
20. Microbial Commons: Governing Complex Knowledge Assets
– Minna Allarakhia46
University of Waterloo, Canada
In talking about the governance of the microbial commons, I will apply a strategic level of analysis and a knowledge perspective. I would like to leave you with three messages:
First, biological knowledge structures are evolving, not only in terms of complexity, but also in terms of their value for future discovery and commercialization. We need to understand what belongs in the commons from a dynamic perspective. What might not have belonged in the commons yesterday may belong in the commons today.
Second, we need to understand clearly the motivation of participants in any commons. It is not just a matter of public sector participants. When my colleagues and I from the University of Waterloo and Wilfrid Laurier University studied 39 open-source initiatives developed after the completion of the Human Genome Project, we found that in many cases private-sector participants were involved, and a few actually catalyzed the formation of those initiatives. We need to understand both the positive and negative consequences of this participation.
Third, we should document these lessons from the commons so that they can be transferred across disciplines and even across markets, as we see researchers from different areas seeking to enter this domain, looking to participate in the commons that are being proposed as well as their own internal commons.
Commencing with the knowledge perspective, the Human Genome Project advanced the view that biological information operates on multiple hierarchical levels and that information is processed in complex networks. It is no longer sufficient to look at just the genomic and proteomic levels. We need to understand the interactions among genes and proteins—how to modulate systems, minimize malfunctions, and optimize for positive functions. From a knowledge perspective, then, biological information has become complementary. Downstream product development relies strongly on upstream research inputs. Furthermore, there is high applicability across biological systems and for our purposes microbial systems.47
Complicating the matter from an intellectual property perspective is that patents can exist at any level of the hierarchy of the research process and, depending on the breadth of those patents they can greatly influence the incentive for follow-on researchers to examine or use elements of such systems. Too broad a patent can inhibit the incentive of users to look at the underlying knowledge assets. So we need to find solutions to manage those incentives, both for first innovators and for follow-on incremental innovators. The proposal of the commons and the liability regime from this symposium is one possible solution.
_____________
46 Presentation slides available at http://sites.nationalacademies.org/xpedio/idcplg?IdcService=GET_FILE&dDocName=PGA_053726&RevisionSelectionMethod=Latest.
47 See Allarakhia. M., Wensley, A. Systems biology: melting the boundaries in drug discovery research [Internet]. In: A Unifying Discipline for Melting the Boundaries Technology Management: Portland, OR, USA: [date unknown] p. 262-274.[cited 2011 Aug 15] Available from: http://ieeexplore.ieee.org/lpdocs/epic03/wrapper.htm?arnumber=1509700.
Turning to motivation, we do need to understand the incentives for participating in the commons. As noted above, our research on 39 initiatives found strong involvement from private-sector participants. There are six classes of incentives:
- The development of a collegial reputation as a reward for working in open science. This is no longer restricted to public-sector participants; private-sector participants want to signal their quality as allies, particularly for downstream product development. Beyond catalyzation of several initiatives in our study by private sector participants, the recent open donation of compounds and continued creation of open source discovery initiatives by several multi-national pharmaceutical organizations such as GSK, Pfizer, Eli Lilly, and Merck, provide evidence of this need to signal quality and openness to further collaboration for downstream development.
- To generate general reciprocity obligations. I mentioned the complexities and the complementarity between knowledge assets. Both public and private sector participants may want to create reciprocal obligations signaling that they are willing to contribute to the commons so that in the future they can access other external knowledge assets. The creation of open patent pools with multiple contributors can signal this reciprocal obligation assuming equitable contributions and fair access terms.
- To influence adoption of a technology or a technology standard through increased diffusion of knowledge. We saw this in our research with the microarray providers participating in the biotech commons in order to influence adoption of their technology as a standard. However, we must note that there may be positive or negative consequences when you influence the adoption of a premature or insufficient technological standard.
- To improve the aggregate performance of an industry in order to increase safety or regulation associated with that industry as we discussed yesterday with reference to microbiological materials and outputs.
- To preempt rivals. We clearly saw this after the mapping of the human genome, when 10 of the world’s largest pharmaceutical companies came together and formed the Single Nucleotide Polymorphism (SNP) Consortium to ensure that rivals would not encroach on this territory and build patent fences around critical areas necessary for future product development.
- To share the risk associated with knowledge production. It is important to examine not only the issue of shared implementation from open-source software development and fair access to technology or biotechnology development, but also the way in which a commons serves to enable collaborative knowledge production. For example, in the pharmaceutical industry, the complexities associated with drug discovery are very intense. The risks are frequently too high and many pipelines for new products are
currently empty. The commons can provide the incentive to share the risk for collaborative knowledge production where there are complexities associated with new knowledge and its association to products.
Furthermore, it is important to understand the incentives to participate as well as to be able to predict when participants may exit from any commons. Here the concept of transition point is of value. The transition point is defined as the point in discovery research when researchers come to believe that unilateral gains from private management of knowledge including appropriation activities are greater than shared gains from open or shared knowledge with the subsequent outcome exit from the commons. Therefore, from a strategic perspective, we need to look at when appropriation will take place—when materials will be removed and no longer deposited.
Because the value of knowledge is changing, we are uncertain at any point what value today’s knowledge will have in terms of discovery and product development. A commons can reduce that uncertainty and make it less likely that premature mistakes will be made— specifically through the enclosure of knowledge, such as occurred with the gene races.
In our research, we sought to analyze 39 open-source initiatives developed after the mapping of the Human Genome Project. We looked at the structure and characteristics of the knowledge at stake. What was being produced? How could you classify or characterize that knowledge? What rules were established to govern both data and materials and for downstream appropriation strategies?
Overall, we have learned the following:
- Participation rules existed in the majority of cases. Consortia had established entry rules, with screening by executive or steering committees. Often commitment policies, membership fees, or large upfront research payments were established or required to enable both cooperation and research.
- Knowledge access policies exist. In the majority of cases, information is released not only to members, but also to the public at large. There were a few initiatives that were somewhat more closed, which shared information only with members that had made upfront monetary commitments.
- There were rules to manage both data and materials. The decision whether to deposit data into an open or a closed repository depended on the knowledge access policy. Where knowledge dissemination was open, peer-reviewed publications and deposit into databases permitted not only the release of data but encouraged the validation of the data or deposits. In the majority of cases, consortia advocated the use of nonexclusive royalty-free licenses for noncommercial use of materials and discovery tools.
- The consortia generally avoided issues related to commercial use and product development. It was assumed that this would be handled at the individual transaction level.
- It was absolutely important to create transparency with regard to motivation. In this case, upfront commitments could provide that clarity i.e. monetary support, human capital support, or open donations of knowledge-based assets.
Motivation for participation given the objectives established by the initiative clearly had an impact upon knowledge dissemination and access as we discovered when comparing the open initiatives to the more closed initiatives in our sample.
How do we apply these observations to the microbial commons? We have limitless capabilities for applying microbial knowledge to the energy and environmental sectors, in the development of alternative biofuels, the generation of biodiesel, and bioremediation. We need to approach this knowledge from a whole systems perspective, however. Hence, we should look at communities of interacting microbes.
Research and applications require integration and analysis of data to discern patterns of communities of microbes. The continued sharing of microbial information will be critical, as will linking literature, databases, and user communities. This is important not only for collaborative discovery, but also for the validation of the data and the results. In addition, given the sophisticated nature of the visualization data that is emerging today, we need to enable the joint representation and standardization of the data. Some of the governance mechanisms we might need to consider are the timing of data deposits, access and use, exemption clauses for noncommercial use, transfer of management from depositors to collective organizations, and commercial use clauses.
Several open access journals, databases, and supporting tools were discussed in this symposium yesterday. I want to add that it is not just a matter of the data, but we also need to have a supporting tool infrastructure in order to create queries to gain benefits from that data and pursue further discovery. For example, the Global Biodiversity Information Facility (GBIF) is an information-based infrastructure for connecting users to a globally distributed network of databases. Here, we use the notion of linking knowledge assets from an information technology infrastructure perspective.
The incentive to share microbial data is also manifested in the private sector. The Helicos BioSciences Corporation has opened up its microbial datasets, as well as a query tool. What is their motivation? We discussed disincentives yesterday, but what would be the incentive in this case? Most likely it is to showcase their genetic analysis system—an attempt to encourage the adoption of their system by displaying the value and the sophisticated nature of their data.
In yesterday’s talks, we discussed the linking of both materials and information in biological resource centers. The goal is to promote common access to biological resources and information services—we see that StrainInfo is providing electronic access to the information about biological materials in repositories.
BioBricks is quite interesting from a materials management perspective. It was developed as a nonprofit foundation by the Massachusetts Institute of Technology, Harvard, and the University of California, San Francisco. With BioBricks we are moving into what I consider the convergence paradigms, as now it is necessary to manage the complexities associated with synthetic biology. BioBricks makes DNA parts available to the public free of charge via MIT’s registry of standard biological parts. This is a collection of approximately 3,200 genetic parts that can be mixed and matched to build synthetic biology devices and systems. The commercial or other uses of these parts are unencumbered—without the assertion of any property rights held by the contributor over users of the contributed materials. However, novel materials and applications produced using BioBricks contributed parts may be considered for protection via conventional property rights.
Beyond the issues of data and materials management, we must also deal with the changing value of knowledge, and we find the commons model being used to manage downstream assets. One example of the latter is the Eco-Patent Commons. This is a project by the World Business Council for Sustainable Development whose mission is to manage a collection of patents pledged for unencumbered uses—even for proprietary purposes in products, processes, and composition of matter—that are directed towards environmentally friendly applications. As of 2008, 100 eco-friendly patents had been pledged by private-sector participants, ranging from manufacturing processes to compounds useful in waste management. What is the motivation there? Perhaps the participants recognize that, in dealing with premature technologies, they need to assure that there will be interoperability between technologies that develop downstream. The AlgOS Initiative is very premature and not particularly coherent yet, but I thought it was worthwhile to mention. It is an open-source initiative seeking ways to produce biodiesel from algae. The group is attempting to aggregate research inputs from a variety of experts in order to arrive at a full-cycle design for biodiesel production from algae, allowing for modification based on the open source software GNU General Public License approach, in which modifications are permitted as long as one complies with the requirements to pass on the source code to any recipients of one’s modifications, to provide them the same freedom to modify it, and to provide notice of those terms of the license.
Finally, in parallel with these other efforts, stakeholders are discussing so-called “green” licensing. Such licensing is directed towards developing countries that are looking to develop green technologies. An international licensing mechanism is under discussion that would have developing countries pay a fee in order to access this technology, while at the same time protecting the innovating firms. It will be interesting to see what form they choose for their fee mechanism. The underlying goal is to allow countries at different stages of development to be able to access the same technology.
Clearly, as knowledge characteristics change, the governance structures may need to change with them. Early on, for example, some experts suggested that data would not be deposited in the commons. Today, there is a question whether tools—pharmaceutical tools or biological tools—should be placed into the commons. Table 20–1 summarizes various examples from the microbial commons, looking at their characteristics and at the various governance strategies that are currently being employed.
| Managing the Microbial Commons | Data | Materials Management | Downstream Assets |
| Example: | MannDB; GBIF; Helicos Microbial Data | WFCC; BioBricks | EcoPatent Commons; AlgOS |
| Knowledge Characteristics: | High complementary, Non-substitutable, High applicability | High complementary, Nonsubstitutable or Substitutable, High applicability | High complementary, Non-substitutable or Substitutable, High applicability |
| Knowledge Governance Strategy: | Open Access; Use of Supporting Open Access Tools | Open Access; MTAs; License Agreements | Non-Assertion Clauses; Green Licensing; GNU-General Public Licenses |
TABLE 20–1 Governing the Microbial Commons
To summarize the pragmatic outcomes, managing knowledge assets has become critical, not only in the systems paradigm, but in the convergence paradigm. Here we see biological sciences, chemical sciences, physical sciences and information sciences increasingly coming together to address the complexities of health product technology, nanotechnology, green technology and energy based technology development. We need to determine what really belongs in the commons and what governance strategies are most appropriate so that researchers in multiple markets can pursue product development opportunities. These goals imply certain policies. The need for greater transparency of motives during knowledge production suggests that there should be an establishment of and commitment to rules regarding knowledge production. Different conventions regarding knowledge dissemination and appropriation imply the need to establish early in a research project what should be disseminated and in what format. Here, our research provides some indication of the commonality of rules for both knowledge production and dissemination. Follow-up research also looks closely at the transition point and its application to varied knowledge assets.
The National Research Council report A New Biology for the 21st Century (2009) advocates large teams converging and varied disciplines working together, whether that is promoted through federal policy or other means. We need to keep in mind that as scientists, information technology professionals, and other experts work together, they each have differing conventions regarding knowledge dissemination and appropriation. Some of them may find value in pure disembodied knowledge. Others may find value and appropriate embodied knowledge with the final goal marketable product development. We need to bring together these disciplines under a common framework, which is what a structured commons can offer to them. Extending our analysis and understanding of knowledge based activities to the convergence paradigm should lend insight into how best to structure a commons with varied disciplinary participants. Consequently, it will be important to analyze new case studies involving open-source innovation that targets the energy and environment sectors in order to look at evolving models of innovation. What types of participants are in those sectors pursuing those models? How do they handle the increased complexities as knowledge assets converge and are increasingly linked together and as new rules and perceptions of value emerge?
Finally, it would be valuable to create a repository of governance strategies for knowledge assets as is currently being undertaken by the BioEndeavor Initiative (www.bioendeavor.net), including any licensing templates or tools, so that we can apply those across commons, across disciplines, and across markets. As new markets choose to develop their own commons, they can have the benefits of the lessons we have learned about managing knowledge based assets and the development of open access journals, open data networks, and the supporting IT infrastructure.
21. Digital Research: Microbial Genomics
– Nikos Kyrpides48
Lawrence Berkeley National Laboratory
I will be talking about the future in microbial genomics: where I would like the future to be, where we are trying to go, and where I think we are going. As of September 2009, there were 4,319 microbial—in particular, archaeal and bacterial—genome projects under way around the world. As shown in Figure 21–1, five big sequencing centers are responsible for more than 50 percent of the world’s production. This is very different from the situation just one year ago, when only two sequencing centers, the Joint Genome Institute (JGI) and the J. Craig Venter Institute (JCVI), were performing more than half of the world’s sequencing. Just in the past year, those two have dropped to well under 50 percent of the world’s production, and the total production has increased by quite a lot. It is possible that this is an indication that we are already seeing the so-called democratization of genome sequencing, as more and more sequencing is taking place in smaller and smaller places, and a number of universities are now buying sequencing machines and starting to produce data.

FIGURE 21–1 Sequencing centers for archaea and bacteria.
SOURCE: http://www.genomesonline.org/
Everybody is talking about the data deluge (Figure 21–2). However, the big question is whether we need more sequencing? My non-microbiologist friends tell me
_____________
48 Presentation slides available at: http://sites.nationalacademies.org/xpedio/idcplg?IdcService=GET_FILE&dDocName=PGA_053955&RevisionSelectionMethod=Latest.
that there are now over 4,000 microbial genomes mapped. Is that enough? When are we going to be happy?
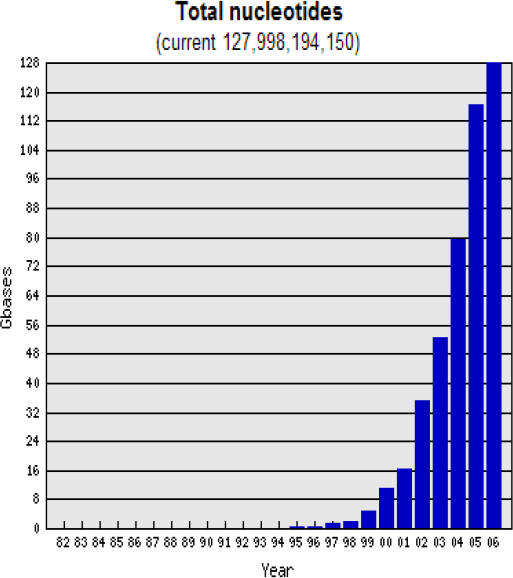
FIGURE 21–2 Total sequencing by year (in billions of base-pairs).
SOURCE: http://www.genomesonline.org/
A number of people believe that the more information we get, the less understanding we get. For bioinformatics, of course, this is not true. There the limiting factor is always the quantity of data. We want more data, and we keep on saying that there is no such thing as enough data—although in the last few months, or perhaps the last year, we have started to question whether we really want to keep saying that.
Of course, the issue is not just a quantitative one; it is also qualitative. We are generating more data, but it is important to look also at the types of data. In 2000, the three phylogenetic groups—Actinobacteria, Firmicutes, and Proteobacteria—accounted for 75 percent of all sequencing projects. Eight years later it was actually worse, with the whole “other phyla” portion covering much less—18 percent. We keep on sequencing more or less the same things. We know already that this is not what we see in nature. There is much greater diversity, and we know that from the rates of occurrence observed
directly in the environment and the known phyla that have no culture representatives at this point.
What do we do about that? And why do we keep on sequencing the same things? By analogy with what we heard yesterday about the brain, up to this point we have been seeing mainly genomics driven by medical applications or by energy and environmental applications, not by to the need to understand the whole diversity of nature. To offer an analogy with the brain, it is as if we do not really want to understand the brain, we just want to cure Alzheimer’s and diseases of the brain. Understanding the brain, which involves doing the fundamental studies, is totally different from learning how to cure its diseases.
As we will see, however, this picture is changing. The way that we address this question is changing from both the academic side and the industry side. Together, industry and scientists have come to realize that we need to go after the uncovered areas even though there may not be any obvious direct applications there.
What should be done about the uncultured majority—the 99 percent of microorganisms that are not able to be cultured with present methods? I am saying “uncultured” instead of “unculturable” because unculturable means we can never culture them, and that may not really be the case. We do not know if we can culture these organisms or not; there have been no systematic efforts to go after the uncultured majority.
One approach would be to go from genomes to metagenomes. Rather than isolating an organism and sequencing it, we will go directly to the community. We will take a pool of the community, sequence the whole pool, and try to understand what is there. This move from genomes to metagenomes will be one of the major transitions in the second decade of genomics.
The field of metagenomics was spearheaded just five years ago by two studies. One was by JGI and the other by JCVI, which collected samples from the Sargasso Sea. Figure 21–3 summarizes what we know about the complexity that exists out there. We can organize different types of metagenomes based on the species complexity.

FIGURE 21–3 Species complexity.
SOURCE: http://www.genomesonline.org/
Very simple environments have just a few organisms; very complex environments, such as the human gut and soil, have several hundred or several thousand different microbes. Some 200 metagenomic projects are now ongoing, and it is likely there are several hundred or a few thousand other metagenomic projects that we do not know about because they are private or because the data and information have not yet been released.
In a simple environment, if we have enough sequencing we can actually cover and assemble the entire genome. As the complexity increases, however, we have more and more of what we call “unassembled reads,” and we do not really know what to do with them. The big challenge here is, How can we study all of these microorganisms?
One way is the ancient Roman approach of “divide and conquer.” Using a technique called binning, researchers attempt to understand the function of a microbial community by determining the individual organism or group of organism it consist of. This is the major tool we have to understand these environments. It does however require having enough isolated genomes—enough reference organisms—to provide a reference for the binning.
This leads us to the second major transition, which is moving from individual genome projects—what we have seen over the past two or three years—to large-scale projects. For its first decade, genomics projects were initiated by a single principal investigator and focused on a single genome. This is changing. A project funded by the Moore Foundation looking at 180 microbes from marine environments was, as far as I know, the first example of one project massively sequencing a large number of microbes. A year later, the Human Gut Microbiome Initiative started. And just a year after that, the National Institute of Health launched the Human Microbiome Project with a goal of sequencing 1,000 microbes isolated from different parts of the human body. This major effort is not targeting specific applications, in the sense of looking for organisms involved in a particular disease or condition. Instead, its goal is to achieve as much coverage on the phytogenetic tree for organisms that we know are living on the human body.
With funding from the Department of Energy, JGI has recently initiated a project called GEBA, the Genome Encyclopedia of Bacteria and Archaea. The goal of this project is to systematically fill in the gaps in sequencing along the bacteria and archaea branch of the tree of life.
Although there have been some projects funded by the National Science Foundation to sequence 10 or 20 different microbes, isolated and identified in diverse parts of the tree, GEBA is the first systematic effort to go after large number of genomes and fill in the gaps in the tree. We started with 150 organisms; now we have a cumulative total of 255. About 60 of them are finished, and another 60 are in the draft stage. The paper describing them will be published soon.
The project is being co-led by Hans-Peter Klenk of DSMZ, the German Resource Center for Biological Materials, which is collaborating with JGI. This project would not have been possible without the contribution of the Culture Collection Center at DSMZ. The main limiting factor in such a project is not sequencing or annotation or downstream analysis, but getting the DNA. So the big breakthrough was partnering with the Culture Collection Center, which has been growing all of the strains and providing them free to JGI.
The project has already produced a number of very important discoveries that will lead to applications, including a number of cellulolytic enzymes. Nonetheless, the main
purpose of the project has always been basic research. We constantly say that we do not even know what is out there, and that is the major driver. We must go into the environment, sequence more organisms, and try to identify novel chemical activities, normal enzymatic activities that exist in nature.
The third major transition that is occurring is moving from populations to single cells. Large-scale genome projects are very good at covering the phylogenetic tree, but their limit is that the organism must be cultured, which represent about 1 percent of all the microorganisms. What can we do for the rest of the microorganisms that cannot be cultured? New technology that allows us to sequence single cells, and therefore bypassing the need for culturing, has been initiated just a few years ago. The important thing is that there are a variety of methodologies of ending up with a single cell, and once you have that, a technology exists to amplify its genome and perform the sequencing.
This is one of the most promising technologies for the near future, but it is not yet where we want it to be. We would like to get a complete genome or an almost complete genome of a single organism, but at this point we get something like 50-70 percent coverage of a genome and often not just a single organism. There is a significant presence of other organisms as well. Still, all of the problems are considered solvable, and we expect that with the next two years, we will have the single cell technology that can give us the entire genome of a single organism.
Once that is done, however, we will have to solve some additional problems. For one thing, this is a single event. You can isolate a cell, you can amplify the DNA, and you can sequence it, but you cannot store it or replicate it, and this leads to a number of issues concerning how to save the information. How can we store the genetic material? In this case we cannot. We isolate the cell, we sequence it, and that is it. It does give us a little information about uncultured organisms, but it is something we cannot repeat.
As I mentioned earlier, we are facing an avalanche of data. By the end of this year we will have more than 1,000 finished genomes. We already have almost 1,000 in draft form, with a total of about 8 million genes. Based on that, we can make some projections about what to expect over the next 15 years.
In five years we will have at least three times as many finished genomes and at least 10 times as many draft genomes, with a total of about 52 million genes. This is a conservative estimate. Assuming a linear increase at the rate presented in a paper by Patrick Chain that just appeared in Science, we will reach these numbers by 2012.
How can we start comparing a million genes against 52 million genes? This is just from the isolate genomes. If we consider the environmental studies, including only what is publicly available; we have approximately 30 million genes. If we add the environmental studies that we know are ongoing and the Human Microbiome Project studies, we expect that we will have at least 300 million genes over the next 3 years. These are really staggering numbers. And even being in bioinformatics, where people generally hope to have too much data, I am starting to worry about the magnitude of this.
So where do we go from here? We will need new technology, since we cannot handle all those data with today’s technology infrastructure. However, we also need conceptual breakthroughs in data processing, data comparison, and data browsing. We will need better ways to present multiple organisms; store and present data, and compute the similarities.
The fourth transition will be going from thousands of genomes to pan-genomes. The term pan-genome was coined a few years ago by Claire Fraser. Here is the issue: In the IMG comparative analysis system we often have a large number of different strains
from the same organism. We have 10 strains of Prochlorococcus marinus, 17 strains of Listeria monocytogenes, and 15 strains of Staphylococcus aureus. Each of these species has a lot of diversity, but most of the genes are the same from strain to strain in a single species. Do we really need to keep all of the instances of the identical genes? We are therefore collapsing all of those different strains of a single species to a single organism, which we call Staphylococcus aureus pan-genome.
Originally, the idea was to do this in order to save both in disc space and computation. However, by doing that throughout the whole phylogenetic genetic tree, we will start getting a totally new picture of microbiology.
A few years ago we thought that all we needed to do in order to understand a microbe was to just sequence a single strain. We now know that is wrong. We need to sequence several different strains. For example, by sequencing different strains of E. coli we find there is significant diversity among the strains. Or consider Staphylococcus aureus. Its genome is 2.7 megabytes, 2,700 genes. If we take all the different strains, but collapse them all to a non-redundant dataset—which means that every unique gene would be counted only once—we have a total of 14,000 genes. This is five times more than the average genome. In the case of Pseudomonas aeruginosa it is much less, a factor of just 1.8.
There are microbes that they are constantly acquiring new genes thus resulting in a much larger pangenome. We call that an open pangenome compared to other organisms that are not so eager to grow their genetic content, which have a close pangenome. This is what we expect to understand if we sequence all of those organisms. For the first time we will have a true understanding of microbial diversity.
The need for a definition of the microbe—the definition of a single microbial cell or single microbial organism—has resulted in a series of paradigm shifts. During the first decades of sequencing, from 1960 to 1990, we were using 16S RNA genes to construct the tree of life, and, accordingly, it looked quite simple (Figure 21–4).
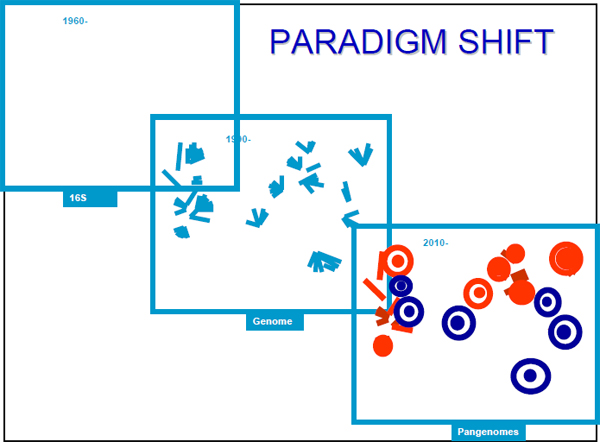
FIGURE 21–4 Changing paradigms for the tree of life.
SOURCE: http://www.genomesonline.org/
For the next 20 years, from 1990 to 2010, we were growing the tree. This is what is happening through the individual genome projects—adding details and branches. We expect that the next 20 years will be the era of pan-genomes, during which we develop a totally different understanding of the definition of the microbes and also of the relationship between the pan-genomes.
At this point I am going to change gears and talk about standards and how everything we have discussed until now is related to genomic standards. The Genomic Standards Consortium (GSC) was initiated by Dawn Field a few years ago with the goal of creating standards for metadata: standards for habitats, for DNA sources, for isolation, and for phenotypes. Its mission is to implement new genomic standards, methods of capturing and exchanging metadata, standardization of metadata collection, and analysis efforts across the wider genomics community.
The consortium’s first major publication, which appeared last year in Nature Biotechnology, was a demonstration by the working group of achieving standards and representing metadata using what is called the Minimum Information about a Genomic Sequence (MIGS) specification. On the GSC website you can find the list of MIGS fields used to specify a sequence, and one of the fields is the source material identifier. This gives the information necessary to find the sequenced organism or the biological material in the culture collection where it has been deposited.
Unfortunately, too few of the sequences from the genome project have been deposited. Of the complete microbial genomes we have so far, 53 percent have been deposited and 47 percent have not. For incomplete genomes it is much worse: Only 35 percent have been deposited. The hope is that since this is ongoing work, the researchers have not yet deposited it, and by the time they release the sequence, they will also deposit it. However, the GSC has emphasized that it should not be left to the discretion of the researchers. The funding agencies and the publications should mandate the deposit of the biological materials, at least at the time of the publication.
It is a different story concerning the release of sequencing data. Sometime around 2001 or 2002, there was a change in the release policy, and the funding agencies—with the Department of Energy (DOE) taking the lead—have been forcing researchers to release the data as soon as possible. From the moment that the scientists get the data, they have three months to release it. This has resulted in an increasing number of genome public releases.
The scientific community does want to have the data as soon as possible, even without an associated publication. The complete genome analysis is not trivial; it requires a tremendous amount of effort and quite often it takes more than a year or two, or even three. We thus expect to see the number of complete genomes put into public databases without a corresponding publication to go up.
It is necessary as well to have at least a minimum amount of information about the organisms that have been sequenced, including the metadata associated to the organisms. For that purpose, the journal Standards in Genomic Sciences was launched in July 2009, with George Garrity as editor-in-chief. The journal, which was funded by Michigan State University, is an open-access, standards-focused publication that seeks to rapidly disseminate concise genome and metagenome reports in compliance with MIGS standards.
The journal provides an easy method for scientists to report their results. They can just download one publication, change the introduction or the information about the
organism, and submit the information about their organism. This allows the community to have access to the additional metadata in addition to the complete genome.
Metadata is not the only interest of the GSC. It also focuses on issues related to data processing, such as sequencing standards, finishing standards, assemblies, and gene predictions, as well as on annotation issues. It is likely that there will be a workshop by DOE early next year that focuses on standards and annotation. A paper just appeared in Science that provides the first standards for genome finishing quality. This resulted from an effort led by Patrick Chain from JGI. And there are similar upcoming efforts that will deal with both gene findings and function predictions.
To sum up: Microbial diversity remains largely uncovered. The vast majority of current genome projects do not cover novel ground. To understand an organism, we need to sequence a reasonable number of closely related strains and incorporate standards.
The question is where do we go from here? How does one use all this information to spearhead or initiate a new project that will address those problems and comply with all the standards on which we have been focusing?
Over the last few months members of the GSC have begun discussing the possibility of launching a global genome sequencing effort for microbes. The idea is to imitate, but on a much larger scale, the Human Microbiome Project effort. By funding different sequencing centers in a single project, NIH has achieved something that seemed almost impossible a few years ago. It has gotten competing sequencing centers to work with each other and share not only metadata, but also pipelines and other resources.
This is one of the things we will try to do by expanding this idea into an international consortium. If you look at a map of who is doing sequencing, based on the number of genome projects per country the United States has complete dominance, although there are many sequencing projects around the world. There are about 20 countries that have significant sequencing efforts. Instead of having a single sequencing center, such as JGI, or even four sequencing centers, as is the case with the Human Microbiome Project, we want to organize a bigger international effort and ask the different countries of the world to contribute to an international genome project.
What will that project be? We want to sequence at least one representative of every cultured microorganism at the Genus level. At this point we have only about 30 percent coverage. This means that we do not have a sequence representative for about 70 percent of all genera—or about 1,500 different genera for which there is a type species. At the species level, the coverage is only about 10 percent. The remaining 90 percent corresponds to about 10,000 species for which we need a sequence representative.
We therefore have an immediate broad goal that cannot be possibly achieved by a single researcher or supported by a single funding agency. This is of global importance, which is why we provide the list of genome targets—the information for the reference points we need in order to support the metagenomic analysis that everybody is doing. For the first time, we will be able to have a reference point for every branch in the tree of life.
Among the Archaea, we have genomes for 86 out of 108 genera and for 98 out of 513 species. So, the remaining genera and species provide us with the immediate targets for the project, but this is really the low-hanging fruit. We are only talking about sequencing already characterized organisms.
It will not be enough, however, simply to sequence type species. Instead, we need to sequence enough strains for each species to fully characterize them and generate a species pan-genome. This is the absolute minimum we need to do in order to have a clear understanding of microbial diversity and what is out there.
Furthermore, there have been only minimal efforts to understand the effects of geographic distribution on species dynamics. We have sequenced at most 30 different strains of the same species from different geographic locations. We need to do this to a much larger extent. This will be another goal of the project.
The key partners in the project will be the GSC, which is definitely the major partner; culture collection centers, which will provide all the biological material for the project; representatives from Grand Challenge projects, including the Genomic Encyclopedia of Bacteria and Archaea, Terragenome, and the Human Microbiome Project; and other participants from large sequencing centers and country members. The response so far has been enthusiastic. A few months ago, there was a meeting of the European Culture Collections Organization, and they all said that they want to contribute. Country members like China, Korea, and Japan have already been invited, and they are very strongly supportive of such a project.
Progress in the future will depend on collaborations across national centers rather than simply between individual researchers. Fortunately, the funding agencies have finally said we will not support you unless you will start working together. This is a critical step, particularly for an area like bioinformatics, where traditionally researchers have thought they could do everything by themselves. Different groups that were competing until now have actually started working together. For the first time we are talking about sharing pipelines, sharing computations, and sharing methods of analysis.
















