore, even for those few large pharmaceutical companies remaining in the field, antibacterial discovery programs are significantly handicapped by economic and regulatory obstructions from within the companies themselves. Antibacterial programs inside pharmaceutical companies compete poorly against other drug development teams (e.g., for cancer, arthritis, dementia, etc.) for internal resources within the companies.
As a result, those few lead antibacterial compounds that are discovered may lack access to adequate capital to complete preclinical development, which may easily cost $5–$10 million to pay for good manufacturing practice-compliant manufacturing, preclinical toxicity studies, filing an Investigational New Drug application, and a first-in-human phase I clinical trial. Millions more dollars must be spent on the several phase I studies typically required to support an adequate drug development program. Costs then escalate by an order of magnitude when proceeding to phase II, which is actually referred to as the “valley of death,” since so many compounds fail to progress beyond this stage because it is so difficult to raise adequate capital to fund such studies. Finally, the average cost of phase III of a clinical development program is $100 million (in fiscal year 2008 dollars) (DiMasi et al., 2003), which of course can only be afforded by large pharmaceutical companies or very rarely by smaller companies that undergo an initial public offering or have unusual access to venture capital funding.
The solution to the poor return-on-investment potential of antibacterial agents is straightforward: the return-on-investment calculation must be modified by enactment of economic incentives. Such incentives must be focused on reducing the cost of developing antibacterial agents and on improving sales linked to antibacterial agents. The rationale for enactment of such economic incentives is the critical public health need for new drugs that can treat organisms resistant to currently available antibacterial agents. Hence, economic incentives should be targeted to developing “priority” antibacterial agents (Spellberg, 2009; Spellberg et al., 2008b), which have been defined as those which treat serious or life-threatening infections caused by antibiotic-resistant bacteria. There is also a specific, urgent need for new oral antibacterial agents, including for the treatment of resistant Gram-negative bacilli.
It is not the intent of this manuscript to provide a detailed description of specific economic incentives that could be effective for improving antibacterial development. Such incentives have been previously discussed in detail (ECDC and EMEA, 2009; Mossialos et al., 2009; Spellberg, 2009; Spellberg et al., 2008b). In brief, incentives that could effectively reduce the cost of developing new antibacterial agents include expansion of basic science and small business grants and contracts focused on discovery and translational development of new antibacterial agents, tax credits to reduce costs of development, and implementation of liability protection for priority antibacterial agents, akin to the Vaccine Injury Compensation Program (VICP), which has effectively facilitated critically needed new vaccine development. Incentives that could effectively improve sales linked to antibacterial agents include patent extensions (either direct or transferable), prolonged market or data exclusivity, guaranteed markets, and prizes. It is critical to emphasize that no single incentive will be capable of reversing the economic disadvantage of antibacterial agents. Rather, a panoply of incentives, capable of appealing to constituents ranging from academic basic and translational investigators, angel and venture capital investors, and both small and large companies, is necessary to overcome the blockades that affect all stages of the R&D cascade (Figure A18-3C).
Other economic forces obstructing antibacterial R&D include (Spellberg, 2009) (1) the large competition from among the >90 antibacterial agents currently on the market in the United States, which diminishes the potential market size of newly approved drugs; (2) the legitimate public health need for thought leaders to discourage use of newly approved antibacterial agents, which typically results in disappointing sales for newly marketed antibacterial agents, in contrast to agents in other drug classes; and (3) the “low-hanging fruit” theory, which states that most of the more readily identifiable and “drug-able” antibacterial agents have already been identified and developed, and that discovery and development of succeeding generations of antibacterial agents will require increasingly sophisticated, expensive scientific methods. These economic forces are far more difficult to reverse with intervention. Hence, the primary policy focus for reversing the economic disadvantage of antibacterial agents should be on improving their return on investment by enacting “push” and “pull” incentives, designed to stimulate preclinical R&D and downstream clinical development, respectively.
The basic scientific complexities of discovery and preclinical development of new antibacterial agents must not be deemphasized, because they are considerable. However, this article focuses on macro policy issues related to clinical development and marketing of antibacterial agents. Scientific issues regarding new antibacterial discovery and preclinical development were the focus of other outstanding presentations at the Institute of Medicine’s April 2010 Forum on Microbial Threats workshop on antimicrobial resistance.
It may be politically unpalatable to pass economic legislation that results in incentives to pharmaceutical companies developing new antibacterial agents.
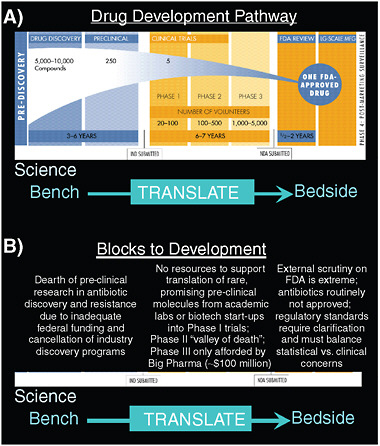
FIGURE A18-3 Schema of the drug development process. (A) The drug approval process takes 10–15 years, during which time 5,000 to 10,000 lead compounds are whittled down to approximately 5 to 10 that make it through preclinical development to enter clinical trials, and on average 1 will receive Food and Drug Administration
Nevertheless, antibacterial agents are a critical public health instrument. Their loss due to the perpetual spread and expansion of antibacterial resistance, and absence of new drug development, would result in catastrophic reversals in advances in modern medicine. Antibacterial resistance will never stop developing. Resistance is the inevitable result of the fundamental and almost incalculable power of microbes to adapt to their environment (Spellberg, 2009). In 2000, Nobel Laureate Dr. Joshua Lederberg wrote that “the future of humanity and microbes will likely evolve as … episodes of our wits versus their genes” (Lederberg, 2000). The extraordinary reality is that in the 10 years since Dr. Lederberg wrote those words, while microbial genes have continued to evolve and adapt, creating ever-increasing resistance, we have stopped using our “wits” to
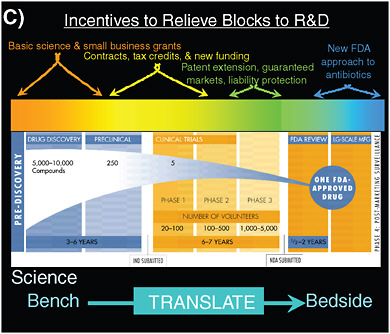
(FDA) approval. (B) Blocks to approval include the lack of preclinical discovery and development due to the exiting of pharmaceutical companies from the field and the inability of academic scientists to get basic science grant funding to do this type of work, inadequate capital to translate rare lead compound discoveries into clinical trials and support development through the phase II “valley of death” and into phase III trials, and inability to get new antibacterial agents approved by the FDA. (C) A panoply of economic incentives are needed to remove the blocks to the research and development (R&D) process for new antibacterial agents.
SOURCE: Figure adapted from “Drug Discovery and Development: Understanding the R&D Process.” (PhRMA, 2007).
keep up with the microbes. Hence, we have fallen behind, and we will stay behind in our race to maintain effective antibacterial agents until we remove barriers to new discovery and development. Responsibly planned economic incentives are necessary. We have no means by which we can make companies discover and develop the antibacterial agents we need; rather we must make companies want to develop those antibacterial agents.
Some have suggested that, since antibacterial resistance is inevitable, we should simply stop bothering to try to develop new antibacterial agents, since they will ultimately become ineffective anyway. Instead, these critics suggest, we should focus on antibiotic stewardship, infection control, and new technologies, such as vaccines and bacteriophages. This is a false choice. Certainly it is true
that resistance is inevitable, and that microbes will demonstrate resistance to any antibacterial agents we develop. Microbes eventually will also become resistant to any bacteriophages we deploy and to any other immunotherapeutic strategy. Furthermore, we will never have 100 percent effective vaccines, and we will never be able to target every possible pathogen with vaccines. Strain and species replacement will also occur, such that infections prevented are replaced by new infections. In short, we definitively do need to have better antibacterial stewardship to slow the spread of resistance, better infection control practices and technologies to reduce the number of infections that occur, more and better vaccines to prevent infections, and immunotherapies as alternative strategies to treating infections. But no amount of these technologies will ever eliminate the need for effective antibacterial agents to treat patients who develop serious or life-threatening infections despite all of the above. Therefore, in addition to focusing on antibacterial stewardship, infection control, and alternative treatment and prevention strategies, we must also establish an infrastructure that can facilitate new antibacterial discovery, development, and modification on an ongoing basis in perpetuity.
Regulatory Obstructions to Antibacterial R&D
The Legacy of Telithromycin
For the last decade, the FDA, as well as the European Medicines Agency, has been reevaluating clinical trial standards that support New Drug Applications leading to approval to market drugs to the public. These changing regulatory standards are the result of (1) a greater understanding of scientific complexities underpinning study design (FDA, 2002; Fleming, 2008; Powers, 2008; Powers et al., 2002; Temple and Ellenberg, 2000) and (2) tremendous political, media, and public pressure applied to the regulatory agencies in the aftermath of highly publicized postmarketing drug failures, such as those seen with rofecoxib (Vioxx®) and, more directly relevant to antibacterials, telithromycin (Ketek®) (Ross, 2007; Shlaes and Moellering, 2008; Soreth et al., 2007).
In particular, telithromycin’s rare but fatal hepatotoxicity in the setting of an approval for treatment of non-life-threatening acute bacterial sinusitis has become a lightning rod for debate regarding the approval of antibacterial agents based on data from “noninferiority” clinical trials. This debate has been exacerbated by the discovery of fraud by site investigators (but not the sponsoring company) who participated in the clinical trial of telithromycin for acute bacterial sinusitis (Ross, 2007; Shlaes and Moellering, 2008; Soreth et al., 2007). Of course, telithromycin was also approved to treat community-acquired pneumonia, a life-threatening infection for which the drug’s efficacy has not been called into question, and where no apparent fraud or questions regarding clinical trial integrity were discovered. It is unfortunate that the approval process for antibacterial agents for life-threatening infections as a whole has broadly fallen victim to questions that
have been raised regarding the use of noninferiority studies to establish efficacy for (typically) non-life-threatening infections, such as acute bacterial sinusitis, acute exacerbations of chronic bronchitis, and acute bacterial otitis media. We must stop allowing the specific problems associated with the telithromycin story, which included a postmarketing safety concern and the unreliability of some of the clinical trial data supporting its use for treatment of sinusitis, to negatively impact our view of the general use of noninferiority clinical trials to determine the efficacy of needed new antibacterial agents for the treatment of serious and life-threatening infections.
The Noninferiority Problem
Complexities in interpreting efficacy results from noninferiority studies have been extensively written about by leading experts (FDA, 2002; Fleming, 2008; Powers, 2008; Temple and Ellenberg, 2000). In brief, a noninferiority study seeks to determine whether an experimental drug is similar in efficacy to a standard comparator drug. Neither the experimental drug nor the comparator drug is directly tested against placebo in the study. Therefore, if the experimental drug is found to be “noninferior” to the comparator drug, there are two possible statistical interpretations: (1) both drugs are superior to placebo for the disease under study, and the experimental drug should be approved by the regulatory agency, OR (2) neither drug is superior to placebo for the disease under study, and the reason why the drugs appear to have similar efficacy is that a similar placebo effect is seen in both arms. Approval of the experimental drug under the latter scenario would result in marketing of an ineffective drug to the public. Regulatory agencies have come to understand that they must carefully scrutinize noninferiority studies to prevent such an occurrence. These points are extensively discussed in guidance documents E9 and E10 from the International Conference on Harmonization (ICH) (EMEA, 1998; FDA, 2001), which serves as a policy basis for regulatory agencies in the United States, Japan, and Europe.
The simplest way for a regulatory agency to ensure that ineffective drugs are not approved as a result of successful noninferiority studies is to make certain that the comparator drug studied is known to be superior in efficacy to placebo. A simple logic flow, akin to the mathematical transitive principle, is the following:
-
If the comparator drug used in a noninferiority study is known to be superior in efficacy to placebo
AND
-
The experimental drug is similar in efficacy to the comparator drug in the noninferiority study
THEN
-
The experimental drug must also be superior in efficacy to placebo.
Hence, as discussed in ICH E9 and E10 guidances (EMEA, 1998; FDA, 2001), experimental drugs should be approved based on noninferiority clinical trials only when the comparator drug can be confidently known to be superior to placebo for the disease under study. The clearest way to know that a comparator drug is superior in efficacy to placebo is based on the availability of previous randomized, placebo-controlled trials relevant to the disease under study.
The desire for previous randomized placebo-controlled trials to support current noninferiority trials for a given disease is entirely logical and reasonable in this context. Unfortunately, it is also the fundamental underpinning for why antibacterial R&D, out of proportion to other drugs, has been so severely impacted by the current regulatory environment. Antibacterial agents were among the first effective drugs used in Western medicine (Thomas, 1983). The first antibacterial agent in the United States was sulfanilamide, which first became available in late 1936 (Northey, 1948). Subsequent sulfa derivatives followed in short order. Penicillin was first used in the United States in 1942 and became widely available post-World War II, in 1946 (Grossman, 2008; Herrell, 1943; Keefer et al., 1943; Lyons, 1943; Spellberg, 2009). Tetracyclines and aminoglycosides were introduced in the late 1940s and early 1950s. All of these agents became available prior to the general understanding and availability of randomized, placebo-controlled studies.
The first randomized, controlled trials were conducted in 1946 (Doll, 1998), already a decade into the antibacterial era. Their widespread adoption into general use took an additional decade. By the time such studies were considered standard for demonstrating efficacy of a medical intervention, most of the classes of antibacterial agents that are available today were already available. It was, therefore, quite sensibly deemed unethical to randomize patients with serious or life-threatening infections to a possibility of treatment with placebo, thereby depriving them of effective therapy.
Of course, in the absence of any data on the efficacy of antibacterial agents, placebo-controlled studies would not only be appropriate, they would be necessary. In 2007, members of the IDSA learned that lack of clarity on the efficacy of antibacterial agents for community-acquired pneumonia had led to considerations about whether placebo-controlled studies should be conducted to determine the magnitude of the benefit of these drugs. In response, the IDSA and FDA agreed to cosponsor a workshop on clinical trial design for community-acquired pneumonia. That 2-day workshop was held in January 2008 (Spellberg et al., 2008c). The possibility of placebo-controlled studies for community-acquired pneumonia was a serious consideration throughout the workshop; on the first day of the workshop alone, the possibility of placebo-controlled trials for community-acquired pneumonia was discussed on more than 20 separate occasions, including
repeated mention by advocates of the concept (FDA, 2008a). The seriousness of the possibility of placebo-controlled trials is underscored by the fact that, even 3 months after the workshop, the FDA specifically asked its Anti-Infectives Drug Advisory Committee to vote on whether such studies could or should be done (FDA, 2008b). That such serious discussions occurred about the use of placebo to study a disease that Osler referred to as “the Captain of the men of death” (Osler, 1901), because it was the leading killer of Americans at the turn of the 20th century, underscores how important it is to balance statistical concerns with a clinical, real-world perspective during consideration of noninferiority trials, and how easy it can be to “lose the forest through the trees.”
A critical component of the efforts preceding, during, and following the IDSA-FDA workshop on community-acquired pneumonia was to delve deeply into the historical literature from the 1920s through the 1950s to determine if any direct comparisons of antibacterial therapy with background medical care had ever been conducted for this disease. On the second day of the workshop, Dr. Singer from the FDA presented her summary of seven historical studies that compared the effect of antibiotics to no therapy in patients with community-acquired pneumonia (Singer et al., 2008). Within several months subsequent to the workshop, 4 additional studies were identified, for a total of 11 studies that compared antibiotics to no treatment for community-acquired pneumonia in adults (Spellberg et al., 2008a). Of these studies, six were historically controlled and five were concurrent controlled studies in which patients were alternated to antibacterial agents plus background medical care versus background medical care alone. In addition, several other studies were identified that exclusively evaluated and confirmed the efficacy of antibacterial agents versus background therapy for pediatric community-acquired pneumonia (Bradley and McCracken, 2008). Every one of these studies demonstrated marked reductions in mortality from pneumonia with antibiotics, and these historical studies—which time had forgotten—explain the absence of any subsequent randomized, placebo-controlled trials for typical community-acquired pneumonia (two randomized, placebo-controlled studies were actually discovered, both of which demonstrated substantial clinical benefit of tetracyclines for mild, Mycoplasma atypical pneumonia in young, healthy military recruits; Kingston et al., 1961; Smilack et al., 1974).
Adding credence to the historical data sets are recent studies confirming the substantial effectiveness of active antibiotic therapy in the setting of comparison of concordant versus discordant therapy (i.e., use of antibiotic against which the etiologic agent is resistant by in vitro testing; Davidson et al., 2002; Dylewski and Davidson, 2006; Endimiani et al., 2005; Ho et al., 2001; Iannini et al., 2007; Kelley et al., 2000; Lonks et al., 2002; Mandell et al., 2007; Musher et al., 2002; Peterson, 2006; Rzeszutek et al., 2004), delayed initiation of therapy versus more rapid initiation of therapy (Houck et al., 2004; Kahn et al., 1990; Meehan et al., 1997), and subtherapeutic exposure to an antibiotic either as a result of inadequate pharmacokinetic and pharmacodynamic (PK-PD) parameters (Ambrose,
2008; File and Schentag, 2008; File et al., 2007) or in vivo drug inactivation (Pertel et al., 2008; Silverman et al., 2005). In summary, four different approaches in the modern era generate results that are in concordance with historical data demonstrating that antibiotics are substantially more effective than no treatment for community-acquired pneumonia. As a result, the FDA Anti-Infective Advisory Committee voted unanimously in April 2008 that placebo-controlled studies of community-acquired pneumonia should not be conducted, and the FDA has clearly indicated the same opinion (FDA, 2009a).
Having avoided the conduct of placebo-controlled studies for community-acquired pneumonia, more recently a similar debate has raged regarding the efficacy of antibacterial agents for complicated skin infections. No randomized, placebo-controlled studies have ever been conducted for antibacterial agents for the treatment of complicated skin infections. Two of the few active controlled studies of antibacterial versus nonantibacterial therapy conducted for complicated skin infections were among the earliest studies of antibacterial agents ever done. In 1937, Snodgrass and Anderson alternated patients with “erysipelas,” which was the term in use at the time for what in the modern era is typically referred to as “cellulitis,” to receive sulfa antibiotics versus ultraviolet (UV) lamp therapy (Snodgrass and Anderson, 1937a, 1937b). UV lamp therapy had become the standard treatment for cellulitis by the 1930s because of its superior efficacy compared to topical ointments and creams (Lavender and Goldman, 1935; Spellberg et al., 2009; Titus, 1934; Ude, 1931). The relatively ineffective nature of the medical therapy available at the time for skin infections is further underscored by the use as a standard medical treatment for patients of (1) a liquid diet and (2) a mandatory liquid paraffin soap-and-water enema, the latter of which was administered on admission to the hospital and subsequently repeated “when necessary.” The relatively ineffective nature of UV lamp therapy is also underscored by the primary outcome measure in the studies, which was the percent of patients who had progressive lesions on subsequent days after initiation of therapy (i.e., percent of patients who got worse on therapy). Not surprisingly, sulfonamides significantly decreased the proportion of patients who had progressive skin lesions and who remained febrile on each subsequent day.
Furthermore, two reports of the annual mortality of cellulitis in the pre- and post-antibiotic era greatly elucidate the power of first sulfonamides and then penicillin therapy for skin infections. The mortality of cellulitis on an annual basis from 1929 to 1936 at Cook County Medical Center in Chicago was an astonishing 10 to 15 percent (Hoyne et al., 1939), which is perhaps 50-fold higher than in the modern era. As soon as sulfanilamide became available in 1937, the mortality rate immediately fell 4-fold (Hoyne et al., 1939). The same approximately half-log reduction in mortality was also seen the year sulfonamides became available in Norway, based on 85 years of data from a national death registry (Madsen, 1973). For the first 55 years, from 1880 through 1935, the mortality rate of cellulitis in Norway was unchanged. Immediately upon avail-
ability of sulfonamides, the mortality dropped by approximately the same 4-fold decline as was seen at Cook County Medical Center over the same period of time. Furthermore, when penicillin became available post-World War II, the mortality rate from cellulitis fell by another 10-fold below the new baseline established by sulfonamides, down to rates by the late 1940s, which are essentially unchanged even today (Madsen, 1973).
To add breadth to these data sets, we systematically reviewed 90 studies of cure and mortality rates of patients with cellulitis, wound infections, and complicated abscesses from 1900 through 1950 (Spellberg et al., 2009). Based on data from >23,000 patients, the overall mortality of cellulitis in the pre-antibiotic era was ~11 percent. That mortality rate was reduced to ~2 percent by sulfonamides, and to ~0.3 percent by penicillin. Penicillin improved the clinical cure rate (defined as alive with resolution of lesions by 28 days in the absence of septic complications or amputation) of cellulitis from 66 percent without antibiotics to 98 percent with penicillin. Penicillin also massively improved the clinical cure rate of infected wounds, from 36 percent without antibiotics to 83 percent with antibiotics. Finally, penicillin improved the cure rate of complicated abscesses (e.g., carbuncles) from 76 percent without antibiotics to 96 percent with antibiotics. Penicillin had to be administered parenterally for this effect to be seen, as the clinical cure rates of patients treated with topical penicillin were not significantly different from patients treated without antibacterial therapy. Collectively these data confirm the very large treatment benefit of antibacterial agents for various types of complicated skin infections.
The Debate Has Shifted from Placebo to What Endpoint to Use?
In the aftermath of the clear establishment of efficacy of antibacterial agents for community-acquired pneumonia and complicated skin infections, the debate has shifted to what the proper endpoints should be in noninferiority studies for these diseases. Since the majority of data establishing the efficacy of antibacterial therapy to placebo/no therapy demonstrated a survival benefit, some have advocated for mortality-only endpoints in clinical trials, particularly for pneumonia (FDA, 2009b). However, given an anticipated mortality rate of 2.5 to 5 percent in clinical trials of community-acquired pneumonia, to power a study based on mortality would require >5,000 patients. Two such studies would have to be conducted to support approval of a drug, meaning that 10,000 patients would have to be enrolled, at the cost of perhaps $500 million. The mortality rates are even lower in skin infection studies, so the sample sizes would have to be even higher. Such studies are not feasible to conduct and will never be conducted.
The IDSA and FDA have summarized the substantial evidence of clinical benefit of antibacterial agents for community-acquired pneumonia (Singer et al., 2008; Spellberg et al., 2008a). Antibacterial agents markedly shorten the duration of symptoms from pneumonia. Furthermore, as described above, there is a
very large clinical benefit of antibacterial agents for complicated skin infections. However, these clinical response differences narrow over time, as some patients not treated with antibacterial agents eventually do resolve their infections and become asymptomatic. The subsequent narrowing of the clinical response differences in historical data sets comparing patients treated with or without active antibacterial therapy has potentially profound implications for modern noninferiority clinical trials. Specifically, if significant narrowing occurred over time, such that the benefit of antibiotics is large at early time points but negligible at later time points, the implication would be that clinical response could not be used as an endpoint in a modern noninferiority study because antibiotics have no long-term clinical benefit. As an alternative compromise, if used as an endpoint, clinical response would have to be assessed at an early time point (i.e., day 2 or 3) while patients were still on treatment for community-acquired pneumonia or complicated skin infections.
Early clinical response analysis may seem a reasonable compromise at first glance; however, it will result in inaccurate adjudication of patients as a success or failure, because (1) patients who are improved sufficiently to be considered a success by 2 to 3 days after initiation of therapy may still progress, relapse, develop septic complications, or even die later during the course of therapy; and (2) patients who are clinically stable or partially improved by day 2 or 3 but not sufficiently so to be considered a success are likely to proceed to resolve their signs and symptoms of infection and survive the infection by the end of therapy and test-of-cure; such patients would be inaccurately adjudicated as a failure based on a day 2 to 3 analysis. Furthermore, acute bacterial infections must be viewed distinctly from chronic illnesses. When the infection resolves, the signs or symptoms should resolve, and baseline clinical status should be restored. Therefore, restoration of the patient’s baseline clinical status is thus the only acceptable goal of antibacterial therapy. To accept an alternative definition of clinical success is to compromise clinical validity and reality in the pursuit of a purist statistical goal that is not grounded in common sense or the reality of life that patients themselves experience. The more appropriate approach is to define the most clinically relevant endpoint and then use the optimal statistical method available to analyze that endpoint.
Three critical points must be emphasized when interpreting historical data comparing clinical response between patients treated with or without antibacterial therapy. First, and foremost, the historical clinical response curves of patients treated with and without antibacterial agents remain widely separated even at later time points. Dr. Singer from the FDA presented data on clinical response of patients treated with sulfa drugs or background therapy at the December 2009 Anti-Infective Drug Advisory Committee (Figure A18-4A) (FDA, 2009b). The maximal separation of the point estimates of clinical response rates between patients treated with sulfonamides versus background medical therapy occurred
on days 3 to 4, with at least a 70 percent absolute difference (i.e., >70 to 80 percent by days 3 to 4 with sulfonamide therapy, and <5 percent without antibacterial therapy). Indeed, the magnitude of this difference narrowed somewhat over the subsequent several days such that, by day 7, >90 percent of patients had clinical response with sulfonamide therapy, versus ~40 percent without antibacterial therapy. However, the smallest estimate of the difference (i.e., the lower bound of the 95 percent confidence interval [CI]) in the clinical response rates between the antibacterial and background therapy groups remained >40 percent at day 7 (Figure A18-4B).
Second, the control curve for this analysis is taken from a large cohort of patients with S. pneumoniae pneumonia treated in the pre-antibiotic era, “all of whom recovered without observed purulent complications” (Bullowa, 1937). That is, dead patients and patients who had purulent complications (e.g., empyema, endocarditis, pericarditis, meningitis, deep tissue abscesses, septic joints, glomerulonephritis) were excluded from the cohort, and only those patients who eventually recovered without any purulent complications were analyzed. Therefore, the clinical response of the control group is artificially high, because those who clinically failed were explicitly excluded (Bullowa, 1937). The difference in clinical response between patients treated with and without antibacterial agents is therefore artificially low, especially more so at later days, as more dead patients and patients with “purulent complications” in the control group are cumulatively excluded from the analysis.
Third, for both community-acquired pneumonia and complicated skin infections, the estimates of converging clinical response over time are based on comparison of sulfonamide antibacterial therapy, not penicillin therapy, to background medical care (Flippin et al., 1939; Meakins and Hanson, 1939; Snodgrass and Anderson, 1937a, 1937b). Yet penicillin therapy was substantially more effective than sulfonamide therapy for both diseases (Austrian and Gold, 1964; Dowling and Lepper, 1951; Finland, 1943; Kassowitz and Muscato, 1952; Madsen, 1973; Spellberg et al., 2008a, 2009). Hence, estimates of clinical response derived from comparisons of sulfa therapy versus no antibacterial therapy are dramatic underestimates of modern antibacterial effectiveness versus no antibacterial therapy. Sulfonamide monotherapy was made largely irrelevant by the advent of β-lactam antibiotics and was formally made obsolete in the late 1960s with the advent of combination sulfonamide plus dihydrofolate reductase inhibitors (e.g., trimethoprim). Understanding of the magnitude of efficacy of modern antibacterial therapy should not be based on the efficacy of a class of drugs that was made obsolete more than half a century ago. This difference is of particular concern for skin infections, where it is known that the mortality of cellulitis was ~10-fold lower with penicillin than with sulfonamides (Madsen, 1973; Spellberg et al., 2009).
As for community-acquired pneumonia, the belief that clinical response curves converge for skin infections between days 3 and 7 of therapy is prob-
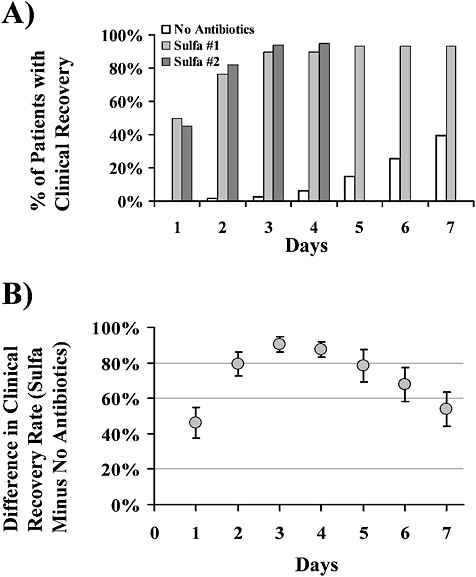
FIGURE A18-4 Improvement in clinical response in patients with community-acquired bacterial pneumonia treated with sulfonamide antibacterial agents versus with standard background medical therapy without antibacterial agents. (A) Percent of clinically responding patients by day post-presentation to the hospital in three cohort studies from 1937 (no antibiotics; Bullowa, 1937) and 1939 (sulfa #1 and 2; Flippin et al., 1939; Meakins and Hanson, 1939). (B) Point estimates (open circles) and 95 percent CI (error bars) of the difference in clinical response rates between patients treated with sulfonamide versus no antibacterial therapy by day.
lematic. The only data sets supporting this concept are the prontosil rubrum or sulfanilamide versus UV lamp therapy studies of Snodgrass and Anderson from 1937 (Snodgrass and Anderson, 1937a, 1937b). Again, the comparison in these studies was between sulfa drugs and no antibacterial therapy, rather than penicillin versus no antibacterial therapy, despite the fact that penicillin was clearly massively superior in efficacy to sulfonamide therapy (Madsen, 1973; Spellberg et al., 2009). Furthermore, the analysis of clinical response was based on the percentage of patients whose skin lesions grew larger on each subsequent day of therapy, not an analysis of resolution of the skin lesions. It is not possible from the data available in the Snodgrass and Anderson studies to determine what proportion of patients in either arm had resolved their skin lesions on any day of therapy, which is the only relevant endpoint in the era of effective antibacterial therapy. A modern drug whose efficacy resulted in stabilization of lesion size by day 3 of therapy would be considered a failed drug, which is unacceptably inferior to the efficacy of other drugs on the market for the treatment of skin infections.
Finally, again the analysis excluded patients who died, and only evaluated survivors. This exclusion of dead patients is extremely important when one accepts that the proper analysis should compare parenteral penicillin, rather than sulfonamides, to no antibacterial therapy, because the mortality rate of patients with cellulitis treated with penicillin was 10-fold lower than patients treated with sulfonamides, and 50-fold lower than patients treated with no antibacterial therapy (Madsen, 1973; Spellberg et al., 2009).
A critical concept to emphasize is that the effect size of antibacterial agents at improving a composite endpoint of alive and clinical response must, by definition, be at least as large as the mortality benefit of the antibacterial agents—dead patients cannot clinically improve, so if there are substantially more dead people in the control (no-antibacterial-therapy) group, there must by definition be substantially fewer treatment successes in that group. Therefore, if one has a reliable estimate of the mortality benefit of antibacterial agents, that estimate can be used as an estimate of the smallest possible composite benefit (alive and clinical responded) of the antibacterial agents, and that estimate can therefore be used to support the use of a composite modern endpoint of alive and clinically responded in a noninferiority study.
To reiterate a point made above, if there is a 97 percent relative reduction, and a 10 percent absolute reduction, in death of patients with cellulitis treated with penicillin versus no antibacterial therapy, then it is impossible for clinical response curves to converge between patients treated with penicillin and no antibacterial therapy—if there are 10 percent more patients dead without antibacterial therapy, there must be at least 10 percent fewer patients cured at end of therapy or test of cure. These facts are reflected in the IDSA analysis of clinical cure for complicated skin infections, where (1) the endpoint was based on objective outcomes, such as resolution of lesions, by day 28 (approximately
the same as the test-of-cure evaluation in modern studies), absence of septic complications, and survival; and (2) the comparison was between parenteral penicillin and no antibacterial therapy (Spellberg, 2009). The IDSA analysis demonstrates a much larger treatment effect for clinical response than has been gleaned from the Snodgrass and Anderson sulfa versus UV lamp therapy studies from 1937. Clinical response curves may narrow somewhat between days 3 and 7 in patients treated with or without active antibacterial therapy, but the difference at day 7 remains so substantial as to enable justification of a noninferiority clinical trial using a composite endpoint of alive and resolved signs and symptoms of infection at end of therapy or test of cure (the advantage of which is capturing relapses).
The dialogue on clinical trial design for community-acquired pneumonia began in 2007. In January of 2008, there was a joint IDSA-FDA workshop on this topic. In April 2008, these issues were formally discussed and voted on at an FDA Advisory Committee meeting. The IDSA subsequently published its societal position paper on this topic (Spellberg et al., 2008a). In early 2009, the FDA released a draft guidance on this topic, which was a remarkable document that cut an insightful balance between an appropriate, substantial increase in regulatory stringency and requirements for the conduct of these studies while still enabling rational, clinical endpoints and feasible study designs. Unfortunately, that guidance document was subsequently withdrawn in the face of statistical criticism. As a result of that criticism, the same data that had been discussed at the April 2008 FDA Advisory Committee were discussed anew in December 2009. As of May 2010, almost 3 years after this discussion began, no guidance on clinical trial designs for community-acquired pneumonia is available.
Meanwhile, only 1 new systemic antibacterial agent has been approved by the FDA in the past 2.5 years, while during the same period of time 6 other anti-bacterial agents have come up before the FDA and been rejected, including the first new oral, broad-spectrum agent for Gram-negative rods since ciprofloxacin was introduced nearly 3 decades ago (faropenem), and 5 drugs with methicillin-resistant Staphylococcus aureus (MRSA) activity (ceftibiprole, oritavancin, dalbavancin, cethromycin, and iclaprim). In the aftermath of these rejections, the lack of clear guidance on how clinical trials should be conducted to FDA’s satisfaction, and a clear sense that such trials will have to be significantly larger and more expensive in the future than ever before, have greatly exacerbated the risk of failure of antibacterial development programs.
The Other Debate: How to Select a Noninferiority Margin?
The ICH E9 and E10 guidance documents (EMEA, 1998; FDA, 2001), and a recent FDA guidance on noninferiority clinical trials (FDA, 2010), describe the
process by which the noninferiority margin (M2) can be selected after knowing the historical effect size of the comparator regimen versus placebo/no therapy (M1). The operating principles are that the noninferiority margin (M2) selected for a clinical trial must (1) be smaller than the historical effect size of the comparator versus placebo/no therapy (M1) and (2) in addition to being smaller than M1, M2 must also preserve a clinically meaningful fraction of M1. In practice, it has commonly been suggested to set M2 so that it is half of M1, preserving 50 percent of the effect size of the comparator drug (Figure A18-5A). More recently, some have begun adding an additional “discount” step, in which the historical effect size (M1) is first cut in half to account for methodological limitations in the data resulting in the calculation of M1. After that discount step, a further 50 percent reduction is applied to “preserve” a clinically meaningful fraction of the 50 percent discounted M1.
These approaches have the danger of being arbitrary and overly conservative, especially in the context of the original estimate of M1 efficacy being generated by the “95/95” method, in which the lower bound of the 95 percent CI of efficacy of the comparator drug is compared to the upper bound of the 95 percent CI of efficacy of placebo/no therapy. The FDA has acknowledged that the 95/95 method is intrinsically highly conservative (FDA, 2010). There is no specific logic or rationale for applying a further 50 percent discount to an M1 calculated based on the 95/95 method, or for requiring preservation of 50 percent of M1 when setting M2, aside from statistical hyperconservatism and the creation of the illusion of mathematical precision. The resulting calculation may appear mathematically precise, but in reality it is merely highly conservative and arbitrary based on a subjective and non-evidenced-based selection of how much to discount M1 to account for methodological issues in calculating M1 and then how much to further reduce the discounted M1 in setting an M2.
It generally has been agreed by clinicians and multiple medical societies that an M2 for mortality should never be larger than 10 percent, because it is never acceptable to approve a drug that results in as much as a 10 percent excess fatality rate than a comparator drug. Therefore, if the mortality treatment effect size (M1) of a comparator drug versus placebo/no therapy is substantially larger than 10 percent, the M2 margin should be 10 percent for mortality. We and others have suggested that a 15 to 20 percent margin may be acceptable for clinical endpoints (nonmortality endpoints), particularly if the experimental drug offers specific advantages over available therapy, such as superior safety, dosing considerations, or, for antibacterial agents, activity against antibiotic-resistant bacteria (FDA, 2002, 2010; Spellberg et al., 2008a, 2009).
One concern raised about setting margins of ≥10 percent is that such a margin appears to suggest that society is willing to approve a drug that is 10 to 15 percent less effective than the comparator drug. Substantial consternation has been expressed in public settings about the possibility that regulatory standards would
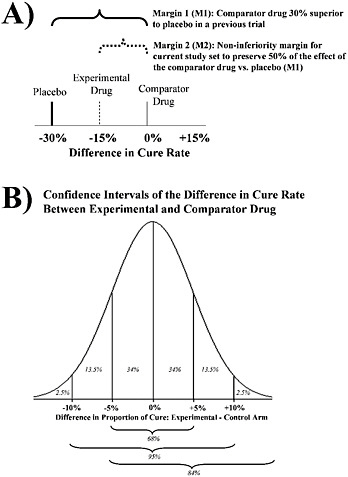
FIGURE A18-5 Determination of noninferiority margins. (A) The M1 margin is the difference in efficacy between the comparator drug versus placebo based on previous studies. The M2 margin is the preplanned noninferiority margin for the current study and is often arbitrarily set to preserve 50 percent of the effect of the comparator drug (i.e., it is set to be 50 percent of the magnitude of M1). (B) Probability distribution of the 95 percent confidence interval (CI) of the difference in efficacy between the experimental and comparator drug from a noninferiority trial. In this example, the point estimate of the difference in efficacy is 0 percent, with a 95 percent CI of −10 percent to +10 percent. The proportion of the overall area under the curve found within each section of the distribution is indicated by italicized percentages within the graph, and the cumulative proportion of the area under the curve is shown below the graph. The critical point is that the true difference in efficacy between the experimental and comparator drug is not equally likely to exist anywhere along the distribution. Rather the true difference is much more likely to exist closer to the point estimate of the difference than at the fringes of the 95 percent CI distribution. In the example shown, there is an 84 percent chance that the experimental drug is no worse than 5 percent less effective than the comparator drug, even though the lower bound of the 95 percent CI of the difference in efficacy of the drugs is −10 percent
be satisfied with approving drugs which are so substantially inferior in efficacy. However, enabling use of a 10 percent (or 15 percent) noninferiority margin in a pivotal phase III clinical trial does not mean that the agency will approve a drug that is 10 percent (or 15 percent) worse than the comparator drug.
The key to interpreting such results is to understand that the difference in efficacy between the two drugs is calculated by generating a 95 percent CI around the point estimate of the difference in efficacy (Figure A18-5B). In this context, the 95 percent CI is a normally distributed probability distribution. Hence the true difference in efficacy between the 2 drugs is 95 percent likely to exist somewhere within the CI distribution. However, the true difference is not equally likely to exist at any point along the CI distribution. Because the CI is normally distributed, the most likely true difference is located close to the center of the CI; the true difference is far less likely to be located at the tails of the CI. This concept can be easily understood visually: in a normal distribution, the central peak is much larger than the tail ends (Figure A18-5B). Indeed, for a 95 percent CI going from +10 percent to −10 percent, there is a 68 percent chance that the true difference in drug efficacy lies within ±5 percent. There is only a 16 percent chance that the true difference in efficacy is ≤−5 percent. This means that for a drug with a point estimate difference versus the comparator of 0 percent with a 95 percent CI of +10 to −10 percent, there is an 84 percent chance that the drug is no more than 5 percent worse than the comparator, and a 50 percent chance that the drug is actually more effective than the comparator.
Furthermore, the FDA often requires that two trials be conducted to support approval of a new drug. Ostensibly the purpose of requiring conduct of two trials is to ensure that the results of the trials are reproducible. Operationally, companies simply run two concurrent trials with identical, or nearly identical, protocols enrolling at different sites. Therefore, beyond the benefit of replication, conduct of two trials enables a pooled analysis to increase sample size and hence increase statistical power. If both trials meet a prespecified −15 percent non-inferiority magin, a pooled analysis of the two trials may well meet a −10 percent noninferiority margin. Requiring that the experimental drug meets noninferiority at a −15 percen margin for both of 2 separate trials, as well as a −10 percent magin for a pooled analysis, rationally can be incorporated into a formal development program to provide additional reassurance to the regulatory agency that an experimental drug is not unacceptably worse than the comparator drug.
Setting a −10 percent noninferiority magin as the maximum difference acceptable for an experimental drug means that one is willing to accept a very small chance that the experimental drug may be 10 percent less effective than the comparator drug, knowing that there is a far greater chance that the drug is much closer in efficacy (or even superior in efficacy) to the comparator drug. But why take any chance that the approved drug could be inferior in efficacy to the comparator drug? As the noninferiority margin shrinks, the required study sample size—and hence study cost and required time to complete enrollment—markedly
increases. If new antibacterial agents are critically needed, it becomes necessary to balance the feasibility of conducting studies against narrowing the desired non-inferiority margin. Patients may be harmed if ineffective drugs reach the market. Patients also may be harmed if they have an infection against which antibacterial agents are ineffective because new drugs are not developed. The key is to create a regulatory path that balances these equally concerning risks.
It is also critical to emphasize that neither the FDA Advisory Committee nor the FDA is obligated in any way to approve a drug simply because it meets its prespecific noninferiority margin in its pivotal study. For example, a drug with a point estimate of −5 percent and a 95 percent CI of −1 percent to −9 percent will have been established to be inferior in efficacy to the comparator regimen, and such a drug is likely not to be approved despite the fact that it met its preplanned non-inferiority margin of −10 percent. Rathe, the FDA considers the totality of the evidence of safety and efficacy when deciding whether or not to approve a drug.
Noninferiority Studies: Where Are We Now?
There is no question that the available data demonstrating antibacterial efficacy are statistically imperfect. They are based on historically controlled as well as rudimentary concurrent controlled studies, the latter of which did not use true randomization and did not employ placebos. Furthermore, the mortality data are more robust than the clinical cure data, and the clinical cure data for skin infections in particular are based on the calculation of weighted averages across single armed cohort studies rather than comparative studies. There is evidence of significant heterogeneity in these reviewed studies, making formal meta-analytical techniques problematic. Yet, despite all of this, the evidence of a large antibacterial treatment effect is overwhelming and is made robust by a concordance of similar effect sizes seen by a number of different types of analyses and comparisons. The effect is confirmed by global analysis of the marked and immediate decline in mortality rates from infections using national death statistics during the first 15 years of the antibacterial era and the plateau effect after that, with minimal further reductions in infectious death despite all subsequent advances in medical therapy, including the invention of modern critical care (Armstrong et al., 1999; Kassowitz and Muscato, 1952).
To quibble over the precise effect size of antibacterial agents, and as a result to refuse to accept clinically relevant, feasible endpoints for modern clinical trials of antibacterial therapy in the face of overwhelming evidence of a large treatment effect, is akin to arguing that we cannot do noninferiority studies of parachutes for prevention of gravitational challenge injury (i.e., jumping out of an airplane) (Smith and Pell, 2003). It is akin to demanding that placebo-controlled studies be done for parachutes in this setting, or failing the feasibility of placebo-controlled studies, demanding that modern studies which
compare one parachute brand versus the other use a mortality endpoint as its primary efficacy measure, despite the miniscule mortality rate that occurs when an effective parachute is used.
The endpoint for modern noninferiority studies of antibacterial agents for serious or life-threatening infections must be a composite of alive and resolution of the specific signs and symptoms which are attributable to the infection. This endpoint should be analyzed at the end of therapy or test of cure, not early during the therapy, where adjudication of clinical success cannot be made absent the further information that occurs during the remainder of the patient’s treatment. Sufficient data are available to justify these endpoints if one accepts that statistical perfection cannot be achieved due to the historical accident of the introduction of antibacterial therapy prior to the availability of randomized, placebo-controlled studies.
Superiority Studies
One alternative perspective is to simply say that noninferiority studies should no longer be conducted for antibacterial agents for serious or life-threatening infections. After all, we want better drugs, not merely “me too” drugs. So why not demand that new drugs that are approved be shown better than current drugs, especially for the treatment of resistant bacteria, which is where our specific societal need is for better treatments?
The patients in whom an experimental antibacterial agent is most likely to achieve superiority to a standard comparator agent are those infected with bacteria resistant to the comparator agent. Ironically, however, these are the same patients who are excluded from enrollment in the study (Rex, 2010). One cannot ethically randomize patients with a serious or life-threatening infection caused by bacteria resistant to an antibiotic to a chance of being treated with that antibiotic, as this would deprive the patient access to alternative effective therapy. Hence, even extremely effective drugs with efficacy against antibiotic-resistant bacteria are unlikely to achieve superiority to a comparator drug, because patients infected with bacteria resistant to the comparator drug cannot be studied in the trial. Noninferiority studies are necessary to enable such new drugs to reach the market, where they can then be used to treat infections caused by antibiotic-resistant bacteria.
One exception to this superiority problem is infections caused by bacteria resistant to all available antibacterial agents (pandrug resistant, or PDR) or those that are extensively drug resistant (XDR; resistant to everything except one or two relatively ineffective antibacterial agents). If the bacteria causing the infection are PDR or XDR, patients ethically can be randomized to relatively or completely ineffective therapy because no alternative, more effective therapy is available. In this setting, superiority studies can be done, and should be done, as emphasized
by the IDSA and several critical care medicine societies (Spellberg and Talbot, 2010).
Our greatest public health need is for development of antibacterial agents with efficacy against PDR or XDR bacteria. Hence, clinical development programs culminating in phase III pivotal superiority studies for infections caused by these bacteria should be encouraged. Yet, current economic and regulatory forces discourage these studies. Specifically, such studies will include in the primary efficacy analysis only infections caused by XDR or PDR Gram-negative bacilli, which will mean screening and initially enrolling many patients for every patient who ends up being evaluable. It will require many sites to capture such patients. Such studies will take a long time to complete and will be very expensive. Furthermore, the resulting study will support an indication only for the treatment of infections caused by XDR or PDR Gram-negative bacilli infections, which is a relatively small market. Hence, the return-on-investment problem will be even greater for companies developing a drug to treat XDR or PDR infections. Specific steps must be taken to encourage these critically needed studies, as elaborated below.
Changes to Encourage Critically Needed New Antibacterial Agents for XDR or PDR Infections
Economic Changes
The relatively small market captured by infections caused by XDR or PDR bacteria will prevent many companies from being interested in developing anti-bacterial agents targeting these pathogens. Economic incentives, as already discussed, are critically needed and should target antibacterial agents with activity against these organisms. No other example more clearly illustrates the critical need for such incentives than the specific societal need for antibacterial agents to treat XDR or PDR bacteria, contrasted with the enormous economic barriers to conducting such clinical development programs. In short, if society wants these drugs, it must act with urgency and determination to fix the economic disincentives, because such drugs are unlikely to be developed under the current economic climate.
Organism-Specific Studies
Historically, the FDA has granted indications to antibacterial agents for the treatment of specific diseases, for example, complicated skin and skin structure infections, community-acquired pneumonia, nosocomial pneumonia, intra-abdominal infections, and so on. Indications have not been granted for the treatment of specific bacteria, such as for the treatment of “antibiotic-resistant Acinetobacter,” MRSA, and so on. In contrast, the FDA has granted indications
to newly approved antifungal agents for the treatment of “invasive candidiasis” and “invasive aspergillosis” rather than specific sites of infection caused by these organisms. This dichotomy between the approval process for antifungal agents and antibacterial agents must be changed.
If companies could enroll a variety of types of infections in clinical trials focusing on XDR or PDR pathogens, it would greatly facilitate the rapidity of capture of evaluable patients, shortening enrollment, speeding conduct of the studies, and making the trials less expensive. Furthermore, the indication granted to the drug would be for the treatment of all of the disease entities studied, expanding the market for the approved drugs, thereby improving the return on investment for the drug. Note, for example, the difference in resulting market size if the approval of a drug is for “the treatment of susceptible Gram-negative bacilli, including XDR/PDR pathogens, causing nosocomial pneumonia, intra-abdominal infection, bacteremia, and meningitis” versus if the approval is for “the treatment of nosocomial pneumonia caused by XDR/PDR pathogens.”
There are critical complexities that have thus far prevented regulatory agencies from approving an antibacterial agent in this manner. However, these complexities are all solvable. The primary complexity is the need to include in the pivotal study only diseases with relatively similar severity, including risk of mortality. For example, the FDA separately approves antifungal agents for the treatment of esophageal candidiasis, which is a non-life-threatening infection, and for the treatment of invasive candidiasis, which is life threatening. Similarly, the agency approves antifungal agents for the treatment of allergic bronchopulmonary aspergillosis (non-life threatening) separately from approval for the treatment of invasive aspergillosis (life threatening). Other non-life-threatening infections, such as pulmonary aspergillomas, are also not included in studies supporting an invasive aspergillosis indication. The parallel procedure for XDR/PDR Gram-negative bacilli infections would simply mean that non-life-threatening infections, such as lower urinary tract infections, superficial wound infections, and so on, would not be included in studies of life-threatening infections, which could instead include bacteremia, meningitis, pneumonia, intra-abdominal infections, deep wound infections (e.g., mediastinitis), and so on.
A second complexity is the need to establish that the drug achieves penetration into the relevant target tissue. For example, drugs that do not penetrate into the central nervous system should not be studied for—or given an indication for—the treatment of meningitis, and drugs with poor pulmonary penetration must not be studied for—or given an indication for—treatment of pneumonia, and so on (unless the drug’s mechanism of action is independent of directly killing bacteria at the site of infection). Fortunately, phase III clinical trials are never planned in a vacuum. Rather, the sponsor must provide a thorough and broad array of “enabling data” (Spellberg and Talbot, 2010; Talbot, 2010) to support the clinical development program. In vitro and in vivo preclinical PK-PD data must confirm that the experimental agent has activity and achieves adequate levels to
treat infections in target tissues. The animal models used must be relevant to the planned clinical program (more on this below). Phase I and II clinical studies should confirm that in humans adequate PK-PD is achieved in patients with the target infections planned for study in the phase III trial.
The failure of tigecycline to achieve noninferiority in its pivotal phase III trial for nosocomial pneumonia is just one recent example illustrating the critical nature of such enabling data. It was discovered after completion of the phase III trial that tigecycline serum levels were as predicted in the enrolled subpopulation of patients with hospital-acquired pneumonia, for which the drug was indeed found to be noninferior to the comparator regiment. However, unexpectedly (because such levels had not been previously determined), the drug’s serum levels were 2-fold lower in patients with ventilator-associated pneumonia, apparently due to increased clearance of the drug in critically ill patients (Ambrose, 2010). As a result, the drug was dosed inadequately in patients with ventilator-associated pneumonia, was inferior in efficacy in that population, and, as a result, was inferior in efficacy to the comparator regimen in the study as a whole. Determination of PK-PD in relevant patient populations prior to conduct of the pivotal study is critically important for planned organism-specific studies.
A third complexity illustrates the depth required for enabling data. Beyond understanding PK-PD, it must be clear that the experimental drug is active as an antibacterial agent at the site of infection. Daptomycin was known to be highly active against Streptococcus pneumoniae, had been shown effective in a guinea pig model of necrotizing pneumonia, and was known to penetrate into the lung. It was logically assumed to be an excellent candidate for the treatment of community-acquired pneumonia. However, the drug was found to be inferior to the comparator regiment in its pivotal phase III studies of community-acquired pneumonia (Pertel et al., 2008; Silverman et al., 2005). The sponsor subsequently studied the drug’s efficacy in a murine model of alveolar pneumonia and found that pulmonary surfactant partially inactivates daptomycin (Silverman et al., 2005). The original guinea pig model had not accurately recapitulated the alveolar nature of community-acquired pneumonia in humans.
A fourth complexity of organism-specific studies deals with how to handle enrollment and evaluation of the primary efficacy population. Should the study only enroll patients after they are confirmed to have the target organisms? Or should the study enroll “all comers” with the target diseases and then exclude patients from the primary efficacy analysis after it is determined that they do not have infections caused by the target organisms? Both approaches are feasible. Enrolling patients only after confirmation that a target pathogen is causing the infection would be more economical. However, given the typical time delay in culture or antigen confirmation of target bacteria, this approach will almost certainly result in antecedent antibacterial therapy being administered prior to patient enrollment. If that therapy was active against the target pathogens, it would greatly
complicate analysis of the efficacy of the study drug and would not be considered acceptable by regulatory agencies. Nevertheless, if only XDR/PDR pathogens are being evaluated, the probability that initial therapy was active might be sufficiently low that regulatory agencies would accept the lack of impact of the previous therapy; sponsors might then prefer this approach for cost reasons.
As mentioned, the alternative approach is to enroll all patients with the target disease and then exclude patients from the primary efficacy analysis population if they are found not to have the target bacteria. This approach will require enrolling many patients for every one that is evaluable, resulting in a more expensive study. However, the study would be far cleaner since no antecedent antibacterial therapy would be administered. Furthermore, when targeting bacteria that are XDR/PDR, enrolled patients determined to have XDR/PDR bacterial infections could be analyzed in a superiority study, while those patients found to have susceptible bacteria could be shunted into a parallel-enrolling noninferiority study. In essence, two distinct trials could be simultaneously operated from the same initial enrollment criteria. This approach would generate a substantial safety database while supporting a superiority clinical trial.
All of the regulatory barriers to organism-specific studies are addressable. We must change our focus and begin allowing study of and approval of antibacterial agents for specific, targeted bacteria. Approving antibacterial agents for the treatment of specific diseases creates perverse marketing and use incentives that are antithetical to effective stewardship of new drugs. By law, companies may only market their drugs for the specific indications for which the drugs are approved. Furthermore, the drugs can only be approved for the specific diseases studied in their pivotal trials. Currently there is no societal need for new antibacterial agents for the treatment of skin infections or community-acquired pneumonia, for which numerous treatment options are available (although this is likely to change in the future, as resistance to current drugs spreads). Yet in past years skin infections and community-acquired pneumonia always have been the first indications sought by companies for their antibacterial agents because historically such studies have been feasible to successfully complete. The nature of the regulatory approval process has encouraged conduct of such studies, and the law has required companies to market drugs for these indications based on completion of such studies.
Tigecycline is an example of the unfortunate, perverse impact of this process of drug approval and marketing. Tigecycline is active against resistant organisms ranging from vancomycin-resistant Enterococcus to highly resistant Gram-negative bacilli, including extended-spectrum β-lactamase-producing Gram-negative organisms, KPC-producing Klebsiella pneumoniae which are resistant to all other antibiotics, and XDR Acinetobacter. To use tigecycline for the treatment of skin infections and community-acquired pneumonia, caused primarily by streptococci and staphylococci, is akin to dropping an atom bomb on an ant. Yet, the vast majority of use of tigecycline in communities is for skin infections and community-
acquired pneumonia, because those are the diseases for which the drug has been approved and hence, by law, for which the company can market the drug. In essence, our regulatory processes and laws inadvertently but implicitly encourage overuse of antibacterial agents where they are not needed, and prevent specific marketing of the drugs where they are needed.
Recently the FDA has made it clear that they are willing to consider approvals for antibacterial agents by organism rather than by disease. The onus is now on both the FDA and sponsors to openly discuss such clinical development programs. The onus is also on the federal government to create economic incentives that can fix the return-on-investment calculation so that sponsors are motivated to conduct such studies and are not deterred from doing so by the relatively small markets that highly resistant infections represent.
Nonprofit Antibacterial Development
A final, outside-the-box approach to developing new antibacterial agents is to provide an alternative option to the for-profit motive. One must be realistic when considering this possibility, since 100 percent of all antibacterial agents in clinical use have been developed by for-profit companies (primarily large companies). Therefore, to establish a nonprofit mechanism to develop new antibacterial agents is to establish a completely new mechanism with no previous track record. One should not view this possibility as replacing the need for economic incentives to stimulate for-profit companies to reenter the antibacterial market. Rather, the nonprofit possibility should be considered a complementary approach.
The primary advantage of a nonprofit entity in developing antibacterial agents is the removal of the disincentive of poor economic return on investment. A nonprofit entity whose mission was to develop new antibacterial agents would be able to focus its development programs on critically needed new antibacterial agents from a societal perspective, irrespective of market size. Novel, cutting-edge development programs could be undertaken to develop drugs for XDR-PDR pathogens even though the market size for those drugs would be relatively small. Furthermore, the nonprofit entity would be content to limit marketing of the drugs postapproval only for critical, societally needed indications, rather than marketing it for the best selling indications. Hence, establishment of a nonprofit entity to develop new antibacterial agents would converge the critical societal need for antibacterial stewardship with the critical societal need for new antibacterial drug development.
A nonprofit entity could very quickly be constructed by tapping into resources existing in pharmaceutical companies. The entity should be considered a public-private partnership, funded in part by both government and possibly donations from industry (more on this below). It should operate as an independent entity, governed by a board of directors and operated by a standard corporate officer structure. Directors should include representatives from government policy agen-
cies (e.g., the Department of Health and Human Services), government scientific agencies (e.g., the National Institutes of Health), academic expert scientists, philanthropy, and the pharmaceutical and biotechnology industry. Representatives from industry would be absolutely essential. Such representatives would be in a position to facilitate bidirectional technology transfer: (1) transferring from industry to the nonprofit entity promising lead compounds which represent too small an eventual market size to be of interest to the source company and (2) later-stage partnership or outlicensures to pharmaceutical companies of compounds developed by the nonprofit entity through phase II trials, in order to conduct the more expensive phase III trials. Furthermore, tapping into the substantial scientific and development expertise in industry would be essential to the success of the nonprofit organization.
By far the largest barrier to establishment of such a nonprofit entity is how to fund it. The capital required would be enormous to facilitate antibacterial approval and would be in the range of hundreds of millions to billions of dollars. Furthermore, a one-time appropriation of funds would likely not be sufficient. There would likely need to be a recurring influx of capital, particularly for the first 5 to 10 years, before the entity was able to generate a revenue stream of its own with which to reinvest in its R&D operation.
The most palatable, rational approach to funding such a nonprofit agency may well be to use the same process by which the highly successful VICP was established. As mentioned previously, the VICP is a government-operated insurance policy for vaccines, which was established due to the recognition that vaccines are a critical public health need, and that lawsuits over vaccine-related injuries were serving as a tremendous barrier to development of new vaccines. The VICP is funded by an extra fee charged during the sale of all vaccines in the United States. Similarly, if a specific “stewardship fee,” or “R&D fee,” was charged against the sale of antibacterial agents in the United States, such funds could be used as a renewable revenue stream to support new antibacterial R&D in a nonprofit entity. Additional mechanisms of funding could include government appropriations and solicited donations from industrial partners. An example of the latter could be that companies could purchase an ex officio position on the board of directors by donating a certain amount of money to fund the nonprofit entity. Similarly, companies could purchase rights to participate as a potential partner in future R&D of promising compounds.
The establishment of a nonprofit entity whose mission is to develop critically needed new antibacterial agents may be a highly promising mechanism to improve the long-term pipeline of such agents. The enormous public health benefit of this approach makes it worth undertaking despite the lack of a track record and the clear barriers that would have to be overcome.
The 10 × ’20 Initiative
In April of 2010, the IDSA formally launched a new campaign called “10 × ’20,” which calls for the development of 10 new antibacterial agents in the next 10 years (i.e., by 2020) (IDSA, 2010). These drugs should be safe and effective (as all drugs should be) and focused on the treatment of highly resistant organisms, currently exemplified by the ESKAPE pathogens (Enterococcus, Staphylococcus aureus, Klebsiella, Acinetobacter, Pseudomonas, and extended-spectrum β-lactamase-producing Gram-negative bacilli, such as Enterobacter and E. coli). This campaign is the result of recognition by the IDSA that society has reached a critical stage of antibacterial resistance and that new drugs are needed on an urgent basis. Successful completion of this campaign will require all levels of constituents to “buy in,” including the White House, Congress, the National Institutes of Health, academic scientists, industry, venture capital, philanthropy, and so on. Already critics have suggested that this call to action is unrealistic and not achievable. I believe those critics would better serve society by pitching in to help convince the required constituents to participate. As mentioned, six antibacterial agents with activity against the ESKAPE pathogens have come before the FDA just in the last several years and failed to gain approval. Approval of a few of those drugs combined with the development of new drugs should be achievable, but only if all levels of constituents pitch in to work together to solve the problem.
We must accept that there will never be a time when we have “enough” antibacterial agents. Resistance will never stop developing. Dr. John Bartlett, one of the giant figures in medicine over the past half century, has said about antibiotics that “the lesson of history is that we need a pipeline” (J. Bartlett to G. H. Talbot, personal communication, March 28, 2008). The overarching concept of the 10 × ’20 initiative should not be taken to mean that after we have developed 10 new drugs we will be finished. Rather, it is a clarion call to action with a short-term goal of developing the needed new drugs by 2020, but a longer-term goal of establishing an infrastructure that will last longer than 10 years, possibly in perpetuity, to continue to support new antibacterial development for future generations.
Why Is This Important?
This manuscript began with a quote from Dr. Lewis Thomas regarding the impact of antibacterial therapy on the practice of medicine. The manuscript closes with quotes from another leading physician of the 20th century, Dr. Walsh McDermott. Dr. McDermott was also a Lasker Award winner and a member of the National Academy of Sciences. Indeed Dr. McDermott served as the first president of the Medical Board of the National Academies, which was the precursor to the Institute of Medicine. In the 1950s Dr. McDermott oversaw a program which was designed to bring modern medicines to highly isolated Navajo Native
American communities in the southwestern United States. In 1960, Dr. McDermott published a manuscript in Science describing this experience. He wrote that “with today’s [antibiotics] it is possible to place in the hands of a barefoot, nonliterate villager more real power to affect the outcome of a … critically ill [patient] … than could have been exerted by the most highly trained urban physician of 25 years ago” (McDermott et al., 1960). And Dr. McDermott would know, since he was a highly trained urban physician of 25 years ago.
Dr. McDermott died in 1981, and shortly before his death he had begun writing his memoirs. In 1982, his friend and colleague, Dr. David Rodgers, published the first chapter of those memoirs in the Johns Hopkins Medical Journal. These are the posthumous words of Dr. McDermott, who practiced medicine prior to antibiotics and experienced the transition first-hand:
It is not too much to state that the introduction of [antibiotics] has represented a force for change in the twentieth century of the same general kind as James Watt’s modification of the steam engine did in the eighteenth. The crossing of the historic watershed could be felt at the time. One day we could not save lives, or hardly any lives; on the very next day we could do so across a wide spectrum of diseases. This was an awesome acquisition of power. (McDermott and Rogers, 1982)
What must Dr. McDermott and Dr. Thomas think if they were alive today to bear witness to the evaporation of that power (Figure A18-1)?
The loss of effective antibacterial therapy will result in a great increase in deaths from infections. It will also greatly impact diverse fields of medicine ranging from surgery, cancer chemotherapy, critical care medicine, and transplantation medicine, all of which are feasible only in the context of effective antibacterial therapy. This issue—the availability of effective antibacterial agents—is not a “lifestyle” issue. It is not a theoretical issue. We are facing a societal crisis of lack of antibiotics that is already resulting in deaths and maiming of patients and will increasingly do so in the coming decades unless precise action is taken on an urgent basis.
The time for endless debate and angst over statistics and politics has passed. Certainly we should continue to gather data and conduct studies to further elucidate the issues of antibiotic development and resistance as much as possible. But calls for “additional data” on this topic should not be allowed to derail the process of fixing the problem. Let additional data be gathered while we fix the problem. The time for action is now.
Acknowledgments
The author would like to offer most sincere gratitude to a number of experts who have provided specific, insightful, and important input on this manuscript,
including John Bartlett, Helen Boucher, John Bradley, Eric Brass, Peter Christensen, Aaron Dane, Barry Eisenstein, David Gilbert, Robert Guidos, Roger Lewis, John Rex, W. Michael Scheld, David Shlaes, George Talbot, and Glenn Tillotson.
Disclosures
Grant support from Gilead, Astellas, Novartis; Consultant: Merck, Pfizer, Basilea, The Medicines Company, Novartis, Cerexa, Wyeth, Trius, Achaogen, GlaxoSmithKline; Speaker’s honorarium: GlaxoSmithKline; Shareholder: NovaDigm Therapeutics, Neutropenia Immunotherapy Solutions.
References
Ambrose, P. G. 2008. Use of pharmacokinetics-pharmacodynamics in a community-aquired pneumonia failure analysis: Implications for future clinical trial study design. Clinical Infectious Diseases 47(Suppl. 3):S225–31.
Ambrose, P. G. 2010. Pharmacokinetic-pharmacodynamic considerations in the design of hospital—acquired or ventilator-associated pneumonia studies: Look before you leap! Clinical Infectious Diseases 51(Suppl. 1):5103–10.
Armstrong, G. L., L. A. Conn, and R. W. Pinner. 1999. Trends in infectious disease mortality in the United States during the 20th century. Journal of the American Medical Association 281(1):61–6.
Austrian, R., and J. Gold. 1964. Pneumococcal bacteremia with especial reference to bacteremic pneumococcal pneumonia. Annals of Internal Medicine 60:759–76.
Bradley, J. S., and G. H. J. McCracken. 2008. Unique considerations for the evaluation of antibacterials in clinical trials of pediatric community-associated pneumonia. Clinical Infectious Diseases 47(Suppl. 3):S241–8.
Bullowa, J. G. M. 1937. Chapter II. The course, symptoms and physical findings. In The management of pneumonias. New York: Oxford University Press. P. 40.
Chemotherapy of meningitis. 1938. Lancet 231(5978):733–4.
Christie, R. V. 1949. Penicillin in subacute bacterial endocarditis. British Medical Journal 2(4634):950.
Davidson, R., R. Cavalcanti, J. L. Brunton, D. J. Bast, J. C. de Azavedo, P. Kibsey, C. Fleming, and D. E. Low. 2002. Resistance to levofloxacin and failure of treatment of pneumococcal pneumonia. New England Journal of Medicine 346(10):747–50.
DiMasi, J. A. 1992. Rising research and development costs for new drugs in a cost containment environment. Pharmacoeconomics 1(Suppl. 1):13–20.
DiMasi, J. A., R. W. Hansen, H. G. Grabowski, and L. Lasagna. 1991. Cost of innovation in the pharmaceutical industry. Journal of Health Economics 10(2):107–42.
DiMasi, J. A., R. W. Hansen, and H. G. Grabowski. 2003. The price of innovation: New estimates of drug development costs. Journal of Health Economics 22(2):151–85.
Doll, R. 1998. Controlled trials: The 1948 watershed. British Medical Journal 317(7167):1217–20.
Dowling, H. F., and M. H. Lepper. 1951. The effect of antibiotics (penicillin, aureomycin, and terramycin) on the fatality rate and incidence of complications in pneumococcic pneumonia. A comparison with other methods of therapy. American Journal of the Medical Sciences 222(4):396–403.
Dylewski, J., and R. Davidson. 2006. Bacteremic pneumococcal pneumonia associated with macrolide failure. European Journal of Clinical Microbiology and Infectious Diseases 25(1):39–42.
ECDC (European Center for Disease Prevention) and EMEA (European Medicines Agency). 2009. The bacterial challenge: Time to react. A call to narrow the gap between multidrug-resistant bacteria in the EU and the development of new antibacterial agents. http://www.ema.europa.eu/pdfs/human/antimicrobial_resistance/53394009en.pdf (accessed May 1, 2010).
EMEA (European Medicines Agency). 1998. E9, statistical principles for clinical trials. http://www.emea.europa.eu/pdfs/human/ich/036396en.pdf (accessed May 19, 2010).
Endimiani, A., G. Brigante, A. A. Bettaccini, F. Luzzaro, P. Grossi, and A. Q. Toniolo. 2005. Failure of levofloxacin treatment in community-acquired pneumococcal pneumonia. BMC Infectious Diseases 5:106.
FDA (Food and Drug Administration). 2001. Guidance for industry. E10, choice of control group and related issues in clinical trials. http://www.fda.gov/downloads/RegulatoryInformation/Guidances/ucm125912.pdf (accessed May 19, 2010).
FDA. 2002. Active control non-inferiority studies: Theory, assay sensitivity, choice of margin. http://www.fda.gov/ohrms/dockets/ac/02/slides/3837s1_02_Temple.ppt (accessed May 19, 2010).
FDA. 2008a. Clinical trial design for community-acquired pneumonia; public workshop. http://www.fda.gov/Drugs/NewsEvents/ucm180241.htm (accessed May 19, 2010).
FDA. 2008b. Questions. Anti-Infective Drugs Advisory Committee, April 1–2. Center for Drug Evaluation and Research. http://www.fda.gov/ohrms/dockets/AC/08/questions/2008-4343q-final.pdf (accessed May 19, 2010).
FDA. 2009a. Guidance for industry. Community-acquired bacterial pneumonia: Developing drugs for treatment. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm123686.pdf (accessed May 19, 2010).
FDA. 2009b. Briefing materials for the December 9, 2009 meeting of the Anti-Infective Drugs Advisory Committee. http://www.fda.gov/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/Anti-InfectiveDrugsAdvisoryCommittee/ucm190755.htm (accessed May 19, 2010).
FDA. 2010. Guidance for industry: Non-inferiority clinical trials. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM202140.pdf (accessed May 19, 2010).
File, T. M., Jr., and J. J. Schentag. 2008. What can we learn from time course of untreated and partially treated community-onset Streptococcus pneumoniae pneumonia? A clinical perspective on superiority and noninferiority trial designs for mild community-acquired pneumonia. Clinical Infectious Diseases 47(Suppl. 3):S157–65.
File, T. M., Jr., S. V. Monte, J. J. Schentag, K. P. Klugman, J. A. Paladino, B. Lavin, V. L. Yu, and M. H. Adelman. 2007. RTI response in the community; observations on the transition state between COPD and CAP. Abstract 1047, 45th Annual Meeting of the Infectious Diseases Society of America (IDSA), San Diego, CA.
Finland, M. 1943. Chemotherapy in bacteremias. Connecticut State Medical Journal 7:92–100.
Fleming, T. R. 2008. Current issues in non-inferiority trials. Statistics in Medicine 27(3):317–32.
Flippin, H. F., J. S. Lockwood, D. S. Pepper, and L. Schwartz. 1939. The treatment of pneumococcic pneumonia with sulfapyridine. Journal of the American Medical Association 112(6):529–34.
Gorlin, R., C. B. Favour, and F. J. Emery. 1950. Long-term follow-up study of penicillin-treated subacute bacterial endocarditis. New England Journal of Medicine 242(26):995–1001.
Grossman, C. M. 2008. The first use of penicillin in the United States. Annals of Internal Medicine 149(2):135–6.
Guest, C. M., and F. F. Harrison. 1948. Acute endocarditis due to Staphylococcus aureus successfully treated with penicillin. American Journal of Medicine 5(6):908–11.
Herrell, W. E. 1943. Further observations on the clinical use of penicillin. Proceedings of the Staff Meetings of the Mayo Clinic 18:65–76.
Ho, P. L., R. W. Yung, D. N. Tsang, T. L. Que, M. Ho, W. H. Seto, T. K. Ng, W. C. Yam, and W. W. Ng. 2001. Increasing resistance of Streptococcus pneumoniae to fluoroquinolones: Results of a Hong Kong multicentre study in 2000. Journal of Antimicrobial Chemotherapy 48(5):659–65.
Houck, P. M., D. W. Bratzler, W. Nsa, A. Ma, and J. G. Bartlett. 2004. Timing of antibiotic administration and outcomes for Medicare patients hospitalized with community-acquired pneumonia. Archives of Internal Medicine 164(6):637–44.
Hoyne, A. L., A. A. Wolf, and L. Prim. 1939. Mortality rates in the treatment of 998 erysipelas patients. Journal of the American Medical Association 113(26):2279–81.
Iannini, P. B., J. A. Paladino, B. Lavin, M. E. Singer, and J. J. Schentag. 2007. A case series of macrolide treatment failures in community acquired pneumonia. Journal of Chemotherapy 19(5):536–45.
IDSA (Infectious Diseases Society of America). 2010. The 10 × ’20 Initiative: Pursuing a global commitment to develop 10 new antibacterial drugs by 2020. Clinical Infectious Diseases 50(8):1081–3.
Is endocarditis lenta always fatal? 1935. Lancet 226:383–4.
ISIS-2 (Second International Study of Infarct Survival) Collaborative Group. 1988. Randomised trial of intravenous streptokinase, oral aspirin, both, or neither among 17,187 cases of suspected acute myocardial infarction: ISIS-2. Lancet 332(8607):349–60.
Kahn, K. L., W. H. Rogers, L. V. Rubenstein, M. J. Sherwood, E. J. Reinisch, E. B.Keeler, D. Draper, J. Kosecoff, and R. H. Brook. 1990. Measuring quality of care with explicit process criteria before and after. Journal of the American Medical Association 264(15):1969–73.
Kassowitz, K. E., and G. H. Muscato. 1952. The long range effect of antibacterial therapy on pneumonia, empyema, bronchiectasis and pulmonary abscess: An analysis of incidence and mortality in 74,489 admissions to a children’s hospital in twenty years. Diseases of the Chest 21(2):161–73.
Keefer, C. S., F. G. Blake, E. K. Marshall, Jr., J. S. Lockwood, and B. W. Wood, Jr. 1943. Penicillin in the treament of infections. Journal of the American Medical Association 122(18):1217–24.
Kelley, M. A., D. J. Weber, P. Gilligan, and M. S. Cohen. 2000. Breakthrough pneumococcal bacteremia in patients being treated with azithromycin and clarithromycin. Clinical Infectious Diseases 31(4):1008–11.
Kerr, A. J. 1955. Subacute bacterial endocarditis. Springfield, IL: Charles C. Thomas.
Kingston, J. R., R. Chanock, M. A. Mufson, L. P. Hellman, W. D. James, H. H. Fox, M. A. Manko, and J. Boyers. 1961. Eaton agent pneumonia. Journal of the American Medical Association 176:118–23.
Lavender, H. J., and L. Goldman. 1935. Facial erysipelas: Evaluation and comparison of specific antiserum and ultraviolet therapy. Journal of the American Medical Association 105:401–3.
Lederberg, J. 2000. Infectious history. Science 288(5464):287–93.
Lonks, J. R., J. Garau, L. Gomez, M. Xercavins, A. Ochoa de Echaguen, I. F. Gareen, P. T. Reiss, and A. A. Medeiros. 2002. Failure of macrolide antibiotic treatment in patients with bacteremia due to erythromycin-resistant Streptococcus pneumoniae. Clinical Infectious Diseases 35(5):556–64.
Lyons, C. 1943. Penicillin therapy of surgical infections in the U.S. Army. Journal of the American Medical Association 123:1007–18.
Madsen, S. T. 1973. Scarlet fever and erysipelas in Norway during the last hundred years. Infection 1(2):76–81.
Mandell, L. A., R. G. Wunderink, A. Anzueto, J. G. Bartlett, G. D. Campbell, N. C. Dean, S. F. Dowell, T. M. File Jr., D. M. Musher, M. S. Niederman, A. Torres, and C. G. Whitney. 2007. Infectious Diseases Society of America/American Thoracic Society consensus guidelines on the management of community-acquired pneumonia in adults. Clinical Infectious Diseases 44(Suppl. 2):S27–72.
McCartney, A. C. 1992. Changing trends in infective endocarditis. Journal of Clinical Pathology 45(11):945–48.
McDermott, W., and D. E. Rogers. 1982. Social ramifications of control of microbial disease. Johns Hopkins Medical Journal 151(6):302–12.
McDermott, W., K. Deuschle, J. Adair, H. Fulmer, and B. Loughlin. 1960. Introducing modern medicine in a Najavo community. Science 131(3395):197–205.
Meakins, J. C., and F. R. Hanson. 1939. The Treatment of pneumococcic pneumonia with sulfapyri-dine. Canadian Medical Association Journal 40(4):333–36.
Meehan, T. P., M. J. Fine, H. M. Krumholz, J. D. Scinto, D. H. Galusha, J. T. Mockalis, G. F. Weber, M. K. Petrillo, P. M. Houck, and J. M. Fine. 1997. Quality of care, process, and outcomes in elderly patients with pneumonia. Journal of the American Medical Association 278(23):2080–4.
Mossialos, E., C. Morel, S. Edwards, J. Berenson, M. Gemmill-Toyama, and D. Brogan. 2009. Policies and incentives for promoting innovation in antibiotic research. London School of Economics and Political Science. http://www.se2009.eu/polopoly_fs/1.16814!menu/standard/file/LSE-ABI%20F-Final.pdf (accessed May 1, 2010).
Musher, D. M., M. E. Dowell, V. D. Shortridge, R. K. Flamm, J. H. Jorgensen, P. Le Magueres, and K. L. Krause. 2002. Emergence of macrolide resistance during treatment of pneumococcal pneumonia. New England Journal of Medicine 346(8):630–31.
National Center for Health Statistics. 1968. Use of vital and health records in epidemiologic research: A report of the United States National Committee on Vital and Health Statistics. DHEW Publication No. (HSM) 73-1265; PHS Publication No. 1000-Series 4-No. 7, March, p. 2. http://www.cdc.gov/nchs/data/series/sr_04/sr04_007.pdf (accessed April 28, 2009).
Northey, E. H. 1948. The sulfonamides and allied compounds. New York: Reinhold.
Osler, W. 1901. Lobar pneumonia. In The principles and practice of medicine, 4th ed. New York: Appelton. P. 108.
Pertel, P. E., P. Bernardo, C. Fogarty, P. Matthews, R. Northland, M. Benvenuto, G. M. Thorne, S. A. Luperchio, R. D. Arbeit, and J. Alder. 2008. Effects of prior effective therapy on the efficacy of daptomycin and ceftriaxone for the treatment of community-acquired pneumonia. Clinical Infectious Diseases 46(8):1142–51.
Peterson, L. R. 2006. Penicillins for treatment of pneumococcal pneumonia: Does in vitro resistance really matter? Clinical Infectious Diseases 42(2):224–33.
PhRMA. 2007. Drug discovery and development: Understanding the R&D process. http://www.innovation.org/drug_discovery/objects/pdf/RD_brochure.pdf (accessed October 26, 2010).
Powers, J. H. 2008. Noninferiority and equivalence trials: Deciphering “similarity” of medical interventions. Statistics in Medicine 27(3):343–52.
Powers, J. H., D. B. Ross, E. Brittain, R. Albrecht, and M. J. Goldberger. 2002. The United States Food and Drug Administration and noninferiority margins in clinical trials of antimicrobial agents. Clinical Infectious Diseases 34(6):879–81.
Projan, S. J. 2003. Why is Big Pharma getting out of antibacterial drug discovery? Current Opinions in Microbiology 6(5):427–30.
Rex, J. 2010. Commercial challenges: Perspectives from big pharma. Slide 14, Institute of Medicine Workshop on Countermeasures for Bioterrorism Pathogens, Rockville, MD.
Ross, D. B. 2007. The FDA and the case of Ketek. New England Journal of Medicine 356(16): 1601–4.
Rzeszutek, M., A. Wierzbowski, D. J. Hoban, J. Conly, W. Bishai, and G. G. Zhanel. 2004. A review of clinical failures associated with macrolide-resistant Streptococcus pneumoniae. International Journal of Antimicrobial Agents 24(2):95–104.
Schmeck, H. M. 1971. Health of nation lags behind scientific gains. New York Times, July 16, 1.
Shlaes, D. M. 2002. Is there hope for the prevention of future antimicrobial shortages—author’s reply. Clinical Infectious Diseases 35:216–7.
Shlaes, D. M. 2003. The abandonment of antibacterials: Why and wherefore? Current Opinions in Pharmacology 3(5):470–73.
Shlaes, D. M., and R. C. Moellering, Jr. 2002. The United States Food and Drug Administration and the end of antibiotics. Clinical Infectious Diseases 34(3):420–2.
Shlaes, D. M., and R. C. Moellering. 2008. Telithromycin and the FDA: Implications for the future. Lancet Infectious Diseases 8(2):83–5.
Silverman, J. A., L. I. Mortin, A. D. Vanpraagh, T. Li, and J. Alder. 2005. Inhibition of daptomycin by pulmonary surfactant: In vitro modeling and clinical impact. Journal of Infectious Diseases 191(12):2149–52.
Singer, M., S. Nambiar, T. Valappil, K. Higgins, and S. Gitterman. 2008. Historical and regulatory perspective on the treatment effect of antibacterial drugs in community acquired pneumonia. Clinical Infectious Diseases 47(Suppl. 3):S216–24.
Smilack, J. D., W. W. Burgin, Jr., W. L. Moore, Jr., and J. P. Sanford. 1974. Mycoplasma pneumoniae pneumonia and clindamycin therapy. Failure to demonstrate efficacy. Journal of the American Medical Association 228(6):729–31.
Smith, G. C., and J. P. Pell. 2003. Parachute use to prevent death and major trauma related to gravitational challenge: Systematic review of randomised controlled trials. British Medical Journal 327(7429):1459–61.
Snodgrass, W. R., and T. Anderson. 1937a. Prontosil in the treatment of erysipelas. A controlled series of 312 cases. British Medical Journal 2(3933):101–4.
Snodgrass, W. R., and T. Anderson. 1937b. Sulphanilamide in the treatment of erysipelas. A controlled series of 270 cases. British Medical Journal 2(4014):1156–9.
Soreth, J., E. Cox, S. Kweder, J. Jenkins, and S. Galson. 2007. Ketek—the FDA perspective. New England Journal of Medicine 356(16):1675–6.
Spellberg, B. 2009. Rising plague: The global threat from deadly bacteria and our dwindling arsenal to fight them. Amherst, New York: Prometheus Books.
Spellberg, B., and G. H. Talbot; for the IDSA, American College of Chest Physicians, American Thoracic Society, and Society of Critical Care Medicine. 2010. Recommended design features of future clinical trials of anti-bacterial agents for hospital-acquired bacterial pneumonia (HABP) and ventilator-associated bacterial pneumonia (VABP). Clinical Infectious Diseases. Suppl. 1: S150–70.
Spellberg, B., J. H. Powers, E. P. Brass, L. G. Miller, and J. E. Edwards. 2004. Trends in antimicrobial drug development: Implications for the future. Clinical Infectious Diseases 38(9):1279–86.
Spellberg, B., G. H. Talbot, E. P. Brass, J. S. Bradley, H. W. Boucher, and D. Gilbert. 2008a. Position paper: Recommended design features of future clinical trials of anti-bacterial agents for community-acquired pneumonia. Clinical Infectious Diseases 47(S3):S249–65.
Spellberg, B., R. Guidos, D. Gilbert, J. Bradley, H. W. Boucher, W. M. Scheld, J. G. Bartlett, and J. Edwards, Jr. 2008b. The epidemic of antibiotic-resistant infections: A call to action for the medical community from the Infectious Diseases Society of America. Clinical Infectious Diseases 46(2):155–64.
Spellberg, B., T. R. Fleming, and D. N. Gilbert. 2008c. Executive summary: Workshop on issues in the design and conduct of clinical trials of antibacterial drugs in the treatment of community-acquired pneumonia. Clinical Infectious Diseases 47(Suppl. 3):S105–7.
Spellberg, B., G. H. Talbot, H. W. Boucher, J. S. Bradley, D. Gilbert, W. M. Scheld, J. E. J. Edwards, and J. G. Bartlett, Antimicrobial Availability Task Force of the Infectious Diseases Society of America. 2009. Antimicrobial agents for complicated skin and skin structure infections: Justification of noninferiority margins in the absence of placebo-controlled trials. Clinical Infectious Diseases 49(3):383–91.
Stewart, W. H. 1968. Areas of challenge for the future. In Symposium schools of public health: Changing institutions in a changing world. Three papers presented on the occasion of the dedication of the Ernest Lyman Stebbins building, September 18, 1968, The Johns Hopkins University School of Hygiene and Public Health.
Talbot, G. H. 2010. Considerations in undertaking a clinical development program for hospital-acquired bacterial pneumonia/ventilator-associated bacterial pneumonia. Clinical Infectious Diseases Suppl. 1:S144–9.
Temple, R., and S. S. Ellenberg. 2000. Placebo-controlled trials and active-control trials in the evaluation of new treatments. Part 1: Ethical and scientific issues. Annals of Internal Medicine 133(6):455–63.
Thomas, L. 1983. The youngest science. Notes of a medicine-watcher. New York: Viking Press.
Titus, N. E. 1934. The treatment of erysipelas by ultra-violet light. British Journal of Physical Medicine 9:150–56.
Trachsler, W. H., G. S. Frauenberger, C. Wagner, and A. G. Mitchell. 1937. Streptococcic meningitis: With special emphasis on sulfanilamide therapy. Journal of Pediatrics 11:248–69.
Ude, W. H. 1931. Erysipelas: Further comparative studies of more recent methods of treatment. Archives of Physical Therapy 12:16–8.
Waring, G. W., and L. Weinstein. 1948. The treatment of pneumococcal meningitis. American Journal of Medicine 5:402–18.
Wenzel, R. P. 2004. The antibiotic pipeline—challenge, costs, and values. New England Journal of Medicine 351(6):523–6.
A19
CHALLENGES IN ANTIMICROBIAL SUSCEPTIBILITY TESTING OF CLINICAL AND ENVIRONMENTAL ISOLATES71
Fred C. Tenover
Cepheid72
Introduction
The primary goal of antimicrobial susceptibility testing (AST) is to help physicians choose the best antimicrobial agent for treating bacterial infections in individual patients (Tenover, 2006). The typical turnaround time required to produce AST results is 26 to 96 hours. This includes 18 to 48 hours for isolation of the bacterial pathogen in pure culture (depending on how fastidious the organism is) and an additional 8 to 48 hours for completion of the AST results. Faster times are associated with use of automated susceptibility testing systems, some of which produce AST results in parallel with bacterial identification in as little as 8 hours (Holland et al., 2009; Jorgensen et al., 2000; Kerremans et al., 2008; Ling et al., 2003; Mirrett and Reller, 1979). Turnaround times are slower for fastidious organisms, such as Streptococcus pneumoniae and obligate anaerobic organisms, which require longer incubation times and are less amenable to rapid testing with automated methods (Jorgensen et al., 2000).
The secondary use of AST data is for guiding empiric therapy for patients
|
71 |
Key words: susceptibility, glycopeptides, β-lactamases, surveillance. |
|
72 |
904 Caribbean Drive, Sunnyvale, CA 94089, Phone: 408-400-4344, Fax: 408-744-1479, E-mail: fred.tenover@cepheid.com. |
with suspected bacterial infections when they are first admitted to the hospital. Empiric therapy is selected by the physician before laboratory tests have identified, or confirmed, the source of infection and the bacterial agent of disease (Hindler and Stelling, 2007). Laboratories typically analyze all of their AST data (which may be a combination of minimal inhibitory concentration [MIC] and disk diffusion results, depending on the organism) and assemble reports that indicate the percentage of organisms, delineated by bacterial species, that are susceptible to the antimicrobial agents that may be used to treat infections. The cumulative reports, which are designated “antibiograms,” are reported at least annually (Tenover and Hindler, 2010). The assembly of the data is guided by recommendations published by the Clinical and Laboratory Standards Institute (CLSI) in document M39 (CLSI, 2009). The accuracy of the AST data for both empiric and definitive therapy is critical to ensure optimal patient outcomes.
The Accuracy of AST Data
The good news about the AST data generated by clinical microbiology laboratories around the world is that proficiency testing studies conducted by the World Health Organization (WHO) in collaboration with the U.S. Centers for Disease Control and Prevention (CDC) over a 10-year period confirm that the majority of data are accurate. This is particularly true for the antimicrobial agents tested against common bacterial species (Chaitram et al., 2003; Tenover et al., 2001). The bad news is that the antimicrobial-resistant organisms that laboratories fail to detect are those that have the greatest impact on patient care, that is, vancomycin-intermediate (VISA) and vancomycin-resistant Staphylococcus aureus (VRSA) (Tenover et al., 1998, 2001; Whitener et al., 2004), Enterobacteriaceae producing extended-spectrum β-lactamases (ESBLs) (Tenover et al., 1999), and Klebsiella pneumoniae isolates that produce carbapenemases (Tenover et al., 2006). Detection of these resistant pathogens continues to challenge many laboratories.
Why is it so difficult for clinical laboratories to detect these specific resistance phenotypes? There are several potential reasons. First, new resistance mechanisms to glycopeptides (as with VISA strains) and β-lactams (as with carbapenemase-producing K. pneumoniae strains) often do not result in high MICs (Tenover, 2006). In contrast to many traditional resistant mechanisms that are easily detected, the MIC results for the novel mechanisms are only a few doubling dilutions higher than the normal MIC values for wild-type isolates and often are in the intermediate range or even at the high end of the susceptible range. Thus, a number of resistant organisms go undetected, particularly when laboratories use breakpoint MIC panels consisting of only three or four dilutions of the antimicrobial agent focused at the susceptible and intermediate breakpoint concentrations.
Breakpoint panels often make it impossible to determine the actual MIC of the antimicrobial agents since so few dilutions are tested (Richter and Ferraro, 2007). Second, the emerging resistance pattern may be the result of a combination of resistance mechanisms (e.g., β-lactamases and porin changes or up-regulation of efflux pumps) that often are difficult to detect using standard laboratory methods (Tenover et al., 2009; Yigit et al., 2001). Third, there is a failure of automated AST systems to adapt quickly to emerging resistance patterns to enable detection of novel resistance mechanisms (Anderson et al., 2007; Juretschko et al., 2007; Tenover et al., 2007). This may be due to the fact that the lowest dilution of the antimicrobial agent tested on the automated AST system is still higher than the MIC value associated with the novel resistant mechanism. Alternately, it may be due to Food and Drug Administration (FDA) requirements for additional testing of new panel configurations before release of the assay modifications. Fourth, many emerging phenotypes require additional screening tests to detect nonsusceptible strains and, due to cost and labor requirements, laboratories often are slow to implement these additional tests (Swenson et al., 2007; Tenover et al., 1998). Finally, there is a greater proportion of resistant bacterial organisms now than there was 10 years ago, making the problem of undetected resistance a more critical problem (Castanheira et al., 2010; Perez et al., 2007; Pitout and Laupland, 2008; Sader et al., 2010).
To improve the detection of emerging resistance, a number of supplementary tests have been introduced into the clinical microbiology laboratory. These include the use of Brain Heart Infusion agar containing 6 μg of vancomycin per ml (BHI-V6) to screen for VISA and VRSA strains, standard and macro Etest methods to detect vancomycin heteroresistance, the cefoxitin disk test for mecA-mediated oxacillin resistance in staphylococci, the D-zone test to detect inducible clindamycin resistance in staphylococci and streptococci, agar and disk diffusion screening tests for high-level aminoglycoside resistance in enterococci (to document potential synergy of aminoglycosides and cell wall active agents), ESBL screening and confirmation tests, and screening tests (such as the modified Hodge test) for the Klebsiella pneumoniae carbapenemase (KPC) and other carbapenemases among Enterobacteriaceae (Holland et al., 2009; Swenson et al., 2007) (Table A19-1). Each of these tests plays an important role in increasing the accuracy of AST reports from the clinical laboratory by detecting subtle resistance mechanisms that may be missed by routine AST methods (Tenover and Hindler, 2010).
Detecting ESBLs and Carbapenemases
During the mid-1990s, it became clear that a number of K. pneumoniae, Escherichia coli, and Proteus mirabilis strains that produced ESBLs demonstrated extended-spectrum cephalosporin MICs that were in the upper end of the
TABLE A19-1 Examples of Supplementary Tests to Identify Resistance Phenotypes
|
Organism Group |
Supplementary Test |
Resistance Mechanism or Purpose |
|
Staphylococcus aureus |
Cefoxitin disk diffusion test |
Identify mecA-mediated oxacillin resistance |
|
Staphylococcus aureus |
Brain Heart Infusion agar containing 6 μg of vancomycin per ml |
Identify vancomycin-intermediate and vancomycin-resistant isolates |
|
Staphylococcus aureus and β-hemolytic streptococci |
D-zone test |
Identify inducible clindamycin resistance in erythromycin-resistant clindamycin susceptible isolates of clinical significance |
|
Enterococci |
Broth microdilution and disk diffusion tests for high-level aminoglycoside resistance |
Identify potential synergy between aminoglycosides and cell wall active agents (β-lactams or vancomycin) |
|
Enterobacteriaceae |
Lower screening breakpoints and clavulanic acid confirmation testing |
Detection of extended-spectrum β-lactamase production |
|
Enterobacteriaceae |
Modified Hodge test |
Detection of carbapenemase production |
|
SOURCES: Adapted from Swenson et al. (2007) and CLSI (2010). |
||
susceptible range (Jacoby and Han, 1996). Patients infected with such strains often failed therapy with extended-spectrum cephalosporins, particularly when these agents were used as monotherapy (Paterson et al., 2001, 2004). Thus, ESBL screening tests were developed to alert the laboratory to strains of these three species that may produce ESBLs (Bradford, 2001). ESBL confirmation tests using clavulanic acid were also developed to differentiate ESBL-producing strains from strains with elevated extended-spectrum cephalosporin MICs due to resistance mechanisms other than ESBL production (Schwaber et al., 2004). Phenotypic detection of ESBL-mediated resistance indicated that the laboratory should change the interpretations of extended-spectrum cephalosporins and penicillins from susceptible to resistant to increase the accuracy of the AST results (Bradford, 2001).
In 2010, the CLSI advocated the use of lower cephalosporin breakpoints to obviate the need for ESBL testing (CLSI, 2010). Part of the impetus to develop lower cephalosporin breakpoints was the rapid dissemination of genes encoding ESBLs, such as the CTX-M β-lactamases (Barlow et al., 2008; Bonnet, 2004), and proficiency testing data showing that laboratories performed poorly when challenged with ESBL-producing strains in blinded susceptibility testing studies (Steward et al., 2000; Tenover et al., 1999). For example, WHO/CDC proficiency
testing data from a survey of 271 laboratories outside of the United States conducted in 2005 with K. pneumoniae American Type Culture Collection (ATCC) strain 700603 (which is the positive quality control organism for CLSI ESBL tests) were discouraging. Only 95 (31 percent) of 271 laboratories surveyed correctly reported the K. pneumoniae isolate as an ESBL producer and changed the interpretations of the penicillins and cephalosporins from susceptible to resistant. Interestingly, 25 percent did not report ESBL production but nonetheless changed the interpretations of the penicillins and cephalosporins from susceptible to resistant, while 3 percent reported the isolate as an ESBL producer but did not change the interpretations of the β-lactams. The remaining 112 laboratories (41 percent) neither reported the strain as an ESBL producer nor changed the interpretations of the β-lactams (Chaitram et al., 2003). Thus, there apparently was a poor understanding of how the supplementary ESBL test results were to be used and reported to improve the accuracy of AST reports.
The newly proposed breakpoints for cephalosporins, which are two to three doubling dilutions lower that the original breakpoints (CLSI, 2010), were developed using microbiologic data (MIC distributions from thousands of organisms), pharmacokinetic/pharmacodynamic models, and outcome data derived from the medical literature of patients treated with a single cephalosporin agent. Novel breakpoint development strategies also were employed (Turnidge and Paterson, 2007). The new breakpoints, which apply to all of the Enterobacteriaceae, should capture the former “susceptible” isolates that produce ESBLs and categorize them as resistant without the need for additional ESBL testing. In some cases, ESBL-producing isolates may test as susceptible to one or more of the extended-spectrum cephalosporins using the new breakpoints. Data provided to support the new breakpoints indicate that infections caused by these isolates should respond to the extended-spectrum cephalosporin therapy. ESBL testing will be reserved for infection control monitoring purposes.
Carbapenem Inactivation Assays for Testing Enterobacteriaceae (modified Hodge test)
Several proficiency testing surveys have shown that clinical laboratories also have a difficult time recognizing carbapenemase-producing Enterobacteriaceae (Steward et al., 2003), primarily because of the inability of automated AST systems to correctly classify the organisms as resistant (Anderson et al., 2007; Tenover et al., 2006). To improve the accuracy of reporting carbapenem results, the CLSI recommended the use of a carbapenem inactivation assay (i.e., the modified Hodge test) (CLSI, 2010). This assay was initially proposed as a means of identifying carbapenemase-producing strains by Yigit et al. (2001), who identified the first KPC-producing strain of K. pneumoniae in the United States. The spotty implementation and problems associated with the interpretation of the modified Hodge test by clinical laboratories led the CLSI to propose lower
alternative breakpoints for the carbapenems for both disk diffusion and MIC testing for the Enterobacteriaceae to obviate the need for the modified Hodge test (CLSI, 2010).
Implementing the New CLSI Breakpoints
The new disk diffusion breakpoints for both carbapenems and cephalosporins can be implemented by clinical laboratories as soon as the laboratory verifies the testing protocol and the medical staff agrees on the implementation of the new breakpoints. Unfortunately, manufacturers of automated MIC systems cannot sell instruments with software that use breakpoints other than those promulgated by the FDA, which are listed in the package label. Thus, to implement the new CLSI MIC breakpoints on automated AST devices in the United States, pharmaceutical manufacturers will have to request new breakpoints from the FDA for the cephalosporins or carbapenems that they manufacture, providing data to support the proposed changes. The FDA then must accept the new breakpoints proposed by the pharmaceutical company and promulgate them before the manufacturers of the AST in vitro device (i.e., the companies that market instruments, such as the BD Phoenix™, MicroScan WalkAway®, Trek Sensititre®, and bioMérieux Vitek® II) can submit their data to the FDA requesting the use of the alternative CLSI breakpoints. Device manufacturers have estimated that the time to convert their AST systems to use the new breakpoints is approximately 3 years, assuming that the pharmaceutical manufactures choose to accept the lower CLSI breakpoints. If this does not occur, some laboratories will continue to have difficulty detecting some ESBL- or carbapenemase-producing strains of Enterobacteriaceae and will likely produce inaccurate results for these critical resistant isolates.
Setting Thresholds for Interventions
The cumulative AST data produced and reported by clinical laboratories constitutes the major source of surveillance data for monitoring changes in bacterial resistance over time (Tenover, 2001). Portions of these data are captured by a number of national and international surveillance systems, including the CDC’s National Healthcare Safety Network (NHSN) (Hidron et al., 2008). What impact, if any, do changes in resistance patterns of bacterial pathogens have on antimicrobial prescribing in the United States?
One successful model of tying interventions to surveillance data is seen with Neisseria gonorrhoeae, the causative agent of gonorrhea, which is not tested routinely by most clinical microbiology laboratories. Gonorrhea is a very common sexually transmitted disease worldwide. In 2008, according to the CDC, 336,742 cases of gonorrhea were reported in the United States, a rate of 111.6 cases per 100,000 population (CDC, 2010a). Until recently, a single oral dose of a fluoroquinolone was effective for treating gonorrhea in most cities in the United States.
In 2007, the CDC withdrew its recommendation to use a fluoroquinolone as primary therapy for gonorrhea in United States since, by that time, the proportion of fluoroquinolone-resistant gonococci (as measured by the CDC’s Gonococcal Isolate Surveillance Program) was greater than 7 percent nationally, a preestablished level for changing therapeutic recommendations (CDC, 2007). Single-dose oral fluoroquinolone therapy was replaced with injectable cephalosporins in most regions of the United States due to the emergence of fluoroquinolone resistance in gonococci. Unfortunately, reports of gonococci resistant to first-line cephalosporins are starting to emerge, which leaves essentially no antimicrobial agents in reserve to treat these infections when cephalosporin resistance becomes more widespread. The CDC notes that there have been reports of four isolates with decreased susceptibility to ceftriaxone in the last several years (CDC, 2010a). Nonetheless, the key issue is that a strategy was designed and implemented that established a threshold for fluoroquinolone resistance that would indicate when a change in therapy was needed.
Such strategies and interventions are not in place for most other bacterial pathogens. For example, thresholds have not been established for emerging resistance among respiratory tract pathogens, urinary tract pathogens, or pathogens associated with a variety of other bacterial diseases (e.g., macrolide resistance in pneumococci, fluoroquinolone resistance in E. coli, cefixime resistance in shigellosis). Nor are there thresholds for critical issues, such as carbapenem-resistant Enterobacteriaceae causing sepsis, where combination therapy or use of colistin or polymyxin as alternate approaches are described. Thus, local resistance reports for key antimicrobial agents listed by organism, such as the periodic cumulative antibiogram reports described earlier, are critical for managing patients that are admitted to U.S. hospitals, particularly where local treatment guidelines have been established (Anderson and Kaye, 2009). Hospitals may consider using their antibiograms to establish their own local thresholds for changing both empiric and definitive therapy for various infections.
Surveillance for Resistance
The NHSN is a voluntary, secure, Internet-based surveillance system conducted by the CDC that gathers data on healthcare-associated infections as well as healthcare personnel safety (Hidron et al., 2008). While rates of device-associated infections among hospitalized patients are usually reported annually, reporting of antimicrobial resistance data by the NHSN currently is intermittent and there are no interventions associated with rising resistance rates. For example, in the most recent NHSN report on resistant microorganisms (2006–2007 data), there was a total of 563 K. pneumoniae infections reported. Of the K. pneumoniae isolates associated with central line-associated bloodstream infections, 452 (80.3 percent) of the isolates were tested for susceptibility to imipenem, meropenem, or ertapenem and 10.8 percent were resistant to at least one of those carbapenem agents
(Hidron et al., 2008). However, there is no indication in the literature that these resistance data, which clearly exceed the level that led to a national intervention for treatment of gonorrhea (10.8 versus 7.0 percent), impacted antimicrobial use for K. pneumoniae bloodstream infections in the United States. Hopefully, in the future, surveillance of the prevalence of antimicrobial-resistant pathogens, in conjunction with surveillance of antimicrobial use, will lead to more judicious antimicrobial use in the United States (Anderson and Kaye, 2009). Such programs, however, are predicated on the assumption that the AST data produced by clinical microbiology laboratories nationwide are accurate.
Using Molecular Methods
The time necessary to produce AST results for organisms isolated from blood, cerebrospinal fluid, lower respiratory tract infections, and other normally sterile body sites (which is often 26 to 96 hours) is often too slow to have a significant impact on anti-infective therapy (American Thoracic Society, 2005; Garnacho-Montero et al., 2010; Kollef, 2008; Morrell et al., 2009). Some laboratories place aliquots of blood from positive blood culture bottles directly into automated AST cards or panels to expedite results (Jorgensen et al., 2000; Kerremans et al., 2008; Ling et al., 2003). Alternately, the use of molecular methods for rapid detection of certain types of resistant organisms directly in clinical samples is becoming more feasible in microbiology laboratories due to the availability of FDA-cleared commercial assays in the United States. Direct detection of MRSA and methicillin-susceptible S. aureus in blood cultures and wounds using commercial polymerase chain reaction tests already occurs in many laboratories (Parta et al., 2009; Stamper et al., 2007; Wolk et al., 2009) and is having a positive impact on antimicrobial usage, especially in reducing the amount of vancomycin use in hospitals. Direct detection of rifampin-resistant strains of Mycobacterium tuberculosis and other drug resistance markers directly in sputum samples (Blakemore et al., 2010; Helb et al., 2010) or using line probe assays on isolated colonies (Akpaka et al., 2008; Evans et al., 2009) also is having a positive impact on treatment of tuberculosis outside of the United States. Pyrosequencing (Arnold et al., 2005) also holds promise for rapid identification of resistant mycobacteria, although these assays currently cannot be used directly on sputum samples.
Microarrays, either to detect bacteria directly in blood samples or to identify resistance determinants, have been described (Cassone et al., 2008; Cleven et al., 2006), but these are primarily for research use at the present time. Implementation in clinical laboratories is limited by the lack of FDA-cleared products. One technology that is emerging for rapid detection of organisms is matrix-assisted laser desorption ionization time-of-flight (MALDI-TOF) mass spectrometry. This technology has been used to identify bacterial species directly in urine and blood samples (Ferreira et al., 2010a, 2010b) and from positive blood culture bottles
(Christner et al., 2010). MALDI-TOF has the potential to identify specific resistant bacteria directly in clinical samples, such as MRSA and carbapenemase-producing K. pneumoniae; however, such assays are still in developmental stages. Other molecular methods for the rapid identification of antimicrobial-resistant bacteria in blood samples have not demonstrated adequate sensitivity and specificity in clinical trials and may require further development. Nonetheless, there are now multiple rapid methods that may improve antimicrobial usage by providing antimicrobial resistance data earlier in the course of infection to aid in optimizing anti-infective therapy.
Monitoring for Resistance in the Environment
A report by D’Costa et al. (2006) noted a high proportion of bacteria in soil samples from several geographic regions that carried multiple antimicrobial resistance determinants, including resistance to newer agents, such as daptomycin and linezolid. Astonishingly, on average, the bacterial isolates showed phenotypic resistance to seven classes of antimicrobial agents. Yet, resistance to daptomycin and linezolid among staphylococci from clinically significant infections remains rare (Jones et al., 2009; Sader et al., 2009). Other investigators have also noted the high prevalence of antimicrobial-resistant organisms in soil (Kummerer, 2004; Martinez, 2008). One may question why more of the soil-related resistance mechanisms, such as daptomycin inactivation by hydrolysis, are not reported from surveys of human pathogens. It is possible that the determinants that encode these mechanisms have not been mobilized out of the soil organisms to human pathogens, or it is possible that the resistance determinants are present but not detected by our current phenotypic AST methods. Surveillance for resistance, for the most part, is limited to clinical microbiology laboratories, which may or may not report unusual resistance patterns to state or federal public health agencies. The lack of recognition of unusual resistance patterns among human pathogens by laboratory staff, combined with the limited resources available to support educating laboratory staff about antimicrobial resistance issues, remains a problem for public health surveillance (Pitout and Laupland, 2008).
Resistance among bacteria colonizing or infecting animals is widely recognized (Furuya and Lowy, 2006). There is surveillance for emerging resistance among foodborne pathogens via CDC’s FoodNet program, but it is limited in scope (CDC, 2010b). Broth microdilution testing is performed on a limited set of bacterial pathogens that are recovered from several food and human sample types in a systematic fashion and the data are analyzed and reported on an annual basis. However, there is no systematic surveillance of resistance among plant pathogens, water-borne organisms, or soil organisms, although resistant organisms are common as shown in several studies (Baquero et al., 2008; Kummerer, 2004; Wright, 2007). For example, limited testing is done for emerging resistance among plant pathogens, such as Erwinia herbicola, which can infect pome fruit
and cause widespread destruction of orchards. Testing is often limited to a few key antimicrobial agents, such as streptomycin and kasugamycin. No systematic surveillance testing of bacteria recovered from soil or wastewater is undertaken. Thus, there are major gaps in our surveillance of bacterial organisms that inhabit much of the biosphere.
Summary
Overall, routine AST methods work well for detecting routine resistance to antimicrobial agents among human pathogens tested in a standard fashion in clinical microbiology laboratories. However, resistance continues to emerge, and commercial AST methods are often slow to adopt the changes necessary to detect new resistance determinants. Detecting cephalosporin and carbapenem resistance among isolates of Enterobacteriaceae using the newly proposed CLSI breakpoints should increase accuracy of susceptibility reports by detecting more organisms with low-level resistance, often due to ESBLs or carbapenemase production, respectively (especially in the United States), but the new breakpoints may take several years to fully implement. Surveillance for the emergence of resistance among bacterial pathogens of humans is modest in scope and is usually not tied to interventions to improve either antimicrobial therapy or stewardship. Surveillance for emerging resistance among foodborne bacteria is even more modest, while surveillance among environmental isolates is sparse at best. Overall, the detection of emerging antimicrobial resistance in bacterial pathogens is a serious challenge for clinical microbiology laboratories, for clinicians who must treat patients, and for public health.
Acknowledgments
I thank Janet Hindler for helpful comments on the manuscript. Disclosures: FCT receives salary and benefits and is a shareholder of Cepheid, a molecular diagnostics company.
References
Akpaka, P. E., S. Baboolal, D. Clarke, L. Francis, and N. Rastogi. 2008. Evaluation of methods for rapid detection of resistance to isoniazid and rifampin in Mycobacterium tuberculosis isolates collected in the Caribbean. Journal of Clinical Microbiology 46(10):3426–28.
American Thoracic Society. 2005. Guidelines for the management of adults with hospital-acquired, ventilator-associated, and healthcare-associated pneumonia. American Journal of Respiratory and Critical Care Medicine 171(4):388–416.
Anderson, D. J., and K. S. Kaye. 2009. Controlling antimicrobial resistance in the hospital. Infectious Diseases Clinics of North America 23(4):vii–viii, 847–64.
Anderson, K. F., D. R. Lonsway, J. K. Rasheed, J. Biddle, B. Jensen, L. K. McDougal, R. B. Carey, A. Thompson, S. Stocker, B. Limbago, and J. B. Patel. 2007. Evaluation of methods to identify the Klebsiella pneumoniae carbapenemase in Enterobacteriaceae. Journal of Clinical Microbiology 45(8):2723–5.
Arnold, C., L. Westland, G. Mowat, A. Underwood, J. Magee, and S. Gharbia. 2005. Single-nucleotide polymorphism-based differentiation and drug resistance detection in Mycobacterium tuberculosis from isolates or directly from sputum. Clinical Microbiology and Infection 11(2):122–30.
Baquero, F., J. L. Martinez, and R. Canton. 2008. Antibiotics and antibiotic resistance in water environments. Current Opinions in Biotechnology 19(3):260–5.
Barlow, M., R. A. Reik, S. D. Jacobs, M. Medina, M. P. Meyer, J. E. McGowan, Jr., and F. C. Tenover. 2008. High rate of mobilization for blaCTX-Ms. Emerging Infectious Diseases 14(3):423–8.
Blakemore, R., E. Story, D. Helb, J. Kop, P. Banada, M. R. Owens, S. Chakravorty, M. Jones, and D. Alland. 2010. Evaluation of the Analytical Performance of the Xpert(R) MTB/RIF Assay. Journal of Clinical Microbiology. 48(7):2495–501.
Bonnet, R. 2004. Growing group of extended-spectrum beta-lactamases: The CTX-M enzymes. Antimicrobial Agents and Chemotherapy 48(1):1–14.
Bradford, P. A. 2001. Extended-spectrum beta-lactamases in the 21st century: Characterization, epidemiology, and detection of this important resistance threat. Clinical Microbiology Reviews 14(4):933–51.
Cassone, M., M. Del Grosso, A. Pantosti, A. Giordano, and G. Pozzi. 2008. Detection of genetic elements carrying glycopeptide resistance clusters in Enterococcus by DNA microarrays. Molecular and Cellular Probes 22(3):162–7.
Castanheira, M., H. S. Sader, and R. N. Jones. 2010. Antimicrobial susceptibility patterns of KPC-producing or CTX-M-producing Enterobacteriaceae. Microbial Drug Resistance 16(1):61–5.
CDC (Centers for Disease Control and Prevention). 2007. Update to CDC’s sexually transmitted diseases treatment guidelines, 2006: Fluoroquinolones no longer recommended for treatment of gonococcal infections. Morbidity and Mortality Weekly Report 56(14):332–6.
CDC. 2010a. Sexually transmitted diseases surveillance, 2008. http://www.cdc.gov/std/stats08/gonorrhea.htm (accessed June 6, 2010).
CDC. 2010b. Preliminary FoodNet data on the incidence of infection with pathogens transmitted commonly through food—10 states, 2009. Morbidity and Mortality Weekly Report 59(14):418–22.
Chaitram, J. M., L. A. Jevitt, S. Lary, and F. C. Tenover. Chaitram, J. M., L. A. Jevitt, S. Lary, and F. C. Tenover. 2003. The World Health Organization’s External Quality Assurance System Proficiency Testing Program has improved the accuracy of antimicrobial susceptibility testing and reporting among participating laboratories using NCCLS methods. Journal of Clinical Microbiology 41(6):2372–7.
Christner, M., H. Rohde, M. Wolters, I. Sobottka, K. Wegscheider, and M. Aepfelbacher. 2010. Rapid identification of bacteria from positive blood culture bottles by use of matrix-assisted laser desorption-ionization time of flight mass spectrometry fingerprinting. Journal of Clinical Microbiology 48(5):1584–91.
Cleven, B. E., M. Palka-Santini, J. Gielen, S. Meembor, M. Kronke, and O. Krut. 2006. Identification and characterization of bacterial pathogens causing bloodstream infections by DNA microarray. Journal of Clinical Microbiology 44(7):2389–97.
CLSI (Clinical and Laboratory Standards Institute). 2009. Analysis and presentation of cumulative antimicrobial susceptibility test data: Approved guideline—Third edition. M39-A3. Wayne, PA: CLSI.
CLSI. 2010. Performance standards for antimicrobial susceptibility testing: Twentieth informational supplement. M100-S20. Wayne, PA: CLSI.
D’Costa, V. M., K. M. McGrann, D. W. Hughes, and G. D. Wright. 2006. Sampling the antibiotic resistome. Science 311(5759):374–7.
Evans, J., M. C. Stead, M. P. Nicol, and H. Segal. 2009. Rapid genotypic assays to identify drug-resistant Mycobacterium tuberculosis in South Africa. Journal of Antimicrobial Chemotherapy 63(1):11–6.
Ferreira, L., F. Sanchez-Juanes, M. Gonzalez-Avila, D. Cembrero-Fucinos, A. Herrero-Hernandez, J. M. Gonzalez-Buitrago, and J. L. Munoz-Bellido. 2010. Direct identification of urinary tract pathogens from urine samples by matrix-assisted laser desorption ionization-time of flight mass spectrometry. Journal of Clinical Microbiology 48(6):2110–15.
Ferreira, L., F. Sanchez-Juanes, I. P. Guerra, M. I. Garcia Garcia, J. E. Sanchez, J. M. Gonzalez-Buitrago, and J. L. Bellido. 2010 (in press). Microorganisms direct identification from blood culture by MALDI-TOF mass spectrometry. Clinical Microbiology and Infection.
Furuya, E. Y., and F. D. Lowy. 2006. Antimicrobial-resistant bacteria in the community setting. Nature Reviews, Microbiology 4(1):36–45.
Garnacho-Montero, J., E. Garcia-Cabrera, A. Diaz-Martin, J. A. Lepe-Jimenez, P. Iraurgi-Arcarazo, R. Jimenez-Alvarez, J. Revuelto-Rey, and J. Aznar-Martin. 2010. Determinants of outcome in patients with bacteraemic pneumococcal pneumonia: Importance of early adequate treatment. Scandinavian Journal of Infection 42(3):185–92.
Helb, D., M. Jones, E. Story, C. Boehme, E. Wallace, K. Ho, J. Kop, M. R. Owens, R. Rodgers, P. Banada, H. Safi, R. Blakemore, N. T. Lan, E. C. Jones-Lopez, M. Levi, M. Burday, I. Ayakaka, R. D. Mugerwa, B. McMillan, E. Winn-Deen, L. Christel, P. Dailey, M. D. Perkins, D. H. Persing, and D. Alland. 2010. Rapid detection of Mycobacterium tuberculosis and rifampin resistance by use of on-demand, near-patient technology. Journal of Clinical Microbiology 48(1):229–37.
Hidron, A. I., J. R. Edwards, J. Patel, T. C. Horan, D. M. Sievert, D. A. Pollock, and S. K. Fridkin. 2008. NHSN annual update: Antimicrobial-resistant pathogens associated with healthcare-associated infections: Annual summary of data reported to the National Healthcare Safety Network at the Centers for Disease Control and Prevention, 2006–2007. Infection Control and Hospital Epidemiology 29(11):996–1011.
Hindler, J. F., and J. Stelling 2007. Analysis and presentation of cumulative antibiograms: A new consensus guideline from the Clinical and Laboratory Standards Institute. Clinical Infectious Diseases 44(6):867–73.
Holland, T. L., C. W. Woods, and M. Joyce. 2009. Antibacterial susceptibility testing in the clinical laboratory. Infectious Disease Clinics of North America 23(4):vii, 757–90.
Jacoby, G. A., and P. Han. 1996. Detection of extended-spectrum beta-lactamases in clinical isolates of Klebsiella pneumoniae and Escherichia coli. Journal of Clinical Microbiology 34(4):908–11.
Jones, R. N., J. E. Ross, J. M. Bell, U. Utsuki, I. Fumiaki, I. Kobayashi, and J. D. Turnidge. 2009. Zyvox Annual Appraisal of Potency and Spectrum program: Linezolid surveillance program results for 2008. Diagnostic Microbiology and Infectious Diseases 65(4):404–13.
Jorgensen, J. H., A. L. Barry, M. M. Traczewski, D. F. Sahm, M. L. McElmeel, and S. A. Crawford... 2000. Rapid automated antimicrobial susceptibility testing of Streptococcus pneumoniae by use of the bioMerieux VITEK 2. Journal of Clinical Microbiology 38(8):2814–8.
Juretschko, S., V. J. Labombardi, S. A. Lerner, and P. C. Schreckenberger 2007. Accuracies of beta-lactam susceptibility test results for Pseudomonas aeruginosa with four automated systems (BD Phoenix, MicroScan WalkAway, Vitek, and Vitek 2). Journal of Clinical Microbiology 45(4):1339–42.
Kerremans, J. J., P. Verboom, T. Stijnen, L. Hakkaart-van Roijen, W. Goessens, H. A. Verbrugh, and M. C. Vos. 2008. Rapid identification and antimicrobial susceptibility testing reduce antibiotic use and accelerate pathogen-directed antibiotic use. Journal of Antimicrobial Chemotherapy 61(2):428–35.
Kollef, M. H. 2008. Broad-spectrum antimicrobials and the treatment of serious bacterial infections: Getting it right up front. Clinical Infectious Diseases 47(Suppl. 1):S3–13.
Kummerer, K. 2004. Resistance in the environment. Journal of Antimicrobial Chemotherapy 54(2):311–20.
Ling, T. K., Z. K. Liu, and A. F. Cheng 2003. Evaluation of the VITEK 2 system for rapid direct identification and susceptibility testing of Gram-negative bacilli from positive blood cultures. Journal of Clinical Microbiology 41(10):4705–7.
Martinez, J. L. 2008. Antibiotics and antibiotic resistance genes in natural environments. Science 321(5887):365–7.
Mirrett, S., and L. B. Reller. 1979. Comparison of direct and standard antimicrobial disk susceptibility testing for bacteria isolated from blood. Journal of Clinical Microbiology 10(4):482–7.
Morrell, M. R., S. T. Micek, and M. H. Kollef. 2009. The management of severe sepsis and septic shock. Infectious Disease Clinics of North America 23(3):485–501.
Parta, M., M. Goebel, M. Matloobi, C. Stager, and D. M. Musher. 2009. Identification of methicillin-resistant or methicillin-susceptible Staphylococcus aureus in blood cultures and wound swabs by GeneXpert. Journal of Clinical Microbiology 47(5):1609–10.
Paterson, D. L., W. C. Ko, A. Von Gottberg, J. M. Casellas, L. Mulazimoglu, K. P. Klugman, R. A. Bonomo, L. B. Rice, J. G. McCormack, and V. L. Yu. 2001. Outcome of cephalosporin treatment for serious infections due to apparently susceptible organisms producing extended-spectrum beta-lactamases: Implications for the clinical microbiology laboratory. Journal of Clinical Microbiology 39(6):2206–12.
Paterson, D. L., W. C. Ko, A. Von Gottberg, S. Mohapatra, J. M. Casellas, H. Goossens, L. Mulazimoglu, G. Trenholme, K. P. Klugman, R. A. Bonomo, L. B. Rice, M. M. Wagener, J. G. McCormack, and V. L. Yu. 2004. International prospective study of Klebsiella pneumoniae bacteremia: Implications of extended-spectrum beta-lactamase production in nosocomial infections. Annals of Internal Medicine 140(1):26–32.
Perez, F., A. M. Hujer, K. M. Hujer, B. K. Decker, P. N. Rather, and R. A. Bonomo. 2007. Global challenge of multidrug-resistant Acinetobacter baumannii. Antimicrobial Agents and Chemotherapy 51(10):3471–84.
Pitout, J. D., and K. B. Laupland. 2008. Extended-spectrum beta-lactamase-producing Enterobacteriaceae: An emerging public-health concern. Lancet Infectious Diseases 8(3):159–66.
Richter, S. S., and M. J. Ferraro. 2007. Susceptibility testing instrumentation and computerized expert systems for data analysis and interpretation. In Manual of clinical microbiology, 9th ed., edited by P. R. Murray, E. J. Baron, J. H. Jorgensen, M. L. Landry, and M. A. Pfaller. Washington, DC: ASM Press. Pp. 245–56.
Sader, H. S., D. J. Farrell, and R. N. Jones. 2009. Evaluation of vancomycin and daptomycin potency trends (MIC creep) against methicillin-resistant Staphylococcus aureus isolates collected in nine U.S. medical centers from 2002 to 2006. Antimicrobial Agents and Chemotherapy 53(10):4127–32.
Sader, H. S., P. D. Fey, D. N. Fish, A. P. Limaye, G. Pankey, J. Rahal, M. J. Rybak, D. R. Snydman, L. L. Steed, K. Waites, and R. N. Jones. 2010. Antimicrobial susceptibility of Gram-positive cocci isolated from skin and skin-structure infections in European medical centres. International Journal of Antimicrobial Agents 36(1):28–32.
Schwaber, M. J., P. M. Raney, J. K. Rasheed, J. W. Biddle, P. Williams, J. E. McGowan, Jr., and F. C. Tenover. 2004. Utility of NCCLS guidelines for identifying extended-spectrum beta-lactamases in non-Escherichia coli and non-Klebsiella spp. of Enterobacteriaceae. Journal of Clinical Microbiology 42(1):294–8.
Stamper, P. D., M. Cai, T. Howard, S. Speser, and K. C. Carroll. 2007. Clinical validation of the molecular BD GeneOhm StaphSR assay for direct detection of Staphylococcus aureus and methicillin-resistant Staphylococcus aureus in positive blood cultures. Journal of Clinical Microbiology 45(7):2191–6.
Steward, C. D., D. Wallace, S. K. Hubert, R. Lawton, S. K. Fridkin, R. P. Gaynes, J. E. McGowan, Jr., and F. C. Tenover. 2000. Ability of laboratories to detect emerging antimicrobial resistance in nosocomial pathogens: A survey of project ICARE laboratories. Diagnostic Microbiology and Infectious Disease 38(1):59–67.
Steward, C. D., J. M. Mohammed, J. M. Swenson, S. A. Stocker, P. P. Williams, R. P. Gaynes, J. E. McGowan, Jr., and F. C. Tenover. 2003. Antimicrobial susceptibility testing of carbapenems: Multicenter validity testing and accuracy levels of five antimicrobial test methods for detecting resistance in Enterobacteriaceae and Pseudomonas aeruginosa isolates. Journal of Clinical Microbiology 41(1):351–8.
Swenson, J. M., J. B. Patel, and J. H. Jorgensen. 2007. Special phenotypic methods for detecting antibacterial resistance. In Manual of clinical microbiology, 9th ed., edited by P. R. Murray, E. J. Baron, J. H. Jorgensen, M. L. Landry, and M. A. Pfaller. Washington, DC: ASM Press. Pp. 1173–92.
Tenover, F. C. 2001. Development and spread of bacterial resistance to antimicrobial agents: An overview. Clinical Infectious Diseases 33(Suppl. 3):S108–15.
Tenover, F. C. 2006. Mechanisms of antimicrobial resistance in bacteria. American Journal of Infection Control 34(5 Suppl. 1):S3–10; discussion S64–73.
Tenover, F. C. and J. Hindler. 2010. Reporting antimicrobial susceptibility testing results. In Antibiogram, edited by P. Courvalin, R. LeClercq, and L. B. Rice. Washington, DC: ASM Press.
Tenover, F. C., M. V. Lancaster, B. C. Hill, C. D. Steward, S. A. Stocker, G. A. Hancock, C. M. O’Hara, S. K. McAllister, N. C. Clark, and K. Hiramatsu 1998. Characterization of staphylococci with reduced susceptibilities to vancomycin and other glycopeptides. Journal of Clinical Microbiology 36(4):1020–7.
Tenover, F. C., M. J. Mohammed, T. S. Gorton, and Z. F. Dembek. 1999. Detection and reporting of organisms producing extended-spectrum beta-lactamases: Survey of laboratories in Connecticut. Journal of Clinical Microbiology 37(12):4065–70.
Tenover, F. C., M. J. Mohammed, J. Stelling, T. O’Brien, and R. Williams. 2001. Ability of laboratories to detect emerging antimicrobial resistance: Proficiency testing and quality control results from the World Health Organization’s external quality assurance system for antimicrobial susceptibility testing. Journal of Clinical Microbiology 39(1):241–50.
Tenover, F. C., L. M. Weigel, P. C. Appelbaum, L. K. McDougal, J. Chaitram, S. McAllister, N. Clark, G. Killgore, C. M. O’Hara, L. Jevitt, J. B. Patel, and B. Bozdogan. 2004. Vancomycin-resistant Staphylococcus aureus isolate from a patient in Pennsylvania. Antimicrobial Agents and Chemotherapy 48(1):275–80.
Tenover, F. C., R. K. Kalsi, P. P. Williams, R. B. Carey, S. Stocker, D. Lonsway, J. K. Rasheed, J. W. Biddle, J. E. McGowan, Jr., and B. Hanna. 2006. Carbapenem resistance in Klebsiella pneumoniae not detected by automated susceptibility testing. Emerging Infectious Diseases 12(8):1209–13.
Tenover, F. C., P. P. Williams, S. Stocker, A. Thompson, L. A. Clark, B. Limbago, R. B. Carey, S. M. Poppe, D. Shinabarger, and J. E. McGowan, Jr. 2007. Accuracy of six antimicrobial susceptibility methods for testing linezolid against staphylococci and enterococci. Journal of Clinical Microbiology 45(9):2917–22.
Tenover, F. C., S. L. Emery, C. A. Spiegel, P. A. Bradford, S. Eells, A. Endimiani, R. A. Bonomo, and J. E. McGowan, Jr. 2009. Identification of plasmid-mediated AmpC beta-lactamases in Escherichia coli, Klebsiella spp., and Proteus species can potentially improve reporting of cephalosporin susceptibility testing results. Journal of Clinical Microbiology 47(2):294–9.
Turnidge, J., and D. L. Paterson. 2007. Setting and revising antibacterial susceptibility breakpoints. Clinical Microbiology Reviews 20(3):391–40.
Whitener, C. J., S. Y. Park, F. A. Browne, L. J. Parent, K. Julian, B. Bozdogan, P. C. Appelbaum, J. Chaitram, L. M. Weigel, J. Jernigan, L. K. McDougal, F. C. Tenover, and S. K. Fridkin. 2004. Vancomycin-resistant Staphylococcus aureus in the absence of vancomycin exposure. Clinical Infectious Diseases 38(8):1049–55.
Wolk, D. M., M. J. Struelens, P. Pancholi, T. Davis, P. Della-Latta, D. Fuller, E. Picton, R. Dickenson, O. Denis, D. Johnson, and K. Chapin. 2009. Rapid detection of Staphylococcus aureus and methicillin-resistant S. aureus (MRSA) in wound specimens and blood cultures: Multicenter preclinical evaluation of the Cepheid Xpert MRSA/SA skin and soft tissue and blood culture assays. Journal of Clinical Microbiology 47(3):823–6.
Wright, G. D. 2007. The antibiotic resistome: The nexus of chemical and genetic diversity. Nature Reviews, Microbiology 5(3):175–86.
Yigit, H., A. M. Queenan, G. J. Anderson, A. Domenech-Sanchez, J. W. Biddle, C. D. Steward, S. Alberti, K. Bush, and F. C. Tenover. 2001. Novel carbapenem-hydrolyzing beta-lactamase, KPC-1, from a carbapenem-resistant strain of Klebsiella pneumoniae. Antimicrobial Agents and Chemotherapy 45(4):1151–61.
A20
MEASURING THE COST OF ANTIMICROBIAL-RESISTANT INFECTIONS: THE FEASIBILITY AND ACCURACY OF ECONOMIC ANALYSIS USING ELECTRONIC MEDICAL RECORD DATABASES
Rebecca R. Roberts,73,77 Linda M. Kampe,74 Ibrar Ahmad,73 Bala Hota,75,77 Edward K. Mensah,76 and Robert A. Weinstein75,77
Introduction
The goal of conducting economic analyses in health is to inform public health and medical decision making. Policy makers are currently working to address the high and rising costs of U.S. health care (Orszag and Ellis, 2007). Complicating these planning efforts, antimicrobial resistance continues to emerge and spread in organisms that infect humans (Avorn et al., 2001; Cosgrove, 2006; Dellit et al., 2007; Goldmann et al., 1996; Graves and McGowan, 2008; IDSA, 2004; Klevens et al., 2007; Maragakis et al., 2008; Spellberg et al., 2008; Streit et al., 2004; Thursky, 2006; Weinstein, 2001; Zaoutis, 2009; Zell and Goldmann, 2007). We recently reported the hospital and societal costs for antimicrobial-resistant infection (ARI) at our hospital (Roberts et al., 2009). We found that just 188 patients with ARI cost our hospital between $18,588 and $29,069 per
patient, resulted in 1,200 to 2,300 excess hospital inpatient days, and doubled patient mortality. Based on the lowest estimate in the sensitivity analysis, the minimum total cost was $13.35 million when converted to 2008 U.S. dollars. However, intensive chart reviews were required to complete that study, resulting in significant delay in calculating these costs and in restriction of analysis to 1 year at a single hospital. As part of healthcare strategic planning, a new national goal has been to expand the implementation of electronic medical records (EMRs) to improve efficiency, minimize errors, and more rapidly conduct large multicenter research trials (Benin et al., 2005; Brant et al., 2006; Cebul, 2008; Dorr et al., 2007; Miller and West, 2007; Onukwugha et al., 2008; Ovretveit et al., 2007; Shcherbatykh et al., 2008; Shekelle et al., 2006; Smith et al., 2009; Tang et al., 2007; Thursky, 2006; Titler et al., 2008). Currently, healthcare facilities differ in their planned use of EMR modalities (Dorr et al., 2007; Greenhalgh et al., 2009; Peabody et al., 2004; Shekelle et al., 2006; Tang et al., 2007). Some may only have administrative, financial, or claims databases, while others have implemented clinical, laboratory, pharmacy, and radiological databases. Our hospital has implemented EMRs and the data have been aggregated in a relational database by the Chicago Antimicrobial Resistance Project (Wisniewski et al., 2003). This presents the opportunity to evaluate the feasibility and accuracy of conducting real-time economic analyses using EMRs rather than laborious chart review. To address the variation in economic data based on using EMRs, we present here an ARI cost analysis using sequential averaging of total cost per individual patient. The goal was to examine the difference in cost analysis results as less precise cost measures were used. We hope this will enable policy makers to weigh the cost of EMR expansion against the need for more accurate and timely cost analyses, which can be used in determining public health policy for disease management and prevention of adverse outcomes such as ARI.
Cost measures have included length of stay, cost per day, hospital bills, cost-to-charge ratios, and hospital reimbursement by third-party payers (Gold et al., 1996; Neumann, 2005; Roberts et al., 2006). When measuring the costs of health interventions, using an average unit cost per hospital day might fail to identify important individual patient differences in resource use or cost. In our prior work, we measured healthcare costs from the hospital perspective using microcosting (Roberts et al., 1997, 1999, 2003, 2006, 2009). Hospital services include length of stay, which is usually measured in days. Because intensive care units (ICUs) are more costly per day than are regular wards or observation units, total costs will differ based on where the patient was treated. The next element in cost measurement is to determine what happened each day while the patient was hospitalized. Significant resources include specialty consultations, bedside procedures, laboratory and radiologic tests, medications given, blood product transfusions, and operating room procedures. Patient factors also matter. For example, patients
with the same diagnosis differ in their severity of illness and number of additional comorbidities. Treatment plans also differ greatly. One patient with metastatic lung cancer may have a new diagnosis and generate high costs due to expensive staging tests and life-prolonging treatments, while another with advanced disease may elect for do-not-resuscitate status and receive only hospice care with fluids and pain medication. EMR databases vary in their ability to measure all of the elements that determine cost results. Studies using only length of stay or claims data may have different results when compared to measurements that include more clinical and cost elements (Jollis et al., 1993). In this project, we compare cost analysis results while using progressively less precise sources of data in the same study sample.
Methods
Overview
This is a secondary analysis of an existing data set, where data were obtained from both EMR sources and a review of the entire written medical record. The methods are fully described in earlier reports (Roberts et al., 1997, 1999, 2003, 2006, 2009, 2010). The data were categorized into cost elements that reflected their electronic availability and contribution to individual variation in total cost. The precision of total cost for each patient was reduced sequentially by subtracting individual cost elements and substituting the average cost for the entire sample. For example, unit costs for each medication and laboratory test for each individual patient were replaced by the daily average pharmacy and laboratory cost for the entire sample of patients. For the purpose of this report, we named the original total “precise cost.” We compared the results for each sequentially less precise total to the original precise cost to quantify any loss of accuracy as progressively less information was used in the measurement. In addition, assessment of severity of illness using the Acute Physiology and Chronic Health Evaluation (APACHE III) score and detection of concurrent healthcare-acquired infection (HAI) required written chart review (Garner et al., 1988; Horan et al., 1992; Knaus et al., 1991). We report cost analyses with and without these elements to determine the effect on results.
For each sequentially averaged total cost, the attributable cost for ARI was estimated using ordinary least squares (OLS) linear regression models and case matching based on propensity scores (Austin, 2008; Kleinbaum et al., 2008; Kurth et al., 2005). The results were then compared to our previously published results using the precise cost (Roberts et al., 2009). Calculations and analyses included all outliers and were completed using SAS (version 9.2, SAS Institute, Inc., Cary, NC) and Microsoft Excel (version 2002, Microsoft Corporation, Redmond, WA). The institutional review board of the hospital deemed this study exempt from review.
Sample Selection
A multistep sample selection process was performed. A random sample of adult patients having greater than five International Classification of Disease, 9th Edition, Clinical Modification (ICD-9-CM) codes at discharge was selected from our urban public hospital. Those hospitalized for burn, trauma, or obstetrical care were excluded. Additional patients with culture-demonstrated ARI were subsequently selected from the same eligible pool, resulting in a total of 1,391 sample patients.
Measurements
All medical costs were measured from the hospital perspective. In the original total cost calculation, EMR sources were used for pharmacy, laboratory, radiology, and length of stay by ward type (ICU, telemetry, and regular ward). Chart review was required for emergency department, consultations, blood product transfusions, bedside procedures (endoscopy, bronchoscopy, cardiac procedures, and hemodialysis), and operating room minutes. For the APACHE III score, EMR sources were used for age and laboratory results. Chart review was used for mental status, urine output, and comorbidities.
Alternate Total Cost and Severity-of-Illness Calculations
The first step was to calculate the average cost per day for each element. Those cost elements (by increasing order of contribution to total daily cost) were emergency department visit, consultations, bedside procedures, radiology, blood product transfusions, pharmacy, laboratory, and location of care (ICU or wards). For surgical patients, the cost for operating room minutes was also a significant contributor to total cost. The cost-averaging process selected was based on (1) the relative contribution to total cost, (2) the importance to cost variation between ARI and non-ARI patients, and (3) the relative ease of data acquisition from the EMR. The original total precise cost added the unit cost for each individual resource used by each patient. In the averaging process, the mean daily cost for each cost element for the entire sample was multiplied by the individual length of stay (LOS) for each patient. For cost 1, the average daily cost for emergency department visits, consultations, bedside procedures, and radiology were averaged for the entire sample and used to replace individual patient costs in those categories. Cost 2 was the same as cost 1, but each individual patient use of blood products was replaced with the sample mean daily cost for blood products multiplied by LOS. Cost 3 was the same as cost 2, with the additional daily averaging of pharmacy and laboratory. Cost 4 was the same as cost 3, except the average daily ward cost for ICU patients, telemetry, and ward patients was averaged and
multiplied by LOS for individual patients. Cost 4 represents LOS multiplied by average cost per day, plus surgical costs. The only additional change in cost 5 was to average all patient surgical procedure costs and substitute that average surgery cost for individual surgical costs, which were based on operating room minutes. The sample average surgery cost used was $7,452.85. Our methods presume that hospital LOS and surgical procedure events would always be available in the EMR.
In addition to resource use for cost measurements, the EMR and written chart were used to determine the highest APACHE III score for each patient in the first 24 hours after admission. The APACHE III score was included in the economic models to control for the cost changes associated with severity of illness. Elements of the APACHE III that were sequentially deleted from the individual patient score and replaced by a sample average were mental status, urine output, vital signs, and laboratory results. Our administrative database included patient diagnosis-related group (DRG) and up to 12 ICD-9-CM codes that could be used to measure comorbidities using the EMR. However, we did not use these codes in the earlier study because they were not yet mapped to diagnoses in our clinical data warehouse. Therefore, chart review was used for comorbidity measurement.
Analysis
In the first methodology, we used OLS linear regression to estimate the cost attributable to ARI (Cody and Smith, 2006; Kleinbaum et al., 2008; Roberts et al., 2003). The goal was to include factors that predicted increased total cost in the economic models. Prior work had identified severity of illness (APACHE III score), ICU care, surgery, and concurrent HAI as cost predictors. In the analysis for all sample patients, model I variables were base intercept, APACHE III score, ICU care, and surgery. The HAI variable was added in model II to control for the cost associated with HAI. Because HAI was not measured electronically, but required chart review, model II represents an increase in precision compared to model I. A second series of OLS models was created without APACHE III scores to determine the value of the severity-of-illness measure in cost prediction. Next, in ICU patients only, sequential averaging of APACHE III scores was done. Those scores were used in an alternate model I for ICU patients using the original precise cost.
The second analytic method compared the cost for ARI patients to the cost of matched non-ARI cases. Matching was done using propensity scores (Austin, 2008; Kurth et al., 2005; Roberts et al., 2009). The variables tested for inclusion in the propensity scores were ICU care, surgery, HAI, and the following comorbidities: any renal disease, renal failure, acute myocardial infarction, congestive heart failure, peripheral vascular disease, stroke, diabetes, diabetes with
complications, any liver disease, hepatic failure, cirrhosis, dementia, collagen vascular disease, chronic obstructive pulmonary disease, cancer, and AIDS. To identify factors that were independently associated with ARI, stepwise logistic regression was used. Only those factors significantly predictive of ARI (P < 0.05) were included in the propensity scores. Each ARI case was matched to a non-ARI case using this propensity score. Student’s t-tests were used to compare the difference in total costs for each ARI versus non-ARI matched pair. The mean differences reported represent the excess cost of ARI.
Results
There were 23,904 patients hospitalized at our facility in the year 2000, and 4,944 met the eligibility criteria. The random sample yielded 1,253 patients. Another 138 patients were identified with ARI from the same eligibility pool, for a total of 1,391 patients. The demographic, comorbidity, mortality, and economic sensitivity analyses are described in our earlier report (Roberts et al., 2009). Table A20-1 shows the average cost per hospital day for each cost element. When arranged in order of contribution to daily average cost in all patients, the input order from least to most important was emergency services, consultations, bedside procedures, radiology, blood products, pharmacy, laboratory, and treatment setting (ICU vs. ward). Examination of the differences between mean daily cost for ARI and non-ARI patients reveals little between-group difference in costs for emergency services, consultations, and bedside procedures. In fact, average daily costs for emergency services and consultations were higher for non-ARI patients. In increasing importance, the largest between-group differences in daily cost were for radiology, pharmacy, blood products, treatment setting, and laboratory.
Table A20-2 shows the results of our cost-averaging sequence. This sequence differed slightly from the measured cost differences between ARI and non-ARI patients because our goal was to examine cost accuracy based on contribution to daily cost, drivers of subgroup cost differences, and availability of data in the EMR. For mean cost in all patients, large differences were seen going from cost 2 to cost 3 (6.9 to 16.8 percent) and going from cost 3 to cost 4 (16.8 to 30.1 percent). Thus, measuring precise pharmacy and laboratory resource use, followed by separately measuring LOS in ICU and wards, were most important in the determination of accurate total hospital costs.
Table A20-3 demonstrates that the first important loss of accuracy in attributable ARI cost estimation occurred at cost 4, where ICU care and ward care were replaced by the sample average daily costs. The relative changes in accuracy when going from cost 4 to cost 5 were small, as the economic models included surgery as a cost predictor. It is also important to note that, compared to the pharmacy, laboratory, and nursing unit location data, adjusting for APACHE III scores did not improve accuracy very much for the measured attributable cost for ARI. The results were similar when the attributable cost for ARI among ICU patients
was separately estimated using progressively less accurate APACHE III scores while using the precise cost and economic model I. The attributable cost for ARI using the full APACHE III and precise cost was $51,459 (standard error 6205). Using the same precise cost in the same economic model while replacing the APACHE III scores for mental status with an average resulted in an attributable ARI cost of $50,824. Substituting sequential APACHE III scores with averaged urine output, vital signs, and laboratory data resulted in attributable ARI costs of $51,037, $50,705, and $52,684, respectively (data not shown).
Table A20-4 shows the cost for specific resistant organism subgroups. Greater underestimation of ARI cost occurred at cost 3 for specific resistant organism subgroups, compared with all ARI patients.
Table A20-5 shows the results from model I for treatment subgroups. For both medical and ICU patients, cost 2 resulted in a larger underestimation compared to precise cost than in the surgery or non-ICU subgroups. In contrast to the other analyses, attributable costs for ARI were overestimated in non-ICU patients for all cost estimates when compared to the precise cost. The results using the second analytic method—case matching based on propensity scores—resulted in underestimation of ARI cost, with significant decreased accuracy at cost 3.
Table A20-6 shows the comparison between ARI and non-ARI patients using case matching by propensity scores. The results compared to the precise cost are similar. The largest loss of accuracy occurred in cost 3.
Conclusion
In summary, as less precise data were included in the cost measurement, progressive underestimation of the attributable cost for ARI resulted. Furthermore, the greater the degree of daily averaging, the greater the underestimation of ARI cost. This was true in all cases except non-ICU patients, a result that would be expected because the more costly ICU patients were included in the average. In most instances, the loss of pharmacy, laboratory, and treatment-setting data had the greatest impact on the magnitude of cost underestimation. This would suggest that cost analysis evaluating the potential healthcare cost savings from ARI prevention should include these data. This especially makes sense now, because laboratory and pharmacy data are increasingly used in healthcare efficiency and quality improvement programs (Benin et al., 2005; Brant et al., 2006; Cebul, 2008; Dorr et al., 2007; Miller and West, 2007; Onukwugha et al., 2008; Ovretveit et al., 2007; Shcherbatykh et al., 2008; Shekelle et al., 2006; Smith et al., 2009; Tang et al., 2007; Thursky, 2006; Titler et al., 2008). For example, EMRs can be used for automating transmission of required reportable diseases and for matching pharmacy and laboratory data to detect medication errors and improve renal function monitoring (Holtorf et al., 2009; Hota et al., 2007; Loonsk, 2004; Smith et al., 2009).
TABLE A20-1 Mean Cost per Day for Individual Hospital Resources and Cumulative Daily Totals
TABLE A20-2 Differences Between Mean Unadjusted Original Precise Cost Based on Charge Review and Sequential Average Costs by Patient Subgroups
TABLE A20-3 Attributable Cost for Antimicrobial-Resistant Infection Using Ordinary Least Squares Regression: Difference Between Precise Cost and Sequential Cost Averages
|
Cost 1: Daily cost for ED, consults, radiology, and bedside procedures were averaged. Cost 2: Daily cost for blood product transfusions were averaged. Cost 3: Daily cost for pharmacy and laboratory were averaged. Cost 4: Daily cost for inpatient regular wards and ICU were averaged. Cost 5: Average cost for all sample patient surgical procedures was substituted for surgical patient actual surgery cost. ARI, antimicrobial- resistant infection; ED, emergency department; HAI, healthcare- acquired infection; ICU, intensive care unit; SE, standard error. |
TABLE A20-4 Resistant Organism Subgroups: Difference Between Precise Cost and Sequential Cost Averages for Ordinary Least Squares Linear Regression
|
Model I Confounders Used in Regression: APACHE III, Surgery, ICU |
MRSA |
|
|
Difference Between Precise Cost and Costs 1–6 |
|
|
|
US$ |
SE |
R2 |
US$ |
% |
|
Precise cost |
18,380 |
2605 |
0.54 |
|
|
|
Cost 1 |
18,191 |
2641 |
0.53 |
189 |
1.03 |
|
Cost 2 |
18,030 |
2523 |
0.53 |
350 |
1.90 |
|
Cost 3 |
15,411 |
2067 |
0.54 |
2,969 |
16.15 |
|
Cost 4 |
14,390 |
1892 |
0.51 |
3,990 |
21.71 |
|
Cost 5 |
13,919 |
1833 |
0.51 |
4,461 |
24.27 |
|
|
VRE |
|
|
|
|
|
Precise cost |
33,944 |
3062 |
0.54 |
|
|
|
Cost 1 |
33,774 |
3104 |
0.53 |
170 |
0.50 |
|
Cost 2 |
30,949 |
2965 |
0.53 |
2,995 |
8.82 |
|
Cost 3 |
25,413 |
2430 |
0.54 |
8,531 |
25.13 |
|
Cost 4 |
24,415 |
2224 |
0.51 |
9,529 |
28.07 |
|
Cost 5 |
23,420 |
2154 |
0.51 |
10,524 |
31.00 |
|
|
AREK |
|
|
|
|
|
Precise cost |
**8,241 |
4075 |
0.54 |
|
|
|
Cost 1 |
***8,028 |
4131 |
0.53 |
213 |
2.58 |
|
Cost 2 |
**8,295 |
3946 |
0.53 |
54 |
0.66 |
|
Cost 3 |
*9,536 |
3234 |
0.54 |
1,295 |
15.71 |
|
Cost 4 |
9,929 |
2960 |
0.51 |
1,688 |
20.48 |
|
Cost 5 |
10,121 |
2867 |
0.51 |
1,880 |
22.81 |
|
|
AIR |
|
|
|
|
|
Precise cost |
48,723 |
8122 |
0.54 |
|
|
|
Cost 1 |
46,691 |
8233 |
0.53 |
2,032 |
4.17 |
|
Cost 2 |
41,093 |
7865 |
0.53 |
7,630 |
15.66 |
|
Cost 3 |
27,028 |
6445 |
0.54 |
21,695 |
44.53 |
|
Cost 4 |
*18,523 |
5899 |
0.51 |
30,200 |
61.98 |
|
Cost 5 |
*15,575 |
5714 |
0.51 |
33,148 |
68.03 |
TABLE A20-5 Treatment Setting Subgroups: Difference Between Precise Cost and Sequential Average Costs When Using Ordinary Least Squares Linear Regression to Estimate the Attributable Cost for Antimicrobial-Resistant Infection
|
Model I Confounders Used in Regression: APACHE III, ICU, HAI |
|
|
|
Difference from Precise Cost |
|
|
|
US$ |
SE |
R2 |
US$ |
% |
|
|
Medical |
|
|
|
|
|
Precise cost |
12,949 |
1815 |
0.42 |
|
|
|
Cost 1 |
12,181 |
1860 |
0.41 |
768 |
5.93 |
|
Cost 2 |
11,024 |
1679 |
0.44 |
1,925 |
14.87 |
|
Cost 3 |
9,300 |
1474 |
0.44 |
3,649 |
28.18 |
|
Cost 4 |
8,678 |
1393 |
0.38 |
4,271 |
32.98 |
|
Cost 5 |
8,711 |
1396 |
0.38 |
4,238 |
32.73 |
|
|
Surgical |
|
|
|
|
|
Precise cost |
33,360 |
7747 |
0.40 |
|
|
|
Cost 1 |
33,270 |
7734 |
0.40 |
90 |
0.27 |
|
Cost 2 |
31,625 |
7597 |
0.39 |
1,735 |
5.20 |
|
Cost 3 |
24,550 |
5793 |
0.39 |
8,810 |
26.41 |
|
Cost 4 |
21,979 |
5016 |
0.34 |
11,381 |
34.12 |
|
Cost 5 |
20,477 |
4718 |
0.31 |
12,883 |
38.62 |
|
Model I Confounders Used in Regression: APACHE III, Surgery, HAI |
|
|
|
Difference from Precise Cost |
|
|
|
Non ICU |
|
|
|
|
|
Precise cost |
3,973 |
886 |
0.41 |
|
|
|
Cost 1 |
3,996 |
963 |
0.38 |
−2 |
−0.5 |
|
Cost 2 |
4,234 |
980 |
0.38 |
−26 |
−6.5 |
|
Cost 3 |
4,995 |
1041 |
0.38 |
−1,02 |
−25.7 |
|
Cost 4 |
6,177 |
1220 |
0.37 |
−2,20 |
−55.4 |
|
Cost 5 |
6,404 |
1202 |
0.41 |
−2,43 |
−61.1 |
|
|
ICU |
|
|
|
|
|
Precise cost |
38,077 |
6946 |
0.35 |
|
|
|
Cost 1 |
36,493 |
6976 |
0.35 |
1,584 |
4.16 |
|
Cost 2 |
33,106 |
6576 |
0.36 |
4,971 |
13.06 |
|
Cost 3 |
24,699 |
5139 |
0.36 |
13,378 |
35.13 |
|
Cost 4 |
20,776 |
4317 |
0.37 |
17,301 |
45.44 |
|
Cost 5 |
19,458 |
4178 |
0.36 |
18,619 |
48.90 |
TABLE A20-6 Differences Between Original Cost and Sequential Cost Averages When Comparing Patients with Antimicrobial-Resistant Infection to Matched Controls
Benefits of EMR
The original study took nearly 6 years to complete. One major benefit will be quicker completion of research and timely dissemination of results. Another benefit was increased internal validity of unit cost measures. The same database sources for laboratory, radiology, pharmacy, and nursing unit LOS were used to measure individual patient resources used and total department outputs used in unit cost calculations. The pharmacy database also included medication costs for each patient and the entire hospital for that study year.
Lessons Learned
Length of stay can be easily miscalculated. For a total length of stay, the correct number is the discharge date minus the admission date, plus one day. We also had a number of patients who changed hospital location several times during their hospital stay. They went from the emergency department to the wards, then worsened and transferred to the ICU, then went back to the wards. Our EMR database collects a daily census report that shows the location of every patient on every nursing unit. When that location changes at the next census 24 hours later, a decision must be made about which ward the patient was on for the majority of that 24 hours. This decision will matter if the cost difference between an ICU and regular ward is great. We recommend using an algorithm that is counterintuitive. If the individual is an ARI patient and is expected to have high costs, use the lower cost ward for that uncertain day. If the patient does not have an ARI, use the higher cost ward. The use of unavoidable bias to refute a study hypothesis will strengthen it (Popper, 1989). For pediatrics, we revised our algorithm because some patients had turned 18 in the study year, but were still only 17 when they were in the hospital. We now use exact birth date rather than birth year.
Continued Challenges
The propensity scores employed comorbidities that were available electronically as ICD-9-CM and DRG codes. We are working to map these administrative codes against clinical diagnoses. Our algorithm for selecting eligible patients missed some burn, trauma, and obstetrical patients that we had intended to exclude. This was corrected using chart review. Some of those patients were outside transfers to our tertiary care facility, while others went to unlikely ward locations due to overcrowding. As we validate the use of DRG, ICD-9-CM or ICD-10-CM, and current procedural terminology (CPT) codes to determine diagnosis and treatment, this will be less of a problem (Elixhauser et al., 1998). It was unexpected that the APACHE III score would have such a minor effect on the accuracy of our cost results. However, others have found APACHE scores to be useful for predicting ICU LOS, while our study used direct LOS measures and only varied the cost-
per-day averages (Zimmerman et al., 2006). HAI are also a high-cost hospital complication and are strongly correlated with antimicrobial resistance (Cummings et al., 2010; Lipsitch et al., 2000; Roberts et al., 2009; Weinstein, 2001; Weinstein et al., 1999). As prevention programs develop, different hospitals may witness varied trends in both infection types (Weinstein et al., 1999). It will be important to differentiate between increased costs due to HAI and increased costs due to ARI. ARI is not a problem to detect electronically in a facility with electronic laboratory data; the future challenge will be to develop sensitive and specific algorithms for detecting patients with HAI (Hota et al., 2007; Trick et al., 2004). Economic model I does not factor out the effect of HAI. Many of the programs that address ARI are also aimed at reducing HAI; model I economic results may be a realistic estimate of cost savings. This also highlights the fact that much of our most useful cost data was from sources that are directly automated, such as laboratory results. The data go directly from a clinical laboratory machine to a health information system with little human interference. The accuracy of data entry and ability to retrieve reliable clinical data for use in research will need further evaluation (Benin et al., 2005; Welker, 2007; Wong et al., 2008).
Our microcosting methods required data entry of all department employee numbers, salaries, benefits, department square footage, vendor, contractual and utility expenditures, and department outputs. Multiple distribution cost allocation algorithms were then written to determine unit costs for resources used. Combining annual expenditure reports or Medicare cost reports and annual department outputs with these algorithms will simplify the unit cost measurement process in the future.
Summary
Accurate assessments of the attributable medical costs for ARI can be made using EMR databases. To maintain accuracy for cost calculations at an individual patient level, the key data elements needed in an EMR were pharmacy use, laboratory testing, location of care on ICUs and wards, and, to a lesser extent, blood product transfusions. The average cost for operating room procedures could be substituted for exact operating room minute costs if the occurrence of any operating room surgery was known and included as a variable in the economic models. Cost elements that required our review of written records—but can now be automated in our EMRs—such as number of consultations, bedside procedures, and APACHE III scores, had far less influence on the final attributable cost results. Our findings present the opportunity to conduct real-time EMR-based cost analyses as part of the evaluation of clinical interventions and prevention programs.
Acknowledgments
This project was funded in part by the Centers for Disease Control and Prevention Chicago Epi-Center grant number 5U01CI000327. None of the authors have any conflicts of interest. We thank Piotr Kieszkowski, Mary Wisniewski, and Yingxu Xiang for their ground-breaking work in constructing the CARP relational database, LaDwyna Williams and her Department of Medical Records for their contributions, and our research associates who ensured these data were collected and accurate: Dr. Fauzia Abbasi, Dr. Ginevra Ciavarella, Dr. Omer Nazeer, Sheena Lee, Nabiha Shamsi, Shawn Prakash, Rohan Shah, and Mark Brodie.
References
Austin, P. C. 2008. A critical appraisal of propensity-score matching in the medical literature between 1996 and 2003. Statistics in Medicine 27(12):2037–49.
Avorn, J. L., J. F. Barrett, P. G. Davey, S. A. McEwen, T. F. O’Brien, and S. B. Levy. 2001. Antibiotic resistance: Synthesis of recommendations by expert policy groups. WHO/CDS/CSR/DRS/2001.10. Boston, Massachusetts: Alliance for the Prudent Use of Antibiotics, World Health Organization. 155 pp.
Benin, A. L., G. Vitkauskas, E. Thornquist, E. D. Shapiro, J. Concato, M. Aslan, and H. M. Krumholz. 2005. Validity of using an electronic medical record for assessing quality of care in an outpatient setting. Medical Care 43:691–8.
Brant, C. A., S. Argraves, R. Money, G. Ananth, N. M. Trocky, and P. M. Nadkarni. 2006. Informatics tools to improve clinical research study implemention. Contemporary Clinical Trials 27(2):112–22.
Cebul, R. D. 2008. Using electronic medical records to measure and improve performance. Transactions of the American Clinical and Climatological Association 119:65–75.
Cody, R. P., and J. K. Smith. 2006. Applied statistics and the SAS programming language, 5th ed. Upper Saddle River, NJ: Pearson.
Cosgrove, S. E. 2006. The relationship between antimicrobial resistance and patient outcomes: Mortality, length of hospital stay, and healthcare costs. Clinical Infectious Diseases 42(Suppl. 2):S82–9.
Cummings, K. L., D. J. Anderson, and K. S. Kaye. 2010. Hand hygiene noncompliance and the cost of hospital-acquired methicillin-resistant Staphylococcus aureus infection. Infection Control and Hospital Epidemiology 31(4):357–64.
Dellit, T. H., R. C. Owens, J. E. McGowan, Jr., D. N. Gerding, R. A. Weinstein, J. P. Burke, W. C. Huskins, D. L. Paterson, N. O. Fishman, C. F. Carpenter, P. J. Brennan, M. Billeter, and T. M. Hooton. 2007. Infectious Diseases Society of America and the Society for Healthcare Epidemiology of America guidelines for developing an institutional program to enhance antimicrobial stewardship. Clinical Infectious Diseases 44(2):159–77.
Dorr, D., L. M. Bonner, A. N. Cohen, R. S. Shoai, R. Perrin, E. Chaney, and A. S. Young. 2007. Informatics systems to promote improved care for chronic illness: A literature review. Journal of the American Medical Informatics Association 14:156–63.
Elixhauser, A., C. Stiener, H. Robert, and R. Coffey. 1998. Comorbidity measures for use with administrative data. Medical Care 36(1):8–27.
Garner, J. S., W. R. Jarvis, T. G. Emori, T. C. Horan, and J. M. Hughes. 1988. CDC definitions for nosocomial infections. American Journal of Infection Control 16(3):128–40.
Gold, M. R., J. E. Siegel, L. B. Russell, and M. C. Weinstein, eds. 1996. Cost-effectiveness in health and medicine. New York: Oxford University Press.
Goldmann, D. A., R. A. Weinstein, R. P. Wenzel, O. C. Tablan, R. J. Duma, R. P. Gaynes, J. Schlosser, and W. J. Martone. 1996. Strategies to prevent and control the emergence and spread of antimicrobial-resistant microorganisms in hospitals: A challenge to hospital leadership. Journal of the American Medical Association 275(3):234–40.
Graves, N., and J. E. McGowan. 2008. Nosocomial infection, the Deficit Reduction Act, and incentives for hospitals. Journal of the American Medical Association 300(13):1577–9.
Greenhalgh, T., H. W. Potts, G. Wong, P. Bark, and D. Swinglehurst. 2009. Tensions and paradoxes in electronic patient record research: A systematic literature review using the meta-narrative method. Milbank Quarterly 87(4):729–88.
Holtorf, A. P., C. McAdam-Marx, D. Schaaf, B. Eng, and G. Oderda. 2009. Systematic review on quality control for drug management programs: Is quality reported in the literature? BMC Health Services Research 9(38). http://www.biomedcentral.com/1472-6968/9/38 (accessed June 8, 2010).
Horan, T. C., R. P. Gaynes, W. J. Martone, W. R. Jarvis, and T. G. Emori. 1992. CDC definitions of nosocomial surgical site infections, 1992: A modification of CDC definitions of surgical wound infections. Infection Control and Hospital Epidemiology 13(10):606–8.
Hota, B., C. Ellenbogen, M. Hayden, A. Aroutcheva, T. Rice, and R. Weinstein. 2007. Community associated methicillin-resistant Staphylococcus aureus skin and soft tissue infections at a public hospital: Do public housing and incarceration amplify transmission? Archives of Internal Medicine 167(10):1026–33.
IDSA (Infectious Diseases Society of America). 2004. Bad bugs, no drugs: As antibiotic discovery stagnates, a public health crisis brews. Alexandria, VA: IDSA.
Jollis, J. G., M. Anuckiewicz, E. R. Delong, D. B. Pryor, L. H. Muhlbaier, and D. B. Mark. 1993. Discordance of databases designed for claims payment versus clinical information. Annals of Internal Medicine 119:844–50.
Kleinbaum, D. G., L. L. Kupper, A. Nizam, and K. E. Muller. 2008. Applied regression analysis and other multivariable methods, 4th ed. Belmont, CA: Duxbury Press.
Klevens, R. M., M. A. Morrison, J. Nadle, S. Petit, K. Gershman, S. Ray, L. H. Harrison, R. Lynfield, G. Dumyati, J. M. Townes, A. S. Craig, E. R. Zell, G. E. Fosheim, L. K. McDougal, R. B. Carey, and S. K. Fridkin; Active Bacterial Core surveillance (ABCs) MRSA Investigators. 2007. Invasive methicillin-resistant Staphylococcus aureus infections in the United States. Journal of the American Medical Association 298(15):1763–71.
Knaus, W. A., D. P. Wagner, E. A. Draper, J. E. Zimmerman, M. Bergner, P. G. Bastos, C. A. Sirio, D. J. Murphy, T. Lotring, and A. Damiano. 1991. The APACHE III prognostic system. Risk prediction of hospital mortality for critically ill hospitalized adults. Chest 100(6):1619–36.
Kurth, T., A. M. Walker, R. J. Glynn, K. A. Chan, J. M. Gaziano, K. Berger, and J. M. Robins. 2005. Results of multivariable logistic regression, propensity matching, propensity adjustment, and propensity-based weighting under conditions of nonuniform effect. American Journal of Epidemiology 163(3):262–70.
Lipsitch, M., C. T. Bergstrom, and B. R. Levin. 2000. The epidemiology of antibiotic resistance in hospitals: Paradoxes and prescriptions. Proceedings of the National Academy of Sciences USA 97(4):1938–43.
Loonsk, J. W. 2004. BioSense—A national initiative for early detection and quantification of public health emergencies. Morbidity and Mortality Weekly Report 53(Suppl.):53–5.
Maragakis, L. L., C. N. Perencevich, and S. E. Cosgrove. 2008. Clinical and economic burden of antimicrobial resistance. Expert Review of Anti-Infective Therapy 6(5):751–63.
Miller, R. H., and C. E. West. 2007. The value of electronic health records in community health centers: Policy implications. Health Affairs (Millwood) 26(1):206–14.
Neumann, P. J. 2005. Using cost-effectiveness analysis to improve health care. Oxford, United Kingdom: Oxford University Press.
Onukwugha, E., C. D. Mullins, and S. DeLisle. 2008. Using cost-effectiveness analysis to sharpen formulary decision-making: The example of tiotropium at the Veterans Affairs Health Care System. Value Health 11(5):980–8.
Orszag, P. R., and P. Ellis. 2007. The challenge of rising health care costs—a view from the Congressional Budget Office. New England Journal of Medicine 357:1793–5.
Ovretveit, J., T. Scott, T. G. Rundall, S. M. Shortell, and M. Brommels. 2007. Improving quality through effective implementation of information technology in healthcare. International Journal for Quality in Health Care 19(5):259–66.
Peabody, J. W., J. Luck, S. Jain, D. Bertenthal, and P. Glassman. 2004. Assessing the accuracy of administrative data in health information systems. Medical Care 42(11):1066–72.
Popper, K. R. 1989. Conjectures and refutations: The growth of scientific knowledge, 5th ed. London, United Kingdom: Routledge & Kegan Paul.
Roberts, R., R. J. Zalenski, E. K. Mensah, R. J. Rydman, G. Ciavarella, L. Gussow, K. Das, L. M. Kampe, B. Dickover, M. F. McDermott, A. Hart, H. E. Straus, D. G. Murphy, and R. Rao. 1997. Costs of an emergency department-based accelerated diagnostic protocol vs. hospitalization in patients with chest pain: A randomized controlled trial. Journal of the American Medical Association 278:1670–6.
Roberts, R. R., P. W. Frutos, G. G. Ciavarella, L. M. Gussow, E. K. Mensah, L. M. Kampe, H. E. Straus, G. Joseph, and R. J. Rydman. 1999. Distribution of variable vs fixed costs of hospital care. Journal of the American Medical Association 281(7):644–9.
Roberts, R. R., R. D. Scott II, R. Cordell, S. L. Solomon, L. Steele, L. M. Kampe, W. E. Trick, and R. A. Weinstein. 2003. The use of economic modeling to determine the hospital costs associated with nosocomial infections. Clinical Infectious Diseases 36(11):1424–32.
Roberts, R. R., L. M. Kampe, M. Hammerman, R. D. Scott, T. Soto, G. G. Ciavarella, R. J. Rydman, K. Gorosh, and R. A. Weinstein. 2006. The cost of care for patients with HIV from the provider economic perspective. AIDS Patient Care and STDs 20(12):876–88.
Roberts, R. R., B. Hota, I. Ahmad, R. D. Scott 2nd, S. D. Foster, F. Abbasi, S. Schabowski, L. M. Kampe, G. G. Ciavarella, M. Supino, J. Naples, R. Cordell, S. B. Levy, and R. A. Weinstein. 2009. Hospital and societal costs of antimicrobial-resistant infections in a Chicago teaching hospital: Implications for antibiotic stewardship. Clinical Infectious Diseases 49:1175–84.
Roberts, R. R., R. D. Scott, II, B. Hota, L. M. Kampe, F. Abbasi, S. Schabowski, I. Ahmad, G. G. Ciavarella, R. Cordell, S. L. Solomon, R. Hagtvedt, and R. A. Weinstein. 2010 (in press). Costs attributable to healthcare-acquired infection in hospitalized adults and a comparison of economic methods. Medical Care.
Shcherbatykh, I., A. Holbrook, L. Thabane, L. Dolovich, for Complete III investigators. 2008. Methodologic issues in health informatics trials: The complexities of complex interventions. Journal of the American Medical Informatics Association 15(5):575–80.
Shekelle, P. G., S. C. Morton, and E. B. Keeler. 2006. Costs and benefits of health information technology. Evidence Report/Technology Assessment No. 132. Prepared by the Southern California Evidence-based Practice Center. AHRQ Publication No. 06-E006. Rockville, MD: Agency for Healthcare Research and Quality.
Smith, D. H., A. C. Feldstein, N. A. Perrin, X. Yang, M. M. Rix, M. A. Raebel, D. J. Magid, S. R. Simon, and S. B. Soumerai. 2009. Improving laboratory monitoring of medications: An economic analysis alongside a clinical trial. American Journal of Managed Care 15(5):281–9.
Spellberg, B., R. Guidos, D. Gilbert, J. Bradley, H. W. Boucher, W. M. Scheld, J. G. Bartlett, J. Edwards, Jr. IDSA. 2008. The epidemic of antibiotic-resistant infections: A call to action for the medical community from the Infectious Diseases Society of America. Clinical Infectious Diseases 46(2):155–64.
Streit, J. M., R. N. Jones, H. S. Sader, and T. R. Fritsche. 2004. Assessment of pathogen occurrences and resistance profiles among infected patients in the intensive care unit: Report from the SENTRY Antimicrobial Surveillance Program (North America, 2001). International Journal of Antimicrobial Agents 24(2):111–8.
Tang, P. C., M. Ralston, M. F. Arrigotti, L. Qureshi, and J. Graham. 2007. Comparison of methodologies for calculating quality measures based on administrative data versus clinical data from an electronic health record system: implications for performance measures. Journal of the American Medical Informatics Association 14(1):10–5.
Thursky, K. 2006. Use of computerized decision support systems to improve antibiotic prescribing. Expert Reviews in Anti-Infective Therapy 4(3):491–507.
Titler, M. G., G. A. Jensen, J. M. Dochterman, X. J. Xie, M. Kanak, D. Reed, and L. L. Shever. 2008. Cost of hospital care for older adults with heart failure: Medical, pharmaceutical, and nursing costs. Health Services Research 43(2):635–55.
Trick, W. E., B. M. Zagorski, J. I. Tokars, M. O. Vernon, S. F. Welbel, M. F. Wisniewski, C. Richards, and R. A. Weinstein. 2004. Computer algorithms to detect bloodstream infections. Emerging Infectious Diseases 10(9):1612–20.
Weinstein, J. W., D. Mazon, E. Pantelick, P. Reagan-Cirincione, L. M. Dembry, and W. J. Hierholzer, Jr. 1999. A decade of prevalence surveys in a tertiary-care center: Trends in nosocomial infection rates, device utilization, and patient acuity. Infection Control and Hospital Epidemiology 20(8):543–8.
Weinstein, R. A. 2001. Controlling antimicrobial resistance in hospitals: Infection control and use of antibiotics. Emerging Infectious Diseases 7(2):188–92.
Welker, J. A. 2007. Implementation of electronic data capture systems: Barriers and solutions. Contemporary Clinical Trials 28:329–36.
Wisniewski, M. F., P. Kieszkowski, B. M. Zagorski, W. E. Trick, M. Sommers, and R. A. Weinstein. 2003. Development of a clinical data warehouse for hospital infection control. Journal of the American Medical Informatics Association 10(5):454–62.
Wong, M. C., J. Y. Jiang, J. L. Tang, A. Lam, H. Fung, and S. W. Mercer. 2008. Health services research in the public healthcare system in Hong Kong: An analysis of over 1 million antihypertensive prescriptions between 2004-2007 as an example of the potential and pitfalls of using routinely collected electronic patient data. BMC Health Services Research 8:138. http://www.biomedicentral.com/1472-6963/8/138 (accessed June 8, 2010).
Zaoutis, T. E. 2009. Antibiotic resistance: Who will pay the bills? Clinical Infectious Diseases 49:1185–6.
Zell, B. L., and D. A. Goldmann. 2007. Healthcare-associated infection and antimicrobial resistance: Moving beyond description to prevention. Infection Control and Hospital Epidemiology 28(3):261–4.
Zimmerman, J. E., A. A. Kramer, D. S. McNair, F. M. Malila, and V. L. Shaffer. 2006. Intensive care unit length of stay: Benchmarking based on Acute Physiology and Chronic Health Evaluation (APACHE ) IV. Critical Care Medicine 34(10):2674–6.
A21
THE ANTIBIOTIC RESISTOME78
Gerard D. Wright79
McMaster University80
Importance of the field: Antibiotics are essential for the treatment of bacterial infections and are among our most important drugs. Resistance has emerged to all classes of antibiotics in clinical use. Antibiotic resistance has, proven inevitable and very often it emerges rapidly after the introduction of a drug into the clinic. There is, therefore, a great interest in understanding the origins, scope and evolution of antibiotic resistance.
Areas covered in this review: The review discusses the concept of the antibiotic resistome, which is the collection of all genes that directly or indirectly contribute to antibiotic resistance.
What the reader will gain: The review seeks to assemble current knowledge of the resistome concept as a means of understanding the totality of resistance and not just resistance in pathogenic bacteria.
Take home message: The concept of the antibiotic resistome provides a framework for the study and understanding of how resistance emerges and evolves. Furthermore, the study of the resistome reveals strategies that can be applied in new antibiotic discoveries.
Keywords: antibiotic resistance, efflux, enzymes, evolution, selection
1.
Introduction
Where does antibiotic resistance come from? Experience from the past 60 years of antibiotic use has proven that there are no ‘irresistible’ antibiotics. In fact, the evidence is quite clear that when antibiotics are deployed in the clinic, resistant organisms emerge quickly (Figure A21-1). Clinicians and drug discoverers have managed this situation using two strategies. The first is through the discovery of new antibiotics from both microbial sources and via synthetic chem-
|
78 |
Reprinted from Wright (2010), The Antibiotic Resistome, Expert Opinion on Drug Discovery, 5(8):1-10, Copyright © 2010, Informa Healthcare. Reproduced with permission from Informa Healthcare. Reprinted with permission from Expert Opinion on Drug Discovery. Requests to reproduce copies of this article should be directed to the Publisher, Informa Healthcare (see www.informahealthcare.com). |
|
79 |
Gerard D. Wright, McMaster University, M.G. DeGroote Institute for Infectious Disease Research, Department of Biochemistry and Biomedical Sciences, Hamilton, ON, L8N 3Z5, Canada, E-mail: wrightge@mcmaster.ca |
|
80 |
M. G. DeGroote Institute for Infectious Disease Research, Department of Biochemistry and Biomedical Sciences, Hamilton, ON, Canada |
istry in order to identify novel chemical scaffolds with antimicrobial activity. The second is through the medicinal chemical alteration of ‘old’, or first generation, antibiotic chemical scaffolds to engineer decreased sensitivity to resistance mechanisms and improve pharmacology.
The first stratagem resulted in the so-called Golden Age of antibiotics, roughly from the implementation of penicillin in the clinic in the early 1940s to the discovery of nalidixic acid, the progenitor of the fluoroquinolone antibiotics, in 1962. During this 2-decade period, the majority of antibiotic chemical scaffolds in current clinical use were discovered including the β-lactams (penicillins and cephalosporins), macrolides, aminoglycosides, tetracyclines, glycopeptides and fluoroquinolones (Figure A21-1) (Fischbach and Walsh, 2009). Overlapping this period and extending into the present day has been a second ‘Golden Age’, this time in medical chemistry. During this period, many of the antibiotic scaffolds were elaborated into better drugs. This continues to be a highly profitable approach to antibiotic discovery and improvement. In some cases such as the
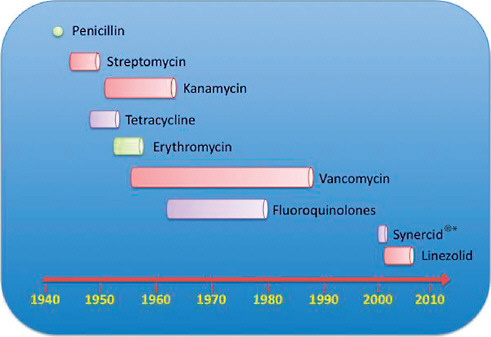
FIGURE A21-1 Antibiotic discovery and resistance. A summary of selected examples of antibiotic entry into clinical practice and the emergence of resistance in pathogens. The beginning of the bar denotes beginning of clinical use and the termination of the bar where resistance in pathogens has become a significant clinical issue. Note that for penicillin (here shown as a circle), resistance pathogens were identified even before the antibiotic was launched into wider clinical use.
*is a trade name for an antibiotic combination of dalfopristin and quinupristin.
cephalosporins, we are now into the fifth generation molecules reflecting the structural changes made to the scaffold to improve activity and avoid resistance over the years.
Yet, every new compound and every new generation of an older scaffold eventually faces the emergence of resistance. This inevitability speaks of a remarkable defense system available to bacteria to counteract the toxic effect of small molecules. Understanding the scope and mechanisms of this system is essential to the discovery of new drugs and the stewardship of new and old antibiotics.
2.
The Antibiotic Resistome
The inevitability of resistance has led us to propose the concept of the antibiotic resistome (Figure A21-2) (Fischbach and Walsh, 2009; D’Costa et al., 2007; Wright, 2007). The resistome consists of the totality of antibiotic resistance genes in pathogens, antibiotic producers and benign environmental bacteria. Additionally, because resistance evolves from precursor genes encoding metabolic or ‘housekeeping’ tasks, the resistome encompasses these genes, which we have termed protoresistance elements (Morar et al., 2009). Finally, resistance to antibiotics at the organism level can arise from the complex interplay of genes and their products arising from exposure to toxic molecules (Breidenstein et al., 2008; Fajardo et al., 2008; Tamae et al., 2008). The resistome, therefore, also includes the intrinsic systems biology of organisms that results in evasion of the activity of antibiotics by an organism.
Because of horizontal gene transfer (HGT) between microbes of diverse species and genera, a parsimonious view of antibiotic resistance is that mechanisms that lead to resistance in one organism, whether or not it is a pathogen, have the potential to emerge in clinically important bacteria. Therefore, understanding of the resistome as a whole and not just as mechanisms that have already emerged in pathogens is vital to the development of new antibiotics and the preservation of those already in clinical use.
Antibiotic resistance can be the result of several general mechanisms. These include: i) alteration of the macromolecular drug target either through chemical modification or by mutation to insensitive variants; ii) protection of the target via the production of immunity proteins; iii) direct chemical modification of the antibiotic generally through the activity of enzyme catalysts; iv) altered transport of the compounds into the cell; or conversely v) increased efflux of the drug out of the cell. Intrinsic insensitivity to antibiotics via physiology or innate genotype should also be included in this survey of resistance mechanisms. These can include the presence of cell structures such as the relatively impermeant outermembrane of Gram-negative bacteria, physiologically quiescent states such as biofilms or spores, as well as the intrinsic redundant networks of genes and proteins that result in (often species-specific) insensitivity to antibiotics. While these mechanisms could be construed less as resistance but more as
FIGURE A21-2 The antibiotic resistome. The resistome comprises of all the genes and their products that contribute to antibiotic resistance. As a result of HGT, and co-optation of chemical mechanisms, the resistome is highly redundant and interlocked. Resistance in the clinic has been the focus of much of the study and literature of the past 7 decades; however, it significantly under represents the resistance capacity of bacteria. High level antibiotic resistance can be found in environmental bacteria (antibiotic producers and many others); furthermore, the presence of intrinsic elements such as efflux systems can also contribute. Ultimately, highly efficient resistance elements are derived from existing biochemical mechanisms, the protoresistome, that serves as a deep reservoir of precursors that can be co-opted and evolved to bone fide resistance elements. The figure, therefore, shows the resistome as a series of concentric circles of different sizes to emphasize the relative number of genes involved and their interrelatedness and ancestral relationship, but does not necessarily suggest a uniform trajectory for all genes. HGT: Horizontal gene transfer.
antibiotic insensitivity because a drug-sensitive target is generally still present, from the perspective of both users and discoverers of antibiotics, the drugs are ineffective and, therefore, we include such a mechanism in the resistome. The resistome, therefore, is a complex array of genes and their functions that directly or indirectly act to block the activity of antibiotics. Adding to the complexity of the situation is that very often resistant strains can harbor several mechanisms, resulting in combinatorial resistance (Figure A21-3), which makes understanding of the contribution of each mechanism to the overall phenotype a challenge and, in practical terms, complicates clinical therapy.
3.
Protoresistance Elements
Regardless of precise molecular mechanism, resistance does not arise spontaneously but must evolve from genes encoding ‘housekeeping’ or metabolic proteins that have little or no contribution to resistance. Nevertheless, through natural selection, such precursor or protoresistance genes (Morar et al., 2009) have given
FIGURE A21-3 Combinatorial resistance. In many cases, antibiotic resistance in organisms is not only the result of a single gene product, but several mechanisms can contribute including alternate transport, the physiological state of the cell (biofilms, growth, etc.) and the presence of modifying enzymes. The diagram shows the possible mechanisms available to confer resistance to antibiotics and the inter-locking lines the known combinations found in resistant bacteria. Not all elements are found in all resistance organisms, but combinations of two or more are common leading to a highly connected chemical-genetic profile that confers high level resistance and is challenging to overcome.
rise to all the highly efficient elements that result in resistance. This is readily apparent for the enzymes that confer antibiotic resistance either through inactivation of drugs or by altering their targets. The determination of protein structure and function has linked resistance elements to their protoresistance progenitors in several instances. In many instances, it is likely that resistance has arisen by co-optation of proteins and enzymes with other functions, and only serendipitous affinity for antibiotics, followed by evolution through natural selection. Examples are discussed below.
Aminoglycoside Inactivating Enzymes
Aminoglycoside antibiotics inhibit bacterial cell growth by binding to the 16S rRNA resulting in a structural change in the aminoacyl–tRNA acceptor site on the ribosome. This is the site of cognate codon–anticodon pairing that is essential to the fidelity of translation. Binding of the aminoglycosides to this region of the ribosome displaces key structural elements in the 16S rRNA that sense correct codon–anticodon paring. This results in an increased error rate in translation and the subsequent production and accumulation of cytotoxic mistranslated proteins. Resistance to aminoglycosides can occur through the expression of a cadre of modifying enzymes: acetyltransferases (AAC), adenylyltransferases (ANT) and kinases (APH). These drug modifications occur on strategic regions on the antibiotic scaffold that play a role in ribosome binding, thereby, reducing the affinity between drug and target resulting in resistance (Wright et al., 1998; Magnet and Blanchard, 2005).
Over the past several years, the determination of aminoglycoside modifying enzyme structures along with elucidation of their molecular mechanisms have revealed that each of the distinct classes, AAC, ANT or APH, share common ancestors with metabolic enzymes with no antibiotic modification capacity. The APH enzymes were among the first linked to high level aminoglycoside resistance in pathogens. The determination of the 3D structures of several APHs has revealed similarity with the Ser/Thr/Tyr class of protein kinases (Hon et al., 1997; Nurizzo et al., 2003; Young et al., 2009; Fong et al., 2010). Furthermore, biochemical studies have shown that APHs use similar chemical mechanisms as protein kinases and in fact have the capacity to catalyze the phosphorylation of protein and peptide substrates (Daigle et al., 1998; Boehr et al., 2001). APHs, therefore, probably have either evolved directly from a protein kinase protoresistance element or APHs and protein kinases share a common ancestor.
Similarly, AACs belong to the GCN5 superfamily of acetyltransferases that include protein acetyltransferases such as histone acetyltransferases (Vetting et al., 2005). As in the case of APHs, 3D protein structure studies reveal kinship between AAC and protein acetyltransferases (Wolf et al., 1998; Wybenga-Groot et al., 1999; Vetting et al., 2002, 2004) and biochemical studies show that AACs have protein modification properties (Wybenga-Groot et al., 1999; Vetting et al.,
2004). Protein acetyltransferases, therefore, are potential protoresistance elements that can evolve into antibiotic resistance enzymes.
ANTs are nucleotidylyl transferases that are important aminoglycoside resistance elements in a number of clinical pathogens (Shaw et al., 1993; Miller et al., 1995). Determination of the 3D structure of ANT(4) revealed structural similarity to DNA polymerase B, which catalyzes mechanistically similar NMP transfer (Pederson et al., 1995). Recently, determination of the structure and mechanism of the clindamycin adenylyltransferase LinB also revealed similarity to ANT and to the metabolic enzymes DNA polymerase B and poly(A) polymerase (Morar et al., 2009). These results suggest that nucleotide polymerases (or their ancestors) are probably protoresistance elements linked to resistance to at least two distinct classes of antibiotics, the lincosamides and aminoglycosides.
Other Antibiotic Resistance Enzymes
Table A21-1 summarizes the structural and functional similarities between several classes of antibiotic resistance enzymes and metabolic and housekeeping elements that could be protoresistance elements giving rise to resistance via natural selection.
Several decades of enzymology and structural biology have shown there is nothing unique about antibiotic inactivation/modification chemistry. Given the vast number of metabolic and housekeeping enzymes in microbes, it is not surprising that Nature has co-opted existing small molecule modifying strategies to inactivate antibiotics. These protoresistance elements are the ultimate origin of resistance genes and understanding of structure and mechanism provides opportunity to chemically tailor new compounds to decrease susceptibility to resistance.
4.
The Environmental Resistome
Antibiotic resistance has generally been the purview of clinical microbiology. It is the emergence of resistance in pathogens and the therapeutic failure of antibiotics in infectious disease medicine that spurs antibiotic discovery and management. While predictable, this focus on resistance in pathogens fails to consider the stunning density of microbes on the planet. There are an estimated 5 × 1030 prokaryotes across the globe (Whitman et al., 1998) and only a very small fraction is associated with human disease. All microbes nevertheless interact with toxic small molecules produced by themselves, their microbial neighbors, plants, animals, local geochemistry and so on (Wright, 2007). This fact predicts that environmental organisms, therefore, should have resistance elements that have evolved as counter-measures to such molecules. The environmental resistome, therefore, should be highly diverse and abundant.
Evidence of this prediction is that opportunistic human pathogens that originate in the environment such as species of Pseudomonas, Stenotrophomonas,
TABLE A21-1 Protoresistance Elements
|
Antibiotic |
Resistance element |
Metabolic or Housekeeping Protein with Similar Structure and Function |
|
Aminoglycosides (streptomycin, kanamycin, gentamicin) |
Kinase Acetyltransferase Adenylyltransferase |
Ser/Thr/Tyr protein kinases GCN5 protein acetyltrasnferases DNA polymerase B, polyA polymerase |
|
β-Lactams (penicillins, cephalosporins) |
Ser-β-lactamases |
Peptidoglycan D, D-carboxypeptidases, peptidoglycan peptidyltransferases Glyoxylase II |
|
|
Metallo-β-lactamases |
|
|
Lincosamides (clindamycin) |
Adenylyltransferase |
DNA polymerase B, polyA polymerase |
|
Streptogramin type A |
Acetyltransferases |
UDP-N-acetylglucosamine O-acetyltransferase |
|
Streptogramin type B |
Lyase |
Muconate-lactonizing enzyme |
|
Vancomycin |
VanA |
D-Ala- D-Ala ligase |
|
Protein structure and function studies have revealed unexpected connections between resistance elements and proteins with other functions. These either may be progenitors of resistance or they share a common ancestor. |
||
Acinetobacter, Burkholderia and others are intrinsically highly drug resistant. In fact, these organisms are often cited as among the most concerning to infectious disease specialists looking for new antibiotics to treat disease (Spellberg et al., 2008; Livermore, 2009). Antibiotic resistance in these opportunistic pathogens of environmental origin is often combinatorial; dominated by multiple efflux pumps, but also including specific enzymes that inactivate particular classes of antibiotics. The genome sequence of Pseudomonas aeruginosa PA01, for example, reveals > 20 efflux pump associated genes and several genes encoding enzymes for resistance to chloramphenicol, aminoglycoside and β-lactam antibiotics (Stover et al., 2000). This arsenal of resistance elements no doubt reflects the evolutionary history of these bacteria within their (often broad) environmental niches, where the ability to evade the activities of toxic molecules produced by a myriad of organisms offers a selective advantage. Such an advantage is perhaps not necessary for common human pathogens such as Staphylococci and Streptococci that have a more restricted host range, and in fact are often human commensals, and as a result are much more intrinsically drug-sensitive.
Another hallmark of many bacteria is the ability to acquire genes via HGT (Barlow, 2009). Microbial genome sequences have revealed the remarkable extent of this phenomenon where the scars of HGT are recognizable in the presence of genes (and pseudogenes) encoding elements required for HGT (transposases, resolvases, etc.), which are often distributed throughout microbial genomes. Frequently, these are physically adjacent in the chromosome to resistance elements (reviewed in D’Costa et al., 2007). HGT can be a recent event as evidenced by the sequencing of the genome of Acinetobacter baumannii strain AYE that showed the acquisition of an 86 kb multi- resistance island containing 45 resistance genes that was absent in the wild-type A. baumannii strain SDF (Fournier et al., 2006).
Environmental bacteria produce many antibiotics and other cytotoxic small molecules. The important question of whether these molecules are synthesized exclusively for their cytotoxic activity, that is, as chemical warfare, is receiving increased scrutiny (Yim et al., 2006, 2007; Linares et al., 2006). Sub-lethal concentrations of many antibiotics have been found to have myriad effects on cellular processes (Yim et al., 2006; Davies et al., 2006; Tsui et al., 2004) and the actual roles of the so-called ‘antibiotics’ that are produced by bacteria are probably much more complex. Whatever their real functions in the ecology of producing bacteria, small molecules with cytotoxic bioactivities are intimately associated with resistance. For example, members of the order Actinomycetales are especially prolific in their ability to synthesize antibiotics. Of course, the capacity to produce antibiotics must co-evolve with resistance for producing organisms to avoid suicide (Cundliffe, 1989). As early as 1973, Benveniste and Davies recognized that resistance mechanisms in aminoglycoside antibiotic producers shared similarities with those found in pathogens. These similarities were extended in later work to include other antibiotics including glycopeptides such as vancomycin.
Vancomycin resistance emerged in enterococci in the late 1980s and was found to be the product of a five-gene cassette that included two regulatory genes and three enzyme encoding genes (Leclerq et al., 1988; Courvalin, 2006). This cassette results in antibiotic inducible reprogramming of intrinsic bacterial peptidoglycan biosynthesis to enrich cell walls with peptidoglycan terminating in D-Alanyl-D-Lactate in place of the canonical D-Alanyl-D-Alanine (Kahne et al., 2005). The latter is the recognition site for the glycopeptide antibiotic such as vancomycin while antibiotics do not bind to the former (Bugg et al., 1991). This complex resistance mechanism and gene cassette is found in glycopeptide antibiotic producers (Marshall et al., 1997, 1998) and non-producing environmental organisms (Hing et al., 2004). Recently, a variant of the van gene cassette has been characterized from the environmental anaerobe Desulfitobacterium halfniense demonstrating genetic variability of glycopeptide resistance (Kalan et al., 2009).
The growing anecdotal evidence that actinomycetes were possible sources of antibiotic resistance elements that share similarity with those found in clinical pathogens prompted us to conduct a systematic study of these organisms from various soils and a survey of their antibiotic resistance profiles (D’Costa et al., 2006). A collection of 480 wild-type actinomycetes was screened against a panel of 21 antibiotics. These included natural products, their semi-synthetic derivatives and completely synthetic molecules. Resistance to all antibiotics was observed and on average each bacterial strain was resistant to seven to eight antibiotics. Analysis of the molecular basis of resistance identified modes of resistance that are shared with clinical pathogens as well as novel mechanisms that so far have not been detected in the clinic. For example, the van gene cluster was found in all five vancomycin resistant strains in the collection, demonstrating that this gene cassette is readily identifiable in the environment. On the other hand, a new mechanism of resistance to the semi-synthetic ketolide antibiotic, telithromycin, was characterized in one strain. Analysis of the inactive drug revealed that antibiotic glucosylation was the mode of resistance (D’Costa et al., 2006).
A subsequent study by Dantas et al. showed that in addition to resistance, it was possible to select environmental organisms that subsist on antibiotics (Dantas et al., 2008). The authors identified bacteria from diverse orders (not just actinomycetes) that used natural products antibiotics such as penicillin G and vancomycin or synthetic antibiotics including ciprofloxacin as sole carbon sources. A subsequent investigation by this group on resistance genes in the human gut and oral microbiomes revealed resistance genes in cultured bacteria and in the metagenome (Sommer et al., 2009). Resistance to a number of classes of drugs was widespread in the bacteria described in this study that looked at two unrelated healthy human subjects.
Functional metagenomic analyses of soils by the Handelsman group has identified a number of resistance genes even in the so-called ‘pristine’ environments not expected to have been exposed to antibiotics of human or agricultural
origin (Allen et al., 2009; Handelsman, 2004). Another metagenomic study of activated sludge identified several bleomycin resistance genes (Mori et al., 2008). Other studies on microbes associated with insects (Allen et al., 2009; Kadavy et al., 2000), animals (Cloud-Hansen et al., 2007; Poeta et al., 2009, 2007; Gilliver et al., 1999) and birds (Bonnedahl et al., 2009; Sjolund et al., 2008; Poeta et al., 2008) demonstrate the wide distribution of microorganisms harboring resistance genes associated with wild animals. There is substantial variation in the frequency of resistance elements in wild animals, however, and there are increased numbers of resistant bacteria in animals that have contact with humans than those with little exposure (Thaller et al., 2010; Osterblad et al., 2001). Not surprisingly then, there is an even larger literature concerning antibiotic resistant organisms associated with farmed animals (Aarestrup et al., 2008a) including poultry (Gyles, 2008), swine (Aarestrup et al., 2008b), cattle (Call et al., 2008) and fish (Cabello, 2006) and often increases in resistant bacteria can be directly correlated with agricultural and aquacultural antibiotic use. Antibiotic exposure then can select low abundance resistant strains in the environment or select for HGT of resistance genes (Baquero et al., 2009).
These studies and our growing understanding of natural product biosynthesis demonstrate that there is nothing particularly special about antibiotics as chemicals. These small molecules, like other primary and secondary metabolites, are part of the natural chemical ecology of the Earth. As such, numerous resistance mechanisms have evolved across microbial genera to either deal specifically with selected antibiotics or classes of antibiotics (for example, inactivating enzymes), or more generally respond to the presence of toxic small molecules, for example, through the expression of broad spectrum efflux proteins. This is analogous to acquired and innate immunity in higher organisms where the innate immune processes are deployed in response to general threats. This contrasts with acquired immunity through antibody production, which provides highly specific and robust immunity. The analogy breaks down, however, in that bacteria can often readily acquire high-level resistance through HGT. The fact that the vast majority of sequenced microbial genomes show evidence of HGT in addition to the presence of resistance genes concretely demonstrates the density of resistance elements in the environment. When considered in the context of a global bacterial population of 5 × 1030, the probability of the emergence of antibiotic resistance in clinically important pathogens becomes a virtual certainty.
5.
The Clinical Resistome
Most clinicians and medical microbiologists restrict their study of antibiotic resistance to clinically important pathogens for good reason. Understanding the mechanisms, dissemination and epidemiology of resistance in pathogenic bacteria is vital to drug use, management and discovery. As suggested above, unlike opportunistic pathogens of environmental origin the bacteria that often
are associated with infection are (or at least were) largely antibiotic-sensitive. There is good evidence that the emergence of resistance in pathogens in many cases is the direct result of natural selection in the clinic. For example, a survey of the Murray collection of enterobacteria (433 stains) collected between 1917 and 1952 showed that while pre-antibiotic use bacterial isolates harbored conjugatitive plasmids capable of HGT, none of them carried resistance genes (Hughes and Datta, 2003). In contrast, following the introduction of antibiotics in a single patient, in a clinical care setting, or across populations, increased prevalence of resistance is the norm and is predictable (e.g., Livermore, 2009; Grayson et al., 1991; Hawkey and Jones, 2009).
Clinical resistance can be the result of selection for single mutants, for example, point mutations in the RNA polymerase gene rpoB that result in rifampin resistance, DNA gyrA and toposiomerase parC that confer fluroquinolone resistance, rpsL and streptomycin resistance. Such mutations usually diminish the productive binding of drug to target. Mutations that result in upregulation of genes can confer resistance and this is not uncommon with efflux mechanisms; alternatively, downregulation of transport proteins such as porins can result in resistance by blocking entry of the antibiotic into the cell. Acquisition of genes and their stable integration into the bacterial chromosome is another form of mutation that leads to resistance. The acquisition of the SCCmec cassette in MRSA is an example of this mechanism (deLencestre et al., 2007). Clonal dissemination of such strains results in resistant populations that can have geographic limits, for example, to distinct healthcare institutions or wards. In such cases, it is the founder strains that can dominate as a result of selective pressures from drug use. Genotyping of bacteria can identify lineages and help to map the natural history of the clinical resistome linked to a specific outbreak, for example.
Alternatively, resistance elements can migrate between strains by HGT resulting in relatively rapid adaptation and radial dissemination of genes into several strains, each of which has the potential to be founders. Genotype analysis in this case can be difficult to interpret. HGT can occur through transformation, transduction or conjugation. Plasmid-mediated HGT has the potential to move resistance genes through microbial populations. It is clear that since the introduction of antibiotics in the 1940s, plasmids have accumulated a greater number of resistance genes and often times these are on transposable elements that facilitate movement of genes into the chromosome (Barlow, 2009). The collection of resistance genes on mobile genetic elements such as a plasmid or transposon means that selection for resistance to one class of antibiotic can inadvertently result in co-selection for genes that confer resistance to other structurally distinct drugs. This mobility and co-selection make the clinical resistome a challenge to map and model.
The source of the resistance genes found on these genetic elements is not known, but the growing understanding of the extent of the environmental resistome as discussed above suggests a strong link. The vast numbers of resis-
tance genes and organisms in the environment are consistent with identifying it as the wellspring of much of the clinical resistome. The similarity of the vancomycin resistance gene cassette in environmental and pathogenic organisms is one example of a likely connection. Another is the link of the CTX-M extended spectrum β-lactamases that are prevalent in clinics across the globe and a reservoir in the environmental bacterium Kluyvera ascorbata (Humeniuk et al., 2002).
6.
The Intrinsic Resistome
Bacteria are not uniformly sensitive or resistant to antibiotics. The presence or absence of genes that contribute to resistance confer species- (and even strain)-specific built-in resistance to drugs. These genes comprise the intrinsic resistome (Breidenstein et al., 2008; Fajardo et al., 2008; Tamae et al., 2008). In some cases, these genes are readily recognizable as members of well-known resistance gene families. For example, Enterococcus faecalis are uniformly insensitive to lincosamide and streptogramin antibiotics as a result of the presence of Lsa efflux protein, which is characteristic of this species (Singh et al., 2002). As noted above, opportunistic pathogens such as P. aeruginosa encode a number of resistance genes, particularly efflux proteins that can provide broad drug insensitivity.
In addition to such well-characterized elements, several systematic studies have revealed a network of genes that contribute to intrinsic resistance. Chemical–synthetic interaction studies have been particularly enlightening. In such experiments, a library of mutants is screened for sensitivity or resistance to an antibiotic at sub-lethal concentrations. Mutants that confer sensitivity to the antibiotic are candidates for the intrinsic resistome. These have potential as targets for new drugs that could potentiate the action of antibiotics. One caveat is that the nature of these studies excludes essential genes from being sampled and, therefore, could underestimate the extent of the intrinsic resistome.
A screen of a P. aeruginosa PA14 transposon mutant library against sublethal concentrations of the fluroquinolone antibiotic ciprofloxacin identified 35 mutants with increased sensitivity to that antibiotic and 79 mutants with decreased sensitivity (Breidenstein et al., 2008). Genes linked to intrinsic resistance included expected efflux systems, but also non-obvious genes such as those involved in DNA repair and replication and the ClpX and ClpP proteases. The former has recently been identified as the target for the acyldepsipeptide antibiotics (Brotz-Oesterhelt et al., 2005) and this offers the possibility of synergistic combinations with fluoroquinolones. In a similar study, two P. aeruginosa transposon mutant libraries were screened against a panel of six antibiotics representative of distinct antibiotic classes (Fajardo et al., 2008). This study found that several mutants increased sensitivity to more than one antibiotic, suggesting a significant lack of discrimination by the genetic networks that protect the cell from toxic molecules. An analogous screen was reported in a transposon mutant library of Acinetobacter baylyi (Gomez and Neyfakh, 2006). This work studied
the impact of 12 antibiotics at subinhibitory concentrations and identified 11 genes with chemical synthetic lethal phenotypes. Several genes were associated with efflux systems and cell wall metabolism; however, like the Pseudomonas screens described above, many genes were unrelated to known antibiotic targets of resistance elements.
Finally, a systematic analysis of the susceptibility of ~4000 single-gene deletion strains of Escherichia coli (the Keio collection; Baba et al., 2006) versus seven antibiotics identified 140 novel synthetic chemical lethal interactions (Tamae et al., 2008). This work was recently updated to cover 22 antibiotics further supporting the complexity of the intrinsic resistome genetic network and at the same time generating a distinctive sensitivity profile that is predictive of antibiotic classes (Liu et al., 2010). This cellular ‘bar code’ has the potential to be applied in antibiotic typing during drug discovery.
7.
Expert Opinion
The concept of the antibiotic resistome is a framework to understand the evolution, origins and genetic complexity of resistance. The majority of the past research on antibiotic resistance focused narrowly on the emergence of resistance in clinical pathogens and its epidemiology. The growing understanding of the molecular mechanisms of resistance along with knowledge of the 3D-structure of these elements, and the fact that many similar resistance genes are found in non-pathogenic organisms help to understand why resistance is so prevalent and emerges so rapidly after antibiotic deployment in the clinic. Furthermore, the resistome concept which reveals the remarkable depth of the gene pool to source resistance and the ease of HGT in bacterial populations explain why resistance-proof antibiotics are a fiction. What is lacking in the field is a thorough understanding of the precise mechanisms of HGT in the environment and more examples of unimpeachable evidence of recent HGT from environmental organism to pathogens.
A response to the pervasiveness of the resistome and inevitability of antibiotic resistance can be despair. Certainly, the challenges of new antibiotic drug discovery are significant (Payne et al., 2007) and the fact that the resistome is so broad is a contributing factor. Nevertheless, antibiotics have proven to be miracle drugs and immensely profitable to the pharmaceutical sector over the past 6 decades despite the fact that the resistome was in existence during this time. There is a lot of room for optimism and understanding the resistome provides new opportunities for drug discovery field.
First, traditional antibiotic discovery from natural and synthetic sources has proven to be successful and should continue. Screening of environmental organisms for resistance to candidate drugs early in the discovery process could help to identify protoresistance and bonafide resistance elements that may eventually emerge in the clinic. This can be used to make strategic decisions in lead opti-
mization or between competing candidates in the preclinical discovery phase. Furthermore, it could identify resistance elements that may emerge in pathogens and thus provide opportunities for diagnostic tests to be used during clinical trials or post approval.
Second, the resistome concept leads naturally to the consideration of combinations of drugs in antibacterial treatment. Combinations of antibiotics are common in infectious disease practice; however, there are relatively few formulated combinations (Synercid® [dalfopristin and quinupristin] and co-trimoxazole are exceptions). Combinations of antibiotics and inhibitors of antibiotic resistance enzymes are thus far limited to three β-lactamase inhibitors (clavulanic acid, sulbactam, tazobactam) formulated with a few penicillins. There is great opportunity to expand this repertoire. In combination with molecular diagnostics that can identify the presence of specific resistance genes, these combinations could be a powerful anti-infective strategy. The revelation of the intrinsic resistome of bacteria presents a number of potential targets for inhibitors of non-essential gene products that could be used in combination with known antibiotics. Such combination strategies could extend the lifetimes of our existing collection of antibiotics for which we have ample understanding of toxicology, pharmacology and so on.
There are significant challenges to this approach including matching of pharmacological profiles of bioactive compounds in any formulated combination drug, not to mention regulatory hurdles and clinical trial design of any combination. However, as the frequency of multidrug resistant bacterial pathogens increases in the healthcare sector and in the community, leveraging our growing understanding of the resistome through the use of drug combinations will become more attractive.
Declaration of Interest
Research in the author’s lab on antibiotic resistance is supported by a Canada Research Chair and the Canadian Institutes Health of Research and the Natural Sciences and Engineering Research Council.
References
Aarestrup FM, Oliver Duran C, Burch DG. Antimicrobial resistance in swine production. Anim Health Res Rev 2008b;9(2):135-48
Aarestrup FM, Wegener HC, Collignon P. Resistance in bacteria of the food chain: epidemiology and control strategies. Expert Rev Anti Infect Ther 2008a;6(5):733-50
Allen HK, Moe LA, Rodbumrer J, et al. Functional metagenomics reveals diverse beta-lactamases in a remote Alaskan soil. ISME J 2009;3(2):243-51
Allen HK, Cloud-Hansen KA, Wolinski JM, et al. Resident microbiota of the gypsy moth midgut harbors antibiotic resistance determinants. DNA Cell Biol 2009;28:109-17
Baba T, Ara T, Hasegawa M, et al. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol 2006;2:2006-8
Baquero F, Alvarez-Ortega C, Martinez JL. Ecology and evolution of antibiotic resistance. Environ Microbiol Rep 2009;1:469-76
Barlow M. What antimicrobial resistance has taught us about horizontal gene transfer. Methods Mol Biol 2009;532:397-411
Boehr DD, Thompson PR, Wright GD. Molecular mechanism of aminoglycoside antibiotic kinase APH(3′)-IIIa: roles of conserved active site residues. J Biol Chem 2001;276(26):23929-36
Breidenstein EB, Khaira BK, Wiegand I, et al. Complex ciprofloxacin resistome revealed by screening a Pseudomonas aeruginosa mutant library for altered susceptibility. Antimicrob Agents Chemother 2008;52(12):4486-91
Bonnedahl J, Drobni M, Gauthier-Clerc M, et al. Dissemination of Escherichia coli with CTX-M type ESBL between humans and yellow-legged gulls in the south of France. PLoS One 2009;4(6): e5958
Brotz-Oesterhelt H, Beyer D, Kroll HP, et al. Dysregulation of bacterial proteolytic machinery by a new class of antibiotics. Nat Med 2005;11(10):1082-7
Bugg TDH, Wright GD, Dutka-Malen S, et al. Molecular basis for vancomycin resistance in Enterococcus faecium BM4147: biosynthesis of a depsipeptide peptidoglycan precursor by vancomy-cin resistance proteins VanH and VanA. Biochemistry 1991;30:10408-15
Cabello FC. Heavy use of prophylactic antibiotics in aquaculture: a growing problem for human and animal health and for the environment. Environ Microbiol 2006;8(7):1137-44
Call DR, Davis MA, Sawant AA. Antimicrobial resistance in beef and dairy cattle production. Anim Health Res Rev 2008;9(2):159-67
Cloud-Hansen KA, Villiard KM, Handelsman J, Carey HV. Thirteen-lined ground squirrels (Spermophilus tridecemlineatus) harbor multiantibiotic-resistant bacteria. J Am Assoc Lab Anim Sci 2007;46(3):21-3
Courvalin P. Vancomycin resistance in gram-positive cocci. Clin Infect Dis 2006;42(Suppl 1): S25-34
Cundliffe E. How antibiotic-producing organisms avoid suicide. Annu Rev Microbiol 1989;43:207-33
Daigle DM, McKay GA, Thompson PR, Wright GD. Aminoglycoside phosphotransferases required for antibiotic resistance are also Serine protein kinases. Chem Biol 1998;6:11-8
Dantas G, Sommer MO, Oluwasegun RD, Church GM. Bacteria subsisting on antibiotics. Science 2008;320(5872):100-3
Davies J, Spiegelman GB, Yim G. The world of subinhibitory antibiotic concentrations. Curr Opin Microbiol 2006;9(5):445-53
D’Costa VM, Griffiths E, Wright GD. Expanding the soil antibiotic resistome: exploring environmental diversity. Curr Opin Microbiol 2007;10(5):481-9
D’Costa VM, McGrann KM, Hughes DW, Wright GD. Sampling the antibiotic resistome. Science 2006;311(5759):374-7
de Lencastre H, Oliveira D, Tomasz A. Antibiotic resistant Staphylococcus aureus: a paradigm of adaptive power. Curr Opin Microbiol 2007;10(5):428-35
Fajardo A, Martinez-Martin N, Mercadillo M, et al. The neglected intrinsic resistome of bacterial pathogens. PLoS One 2008;3(2):e1619
Fischbach MA, Walsh CT. Antibiotics for emerging pathogens. Science 2009;325(5944):1089-93
Fong DH, Lemke CT, Hwang J, et al. Structure of the antibiotic resistance factor spectinomycin phosphotransferase from Legionella pheumophila. J Biol Chem 2010;285(13):9545-55
Fournier PE, Vallenet D, Barbe V, et al. Comparative genomics of multidrug resistance in Acinetobacter baumannii. PLoS Genet 2006;2(1):e7
Gilliver MA, Bennett M, Begon M, et al. Antibiotic resistance found in wild rodents. Nature 1999;401(6750):233-4
Gomez MJ, Neyfakh AA. Genes involved in intrinsic antibiotic resistance of Acinetobacter baylyi. Antimicrob Agents Chemother 2006;50(11):3562-7
Grayson ML, Eliopoulos GM, Wennersten CB, et al. Increasing resistance to beta-lactam antibiotics among clinical isolates of Enterococcus faecium: a 22-year review at one institution. Antimicrob Agents Chemother 1991;35(11):2180-4
Gyles CL. Antimicrobial resistance in selected bacteria from poultry. Anim Health Res Rev 2008;9(2):149-58
Handelsman J. Metagenomics: application of genomics to uncultured microorganisms. Microbiol Mol Biol Rev 2004;68(4):669-85
Hawkey PM, Jones AM. The changing epidemiology of resistance. J Antimicrob Chemother 2009;64(Suppl 1):i3-10
Hon WC, McKay GA, Thompson PR, et al. Structure of an enzyme required for aminoglycoside antibiotic resistance reveals homology to eukaryotic protein kinases. Cell 1997;89(6):887-95
Hong HJ, Hutchings MI, Neu JM, et al. Characterization of an inducible vancomycin resistance system in Streptomyces coelicolor reveals a novel gene (vanK) required for drug resistance. Mol Microbiol 2004;52(4):1107-21
Hughes VM, Datta N. Conjugative plasmids in bacteria of the ‘pre-antibiotic’ era. Nature 1983;302(5910):725-6
Humeniuk C, Arlet G, Gautier V, et al. Beta-lactamases of Kluyvera ascorbata, probable progenitors of some plasmid-encoded CTX-M types. Antimicrob Agents Chemother 2002;46(9):3045-9
Kadavy DR, Hornby JM, Haverkost T, Nickerson KW. Natural antibiotic resistance of bacteria isolated from larvae of the oil fly, Helaeomyia petrolei. Appl Environ Microbiol 2000;66(11):4615-9
Kahne D, Leimkuhler C, Lu W, Walsh C. Glycopeptide and lipoglycopeptide antibiotics. Chem Rev 2005;105(2):425-48
Kalan L, Ebert S, Kelly T, Wright GD. Noncanonical vancomycin resistance cluster from Desulfitobacterium hafniense Y51. Antimicrob Agents Chemother 2009;53(7):2841-5
Leclercq R, Derlot E, Duval J, Courvalin P. Plasmid-mediated resistance to vancomycin and teicoplanin in Enterococcus faecium. N Engl J Med 1988;319(3):157-61
Linares JF, Gustafsson I, Baquero F, Martinez JL. Antibiotics as intermicrobial signaling agents instead of weapons. Proc Natl Acad Sci USA 2006;103(51):19484-9
Liu A, Tran L, Becket E, et al. Antibiotic sensitivity profiles determined with an Escherichia coli gene knockout collection: generating an antibiotic bar code. Antimicrob Agents Chemother 2010;54(4):1393-403
Livermore DM. Has the era of untreatable infections arived? J Antimicrob Chemother 2009;64(Suppl 1):i29-36
Magnet S, Blanchard JS. Molecular insights into aminoglycoside action and resistance. Chem Rev 2005;105(2):477-98
Marshall CG, Broadhead G, Leskiw BK, Wright GD. D-Ala-D-Ala ligases from glycopeptide antibiotic-producing organisms are highly homologous to the enterococcal vancomycin-resistance ligases VanA and VanB. Proc Natl Acad Sci USA 1997;94(12):6480-3
Marshall CG, Lessard IA, Park I, Wright GD. Glycopeptide antibiotic resistance genes in glycopeptide producing organisms. Antimicrob Agents Chemother 1998;42(9):2215-20
Miller GH, Sabatelli FJ, Naples L, et al. The most frequently occurring aminoglycoside resistance mechanisms-combined results of surveys in eight regions of the world. J Chemother 1995;7(Suppl 2):17-30
Morar M, Bhullar K, Hughes DW, et al. Structure and mechanism of the lincosamide antibiotic adenylyltransferase LinB. Structure 2009;17(12):1649-59
Mori T, Mizuta S, Suenaga H, Miyazaki K. Metagenomic screening for bleomycin resistance genes. Appl Environ Microbiol 2008;74(21):6803-5
Nurizzo D, Shewry SC, Perlin MH, et al. The crystal structure of aminoglycoside-3′-phosphotransferase-IIa, an enzyme responsible for antibiotic resistance. J Mol Biol 2003;327(2):491-506
Osterblad M, Norrdahl K, Korpimaki E, Huovinen P. Antibiotic resistance. How wild are wild mammals? Nature 2001;409(6816):37-8
Payne DJ, Gwynn MN, Holmes DJ, Pompliano DL. Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nat Rev Drug Discov 2007;6(1):29-40
Pedersen LC, Benning MM, Holden HM. Structural investigation of the antibiotic and ATP-binding sites in kanamycin nucleotidyltransferase. Biochemsitry 1995;34:13305-11
Poeta P, Radhouani H, Pinto L, et al. Wild boars as reservoirs of extended-spectrum beta-lactamase (ESBL) producing Escherichia coli of different phylogenetic groups. J Basic Microbiol 2009;49(6):584-8
Poeta P, Costa D, Igrejas G, et al. Characterization of vanA-containing Enterococcus faecium isolates carrying Tn5397-like and Tn916/Tn1545-like transposons in wild boars (Sus Scrofa). MicrobMicrob Drug Resist 2007;13(3):151-6
Poeta P, Radhouani H, Igrejas G, et al. Seagulls of the Berlengas natural reserve of Portugal as carriers of fecal Escherichia coli harboring CTX-M and TEM extended-spectrum beta-lactamases. Appl Environ Microbiol 2008;74(23):7439-41
Shaw KJ, Rather PN, Hare RS, Miller GH. Molecular genetics of aminoglycoside resistance genes and familial relationships of the aminoglycoside-modifying enzymes. Microbiol Rev 1993;57:138-63
Singh KV, Weinstock GM, Murray BE. An Enterococcus faecalis ABC homologue (Lsa) is required for the resistance of this species to clindamycin and quinupristin-dalfopristin. Antimicrob Agents Chemother 2002;46(6):1845-50
Sjolund M, Bonnedahl J, Hernandez J, et al. Dissemination of multidrug-resistant bacteria into the Arctic. Emerg Infect Dis 2008;14(1):70-2
Sommer MO, Dantas G, Church GM. Functional characterization of the antibiotic resistance reservoir in the human microflora. Science 2009;325(5944):1128-31
Spellberg B, Guidos R, Gilbert D, et al. The epidemic of antibiotic-resistant infections: a call to action for the medical community from the Infectious Diseases Society of America. Clin Infect Dis 2008;46(2):155-64
Stover CK, Pham XQ, Erwin AL, et al. Complete genome sequence of Pseudomonas aeruginosa PA01, an opportunistic pathogen. Nature 2000;406(6799):959-64
Tamae C, Liu A, Kim K, et al. Determination of antibiotic hypersensitivity among 4,000 single-gene-knockout mutants of Escherichia coli. J Bacteriol 2008;190(17):5981-8
Thaller MC, Migliore L, Marquez C, et al. Tracking acquired antibiotic resistance in commensal bacteria of Galapagos land iguanas: no man, no resistance. PLoS One 2010;5(2):e8989
Tsui WH, Yim G, Wang HH, et al. Dual effects of MLS antibiotics: transcriptional modulation and interactions on the ribosome. Chem Biol 2004;11(9):1307-16
Vetting MW, de Carvalho LPS, Yu M, et al. Structure and functions of the GNAT superfamily of acetyltransferases. Arch Biochem Biophys 2005;433(1):212-26
Vetting MW, Hegde SS, Javid-Majd F, et al. Aminoglycoside 2′-N-acetyltransferase from Mycobacterium tuberculosis in complex with coenzyme A and aminoglycoside substrates. Nat Struct Biol 2002;9(9):653-8
Vetting MW, Magnet S, Nieves E, et al. A bacterial acetyltransferase capable of regioselective N-acetylation of antibiotics and histones. Chem Biol 2004;11(4):565-73
Whitman WB, Coleman DC, Wiebe WJ. Prokaryotes: the unseen majority. Proc Natl Acad Sci USA 1998;95(12):6578-83
Wolf E, Vassilev A, Makino Y, et al. Crystal structure of a GCN5-related N-acetyltransferase: Serratia marcescens aminoglycoside 3-N-acetyltransferase. Cell 1998;94:439-49
Wright GD. The antibiotic resistome: the nexus of chemical and genetic diversity. Nat Rev Microbiol 2007;5(3):175-86
Wright GD, Berghuis AM, Mobashery S. Aminoglycoside antibiotics. Structures, functions, and resistance. Adv Exp Med Biol 1998;456:27-69
Yim G, Wang HH, Davies J. The truth about antibiotics. Int J Med Microbiol 2006;296(2-3): 163-70
Yim G, Wang HH, Davies J. Antibiotics as signalling molecules. Philos Trans R Soc Lond B Biol Sci 2007;362(1483):1195-200
Yim G, de la Cruz F, Spiegelman GB, Davies J. Transcription modulation of Salmonella enterica serovar Typhimurium promoters by sub-MIC levels of rifampin. J Bacteriol 2006;188(22):7988-91
Young PG, Walanj R, Lakshmi V, et al. The crystal structures of substrate and nucleotide complexes of Enterococcus faecium aminoglycoside-2″-phosphotransferase-IIa [APH(2″)-IIa] provide insights into substrate selectivity in the APH(2″) subfamily. J Bacteriol 2009;191(13):4133-43

























































































































































































































































































































































