
The world has changed rapidly in the past 30 years, and it will continue to change for the foreseeable future. Some of these changes are evident to any keen observer. Globalization of infectious agents over recent decades has contributed to the well-publicized spread of HIV, avian influenza, SARS, and multi-drug resistant tuberculosis. The globalization of the world’s food and drug supplies is less obvious, perhaps because it has been so rapid and less dramatic to the average consumer.
International commerce is a reality of modern food production and medical product manufacture. The U.S. Food and Drug Administration (FDA) oversees 20 million import lines, including close to seven million import lines for medical devices alone (Figure 1-1), a three-fold increase in regulated imports from a decade ago (Figure 1-2a) (Gill, 2011). Around 85 percent of the seafood, 39 percent of the fruits and nuts, and 18 percent of the vegetables that Americans buy come from abroad, as do 80 percent of active pharmaceutical ingredients (API) and 40 percent of finished drugs (Figure 1-2b) (Pew Health Group, 2011; Jerardo, A., pers. comm.).
Even to say that American foods and drugs come from abroad is an oversimplification. Prepared foods and fixed-dose pharmaceuticals, end products in the marketplace, are themselves mixtures of dozens of ingredients, often each one from a different country, prepared and repackaged by intermediaries around the world before their final sale. Modern supply chains are complex and reach every corner of the globe. Figure 1-3 shows the path tunafish may travel to reach an American supermarket, and Figure 1-4 describes a modern drug supply chain and the many potential points of vulnerability. As the FDA’s Pathway to Global Product Safety and
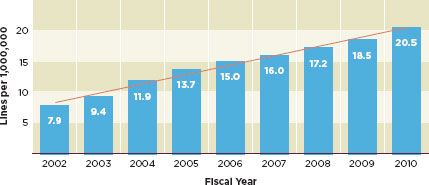
FIGURE 1-1
Imports of regulated products increased nearly three-fold between 2002 and 2010.
SOURCE: Gill, 2011.
Quality pointed out, the distinction between foreign and domestic producers is no longer clear (FDA, 2011b).
For the same reasons, through international travel and trade, the health and safety of the American public is now intricately linked to the health and safety of people around the world. Recognition of this reality is the cornerstone of the Department of Health and Human Services’ (HHS’s) 2011 Global Health Strategy. Secretary of Health Kathleen Sebelius called for the new strategy to guide the Department in realizing its own goals and those of other countries, explaining that “only through … multiple and collaborative efforts will [HHS] truly make a mark by improving global health” (HHS, 2011, p. 3). The strategy lays out three interconnected goals for HHS, the parent agency of the FDA. They are protecting and promoting the health of Americans through global health action; providing leadership and technical expertise to improve global health; and advancing U.S. interests through global health action (HHS, 2011). (See Figure 1-5.)
The Global Health Strategy marks a departure from the traditional conception of HHS and its agencies, including the FDA, as purely domestic organizations with an almost exclusively domestic focus. For the FDA in particular, the new HHS strategy aims to “strengthen regulatory capacity on a global basis. Extending … surveillance, regulatory, and program activities beyond the U.S. borders enables more effective protection of Americans’ health through improving the health of the world’s population” (HHS, 2011, p. 19). This is consistent with the evolving scope of the FDA’s work presented in the Pathway to Global Product Safety and Quality. This report explains the changes the FDA sees as necessary to “transform itself from a domestic agency operating in a globalized world to a truly global
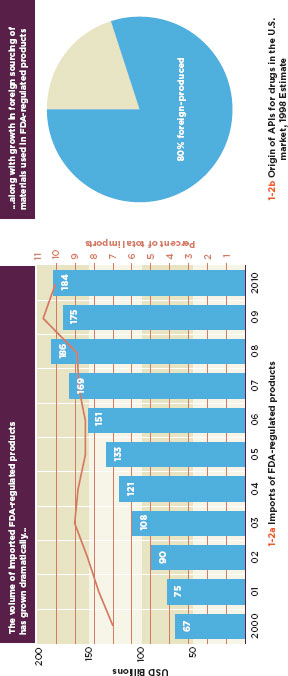
FIGURE 1-2
Increases in global trade are increasing the exposure of U.S. consumers to foreign products and source materials.
SOURCE: FDA, 2011b.
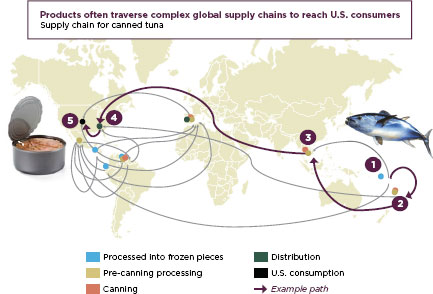
FIGURE 1-3
Canned tuna travels the global supply chain before reaching American tables.
SOURCE: FDA, 2011b.
agency fully prepared for a regulatory environment in which product safety and quality know no borders” (FDA, 2011b, p. 3). To this end, the FDA plans to work more with its counterpart agencies to create global coalitions of regulators; to develop international data-sharing systems; to expand its intelligence system; and to work with public and private third parties and industry to increase the returns on their mutual efforts (FDA, 2011b).
THE CHANGING FACE OF THE FDA
According to its website, the FDA “is responsible for protecting the public health by assuring the safety, efficacy and security of human and veterinary drugs, biological products, medical devices, our nation’s food supply, cosmetics, and products that emit radiation” (FDA, 2010). Because of globalization, its responsibilities now require more international work. The changes put forth in Pathway to Global Product Safety and Quality and the HHS Global Health Strategy are the culmination of a gradual shift in the FDA’s way of operating, the ramifications of which might not yet be widely recognized, as the American regulatory system moves from mainly reacting to crises to preventing them (Olson, 2011). The 2011 Food Safety
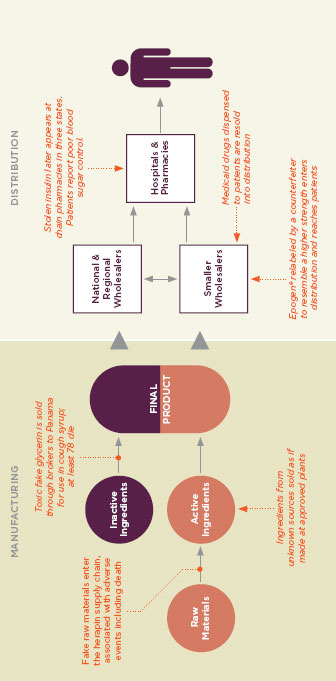
FIGURE 1-4
In complex global drug supply chains, there are many opportunities for unsafe products to be introduced before reaching the consumer. Figure reprinted with permission from the PEW Health Group.
SOURCE: PEW Health Group, 2011.
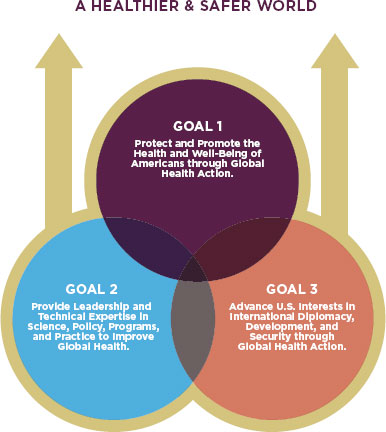
FIGURE 1-5
The HHS’s Global Health Strategy lays out three interconnected goals.
SOURCE: HHS, 2011.
Modernization Act improves the FDA’s ability to prevent and respond to outbreaks and gives the FDA the authority to recall foods (Stewart and Gostin, 2011). The act also aims to improve the safety of imported foods by requiring importers to verify that their plants overseas adhere to U.S. safety standards, and by establishing a way for qualified third parties to certify that producers abroad meet quality standards (FDA, 2011b). The act also increases the number of foreign inspections required of the FDA to 600 in 2011, doubling every year for the next 5 years after that (FDA, 2011b).
The FDA is also under pressure to increase the number of foreign
inspections it does of medical product manufacturers. In 2008 the Government Accountability Office (GAO) estimated that at the rate FDA was inspecting drug factories overseas, it would take 13 years to inspect every exporter once (GAO, 2008a). A complementary study reported that high-risk medical device factories were inspected once every 6 years and medium-risk devices about once every 27 years (GAO, 2008b). The FDA responded by creating a group of U.S. staff who exclusively inspect manufacturers overseas and by placing inspectors in the FDA’s foreign offices (GAO, 2010a). More recent analyses suggest that improvements to the agency’s import databases would allow for more efficient management of inspections abroad (GAO, 2010a, 2011). The FDA’s current data management system is often criticized; a recent New York Times editorial called it “antiquated” and a 2007 FDA Science Board review found it “obsolete” (Dangerous imports, 2011; FDA Subcommittee on Science and Technology, 2007; GAO, 2011).
The FDA’s public image is mostly a function of infrequent and controversial debates. The FDA suffers from what former Deputy Commissioner Joshua Scharfstein calls “competing narratives” of its work and mission (Sharfstein, 2011). To some observers, the agency’s new emphasis on working across borders and markets to advance the emerging field of regulatory science may be at odds with a conservative understanding of the agency as a domestic enforcer of product safety regulations. The FDA commissioned this report to advance its global mission and promote the necessity of working across borders for product safety.
CHARGE TO THE COMMITTEE
This report, the HHS Global Health Strategy, and the FDA’s Pathway to Global Product Safety and Quality draw on a common implicit conceptual framework: no country protects its citizens by working alone. In July 2010 the FDA Office of International Programs wrote to the Institute of Medicine (IOM) and provided background on the challenges it faces in improving food and drug regulation in developing countries (see Appendix H). This background, which complemented the statement of task, emphasized that the FDA was seeking assistance in developing a broad global health and development vision for the agency. Beyond the traditional statutory food and drug safety mission focused on protecting the U.S. population, the FDA request noted that, “equally important, strengthening regulatory capacity in the developing world will reap tremendous benefits for the health and quality of life of individuals and communities in those countries. Stronger regulatory systems in other countries can help to bolster current U.S. government (USG) investments being made in public health and development, e.g. through the President’s Global Health Initiative and
USG agencies, as well as contributions through multilateral organizations, and the broader global health and development community. These efforts increasingly embrace the principles of health systems strengthening, government ownership, and universal coverage.” (See Appendix H.)
With this request, the FDA asked the IOM to convene a committee of international experts to identify the core elements that should be common among regulatory systems in developing countries, to explain the main gaps in developing country regulatory systems, and to recommend a strategy for the FDA and other stakeholders to work with regulators around the world to improve product safety.
In the course of discussing the initially proposed statement of task with the FDA, there was concern that because the term “developing countries” includes a heterogeneous group of about 150 low- and middle-income countries, the committee’s data gathering needed to be focused on a more manageable sample. Thus for the purposes of this study it was agreed that emphasis in formulating cross-cutting insights would be based on looking at commonalities found in a sample of five or six countries that currently are or are expected to soon become major pharmaceutical and agricultural trading partners with the United States (e.g., Mexico, Brazil, South Africa, India, Thailand, and China). Language to that effect was added to the statement of task originally proposed by the FDA. Box 1-1 shows the final statement of task for this study.
BOX 1-1
Statement of Task
The FDA has requested that the Institute of Medicine convene a consensus study to assist the FDA in:
(A) Identifying the core elements of needed pharmaceutical, biologics, medical device, and food safety regulatory systems development in developing countries; and in
(B) Prioritizing these needs and recommending a strategic approach to the FDA’s moving forward to address regulatory capacity needs in the context of globalization.
In addition to identifying and prioritizing the core elements of regulatory systems development, the consensus study would also identify:
(C) Potential areas in which progress could be made in a 3- to 5-year time frame;
(D) Priorities for FDA engagement;
(E) Areas to which others (bilateral donors, development banks, foundations, academia, industry, and non-governmental organizations) are best suited to contribute; and
(F) How the FDA might best “partner” with these other institutions to bring to their efforts the expertise that the FDA has in an effort to leave a more sustainable “footprint” from both their and the FDA resource commitments.
Specific questions to be explored by the Consensus Study Committee shall at least include:
1. What critical issues do developing country regulatory authorities face, and how are they prioritized?
2. In what ways do they participate in standard-setting processes, organizations, and harmonization efforts?
3. What issues do they face in utilizing/implementing standards in a sustainable way?
4. What are the core elements of their regulatory systems, and are there others that should be considered?
5. What are the major gaps in systems, institutional structures, workforce, and competencies?
6. In what ways could those gaps be addressed?
7. In what ways could the U.S. FDA help address those gaps?
8. In what ways could others (as delineated above) help meet those gaps?
9. In what ways could the FDA partner with others to help meet those gaps?
10. What recommendations have already been put forward to strengthen regulatory systems?
11. What obstacles exist to implement those recommendations?
12. What steps could be taken to remove those obstacles?
13. What incentives and controls would be needed to support efforts?
Given that “developing countries” include a heterogeneous group of about 150 low- and middle-income countries, for the purposes of this study emphasis will be given to understanding in some depth the issues for a limited number of countries that currently are or are expected to soon become major pharmaceutical and agricultural trading partners with the United States (e.g., Mexico, Brazil, South Africa, India, Thailand, and China).
To address this task the IOM brought together the Committee on Strengthening Core Elements of Food and Drug Regulatory Systems in Developing Countries with expertise in global public health, pharmaceutical science and practice, agricultural science and practice, food safety, product quality assurance, risk assesment and risk management, supply chain management, globalization and trade, information technology, medical product regulation, food regulation, regulatory agency leadership, and regulatory or international health law. Box 1-2 describes the committee’s process and its domestic and international workshops.
Both the Food Safety Modernization Act and the FDA’s Pathway to Global Product Safety and Quality emphasize collaboration between the FDA and its counterpart agencies abroad (Tanne, 2011). To work with these agencies, it is important to first understand how regulatory systems work and the challenges regulators face in the world’s emerging manufacturing nations. The FDA and other stringent regulatory authorities have a stake in building the regulatory infrastructure and workforce in these countries. It is likely, however, that improving food and medical product regulation overseas will require costly investments that the FDA is neither authorized nor funded to make. Nor should the FDA, or any one agency, shoulder the burden of building capacity in low- and middle-income countries’ systems alone. Problems resulting from the globalization of the food and medical products supply require global solutions.
The FDA commissioned this study with the frank admission that its methods of ensuring product safety, inspections at factories and ports of entry, are inadequate when regulated products arrive at 300 different ports of entry from over 300,000 factories in 150 different countries (FDA, 2011b). There are also fundamental flaws with a plan to catch violators by inspecting consignments at random. Ensuring the safety of food and medical products imported from around the world is a difficult task, and one that the FDA has executed fairly successfully so far. There is no reason to believe that its luck will hold over the next 10 years without substantive improvements in the capacity of its counterpart agencies abroad.
The committee’s first task was to review the statement of task with the study sponsors from the FDA Office of International Programs. It did this in an open discussion at the first committee meeting. At this meeting, Ms. Mary Lou Valdez, Associate Commissioner for International Programs, explained to the committee that the statement of task is not “suggesting the committee do individual assessments of countries, but to look more widely at the [regulatory] landscape…. What are [the] essential elements of any system? … What key competencies are essential with any viable regulatory system?” The committee and the sponsors discussed a vision for a report describing the commonalities across low- and middle-income countries, identifying the common problems and a general strategy for
BOX 1-2
The Committee Process
In February 2011, the Institute of Medicine formed a 12-person committee to complete the task given by the FDA. See Appendix F for committee member biographies.
The full committee met in March, July, and October of 2011 to hear from outside speakers and make its recommendations for this report. At the March meeting, the committee heard from eight senior staff at the FDA, including Commissioner Hamburg; it also heard from 11 others, including a representative of the Mexican Ministry of Health. In July the committee heard from an information systems expert and had a public phone call with Anvisa, the Brazilian food and drug regulatory authority. The October meeting was entirely closed to the public. See Appendix E for meeting agendas.
In addition, travel delegations made up of committee members and IOM staff traveled to China, Brazil, South Africa, and India to meet with regulators, representatives of regulated industry, academics, and health and development workers. In China, Brazil, and South Africa the travel delegation had 1 day in each country of open public workshops and 1 day of small group meetings or office visits. In India, the travel delegation did not have a large public workshop, but instead had 3 days of small group meetings and office visits. The India meeting had originally been planned to coincide with the China meeting, but visas for most of the travel delegation were withheld for many weeks. The trip had to be canceled on short notice and rescheduled.
All the overseas workshops were open to the public. Over the course of 10 days of meetings abroad the travel delegations met with 140 stakeholders, including 21 U.S. government staff posted overseas and 46 regulators from China, India, Bangladesh, Thailand, Brazil, Uruguay, Peru, Chile, Tanzania, South Africa, and the African Union. See Appendix E for travel meeting agendas.
At workshops overseas, committee chair Dr. Jim Riviere’s opening remarks noted that the committee was not going to suggest ways to restructure any one country’s regulatory system or to describe any country’s shortcomings, but to identify common problems and common solutions across a range of low- and middle-income countries.
Public testimonies and information provided to the committee by various stakeholders informed its deliberations, the content of this report, and the committee’s recommendations to the FDA and other organizations as to how they can build capacity for food and medical product regulation around the world. After hearing public testimony and identifying the main product safety problems in developing countries, the committee and IOM staff examined these problems in the published literature, including scientific studies, commentaries, new articles, and books. This literature review gave depth to the committee’s findings and more context to its conclusions.
solving them. It was clear to all parties that the statement of task does not request analysis of specific regulatory codes in different countries. Instead it requires a high-level analysis across food and medical product lines in a broad cross-section of countries. Ms. Valdez, her colleague Dr. Katherine Bond, and Commissioner Margaret Hamburg explained that one of the main goals of this study, from the sponsor’s perspective, was to integrate the strengthening of regulatory systems building into the global public health and economic development agenda.
Global public health is a broad field, and many organizations are working in it. Implicit in the statement of task is the recognition that the FDA budget can barely fund its own activities; it cannot be a donor agency. However, this report aims to help the FDA target its capacity building efforts and to lay out a strategy that the U.S. government, other governments, universities, development banks, and nongovernmental organizations (NGOs) can use to ensure safe food and medical products around the world.
This is a complicated problem, but by no means a new one. In its analysis of the core elements of a food and medical products regulatory system, the committee gained perspective by first considering how one gold-standard regulatory agency evolved over the past century. The lessons learned from looking at the history of the FDA give a good foundation for understanding the building of similar agencies in developing countries.
THE BUILDING OF A MODERN REGULATORY AGENCY
For much of the past century, Americans have taken safe food and drugs for granted, but this was not always so. Quality assurance is a relatively modern concept and not one applied to foods or medicines until after the Second World War, when technological improvements spurred the growth of the manufacturing sector (ASQ, 2012). But starting in the late 19th century, industrialization encouraged migration from rural to urban areas, and country people who had once raised their own food were obliged to buy it. Swindlers flourished in the anonymity of city life. Under the leadership of Harvey Wiley, the Department of Agriculture found and reported on endless cases of food adulteration: bleaches and dyes in molasses, charcoal in ground pepper, and metal salts in canned foods, to name a few (Barkan, 1985; Law and Libecap, 2004). Lead, copper, and mercury salts were used to color candy (Jackson, 2009), and patent medicines, many marketed for children, commonly contained lethal doses of opiates (Finch, 1999). Some eastern states took legal action to prevent fraud, and sparsely populated western ones became a dumping ground for spurious products (Kane, 1964).
The public distrusted manufacturers, but was ill-equipped to judge the
quality of foods and drugs at purchase (Law and Libecap, 2004). A tangible anxiety resonated on the pages of period journals, and readers responded. A Ladies Home Journal letter-writing campaign petitioned President Theodore Roosevelt, his cabinet, and members of Congress for food and drug safety laws (Barkan, 1985). Concerns about product quality also took an economic toll. American exports were less competitive in Europe, where many countries had food and drug regulatory systems in place (Barkan, 1985). Public opinion was shifting in favor of regulation, and Upton Sinclair’s 1906 novel The Jungle, with its horrific depictions of the Chicago meat packing industry, perhaps did more than anything else to push government to act. Only 4 months after the book’s publication, during which time President Roosevelt sent inspectors to verify Sinclair’s portrayal of stock yards and slaughterhouses, Congress passed the Pure Food and Drug Act and the Meat Inspection Act, banning food adulteration, deceptive statements on labels, and the interstate sale of adulterated foods (Jackson, 2009).
Around the same time, medicine was also changing. In 1901, a 5-year-old girl died from tetanus in a St. Louis hospital (Junod, 2002). The infection was traced back to a milk wagon horse. Horse blood serum antitoxin was then widely used to treat diphtheria, but there were no controls over its production or requirements for batch testing. When this horse contracted tetanus, its contaminated serum killed 13 children in total (Bren, 2006). As a result, in 1902, Congress passed the Biologics Control Act, which man-

Nineteenth-century patent medicines often contained opiates, stimulants, or alcohol.
dated that producers be licensed for the manufacture and sale of vaccines, serums, and antitoxins (Bren, 2006).
In 1910 the Flexner Report introduced guidelines for medical school accreditation to the United States, requiring doctors to train in anatomy, physiology, and laboratory science and to complete a 2-year hospital internship (Beck, 2004; Flexner, 1910). Standards for accreditation and licensure made the distinctions between medical doctors and quacks clear even to uneducated patients. Over time, the practice of medicine grew more tied to the prescription of controlled drugs. A growing public concern with addiction motivated the Harrison Narcotic Act of 1914, which gave the authority to distribute narcotic drugs and cocaine to licensed physicians only (Spillane and McAllister, 2003).
Legal provisions for food and drug safety still fell short, however. By 1931 the newly formed U.S. Food and Drug Administration (FDA) did not have the authority to inspect farms or factories, and politicians were loath to expand its mandate fearing accusations of socialism (Temin, 1978). The political climate was tense in the early years of the antimicrobial revolution, when the S.E. Massengill Company responded to demand for a liquid sulfa drug preparation with the so-called Elixir Sulfanilamide, a solution of caramel, raspberry extract, water, and the drug sulfanilamide dissolved in diethylene glycol (Ballentine, 1981; Wax, 1995). More than 100 people died from taking it, many of them children who could not swallow the alternative tablet drug (Ballentine, 1981). The Federal Food, Drug, and Cosmetics Act passed in the emotional aftermath of the mass poisoning required drug manufacturers to prove safety before releasing a medicine for sale.
The FDA grew slowly, often in spurts inspired by egregious industry negligence (Figure 1-6). Social changes also drove the need for a government food and drug regulator. Rural to urban migration continued throughout the 20th century and the Great Depression years before the passage of the Federal Food, Drug, and Cosmetics Act saw distinct migration patterns. African Americans left the Jim Crow South for jobs in northern cities, many in the Chicago meat-packing houses, and small landholders whose ill-advised farming practices devastated Oklahoma and surrounding states traveled west to California looking for work as day laborers. America changed in the 20th century: diet and marketing patterns changed, medicine changed, and pharmacology exploded. Designing a regulatory system to adapt to these changes was difficult and expensive, especially during the hard times of the world wars and the Depression.
Similar Changes and Similar Hurdles in Emerging Economies
Now more than 150 other countries are facing the same problems, but globalization has magnified them 10-fold. Every week, 1.5 million people
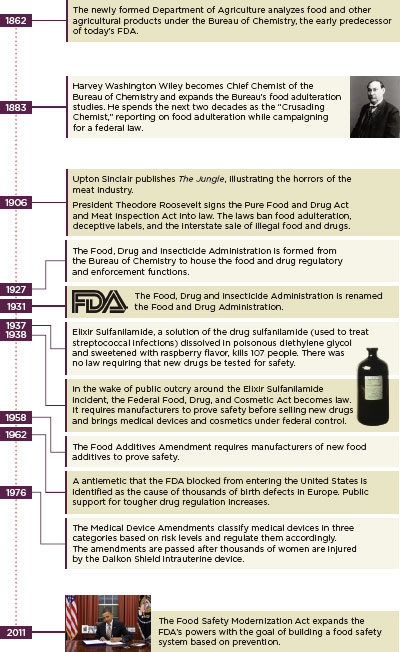
FIGURE 1-6
The evolution of regulation: History of the U.S. Food and Drug Administration.
leave the countryside for urban centers (FDA, 2011b), and today’s migratory workers are as likely to find work in foreign cities as in the provincial capital. Doctors around the world train against similar standards and want to treat their patients with modern medicines. Donor organizations sometimes supply these drugs, but some are sold at market prices. Firms in middle-income countries export medicines cheaply when tariffs allow. Even vaccines, though exceedingly complicated to manufacture, are available around the world, in part because of a combination of technological sophistication and low overhead in India, Indonesia, and Brazil.
The global recession has hit poor countries hard. By World Bank estimates, another 90 million people live on less than $1.25 a day because of the 2008-2009 financial crisis (UN, 2009; World Bank, 2010). The added financial stress came at a bad time for countries transitioning away from a state-controlled economy. Some post-colonial and former communist country governments see a tension between embracing free market capitalism and wanting a government check on industry.
Many of the problems are logistical. Packaged and prepared foods are popular even in traditional societies (Unnevehr, 2007), and these foods are vulnerable to contamination; microbes on only one ingredient can easily contaminate a large amount of food during processing. It is difficult to sanitize equipment without plentiful, clean water (Bester, 2011). It is also hard to enforce sanitary standards when these standards are at odds with prevalent ideas about hygiene.
Fraud is another old problem aggravated by globalization. Fake medicines are a lucrative business worth between $75 and $200 billion a year (Poison pills: Fake drugs, 2010). The trade in them is worst in countries with weak regulatory and law enforcement systems (Siva, 2010). In 2008 New York Times reporters Walt Bogdanich and Jake Hooker won the Pulitzer Prize for their investigative series, “A Toxic Pipeline,” that tracked adulterated medicine from China to Panama, where over-the-counter cough syrup mixed with diethylene glycol killed hundreds (Bogdanich and Hooker, 2007; The Pulitzer Prizes, 2008). The same network fed solvents to Haiti, India, Nigeria, Argentina, and Bangladesh, killing thousands in countries where the poor die at home, without seeking medical care, outside the reach of surveillance and reporting systems (Bogdanich et al., 2007). Bogdanich and Hooker reported on a crisis similar to the aforementioned Elixir Sulfanilamide tragedy in everything but scale; they found that distance and delay made it almost impossible to trace toxic adulterants through international webs of forged certificates and missing receipts. For criminals dodging accountability, it is easy to hide in the global village.
MODERN FOOD AND DRUG CONTAMINANTS TRAVEL FAR AND FAST
In January 2008 the Centers for Disease Control and Prevention (CDC) investigated a spike in reported severe allergic reactions among dialysis patients taking heparin, a blood thinner sold by Baxter International. The CDC and the FDA traced the problem to over-sulfated chondroitin sulfate in the heparin active ingredient from China (GAO, 2010b). The adulterated heparin mimicked the properties of the authentic drug in standard screening tests, though it cost roughly 100 times less to manufacture (Pew Health Group, 2011). Neither Baxter nor the FDA was ever able to pinpoint the exact source of the adulteration (GAO, 2010b; Pew Health Group, 2011).
In a report to Congress, the GAO concluded that the FDA handled the crisis well, taking speedy and appropriate action to protect the American public from contaminated heparin (GAO, 2010b). The report identified limitations in the FDA’s ability to inspect and investigate heparin producers in China (GAO, 2010b). Although a clear weakness in the FDA’s reaction to crisis, the inability to quickly inspect foreign firms is not entirely within its control. The FDA did not have staff permanently stationed in China until November 2008 (FDA, 2011a). Even if it had, and even if its entire inspectorate devoted itself only to inspecting the workshops making heparin in China, it would still have been nearly impossible to identify the source of the problem. Sellers of the contaminated product made between $1 and $3 million by adding over-sulfated chondroitin sulfate to the active ingredient (Villax, 2008). The prospect of such payoffs will surely continue to motivate criminal behavior.
The American public became more aware of its vulnerability after the heparin incident, though a Harris poll suggested public confidence in the FDA had been waning much earlier (Harris Interactive, 2008). In March 2007, the FDA recalled 60 million packages of pet food containing Chinese wheat gluten tainted with melamine, a cheap additive that mimics protein in testing (Barboza and Barrionuevo, 2007). In September 2008, the same adulterant was found in the Chinese domestic milk supply. An estimated 300,000 children were sickened from the contaminated milk, many suffering permanent kidney damage (Branigan, 2008). The enormity of the dairy companies’ actions drew public attention to weaknesses in the Chinese food industry (Branigan, 2008; Sternberg, 2008). In an interview with Voice of America, WHO food safety scientist Peter Ben Embarek attributed much of the problem to a lag between private-sector production capability and the public sector’s ability to regulate (Schlein, 2008).
REPORT STRUCTURE
This report aims to identify ways to protect the safety of the food and medical product supply around the world. This includes protecting U.S. consumers from nefarious suppliers and poorly controlled imports and building regulatory capacity in low- and middle-income countries. This report will describe ways to work toward standards that will protect both foreign and domestic markets.
There are manifold gaps in the public sector’s ability to regulate food and medical products, both in developing and developed countries. This report examines these gaps using examples from specific countries to illustrate common trends. The report presents a strategy for bridging gaps in developing country regulatory systems and ways in which the U.S. government and other stakeholders can work together to bridge these gaps.
This report responds to the statement of task by first describing, in Chapter 2, the core elements of food and medical product regulatory systems as well as the minimal elements of a functional system in a low- or middle-income country. Chapter 2 also describes the common elements in food, drug, and medical device regulatory systems across countries. Chapter 3 summarizes the critical issues regulators in developing countries face. These issues fall into the main categories of adherence to standards, controlling supply chains, problems with infrastructure, legal problems, workforce development, institutional fragmentation, surveillance, communication, and the lack of political will. Chapter 4 lays out the committee’s strategic approach to bridging the gaps in developing country regulatory systems. Chapter 5 contains the committee’s recommendations to various international organizations and outlines partnerships the FDA could have with these stakeholders. Chapter 6 recommends domestic action that could improve the capacity of food and medical product regulation around the world. Chapter 7 discusses which of the recommendations can be implemented in 3 to 5 years and the report’s consistency with the objectives outlined in the Global Health Strategy.
REFERENCES
ASQ (American Society for Quality). The history of quality: Overview. http://asq.org/learn-about-quality/history-of-quality/overview/overview.html (accessed April 2, 2012).
Ballentine, C. 1981. Taste of raspberries, taste of death: The 1937 elixir sulfanilamide incident. FDA Consumer Magazine. http://www.fda.gov/AboutFDA/WhatWeDo/History/ProductRegulation/SulfanilamideDisaster/default.htm (accessed October 25, 2011).
Barboza, D., and A. Barrionuevo. 2007. Filler in animal feed is open secret in China. New York Times, April 30.
Barkan, I. D. 1985. Industry invites regulation: The passage of the Pure Food and Drug Act of 1906. American Journal of Public Health 75(1):18-26.
Beck, A. H. 2004. The Flexner Report and the standardization of American medical education. Journal of the American Medical Association 291(17):2139-2140.
Bester, A. 2011. The importance of food safety in Africa. How We Made It in Africa. http://www.howwemadeitinafrica.com/the-importance-of-food-safety-in-africa/5043/ (accessed June 14).
Bogdanich, W., and J. Hooker. 2007. From China to Panama, a trail of poisoned medicine. New York Times, May 6.
Bogdanich, W., J. Hooker, H. Kumar, A. Giridharadas, and J. Ali Manik. 2007. As FDA tracked poisoned drugs, a winding trail went cold in China. New York Times, June 17.
Branigan, T. 2008. Chinese figures show fivefold rise in babies sick from contaminated milk. Guardian, December 2.
Bren, L. 2006. The road to the biotech revolution: Highlights of 100 years of biologics regulation. FDA Consumer Magazine. http://www.fda.gov/AboutFDA/WhatWeDo/History/FOrgsHistory/CBER/ucm135758.htm (accessed October 25, 2011).
Dangerous imports. 2011. New York Times, June 25.
FDA (Food and Drug Administration). 2010. About FDA: What we do. http://www.fda.gov/aboutfda/whatwedo/default.htm (accessed March 1, 2012).
———. 2011a. FDA’s international posts: Improving the safety of imported food and medical products. http://www.fda.gov/forconsumers/consumerupdates/ucm185769.htm#TheChinaOffice (accessed November 22, 2011).
———. 2011b. Pathway to global product safety and quality. Washington, DC: U.S. Food and Drug Administration.
FDA Subcommittee on Science and Technology. 2007. FDA science and mission at risk: Report of the Subcommittee on Science and Technology. Washington, DC: FDA.
Finch, L. 1999. Soothing syrups and teething powders: Regulating proprietary drugs in Australia, 1860-1910. Medical History 43(1):74-94.
Flexner, A. 1910. Medical education in the United States and Canada. New York: The Carnegie Foundation for the Advancement of Teaching.
GAO (Government Accountability Office). 2008a. Drug safety: Better data management and more inspections are needed to strengthen FDA’s foreign drug inspection program. GAO-08-970. Washington, DC: GAO.
———. 2008b. Medical devices: Challenges for FDA in conducting manufacturer inspections. GAO-08-428T. Washington, DC: GAO.
———. 2010a. Drug safety: FDA has conducted more foreign inspections and begun to improve its information on foreign establishments but more progress is needed. GAO-10-961. Washington, DC: GAO.
———. 2010b. Food and Drug Administration: Response to heparin contamination helped protect public health; controls that were needed for working with external entities were recently added. GAO-11-95. Washington, DC: GAO.
———. 2011. Drug safety: FDA faces challenges overseeing the foreign drug manufacturing supply chain. GAO-11-936T. Washington, DC: GAO.
Gill, L. 2011 March 2. Addressing the challenges of medical device safety in a global environment. Presented at Strengthening Core Elements of Regulatory Systems in Developing Countries: Meeting One, Institute of Medicine, Washington, DC.
Harris Interactive. 2008. Confidence in FDA hits new low, according to WSJ.com/Harris Interactive study. http://www.harrisinteractive.com/vault/Harris_Interactive_News_2008_04_22.pdf (accessed November 30, 2011).
HHS (Department of Health and Human Services). 2011. The global health strategy of the U.S. Department of Health and Human Services. Washington, DC: HHS.
Jackson, L. S. 2009. Chemical food safety issues in the United States: Past, present, and future. Journal of Agriculture and Food Chemistry 57(18):8161-8170.
Junod, S. W. 2002. Biologics centennial: 100 years of biologics regulation. Update (November-December). http://www.fda.gov/aboutfda/whatwedo/history/productregulation/?selectionsfromfdliupdateseriesonfdahistory/ucm091754.htm (accessed October 25, 2011).
Kane, R. J. 1964. Populism, progressivism, and pure food. Agricultural History 38(3):161-166.
Law, M. T., and G. D. Libecap. 2004. The determinants of Progressive Era reform: The Pure Food and Drug Act of 1906. Cambridge, MA: National Bureau of Economic Research.
Olson, E. D. 2011. Protecting food safety: More needs to be done to keep pace with scientific advances and the changing food supply. Health Affairs 30(5):915-923.
Pew Health Group. 2011. After heparin: Protecting consumers from the risks of substandard and counterfeit drugs. Washington, DC: Pew Health Group.
Poison pills: Fake drugs. 2010. Economist, September 4.
Schlein, L. 2008. China’s melamine milk crisis creates crisis of confidence. Voice of America, September 26.
Sharfstein, J. M. 2011. The FDA—a misunderstood agency. Journal of the American Medical Association 306(11):1250-1251.
Siva, N. 2010. Tackling the booming trade in counterfeit drugs. Lancet 376:1725-1726.
Spillane, J., and W. B. McAllister. 2003. Keeping the lid on: A century of drug regulation and control. Drug and Alcohol Dependence 70(3, Suppl 1):S5-S12.
Sternberg, J. 2008. Notes on a milk scandal. Wall Street Journal, October 10.
Stewart, K., and L. Gostin. 2011. Food and Drug Administration regulation of food safety. Journal of the American Medical Association 306(1):88-89.
Tanne, J. H. 2011. FDA seeks global partners to improve safety of imported food, medicines, and devices. BMJ 342(d4106).
Temin, P. 1978. Working paper: The origin of compulsory drug prescription. Cambridge, MA: Massachusetts Institute of Technology.
The Pulitzer Prizes. 2008. The 2008 Pulitzer Prize winners: Investigative reporting. http://www.pulitzer.org/citation/2008-Investigative-Reporting (accessed December 20, 2011).
UN (United Nations). 2009. Rethinking poverty: Report on the world social situation 2010. New York, NY: UN.
Unnevehr, L. J. 2007. Food safety as a global public good. Agricultural Economics 37(Suppl 1):149-158.
Villax, G. 2008. Business of counterfeit heparin and its implications. Paper presented at 3rd EFCG Pharma Business Conference, Lisbon, Portugal, May 29.
Wax, P. M. 1995. Elixirs, diluents, and the passage of the 1938 Federal Food, Drug and Cosmetic Act. Annals of Internal Medicine 122(6):456-461.
World Bank. 2010. Impact of the global financial crisis on fragile and conflict-affected countries. http://siteresources.worldbank.org/INTLICUS/Resources/ImpactofFinancialCrisisFactSheetSept28.pdf.




















