
7
Presentation by Thomas Fleming: Biomarkers and Surrogate Endpoints in Chronic Disease
Thomas Fleming provided a keynote presentation on the critical issues involved in the validation of surrogate endpoints. In his introduction of the speaker, John R. Ball noted that Dr. Fleming’s work, and in particular, his publication with David DeMets (Fleming and DeMets, 1996), was influential to the committee and its recommendations.
Dr. Fleming began his talk by returning to a topic raised in discussion following presentations by stakeholders from industry: Is the report going to have a chilling effect on biomarker research and application? In his experience, such problems have resulted from “not having some consensus, both from a regulatory and scientific perspective, as to what it is that we need to show.” Therefore, he believes the report should actually counter chilling effects. More importantly, interest in the report should be focused on asking whether it offers “enlightenment about how to enhance providing the public an informed choice, as well as more enhanced, reliable evidence about benefit to risk.” In that regard, he said, he was very impressed with what the report had achieved “in taking on this complicated set of issues.”
A CORRELATE DOES NOT A SURROGATE MAKE
Dr. Fleming focused on two main issues, the first of which he described as “digging deeper” into the reasoning behind his statement that “a correlate does not a surrogate make.” He identified three main criteria for choosing endpoints in clinical trials in order to best determine
an intervention’s benefit relative to its risk: sensitivity, measurability/interpretability, and clinical relevance. He illustrated the first criterion, sensitivity, with the choice of an endpoint for the trial of an analgesic for pain in preterminal cancer patients. Survival is critically important to such patients, he noted, but pain relief is the most sensitive measure of efficacy for this intervention.
The second criterion involves both measurability and interpretability. Dr. Fleming illustrated poor measurability with a hypothetical study requiring monthly liver biopsies. If such a study were conducted, it would not be acceptable to many patients and clinicians, and “you’re not going to retain patients very long.” He noted that interpretability is also important in selecting endpoints in clinical trials, and he provided some thoughts on composites of disease. One composite of disease, the combination of cardiovascular death, stroke, and myocardial infarction (MI), is interpretable because each of these conditions results in irreversible morbidity and mortality. However, interpretation becomes much more complicated if putative surrogate elements, such as “asymptomatic distal deep vein thrombosis,” are added to the composite (as they often are in studies of knee or hip replacement, he noted).
Clinical relevance of the endpoint is the ultimate criterion for its acceptance, according to Dr. Fleming. He referenced Robert Temple’s definition of a clinical endpoint: “a direct measure of how a patient functions, feels, and survives,” which is also reflected in the Biomarkers Definitions Working Group and the Institute of Medicine (IOM) committee’s reports (Biomarkers Definitions Working Group, 2001; IOM, 2010). Each of these attributes are difficult to measure, he acknowledged. Survival takes a long time to assess in many settings, and patient feelings and function are often based on patient-reported outcomes (PROs), which “can be very difficult to validate, often can have missing data, require blinding, and have a multiplicity associated with them,” he said. “It’s very tempting to look at objective alternative biomarkers.”
A common approach to finding such biomarkers is to identify one that is correlated with the desired clinical endpoint, show an effect in the biomarker, and make the “leap of faith” that this biomarker does, in fact, translate to clinical benefit, Dr. Fleming said. “Unfortunately,” he added, “that’s often not the case.” He proceeded to discuss the various reasons why a biomarker might fail as a surrogate endpoint, each of which are illustrated diagrammatically in Figure 7-1.
The first reason for a biomarker to fail as a surrogate endpoint is that the biomarker does not lie in the causal pathway by which the disease influences the clinical endpoint, so an intervention’s effect on the biomarker will not provide a reliable estimate of the intervention’s clinical efficacy (as shown in Figure 7-1A), Dr. Fleming said. An example of this
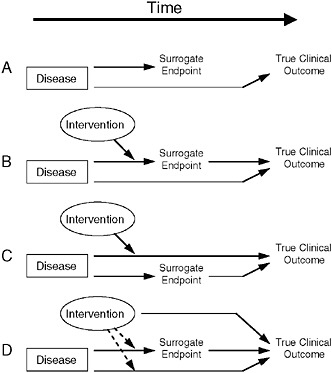
FIGURE 7-1 Reasons for failure of biomarker (surrogate) endpoints. (A) The surrogate is not in the causal pathway of the disease process. (B) Of several causal pathways of disease, the intervention affects only the pathway mediated through the surrogate. (C) The surrogate is not in the pathway of the intervention’s effect or is insensitive to its effect. (D) The intervention has mechanisms of action independent of the disease process.
NOTE: Dotted lines = mechanisms of action that might exist.
SOURCE: Fleming and DeMets, 1996. Adapted, with permission, from the Annals of Internal Medicine. Copyright 1996 by the American College of Physicians.
case occurs in mother-to-child transmission of HIV. In pregnant women with HIV, there is a very strong negative correlation between maternal CD4 (helper T-cell) count and likelihood of HIV transmission to the infant, he said. Thus, one might suppose it would be useful to give the mother interleukin-2 (IL2) late in pregnancy in order to raise her CD4 count closer to normal levels. However, doing so has no effect on transmission because CD4 count is not part of the causal mechanism for HIV transmission.
Many diseases have biomarkers that are not in their pathophysiologic pathways, Dr. Fleming noted. Examples include carcinoembryonic antigen, a biomarker for ovarian cancer, and prostate-specific antigen (PSA); such biomarkers are useful for disease diagnosis and assessing prognosis,
he said. However, in the case of prostate cancer, he noted that controversy has arisen as to whether PSA levels should dictate the type of intervention undertaken, and also as to the value of ongoing assessment of prognosis for this typically latent disease. Correlation with a clinical endpoint is all that is needed for a biomarker to be useful in detecting disease and assessing prognosis, Dr. Fleming concluded. However, a biomarker used as a surrogate endpoint must lie in the pathophysiologic causal pathway of the disease.
A second scenario for biomarker failure as a surrogate endpoint occurs when multiple pathways influence outcome. If the intervention only affects the disease pathway through the biomarker (Figure 7-1B), a false positive can result, Dr. Fleming said. Conversely, if the intervention only affects pathways other than the one including the biomarker, the result would be a false negative (Figure 7-1C).
An example of the latter case involved chronic granulomatous disease—a condition that occurs in children who have a compromised immune system, resulting in serious infections, according to Dr. Fleming. Researchers considered treating this disease with gamma-interferon to increase bacterial killing. In fact, he said, bacterial killing was going to be used as the endpoint, “because we did not want to randomize and expose half of these children to three injections of placebo per week to gamma-interferon.” However, due to concerns this surrogate endpoint might be misleading, a 12-month trial with interim analyses was conducted (Gallin et al., 1991). Early results of this trial were persuasive, yielding a 70 percent reduction in the clinical endpoint, but no effect at all on the biomarker, Dr. Fleming said. Gamma-interferon did indeed provide clinical benefit to affected children, perhaps by killing bacteria at an undetectable level, or through some other means, such as by increasing antibiotic uptake, he speculated.
Another example involves vancomycin-resistant enterococci (VRE) infections in the gastrointestinal (GI) tract. Patients with these infections are at considerable risk for bloodstream infections due to VRE, said Dr. Fleming. However, because more than 1,000 patients would be required to test the effect of an antimicrobial on that endpoint, researchers were interested in using decolonization of the GI tract by VRE as a biomarker for infection clearance. He noted that this biomarker has several flaws: it fails to take into account VRE colonization outside the GI tract, such as in or on the skin, and also for the magnitude and duration of antibiotic effect needed for protection. “If VRE GI levels are reduced to lower than detectable levels, it doesn’t mean eradication,” he said. “It’s entirely possible that … you would still have risk of bloodstream infections.” He also said that the method of quantification of VRE from fecal samples may not have adequately captured the extent of colonization. In addition to uncertain-
ties regarding the magnitude of effect on this biomarker that is needed for protection, there also is lack of clarity regarding how long VRE has to be cleared from the GI tract in order to reduce risk. The VRE decolonization biomarker also is unable to capture unintended effects of antimicrobials, such as suppressing the immune system or altering the composition of the GI flora, both of which could lead to opportunistic infections by other microbes. Indeed, he said, “there are many aspects of the ultimate effect of antimicrobials on the clinical endpoint that may not be captured by the biomarker [VRE decolonization].”
Even for interventions that affect all causal pathways leading to a clinical endpoint, there are potential off-target effects through which the intervention can directly influence the true clinical endpoint, and which the biomarker does not capture (Figure 7-1D), according to Dr. Fleming. One such scenario is the suppression of cardiac arrhythmia post-MI with ecainide or flecainide, which in a placebo-controlled trial were shown to triple the death rate among patients (Bigger, 1986; Cardiac Arrhythmia Suppression Trial [CAST] Investigators, 1989; Echt et al., 1991; Mukharji et al., 1984; Ruberman et al., 1977). There are many more such examples among treatments for cardiac arrhythmia (for example, quinidine, lidocaine) and among agents that improve cardiac output and ejection fraction (for example, milrinone, flosequinan), all of which were found to increase patient death rates.
Regarding cholesterol, Dr. Fleming noted that a meta-analysis of 50 trials of early generation agents for lipid-lowering—diet, lovastatin, fibrates, resins, and hormones—found that these agents produced a 10 percent reduction in low-density lipoprotein cholesterol (LDL-C), but no overall impact on overall survival (coronary heart disease [CHD]-related death was reduced, but non-CHD related deaths were increased) (Gordon, 1994). Later, the more potent statins produced a 30 percent reduction in LDL-C, and researchers began to see a relationship where such reduction in LDL-C predicted an effect on overall survival, he said. However, “as we’ve heard, when we then introduced torcetrapib with atorvastatin to achieve increases in HDL-C [high-density lipoprotein cholesterol] as well as reductions in LDL-C, this study was terminated early, surprisingly, with increased death and increased rates of CHD death, MI, and stroke” (Barter et al., 2007).
These results raise the important issue of bridging, Dr. Fleming said. This occurs when a biomarker that is a valid surrogate for a clinical endpoint—as effects on lipids were for the effects of atorvastatin on death, stroke, and MI—is proposed as a surrogate endpoint for a new intervention with a potentially different mode of action (for example, torcetrapib). In that case, the effect of that new intervention on the surrogate may not reliably predict the clinical endpoint, he emphasized.
Paula Trumbo later asked whether this concern applied equally to
surrogate endpoints validated for drugs that might be used to support a health claim for a food. Dr. Fleming constructed such a scenario—involving blood pressure effects that had been validated in a hypertensive setting for several classes of drugs—proposed as a biomarker for foods. “If I had my ideal, I’d like to validate each biomarker for classes of agents,” he said, adding that he might be more willing to accept such an extrapolation if the same putative mechanisms were involved in both food and drug effects. One could say that foods are less likely to produce adverse (off-target) effects than drugs, he continued, but one could also argue that drugs might be more likely than foods to have broad positive (on-target) effects, and not just on the causal pathway measured by the biomarker.
VALIDATING SURROGATE ENDPOINTS
To introduce his second main topic, the validation of surrogate endpoints, Dr. Fleming showed an attempt to use hematocrit levels as a surrogate endpoint for the risks of death and MI due to end-stage renal disease (Besarab et al., 1998). Standard-dose epogen, an erythropoiesis-stimulating agent (ESA), had been shown to partially normalize hematocrit levels, he said. Thus, a randomized trial was performed against more aggressive use of ESAs in patients with end-stage renal disease to see whether complete normalization of hematocrit would reduce their risk of death and MI. He noted that analyses of data in the standard-dose and in the high-dose arms indicated that every 10-point increase in hematocrit was associated with a 30 percent reduction in the relative risk of death. Since high-dose Epogen raised hematocrit by an average of nearly 10 points relative to standard-dose Epogen, one would expect that high-dose Epogen would reduce the death rate at least 25 percent as compared with the standard dose. However, nearly the opposite occurred: death rates of high-dose recipients increased 30 percent over those receiving the standard dose. “We now understand [these excess deaths] to be due to off-target effects, likely based in part on a thrombosis off-target mechanism,” he said.
“A valid surrogate is one where the effect of the intervention on the surrogate is reliably telling us what the effect of the intervention is on the clinical endpoint,” Dr. Fleming said. One setting in which a valid surrogate is being sought is type-2 diabetes. Hemoglobin A1c is a standard biomarker for blood sugar control. If an intervention reduced this biomarker by half a percent over a 6-month period, he asked, could we say that such an intervention would be effective in mitigating the long-term risk for microvascular and macrovascular complications in type 2 diabetes? Dr. Fleming noted that when this has been tried, the following major adverse effects have occurred:
-
In the case of troglitazone, increased serious hepatic risks caused the drug to be taken off the market.
-
Peroxisome proliferator-activated receptor agonists (muraglitazar and rosiglitazone) appeared to increase risk of death, stroke, and MI.
-
The ACCORD trial found that an aggressive strategy to normalize hemoglobin A1c led to an increase in mortality.
Given these results, Dr. Fleming asked how one could elucidate effects on hemoglobin A1c that reliably predict clinical benefit? The Prentice criteria provide guidance, he said: first, the potential surrogate needs to be a correlate; second, the surrogate endpoint must fully capture the net effect of the intervention on all mechanisms that influence the clinical outcome. To determine circumstances in which hemoglobin A1c can act as a surrogate for type 2 diabetes, one can first examine the effect of the intervention on the clinical endpoint, such as the rate of cardiovascular death, stroke, and MI, he said. Then a statistical model, the Cox proportional hazards model, can be used to determine the proportion of net treatment effect explained by the surrogate endpoint. The key question is whether the effect of treatment on the clinical endpoint is fully captured by how treatment affects hemoglobin A1c levels.
Dr. Fleming emphasized that this question can best be answered by meta-analyses of many clinical studies in order to obtain sufficient evidence to determine whether or not the treatment’s effect on the clinical endpoint is being fully captured by the effects on the biomarker. However, determination of the net effect of an intervention on a surrogate endpoint does not exclude the possibility that the intervention produces off-target effects on the clinical endpoint, he noted.
If an intervention for type 2 diabetes provided a 20 percent reduction in the rate of cardiovascular death, stroke, and MI, and that level effect exactly matched what is predicted based on its effect on hemoglobin A1c, it does not mean that is the only way that treatment influenced outcome, Dr. Fleming said. The intervention may have provided additional benefits via other causal pathways. Such off-target benefits could also be offset by adverse off-target effects that raise the risk of serious cardiovascular complications. Under these circumstances, the net effects on the surrogate and clinical endpoints would appear to be identical, but the biological reality would be very different. Echoing a point made by Victor De Gruttola in earlier discussion, he noted that “you can never discern whether the effect on the biomarker is capturing the totality of the effects [because] the effects are always going to be confounded … you are only able to assess the net effect.”
In subsequent discussion, Richard Kuntz asked if these confounders could be reduced by intentionally studying the effects of different interventions on a biomarker. Dr. Fleming replied that if many different classes of interventions with diverse mechanisms produced the same effect on an endpoint of interest, that evidence would support the biomarker’s validity. However, he said such evidence appeared to support lipid effects as a surrogate for CHD, until the effects of torcetrapib were understood. “Thank goodness we had clinical endpoint studies that reflected the totality of the effect,” he said.
Such cases make clear that “biology does matter,” Dr. Fleming said. “From the clinical perspective, it is key to have a comprehensive understanding of the causal pathways of the disease process, and of the off-target as well as the on-target effects of the intervention,” he said. He noted that decades ago, it was generally thought that better statistics were needed to be able to validate surrogate endpoints. Today, he said, the evidence suggests that we need a richer clinical understanding of the disease processes and of the mechanisms of the intervention. Because it is “almost impossible” to obtain a complete biological understanding of a complex chronic disease, meta-analyses of clinical data provide the best route to such insights, he said.
One example of this is in the adjuvant colon cancer setting, when cancer is detected early enough to permit curative surgery. Among patients whose excised tissue contains positive nodes, about half will experience recurrence of cancer, due to microscopic undetected residual disease, and will die within 5 years, Dr. Fleming said. Therefore, patients with positive nodes receive chemotherapy to eradicate residual disease. He described a meta-analysis of 18 randomized clinical trials that was used to determine if delaying or reducing recurrence of disease is a valid surrogate for improving survival (Alonso and Molenberghs, 2008). The results suggested that to the extent that an intervention delayed or reduced recurrence, it had a proportionate effect on survival, he said. While this evidence supports the validity of the proposed surrogate endpoint, it also demonstrates that “surrogates tend to work best where you need them the least,” Dr. Fleming said. In this case, the surrogate appears to be so close to the clinical endpoint as to provide modest advantage to its use.
In the HIV setting, Dr. Fleming said it is important to understand when a biomarker, such as viral load, may be used as a surrogate endpoint. For example, if viral load is lowered to undetectable levels in patients for 1 year, he said “it is probably going to be a pretty reliable surrogate for the ability to influence symptomatic AIDs-defining events and death if it is used in individuals with CD4 counts below 150.” However, such a surrogate would have much greater clinical utility if it could be used to determine whether interventions to reduce viral load would
improve the long-term prognosis for newly infected people. Dr. Fleming said this is a much more complicated question.
A BIOMARKER HIERARCHY
Dr. Fleming has developed a four-level hierarchy for outcome measures, depending on the levels of evidence available:
-
True clinical efficacy measures;
-
Validated surrogate endpoints;
-
Nonvalidated surrogate endpoints that are “reasonably likely to predict clinical benefit”; and
-
Correlates that are solely a measure of biological activity (Fleming, 2005).
Dr. Fleming added that stroke might be a surrogate endpoint for overall survival in patients with atrial fibrillation, yet it also is a true clinical efficacy measure in that setting. He said that validated surrogate endpoints, the second level in the hierarchy, are relatively rare and include the earlier example of colon cancer recurrence and survival, as well as blood pressure, as a surrogate for clinical endpoints in antihypertensive interventions. Both of these surrogates were validated based on large amounts of data from multiple clinical trials, and they were validated for specific types of interventions used in those trials and for specific clinical endpoints, he said. In addition, the magnitude of each intervention’s effect on the surrogate endpoint accurately predicted its effect on the clinical endpoint.
Most biomarkers occupy the two lowest levels of Dr. Fleming’s hierarchy: those that are “reasonably likely to predict clinical benefit” and those that merely correlate with clinical benefit. Dr. Fleming emphasized that the definition “reasonably likely to predict clinical benefit” is an important distinction from a regulatory perspective because the accelerated approval process can be used in such settings. Biomarkers that attain the third level have the following attributes (beyond correlation with the clinical endpoint) according to Dr. Fleming:
-
They accurately capture the treatment’s effect on the predominant mechanism through which the disease process induces clinical risks.
-
They are likely to capture large treatment effects on the clinical endpoint.
-
They make predictions consistent with the net effect of an intervention on the clinical endpoint.
-
They produce a target effect that is sufficiently strong and durable to enable them to predict meaningful benefit.
Biomarkers that correlate with clinical endpoints, but cannot predict them with reasonable likelihood, still may have many important uses, Dr. Fleming said. These include diagnosis of disease and assessing prognosis (as is the case for PSA); informing patient-specific therapeutic strategies (for example, adapting treatment for patients with pneumonia based on their body temperature, a biomarker for clinical benefit); serving as a primary endpoint for proof-of-concept or screening trials; and as an additional supportive measure of biological activity in phase III clinical trials. None of these uses is controversial, he pointed out; what is controversial, is using a biomarker as a surrogate endpoint.
Dr. Fleming also briefly discussed another purpose for biomarkers, patient enrichment, which is a strategy to identify and select patients who are likely to respond to a given intervention, such as a targeted therapeutic. Dr. Fleming characterized this use as very complicated, and noted that enrichment is typically used when the “key mechanisms of treatment effect on the causal factors of the disease process are specific to a targeted population.” Examples of this include trastuzumab for patients whose breast cancers overexpress HER2, as well as cetuximab treatment for nonmutated k-ras tumors in colorectal cancer. In these types of situations, he noted, validation is particularly complex, as it is necessary to confirm that the “enriched” population defined using the biomarker responds differently than the general population; in addition, a robust assay must be able to define the target population.
John A. Wagner asked Dr. Fleming to comment on the variable utility of clinical endpoints, since some clinical endpoints suffer from many of the same measurement issues that biomarkers and surrogate endpoints do, as well as similar unintended consequences. For example, a PRO can measure how depressed patients feel, but it cannot capture the effect of a treatment on blood pressure or on suicide. Thus, he said, “I think it’s important to take a bit of a look at the big picture for endpoints in general, and not just surrogate endpoints.”
“My sense, and what the [IOM report] clearly points out, is the goal of clinical research is to enhance benefit to risk for the public,” said Dr. Fleming. “That means basically improving how a patient feels, functions, and survives.” Patients do not take therapy to alter their biomarkers; rather “they take therapy specifically to alter their clinical risk,” he said.
However, he agreed with Dr. Wagner that some PROs may not capture important outcomes to patients, and said it is critically important to define endpoints that can reflect what matters to patients. Dr. Fleming reemphasized the need to select endpoints that are sensitive, clinically
relevant, and ideally, as comprehensive as possible. In life-threatening situations, Dr. Fleming said that survival often is the best endpoint to use, and there are many advantages to its use, since it is the easiest endpoint to validate and capture fully. In non-life-threatening situations, survival would not be the endpoint of choice, said Dr. Fleming. However, “in any setting, there could be clinically tangible effects that aren’t captured by the primary endpoint,” he added. “Therein lies the primary value of secondary endpoints … [of which] there should be a small number, because you otherwise run the risk of exploring the data and looking for those things that make you feel better about benefit to risk.”
CONCLUDING THOUGHTS
Dr. Fleming addressed the consequences of relying on biomarkers as surrogate endpoints in the regulatory setting. He noted that natalizamab, a drug for multiple sclerosis, was given accelerated approval because it reduced the rate of relapse within a year; however, it was later associated with increased risk for progressive multifocal leukoencephalopathy (PML), a rare but serious brain infection. Two previously discussed treatments for type 2 diabetes—muraglitazar and rosiglitazone—were evaluated for full approvals on the basis of their effects on hemoglobin A1c, he added. These drugs have been associated with increased risk for death, stroke, and MI. These examples demonstrate that “when we’re using surrogates, we’re not only getting less reliable evidence about efficacy, we’re also getting less reliable evidence about safety,” Dr. Fleming said. Furthermore, because “everything is benefit-to-risk, the more limited information you have about the level of efficacy, the less resilient you are” when safety issues emerge. In the case of natalizamab, had trials been conducted that established beneficial effects in delaying clinical progression to walking with a cane or to being wheelchair bound, such effects on measures of irreversible morbidity would have provided much greater confidence in use of the agent even when evidence of PML emerged.
With regard to foods, Dr. Fleming noted that it is important to understand the net effect of an entire food on both biomarkers and clinical endpoints. Thus, he said, if the food in question has more than one experimental ingredient, it would be important to understand how each ingredient affects relevant biomarkers and clinical endpoints.
Addressing specific questions posed to him by the committee, Dr. Fleming revisited why it is important that each surrogate endpoint be evaluated on a case-by-case basis, and he discussed why a biomarker cannot be deemed a generic surrogate for a disease. “We have talked about several reasons why the generalizing of surrogates can be problematic,” he said. He illustrated this with the following scenario: an intervention is
known to effectively reduce low-density lipoprotein cholesterol (LDL-C), and investigators want to use LDL lowering as a surrogate endpoint for death or MI. The new intervention has a similar effect on LDL, but it has far weaker effects on other positive mechanisms as compared with the original intervention. This could be because the original intervention not only reduced LDL, but also positively affected triglycerides and high-density lipoprotein cholesterol (HDL-C). The new intervention appears more beneficial than it is if investigators solely judge it by the effect on LDL, because biomarkers used as surrogate endpoints may not take into effect the multiple causal pathways involved in a disease process, he said.
Conversely, the new intervention could have unintended adverse effects such as increasing blood pressure through the angiotensin-renin system. Then, the effect on the lipid-based biomarker does not represent the totality of effects. “That is in fact what we saw when we looked at torcetrapib,” he said. “Fortunately we recognized that torcetrapib and atorvastatin … [produced] an adverse [net] effect because we had clinical endpoint studies.”
Dr. Fleming also reminded the audience that the magnitude and duration of the effect of the intervention matters. For example, an intervention that has a modest effect on LDL-C may not produce a clinical benefit. On the other hand, “we’ve also seen with some of the surrogates, that if the effect is particularly profound, more isn’t always better,” he said. Examples of this scenario include hematocrit normalization with ESAs, reducing hemoglobin A1c in type 2 diabetes, and large reductions in blood pressure (Staessen et al., 2003).
In response to the committee’s question—how biomarker evaluation effects the public—Dr. Fleming replied that biomarkers are of great interest because they allow for timely assessment of interventions. However, he added, “it is critically important that [assessments] not just be timely, but reliable.” The ultimate goal of these assessments is not to give the public more choices but rather more informed choices, he said. In that regard, he described the report as “very enlightened” in its discussion of the steps involved in biomarker evaluation: validation, qualification, and utilization. In undergoing such evaluation, he said it happens more frequently than one might expect that the effect of an intervention on a biomarker fails to accurately predict its effect on a clinical endpoint.
“It is not so much the things we don’t know that get us into trouble; it’s the things that we do know that aren’t so,” said Dr. Fleming. He added that for the public, the most problematic aspect of biomarker use results when biomarkers that are not truly validated give us the impression that we understand a treatment effect when we do not.












