
THE SCIENCE AND APPLICATIONS OF MICROBIAL GENOMICS: PREDICTING, DETECTING, AND TRACKING
NOVELTY IN THE MICROBIAL WORLD
Over the past several decades, new scientific tools and approaches for detecting microbial species have dramatically enhanced our appreciation of the diversity and abundance of the microbiota and its dynamic interactions with the environments within which these microorganisms reside. The first bacterial genome2 was sequenced in 1995 and took more than 13 months of work to complete. Today (2012), a microorganism’s entire genome can be sequenced in a few days. Much as our view of the cosmos was forever altered in the 17th century with the invention of the telescope (Nee, 2004), these genomic technologies, and the observations derived from them, have fundamentally transformed our appreciation of the microbial world around us.
Nucleic acid sequencing technologies now provide access to the previously “unculturable”—and thus, undetected—microorganisms that comprise the majority of microbial life. Rapid and inexpensive sequencing platforms make it
__________________
1 The planning committee’s role was limited to planning the workshop, and the workshop summary has been prepared by the workshop rapporteurs (with the assistance of Pamela Bertelson, Rebekah Hutton, and Katherine McClure) as a factual summary of what occurred at the workshop. Statements, recommendations, and opinions expressed are those of individual presenters and participants, and are not necessarily endorsed or verified by the Institute of Medicine, and they should not be construed as reflecting any group consensus.
2 For the purposes of this summary, the genome is defined as the complete set of genetic information in an organism. In bacteria, this includes the chromosome(s) and plasmids (extrachromosomal DNA molecules that can replicate autonomously within a bacterial cell) (Pallen and Wren, 2007).
commonplace to sort through the genomes of dozens of strains of a single microbial species or to conduct “metagenomic” analyses of vast communities of the microbiota from a wide variety of environments. These technical advancements and concurrent investments in the fields of microbial ecology, evolution, forensics, and epidemiology have transformed our ability to use genomic sequence information to explore the origins, evolution, and catalysts associated with historical, emergent, and reemergent disease outbreaks. The ability to “read” the nucleic acid sequence of microbial genomes has provided important insights into this previously hidden, unculturable world by revealing the vast diversity and complexity of microbial life around us, and their myriad interactions with their abiotic and biotic environmental niches.
Recent examples of the use of “whole genome” sequencing to investigate outbreaks of emerging, reemerging, and novel infectious diseases illustrate the potential of these methods for enhancing disease surveillance, detection, and response efforts. Using slight sequence differences between isolates to discriminate between closely related strains, investigators have tracked the evolution of isolates in a disease outbreak, traced person-to-person transmission of a communicable disease, and identified point sources of disease outbreaks. When genomic information about related strains or past disease outbreaks is available, the genome sequence of outbreak strains has proved useful in identifying factors that may contribute to the emergence, virulence, or spread of pathogens, as well as in speeding diagnostic tool development. In a recent development, fast genome sequencing was used to halt the spread of a methicillin-resistant Staphylococcus aureus (MRSA) infection in a neonatal ward in a hospital in Cambridge, United Kingdom (Harris et al., 2012)
Statement of Task
On June 12 and 13, 2012, the Institute of Medicine’s (IOM’s) Forum on Microbial Threats convened a public workshop in Washington, DC, to discuss the scientific tools and approaches being used for detecting and characterizing microbial species, and the roles of microbial genomics and metagenomics to better understand the culturable and unculturable microbial world around us.3 Through invited presentations and discussions, participants examined the use of microbial genomics to explore the diversity, evolution, and adaptation of microorganisms in
__________________
3 A public workshop will be held to explore new scientific tools and methods for detecting and characterizing microbial species and for understanding the origins, nature, and spread of emerging, reemerging, and novel infectious diseases of humans, plants, domestic animals, and wildlife. Topics to be discussed may include microbial diversity, evolution, and adaptation; microbial genomic, epidemiology, and forensic tools and technologies; infectious disease detection and diagnostic platforms in clinical medicine, veterinary medicine, plant pathology, and wildlife epidemiology; development of microbial genomic and proteomic databases; and strategies for predicting, mitigating, and responding to emerging infectious diseases.
a wide variety of environments; the molecular mechanisms of disease emergence and epidemiology; and the ways that genomic technologies are being applied to disease outbreak trace back and microbial surveillance. Points that were emphasized by many participants included the need to develop robust standardized sampling protocols, the importance of having the appropriate metadata (e.g., the sequencing platform used, sampling information, culture conditions), data analysis and data management challenges, and information sharing in real time.
Organization of the Workshop Summary
This workshop summary was prepared by the rapporteurs for the Forum’s members and includes a collection of individually authored papers and commentary. Sections of the workshop summary not specifically attributed to an individual reflect the views of the rapporteurs and not those of the members of the Forum on Microbial Threats, its sponsors, or the IOM. The contents of the unattributed sections of this summary report provide a context for the reader to appreciate the presentations and discussions that occurred over the 2 days of this workshop.
The summary is organized into sections as a topic-by-topic description of the presentations and discussions that took place at the workshop. Its purpose is to present information from relevant experience, to delineate a range of pivotal issues and their respective challenges, and to offer differing perspectives on the topic as discussed and described by the workshop participants. Manuscripts and reprinted articles submitted by some but not all of the workshop’s participants may be found, in alphabetical order, in Appendix A.
Although this workshop summary provides a distillation of the individual presentations, it also reflects an important aspect of the Forum’s philosophy. The workshop functions as a dialogue among representatives from different sectors and disciplines and allows them to present their views about which areas, in their opinion, merit further study. This report only summarizes the statements of participants over the course of the workshop. This summary is not intended to be an exhaustive exploration of the subject matter, nor does it represent the findings, conclusions, or recommendations of a consensus committee process.
GLIMPSES OF THE MICROBIAL WORLD
Microbiologists investigate a largely hidden world, laboring to understand the structure and function of organisms that are essentially invisible to the naked eye. Critical methodological advances—from microscopy through metagenomics—have made the staggering diversity of the microbial worlds on this planet easier to study and have brought them into focus (Table WO-1). Over the past several centuries, these approaches have provided ever-expanding views of the extraordinary organismal, metabolic, and environmental diversity of microorganisms.
TABLE WO-1 Some Major Methods for Studying Individual Microbes Found in the Environment
|
Method |
Summary |
Comments |
|
Microscopy |
Microbial phenotypes can be studied by making them more visible. In conjunction with other methods, such as staining, microscopy can also be used to count taxa and make inferences about biological processes. |
The appearance of microbes is not a reliable indicator of what type of microbe one is looking at. |
|
Culturing |
Single cells of a particular microbial type are grown in isolation from other organisms. This can be done in liquid or solid growth media. |
This is the best way to learn about the biology of a particular organism. However, many microbes are uncultured (i.e., have never been grown in the lab in isolation from other organisms) and may be unculturable (i.e., may not be able to grow without other organisms). |
|
rRNA-PCR |
The key aspects of this method are the following: (a) all cell-based organisms possess the same rRNA genes (albeit with different underlying sequences); (b) PCR is used to make billions of copies of basically each and every rRNA gene present in a sample; this amplifies the rRNA signal relative to the noise of thousands of other genes present in each organism’s DNA; (c) sequencing and phylogenetic analysis places rRNA genes on the rRNA tree of life; the position on the tree is used to infer what type of organism (a.k.a. phylotype) the gene came from; and (d) the numbers of each microbe type are estimated from the number of times the same rRNA gene is seen. |
This method revolutionized microbiology in the 1980s by allowing the types and numbers of microbes present in a sample to be rapidly characterized. However, there are some biases in the process that make it not perfect for all aspects of typing and counting. |
|
Shotgun genome sequencing of cultured species |
The DNA from an organism is isolated and broken into small fragments, and then portions of these fragments are sequenced, usually with the aid of sequencing machines. The fragments are then assembled into larger pieces by looking for overlaps in the sequence each possesses. The complete genome can be determined by filling in gaps between the larger pieces. |
This has now been applied to over 1,000 microbes, as well as some multicellular species, and has provided a much deeper understanding of the biology and evolution of life. One limitation is that each genome sequence is usually a snapshot of one or a few individuals. |
SOURCE: Eisen (2007).
There are three recognized domains of life: the Archaea, the bacteria, and the eukarya. Microorganisms are now recognized as the primary source of diversity for life on Earth and its inhabitants (Figure WO-1). Even more astonishing, perhaps, is what still remains to be discovered about the microbiota on this planet. As Fraser et al. (2000) have observed, “The genetic, metabolic and physiological diversity of microbial species is far greater than that found in plants and animals. The diversity of the microbial world is largely unknown, with less than one-half of 1% of the estimated 2–3 billion microbial species identified [emphasis added].” Moreover, while there are well over 10 million species of “known” bacteria only a few thousand have been formally described (Eisen, 2007). With the advent of genomic technologies, we are entering a new era of scientific discovery that holds great promise for revealing the breadth of diversity and depth of complexity inherent to the microbial world.
From Animalcules to Germs
Until just over 300 years ago, the microscopic world that we share the planet with was largely unseen and unknown. In the 17th century, Antonie van Leeuwenhoek provided the first detailed glimpses of the “animalcules” in the microbial world when he developed viewing techniques and magnifying lenses with sufficient power to see microorganisms. Van Leeuwenhoek obtained these organisms, as illustrated in Figure WO-2, from a variety of environmental sources, ranging from rain and pond water to plaque biofilms scraped from teeth. Their simple morphologies prevented the precise identification and classification of these organisms, but through detailed descriptions and illustrations in his letters to the Royal Society of England, van Leeuwenhoek brought the invisible world of microscopic life forms to the attention of scientists (Handlesman, 2004).
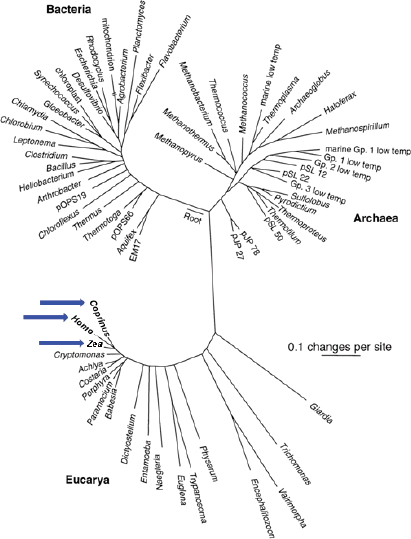
FIGURE WO-1 Universal tree of life based on a comparison of nucleic acid (RNA) sequences found in all cellular life (small subunit ribosomal RNA). “A sobering aspect of large-scale phylogenetic trees such as that shown in Figure WO-1 is the graphical realization that most of our legacy in biological science, historically based on large organisms, has focused on a narrow slice of biological diversity. Thus, we see that animals (represented by Homo), plants (Zea), and fungi (Coprinus) (see blue arrows) constitute small and peripheral branches of even eukaryotic cellular diversity” (Cracraft and Donoghue, 2004).
NOTE: The scale bar corresponds to 0.1 changes per nucleotide position.
SOURCE: From Pace, N. R. 1997. A Molecular View of Microbial Diversity and the Biosphere. Science 276:734-740. Reprinted with permission from AAAS.

Careful observation of microorganisms by scientists such as Louis Pasteur revealed the connections between microorganisms and practical phenomena. The production of beer and vinegar, for example, depended upon the presence of yeast for the conversion of sugar to alcohol and the fermentation of alcohol into acetic acid, respectively. Until the development of standardized culturing techniques in the late 19th century researchers could do little more than observe these creatures as a mixture of organisms in complex matrices. Pasteur also examined the connections between microorganisms and diseases of plants, animals, and humans, becoming an early proponent of the “germ theory” of disease (de Kruif, 1926).
In 1884, Robert Koch and Friedrich Loeffler formalized the germ theory of disease by outlining a series of tests designed to determine whether a specific microorganism was the causative agent of a specific disease. These tests, known as Koch’s postulates (Box WO-1), required the isolation and propagation of “pure cultures” of microorganisms. Koch initially applied these tests to establish the infectious etiology of anthrax and tuberculosis (de Kruif, 1926). Using these techniques, researchers could conduct experimental investigations of specific microorganisms under controlled conditions.
Our current understanding of microbe–host interactions have been influenced by more than a century of research, sparked by the germ theory of disease and rooted in historic notions of contagion that long preceded the research and intellectual syntheses of Pasteur and Koch in the 19th century (Lederberg, 2000). The success of this approach to the identification of the microbial basis of disease launched generations of “microbe hunters” who began a systematic search for disease-causing microbes that could be isolated and cultured under controlled laboratory conditions. Their work set a new course for the study and treatment of infectious disease-causing organisms. The “power and precision” of their studies
1. The parasite occurs in every case of the disease in question and under circumstances that can account for the pathological changes and clinical course of the disease.
2. The parasite occurs in no other disease as a fortuitous and nonpathogenic parasite.
3. After being fully isolated from the body and repeatedly grown in pure culture, the parasite can induce the disease anew.
SOURCES: Fredericks and Relman (1996), Koch (1891), and Rivers (1937).
using pure culture established these methods as the standard laboratory microbiology technique (Lederberg, 2000). At the same time, this disease-centric approach to microbe discovery has, for the past century and a half, not only influenced our collective perceptions of what microbes do “to” rather than “for” their hosts but also biased the database of the tree of life to one that, until relatively recently, has been focused almost entirely on disease-causing, culturable microorganisms.
This pathogen-centric bias attributed disease entirely to the actions of invading microorganisms, thereby drawing battle lines between “them” and “us,” the injured hosts (Casadevall and Pirofski, 1999). Although it was recognized in Koch’s time that some microbes did not cause disease in previously exposed hosts (e.g., milk maids who had been exposed to cowpox did not become infected with smallpox), the fact that his postulates could not account for microbes that did not cause disease in all hosts was not generally appreciated until the arrival of vaccines and the subsequent introduction of immunosuppressive therapies in the 20th century (Casadevall and Pirofski, 1999; Isenberg, 1988). By then, the paradigm of the systematized search for the microbial basis of disease, followed by the development of antimicrobial and other therapies to eradicate these pathogenic agents, had been firmly established in clinical practice.
THE CULTIVATION BOTTLENECK, GENOMICS,
AND THE UNIVERSAL TREE OF LIFE
In the 1950s and 1960s this focus on a few easily cultured organisms produced an explosion of information about microbial physiology and genetics that overshadowed efforts to understand the ecology and diversity of the microbial world (Pace, 1997). As the workhorses of the emerging field of molecular biology bacteria, such as Escherichia coli and Bacillus subtilis and their viruses (bacteriophages) became perhaps some of the best characterized microorganisms
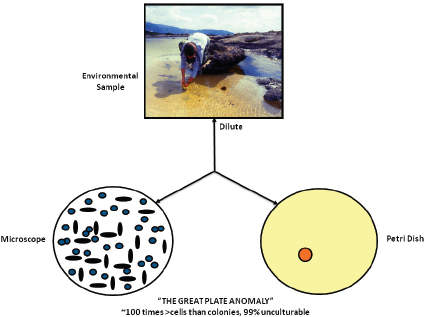
FIGURE WO-3 The great plate count anomaly.
SOURCE: Lewis (2011). Figure by Kim Lewis, Courtesy of Moselio Schaechter, Small things Considered, The Microbe Blog.
in biological research. While a rich source of discovery and knowledge, this focus on readily cultured organisms limited most researchers’ appreciation of the diversity and ubiquity of microbial life.
The predisposition toward discovery, isolation, and characterization of microorganisms that could be readily cultured4 in the laboratory is known as the “cultivation bottleneck” and is evident in the substantial difference in population counts of microorganisms present in a sample depending on whether they are conducted using microscopy or culturing techniques—a phenomenon known as the “great plate anomaly” (see Figure WO-3). This difference is attributed to the fact that the vast majority of microorganisms, 99 percent by some estimates, cannot be isolated and cultured5 using standard laboratory techniques (Handelsman, 2004).
_______________________
4 The ease of isolation and culturing of certain organisms reflects an organism’s ability to grow rapidly into colonies on high-nutrient artificial growth media, typically under aerobic conditions. This had led some to characterize these species as the “weeds” of the microbial world (Hugenholtz, 2002).
5 Microorganisms may be unculturable because of the inability to replicate important nutritional or environmental requirements for growth, including the services provided by other microorganisms that may be present in natural settings.
SEQUENCE-BASED DETECTION AND DISCOVERY
Pace and colleagues (1985) used sequence-based methods to investigate the composition of all constituents of the microbial biosphere. These culture-independent surveys led to the discovery of previously unknown and diverse lineages of organisms from habitats across the Earth, including bacterial and parasitic pathogens in the human body (Handelsman, 2004; Pace, 1997; Relman et al., 1990; Santamaria-Fries et al., 1996). The polymerase chain reaction6 (PCR) technique, developed in 1983 by Kary Mullis, aided these studies by allowing researchers to easily amplify single copies of a particular DNA sequence into thousands or millions of copies. This advance enabled investigators to rapidly and comprehensively catalog the diversity of life forms in the microbial world. Initial molecular phylogeny studies demonstrated that this “unseen world” of microorganisms could be studied and confirmed that the number of organisms represented in the unculturable world far exceeded the size of the culturable world.
While culture-based techniques remain the gold standard for disease detection, outbreak investigations, and infectious disease epidemiology, over the past several decades a range of sequence-based methods—including broad-range PCR, high-throughput sequencing technologies, microarrays, and shotgun metagenomics—have been applied to improve the detection and discovery of pathogens and other microorganisms. rRNA gene sequences may also be used to phylogenetically identify microbes that are otherwise uncharacterizable by other methods and approaches.
Broad-Range PCR
Some conserved genes and their encoded molecules have properties that render them useful as “molecular clocks.” These conserved genes, such as the 16S rRNA gene in bacteria, can be amplified from any member of a phylogenetic group using consensus primers.7 The sequences of the amplified, intervening gene regions with variable composition are then determined, in order to identify known or previously uncharacterized members of the group, and their evolutionary relationships to all other organisms revealed. This approach has been used to discover previously uncharacterized bacterial, viral, and parasitic pathogens (Nichol et al., 1993; Relman, 1993, 1999, 2011; Relman et al., 1990, 1992).
_______________________
6 The polymerase chain reaction (PCR) is a biochemical technology in molecular biology that amplifies a single or a few copies of a piece of DNA across several orders of magnitude, generating thousands to millions of copies of a particular DNA sequence.
7 Primers whose sequences are found in all known, and presumably unknown, members of the group.
High-Throughput Sequencing Technologies8
Nucleic acid sequencing technologies have dramatically enhanced our understanding of the diversity of the microbiota and their dynamic interactions with the environments they reside in. The genomes of thousands of organisms from all three domains of life, as well as those of quasi-life forms such as viruses, have been sequenced. Metagenomics has taken this approach a step further by cataloging the genomic components of microbes living in complex environmental matrices, from soil samples, to the ocean, coral reefs, and the human body (Mardis, 2008). The conventional or first-generation technology of automated Sanger sequencing produced all of the early microbial sequence data. Next-generation9 sequencing technologies, which were introduced in 2005, have decreased the cost and time necessary for sequence production.
Sequence data have been used for a number of applications, including:
• De novo assembly of entire genomes to produce primary genetic sequences and to support the detailed genetic analysis of an organism.
• Whole genome “resequencing” for the discovery of variants that differ in sequence to known genome sequences of a closely related strain.
• Species classification and the identification of predicted coding sequences and novel gene discovery in genomic surveys of microbial communities (metagenomics).
• “Seq-based” assays that determine the sequence content and abundance of mRNAs, non-coding RNAs, and small RNAs (collectively called RNA-seq); or measure genomewide profiles of DNA-protein complexes (ChIP-seq), methylation sites (methyl-seq), and DNase I hypersensitivity sites (DNase-seq) (Metzker, 2010).
Microarrays
Microarray technology runs the gamut from assays that contain hundreds to those containing millions of probes. Probes can be designed to distinguish differences in sequence variation that allow for pathogen speciation, or to detect thousands of agents across the tree of life. Arrays comprising longer probes (e.g., > 60 nucleotides) are more tolerant of sequence mismatches and may detect agents that have only modest similarity to those already known. Two longer probe array platforms are in common use for viral detection and discovery: the
_______________________
8 These are large-scale methods to purify, identify, and characterize DNA, RNA, proteins, and other molecules. These methods are usually automated, allowing rapid analysis of very large numbers of samples. http://www.learner.org/courses/biology/glossary/through_put.html (accessed November 13, 2012).
9 As more advanced technologies are introduced, these technologies are sometimes referred to as “second generation” technologies. Nearly all current sequencing is “next generation” (i.e., not Sanger methodology).
GreeneChip and the Virochip. Although they differ in design, both employ random amplification strategies to allow a relatively unbiased detection of microbial targets.
Shotgun Metagenomics
In 1995, The Institute for Genomic Research (TIGR) used a “shotgun sequencing” strategy coupled with Sanger sequencing and advanced bioinformatics methods to produce the first whole genome sequence of a free-living organism, Haemophilus influenzae10 (Fleischmann et al., 1995). Shotgun sequencing refers to the fragmentation of an organism’s genome into small pieces that can then be sequenced in parallel using automated sequencing platforms. It is now used routinely for producing whole genome sequences. Individual sequence fragments are then additively assembled into larger units (known as “contigs”) of the genome. The resulting “draft” typically represents more than 99 percent of the genome (Pallen and Wren, 2007). Draft sequence data may be sufficient for surveying species and metabolic diversity in communities of microorganisms that cannot be grown in culture, or for comparative studies if a complete sequence is available for a closely related strain or species and can be used to order and orient contigs (Fraser et al., 2002).
Finishing a genome-sequencing project is a costly and time-consuming process in which gaps in the assembly are closed and sequence errors are resolved. For this reason, many sequences are left in draft form (MacLean et al., 2009). Finished sequences provide complete genomic information, including the overall organization of a genome and the presence of particular genes on plasmids versus chromosomes (Fraser et al., 2002).
Improvements in sequencing methods and the development of automated systems have contributed to significant decreases in the cost and time it takes to produce a completed genome. The genome of Haemophilus influenzae Rd required 13 months of work. Today, draft bacterial genomes can be sequenced in days. In addition, the cost of sequencing the human genome has dropped by three orders of magnitude, from about $1 million per genome to about $1,000 (JASON, 2010; Figure WO-4). Over the past several decades these advances have led to a proliferation of genome sequencing projects of bacteria, eukaryotes, and of entire microbial communities (metagenomes) that have resulted in a number of completed genomes for a variety of microorganisms (Figure WO-5).
_______________________
10 The Haemophilus influenzae genome was selected for its genome size (1.8 million base pairs), which was typical for bacteria, its G + C base composition (38 percent) was close to that of the human genome, and the fact that a physical clone map did not exist.
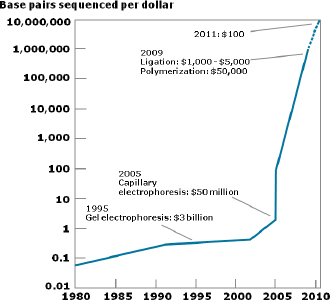
FIGURE WO-4 The improvements in DNA sequencing efficiency over time. Costs excludes equipment and personnel.
SOURCE: JASON (2010).
Viral Diversity Discovery
Studies of viral diversity and genomics have only recently come into their own. Because there is no single gene that is common to all viral genomes, “total uncultured viral diversity cannot be monitored using approaches analogous to ribosomal DNA profiling” (Edwards and Rohwer, 2005). The introduction of high-throughput sequencing and metagenomic analyses are now providing insights into the composition and diversity of cultured viral species and environmental viral communities. These analyses are still limited by current capacity to match sample sequences to sequences stored in databases, but the initial efforts have demonstrated that we have only begun to scratch the surface of virus discovery (Lipkin, 2010; Figure WO-6).
MICROBIOLOGY IN THE POST-GENOMIC ERA
As of mid-2011, complete genome sequences had been published for 1,554 bacterial species (the majority of which are pathogens), 112 archaeal species, and 2,675 virus species. Within these species, sequences exist for tens of thousands of strains; there are approximately 40,000 strains of flu viruses and more than
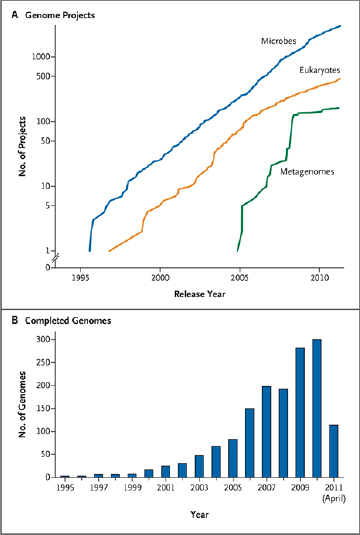
FIGURE WO-5 Genome projects and complete genomes since 1995. Panel A shows a cumulative plot of the number of genome projects (involving microbial [bacterial and archaeal], eukaryotic, and viral genomes) and metagenome projects, according to the release year at the National Center for Biotechnology Information since 1995. Panel B shows the number of completed microbial genome sequences according to year (the most recent data were collected on April 21, 2011).
SOURCE: Relman (2011). From The New England Journal of Medicine, David A. Relman, Microbial Genomics and Infectious Diseases, 365, 347-357. Copyright © 2011 Massachusetts Medical Society. Reprinted with permission from Massachusetts Medical Society.
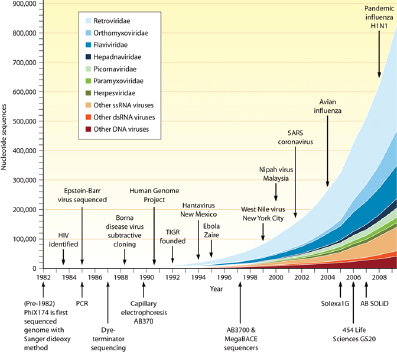
FIGURE WO-6 Growth of the viral sequence database mapped to seminal discoveries and improvements in sequencing technology.
SOURCE: (2010) Copyright © American Society for Microbiology, Lipkin, W. I.(2010). Microbe Hunting. Microbiology and Molecular Biology Reviews 74 (3):363-377:doi:10.1128/MMBR.00007-10. Reproduced with permission from American Society for Microbiology.
300,000 strains of HIV, for example (Relman, 2011). As the fidelity and resolution of nucleic acid sequencing technologies have improved, so has the ability of investigators to explore the diversity and predicted function of microorganisms and the composition and dynamics of the communities they form. These advances offer the hope that we can one day channel some of the activities of microorganisms for improvements to the health and well-being of plants, animals, humans, and ecosystems.
USE OF WHOLE GENOME SEQUENCING
IN OUTBREAK INVESTIGATIONS
Recent examples, discussed below, of the use of whole genome sequencing to investigate outbreaks of emerging, reemerging, and novel infectious diseases
illustrate the potential of these methods for enhancing disease surveillance, detection, and response efforts. Using slight sequence differences between isolates to discriminate between closely related strains, investigators have tracked the evolution of isolates in a disease outbreak; traced person-to-person transmission; and identified point sources of disease outbreaks. When genomic information about related strains or past disease outbreaks is available, the genome sequence of outbreak strains has proved useful in identifying factors that may contribute to the emergence, virulence, or spread of pathogens, as well as in speeding diagnostic tool development. For example:
• Investigators used genomic sequencing to investigate, and find the source for, the cholera outbreak in Haiti in 2010, a disease that had been absent from the island of Haiti for almost a century. Twenty-four Vibrio cholerae isolates from Nepal were found to belong to a single monophyletic group that also contained isolates from Bangladesh and Haiti. These findings (Hendriksen et al., 2011) supported the epidemiological conclusion that cholera was introduced into Haiti by soldiers from Nepal, who served as United Nations’ peacekeepers in the aftermath of the 2010 earthquake (Chin et al., 2011; Frerichs et al., 2012; Piarroux et al., 2011).
• The Black Death, which swept through Europe in the 14th century, was one of the most devastating pandemics in human history. In order to investigate the origins of this pandemic, investigators compared the genomes of today’s bubonic plague bacteria (Y. pestis), obtained from plague-endemic countries, to “plague” obtained from victims who were buried in mass graves in the 14th century. These investigations were able to confirm that Y. pestis was the cause of the Black Death and that it originated from China, more than 1,000 years ago (Bos et al., 2011; Haensch et al., 2010; Morelli et al., 2010).
• Some strains of MRSA are resistant to almost all commonly available antibiotics. Through sequencing and comparing the genomes of MRSA, researchers have been able to trace the origins of this “superbug” to Europe in the 1960s, tracked its global spread, and established a previously unknown link among five patients from a single hospital in Thailand (Harris et al., 2010).
• The 2011 European outbreak of E. coli O104:H4 (discussed by Pallen on pages 86-87) was the deadliest outbreak of food poisoning on record. Thousands were sickened and more than 50 died, many due to a deadly complication of this food-borne infection that can lead to hemolytic uremic syndrome. Comparison of the genomic sequences of the outbreak strain and 11 related strains of E. coli revealed the presence of an unusual combination of virulence factors, which may help to account for the high frequency of hemolytic uremic syndrome associated with this outbreak (Scheutz et al., 2011).
• A 2011 outbreak of a highly drug-resistant strain of Klebsiella pneumoniae proved extremely difficult to treat. By comparing the genome of the outbreak strain to the genomes of 300 previously isolated strains of K. pneumoniae, researchers were able to identify a stretch of DNA that was unique to the outbreak strain. These sequences were then used to develop a rapid diagnostic test for screening patients for this dangerous pathogen (Kumarasamy et al., 2010).
Microbes and Human History
The workshop opened with keynote remarks by Paul Keim of Northern Arizona University, who observed that we are moving toward studying microbial diversity on unprecedented scales, using novel methods that we have never had before (Dr. Keim’s contribution to the workshop summary report may be found in Appendix A, pages 207-229). According to Keim, our understanding of microbial diversity has been severely biased because of our inability to culture the vast majority of microorganisms. This means that what we know about microorganisms, and microbiology generally, comes from a very, very, small subset of the microbial universe. Moreover, we have a very anthropocentric view of the microbial world and tend to focus on those microorganisms that cause illness or death in people. Non-human disease reservoirs are very important in disease ecology, but they are often difficult to identify and study because of their sometimes cryptic and transient nature within their “host” environments—making sampling extremely difficult. It is hoped that the use of whole genome sequencing will expand our understanding of the evolution and population structure of all microbes, including pathogens.
Keim discussed Yersinia pestis, the causative agent of plague,11 as an example of how the emergence of a highly fit microbial clone can alter human history, and how population genetics can help us understand this disease. The dogma has been that there were three major pandemics of plague (reviewed in Perry and Fetherston, 1997, and illustrated in the plague map in Figure WO-7, Morelli et al., 2010).
The first—the “Plague of Justinian”—spread across the eastern Mediterranean and parts of the Middle East and Central Asia from AD 547 to 767, decimating the Byzantine Empire with population losses estimated to be 50 to 60 percent. The second pandemic, referred to as “the Black Death,” began in the Middle Ages and persisted into the 19th century, spanning North Africa, Europe, and parts of Asia. Keim noted that an estimated 17 to 28 million people, or 30 to 40 percent of the European population, died as a result of successive waves of this pandemic. The third pandemic began in the late 1850s and continues to this day. Starting in China and initially spread by steamships, this pandemic has been
_______________________
11 The Black Death.
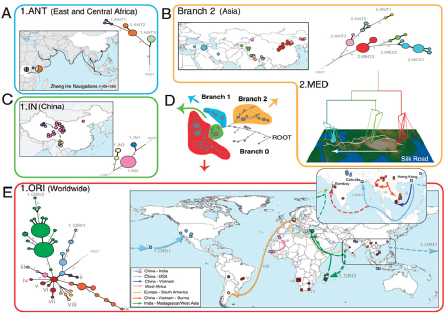
FIGURE WO-7 Postulated routes of spread. Panel A: Sources of 1.ANT isolates in Central Africa and routes of navigation (black lines) by Zheng He from China to Africa in 1409–1433. Panel B: Upper left—sources of isolates from branch 2 superimposed on a map of Asia. Lower right—sources of 2.MED isolates superimposed on a topographical map of the Silk Road (white lines; 200 BC–AD 1400). Panel C: Direction of spread of 1.IN nodes within China from northwest to south. Panel D: A condensed MSTree. Panel E: Postulated routes of migration of 1.ORI since 1894. ii–ix—Radiations with few isolates.
SOURCE: Morelli et al. (2010). Reprinted by permission from Macmillan Publishers Ltd: NATURE GENETICS. Morelli, G., Y. et al. 2010. Yersinia pestis genome sequencing identifies patterns of global phylogenetic diversity. Nature Genetics 42(12):1140-1143, copyright 2010.
responsible for millions of deaths worldwide. Modern hygiene (e.g., rat control) and antibiotics have largely controlled—but have not eradicated—this pandemic.
Plague Ecology
Basic plague ecology involves a bacterial pathogen, Y. pestis, that moves back and forth between a warm-blooded host (almost always rodents) via an arthropod flea vector. On a larger scale, Keim explained that plague ecology involves different hosts and different vectors at different times (Figure WO-8).
Y. pestis continues to evolve out of sight, for decades or even centuries, in a “reservoir” or “cryptic” phase called the enzootic cycle. Sampling during an epizootic cycle or during human pandemics provides evidence for the changes occurring in the reservoir phase. Outbreaks of plague in other “indicator” species (generally rodents) occur during epizootic cycles. Other species, including humans, are also part of the complex ecology of plague. Phenotypic manifestation in humans can be bubonic, septicemic, or pneumonic. Pneumonic plague is highly contagious via respiratory aerosols. Study of the enzootic cycle is extremely difficult; however, sampling during an epizootic cycle or during human pandemics provides evidence for the changes occurring in the reservoir phase.
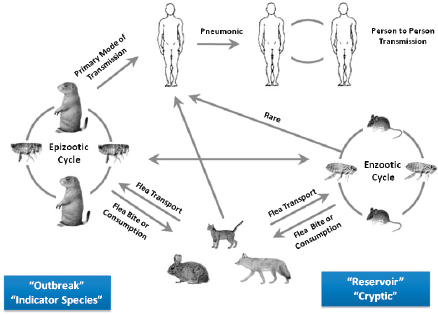
FIGURE WO-8 Plague ecology.
SOURCE: Gage and Kosoy (2005).
A 1951 publication by Devignat first linked the three historical pandemics with three different metabolic phenotypes, or biovars of Y. pestis (antigua, mediaevalis, and orientalis) defined by their ability to ferment glycerol and reduce nitrate (Devignat, 1951). These phenotypes are the result of successive losses of function and, as discussed below, there is no real concordance with phylogenetic12 information that is now available.
Y. pestis is a relatively young, recently emerged, organism. Single nucleotide polymorphisms (SNPs)13 in the core genome are better than 99.9 percent conserved, consistent with clonal propagation. Keim explained that Y. pestis generates diversity by accumulating mutations in a sequential fashion over time. One can select for these mutations in order to assemble a phylogenetic reconstruction of the organism’s history.
Keim cited the collaborative work of Achtman et al. (2004) and Morelli et al. (2010), who used whole genome sequencing and SNP typing to develop a phylogeny for Y. pestis. Their analysis demonstrated that Y. pestis emerged from Y. pseudotuberculosis and acquired new genes in order to become a highly fit clone (Achtman et al., 2004; Morelli et al., 2010). Instead of the antigua, mediaevalis, and orientalis biovar structure, they offer a new type of structure that provides a detailed, high-resolution population genetics map of Y. pestis based on an analysis of 933 SNPs from 282 carefully selected isolates representing the diversity of Y. pestis across the globe (Figure WO-9). Their conclusion is that Y. pestis originated in China and has reemerged from the region in a series of pandemics—more than just three.
The cause of the second plague pandemic in the Middle Ages remains controversial, with some speculating that the cause was not Y. pestis but some other organism(s). Keim cited the work of Bramanti and collegues (Haensch et al., 2010) who studied ancient DNA samples taken from victims of the Black Death buried in mass graves in sites across Europe. They concluded that distinct clones of Y. pestis were in fact associated with the Black Death, and that there were multiple, distinct, waves of Y. pestis coming out of China during the Middle Ages.
A more recent study by Bos and colleagues (2011) reconstructed the ancient genome of Y. pestis from DNA samples obtained from plague victims buried in mass graves that were known to be used from 1348 through 1350 in London. Their findings were similar to the earlier work of Bramanti (Haensch et al., 2010) and consistent with the idea that the Black Death during the Middle Ages was a series of epidemics. Keim noted that only a very small number of SNPs differ
_______________________
12 The study of evolutionary relationships among groups of organisms (e.g., species, populations), which is discovered through molecular sequencing data and morphological data matrices.
13 SNPs are DNA sequence variations that occur when a single nucleotide (A, T, C, or G) in the genome sequence is altered.
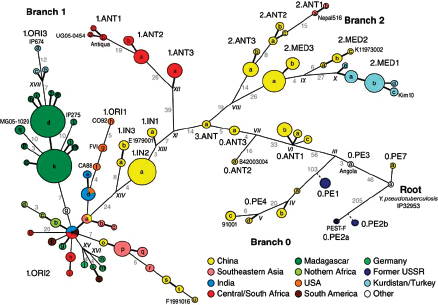
FIGURE WO-9 Fully parsimonious minimal spanning tree of 933 SNPs for 282 isolates of Y. pestis colored by location.
NOTE: Large, bold text: Branches 1, 2 and 0. Smaller letters: populations(e.g., 1.ORI3). Lowercase letters: nodes(e.g., 1.ORI3.a). Strain designations near terminal nodes: genomic sequences. Roman numbers: hypothetical nodes. Grey text on lines between nodes: numbers of SNPs, except that one or two SNPs are indicated by thick and thin black lines, respectively. Six additional isolates in 0.PE1 and 0.PE2b (blue dashes) were tested only for selected, informative SNPs.
SOURCE: Morelli et al. (2010). Reprinted by permission from Macmillan Publishers Ltd: NATURE GENETICS. Morelli, G., Y. et al. 2010. Yersinia pestis genome sequencing identifies patterns of global phylogenetic diversity. Nature Genetics 42(12):1140-1143, copyright 2010.
between the whole genome sequence from the 14th-century plague and what was observed by Morelli.
The third pandemic, which is ongoing, arrived in North America by first appearing in Hawaii in 1899, and later through mainland port cities. From localized outbreaks of rat-borne plague beginning in the port cities of the West Coast in the early 1900s, Y. pestis then spread to native ground squirrels and became ecologically established across the American West and migrated east through the mid-1940s (Link, 1955; Pollizter, 1951). Capitalizing on whole genome sequences and SNPs from U.S. isolates of Y. pestis, Keim concluded that plague in the United States is likely the result of a single introduction from nonnative rodents (i.e., rats on ships) to native rodents.
The population structure of Y. pestis in the United States suggests introduction through a genetic bottleneck, followed by radiation of different lineages across the landscape in a strictly clonal fashion. There do not appear to be adaptive benefits for any given lineage, and there was no sequential wave across the landscape. Rather, the transmission pattern across North America is complex and suggests that some places in North America were colonized more than once, that diverse populations coexisted in the same geographical location, and that dispersal was initially west to east but with some east to west reintroductions.
Studying Y. pestis Evolution in Real Time
Keim and colleagues investigated a plague outbreak in colonial ground squirrels (prairie dogs) that occurred over the course of several months around Flagstaff, Arizona, in 2001, and developed a mutation-rate-based model for assessing plague transmission patterns in real time in order to better understand how plague spread so quickly in the United States (Girard et al., 2004). By collecting fleas from the prairie dog holes in plague-infested areas, Keim’s group was able to directly genotype Y. pestis from DNA extracted from flea vectors without the need for culturing the organism. Studying variable number tandem repeats (VNTRs),14 Keim’s research team developed a phylogenetic tree for Y. pestis in Arizona that suggests that plague entered the state in the late 1930s or early 1940s, swept across the landscape from west to east, and became established in rodent reservoirs where it continues to coevolve with its vector and mammalian host(s) to the present day. In certain years, plague emerges, causing epidemics in prairie dog colonies, resulting in rapid geographic dispersal of Y. pestis. Interestingly, phylogenetically distinct types of Y. pestis were observed in different geographically clustered reservoirs in the Flagstaff area, resulting in a star phylogeny (many short branches off of a single node, rather than a dichotomous tree).
_______________________
14 VNTRs are short nucleotide sequences that are present in multiple copies at a particular locus in the genome. The number of repeats can vary from individual to individual, making analysis of VNTRs useful for subtyping of microorganisms.
Plague is also endemic in the highlands of Madagascar, resulting in at least 100 human cases of plague each year. Plague first arrived in Madagascar in the late 1800s, and outbreaks in rats and humans occurred in the port city of Mahajanga in the early 1900s. Plague moved to the highlands and became ecologically established around 1926, and it did not recur in Mahajanga for more than 60 years, reappearing in 1991. Genetic analysis suggests that plague was reseeded in Mahajanga from one of the endemic foci in the highlands. Analysis of hypervariable sequences, whole genome sequences, and SNPs from about 40 samples from Mahajanga revealed an unusual, linear phylogeny (in contrast to the star radiation observed in Arizona), suggesting multiple introductions (Vogler et al., 2011).
Although the SNPs in the ancient Y. pestis DNA from the Black Death and in DNA from the current plague are different, Y. pestis remains very effective at killing. As such, Keim suggested that pandemicity has less to do with the organism’s pathogenicity, and more to do with the ecological situation it found itself in. Clonal propagation can reseed a reservoir, and it can also lead to a massive increase in the number of organisms, dispersal, and an increase in fitness. As a clonal pathogen, Y. pestis is not taking in genetic material, but Keim suggested that perhaps clonal organisms are contributing to the diversity of the ecosystem. Keim also observed that the saprophytic soil organism, Burkholderia pseudomallei15 has a set of genes that encode for fimbriae that appear to have been horizontally transferred from a Yersinia-like organism.
Microbial Forensics
Forensic evidence is a continuum, and the quality of information, potential errors, and uncertainty influence the power of and confidence in the analysis, interpretation, and inferences made. Microbial forensics is not a new discipline; epidemiologists have been practitioners of the science and art of forensic microbiology since at least the 19th century, identifying the agent, exposed population, the source of exposure, and the extent of contamination, with the goal of disease identification, containment, and treatment of ill populations. The development of genomic sequencing technologies and platforms was stimulated in part by the law enforcement communities to apply these tools and approaches for use in forensic analysis. Speaker Bruce Budowle of the University of North Texas Health Science Center defined microbial forensics as the analysis of evidence from an act of bioterrorism, biocrime, or inadvertent microorganism/toxin release for attribution purposes (Dr. Budowle’s contribution to the workshop summary report may be found in Appendix A, pages 117-133). A microbial forensic investigation, according to Budowle, is more about attribution, determining the agent, source,
_______________________
15 An organism on the U.S. Department of Health and Human Services select agent list. For a complete list of select agents as of 2012, see www.selectagents.gov (accessed November 1, 2012).
and perpetrator, and interpreting and presenting evidence to investigators, the courts and policy makers. In addition, evidence can be used to eliminate certain sources. Traditional trace evidence including DNA, hairs, fibers, and fingerprints may also be involved.
Any infectious agent may be deployed offensively as a biological weapon against a suitable living or nonliving target. According to Budowle, there are more than 1,000 agents—bacteria, viruses, fungi, and protozoa—that are known to infect humans, plants, and animals, along with emerging pathogens and potentially bioengineered organisms. A forensic investigation seeks to gather as much information as possible about the threat agent and compare it to known samples to characterize the organism and/or its processing (e.g., engineering, production, geolocation, date) and delivery. Microbial genomics, including phylogenetics, can help to narrow the focus of the investigation.
Challenges
Source attribution for a “biocrime” requires more circumspection than predicting a source for research purposes. Budowle raised some concerns about interpretation, which hinges on the sensitivity and reliability of the analysis. Missing data are also a concern, and inferences must be made in situations where there is vast uncertainty. Budowle, and others throughout the workshop, emphasized the need for appropriate databases. A particular challenge for forensics is that when a case is under scrutiny in court, it may only be possible to say that isolates are closely related, and experts for the opposition may challenge that the references or databases are insufficient or inappropriate.
With regard to technology limitations, Budowle noted that not all microbial forensic evidence is suitable for genetic analysis using next-generation sequencing. Some samples will be limited in quantity, highly degraded, and/or contaminated. A challenge is to extract as much genetic information as possible from limited materials and nonviable organisms. In forensics, sequencing errors will inflate differences between samples creating a degree of uncertainly. As such, defining and quantifying the error rates associated with the sequencing platform and chemistry are critically important. Again, the quality of sequence data and the results of bioinformatics analyses must be as high as possible.
Budowle also emphasized the need for standard reference and test materials. Today’s databases will be the test panels and reference samples of tomorrow, but some data are woefully inadequate. As sequencing capability moves into application-oriented laboratories, we need to consider quality control, and perhaps something along the lines of proficiency testing, to ensure high-quality data in databases. Best practices need to be established regarding what qualifies as a reference sample, as well as standards for preparation, validation, and characterization (including metadata). It is not clear where the responsibility lies for generating such standard reference materials, because there are numerous stakeholders (Table WO-2).
TABLE WO-2 Interests in Reference Collections and Management
|
Agency or Organization |
Role in National Biodefense System |
Interests Related to Pathogen Collections |
|
DHS |
R&D in detection and microbial forensics; central laboratory role in microbial forensics investigations |
Research access to isolates for assay development, validation, and other R&D; Archives for comparisons to forensic samples |
|
FBI (DOJ) |
Investigation and prosecution of crimes using biological agents |
Quickly identifying possible sources for a pathogen used in an attack; development of standards and controls |
|
CDC (HHS) |
Epidemiological investigation of disease outbreaks; administrator of the Select Agent Program |
Tracking endemic strains and identifying sources of outbreak strains |
|
NIAID (HHS-NIH) |
Understanding disease mechanisms and host-pathogen interaction; development of treatments and diagnostic assays |
Research access to isolates for studies of pathogenicity mechanisms, host-pathogen response, and other R&D related to medical countermeasures |
|
FDA (HHS) |
(With CDC) investigation of food-borne disease outbreaks |
Tracking endemic strains and identifying outbreak strains and their sources |
|
USDA |
Plant and animal pathogens |
Disease tracking, identifying outbreak strains and their sources, developing treatments and vaccines |
|
AFMIC, DTRA, Services (DOD) |
Military force protection |
Tracking foreign disease that may impact military operations; medical countermeasures |
|
ATCC BEI Resources SDO |
Biomaterials resource |
Development of biothreat and EID standard biomaterials that meet ISO and ANSI guidelines |
NOTE: DHS, Department of Homeland Security; FBI, Federal Bureau of Investigation; DOJ, Department of Justice; CDC, Centers for Disease Control and Prevention; HHS, Department of Health and Human Services; FDA, Food and Drug Administration; USDA, U.S. Department of Agriculture; AFMIC, Armed Forces Medical Intelligence Center; DTRA, Defense Threat Reduction Agency; DOD, Department of Defense; ATCC, American Type Culture Collection; SDO, Standards Development Organization; EID, emerging infectious disease; ISO, International Organization for Standardization; ANSI, American National Standards Institute.
SOURCE: Budowle (2012).
Forensics Case Example: The Amerithrax Investigation
Claire Fraser of the Institute for Genome Sciences provided an overview of the genomic approaches used in the Amerithrax investigation.16 Fraser reminded
_______________________
16 Amerithrax is the case name assigned by the Federal Bureau of Investigation (FBI) to the 2001 anthrax letter attacks.
participants that in 2001 there was only one sequencing platform available (the Sanger 3730), the cost for sequencing was $200,000 to $300,000 per genome, and it took nearly a year to completely sequence a genome. While the technology has changed dramatically over the past decade, our understanding of the dynamics of microbial genomes is still quite limited.
In collaboration with the overall FBI Amerithrax investigation, the goal of the scientific investigation, according to Fraser, was to explore whether a genomics-based approach could be used to attribute the spore preparation of a genetically homogeneous species (used in the letters) to a potential source (Keim et al., 1997; Rasko et al., 2011; Read et al., 2002). Could genetically unique features be identified using traditional DNA sequencing-based analysis that would be useful for the purposes of attribution? Fraser reiterated the point made by Budowle that attribution falls on a continuum; it may not be an exact match, and exclusion may also be important.
The starting material was the Bacillus anthracis spore preparations obtained from the letters that were mailed to the office of Senator Leahy and to the New York Post, supplemented by related material obtained from other sources including the Hart Senate Office Building, postal workers from the Brentwood (Washington, DC) post office facility, and people exposed to anthrax spores in the Hart Senate Office Building. Gross examination of the physical characteristics of the samples suggested there were at least two different preparations. It became quickly apparent that traditional genotyping methods were not achieving sufficient resolving power for the purposes of this investigation. Based on VNTR analysis by the Keim laboratory, it appeared that all of the isolates initially collected as part of this investigation were the Ames strain of B. anthracis.17
At the same time, TIGR was in the final stages of assembling the first genome sequence of B. anthracis and was asked to partner with the FBI and other laboratories to determine whether the complete genome sequence would be useful for purposes of attribution. With funding from the National Science Foundation, TIGR began sequencing a colony of B. anthracis recovered from the spinal fluid of Mr. Robert Stevens of American Media in Florida, the first victim to die as a result of exposure to anthrax spores mailed to him (referred to as B. anthracis Florida). Because no appropriate reference strain was available, TIGR also sequenced the genome of the B. anthracis Ames ancestral strain. (Fraser explained that the strain TIGR had initially sequenced was obtained from a facility in the United Kingdom that had been cured of its two virulence plasmids—pXO1 and pXO2—making it an inappropriate reference strain for comparative purposes.)
According to Fraser, SNP analysis of the reference B. anthracis Ames ancestor and the Florida isolate found no differences in more than 5 million base pairs assessed. Similarly, no polymorphisms were found when the wild-type isolates
_______________________
17 The Ames strain was originally isolated from a dead Beefmaster heifer in Texas in 1981. It quickly became a standard laboratory research strain used worldwide for vaccine challenge studies.
from the letters to the New York Post and Senator Leahy’s office were compared to the reference Ames ancestor sequence (Rasko et al., 2011). While B. anthracis is highly monomorphic, Fraser noted that it was somewhat surprising to find absolutely no sequence differences, and it raised questions about whether genomics would be useful in the investigation after all.
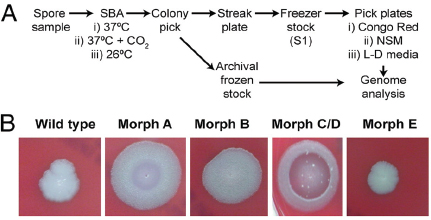
At the same time, researchers at the U.S. Army Medical Research Institute of Infectious Diseases (USAMRIID) began to notice some B. anthracis colonies with distinct, and apparently heritable, morphology as the spore preparations from four anthrax-laced letters were passaged in culture. Examination of the colonies formed on sheep blood agar (SBA) resulted in the identification of four distinct morphological variants (morphotypes)—designated A, B, C/D, and E—from each of the material analyzed (Rasko et al., 2011). These morphotypes are illustrated in Figure WO-10.
According to Fraser, these phenotypic variants were all found to be altered with regard to their ability to sporulate under different conditions, potentially linking the events in New York and Washington. This new information inspired a population genomics approach to this investigation. Could the population composition (rather than the wild-type) be used to make a match?
Morphologic variants from the Leahy and New York Post letters were sequenced and compared to both wild-type and the Ames ancestor strains. Morphological variant A was the most different from the wild-type in terms of sequence, although the sequence variability represented a very small portion of the genome.
Fraser noted that it was initially believed the sequence was identical to the wild-type. However, using paired-end sequencing, it was possible to look at mate pairs that were present in the assembly at distances that would not be expected based on the insert sizes that went into the cloned library. This led to the discovery of a number of chimeric reads in the assembly that ended up being a tandem duplication, and exhaustive PCR analysis was done to establish confidence in the finding. Analysis of three morphotype A mutants present in all the letters showed that while they were identical phenotypically, they were genetically different.
Isolates of morphological variant B have SNPs in a sporulation gene, variants C/D have two sequence variants in a histidine kinase sensor gene (C has a SNP, and D has an indel18), and opaque variants have an indel of either 9 or 21 base pairs in a response regulator gene (i.e., all are mutations along the sporulation pathway). Fraser went on to explain that each of these genetic variants was converted into a quantitative PCR assay and used to screen a repository of nearly 1,100 samples collected by the FBI. All four of these mutations were found in a sample from a single source, a flask at USAMRIID labeled RMR-1029. Other samples that also contained the four mutations could trace their provenance back to RMR-1029. An assortment of other samples were also found to have subsets of these four mutations, but not all of them. RMR-1029 was a heterogeneous mixture cultivated for vaccine trials in the late 1990s and flasks were stored at USAMRIID in Maryland and the Dugway Proving Ground in Utah.
In summary, it was population genomics that provided the unique signature that facilitated attribution in the Amerithrax case. The minor subpopulation was unique to the spore preparations recovered from the letters. Fraser noted that these polymorphisms were used to screen batch cultures, not single B. anthracis colonies on a plate, which was very different from all of the clonal genome projects that had been completed to that point in time.
Jumping forward a decade, what might be different today in the era of metagenomics? Clearly, the process could occur more quickly and at much lower cost. With current technologies investigators would be working with shorter sequence reads and, Fraser added, it is not possible to know if the gene duplication would have been as easy to identify from short reads as it was from the 800-plus-base-pair Sanger reads that were used at the time. What would community-level analysis with very deep coverage provide versus what was done by looking at single colonies? Morphotype A was present in all RMR-1029 samples, but not necessarily at the same low level of abundance. What does it mean in terms of being able to say that something is the same or not? Fraser and Budowle emphasized that a significant gap, that still persists today, is the lack of appropriate, standardized, criteria (thresholds, confidence limits, etc.) that would lead one to conclude that a given microbial sample was or was not derived from the same source, to answer the question of what makes a match with confidence.
_______________________
18 Indel refers to an insertion or deletion mutation.
Fraser noted that the experience with B. anthracis is in no way generalizable to other pathogens or potential agents of bioterrorism. Had it been an organism with horizontal gene transfer and genome rearrangements over periods of time, it would likely have been in a very different situation. Budowle added that the Amerithrax forensic investigation was somewhat unique because as many samples as could be collected from the letter attacks were collected, and more inferences could be made in this case than might be possible in many other cases.
Microbial Evolution: Studying Genomes, Pangenomes, and Metagenomes
The ability to sequence and compare whole genomes of many related microorganisms has prompted a deeper understanding of the biology and evolution of microorganisms. The completeness of finished microbial genomes is particularly powerful in a comparative context. Differences in genomic content such as the presence or absence of genes or changes in gene order or sequence, from SNPs to large indels,19 may have important phenotypic consequences. The comparison of multiple, related genome sequences offers insights into an organism’s evolutionary history—including the relative importance that natural selection attaches to specific gene functions (Eisen et al., 1997; Fraser-Ligget, 2005).
Comparative analyses have revealed that the microbial genome is a dynamic entity shaped by multiple forces including gene loss/genome reduction, genome rearrangement, expansion of functional capabilities through gene duplication, and acquisition of functional capabilities through lateral or horizontal gene transfer, as shown in Figure WO-11 (Fraser-Liggett, 2005). Three main forces shape bacterial genomes: gene gain, gene loss, and gene change. All three can take place in a single bacterium. Some of the changes that result from the interplay of these forces are shown in the following illustration. Several natural processes carry genetic information from one species to another. DNA can be transported by viruses (transduction), via bacterial mating (conjugation), and through the direct uptake of DNA from the environment (transformation). Genes that must function together are transferred together as genomic islands (e.g., pathogenicity islands) (Hacker and Kaper, 2000).
The frequent gain and loss of genomic information exhibited by many bacterial species makes it difficult to trace bacterial phylogenies and has strained the species concept (Bentley, 2009). Among genetically variable bacterial species, it is clear that a single strain rarely typifies an entire species. Instead, researchers sequence multiple strains of a species to compile the “pan-genome” or global gene repertoire of a bacterial species (Medini et al., 2005) (Figure WO-12). The pan-genome can be divided into three elements: the core genome (housekeeping genes shared by all strains); a set of strain-specific genes that are unique to various isolates; and a set of dispensable genes that are shared by some but not
_______________________
19 Insertions and deletions.
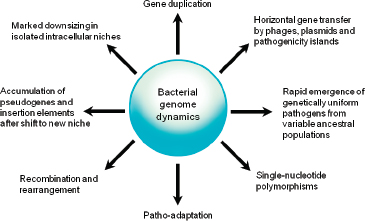
FIGURE WO-11 Bacterial genome dynamics.
SOURCE: Pallen and Wren (2007). Reprinted by permission from Macmillan Publishers Ltd: NATURE. Pallen, M. J., and B. W. Wren. 2007. Bacterial pathogenomics. Nature 449:835-842, copyright 2007.
all isolates. These latter, dispensable sequences are associated with high rates of nucleotide sequence variability and contribute to phenotypic diversity within bacterial populations (Medini et al., 2008).
The comparison of eight genomes from strains of group B Streptococcus (GBS) found an average of 1,806 genes in the core genome and 439 dispensable or strain-specific genes. Based on these data, models predict that the GBS pan-genome is “open,” with unique genes continuing to emerge even after hundreds or thousands of genomes are sequenced. Indeed, many bacterial species have extensive genetic diversity, with an average of 20 to 35 percent of genes being unique for a single strain. In contrast, as discussed by Fraser, other organisms appear to be monomorphic—with a “closed” pan-genome. In the case of B. anthracis four genome sequences completely characterize the species (Medini et al., 2008).
A species’ pan-genome likely reflects selective pressure to adapt to specific environmental conditions. Species with an open pan-genome typically “colonize multiple environments and have many ways of exchanging genetic material.” By contrast, monomorphic species with a closed pan-genome “live in isolated niches and have a low capacity to acquire foreign genes” (Medini et al., 2008). In natural settings, bacteria and other microorganisms interact with each other and with their surroundings to form complex communities that occupy diverse environmental niches. The shuttling of genes between species via horizontal gene transfer (HGT) plays an important role in a species’ ability to adapt to environmental change. As discussed in the section that follows, ecological factors may strongly influence the acquisition and loss of genes via HGT (Smillie et al., 2011). The pan-genome concept suggests the presence of a large microbial gene pool in the environment
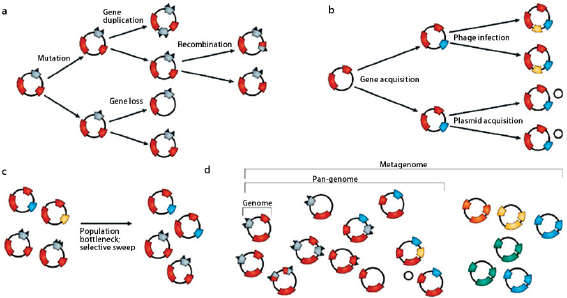
FIGURE WO-12 Molecular evolutionary mechanisms that shape bacterial species diversity: one genome, pan-genome, and metagenome. Intraspecies (a), inter-species(b), and population dynamic(c) mechanisms manipulate the genomic diversity of bacterial species. For this reason, one genome sequence is inadequate for describing the complexity of species genera and their inter-relationships. Multiple genome sequences are needed to describe the pan-genome, which represents, with the best approximation, the genetic information of a bacterial species. Metagenomics embraces the community as the unit of study and, in a specific environmental niche, defines the metagenome of the whole microbial population (d).
SOURCE: Medini et al. (2008). Reprinted by permission from Macmillan Publishers Ltd: NATURE REVIEWS MICROBIOLOGY. Medini, D. et al. 2008. Microbiology in the post-genomic era. Nature Reviews Microbiology 6: 419-430, copyright 2008.
that drives microbial evolution, with HGT providing microorganisms with rapid access to genetic innovation. HGT can enable beneficial traits (such as resistance to antimicrobial drugs or other environmental perturbations) to spread through entire populations (Medini et al., 2005, 2008).
Pathogenomics
The vast majority of microbes do not produce overt illness in their hosts, but may, instead, establish themselves as persistent colonists that can be described as either low-impact parasites (e.g., causes of asymptomatic infection), commensals (i.e., organisms that “eat from the same table,” deriving benefit without harming their hosts), or symbionts (establishing a mutually beneficial relationship with the host) (Blaser, 1997; Merrell and Falkow, 2004). These states, while separate, represent part of a continuum extending to pathogenesis and disease that may be occupied at any point by a specific microbial species through the influence of environmental and genetic factors (Casadevall and Pirofski, 2000, 2002, 2003). Persistent colonization of a host by a microbe is rarely a random event; such coexistence depends upon a relationship between host and microbe that can be characterized as a stable equilibrium (Blaser, 1997).
Over the course of the past century, the identification of increasing numbers of microbial pathogens and the characterization of the diseases they cause have begun to reveal the extraordinary complexity and individuality of host–microbe relationships. In the case of microbes that cause persistent, asymptomatic, infection, physiological, or genetic changes in either host or microbe may disrupt this equilibrium and shift the relationship toward pathogenesis, resulting in illness and possibly death for the host (Merrell and Falkow, 2004). As a result, it has become exceedingly difficult to identify what makes a microbe a pathogen.
Virulence as an Emergent Property
The question of “why some microbes cause disease and others do not” has puzzled microbiologists for centuries. Genomics is a new and useful tool for exploring this question, but it has its limitations, according to Arturo Casadevall of the Albert Einstein College of Medicine (Dr. Casadevall’s contribution to the workshop summary report may be found in Appendix A, pages 134-140). As illustrated by several examples discussed by Casadevall in his prepared remarks, the complexity of host–microbe interactions complicates researchers’ ability to link genotypic information with phenotypic expression of this genomic information. This complexity has important implications for the development of predictive tools to identify microbial threats.
Research associating certain microorganisms with activities that could be beneficial or harmful to human health has lead scientists to a central question: are pathogenic microbes inherently different from nonpathogens? Most
microbiologists in the early 20th century reasoned that pathogenic—disease-causing—microbes must differ from nonpathogenic microorganisms in the expression of traits associated with virulence. Others, including the Belgian immunologist Jules Bordet,20 argued that there could be no difference. He based his argument on two observations of the context dependence of virulence:
1. The same organism can exist in both virulent and nonvirulent states. For example, isolates of Neisseria meningitidis associated with a meningococcal epidemic lose virulence when maintained in laboratory culture and regain virulence after passage21 through a mouse that resulted in the selection for characteristics that allowed survival in the mammalian hosts and thus reenabled virulence.
2. In an infected but immunized host, a pathogenic organism exists in a nonpathogenic state.
Indeed, during the early 20th century, many common infectious diseases disappeared as a result of immunization, and microorganisms that were not previously considered pathogenic were increasingly associated with disease later in the century. The microbes did not change, noted Casadevall, “what happened was that we changed the host.”
Casadevall went on to emphasize that the concept of a “pathogen” is flawed, because it assumes that pathogenicity is an intrinsic, immutable characteristic of a microorganism. Neither pathogenicity nor virulence is an independent microbial property; according to Casadevall both are characteristics that are expressed only in a susceptible host. Labeling a microbe a “pathogen” endows it with properties that are not its own. In Casadevall’s view, there are only “microbes” and “hosts”—what is truly important is the outcome of interactions between the microbe and its host environment(s).
Casadevall and his colleague Liise-anne Pirofski developed the “damage-response framework” to provide an integrated theory that accounts for the contribution of both the host and the pathogen to pathogenesis (Casadevall and Pirofski, 2003). Within this framework, a pathogen is defined as a microorganism that is capable of causing disease, and pathogenicity is the capacity of a microbe to cause damage in a host. The damage-response framework defines a virulence factor as a microbial component that damages the host and virulence as the relative capacity of a microbe to cause damage in a host (Casadevall and Pirofski, 1999). Damage is thus an expression of microbe–host interactions, which for most host–pathogen interactions, can be graphed as a parabola, as illustrated in
_______________________
20 Jules Bordet was awarded the Nobel Prize in physiology or medicine in 1919 for his discoveries relating to immunity.
21 In microbiology, “passage” refers to the successive transfer of cultures of microorganisms across various nutrient mediums or the reinoculation of one animal with pathogenic microbes from another infected animal.
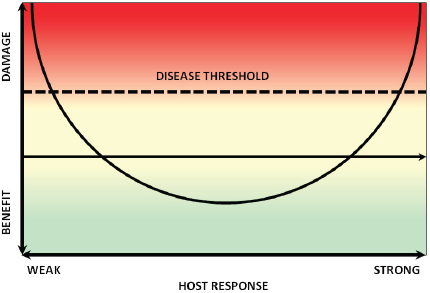
FIGURE WO-13 Damage-response framework and the case of S. cerevisiae.
SOURCE: Casadevall (2012).
Figure WO-13 (Casadevall and Pirofski, 2001). As an individual becomes immunosuppressed, damage can occur, and once a certain threshold is reached, disease may occur. The same organism might also elicit an untoward immune response resulting in disease (Casadevall and Pirofski, 2003).
For example, Saccharomyces cerevisiae, commonly used in baking and brewing, can cause disease in immunocompromised patients with HIV; vaginitis in normal women that is indistinguishable from candidiasis; and lung nodules in bakers as a result of hypersensitivity responses in the lung. S. cerevisiae cannot be defined as food, commensal organism, opportunistic pathogen, or primary pathogen without taking into account the host. As noted by Casadevall, a reductionistic approach—whether the microbe-centric view of many microbial geneticists, who focus on virulence factors, or the host-centric view of many immunologists, who focus on factors affecting host susceptibility—provides an incomplete picture of this continuum of outcomes.
Host- vs. environment-acquired microbes The diversity of possible outcomes associated with many host–microbe interactions is also evident when one considers virulence factors associated with microbes acquired from another host or directly from the environment. Organisms acquired from another host include all viruses, many parasites, most bacteria, and a few fungi. These are generally communicable diseases with a limited host range. These organisms are not free
living, and there is likely to be selective pressure on the microbe to coexist with the host. Disease often results from the disruption of the host–microbe relationship (Casadevall and Pirofski, 2007).
Environmentally acquired microbes include bacteria, fungi, and some parasites. They are not communicable, have a very broad host range, and are free living. The selective pressures in the environmentally acquired microorganisms for causing disease are unknown. Because they have no host requirement for survival, these are the only organisms that are known to cause extinction.22 Disease often manifests in hosts with impaired immunity, or when there are large microbial inocula (Casadevall and Pirofski, 2007). The fungus Cryptococcus neoformans is an example of a soil-dwelling, environmentally acquired” microorganism that infects a wide range of hosts—including plants, animals, and humans—but only rarely causes disease. Casadevall noted that “everyone in this room is infected [with C. neoformans], but you have a one-in-a-million chance of getting disease unless you become immunosuppressed.” The virulence of C. neoformans is complex, explained Casadevall, because the organism did not evolve to cause disease in these hosts. Instead, this organism was selected for properties that allowed it to survive in the soil, and “by the luck of the draw” it happens to have the traits necessary to cause disease in some hosts. Rather than being a special property of only certain microorganisms, virulence is an emergent property.
The challenges of studying an emergent property Casadevall defined an emergent property as a novel property that unpredictably comes from a combination of two simpler constituents—in essence the “whole is greater than the sum of its parts” (Casadevall et al., 2011). In this case, a host and a microbe are the components, and the novelty may be expressed as either virulence and pathogenicity, mutualism, commensalism, or even the death of either party.
Emergent properties abound in the natural world, and while they can be understood after the fact, emergence is not reducible or predictable. We understand the structure of water, for example, and we can explain surface tension when we see it, but we cannot predict surface tension from individual water molecules. We understand the physics of small particles, but we cannot predict sand dunes. Emergent properties cannot be reduced to either component. In this regard, Casadevall suggested that research focused on either the host or the microbe may produce interesting results, but it may not be relevant to understanding outcomes of host–microbe interactions.
To illustrate the limits of a reductionistic approach to understanding virulence, Casadevall described recent research on C. neoformans. Applying a mathematical model to compute the relative contribution of microbial virulence factors, Casadevall estimated that the majority of cryptococcal virulence in mice
_______________________
22 The environmentally acquired fungi Geomyces destructans and Batrachochytrium dendrobatidis, for example, currently pose a significant threat to populations of New World bats and amphibians worldwide (IOM, 2011).
can be attributed to the C. neoformans capsule and cell wall melanin (McClelland et al., 2006). Casadevall then evaluated the relative virulence of C. neoformans and two closely related cryptococcal species in Galleria mellonella moth larvae. As expected, C. neoformans was pathogenic in this system. However, the cryptococcus species that has a capsule and makes melanin was not pathogenic, and the cryptococcus species that has no capsule and no melanin was pathogenic in the larvae.
Casadevall challenged workshop participants to consider whether virulence is a chaotic system—like weather. If it is, then the outcome of host–microbe interactions, including virulence, may be inherently unpredictable. There are limits to what we can know and predict through reductionism (i.e., through a focus only on one component or the other, the host or the microbe). Still, despite emergence and potentially chaos, progress is still possible. Casadevall noted that the use of computer analysis has improved the accuracy of weather forecasts. Looking ahead, Casadevall suggested that the focus of future research should be on developing probabilistic models for host–pathogen interactions.
Microbial Genomics: Epidemiology and the
Mechanisms of Disease Emergence
Comparative genomics has helped to inform our understanding of host– microbe interactions from that of a “war metaphor” (“the only good bug is a dead bug”) to one that places these interactions into a broader ecological and evolutionary context. These insights have profound implications for detecting, diagnosing, and anticipating infectious disease emergence, including:
• Bacterial genome sequence data have challenged the simplistic views that pathogens can be understood solely by identifying their virulence factors, and that pathogens often evolve from “nonpathogenic” organisms through the acquisition of virulence genes from plasmids, bacteriophages, or pathogenicity islands. Metagenomic surveys conducted in diverse environments have improved our understanding of the biodiversity and biogeography of microbes and have underscored the important role of environmental factors in disease emergence and spread.
• Simply studying a pathogen without understanding its biotic and abiotic environmental contexts may lead to false confidence in our ability to detect it. Microbial detection will be most effective if there is sufficient basic scientific information concerning microbial genetics, evolution, physiology, and ecology.
• Likewise, strain sub-typing will be difficult to interpret if we do not understand some of the basic evolutionary mechanisms and population diversity of pathogens and nonpathogens alike (JASON, 2009).
The following case examples explore some of the different kinds of systems in which genomics has been used to study microorganisms or microbial communities associated with disease. These studies have revealed important insights into the mechanisms of variation and genome change as well as the role of host– microbe–environment interactions on the evolution and adaptation of pathogens and nonpathogens alike.
Comparative Genomics: E. coli Including Shigella
Speaker David Rasko of the University of Maryland School of Medicine discussed how comparative genomics can be used to improve diagnostic methods. According to Rasko whole genome sequence analysis has rapidly advanced researchers’ understanding of pathogenic variants of E. coli—an organism that has been intensely studied for almost 40 years.
Diarrheagenic E. coli (DEC) are food-borne and water-borne pathogens associated with approximately 300,000 deaths annually, primarily in the developing world. Pathogenic variants of DEC (referred to as pathovars or pathotypes) exhibit diverse characteristics and pathogenic mechanisms, only some of which have been well characterized (Figure WO-14).
With well over 100 serotypes, the phylogeny and evolution of DEC is diverse and complex. Even though E. coli has been studied for decades, Rasko noted that current diagnostic and typing methods are inadequate and there are no approved vaccines. Genomic analyses provide new ways to characterize organism diversity and to identify novel virulence factors—information that will enhance methods for outbreak and strain identification and current understanding of pathogen emergence.
The genome structure of E. coli is highly conserved, yet between 20 to 25 percent of the DNA in any strain can be novel (i.e., 1.5 megabases of the total 5 megabase). The widely used technique for categorizing E. coli strains according to sequence similarity—multi-locus sequence typing (MLST)—compares a subset of sequence from each strain’s genome (~3,400 bases). As noted by Rasko, the resulting phylotypes23 are not well resolved, and pathotypes are not restricted to any one phylotype.
Whole genome sequencing allows researchers to compare a much larger proportion of the genome (2.3–2.7 million bases) and provides greater discriminating power than MLST. The greater resolution produced by whole genome sequencing is dependent upon the quality and number of genomes and isolates to which strains can be compared. Whole genome phylogenies are more robust and clearly distinguish each phylotype. Rasko added that there is practically no cost difference between MLST and whole genome phylogeny. Rasko underscored the importance of this “context” by noting that when there was an outbreak of
_______________________
23 Classification of an organism by its evolutionary relationship to other organisms.
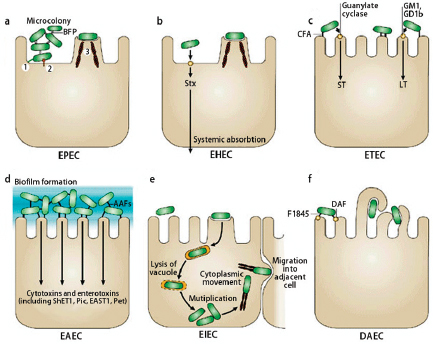
FIGURE WO-14 Six distinct pathogenic variants of diarrheagenic E. coli. Panel a, Enteropathogenic E. coli (EPEC) form microcolonies via the association of bundle-forming pili (BFP) (1), bind to the small intestinal epithelium (2), and cause the reorganization of cytoskeletal actin, pedestal formation, and attaching-and-effacing lesions (3). Panel b, Enterohemorrhagic E. coli (EHEC) infect the large intestine, form attaching-and-effacing lesions, and release Shiga toxin (Stx). Panel c, Enterotoxigenic E. coli (ETEC) release heat-stable toxin (ST) and heat-labile toxin (LT) into host cells. Panel d, Enteroaggregative E. coli (EAEC) form a biofilm on HEp-2 cells in vitro; however, they do not have a common virulence factor that can be used to identify the entire group. Panel e, Enteroinvasive E. coli (EIEC), including Shigella, are the only E. coli that grow intracellularly. Panel f, Diffusely adhering E. coli (DAEC) have one adhesion factor that has been characterized thus far, but other mechanisms of pathogenesis are not yet known.
SOURCE: Reprinted from Kaper et al. (2004). Reprinted by permission from Macmillan Publishers Ltd: NATURE REVIEWS MICROBIOLOGY. Kaper, J. B., J. P. Nataro, and H. L. Mobley. 2004. Pathogenic Escherichia coli. Nature Reviews Microbiology 2(2):123-140, copyright 2004.
a rare strain of E. coli O104:H4—first identified in northern Germany in May 2011—“because we had a good sequence database and collection of isolates we could very quickly and accurately place the [E. coli O104:H4] outbreak as being an enteroaggregative E. coli that had acquired Shiga toxin, [rather than] an enterohemorrhagic E.coli.”
Case example Attaching-and-effacing E. coli include the pathotypes enteropathogenic E. coli (EPEC) and enterohemorrhagic E. coli (EHEC) (see Figure WO-14 on page 38). According to Rasko, “These groups are lumped together because the community has had difficulty over the past 20 to 30 years actually defining each of these pathotypes distinctively.” This diverse group of E. coli has a large number of phage and is variable in the presence of Shiga toxin. They are defined by their common locus of enterocyte effacement (LEE), which codes for the type III secretion system24 and other elements involved in pedestal formation and the development of attaching-and-effacing lesions.
In order to develop more accurate diagnostics for this group of E. coli, Rasko’s lab sequenced and aligned 136 genomes (113 attaching-and-effacing strains and 23 reference isolates spanning all of the pathotypes) to create a whole genome phylogeny. Rasko noted that while virulence factors have been the basis for isolate typing for the past 30 to 40 years, it is now clear that these characteristics do not always match the genome phylogenies (Figure WO-15). Rasko went on to observe that there is a mismatch between phylogeny based on the core genome, and phylotype based on virulence factors when characterizing which strains produce Shiga toxin (indicative of EHEC) and which have a bundle-forming pilus (indicative of EPEC).
In evaluating phylogenetic clusters in comparison to virulence factors, Rasko found that the majority of attaching-and-effacing strains contain the LEE pathogenicity island. Rasko observed that the LEE regions encoding the type III secretion system were highly conserved; the areas coding for the secreted effectors were more variable. Isolates can be loosely grouped based on secreted effectors. Rasko is now investigating possible associations between those differences and virulence or disease severity. Interestingly, there is genome conservation and virulence factor conservation even though the majority of these secreted effectors are not encoded by the LEE region, but rather on other phage in the genome.
Using whole genome alignments to identify novel genome features and possible biomarkers While comparative methods are adequate for the pairwise comparison of a limited number of genomes, identifying novel regions of interest across hundreds of genomes presents a challenge. To help address this analytical
_______________________
24 A protein appendage found in several Gram-negative bacteria. In pathogenic bacteria, the needle-like structure is used as a sensory probe to detect the presence of eukaryotic organisms and secrete proteins that help the bacteria infect them.
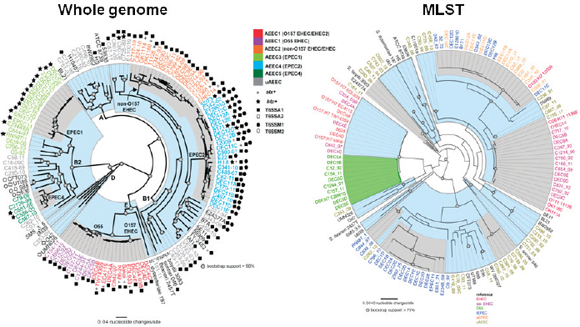
FIGURE WO-15 Inconsistency of typing with whole genomes or MLST.
SOURCE: Rasko(2012).
weakness, Rasko is using what he terms “genomic epidemiology” to look for unique genome signatures that are present in EHEC but not EPEC, or vice versa. This is based on alignment of the genomes, he explained, and is gene independent. Out of the genome features that were identified as being different between the groups, about 10 percent were known virulence factors unique to one of the groups. Functional analysis of the previously unknown and unique features is under way.
Genomic information is also informing the development of group-specific biomarkers. Detection of virulence factors using PCR is commonly used to identify an isolate. Rasko explained that virulence factors are often not the best identification markers. All attaching-and-effacing E. coli isolates will have genes for components of the type III secretion system, while the presence of genes for other virulence factors (Shiga toxin 1 and 2, bundle-forming pilus) is extremely variable. In addition, virulence factors tend to be mobile. Novel genome regions that are unique to a specific group provide a more effective way to rapidly identify the phylotype of an isolate using PCR. In his presentation, Rasko shared an algorithm for the identification of different attaching-and-effacing pathogenic E. coli (Figure WO-16).
The primary obstacle for developing diagnostics, vaccines, and therapeutics for EPEC, according to Rasko, is characterizing the pathogen beyond serotype and virulence factors. While the sequencing of isolates is relatively easy, there is a need for case-control studies with well-defined parameters, and patient and isolate metadata to inform the analysis and interpretation of genomic data. In addition, observed Rasko, our understanding of the population structure of these organisms is woefully inadequate to the task at hand. For example, how many distinct EPEC isolates are within one individual? What is the rate of variation within that host and within the environment? The development of rapid diagnostics requires appropriate comparison to close relatives, Rasko concluded, including both pathogens and commensals (the majority of E. coli in the gut are not pathogens). Rasko concurred with Casadevall that virulence is only expressed in a susceptible host, and he reiterated the importance of looking at population structure. We can develop probabilistic models in terms of whether an organism is likely to cause disease, he said; whether or not it does is entirely contextual.
Signatures of Selection and Transmission: Staphylococcus aureus,
Streptococcus pneumoniae, Vibrio cholerae
The limitations of current typing methods—such as MLST—to discriminate between closely related organisms poses a challenge to tracking the transmission and adaptation of very recently evolved strains of pathogenic bacteria. Julian Parkhill of the Sanger Institute in Cambridge (UK) reviewed several examples of how high-throughput genomics can provide the higher-resolution view required in order to better understand the epidemiology and microevolution of strains that
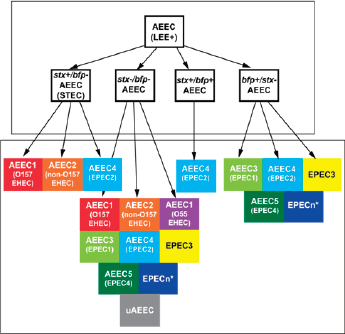
FIGURE WO-16 Algorithm for the identification of attaching-and-effacing E. coli (AEEC) pathogens. Classically, LEE-positive isolates are assayed for the presence of other virulence factors and subgrouped accordingly (top box). The introduction of phylogenetic markers allows for more rapid and accurate subgrouping (bottom box).
NOTE: stx, Shiga toxin gene; bfp, bundle-forming pilus structural gene; STEC, Shiga toxin-producing E. coli.
SOURCE: Rasko (2012).
have evolved and spread within the past 30 to 40 years (Dr. Parkhill’s contribution to the workshop summary report may be found in Appendix A, pages 257-269).
Parkhill noted the multiple evolutionary processes that have an effect on bacterial genomes—including random mutation, homologous DNA exchange, acquisition and loss of genes, genetic drift, and Darwinian selection. Parkhill emphasized that these processes are acting simultaneously on different time scales with different strengths. Whole genome data help researchers to distinguish between the effects of these different processes and to identify underlying signatures of selection and transmission.
Identifying signatures of selection Parkhill discussed one strain—sequence type 239 (ST239)—of the globally important human pathogen methicillin-resistant Staphylococcus aureus (MRSA). This multi-antibiotic-resistant strain
arose in Europe and, later, spread globally (Harris et al., 2010).25 According to Parkhill, although 63 different isolates of this strain are indistinguishable by current typing techniques (including MLST and pulsed-field gel electrophoresis), whole genome data has revealed a great amount of variation—with 4,310 SNPs in the core sequence alone.
Parkhill observed that the isolates’ SNPs were almost entirely random, which would suggest the absence of immediate selection. SNPs are randomly distributed across the genome and exhibit a random rate for acquisition over time. Parkhill’s group constructed a maximum likelihood phylogeny based on the 4,310 sites in the core genome of ST239 that contained one or more SNPs. Charting root-to-tip distance against the year of strain isolation suggested that this strain arose in the late 1960s (corresponding with the first administration of methicillin in 1959 and the subsequent emergence of MRSA in Europe). Parkhill noted that the rate of SNPs acquisition of approximately 3×10–6 per site per year is about 1,000 times faster than the accepted mutation rate of bacterial SNPs; it is, however, consistent with what is typically found in studies of very recently evolved organisms (Croucher et al., 2011; Mutreja et al., 2011).
Researchers use the ratio of nonsynonymous changes to synonymous changes (dN/dS) as a measure of selection. For ST239 the ratio is close to 1 (0.68).26 Parkhill noted that in this case the rate is very close to 1, not because there is neutral selection, but because selection has not had time to act. Subsequent studies provided insights into how the rate of change, and therefore the rate of selection, varied with time (as measured by similarity between genomes) and for different regions of the genome (Castillo-Ramirez et al., 2011). As described by Parkhill and depicted in Figure WO-17, “The more closely related the genomes are, the closer the dN/dS ratio is to 1. That means that as the SNPs occur, there is no selection going on. dN/dS is 1 simply because nonsynonymous changes and synonymous changes are occurring randomly at the same rate.” Over time, Parkhill continued, selection starts to act. “You can see selection acting as the dN/dS falls away.” Parkhill emphasized that the rate of change is different between the core and the noncore genes. The noncore genes may contain mobile elements or DNA that has been exchanged with other strains. This has two effects; the changes are effectively “older,” which increases the likelihood that mutations have undergone selection, and the changes are drawn from a larger effective population size, which increases the effectiveness of selection.
_______________________
25 “By analyzing whole genome data of a collection of MRSA ST239, we have gained new insights into fundamental processes of evolution in an important human pathogen. By creating a precise and robust phylogeny for the collection, we now have a highly informative perspective on the evolution of the clone” (Harris et al., 2010).
26 Synonymous changes are considered “silent” because the base change within an exon of a gene coding for a protein does not result in changes to the protein’s amino acid sequence. Nonsynonymous changes result in altered amino acid sequences. A dN/dS ratio greater than 1 suggests diversifying (or disruptive) selection, while a ratio below 1 is associated with purifying (or stabilizing) selection, and a ratio close to 1 indicates neutral selection.
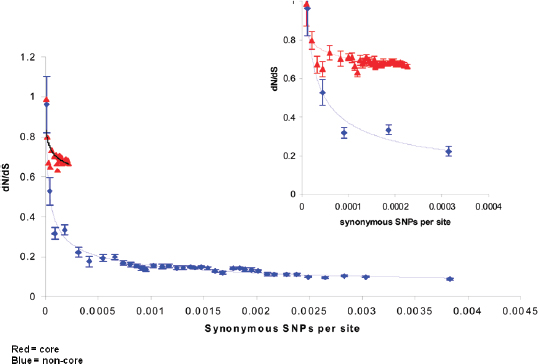
The random nature of mutation in the core genome allowed Parkhill to use these sequences to build a robust and congruent phylogenetic tree. In so doing, he was also able to identify a subset of sites that were “acting unusually,” suggesting that they were under selection. Of the 4,310 SNPs in the tree, 38 loci were homoplasic, that is, they appeared independently on different branches as a result of convergent evolution (Harris et al., 2010). Parkhill explained that the lack of recombination in the tree overall suggests that these homoplasies are under recent and very strong selection. Eleven of these loci correspond to known drug resistance mutations suggesting an association with antibiotic use in clinical practice. When most SNPs are random and overall recombination is moderate, homoplasy works very well as an alternative to dN/dS and other classical measures to identify selection.
Parkhill noted the use of homoplasy to identify selective pressures acting on isolates of other pathogenic bacteria, including Mycobacterium tuberculosis and Clostridium difficile. “By looking, not for dN/dS or all those classical measures of selection, but for things that don’t fit the tree,” Parkhill said, researchers can see selective pressures such as drug resistance and compensatory mutations, as well as evidence of selection on surface proteins, two-component sensor/regulators, and other genes that are likely to be under diversifying selection from host immune pressure.
Parkhill also described an approach that makes it possible to use homoplasy to identify selection in organisms where recombination is very common such as Streptococcus pneumoniae.27 Because they are derived from other strains of S. pneumoniae, recombined regions are representative of the wider species, noted Parkhill, and therefore have an older date of origin. Constructing a coherent, consistent phylogenetic tree of the multi-drug resistant clone of S. pneumoniae (sequence type 81 and serotype F) that emerged in the 1970s required the identification and elimination of recombined regions so that only the vertically transmitted, random point mutations remained. Parkhill and colleagues were then able to put the recombination events back onto the tree to see where, when, and why they occurred (for more information see Croucher et al., 2011).
Identifying transmission Parkhill explained that the development of a congruent phylogenetic tree—such as that developed using the S. aureus ST329 data set—can also be used to define transmission events—from intercontinental transmissions over the course of decades to person-to-person transmission within a
_______________________
27 “The ability to distinguish vertically acquired substitutions from horizontally acquired sequences is crucial to successfully reconstructing phylogenies for recombinogenic organisms such as S. pneumoniae. Phylogenies are, in turn, essential for detailed studies of events such as intercontinental transmission, capsule type switching, and antibiotic-resistance acquisition. Although current epidemiological typing methods have indicated that recombination is frequent among the pneumococcal population, they cannot sufficiently account for its impact on relations between strains at such high resolution” (Croucher et al., 2011).
hospital; a robust tree even allows researchers to identify ongoing transmission events (Harris et al., 2010).
Parkhill concluded his presentation by demonstrating how whole genome approaches have clarified the origin and international spread of a globally important pathogen, Vibrio cholerae. The El Tor strain of this pathogen is associated with the seventh cholera pandemic that originated in the early 1960s and persists today—including the 2010 Haitian cholera outbreak. Before whole genome sequencing, cholera typing was done on the basis of the presence or absence, or variable sequences, of mobile elements. This approach identified a superficial diversity among cholera strains when, in fact, the core genome—excluding possible recombination events—forms a robust transmission tree (Mutreja et al., 2011).
For example, SXT is a mobile element that encodes multi-drug resistance. Looking only at the tree generated with the core genome, its presence in the El Tor chromosome appears to have resulted from a single acquisition. However, generating a tree using 3,000 SNPs in the 60 kb SXT element itself, reveals that the phylogeny of the element is different from that of the core genome. This phylogeny also reveals an apparent rate of SNP accumulation in the SXT element that is 100 times greater than that of the core El Tor genome. Parkhill noted that this elevated rate of mutation acquisition is “very, very unlikely to be true.” Rather, this result suggests that the SXT element is much older and is evolving outside of El Tor. Indeed, comparing the SXT tree with a tree based on the core El Tor genome, novel versions of the SXT element appear to have entered the El Tor chromosome at least five times (Mutreja et al., 2011). “Trying to understand the transmission of cholera based on [the analysis of] mobile elements like SXT is doomed to failure,” Parkhill said, “because this approach is reporting the movements of older elements in and out of the organism’s genome, rather than the phylogeny of the core genome, which should represent the ancestry of the organism itself. To understand transmission, we need to strip out all of these mobile elements and recombinations and look only at the core genome.”
The consistent rate of SNP accumulation of the core genome among isolates also illuminated the origin and spread of El Tor as part of the seventh cholera pandemic. The El Tor core genome tree shows three groups of isolates, which correlate with three different time periods. When coupled to the physical origin of each isolate, Parkhill said, you can see that over the past 40 to 50 years, “three independent waves [of El Tor strains] have spread around the world from a single location, almost certainly around the Bay of Bengal” (Mutreja et al., 2011). The El Tor strain of V. cholerae has been introduced multiple times into different parts of the world. New outbreaks are derived from the trunk of the tree, then expand and die out (Figure WO-18) (Mutreja et al., 2011).
In summary, Parkhill said that different processes are acting on different time scales with different strengths. Whole genome sequencing allows investigators to identify and separate out the varying effects of mobile elements, recombination, and point mutation. The resulting high-resolution phylogenies can provide
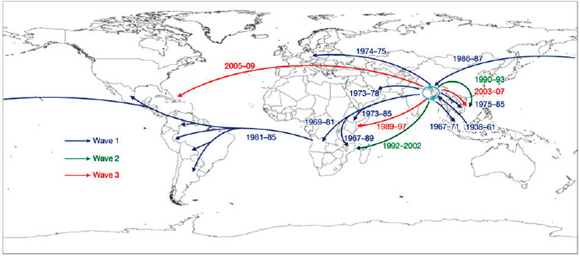
FIGURE WO-18 V. cholerae—repeated global transmission. Transmission events inferred from the phylogenetic tree of the seventh pandemic lineage of V. cholerae based on SNP differences across the whole core genome, excluding probable recombination events.
SOURCE: Mutreja et al. (2011). Reprinted by permission from Macmillan Publishers Ltd: NATURE. Mutreja, A. et al. 2011. Evidence for several waves of global transmission in the seventh cholera pandemic. Nature 477(7365):462-465, copyright 2011.
the basis for important epidemiological and phenotypic analyses and insights to illuminate patterns of gene selection and transmission.
Intra-Host Diversity, Selection, and Evolution: Influenza Virus
Investigators are also exploiting next-generation sequencing approaches to understand how viral genetic diversity changes within a host and during transmission between hosts. Most of what is known about the intra-host diversity of viruses is derived from research on viruses that cause persistent infections (e.g., HIV, hepatitis C virus), where accumulation of replication errors and recombination over time leads to high levels of diversity. Speaker Elodie Ghedin of the University of Pittsburgh School of Medicine elaborated on her interest in intra-host diversity, selection, and evolution of influenza viruses, which cause acute infections (Dr. Ghedin’s contribution to the workshop summary report may be found in Appendix A, pages 151-165). Her work focuses on the following questions:
• Does natural selection occur within individual hosts?
• How big is the population bottleneck at transmission?
• What is the extent of mixed infection?
• What is the mutational spectrum within an individual host?
• What is the fitness distribution of these mutations?
Influenza is a negative strand RNA virus with an eight-segmented genome. Coinfection of a host cell with different strains can result in reassortment of the segments and a mixed population. Ghedin noted that during the H1N1 influenza pandemic, researchers were able to track some of the segments to two different types of swine flu. Whole genome sequencing of influenza generally produces a consensus sequence with one genome that is representative of the strain an individual host carries. In the case of influenza, even though the strain could have high diversity, according to Ghedin you are only looking at what is dominant, so you just have a consensus of each of the segments. The use of next-generation sequencing and methodologies will be important to capture this diversity, Ghedin said.
Influenza can adapt to its host after transmission, or changes may occur before transmission, facilitating movement into a different host species. Immune status of the host (e.g., whether the host is immunocompromised or vaccinated) can also influence the evolution and co-adaptation of a virus. In an immunocompromised host, influenza infection persists well beyond the typical 5 to 7 days, in some cases as long as 5 to 6 months. In the case of a persistent infection, what happens to viral diversity?
Ghedin described the case of an immunocompromised boy infected with pandemic H1N1 influenza virus for 35 days. He was treated with Tamiflu (an antiviral neuraminidase inhibitor) on day 2, and at some time between when viral
RNA was sampled and sequenced on days 6 and 13, a Tamiflu-resistant variant had emerged. This variant may have been present in the population before treatment was started, or it could have been the result of a de novo mutation. Current thinking is that because the drug-resistance mutation also reduces viral fitness, the emergence of resistance is more likely to result from a de novo mutation that is favored under drug pressure. Still, as Ghedin pointed out, the use of consensus sequences does not allow researchers to distinguish between these two possibilities.
In contrast to consensus sequencing, deep sequencing identifies minor variants (above the background of error) and reports the percentage of reads that have the dominant or minor codons at a given position. This approach is illustrated in Figure WO-19, which depicts the alignment of short sequence reads from a sample to the gene sequence of interest—in this case the neuraminidase gene sequence of influenza. Whereas consensus assembly would read the codon at position 275 as CAC (coding for histidine), deep sequencing reveals that some sequences read TAC (coding for tyrosine), which is a codon associated with drug resistance. In the case of this boy, the variant codon (TAC) was present in a very
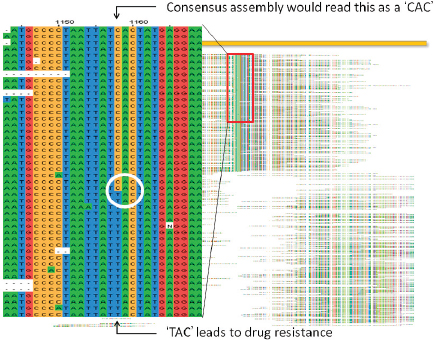
FIGURE WO-19 Next generation sequencing and new methodologies will help to capture intra-host diversity.
SOURCE: Ghedin (2012).
low percentage of reads in samples taken at day 1 (pre-treatment) and day 6; by day 13, this variant was the dominant codon.
When the 2009 H1N1 influenza A virus emerged in the United States, there were multiple clades of viral strains that, while antigenically similar, were clearly clustered by geographic region. As Ghedin observed, “We know a lot about the circulation of different strains, but we know very little about what is actually carried by individuals.” Ghedin’s research has also explored whether the variants observed in a patient resulted from a de novo mutation in the wild-type or from a mixed infection in which the individual is infected with multiple variants either simultaneously or sequentially.
By the second wave of the pandemic, there was a complete mixing geographically, and a single viral clade became dominant across the country, while others appeared to fade away. This is a typical pattern, Ghedin noted. She noted that given this observed tendency toward one dominant viral clade, mixed infections28 (which can occur and are an important driver of genotypic diversity) are generally considered to be rare (Ghedin et al., 2011).
Consensus sequencing of samples taken from an immunosuppressed patient during the second wave of the pandemic showed that the patient’s virus was drug sensitive on both day 1 (pre-treatment) and on day 14 (after antiviral treatment early in infection). Deep sequencing, however, identified three distinct variants. Partial reconstruction of sequence reads into different viral genomes placed two variants into two phylogenetically distinct clades of the pandemic H1N1/2009 virus, a result that strongly suggests a mixed infection (Ghedin et al., 2011). Ghedin concluded that “when we see strains disappear, it doesn’t mean they are completely gone. They can still be present at a low level and may lead to emergence [of new phenotypes].”
Having identified the presence of multiple viral variants within an individual, Ghedin sought to examine how transmission affected diversity. She cited a clear case of transmission of influenza from a son to his father in which the father was prophylactically treated with Tamiflu at the same time the son started therapeutic treatment (Baz et al., 2009). Six days after the start of his Tamiflu treatment, the father had a completely resistant virus. Deep sequencing revealed that more than 2 percent of the son’s viral population was drug-resistant; further analysis demonstrated that multiple variants were being transmitted (i.e., the transmission bottleneck was not especially narrow) (Ghedin et al., 2012). While some variants are lost at transmission, others attain a higher frequency in the recipient.
Ghedin suggested that this improved understanding of the role of transmission in shaping the genetic diversity of influenza may help to refine influenza transmission models. In contrast to consensus sequences, deep sequencing can provide the fine-grained genomic mapping needed to identify significant changes
_______________________
28 Infection in which the individual is infected with multiple variants either simultaneously or sequentially.
in the distribution in the intra-host population. These approaches may also help to reconstruct chains of transmission for an epidemic—insights which may inform future response strategies.
Evolution of Novelty and Pathogenicity: Chytrid Fungal Pathogen of
Amphibians
Novel microbial pathogens do not just appear, they evolve, according to speaker Erica Bree Rosenblum of the University of California, Berkeley (Dr. Rosenblum’s contribution to the workshop summary report may be found in Appendix A, pages 291-311). Genomics, she continued, can provide a useful tool for studying a pathogen’s evolutionary history. Rosenblum’s presentation described an emerging fungal pathogen of amphibians—Batrachochytrium dendrobatidis—and underscored the importance of studying novelty at multiple levels, because a pathogen may be novel in multiple ways including changes in geographic range, host range, virulence, function, etc.. She noted that because of recent technological advances, genomic studies can now be conducted for time-critical studies in non-model species—even in ecological systems where there has not been much previous research.
Bd and amphibian declines Batrachochytrium dendrobatidis, referred to as Bd, is a chytrid fungus that has been implicated in worldwide declines and possible extinctions of amphibian populations. With the exception of Antarctica, Bd occurs on every continent, infecting more than 500 species of amphibians. Genetic and spatial-temporal data demonstrate that Bd is a novel, emerging, pathogen that has quickly spread around the world. Data from many parts of the world trace Bd’s arrival and subsequent spread in a wave-like fashion throughout amphibian populations (Lips et al., 2006). Rosenblum noted the rejection of an early hypothesis that Bd had long existed as a commensal organism and that this relationship had changed due to shifts in the environment.
Chytrids are basal fungi, and almost all known chytrids are saprobes—organisms that live on decaying organic matter in leaf litter or aquatic environments. Bd is unique among chytrids because it is the only species known to infect vertebrates. Rosenblum noted that there appears to have been a very recent change in Bd that allows it to exploit this new host niche. Bd kills frogs by disrupting the structure and function of their skin such that important skin functions, including osmoregulation and electrolyte balance, are compromised. Recent studies suggest that Bd may also have an immune evasion or suppression strategy, and there is speculation that Bd may also release toxins (Rosenblum et al., 2009, 2012).
Although Bd was discovered and described more than 10 years ago, several persistent unanswered questions remain about the origin and spread of Bd and the interaction between Bd and its amphibian hosts. Simply stated: where did it come
from, and, what makes it so deadly? Rosenblum noted that ecological analyses alone cannot answer many of the questions about Bd and, as yet, Bd cannot be manipulated for cellular and molecular analyses. Rosenblum is using comparative and functional genomics to try to understand Bd evolution and pathogenicity.
Using genomics to understand evolutionary novelty of Bd Rosenblum first explored novelty at the phylogenetic level looking for important genetic or functional variation within isolates from around the world, which are collectively described under the single species name, Bd. Sequencing of the genomes of these 28 Bd isolates, as well as the genome of the closest known nonpathogenic chytrid, produced more than 100,000 SNPs that could be used for understanding the evolutionary history of Bd.
Given the rapid spread of Bd around the world and its presumed recent origin, Rosenblum said she expected samples from all over the world to appear all mixed up along very short branches, or a tree reflecting a linear progression from a basal group to subsequent radiations into new geographic or host-specific clusters. Instead, the evolutionary history of Bd was much more complex. As illustrated in Figure WO-20, there is no clear point-source for the origin of Bd or any linear history of how it has spread around the world. The absence of geographic or host-specific population structure confirms the rapid spread and broad host range of Bd. Yet the tree has more structure than expected, exhibiting two highly divergent lineages: a basal lineage with isolates from Latin America and a large clade with significant global diversity. Rosenblum suspects that with more geographic sampling, Bd will continue to look more like a “they” than an “it.”
Using genomics to understand functional novelty of Bd As previously noted, Bd exhibits functions that no other chytrid has acquired. Comparative chytrid genomics has revealed that while most fungal genomes had one or several copies of genes from different protease gene families, the Bd genome had massive expansions of protease gene families—dozens of fungalysin metallopeptidases and serine protease genes as just two examples (Rosenblum et al., 2008). This is interesting, Rosenblum explained, because these two protease gene families have been implicated as potential pathogenicity factors in other fungal pathogens that infect vertebrates including for example, tinea and ringworm, as well as other dermatophytic fungi that infect vertebrate skin. To follow up on these observations Rosenblum again used genomic approaches to look for functional and evolutionary evidence that these proteases may have been important to Bd’s transition from saprobe to amphibian pathogen. Functional genomics studies of the presence or absence of gene expression at different life stages or in response to different nutrient conditions (nutrient broth versus frog skin) have identified gene copies of particular interest—those that show higher levels of expression in the host tissue and during life stages in which Bd is known to infect amphibians (Rosenbaum et al., 2012).
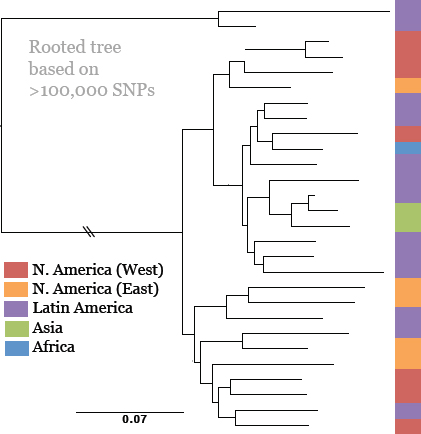
Rosenblum and colleagues compared the genomes of Bd and its closest relative, the nonpathogenic saprobe Homolaphlyctis polyrhiza, in order to investigate whether the protease gene family expansions accompanied the evolution of pathogenicity. Results suggested these gene family expansions are recent, and generally Bd specific—for example, Bd was found to have 38 fungalysin peptidase gene family copies where H. polyrhiza has 5 (Joneson et al., 2011). This study also revealed that Bd has 62 Crinkler-like proteins, which are proteins that have never before been described in any fungal species. What is interesting
about this observation, according to Rosenblum, is that the closest Bd relative that has Crinklers are the oomycetes, in which they [Crinklers] are believed to be virulence effectors.
Rosenblum concluded that genomic approaches have been invaluable in understanding the complex history of Bd and identifying its evolutionary transition points. Genomic approaches have also assisted in developing hypotheses about functional aspects of Bd’s lifestyle. Using these genomic tools and approaches to Bd has revealed that this fungal vertebrate pathogen has a much more complex and deeper evolutionary history than had been previously appreciated. These same approaches, according to Rosenblum, may also help investigators identify key transition points in the evolutionary history of this pathogen.
Microbial Communities in Coral Health and Disease
Corals are very simple animals that harbor diverse microorganisms in and on their tissues—including archaea, bacteria, viruses, and zooxanthellae (Figure WO-21). Endosymbiotic dinoflagellate algae (Symbiodinium, or more commonly, Zooxanthellea) live within the endothelial tissue of the coral. The symbiotic relationship between the coral animal and this single-celled algae has been well studied, but far less is known about the bacterial communities that are associated with the algae and the coral, explained speaker Kim Ritchie of the Mote Marine Laboratory (Dr. Ritchie’s contribution to the workshop summary report may be found in Appendix A, pages 269-290).
Molecular methodologies and metagenomic sequencing confirm the broad diversity of bacterial species associated with corals. Some bacteria—Vibrio species, Serratia species, Aspergillus sydowii, Aurantimonas coralicida—are opportunistic pathogens that have been implicated in coral diseases when the coral communities are stressed. Other bacteria may provide beneficial services to the corals. Ritchie reviewed current research using culture-based approaches that explore the nature and specificity of coral-bacterial associations, and the ways that these associations are maintained over time.
Bacteria and early life stages of coral Ritchie also reviewed recent research exploring bacterial colonization of developing coral tissue and the role of bacterial biofilms in this symbiotic association. There are two general types of corals; broadcast spawners and brooding corals. Broadcast spawners produce eggs and sperm that are externally fertilized, and the eggs therefore acquire Symbiodinium algae, horizontally from the water column. Brooding corals have internal fertilization and release planula larvae that already have the parent Symbiodinium. Bacterial colonization of most corals (both brooders and broadcast spawners) occurs during planula larvae or postlarval settlement stages29 much like the Hawaiian
_______________________
29 According to Ritchie, an exception is the Caribbean coral Porites astreoides, in which bacteria are passed directly from parent to offspring (vertical transmission).
FIGURE WO-21 Interactions within coral reef communities. Panel a, Healthy coral reefs support a diverse ecosystem, as shown here by a healthy Montastraea faveolata colony (H) and a dead colony (D). Panel b, A coral colony comprises genetically identical polyps that are connected by a living tissue. Panel c, Functionally and taxonomically diverse microbial communities are found on the coral surface, within the gastrodermal cavity and in the intracellular spaces of the polyp. Coral-associated bacteria (B) are thought to supply the host with fixed nitrogen. Bacteria produce antimicrobials with a potential function in host defense. Symbiotic dinoflagellates (Symbiodinium spp., [S]) are housed within cells of the ectoderm. Symbiodinium provides photosynthate to its animal host, which then supplements its diet by feeding on plankton. In addition to satisfying the energy needs of the coral animal host, the photosynthate from Symbiodinium is converted into glycoprotein-rich mucus (M) within specialized cells of the polyp, called mucocytes. Mucus is then excreted onto the coral surface. Corals actively manipulate their associated microbial communities, and the mucus contains nutrients that support growth of bacteria and eukaryotes. Antimicrobials in coral mucus may also contribute to the coral microbiome. Coral lectins (L) bind and coagulate bacteria. Populations of coral-associated bacteria are also controlled by phages (P)—viruses of bacteria that may function as biocontrol agents for opportunistic pathogens of corals.
NOTE: Reproduced with permission from Jonathan Onufryk (a) and Erich Bartels (b). Schematic drawing (c) not to scale.
SOURCE: Teplitski and Ritchie (2009). Reprinted from Trends in Ecology & Ecolution, 24, Max Teplitski and Kim Ritchie, How feasible is the biological control of coral diseases? 24(7):378-385, Copyright 2009, with permission from Elsevier.
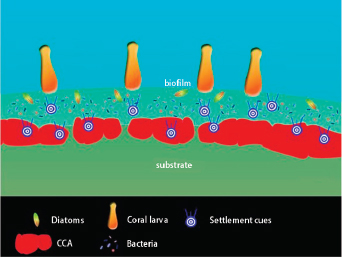
FIGURE WO-22 Microbial biofilms are necessary for larval settlement. Crustose coralline algae (CCA) and biofilm microbial communities facilitate attachment and settlement of coral larvae via inductive compounds (settlement cues) produced by the CCA or by recruiting specific bacteria that release these cues.
SOURCE: Sharp and Ritchie (2012). Sharp, K. H., and K. B. Ritchie. 2012. Biological Bulletin 174:319-329. Used with permission from the Marine Biological Laboratory, Woods Hole, MA.
bobtail squid Euprymna scolopes forms a persistent association early in its life cycle with the Gram-negative luminous bacterium Vibrio fischeri (McFall-Ngai et al., 2012; Sharp and Ritchie, 2012; Sharp et al., 2010).
According to Ritchie, Roseobacter clades are present in the early life stages of many corals. Their consistent detection in seawater during coral spawning suggests their potential importance for mediating larval settlement and survival. Ritchie cited a recent study that suggests that coral larvae need a microbial bio-film on some type of substrate, preferably calcium, for settlement—as illustrated in Figure WO-22 (Sharp and Ritchie, 2012). Roseivivax (in the Roseobacter clade) and Marinobacter were among the bacteria found to encourage coral larval settlement.
Ritchie also reviewed the recent finding that all of the Symbiodinium-associated roseobacters tested produced gene transfer agents (GTAs) (McDaniel et al., 2010, 2012; Paul et al., in review). Ritchie explained that GTAs resemble bacteriophage, packaging random pieces of host DNA and transferring them to other bacteria (McDaniel et al., 2010). Interestingly, gene transfer via GTAs was found to be 100 million times higher in the reef environment than in open oceans and in Tampa Bay. There are also more roseobacters in the reef environment,
Ritchie added, particularly during coral spawning, and experiments suggest that Roseobacter and gene transfer agents may increase larval settlement.
Microbial community regulation of coral disease development When stressed by environmental conditions—such as elevated sea surface temperatures—corals often “bleach.” As corals begin to bleach, the population of Vibrio species that are normally associated with healthy corals and their symbiotic algae increase while the population of other types of bacteria decline. Studies by Ritchie to explore the coral bleaching phenomenon have revealed that the surface mucus of healthy elkhorn coral, Acropora palmata, may be a potent defense against disease. In vitro, it inhibits the growth of a range of potentially invasive microorganisms. Ritchie identified a number of antibiotic-producing bacterial species that were isolated from surface mucus that could contribute to this defense. Mucus collected during a bleaching event (i.e., when the coral were unhealthy or stressed) were found to lack this antimicrobial activity (Ritchie, 2006).
By sampling corals monthly, Ritchie discovered that when sea surface temperatures increase, the population of antibacterial-producing bacteria decreases, while the population of potentially pathogenic bacteria (including Vibrio species) increases (Ritchie, 2006). A mathematical model based on these findings suggests that there is a lag time in coral recovery following a warming event (Mao-Jones et al., 2010). Consistent with this prediction, Ritchie has detected overgrowth and persistence of different types of Vibrio species in the coral mucus long after environmental conditions were once again favorable for healthy coral populations and their associated microbiota. This lag in response and recovery, or hysteresis, may explain why corals are more susceptible to disease and bleaching both during and after ocean warming events, Ritchie said.
Ritchie used both culture-based and molecular methods to identify bacteria associated with a diverse collection of Symbiodinium samples. Bacteria isolated were primarily members of three bacterial groups: the Roseobacter clade, marinobacters (oceanospirillates group), and the Cytophaga-Flavobactrium-Bacteroides group. Emerging evidence suggests that Roseobacters are present in the early life stages of corals and are abundant on coral reefs when coral spawn. Simple laboratory experiments have revealed that roseobacters produce antimicrobial compounds, enhance the growth of Symbiodinium, and reverse symptoms of disease caused by the putative coral pathogen, Serratia marcescens (Krediet et al., in press; Ritchie, 2011). Together, these data suggest that roseobacters are likely to be critical in the maintenance of a healthy coral ecosystem.
Many of the beneficial bacteria isolated by Ritchie produce compounds associated with a cell-to-cell communication system used by a variety of bacterial species to mediate host–microbe interactions. These signals have been shown to be produced in situ on the surface of corals and to inhibit swarming and biofilm formation by coral pathogens (Alagely et al., 2011). Ritchie is using the polyp anemone, Aiptasia pallida, as a model system (there are no adequate coral
models) to explore interactions between bacterial community members, their coral hosts, and potential pathogens. As reported by Sharp and Ritchie (2012), Aiptasia pallida presents an opportunity to integrate a model systems approach with novel technologies from the “omics age” to learn more about multipartner interactions in corals in a moment of great environmental change.
METAGENOMICS
Exploring Microbial Diversity
As emphasized throughout the workshop, microbial genomics has supported the exploration of the vast diversity of the unseen microbial world. During the past decade, the scope and scale of these studies have increased from studies of low-complexity microbial communities (such as those found in acid mine drainage biofilms) to broad surveys of how complex microbial communities vary across space and time (Hugenholtz and Tyson, 2008) (Figure WO-23). The Human Microbiome Project, for example, seeks to map microbial communities associated with the different environments on and in the human body (e.g., gut, mouth, skin, vagina), track how these communities differ by individual and his or her health status, and identify how the microbiota (individual/community) contributes to states of health and disease (Turnbaugh et al., 2007). The Earth Microbiome Project is a metagenomics survey that will collect natural samples and analyze microbial communities from around the world; it is anticipated that 200,000 environmental samples will be collected from across the various biomes of Earth and sequenced for taxonomic and functional analysis (Gilbert and DuPont, 2011).
Metagenomic studies have provided insights into the rich and untapped genetic potential of microorganisms, the functional (metabolic) potential of microbial communities, and the structure (species richness and distribution) of communities in a wide range of environments. Examples include:
• The analysis by Venter et al. (2004) of seawater from the Sargasso Sea resulted in the identification of more than 1.2 million new genes, including more than 700 new rhodopsin-like photoreceptors. These proteins are now thought to be a major source of energy flux in the world’s oceans (Hugenholtz and Tyson, 2008).
• Tyson et al. (2004) were able to reconstruct two almost complete genome sequences of Leptospirillum group II and Ferroplasma type II and the partial sequence of three other species from a low-complexity acid mine drainage biofilm. Genome analysis for each organism revealed specified pathways for carbon and nitrogen fixation and energy generation. More recently, Denef and Banfield (2012) analyzed samples taken at this site over a 9-year period to measure the evolutionary rate of free-living
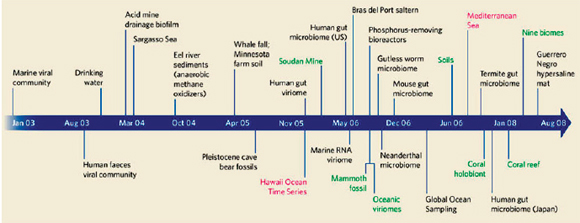
FIGURE WO-23 Timeline of sequence-based metagenomic projects showing the variety of environments sampled since 2003. The oceanic viromes(all viruses in a habitat)(August 2006) were from the Sargasso Sea, Gulf of Mexico, coastal British Columbia, and the Arctic Ocean. The nine biomes (March 2008) were stromatolites, fish gut, fish pond, mosquito virome, human-lung virome, chicken gut, bovine gut, and marine virome. The different technologies used are dye-terminator shotgun sequencing (black), fomid library sequencing (pink), and pyrosequencing (green).
NOTE: Graphic based on data sets represented at www.genomesonline.org.
SOURCE: Hugenholtz and Tyson(2008). Reprinted by permission from Macmillan Publishers Ltd: NATURE. Hugenholtz, P. and G. W. Tyson. 2008. Metagenomics. Nature 455:481-483, copyright 2008.
microorganisms in the wild. Such studies will illuminate how ecologic and evolutionary forces interact to shape microbial population dynamics.
• A recent effort to catalog the genes of the adult human gut microbiome suggested that the genes contained within the gut flora outnumber those contained within our own genome by 150-fold. An individual likely harbors at least 160 bacterial species in his or her lower digestive tract (Qin et al., 2010). These communities appear to be carefully calibrated enterprises, and the disruption of this delicate balance may contribute to a variety of diseases including obesity, autoimmune diseases, and asthma (Ley et al., 2008).
• Tringe et al. (2008) used a gene-centric analysis to assess DNA isolated from highly complex and nutrient-rich environments: soil and three isolated deep-sea “whale fall” carcasses. Comparisons of gene abundance may provide habitat-specific fingerprints that reflect known characteristics of the sampled environment and “hint at certain nutrition conditions, novel genes, and systems contributing to a particular life style or environmental interactions.”
Workshop speakers elaborated on several current metagenomics projects that seek to provide insights into the interactions among microorganisms within a community.
Human-Associated Microbial Communities: Links to Health and Disease
Just as microbes colonize the bobtail squid’s light organ shortly after hatching, microbes colonize the human body internally and externally during its first weeks to years of life and become established in relatively stable communities in a variety of microhabitats (Dethlefsen et al., 2007). Research to date suggests that the site-specific microbial communities—known as microbiota or microbiomes30—that inhabit the skin, intestinal lumen, mouth, teeth, and so on of most individuals contain characteristic microbe families and genera. The species and strains of microbes present on or in any given individual may be as unique as a fingerprint (Dethlefsen et al., 2007). The microbiota of other terrestrial—and possibly aquatic—vertebrates are dominated by microbes that are related to, but distinct from, those found in humans. This suggests that host species and their microbial colonists have uniquely coevolved with and adapted to one another.
As discussed previously, the complexity of the human microbiome is astounding. Each community has its own unique structure and ecosystem that is
_______________________
30 The term microbiome is attributed to the late Joshua Lederberg, who suggested that a comprehensive genetic view of the human as an organism should include the genes of the human microbiome (Hooper and Gordon, 2001). Because most of the organisms that make up the microbiome are known only by their genomic sequences, the microbiota and the microbiome are from a practical standpoint largely one and the same (IOM, 2009).
shaped by and actively influences the habitat within which it resides. As speaker George Weinstock of the Genome Institute at Washington University observed, “in all microbiomes, the organisms are talking to each other. They are talking to their hosts; they are doing things to the environment” (Dr. Weinstock’s contribution to the workshop summary report may be found in Appendix A, pages 357-378). He noted that their dynamic nature makes them challenging to study, but that understanding these dynamics may shed light on “the emergent and amazing properties that microbiomes bestow on their hosts and environments.” According to Weinstock, “the fundamental goal of human microbiome research is to measure the structure and dynamics of microbial communities, the relationships between their members, what substances are produced and consumed, the interaction with the host, and differences between healthy hosts and those with disease” (Weinstock, 2012a).
He offered four vignettes from research associated with the Human Microbiome Project to illustrate the challenges and insights emerging from studies of variation in the human microbiome:
• discovery of new bacterial taxa (using 16S rRNA sequence variation)
• discovery of new strains of known taxa (looking for within species variation)
• health versus disease (variation of community structure)
• tracking organisms (genetic variation in populations)
Discovery of new bacterial taxa The Human Microbiome Project has generated a massive amount of 16S rRNA gene sequence data.31 16S rRNA differs for each bacterial species (Weinstock, 2012a). A bacterial species is hard to define, but it is often thought of as organisms with 16S rRNA gene sequences having at least 97 percent identity—an operational taxonomic unit (OTU) (Weinstock, 2012a).
In order to identify organisms in the human body that had not been described before, Weinstock chose 11 subjects from the Human Microbiome Project and analyzed their stool samples for sequences that had < 97 percent identity to any known 16S rRNA sequence.32 Stringent criteria were applied to subject selection33 and analysis of sequence data to distinguish truly novel sequences from artifacts (e.g., sequencing errors, chimeric sequences). After validation of the candidate
_______________________
31 The 16S rRNA gene is used for phylogenetic studies because it is highly conserved between different species of bacteria and archaea. In addition to these, mitochondrial and chloroplastic rRNA are also amplified.
32 The Human Microbiome Project has sampled 18 body sites in 300 subjects.
33 In response to a question from a Forum member, Weinstock elaborated on the criteria for what a healthy individual was and inclusion/exclusion criteria for this study. Exclusion criteria included everything from not having had an antibiotic treatment for a period of time prior to sample collection to not having rashes and having most of your teeth. Three days before sample collection, the subjects were also given packs of soap and toothpaste so that the microbiome being sampled was the microbiome of humans who use these products and who [are otherwise healthy].
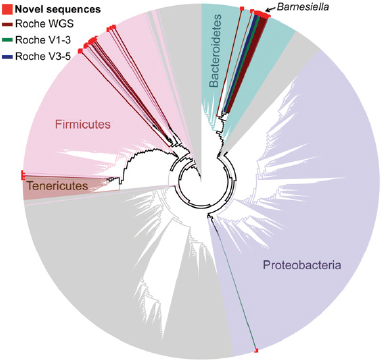
FIGURE WO-24 Novel taxa are found in three of the major phyla associated with human stool samples.
SOURCE: Wylie et al. (2012a).
taxa by sequencing with multiple platforms and approaches, 26 sequences remained as candidate novel OTUs (i.e., genetically distinct groups of microbes). As illustrated in Figure WO-24, taxonomic analysis suggested that they are most likely new genera or species that are most similar to uncultured bacteria in the phyla Firmicutes, Bacteroidetes, and Proteobacteria. Further analysis of stool sample data from100 additional Human Microbiome Project participants demonstrated that most of the novel taxa were found in multiple individuals sampled at two different geographic locations (Wylie et al., 2012a).
Discovery of new strains of known taxa Novel taxa remain to be discovered, Weinstock observed. In this survey, novel taxa were low in abundance within individuals, but were found in multiple people. The novel taxa identified are related, but not identical to, previously identified organisms, and are most similar to uncultured organisms. Looking for variation within species has led to the discovery of new strains of known species of bacteria and the association of these new strains with disease phenotypes.
Weinstock described a study, in collaboration with Huiying Li at the University of California, Los Angeles, that looked for a correlation between the composition of the human microbiome and acne (Fitz-Gibbon et al., 2013). Samples taken from the epithelial skin pores of the noses of 101 participants—half with and half without acne—were sequenced and cultured. 16S rRNA gene sequencing revealed similar community structure on a species level, with samples from both groups dominated by Propionibacterium acnes. This observation is illustrated in Figure WO-25.
Full length sequencing of more than 31,000 16S rRNA gene clones allowed investigators to take a more detailed examination of community structure, which revealed variation at the subspecies level. Weinstock presented data for the 10 most abundant ribotypes. Compared against the most abundant strain, the sequence of the 16S rRNA gene of the other 9 strains differed by one or two nucleotides. Six of those nine were clearly more abundant in individuals with acne than those without (84 to 100 percent of clones with these ribotypes came from participants with acne). These 10 ribotypes clustered into five microbiome types, two that were primarily found in acne patients and three that were found in both groups. Whole genome sequencing of 71 cultured strains showed that two ribotypes highly associated with acne are distinct because they have two chromosomal regions that lack the other ribotypes (Figure WO-26). In addition, these two ribotypes have plasmid DNA.
Genomewide SNP clustering of the 71 P. acnes strains demonstrated that strains that tend to be associated with acne cluster together, as do the strains that tend to be associated with healthy people. Weinstock remarked that these results were striking; the identification of a one- or two-nucleotide difference in 16S rRNA has led to the discovery of what appear to be potentially pathogenic strains of P. acnes and regions of the P. acnes genome that might be relevant to acne pathogenesis. Experiments are now being designed to test this intriguing hypothesis. Weinstock further observed that subspecies-level variation may be a widespread driver for other diseases, such as bacterial vaginosis, in which clinical presentation of disease does not always show a relationship with community variation at the species level.
Health vs. disease: Variation between communities A primary interest of the Human Microbiome Project is defining changes in a microbiome that are associated with disease. However, the intrinsic variation raises several important questions related to study design:
• How does one approach hypothesis testing, and power and sample size determination?
• What kind of a distribution best fits metagenomic data?
• How do you derive a number or a metric that conveys how similar or different two people are?
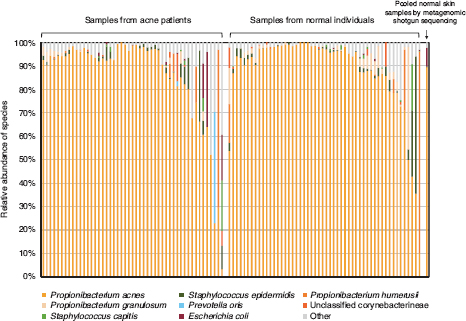
FIGURE WO-25 P. acnes is dominant in pilosebaceous units in both acne patients and individuals with normal skin. By 16S rDNA sequencing, P. acnes sequences accounted for 87 percent of all the clones. Species with a relative abundance > 0.35 percent are listed in order of relative abundance. As shown on the far-right column, species distribution from a metagenomic shotgun sequencing of pooled samples from normal individuals confirmed the high abundance of P. acnes in pilosebaceous units.
SOURCE: Fitz-Gibbon et al. (2013). Reprinted by permission from Macmillan Publishers Ltd: [JOURNAL OF INVESTIGATIVE DERMATOLOGY] (Fitz-Gibbon et al., 2013), copyright(2013).
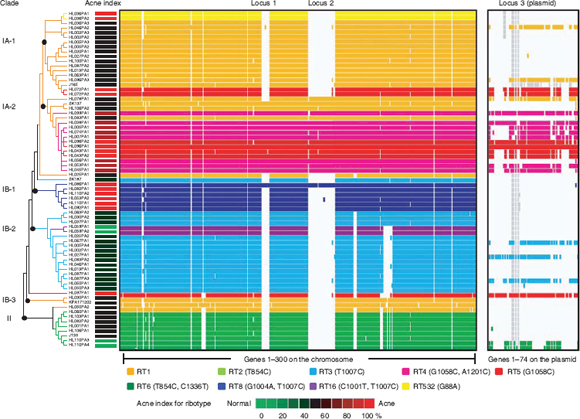
FIGURE WO-26 Genomic differences at the subspecies level (P. acnes). Genome comparison of 71 P. acnes strains shows that the genomes of RT4 and RT5 are distinct from others. Two chromosomal regions, loci 1 and 2, are unique to clade IA-2 and one other genome HL086PA1. Clade IA-2 consists of mainly RT4 and RT5 that are highly enriched in acne. The presence of a plasmid (locus 3) is also characteristic of RT4 and RT5. Each row represents a P. acnes genome colored according to the ribotypes. Rows are ordered by the phylogeny calculated based on the SNPs in P. acnes core genome. Only the topology is shown. The clades were named based on their recA types (IA, IB, and II). Columns represent 16 predicted open reading frames (ORFs) in the genomes and are ordered by ORF positions along the finished genome HL096PA1, which encodes a 55 Kb plasmid. Only the first 300 ORFs on the chromosome (on the left) and all the ORFs on the plasmid (on the right) are shown. The colored plasmid regions represent genes on contigs that match exclusively to the HL096PA1 plasmid region. The genes that fall on contigs that clearly extend beyond the plasmid region are likely to be chromosomally located and are colored in grey. Acne index for the ribotypes was calculated based on the percentage of clones of each ribotype found in acne.
SOURCE: Fitz-Gibbon et al. (2013). Reprinted by permission from Macmillan Publishers Ltd: [JOURNAL OF INVESTIGATIVE DERMATOLOGY] (Fitz-Gibbon et al., 2013), copyright (2013).
Weinstock reviewed the Dirichlet-multinomial distribution for modeling and comparing species abundance distributions in human microbiota samples. This approach allows for quantification of community similarity by overdispersion (Figure WO-27), a metric that can be used to factor variation in community structure between healthy subjects (i.e., normal variation) and variation between states of health and disease into study design (La Rosa et al., 2012).
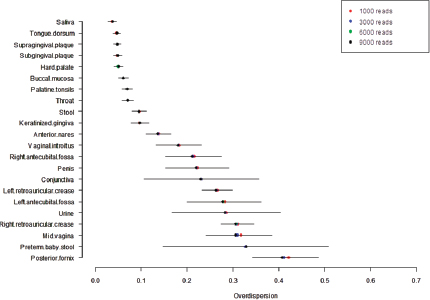
Understanding the intrinsic variation in any two samples is essential for designing meaningful comparative studies. Weinstock noted that investigators are just beginning to define different classes of communities within a body site; such as high and low bacterial communities of lactobacillus in the vagina.34 A somewhat controversial area in human microbiome research, Weinstock said, is whether individuals or groups of people can be uniquely identified by their gut microbiomes. Three distinct gut community “enterotypes” have been proposed to group people and communities based on the predominance of one of three classes of organisms, Ruminococcus, Bacteroides, or Prevotella. Weinstock cautioned that for such community classifications, “time will tell whether these really reflect some kind of difference in people.” He noted however, that “we certainly know how to do an analysis that teases [apart] differences and allows you to test certain [hypotheses].”
Weinstock offered two examples of studies of microbial community variation associated with states of “disease” and “health.” The first, a collaboration
_______________________
34 According to Weinstock, Jacques Ravel, Larry Forney, and others who have investigated this for a long time think there may be as many as five different community classes of the vaginal [microbiome].
with Homer Twigg of the Indiana University Medical Center in Indianapolis, was a longitudinal study of the effect of HIV on the lung microbiome, which suggests that variation between these communities increases with HIV infection and is reduced when patients are treated with highly active antiretroviral therapy (HAART). These observations are illustrated in Figure WO-28. After HIV is removed by antiretroviral therapy, it takes a long time for the disturbed microbiome to return to a healthy equilibrium state and for the host to re-establish immunological homeostasis. Interestingly, Tropheryma whippelei (the causative agent of Whipple’s disease, a rare gastrointestinal malabsorptive condition) was the dominant organism in lung samples during HIV infection. The significance of this finding is yet to be determined, but according to Weinstock, it underscores how little we know about the microbiome, and the potential for metagenomic analyses to generate new insights into human health and disease.
The second study discussed by Weinstock surveyed babies 36 months of age or younger that had been admitted to the hospital emergency room with a high fever (Wylie et al., 2012b). Standard diagnostic tests could not identify a causative agent in more than 30 percent of these patients. An analysis of nasal and plasma samples obtained from both febrile and afebrile children using shotgun
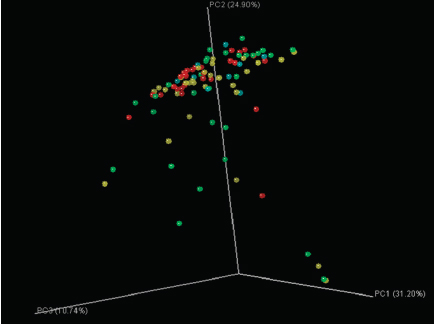
FIGURE WO-28 Change in HIV communities with HAART.
SOURCE: in collaboration with Homer Twigg of the Indiana University Medical School in Indianapolis (unpublished).
sequencing35 demonstrated that, while numerous DNA and RNA viruses were detected in all patients, virus sequences were significantly more abundant in febrile children. Weinstock highlighted the intriguing result that healthy (or at least asymptomatic) children harbor many viruses, that these viruses have also been detected in nasal, vaginal, gastrointestinal, skin, and oral samples from healthy adults, and these infections are stable (i.e., not transient).
Tracking organisms: Genetic variation in populations Weinstock discussed insights emerging from research on genetic variation in metagenomic populations. Pooling of stool sample sequence data from the Human Microbiome Project and MetaHIT36 (together totaling 252 samples obtained from 207 people) created a massive amount of sequence information on the gut microbiome. When aligned with a reference set of approximately 1,500 finished bacterial genomes, this data set produced high-quality sequence information for 101 bacterial species, which could be further studied for genetic variation (Schloissnig et al., 2013).
This project has now created a catalog of how these 101 different species may fluctuate in different individuals, Weinstock explained, identifying 10.3 million SNPs, 1.1 million short insertions and deletions, and more than 4,000 structural variants in these species. Phylogenetic distribution and abundance of the 101 samples in the combined Human Microbiome Project-MetaHIT cohort were consistent with the usual community structure observed for the gut. By assessing the presence or absence of specific SNPs in samples collected from the same patient at different times, researchers have been able to show that strains present in an individual were fairly stable over time—leading to the conclusion that a person has his or her own, unique, microbial communities that are distinct from the strains from another individual.
In another study, SNP analysis of samples from three different oral sites—buccal mucosa, supragingival plaque, and the tongue dorsum, revealed site-specific strains of Streptococcus oralis. An analysis of the community structure of the mouth may simply identify S. oralis as a member of the mouth microbiota. It appears, however, that different strains of S. oralis reside in different sites—distinct ecological niches—in the mouth. Weinstock reiterated the importance of subspecies variation with respect to the pathogenicity of P. acnes, and wondered what else might be different between these three strains of S. oralis and what role those differences might play in oral health and disease.
_______________________
35 “Shotgun sequencing” randomly shears genomic DNA into small pieces that are cloned into plasmids and sequenced on both strands, thus eliminating the BAC (bacterial artificial chromosome) step from the HGP’s (Human Genome Project’s) approach. Once the sequences are obtained, they are aligned and assembled into finished sequence.
36 MetaHIT, or Metagenomics of the Human Intestinal Tract, is a academic-industry consortium funded by the European Commission to study the association of bacterial genes in the human intestinal microbiota with human health and disease. See www.metahit.eu.
Phylogenetic and Functional Diversity in Deep-Sea Microbial Communities
Occupying 80 percent of our biosphere, the deep ocean37 remains a largely unexplored terra incognita. Located at an average depth of 4,000 meters (~2.5 miles) below the sea surface, the deep-sea floor includes the largest mountain range on earth—the 50,000 mile long mid-ocean ridge system—and is the site of ~90 percent of the volcanic activity on the planet. According to speaker Peter Girguis of Harvard University, the ocean is also dominated by microorganisms. About 50 percent of the oxygen in Earth’s atmosphere—as well as about 50 percent of Earth’s primary production—is produced by marine photosynthetic microbes. Marine microbes also play a major role in other biogeochemical cycles critical to our biosphere. The grand challenge for investigators interested in environmental microbes, Girguis said, is linking microbial identity to functional potential and activity, including elucidating the role a given microbe plays in global biogeochemical cycles.
The deep-sea floor influences Earth’s heat and energy budgets, primarily through hydrothermal circulation. The ocean circulates through the crust and underlying sediments on the sea floor, known collectively as the ocean crustal aquifer.38 With an estimated 1029 microbes in ocean sediments alone, the crustal aquifer harbors an extensive, and very poorly understood, community of microorganisms (Whitman et al., 1998). These microbes are thought to play a major role in marine biogeochemical cycles that influence the entire ocean, such as metal cycling.
Much of the fluid circulating through the crustal aquifer is discharged at deep-sea hydrothermal vents. These vents are prominent features along mid-ocean ridges, and emit hot, chemically reduced fluids. The buoyant fluid emerges through the crust, and metals precipitate to form chimneys or “smokers.” Temperatures range from 4 degrees Celsius in ambient water to more than 350 degrees Celsius inside smokers. Pressure at vents is around 250 atmospheres, or 4,000 psi. Vent fluid contains millimolar concentrations of hydrogen sulfide and high concentrations of heavy metals such as arsenic, and it has an acidic pH (similar to vinegar) (Figure WO-29). According to Girguis, hydrothermal vent water is “pretty hostile stuff” that can melt plastics, dissolve tin and pewter, char wood, and melt glass. “But, what is amazing” he continued “is that the communities around these hydrothermal vents are immense in terms of their biomass. These are highly productive communities.” Indeed, chemoautotrophic39 microorganisms flourish around these vents and support abundant communities of flora and fauna, reaching densities comparable to those of rainforests.
_______________________
37 Deeper than 1,000 meters.
38 The entire volume of this aquifer is expelled every 2,000 to 5,000 years, and the entire ocean circulates through this aquifer every 70,000 to 200,000 years (Fisher and Von Herzen, 2005).
39 Organisms that use energy derived from the oxidation of inorganic compounds to fix carbon.

FIGURE WO-29 Hydrothermal vents host substantial endolithic microbial communities.
SOURCE: Girguis (2012); Schrenk et al. (2003).
The basis of the food chain at hydrothermal vents is chemoautotropic microbes, but according to Girguis, “we don’t really know who is doing what, and how they are doing it.” Studying microbes in the laboratory is hampered by the fact that more that 99 percent of known microorganisms have eluded cultivation. For the study of deep-sea organisms specifically, it is difficult to replicate the conditions of the deep-ocean trenches on Earth’s surface in order to conduct in situ chemical measurement and experiments. Girguis has designed novel instruments to collect contextual metadata in these remote sites (such as underwater mass spectrometers to look at dissolved gases). Measuring activity at the microbial scale (such as, microbial influence on pH, temperature, CO2) is a slow and tedious process, and the nature, quality, and abundance of contextual data available to link specific microbes to biogeochemical processes varies widely. As such, there is an “impedance mismatch” between the volume and nature of data available from high-throughput sequencing technologies and the volume and nature of the contextual data obtained from geochemical sensors. Omics-derived data that inform us about who is there and what they are potentially capable of doing, according to Girguis, will be helpful in guiding the design and conduct of geochemical studies.
One paradox in vent microbial ecology is that the rate of sulfide oxidation in chimneys is higher than expected given the limited concentration of oxidants in vent effluents. As it turns out, metal sulfides in chimney walls (e.g., chalcopyrite, pyrite, wurtzite) are highly conductive, and vent-dwelling microbes are capable
of extracellular electron transfer, a process of shuttling electrons from anaerobic respiration inside the cell to solid phase insoluble remote oxidants outside the cell. In essence, the anaerobes are benefiting from electrical continuity to oxygen as an oxidant, without being in chemical continuity with that oxygen. Further studies suggest that, in fact, vent microbes are using extracellular electron transfer to both donate and accept electrons. Microbes are essentially sharing electron equivalents through the conductive vent matrix.
Girguis described studies of microbial extracellular electron transfer using “microbial fuel cells” that mimic mineral oxidants. In a lab-based artificial vent, he measured the amount of current across electrodes in vent fluid and in oxygenated seawater, which in the absence of bacteria was minimal. Inoculation of the vent fluid with microbes collected from hydrothermal vents resulted in a substantial, measurable increase in current, which Girguis noted, is continuous and sustained (over 6 months thus far). Pyrite in the system acts as a conductor and supports the establishment of a lush and diverse microbial community that is representative of what is seen in an active hydrothermal vent community freshly recovered from the sea floor (Schrenk et al., 2003; Takai et al., 2003) (Figure WO-30). In the absence of electrical continuity, the minimal community that forms on pyrite bears a greater resemblance to what is found on extinct sulfides (Sylvan et al., 2012). Girguis noted, “our literature [is] unfortunately populated with a lot of data that may not actually represent the conditions that the microbes

FIGURE WO-30 Electrical continuity yields diverse representative community.
SOURCE: Girguis (2012).
are seeing in situ” because many researchers have studied sulfides from the sea floor without electrical continuity to oxygen.
Metagenomics suggests that microbial communities formed in the presence of electrical continuity have the potential for sulfide oxidation, hydrogen oxidation, and carbon fixation. Experiments conducted in the presence of electrical continuity result in substantially more carbon fixation—which rectifies the discrepancies seen in previous biogeochemical measurements. Using shotgun metagenomics to interrogate communities recovered from the interior and exterior of sulfide deposits has led to the identification and isolation of iron oxidizers that are accepting electrons and using it to fix carbons. Essentially, Girguis noted, these microbes are sharing electron equivalence through a conductive matrix—a finding that has reshaped the way we think about the communities growing at these hydrothermal vents.
From a biogeochemical perspective, extracellular electron transfer enables microbes to access remote oxidants, stimulates primary productivity, and influences local alkalinity. Extracellular electron transfer also reshapes the notion of anaerobic metabolism, Girguis said. Although these organisms are anaerobes (and do not grow in the presence of oxygen) it is clear they are coupled to the availability of remote oxidants via extracellular electron transfer. Girguis and collaborators are currently focused on characterizing the phylogenetic and functional diversity of microbes living in hydrothermal vents around the world.
Community Ecology and Adaptation to the Environment—Use of Genomic Approaches to Study Evolution in Real Time
Comparative analyses of microbial genomes have demonstrated that the microbial genome is a dynamic entity shaped by multiple forces including gene loss/genome reduction, genome rearrangement, expansion of functional capabilities through gene duplication, and acquisition of functional capabilities through lateral or horizontal gene transfer (HGT) (Fraser-Liggett, 2005). Several natural processes carry genetic information from one species to another. DNA can be transported by viruses (transduction), via bacterial mating (conjugation), and through the direct uptake of DNA from the environment (transformation). Genes that must function together are transferred together as genomic islands (e.g., pathogenicity islands) (Hacker and Kaper, 2000).
While microorganisms might superficially appear the same according to 16S gene sequence profiles, specific SNPs in the genome and particular genes that are acquired by bacteria can contribute to fine-scale diversity and adaptation to environments. Horizontal gene transfer studies have identified DNA that has been moved across different organisms in the environment quite recently as well as more than a million years ago. In his prepared remarks, Eric Alm of the Broad Institute described two microbial ecology projects that study how bacteria adapt to their environment in real time.
Global network of horizontal gene transfer The first project Alm described involved the horizontal transfer of nearly identical genes from one species of bacteria into an entirely different species. Alm was inspired by a study describing the transfer of a gene for an enzyme that breaks down algal polysaccharides. The gene appears to have transferred from the algae itself, to marine bacteria, to the gut microbiota of Japanese individuals (potentially through the consumption of an edible seaweed) (Hehemann et al., 2010). In each case, Alm noted, the acquisition of this gene provided a selective advantage to the recipient host.
The Alm laboratory searched GenBank and found nearly 11,000 different proteins for which the genes were close to 100 percent identical in two totally different bacterial species (suggesting recent transfer of the DNA). Even more striking, Alm said, was that despite screening equal amounts of environmental and human-associated bacteria, the vast majority of pairs of bacteria that shared a nearly identical gene were isolated from sites on the human body. The highest rates of transfer were between bacteria isolated from the same body part (albeit on two different individuals), that is, ecologically similar but physically separated sites (Figure WO-31). Horizontally transferred genes for antibiotic resistance are a particular concern, and there is evidence that transfer does occur between humans, the foods they consume, and the livestock they raise (Smillie et al., 2011).
Sympatric speciation Alm explained that there are two conflicting, empirically based observations for how sympatric speciation occurs.40 The “ecotype” model hypothesizes that within a particular ecological niche, one bacteria will acquire a selective advantage and out compete everything else within, but not outside, that niche. Over time this type of clonal expansion leads to the tree structure that is commonly observed when looking at bacterial diversity (Gevers et al., 2005; Lipsitch et al., 2009). The “gene-centric” model states that there are environment-specific genes that are independent of species (Coleman and Chisholm, 2010).
It had been assumed that sympatric speciation was only theoretically possible when the number of adaptive loci was relatively small (Kondrashov and Mina, 1986). Alm pointed to ecological differentiation in animals to illustrate this point. In parts of Western Africa there are two variants of the malaria vector, Anopheles gambiae, that coexist in the same place and at the same time, differing by only a very few select loci in the genome (White et al., 2010).
To study ecological differentiation in microbial communities, Alm sequenced Vibrionaceae strains from coastal bacterioplankton together with Martin Polz at the Massachusetts Institute of Technology (Hunt et al., 2008) (Figure WO-32). They identified one clade of Vibrio that differentiated phylogenetically into 15 clusters that inhabited different parts of the water column. Upon sequencing chromosome 141 from 20 closely related strains in one particular cluster, it was
_______________________
40 Speciation in the absence of physical barriers to genetic exchange between incipient species.
41Vibrio have two circular chromosomes.
found that these strains partitioned further into two distinct, ecologically oriented groups. The sequence of chromosome 2, however, suggested significant recombination had occurred, which did not support the ecotype theory.
Alm concluded that most mutations across the genome supported an ecological tree, although there is a history of rampant recombination within and between
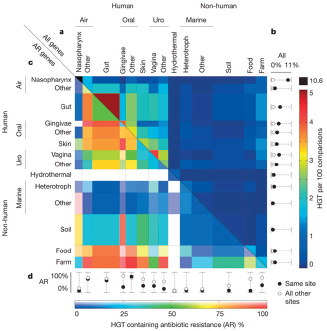
FIGURE WO-31 HGT is ecologically structured by functional class and at multiple spatial scales. The frequency of transfer between different environments is shown for all functional groups (Panels a, b) and for antibiotic resistance (AR) genes only (Panels c, d). Box widths indicate the number of genomes from each environment. Panel a, When all genes are considered (upper half), human isolates form a block of enrichment (upper left). Panel b, For every environment examined we observe more transfer within the same environment (black dots) than between environments (white dots). Panel c, The fraction of gene transfers that includes at least one AR gene for each environment. Statistical uncertainty in the proportion of AR transfer is indicated by decreased color saturation (see Methods). Panel d, AR genes comprise a significantly higher fraction of observed HGT between different environments (white dots) relative to that within the same environment (black dots) in contrast to Panel b.
NOTE: Uro, urogenital.
SOURCE: Smillie et al. (2011). Reprinted by permission from Macmillan Publishers Ltd: NATURE . Smillie, C. S. et al. 2011. Ecology drives a global network of gene exchange connecting the human microbiome. Nature 480(7376):241-244, copyright 2011.
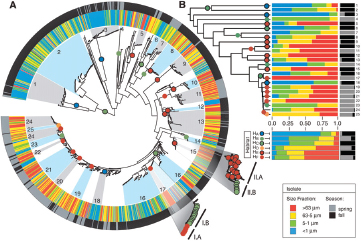
FIGURE WO-32 Season and size fraction distributions and habitat predictions mapped onto Vibrionaceae isolate phylogeny inferred by maximum likelihood analysis of partial hsp60 gene sequences. Projected habitats are identified by colored circles at the parent nodes. Panel A, Phylogenetic tree of all strains, with outer and inner rings indicating seasons and size fractions of strain origin, respectively. Ecological populations predicted by the model are indicated by alternating blue and gray shading of clusters if they pass an empirical confidence threshold of 99.99 percent (see supporting online material for details). Panel B, Ultrametric tree summarizing habitat-associated populations identified by the model and the distribution of each population among seasons and size fractions. The habitat legend matches the colored circles in (A) and (B) with the habitat distribution over seasons and size fractions inferred by the model. Distributions are normalized by the total number of counts in each environmental category to reduce the effects of uneven sampling. The insets at the lower right of (A) show two nested clusters (I.A and I.B and II.A and II.B) for which recent ecological differentiation is inferred, including habitat predictions at each node. The closest named species to numbered groups are as follows: G1, V. calviensis; G2, Enterovibrio norvegicus; G3, V. ordalii; G4, V. rumoiensis; G5, V. alginolyticus; G6, V. aestuarianus; G7, V. fischeri/logei; G8, V. fischeri; G9, V. superstes; G10, V. penaeicida; G11 to G25, V. splendidus.
SOURCE: Hunt et al. (2008). From Hunt, D. E., L. A. David, D. Gevers, S. P. Preheim, E. J. Alm, and M. F. Polz. 2008. Resource partitioning and sympatric differentiation among closely related bacterioplankton. Science 320(5879):1081-1085. Reprinted with permission from AAAS.
ecological groups, and just a few highly divergent, habitat-specific loci drive the genome-wide ecological signal (as originally predicted by Kondrashov in 1986). Alm also found evidence for the recent emergence of barriers to homologous recombination between habitats (i.e., bacteria tend to recombine with others from the same environment).
Based on this work, Alm and colleagues proposed a new model for sympatric speciation in bacteria (see Figure WO-33) that reconciles the conflicting ecotype and gene-centric models described earlier (Shapiro et al., 2012). The new model begins with an ancestral population in which there was significant homologous recombination. At some point a new niche arose and a small number of alleles conferred fitness to one niche and not the other. Those alleles then selectively swept through each of those smaller populations.
If one were to sample a random housekeeping gene that is not one of these very few loci that contribute to fitness, there would be no clear ecological patterns in the phylogenetic tree (sample t1 in the figure). Over long periods of time, if these gene pools remain separate, then there will be habitat-specific clusters that emerge (sample t2 in the figure).
Cataloging and Characterizing the Earth’s Microbiome
The Earth Microbiome Project seeks to provide a systematic characterization of microbial life on Earth, cataloging and comparing the microbial diversity across all Earth environments (e.g., air, soil, water, humans). Because the majority of Earth’s microbes are at present not readily cultivable, the Earth Microbiome Project takes a metagenomic approach to tap into what speaker Jack Gilbert of the Argonne National Laboratory called the “great dark biosphere,” extracting and sequencing total DNA from environmental samples (Figure WO-34) (Dr. Gilbert’s contribution to the workshop summary report may be found in Appendix A, pages 166-188).
The Earth Microbiome Project involves more than 120 collaborators across more than 50 institutions in 25 countries. Gilbert noted that unlike the Human Microbiome Project, the Earth Microbiome Project is not very well funded at the moment. Beyond funding, one of the biggest technical challenges is collecting samples from around the globe. Obtaining samples from the Peruvian Amazon, the bottom of the Marianas Trench, the air above Colorado, and from humans across a vast swath of Africa requires a network of collaborators who have access to those samples. More than 50 researchers have pledged more than 60,000 samples to the project. Samples are selected on the basis of their position in environmental gradients—those chemical, physical, and biological gradients
FIGURE WO-34 Metagenomic approach to studying the Earth’s microbiome. Total DNA is extracted from an environmental sample, in this case, a marine microbial community. Taxonomy and protein function are inferred from DNA sequences.
SOURCE: Gilbert (2012) from Gilbert and Dupont (2011). Republished with permission of Annual Reviews, from Microbial metagenomics: Beyond the genome, Gilbert, J.A and Dupont C.L., 3, 2011; permission conveyed through Copyright Clearance Center, Inc.
that define the niche space within which bacteria, archaea, microbial eukaryotes, and viruses exist. Rich contextualization of that environment at the time of sample collection is important, including, for example, nutrient concentrations, pH, temperature, host, time of collection, latitude and longitude, and any other environmental, contextual data that can provide clues as to why that community exists where it exists, and why it exhibits a particular functional phenotype.
Gilbert provided an overview of some of the analyses that have been done to date on approximately 15,000 samples. By cataloging microbes across specific environmental gradients (in extremes of temperature, extremes of nutrient availability, extremes of pressure and light availability, etc.) it is possible to systematically catalog that community in a much more robust fashion. Sequences from the Earth Microbiome Project map to 82 percent of the Greengenes tree of life (compared to the Human Microbiome Project, which has thus far cataloged 17 percent of the tree). Gilbert emphasized that this is just what can be matched against what is already known from surveys of 16S rRNA genes. This known diversity accounts for only 5 or 6 percent of the total diversity cataloged in the Earth Microbiome Project. In the first 10,000 samples analyzed, ~600,000 bacterial OTUs, or putative bacterial species, were identified.
Gilbert observed that the environment with the most diverse microbial community composition sampled thus far is oil spill sediments in the Gulf of Mexico, followed by stream water, and soil. Interestingly, the insect microbiome (bacteria in ant and termite intestines) was the least diverse environment, but contained the greatest number of new species identified. If we want to catalog more new bacterial species, Gilbert said, we should be looking to those particular environments with the most novelty, as opposed to environments that are already the subject of systematic surveys such as ice cores, marine water, and human environments.
Genomic environmental monitoring The Genomic Observatories network was described by Gilbert as “a network of sites working to generate genomic observations that are well-contextualized and compliant with global data standards.” Observatories will participate in long-term monitoring of environments across the globe, applying genomic technologies to monitor microbial, vertebrate, invertebrate, and eukaryotic diversity and interactions.
Gilbert reviewed one project exploring the seasonal microbial community structure in the English Channel. Researchers conducted microbial 16S rRNA gene surveys, followed by shotgun metagenomic and shotgun metatranscriptomic surveys to understand function, every month for 6 years—a total of 72 time points. Gilbert then developed a model to predict community structure from environmental parameter metadata (e.g., temperature, nitrate concentration, pH), predict functional metabolism from that community structure, and to then predict how that functional metabolism influences environmental parameters. The resulting positive feedback loop can be used for microbial forecasting (Larsen et al., 2012). The model takes into account biological interactions; if one particular
organism increases its relative abundance, then it will also have an impact upon the relative abundance of another organism. Gilbert described how he was able to extrapolate from the information gleaned from the 6-year survey of one location in the English Channel to predict the relative abundance of more than 100,000 different bacterial species at any given time and location in this environment. This extrapolated predictive capability may then be used to refine the sampling strategy.
The next step in these studies is what Gilbert referred to as “predictive relative metabolic turnover,” in which functional dynamics of the predicted community are inferred from the metagenome and metatranscriptome information—for example, how that community responds to environmental change (Larsen et al., 2011). From the predicted functional ability of a community, we can predict its metabolic capacity. The ultimate goal is to study how changes in metabolism feed back and influence the environment within which the organisms are found.
Looking at CO2 turnover within the English Channel environment over a 24-hour period, for example, Gilbert and colleagues saw an impressive 94 percent correlation between their ability to predict CO2 generation or consumption as a semi-quantitative measure, and the quantitated, analyzed, and observed flux of CO2 from the channel surface as reflected in the UNESCO Surface Ocean CO2 atlas. These observations are depicted in Figure WO-35.
Other microbiome projects Gilbert sampled different environments on a couple (e.g., palm, heel, inside of nose) and their home including the kitchen counter, floor, and light switch; bedroom floor; front door knob; and bathroom door knob. These data are presented in Figure WO-36.
Gilbert went on to discuss several environment-specific microbiome projects under way, including the Home Microbiome Study that seeks to understand how humans interact with the microbes on their skin and the microbes in their home. We live in our spaces, but those spaces are also living, Gilbert said. When a person moves into a new house, does the person adopt the microbiome that already exists in the house, or does the house adopt the microbiome of its new inhabitant? Or perhaps there is a new state where the microbiomes of both house and inhabitant are modified?
He found that the microbiomes of the feet of both the man and the woman were very heavily dominated by Staphylococcaceae. When they moved in, the solid oak bedroom floor was dominated with Mycoplasmataceae; however, after 6 days the floor became repopulated with Staphylococcaceae. While bedroom floors reflect their inhabitants, kitchen countertops in general look very similar across houses because they are constantly in a state of dynamic flux due to frequent cleaning. Gilbert added that people living together tend to have similar microbiota.
The final project that Gilbert discussed was the Hospital Microbiome Project that is just getting under way. This study is taking advantage of a new hospital
pavilion being built at the University of Chicago to try to understand what happens to the microbiome of the hospital when the humans move in. Samples will be collected before the hospital opens in early 2013, and then every day for a year across patient rooms, nursing stations, hospital corridors, hallways, and people, including patients and staff. The goal is to better understand the dynamics of microbial populations over space and time and how these populations may lead to nosocomial infections.
PRACTICAL APPLICATIONS OF GENOMIC TECHNOLOGIES
Applications of microbial genomics, such as those discussed throughout the workshop, have expanded researchers’ appreciation for the biology of microorganisms including their organismal, metabolic, and environmental diversity; the structure of microbial populations over space and time; the evolution of microbial species; and the acquisition of novel virulence factors and pathogenicity islands. These areas of inquiry are providing important insights into the “ground rules” of
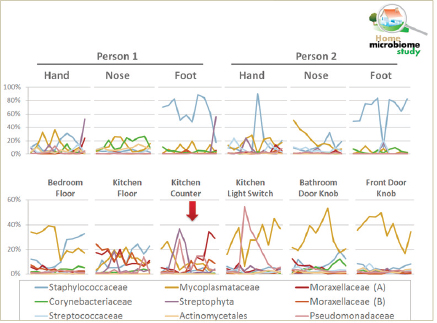
FIGURE WO-36 We change the microbial community of a house.
SOURCE: Gilbert (2012).
pathogen evolution and will inform the development of a “versatile platform for developing new responses to infectious disease” (Lederberg, 2000).
Tools for Microbial Detection, Surveillance, and Response
Although still very much a research enterprise at this stage, microbial genomics has great potential for practical application. As illustrated in Figure WO-37, microbial genomics is becoming an increasingly important tool for a wide range of applications. Relman has previously noted that, “[d]ifferences in the sequence and structure of genomes from members of a microbial population reflect the composite effects of mutation, recombination, and selection. With the increasing availability of genome sequences, these effects have become better characterized and more effectively exploited in order to understand the history and evolution of microbes and viruses and their occasionally intimate relationships with humans.
The resulting insights have practical importance for epidemiologic investigations, forensics, diagnostics, and vaccine development” (Relman, 2011).
A genome sequence facilitates the development of a variety of tools and approaches for understanding, manipulating, and mitigating the overall effect of a microbe. The sequence provides insight into the population structure and
FIGURE WO-37 Microbial genomics and tool development.
SOURCE: Relman (2011). From New England Journal of Medicine, David A. Relman, Microbial Genomics and Infectious Diseases, 365(4):347-357. Copyright ©2011 Massachusetts Medical Society. Reprinted with permission from Massachusetts Medical Society.
evolutionary history of a microbe for epidemiologic investigation, information that could be used to develop new diagnostic tests and cultivation methods, new targets of drug development, and antigens for vaccine development (Relman, 2011).
Microbial Genomics as a Frontline Public Health Tool
Understanding how a pathogen spreads from person to person or through populations is essential to developing an effective public health response. While we understand a great deal about the means of transmission for some pathogens—measles and influenza viruses are spread on droplets, HIV is a blood-borne disease, and West Nile virus is a vector-borne viral disease—public health epidemiologists do not yet fully understand how pathogens spread from person to person, from community to community, over space and time, and how underlying social network structures shape the movement of pathogens, said speaker Jennifer Gardy of the British Columbia Centre for Disease Control, Vancouver, Canada (Dr. Gardy’s contribution to the workshop summary report may be found in Appendix A, pages 141-150).
Field epidemiology, or traditional “shoe leather” epidemiology, involves interviewing people—the “host”—and looking for commonalities in social contacts, attendance at particular locations, contact with foods, or other behaviors that might explain the appearance and spread of disease. Molecular epidemiology, by contrast, essentially interviews the pathogens, Gardy said, through serotyping and genetic fingerprinting, including the use of restriction fragment length polymorphism analysis and multilocus sequence typing.
Tuberculosis: Whole genome sequencing vs. genetic fingerprinting One of the problems with the current standard for outbreak investigations, which uses a combination of field epidemiology and genetic fingerprinting, is that different molecular epidemiology techniques have different resolutions, which influence the perception of an outbreak.
Recalling investigations of tuberculosis (TB) outbreaks in British Columbia, Gardy noted that when the provincial public health laboratory switched from using 12-loci MIRU-VNTR (a fingerprinting technique used for Mycobacterium tuberculosis) to 24-loci MIRU-VNTR (a higher resolution tool), some clusters of cases that appeared at first to be related were actually unrelated. Such differences influence the perceptions of how cases might be related to each other and how an outbreak is reconstructed, Gardy said. Another issue with genetic fingerprinting methods is that the order of transmission is not always easy to infer (i.e., who infected whom and when).
Even using the best genetic fingerprinting techniques, only a very small fraction of the genome is being sampled. For the average bacterium, noted Gardy, MLST examines short fragments of seven housekeeping genes, essentially 0.03 percent of the available DNA. She continued, “that means you are ignoring 99.97 percent of its genome that contains informative and interesting variation that might give you a high-resolution view of an outbreak and that might actually be able to tell you about the order of transmission.” The whole genome sequence has often been referred to in molecular epidemiology as “the ultimate genotype.” With next-generation genome sequencing technology, molecular epidemiologists may be able to sequence numerous isolates from an outbreak quickly and cheaply.
An emerging discipline of “genomic epidemiology” is employing whole genome sequences from outbreak isolates to track person-to-person spread of an infectious disease. Even in the space of a few days or a week, pathogens are measurably evolving and accruing mutations, and there can be enough informative variation (i.e., SNPs) to be able to distinguish isolates from each other. Whole genome sequencing, combined with information about the social or other relationships between cases, can facilitate “visualizing” the actual transmission events.
Gardy noted that this approach has been used twice so far in British Columbia for TB outbreaks (Gardy et al., 2011). In one example, 24-loci molecular fingerprinting suggested that all of the outbreak isolates were identical to each other. The social contact network for the extremely close-knit community hinted
at the source case of the outbreak; however, after the first two cases or so, there were too many possibilities for transmission pathways for any one person’s infection. Even though all the isolates had the same mycobacterial interspersed repetitive unit-variable number tandem repeat (MIRU-VNTR) fingerprint, there was enough variation observed in whole genome sequencing to divide the isolates into two distinct phylogenetic clades, making that social network more amenable to interpretation (Figure WO-38).
The second TB outbreak discussed by Gardy was a location-based outbreak. The affected individuals did not necessarily have social connections to each other, but they all shared attendance at common locations—homeless shelters in the interior region of British Columbia. In addition, many of the strains isolated were resistant to low levels of isoniazid—the frontline antibiotic for the treatment of tuberculosis.
Whole genome sequencing suggested an early wave in the Vancouver area, including an individual in whom it is hypothesized that the resistance mutation arose, followed by the first wave of transmission associated with one of the shelters, then a second wave of infection at the second shelter that was likely seeded by one individual showing one very characteristic mutation. Gardy noted that the uses of microbial genomics described in her presentation are largely confined to the research environment, and that there is a long way to go before such an approach becomes a clinically validated technique used in reference laboratories. Gardy emphasized that in order to move forward with whole genome sequencing as a molecular epidemiology tool, it is important to remember that genomic data must be interpreted in the context of epidemiological and clinical data. It is not possible to reconstruct an outbreak from genome sequence alone. Gardy concluded that public health has the registries and the clinical expertise to complement the genome sequencing research, and collaboration and data sharing is key to maximizing the value of genomic data sets.
Microbial Diagnostics and Genomic Epidemiology
Speaker Mark Pallen of the University of Birmingham (UK) assessed the state of diagnostic microbiology by saying “We are now in the 21st century, but most of the time we are relying on 19th-century techniques” (Dr. Pallen’s contribution to the workshop summary report may be found in Appendix A, pages 238-256). As illustrated by two case studies of genomic epidemiology of Gram-negative pathogens, Acinetobacter and E. coli, genomics provides a way to do things a bit differently and perhaps better.
Acinetobacter: Defining species, transmission, and resistance Pallen first described how he used whole genome sequencing of Acinetobacter baumannii isolates to detect differences between isolates within an outbreak and to determine chains of transmission. A. baumannii is a Gram-negative bacillus associated with
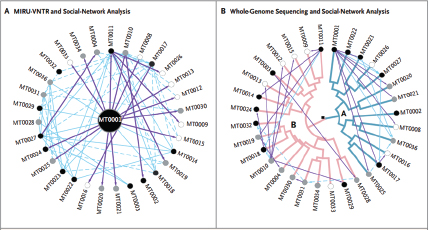
FIGURE WO-38 Putative transmission on networks constructed from genotyping data versus whole genome data for 32 patients. Genotyping data from analyses of mycobacterial interspersed repetitive unit-variable number tandem repeats (MIRU-VNTRs) were used in Panel A, and whole genome data were used in Panel B. Each panel shows patients (identified by case number) represented by circles colored according to smear status and clinical presentation as an index of infectivity: black circles indicate smear-positive pulmonary disease, gray circles smear-positive miliary disease or smear-negative pulmonary disease, and white circles smear-negative extrapulmonary disease. The cases are connected by arrows on the basis of reported social relationships representing plausible transmission attributable to a single case (purple arrows) or multiple potential sources of transmission (light blue lines), with dashed arrows indicating moderately infective patients and solid lines highly infective patients. In Panel A, the case with the earliest symptom onset was MT0001 (center), and the second-earliest case is shown at the 12 o’clock position, with the remaining cases listed in the clockwise direction in order of increasing time since symptom onset. When a clonal outbreak is assumed, the epidemiologic interpretation of the data suggests that most transmission events can be traced to the source case, MT0001. In Panel B, the cases are shown according to circular dendrograms based on whole genome data reflecting the tuberculosis lineage (A in blue and B in pink). This network provides a more accurate picture of transmission, with transmission restricted to each lineage, facilitating epidemiological interpretation of the underlying social-network data and revealing the role of the second and third source cases (MT0010 and MT0011).
SOURCE: Gardy et al. (2011). From New England Journal of Medicine, Jennifer L. Gardy, et al., Whole-Genome Sequencing and Social-Network Analysis of a Tuberculosis Outbreak, 364, 730-739. Copyright ©2011 Massachusetts Medical Society. Reprinted with permission from Massachusetts Medical Society.
wound infections, ventilator-associated pneumonia, and bacteremia. Isolates are generally multi-drug resistant, with colistin and tigecycline the only reserve antibiotic agents in many cases. A. baumannii has been isolated from military personnel returning from Iraq and Afghanistan, and there have been cases of transmission from military to civilian patients in shared health care facilities.
Isolates from an outbreak in Birmingham Hospital, in 2008, were classified as indistinguishable by standard typing methods. Using whole genome sequencing of six isolates, Pallen’s team was able to identify three loci that were SNP variations between isolates (Lewis et al., 2010). Pallen then developed and validated a genotyping scheme based on those SNPs and was able to map different aspects of the outbreak over space and time.
In collaboration with colleagues in London, Pallen found significant differences between A. baumannii isolates collected from a patient before and after tigecycline therapy. Prior to therapy the strain was drug-sensitive; following treatment it became drug-resistant. Whole genome sequencing of the post-treatment isolate detected 18 SNPs that were absent from the sequence of the pre-treatment isolate. Nine of the SNPs were non-synonymous (i.e., the change resulted in translation to a different amino acid), and one of them was actually in a gene called adeS, which is known to be part of a pair of genes that encode a two-component regulatory system that is involved in the resistance in this particular pathogen to tigecycline (Hornsey at al., 2011). In addition, there were three contigs in the pre-treatment isolate that were not found in the post-treatment isolate. It is often assumed that developing resistance involves acquisition of DNA. In this case, moving toward resistance was actually associated with a loss of DNA. One deletion resulted in a truncated mutS gene that is involved in DNA repair. Pallen hypothesized that loss of DNA repair led to an increase in mutation rate and primed the isolate to then acquire antibiotic resistance.
Based on phylogenetic studies, Pallen speculated that, for Acinetobacter species, it should be possible to define species by genome sequence alone, without the need for any phenotypic testing or any other screening methods like DNA-DNA hybridization to define a species. 16S sequences, which are commonly used as a taxonomic marker, were not capable of delineating the accepted species within the Acinetobacter genus. A core genome phylogenetic tree, however, was consistent with the currently accepted taxonomy and also identified three misclassifications within strains and strain collections. Pallen noted that phylogenetics can be very processor- and time-intensive. However, the use of the “average nucleotide identity” approach quickly delivered results that were consistent with the traditional and phylogenetic classifications (Hornsey et al., 2011).
E. coli: Crowdsourcing the genome To illustrate the power of making data publicly available, Pallen described how the use of social media such as blogging and Twitter can augment the usual channels of scientific discovery and academic discourse. An outbreak of more than 4,000 cases of E. coli O104:H4 in Germany
in the summer of 2011 led to more than 50 deaths. This E. coli outbreak was characterized by a very high risk of hemolytic uremic syndrome (a complication of Shiga toxin–producing E. coli) and was ultimately linked to the consumption of sprouted fenugreek seeds.
Pallen’s collaborators in Hamburg sent DNA from an outbreak isolate to BGI42 for sequencing; BGI subsequently released the sequence data from five sequencing runs on the Ion Torrent platform into the public domain. Within 24 hours of release of the data, Nick Loman in Pallen’s research group had assembled the genome and, through his blog post and Twitter account, called upon the bioinformatics community to analyze these data. Within 2 days, the genome had been assigned to an existing lineage of E. coli; within 5 days, additional analyses had given rise to a strain-specific diagnostic test. Within a week there were more than two dozen reports filed on the biology and evolution of this strain on an open-source wiki. Pallen underscored that contributors from around the world43 to this example of open-source genomics were “not professional public health people, for the most part … they were people just doing this out of interest and good will … and because there was a clear and present need for it.”
Tweets and blog posts, however, are no substitute for a peer-reviewed publication, noted Pallen. In collaboration with his German and Chinese collaborators, Pallen and his colleagues wrote up a case study of a family outbreak of O104:H4, which included a description of community research efforts that were coordinated via social media as well as confirmation of all reported analyses (Rohde et al., 2011). Pallen noted that this was just one example of a crowdsourced research project (e.g., www.crowdsourcing.org or www.ancientlives.org). Although everyday science is not likely to open its laboratory notebooks and share everything all the time, this approach could be useful and appropriate in times of a public health emergency.
This collaborative approach raises the question of what constitutes “published” for the purposes of avoiding duplicate publication of original material (the Ingelfinger rule). All of the sequence data and analysis had already been placed into the public domain (through Twitter and blogs) prior to submission of these research results to the New England Journal of Medicine, yet the manuscript was accepted for publication (Rohde et al., 2011).
_______________________
42 BGI (formerly known as the Beijing Genomics Institute) is a premier genomics and bioinformatics center with facilities around the world.
43 “If you just look at the names … you can see the variety of input…. You have English names, Spanish names, Chinese names, Muslim names, Jewish names…. People from around the world, from multiple continents, were actually involved in this activity.”
Metagenomic Screening for Novel Viruses
David Wang of Washington University in St. Louis, focused his remarks on how the application of viral metagenomics to outbreaks and other settings may provide new directions for investigations (Dr. Wang’s contribution to the workshop summary report may be found in Appendix A, pages 311-338). Wang shared two case examples of the application of viral metagenomics: how the investigation of outbreaks can provide new directions for research on viruses in infectious disease, and how viruses may potentially be used as probes to elucidate fundamental mechanisms of innate immunity.
Application of genomics to diarrheal diseases Diarrheal diseases are a significant cause of morbidity and mortality worldwide. Nearly 2 million children die from diarrheal diseases annually, primarily children under the age of 5 in the developing world, who succumb to dehydration in the absence of medical interventions. In the developed world, fatalities from diarrheal diseases are almost nonexistent. Major viral causes of diarrhea include norovirus, rotavirus, adenovirus, and astrovirus. A number of other viruses have also been implicated in acute diarrhea; however, 40 percent of all cases of acute diarrhea have no known etiologic agents. According to Wang, viral metagenomics is a pivotal strategy for detecting potential pathogens in these unknown cases.
Wang described his efforts to identify novel viruses in stool samples from children with clearly diagnosed acute diarrhea that were negative in conventional assays for major known diarrheal viruses. Metagenomic screening provided evidence for many novel viruses, including a new astrovirus.44 Wang noted that previously, only one species of human astrovirus with eight closely related serotypes (10 percent amino acid variation between serotypes) had been defined. Wang recovered seven astrovirus sequence reads from one sample that mapped to two different loci in the genome (Finkbeiner et al., 2008a). The complete genome of the virus was painstakingly sequenced. Phylogenetic analysis revealed that the newly detected astrovirus was highly divergent—exhibiting less than 60 percent amino acid identity. This was clearly a distinct astrovirus, Wang concluded, not a new serotype of the previously described human astrovirus (Finkbeiner et al., 2008b).
There are many unanswered questions about the role of novel astroviruses in human disease; first and foremost, are they in fact a causative agent of acute human diarrhea, or do they cause disease outside the gastrointestinal tract and are simply shed or transmitted by a fecal–oral route? Could this astrovirus be a
_______________________
44 Astroviruses are small, single-stranded, positive sense RNA viruses, 6 to 8 kb in length. They cause diarrhea in humans and other animals. When Wang’s group did this study, there was essentially one species of human astrovirus that had been previously defined. The first member of the species was discovered in 1975. Subsequently, eight closely related viral serotypes have been described. At the amino acid sequence level, they vary by about 10 percent from each other.
commensal or a symbiotic part of the human virome? Or was the virus simply ingested by the child and passaged through the gut, having nothing to with human infection? Wang shared preliminary data that suggest that 100 percent of the normal healthy general population is seropositive for this astrovirus (i.e., has been infected at some point in time). Wang noted that viral metagenomic strategies can be used to identify candidate agents but that these leads must undergo a very long process of classic biological workup in order to understand “what the relevance is, what the role is, what this virus is doing—either in a particular set of candidate diseases or even in other diseases that we haven’t thought about yet.”
Wang described the application of next-generation sequencing to the investigation of an outbreak of acute gastroenteritis in a daycare center. Twenty-six children and teachers became ill over the course of several weeks, and the center was closed. Conventional testing was negative for all known enteric pathogens. Wang sequenced six fecal samples that were available and found another novel astrovirus (see Figure WO-39). He pointed out that while sequencing the entire genome of the novel astrovirus described above took several months, by using next-generation technologies this genome was sequenced in 10 hours. Real-time PCR demonstrated that this novel astrovirus—AstV-VA1—was present in high titer in three out of the six samples available from this outbreak. Wang reiterated that it is not known if this is, in fact, the causal agent of this outbreak, but it certainly generates new hypotheses.
An evaluation of the global diversity of viruses will provide researchers with important genomic libraries for future studies of novel viruses associated with disease outbreaks. Wang and colleagues are now evaluating more than 1,000 stool samples collected from patients with diarrhea or from healthy children, shared by collaborators from around the world. Numerous new viruses have been found, including six additional astroviruses. They are also taking a shotgun metagenomics approach to sequencing untreated raw sewage collected from different sites, integrating environmental metagenomics with clinical metagenomics. Data thus far show incredible diversity in terms of viruses. Wang reported that “the number of viruses that we knew about historically, which were mostly guided by culture-based methods, vastly underestimates the amount of virus that is in any given specimen or niche.”
Using genomics to identify viruses that infect the nematode, Caenorhabditis elegans C. elegans is a genetically tractable model organism that has been used for more than 40 years in developmental biology and neuroscience research investigations. Many fundamental discoveries that were initially made in C. elegans have now been translated to mammals and humans. Current thinking is that C. elegans, as a primitive eukaryote with no known adaptive immunity and distinct innate immunity, may be a robust system to study host–virus interactions and identify novel antiviral immune pathways. The challenge is that no virus has ever been described that naturally infects C. elegans.
FIGURE WO-39 Phylogenetic analysis of novel astrovirus VA145 identified in an outbreak of gastroenteritis. stV-VA1 present in 3 out of 6 samples in outbreak.
SOURCE: Finkbeiner et al. (2008b).
Much of the work in C. elegans has been conducted using a reference laboratory strain that is not exposed to any natural pathogens.To identify natural viral pathogens of C. elegans, Wang sampled worms in collaboration with Marie-Anne Félix, a nematode ecologist, who collected wild isolates of C. elegans and Caenorhabditis briggsae from rotting fruits obtained in French orchards. Félix identified several worm isolates for further investigation that appeared sick (i.e., having an unusual intestinal cell morphology) and did not respond to treatment with antibiotics. Multiple isolates appeared to be infected by viruses, and sequencing identified three novel viruses, one from a wild C. elegans strain and two from a wild C. briggsae strain (Félix et al., 2011). The three viruses are distinct but related to each other, and they are distantly related to nodaviruses (i.e., positive sense RNA viruses that are significant pathogens of fish and also infect insects). The laboratory C. elegans strain could also be infected with virus isolated from the wild nematodes. The naïve worms developed the disease phenotype, fulfilling Koch’s postulates.
Viral infection of C. elegans strains that had a defective RNAi pathway—a well-established antiviral mechanism in plants and insects—resulted in 50- to 100-fold greater accumulation of viral RNA in the mutant strains than in the wild-type C. elegans strain. Immunofluorescence assays using antibodies against
_______________________
45 Astrovirus VA1 is most closely related to ovine astrovirus.
a viral polymerase showed that the virus primarily infects cells of the nematode intestine.
These are the first data to clearly demonstrate in nematodes that RNAi is antiviral using a bona fide virus infection, Wang explained. More importantly, this demonstrates the feasibility of using the nematode as a model animal system for probing host–virus interactions and for capitalizing on the genetics that have already been worked out in the nematode system as a tool to identify novel modalities of antiviral immunity. Specifically, one can take mutants with defined deletions in genes and obtain phenotypes that are quantifiable as increased viral replication (Félix et al., 2011). Wang emphasized that viral metagenomics provide a useful starting point for biological investigations and that numerous biological questions can now be explored due to this revolution in genomic sequencing technology.
Assessing Abundance: The Rare Biosphere vs. Sequencing Errors
There are many similarities between bacterial communities across samples and environments. Speaker Susan Huse of the Marine Biological Laboratory in Woods Hole46 said that profiles of microbial communities generated by sequencing of 16S rRNA hypervariable regions consistently have a small number of highly abundant organisms and a large number of low-abundance organisms (the “rare47 biosphere”) (Dr. Huse’s contribution to the workshop summary report may be found in Appendix A, pages 188-207). A persistent question is how to determine what is truly rare in a complex mixture and what is sequencing error. For some projects, researchers address this question by simply eliminating all of the low-abundance organisms from the analysis. This approach is obviously not appropriate when studying diversity.
Huse described analysis of the relative abundance of the microbiota in samples from the Human Microbiome Project (Huse et al., 2012). Sequencing 16S rRNA hypervariable regions of 210 stool samples, for example, shows that distribution of abundance varies from patient to patient. Organisms that are present in all patients may be relatively rare in some and highly abundant in others. Bacteroides, for example, is the most abundant genus in the stool samples sequenced. Some patient samples contained almost no Bacteroides, while others contained nearly 100 percent Bacteroides. Huse noted that these results “speak to the importance of not throwing away those rare organisms in these patients, because it’s part of a pattern with other patients, where it is more abundant.”
A graph of the relative abundance of sequences for a variety of different stool samples shows how widely the observed rare biosphere can vary (Figure WO-40). This does not mean that everything that is rare is true, but rather that everything
_______________________
46 Dr. Huse has since relocated to Brown University.
47 For the purposes of this talk, Huse defined rare as less than 0.1 percent.
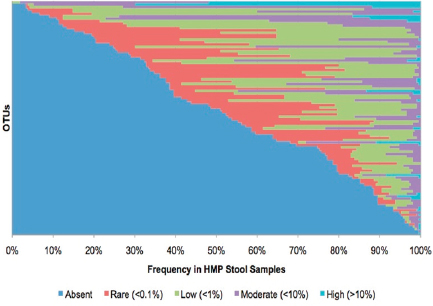
FIGURE WO-40 Distribution of OTU relative abundances across 210 Human Microbiome Project stool samples.
SOURCE: Huse (2012). Data from VandeWalle et al. (2012).
that is abundant can be rare somewhere else. Rather than throwing these data away, Huse noted that researchers need better ways of finding singletons or rare organisms and seeing them in a context of other samples where they are not so rare.
As Huse noted, there is no universal definition of “rare.” For microorganisms, rare is not one or two, but tens of thousands, and even millions of cells. Huse observed that one estimate is that there are 1×108 bacterial cells in a milliliter of seawater. If “rare” is as low as 0.01 percent of a sample, then rare is about 10,000 cells in a milliliter of seawater. If the estimate is that there are 1×1010 bacterial cells in 1 gram of stool, then rare is 1 million cells in a 1 gram sample.
To be able to compare across samples and to identify truly rare organisms in a sample, researchers need to minimize errors and compensate for sequencing quality limitations. Huse suggested that before sequencing begins, a variety of primers should be designed with diverse affinity and minimal bias. If a primer has low affinity to some organisms, they may appear to be part of a rare biosphere, when in fact there was bias against them. The use of proofreading enzymes is important, as is catching PCR errors via sequencing replication, to reduce the impact of early rounding errors. Huse also cautioned that low microbial biomass
samples can be confounded by contaminants. The use of samples that have undergone whole genome amplification can also dramatically alter results.
Once obtained, data must be filtered based on the quality of the sequencing. Huse advised “when in doubt, leave it out,” especially when working with 16S rRNA amplicons. The focus is on sequence quality, not simply removing things that are low abundance—because that will bias against truly low-abundance organisms. Quality filtering of next-generation sequencing reads could include, for example, omitting reads with Ns (unspecified bases), with inexact matches to primers, with low-quality scores, or that do not meet a minimum length. Also useful are stringent chimeric filtering (to remove chimeras from PCR amplification and sequencing errors) and paired-end reads with overlap, in order to eliminate overlapping sequences that are not identical. The next step is to analyze the data at the taxonomic level, filter out anything that is not bacteria (e.g., archaeans, organelles), and aggregate similar sequences.
Because not all species have been named, another common approach is to aggregate the 16S sequences into groups of similar sequences, or OTUs. Huse noted, however, that results often vary by clustering method. Huse described a new clustering method involving a pre-clustering step that improves prediction of OTUs (Huse et al., 2010). Regardless of the platform used, and the quality filtering applied, there will be errant OTUs remaining, however rare the biosphere. Huse discussed several metrics to estimate diversity, including Shannon and Simpson diversity estimates for alpha diversity (within the community) and Jaccard, Bray-Curtis, and Morisita-Horn for beta diversity (across communities).
Huse explained that if a sequence is abundant elsewhere, or is present elsewhere, and follows an ecologically meaningful pattern, then it is highly likely that it is a true rare sequence and not an error. As an example, Huse described two Acinetobacter V6 ribosomal sequence tags that differed by a single nucleotide and demonstrated an ecological balance based on season (Figure WO-41). Researchers that only had samples from one time point might have thrown out the low-abundance taxa as an error and missed this potentially important pattern. Huse noted that her group always keeps rare sequences that cannot otherwise be identified as errors until all of the data can be analyzed for broad ecological patterns.
An Approach to Analyzing Metagenomic Data: Inferring Function with MG-RAST
As metagenomics enters into the era of “big data,” the research community needs to find ways to drop the costs of data analysis; integrate the terabytes of data being generated; and make these data comparable, said speaker Folker Meyer of the Argonne National Laboratory (Dr. Meyer’s contribution to the workshop summary report may be found in Appendix A, pages 230-238). Without doing so, these data will essentially be lost.
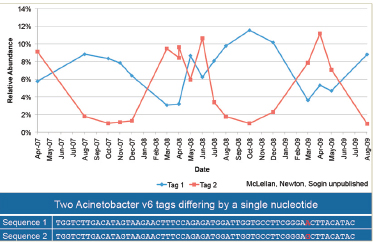
FIGURE WO-41 Two Acinetobacter V6 tags differing by a single nucleotide and demonstrating a seasonally balanced abundance.
SOURCE: Huse (2012). Data from VandeWalle et al. (2012).
Meyer illustrated the challenges of analyzing metagenomic data by reviewing his work with Metagenomics RAST (MG-RAST), an open-source, high-performance computing server for metagenomic sequence analysis (Meyer et al., 2008). Developed in 2007 as a simple tool that automated several procedures for data analysis, MG-RAST immediately attracted more than 100 user groups and 100 data sets. As of May 2012, MG-RAST included 49,600 data sets (metagenomes) submitted by users. Since its launch in 2007, more than 14 terabase pairs (or 1012 base pairs) of data and more than 120 billion sequences have been analyzed in the system. Meyer explained that user-submitted sequence data undergoes normalization and intensive quality analysis and are mapped against known annotations in numerous data sources (e.g., SEED [Overbeek et al., 2005], KEGG48 [Kanehisa, 2002], COG49 [Tatusov et al., 2003]) to predict features of interest (e.g., genes). The system further transforms these data into phylogenetic and functional profiles, which are useful for sample comparison.
MG-RAST requires the submission of metadata along with sequence data, and Meyer underscored the critical importance of metadata—where the data are from, what procedures were used in data generation, and how the data were recorded—to the interpretation of results. Meyer cited functional and taxonomic comparisons of 1,606 Human Microbiome Project shotgun metagenomes against
_______________________
48 Kyoto Encyclopedia of Genes and Genomes.
49 Clusters of orthologous groups of proteins.
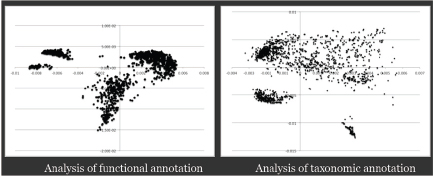
FIGURE WO-42 Principal coordinates analysis of 1,606 Human Microbiome Project shotgun metagenomes.
SOURCE: Meyer (2012).
known annotations that resulted in two different principal coordinates analysis (PCoA) outputs (Figure WO-42).
Outlier groups for both comparisons reflect the different sequencing technologies and platforms used for different samples (Figure WO-43). Meyer pointed out that this finding is only possible because there are metadata regarding the sequencing technology for the majority of the samples.
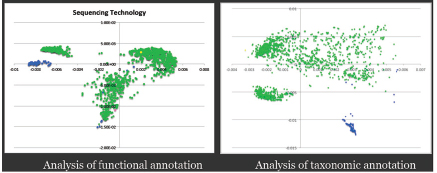
Quality control for de novo sequencing Data quality is tightly correlated to the ability to analyze and compare sequences, especially on automated platforms such as MG-RAST. Next-generation sequencing often produces “noisy” data. However, unlike the more mature field of phylogenetic surveys, which has developed “de-noising” approaches (discussed by Huse), the field of shotgun metagenomics does not have any “vendor-neutral” way to characterize data quality. Novel quality control approaches are needed to ensure the integrity of comparison between metagenomic datasets.
Meyer described a novel method of “duplicate read inferred sequencing error estimation” (DRISEE) that uses PCR artifacts in replicate reads to develop error profiles for shotgun metagenomic sequencing data sets (Keegan et al., 2012). He noted that when his group has used DRISEE, it has demonstrated that different experiments, and different samples in a single experiment, exhibit unique error profiles. He noted that this phenomenon was not related to the sequencing hardware but rather to operator error; some sequencing centers consistently produce high-quality data while others are more highly variable. Echoing Huse, Meyer noted that “errors are much, much higher in our estimate than what the vendors typically tell us they are.” He concluded that “we as a community need to find a way to look at errors, at error profiles, [so we know when to] discard data sets or redo experiments.”
Predicting features Meyer described two basic methods for predicting protein coding features from genome sequence data: (1) statistics-based approaches using codon usage and (2) similarity-based approaches using BLASTX searches. He noted that it is difficult to find novel proteins, genomic islands, and horizontally transferred genes using a statistics-based approach. Similarity-based approaches require substantial computational time, often weeks on multiple machines, and are correspondingly expensive (about 10 times the cost of generating the sequence data). In addition, novel proteins will not be identified through similarity searching. Again, novel computational approaches are needed.
Meyer and colleagues evaluated the reading frame prediction accuracy of five current gene computation algorithms—FragGeneScan, MetaGeneAnnotator, MetaGeneMark, Prodigal, and Orphelia. They found the tools were comparable in making predictions based on error-free sequence. When these tools were applied to simulated data sets containing errors, there were notable differences in performance (Trimble et al., 2012). Meyer concluded, “Even in the best of all cases, we only hit 60 percent of all possible gene features that we can see in our data.”
Annotation and reproducibility There are multiple annotation databases that can be used to find similarities between sequencing data and known proteins. As such, MG-RAST supports a number of different databases to annotate function. Despite identical processing of data, choosing a different database as your basis for comparison significantly changes results, raising questions of data comparability using a different pipeline.
To address a similar question about result reproducibility from the same setting, Meyer presented data suggesting that results obtained from shotgun metagenomics experiments were reproducible. He also referred to the work of Zhou who found that analyses of 16S rRNA amplicons were not reproducible (Zhou et al., 2011). Meyer suggested that this is because abundance was not taken into account.
Assembly Metagenome assembly should be relatively straightforward, but it is fraught with challenges regardless of the assembly tool used. Meyer noted that algorithms designed to assemble clonal genomes cannot handle the subspecies or strain variation present in metagenomic samples. Moreover, he found that varying the size of the overlap that the assembler is looking for (the k-mer size) changed the results dramatically. Using short k-mers resulted in many short contigs, while long k-mers led to a few long contigs. According to Meyer, this is a problem because most metagenomic studies “picked an arbitrary point in that [k-mer] space, declared it to be true, and submitted that data.” This suggests that “everything we have assembled and deposited in terms of metagenomics needs to be revisited, and we frequently do not have access to the raw reads.”
If assembly does work, and the data set is of high quality, metagenomic data can support new types of research. Analyses can produce genomes that serve as a reference strain for mapping all of the reads in a metagenome, allowing investigators to conduct population genomic and genetic studies on single metagenomes—all for less than $1,000 (Meyer et al., in preparation).
The analysis bottleneck Computing costs of sequence analysis far outsize those associated with generating metagenomic sequencing data (Figure WO-44). Although new analytical tools are emerging, new approaches to data sharing and storage, as well as to building and maintaining community resources, will be needed (Thomas et al., 2012). Meyer emphasized that although the community relies upon curated databases of existing knowledge, annotation and reference databases are not well funded. Currently metagenomic data are not centralized; most are stored in private resources (e.g., the RAST server at Argonne National Laboratory) rather than in public resources such as GenBank. There need to be easier ways for the community to deposit and openly access data deposited and curated in central locations.
Meyer suggested that the challenge is so substantial that “[the field] needs to change the way we do business.” In particular, Meyer called for standards that would support data integration and exchange. The community could also identify and make available “gold standard” data sets. Ideally, this would mean that data sets would only need to be analyzed once, and a common archive could be established to distribute the raw data and analytical results for further study (Desai et al., 2012).
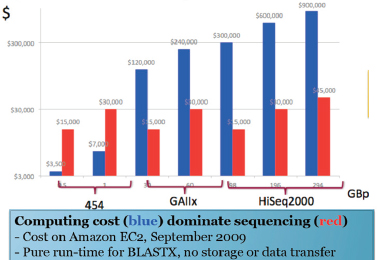
FIGURE WO-44 The costs associated with data analysis are becoming the bottleneck for metagenomic analyses.
SOURCE: Meyer (2012). Adapted from Wilkening et al. (2009).
Moving Forward: Challenges and Opportunities
While genomics has introduced a new era in microbiology with significant implications for improving our ability to detect, diagnose, treat, and even anticipate infectious disease emergence, there exist many challenges and caveats that must be addressed before these technologies will find widespread adoption and use beyond the research laboratory. Sequencing technologies that have supported this revolution continue to evolve, and while this evolution leads to ongoing advancement of the field, it also creates a moving target with new challenges. For Sanger sequencers, sequence production was the rate-limiting step. Next-generation technologies that became available beginning in 2005 are radically different from Sanger-based technology;50 they are “massively parallel” systems that produce “sequence information from hundreds of thousands to hundreds of millions of DNA molecules simultaneously” (Mardis, 2011). Each new platform has struck a different balance between cost, read length, data volume, and rate
_______________________
50 Although the biochemistry of each platform is different, next-generation sequencing technologies feature simpler sample preparation steps, dispense with the need to create libraries of cloned sequences in bacteria, and rely on parallel, cyclic interrogation of sequences of clonal amplicons of DNA. An orchestrated series of repeating steps are performed and detected automatically (Mardis, 2011).
of data generation, but all produce far more sequence reads per instrument run than capillary sequencers and at significantly lower cost (Figure WO-45) (Mardis, 2011). The massive influx of data from next-generation sequencing technologies, with shorter read lengths and different error profiles, has also brought significant challenges to their analysis (Mardis, 2011).
Data Quality, Comparability, and Analysis
The explosion of sequencing technologies and applications is generating more results than ever, but it is unclear what these data may mean. Workshop speakers and participants discussed some of the key challenges in this rapidly advancing field, and potential methods to address them, focusing on the core themes of data quality and analysis.
Comparability Across Sequencing Platforms and Technologies
Perhaps the most formidable challenge for investigators using different sequencing technology platforms is data comparability across methods and
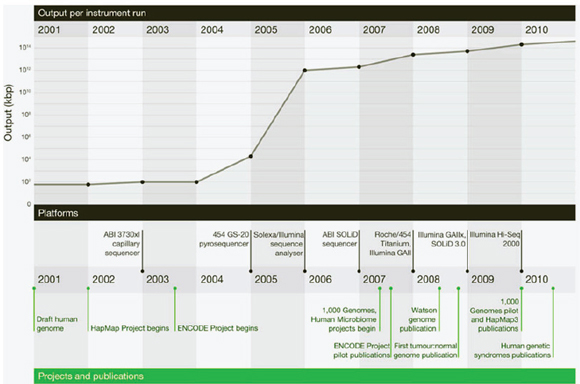
FIGURE WO-45 Changes in instrument capacity over the past decade, and the timing of major sequencing projects. Top panel, Increasing scale of data output per run plotted on a logarithmic scale. Middle panel, Timeline of major milestones in massively parallel sequencing platform introduction and instrument revisions. Bottom panel, timing of selected projects and milestones.
SOURCE: Mardis (2011). Reprinted by permission from Macmillan Publishers Ltd: NATURE. Mardis, E.R. 2011. A decade’s perspective on DNA sequencing technology. Nature 470:198-203, copyright 2011.
approaches. Each sequencing platform uses a unique set of protocols for template preparation, amplification and sequencing, imaging, and data analysis (genome alignment and assembly methods) that determine the type and quality of data produced, and such protocols may influence the strength of the “signal” versus the “noise” and overall accuracy of these data.51 Although each manufacturer provides quality scores and accuracy estimates, there is no consensus that a “quality base from one platform is equivalent to that from another platform” (Metzker, 2010). The cross-platform comparability of data also has implications for the reproducibility of research results.
As sequencers have become much smaller and less expensive, more and more laboratories are starting to perform their own sequencing. Benchtop sequencers are poised to revolutionize microbiology, but platforms do have strengths and weaknesses (see Loman et al., 2012a). In his presentation, Pallen described sequencing an isolate from the German E. coli O104:H4 outbreak on three different platforms and concluding that all three were “fit for the purpose,” although each had particular issues; both the Ion Torrent PGM and the 454 GS Junior produced homopolymer-associated indel errors (Loman et al., 2012b). Pallen emphasized it is not only the sequencer used, but also the analytical approaches, particularly the assemblers employed, that may contribute to divergent results. None of these sequencing platforms or assemblers gives perfect results.
Training Gilbert observed that in addition to concordance between platforms, there are also issues of concordance within a platform. The same sample run multiple times on the same platform can give disparate results depending upon the technician/operator, and data quality is often more dependent on the people doing the sequencing than on the platform itself.
In addition, the adoption of these technologies to enable clinical applications will also require the development of a “gold standard” approach to sequencing, including training of new staff, or training of staff on new equipment. Good training is essential in order to ensure comparable results. Most training takes place on the job, and Gilbert noted that a laboratory can lose two or three sequence runs because the genomic libraries were constructed inappropriately. These types of errors can reduce customer confidence and set the laboratory back in terms of time and money. One approach is to send laboratory staff to a major sequencing center for training, and Weinstock observed that laboratories that purchase next-generation sequencing instruments often send their core facility person to the Genome Institute at Washington University in St. Louis, Missouri to learn the protocols.
_______________________
51 Errors may include insertion and deletion errors; mismatches/substitutions; and underrepresentation of certain regions—such as AT- or GC-rich repeat regions that reduce the accuracy of sequence reads.
Communication The sharing of experiences by sequencing community members is also important for fostering high-quality results across platforms and laboratories. Pallen noted that when his laboratory first acquired the 454 sequencer, an experienced postdoctoral fellow who began to use it obtained poor results. Pallen’s first impulse was to blame the fellow. In discussing this problem with another laboratory, Pallen learned that the other laboratory was also obtaining poor results on a machine they had used for 18 months. Inasmuch as this machine was being operated by the same technicians, it soon became clear that, in fact, the source of the problem was “bad” reagents rather than operator error.
Many workshop participants agreed that there is a need for better communication among the platform user community. There is no central location to report and track problems with sequencing systems (along the lines of the Food and Drug Administration website for reporting adverse reactions to drugs). Users resort to informal communication media, such as Twitter, blogs, and community forums like SEQanswers to share and obtain information about sequencing issues. It was pointed out that manufacturers’ websites generally have a page where problems can be posted and seen by all users. While posting an issue on a manufacturer’s webpage garners a response from a representative to help solve the particular issue, it does not necessarily help to promote discussion within and across the community. Instead it is an approach to solving issues one laboratory at a time, for what may, in fact, be a community-wide problem.
Regulatory issues From a regulatory perspective, sequencing is primarily a research activity. A laboratory that performs sequencing that will be used in clinical medicine must first be approved under the Clinical Laboratory Improvement Amendments (CLIA). In addition, diagnostic kits must have approval from the FDA. In this regard, it is not clear whether sequencers used for diagnostics will be classified as Class III medical devices. Weinstock noted that the Genome Center at Washington University in St. Louis, Missouri, is currently seeking CLIA approval.
Data Quality: Careful Sample Preparation and Results Filtering
Much of data quality resides in careful planning before sequencing, including primer design and sample preparation, and quality filtering of raw sequence reads before assembly and analysis—approaches discussed earlier by Huse and Meyer. With regard to planning, participants noted that multiple samples are commonly assayed in a single sequencing run, and there exists the potential for data mix-ups due to bar code errors. Huse explained that if bar codes are within one base pair of one other, there can be sequencing errors that take data bioinformatically from one sample to another.
Weinstock suggested that for microbial genomics, the goal is to routinely be able to produce a perfect genome. The only sequences that are ambiguous in such a perfect genome are bases that are programmed to change; for example, in
the process of culturing the organism to harvest DNA for sequencing there will be a certain amount of variation (e.g., antigenic variation). Obtaining the perfect genome requires the use of multiple sequencing platforms for data comparability.
Weinstock emphasized data quality over quantity. One approach is to prepare sequence read pairs that overlap in the variable region of the 16S rRNA and to only use sequences where there is complete agreement over the entire length. This means that a lot of sequence data are discarded, but what remains is unlikely to have errors. Speaker Huse noted that if one is not careful with quality filtering, and only relies on a perfect overlap for example, then it is possible to end up with a range of perfect, but incorrect reads. Pallen emphasized the importance of human involvement and the expertise of the microbiologist looking at the sequence data to make observations, find errors, and draw conclusions. Participants also discussed the importance of training in this regard as well as the performance of the sequencing platforms.
Gilbert noted the situation is somewhat different for high-throughput metagenomic community analysis. The primary interest in doing a 16S rRNA survey is screening the samples to search for emergent properties in the ecological patterns that exist between samples. If there are patterns of interest, then the system can be characterized using more rigorous techniques. Weinstock concurred that 16S rRNA sequencing is mainly to compare organisms, and there is a tolerance for errors because it is possible to get a database hit without necessarily having a perfect sequence (as long as errors are consistent and do not introduce biases).
Sample collection and preparation Throughout the workshop presentations, the need for improvement in sample collection and preparation was repeatedly discussed and highlighted. Weinstock noted that as the capacity for sequencing increases, the challenge for laboratories is how to handle much larger numbers of samples in order to take advantage of the available sequencing capacity.
Another challenge is the volume of sample needed, and participants discussed how it would be helpful to have sequencing platforms that require much lower amounts of nucleic acid input. It was noted that there are new library preparation instruments in development that use nanochannel arrays. Reducing the amount of nucleic acid needed for sequencing would allow researchers to take advantage of sequencing technologies for samples with very small amounts of material (Woyke et al., 2010). One example of a nanopore sequencing technology under development is a portable, disposable, single molecule analysis device (Figure WO-46) (Pennisi, 2012).
Improvements in sampling and the recovery of genomes from environmental samples will be particularly relevant to the analysis of samples from infectious disease outbreaks. The rapid sequencing of samples from the deadly epidemics of cholera in Haiti in 2010 and E. coli O104:H4 in Germany in 2011 (discussed earlier by Pallen) suggest that it may soon be feasible for clinicians to access the genomic content of an outbreak strain in close to real time (Otto, 2011). In this

FIGURE WO-46 Prototype of a nanopore sequencing technology currently under development.
SOURCE: Oxford Nanopore Technologies.
regard, Budowle suggested that a role for government is to develop sampling strategies, modeling outbreak and biocrime scenarios to determine the best approaches to gather information to enable rapid inferences (rather than focusing on technology or software, which industry is already working on).
Keim noted that the ability to collect and bring samples home from overseas—such as from China and Russia—has become much more difficult since the terrorist attacks of 9/11, because of changes in both U.S. and foreign regulations on the movement, export, and import of microbiological samples. The strategy now is to engage the international community and raise its technological capabilities and standards so that advanced genomics may be reliably performed locally. While we may not be able to transport organisms, SNP genotypes can be transferred instantaneously around the world via the Internet.
Standards for Data Sharing and Analysis
To date, most laboratories have deposited sequences in public sequence repositories. As the amount of sequencing data continues to grow, consideration is needed of how to usefully record sequence data as well as experimental metadata. The Genomics Standards Consortium52 and the Sanger Institute have proposed standards for the description of genome sequences, including information about the origin, pathogenicity, host, growth conditions, estimated genome size, and
_______________________
52 The genomic standards consortium is a consortium of groups, including the DNA Data Bank of Japan, the European Bioinformatics Institute, the European Molecular Biology Laboratory, the Joint Genome Institute, and the National Center for Biotechnology Information.
characteristics related to growth, habitat, taxonomy, and genetics (MacLean et al., 2009). Standards development for metadata will be critical for comparative analysis of large metagenomic datasets.
As discussed earlier, it has become feasible for individual laboratories to undertake sequencing projects rather than sending samples to a few large genome centers. Current large-scale genome and metagenome sequencing projects are deploying multiple platforms and different sequencing chemistries in parallel. The additional challenge created by so many sequencers is the sheer volume of data. The production of billions of sequence reads places substantial demands on the existing information technology infrastructure in terms of data transfer, storage, quality control, computational analysis, and information management systems for sample tracking and process management (Metzker, 2010).
Investigators must increasingly struggle with the question of how relevant information will be most effectively extracted from the massive amounts of genomic data being generated, and what other methods, tools, data, and analytical approaches may be needed in order to effectively interpret and interrogate these data for a variety of applications (Mardis, 2011; Relman, 2011). There is a need for better bioinformatics algorithms, including assembly algorithms, that can integrate data from diverse sources; access to faster computing capabilities; larger and more efficient data storage devices; and careful capture of important metadata (e.g., date and location of isolation, culture passages in the laboratory, patient medical information).
Workshop participants discussed the challenges of maintaining curated data sets, particularly the lack of funding for this purpose. One option suggested was a “broker ecosystem” where one broker for metagenomics would have stable funding for a number of years and would provide services to the community (e.g., curate data, metadata, act as chaperone for the data of the specific communities). Another suggestion discussed was to develop applications that take a first pass at the data stream to classify it in some way (e.g., taxonomic, gene family, etc.) prior to comparisons with databases (rather than constant, expensive, all-versus-all BLAST searches)—more of an iterative, semi-distributed, and then semi-centralized analysis pipeline. The problems surrounding lack of data sharing were also noted. Developers are creating algorithms based on 5-year-old data from old sequencing technology. If data were available in a timely fashion to research groups, algorithms developed would be more relevant to the current data emerging from next-generation technologies.
Useful Metagenomic Data Sets: Experimental Design and Collection of Metadata
“For the data sets produced for metagenomic studies, the size of data sets, their heterogeneity, and lack of standardization for both metadata and gene descriptive data present significant challenges for comparative analyses” (DeLong,
2009). Gilbert et al. (2011) noted the need to balance observational studies with carefully designed experimental approaches. The authors cite the Global Ocean Sampling expedition as an example of an early and groundbreaking observational study that provided an unprecedented snapshot of the diversity and heterogeneity in naturally occurring microbial populations (Gilbert and DuPont, 2011; Rusch et al., 2007). The study collected 41 different samples from a wide variety of aquatic habitats from more than 8,000 km and resulted in 7.7 million sequencing reads.
The resulting data set is one of the largest metagenomic data sets ever collected, comprising more than 6.12 million predicted proteins including representatives from all previously known families of microbial proteins and 1,700 new ones (Hugenholtz and Tyson, 2008). This data set has, however, been criticized “for poor experimental design and the absence of appropriate metadata necessary for analysis of the influence of environment on microbial diversity” (Gilbert et al. 2011). As metagenomics moves from the description of apparent diversity to the genuine description of complexity and function, carefully designed experimental approaches are needed to deliver the true potential of metagenomics (Knight et al., 2012).
Improving the Genome Knowledge Base: Diversity and Meaningful Annotation
Annotation is the process of assigning meaningful information, such as the location or function of genes, to raw sequence data. Reliable and consistent annotations are thus essential for the analysis and interpretation of genome data (Berglund et al., 2009). Insights from genomics, such as functional prediction, depend upon the accessibility of existing, well-annotated gene sequences. One obstacle to the use of existing annotations is the bias in the genome and protein knowledge bases. Organisms selected for genomic sequencing are culturable, numerically dominant in habitats of interest (e.g., the human body), and predominantly associated with (human) disease (Relman, 2011).
To be useful, whole genome sequence data must be annotated with functional information and gathered from a diverse array of microorganisms. To aid the characterization of the human microbiota, for example, one of the goals of the human microbiome project is to sequence 3,000 bacterial genomes as a reference set, as well as other genomes from fungi and eukaryotic organisms (The Human Microbiome Jumpstart Reference Strains Consortium, 2010). As illustrated by Wu et al. (2009), a considerable amount of phylogenetic “dark matter” remains unsampled (Figure WO-47).
Whole genome sequence information is available for a small subset of the known phylogenetic diversity of bacteria and archaea (based on unique SSU rRNA gene sequences). Here the authors of the Genomic Encyclopedia of Bacteria and Archaea (GEBA) depict four subsets of phylogenetic diversity: organisms with sequenced genomes pre-GEBA (blue), the GEBA organisms (red), all cultured organisms (dark grey), and all available SSU rRNA genes (light grey).
FIGURE WO-47 Phylogenetic “dark matter” left to be sampled.
SOURCE: Wu et al. (2009). Reprinted by permission from Macmillan Publishers Ltd: NATURE. Wu, D. et al. 2009. A phylogeny-driven genomic encyclopaedia of Bacteria and Archaea. Nature 462:1056-1060, copyright 2009.
For each subtree, taxa were sorted by their contribution to the subtree phylogenetic diversity, and the cumulative phylogenetic diversity was plotted from maximal (left) to the least (right). The inset magnifies the first 1,500 organisms. Comparison of the plots shows the phylogenetic dark matter left to be sampled (Wu et al., 2009).
Looking Forward
“Once the diversity of the microbial world is catalogued, it will make
astronomy look like a pitiful science.”
—Julian Davies, Professor Emeritus,
Microbiology and Immunology,
University of British Columbia
In order to maximize the discovery and characterization of new gene families and their associated novel functions, Wu et al., (2009) suggest that phylogenetic considerations should guide the selection of genomes for sequencing. By focusing on the novelty of an organism (highly divergent lineages of bacteria or archaea that lack representatives with sequenced genomes) the Genomic Encyclopedia of Bacteria and Archaea (GEBA) project seeks to “provide a phylogenetically balanced genomic representation of the microbial tree of life” (Wu et al., 2009). Sequencing the genomes of 1,520 phylogenetically selected isolates could include half of the phylogenetic diversity represented by known cultured bacteria and
archaea. The sequencing of an additional 9,218 genome sequences from currently uncultured species could capture 50 percent of this subset of recognized diversity. According to Wu et al. (2009), “such an undertaking will require the development of new approaches to culturing or processing of multi-species samples using methods such as … physical isolation of cells from mixed populations followed by whole genome amplification methods.”
The field of microbiology has made tremendous strides over the past several decades in describing the microbial world glimpsed for the first time just 300 years ago. Comparative genomic studies of bacterial species and metagenomic analyses of microbial communities to date have revealed how vastly we have underestimated the diversity, variability, and complexity of the microbial world. Microbial genomics offers the potential to efficiently characterize the vast cosmos of microbial diversity and rewrite the microbial community’s tree of life. Indeed, with the proliferation of culture-independent technologies and generation of enormous quantities of raw genomic sequences of microorganisms from diverse settings, the field of microbiology now suffers an “embarrassment of riches.” As observed in a recent editorial in Nature Reviews Microbiology (Editorial, 2011), “[t]he scale of life in the microbial world is such that amazing numbers become commonplace. These numbers can be sources of inspiration for those in the field and used to inspire awe in the next generation of microbiologists.”
Achtman, M., G. Morelli, P. Zhu, T. Wirth, I. Diehl, B. Kusecek, A. J. Vogler, D. M. Wagner, C. J. Allender, W. R. Easterday, V. Chenal-Francisque, P. Worsham, N. R. Thomson, J. Parkhill, L. E. Lindler, E. Carniel, and P. Keim. 2004. Microevolution and history of the plague bacillus, Yersinia pestis. Proceedings of the National Academy of Sciences USA 101(51):17837-17842.
Alagely, A., C. J. Krediet, K. B. Ritchie, and M. Teplitski. 2011. Signaling-mediated cross-talk modulates swarming and biofilm formation in a coral pathogen Serratia marcescens. International Society for Microbial Ecology Journal 5(10):1609-1620.
Alm, E. 2012. Session I: The Application of Computational/Theoretical and Experimental Approaches to Study the Evolution of Microorganisms. Paper presented at the Forum on Microbial Threats Workshop, The Science and Applications of Microbial Genomics, Washington, DC, Institute of Medicine, Forum on Microbial Threats, June 12.
Baz, M., Y. Abed, J. Papenburg, X. Bouhy, M. E. Hamelin, and G. Boivin. 2009. Emergence of oseltamivir-resistant pandemic H1N1 virus during prophylaxis. New England Journal of Medicine 361(23):2296-2297.
Bentley, S. 2009. Sequencing the species pan-genome. Nature Reviews Microbiology 7:258-259.
Berglund, E. C., B. Nystedt, S. G. E. Andersson. 2009. Computational resources in infectious disease: Limitations and challenges. PLoS Computational Biology 5(10):e1000481.
Blaser, M. 1997. Ecology of Helicobacter pylori in the human stomach. Journal of Clinical Investigation 100(4):759-762.
Budowle, B. 2012. Session IV: Microbial Forensics. Paper presented at the Forum on Microbial Threats Workshop, The Science and Applications of Microbial Genomics, Washington, DC, Institute of Medicine Forum on Microbial Threats, June 13.
Bos, K. I., V. J. Schuenemann, G. B. Golding, H. A. Burbano, N. Waglechner, B. K. Coombes, J. B. McPhee, S. N. DeWitte, M. Meyer, S. Schmedes, J. Wood, D. J. Earn, D. A. Herring, P. Bauer, H. N. Poinar, and J. Krause. 2011. A draft genome of Yersinia pestis from victims of the Black Death. Nature 478(7370):506-510.
Casadevall, A. 2012. Session III: Virulence as an Emergent Property. Paper presented at the Forum on Microbial Threats Workshop, The Science and Applications of Microbial Genomics, Washington, DC, Institute of Medicine, Forum on Microbial Threats, June 13.
Casadevall, A., and L. A. Pirofski. 1999. Host-pathogen interactions: Redefining the basic concepts of virulence and pathogenicity. Infection and Immunity 67(8):3703-3713.
———. 2000. Host-pathogen interactions: Basic concepts of microbial commensalism, colonization, infection, and disease. Infection and Immunity 68(12):6511-6518.
———. 2001. Host-pathogen interactions: The attributes of virulence. Journal of Infectious Diseases 184(3):337-344.
———. 2002. The meaning of microbial exposure, infection, colonization, and disease in clinical practice. Lancet Infectious Diseases 2(10):628-635.
———. 2003. The damage-response framework of microbial pathogenesis. Nature Reviews Microbiology 1(1):17-24.
———. 2007. Accidental virulence, cryptic pathogenesis, martians, lost hosts, and the pathogenicity of environmental microbes. Eukaryotic Cell 6:2169-2174.
Casadevall, A., F. C. Fang, and L. A. Pirofski. 2011. Microbial virulence as an emergent property: Consequences and opportunities. PLoS Pathogens 7(7):e1002136.
Castillo-Ramírez, S., S. R. Harris, M. T. Holden, M. He, J. Parkhill, S. D. Bentley, and E. J. Feil. 2011. The impact of recombination on dN/dS within recently emerged bacterial clones. PLoS Pathogens 7(7):e1002129.
Chin, C. S., J. Sorenson, J. B. Harris, W. P. Robins, R. C. Charles, R. R. Jean-Charles, J. Bullard, D. R. Webster, A. Kasarkis, P. Peluso, E. E. Paxinos, Y. Yamaichi, S. B. Calderwood, J. J. Mekalanos, E. E. Schadt, and M. K. Waldor. 2011. The origin of the Haitian cholera outbreak strain. New England Journal of Medicine 364(1):33-42.
Coleman, M. L., and S. W. Chisholm. 2010. Ecosystem-specific selection pressures revealed through comparative population genomics. Proceedings of the National Academy of Sciences USA 107(43):18634-18639.
Cracraft, J., and M. J. Donoghue. 2004. Assembling the Tree of Life. New York: Oxford University Press.
Croucher, N. J., S. R. Harris, C. Fraser, M. A. Quail, J. Burton, M. van der Linden, L. McGee, A. von Gottberg, J. H. Song, K. S. Ko, B. Pichon, S. Baker, C. M. Parry, L. M. Lambertsen, D. Shahinas, D. R. Pillai, T. J. Mitchell, G. Dougan, A. Tomasz, K. P. Klugman, J. Parkhill, W. P. Hanage, and S. D. Bentley. 2011. Rapid pneumococcal evolution in response to clinical interventions. Science 331(6016):430-434.
de Kruif, P. 1926. Microbe hunters. Orlando, FL: Harcourt, Inc.
DeLong, E. F. 2009. The microbial ocean from genomes to biomes. Nature 459:200-206.
Denef, V. J., and J. F. Banfield. 2012. In situ evolutionary rate measurements show ecological success of recently emerged bacterial hybrids. Science 336:462-466.
Desai, N., D. Antonopoulos, J. A. Gilbert, E. M. Glass, and F. Meyer. 2012. From genomics to metagenomics. Current Opinion in Biotechnology 23(1):72-76.
Dethlefsen, L., M. McFall-Ngai, and D. A. Relman. 2007. An ecological and evolutionary perspective on human-microbe mutualism and disease. Nature 449:811-818.
Devignat, R. 1951. Varieties of Pasteurella pestis; new hypothesis (in French). Bulletin of the World Health Organization 4(2):247-263.
Dobell, C. 1932 (reisssued 1960). Antony van Leeuwenhoek and his “Little Animals.” New York: Harcourt, Brace and Company.
Editorial. 2011. Microbiology by numbers. Nature Reviews Microbiology 9:628.
Edwards, R. A., and R. Rohwer. 2005. Viral metagenomics. Nature Reviews Microbiology 3(6): 504-510.
Eisen, J. A. 2007. Environmental shotgun sequencing: its potential and challenges for studying the hidden world of microbes. PLoS Biology 5(3):0384-0388.
Eisen, J. A., D. Kaiser, and R. M. Myers. 1997. Gastrogenomic delights: A movable feast. Nature Medicine 3(10):1076-1078.
Félix, M. A., A. Ashe, J. Piffaretti, G. Wu, I. Nuez, T. Bélicard, Y. Jiang, G. Zhao, C. J. Franz, L. D. Goldstein, M. Sanroman, E. A. Miska, and D. Wang. 2011. Natural and experimental infection of Caenorhabditis nematodes by novel viruses related to nodaviruses. PLoS Biology 9(1):e1000586.
Finkbeiner, S. R., A. F. Allred, P. I. Tarr, E. J. Klein, C. D. Kirkwood, and D. Wang. 2008a. Metagenomic analysis of human diarrhea: Viral detection and discovery. PLoS Pathogens 4(2):e1000011.
Finkbeiner, S. R., C. D. Kirkwood, and D. Wang. 2008b. Complete genome sequence of a highly divergent astrovirus isolated from a child with acute diarrhea. Virology Journal 5:117.
Finkbeiner, S. R., Y. Li, S. Ruone, C. Conrardy, N. Gregoricus, D. Toney, H. W. Virgin, L. J. Anderson, J. Vinje, D. Wang, and S. Tong. 2009. Identification of a novel astrovirus (Astrovirus VA1) associated with an outbreak of acute gastroenteritis. Journal of Virology 83(20):10836-10839.
Fisher, A. T., and R. P. Von Herzen. 2005. Models of hydrothermal circulation within 106 Ma seafloor: Constraints on the vigor of fluid circulation and crustal properties, below the Madeira Abyssal Plain. Geochemistry Geophysics Geosystems. 6: Q11001. doi:10.1029/2005GC001013. doi:10.1029/2005GC001013.
Fitz-Gibbon, S., S. Tomida, B. H. Chiu, L. Nguyen, C. Du, M. Liu, D. Elashoff, M. C. Erfe, A. Loncaric, J. Kim, R. L. Modlin, J. F. Miller, E. Sodergren, N. Craft, G. M. Weinstock, and H. Li. 2013. Propionibacterium Acnes strain populations in the human skin microbiome associated with acne. Journal of Investigative Dermatology doi: 10.1038/jid.2013.21. [Epub ahead of print].
Fleischmann, R. D., M. D. Adams, O. White, R. A. Clayton, E. F. Kirkness, A. R. Kerlavage, C. J. Bult, J. F. Tomb, B. A. Dougherty, J. M. Merrick, K. McKenney, G. Sutton, W. FitzHugh, C. Fields, J. D. Gocayne, J. Scott, R. Shirley, L. I. Liu, A. Glodek, J. M. Kelley, J. F. Weidman, C. A. Phillips, T. Spriggs, E. Hedblom, M. D. Cotton, T. R. Utterback, M. C. Hanna, D. T. Nguyen, D. M. Saudek, R. C. Brandon, L. D. Fine, J. L. Frichman, J. L. Fuhrmann, N. S. M. Geoghagen, C. L. Gnehm, L. A. McDonald, K. V. Small, C.M. Fraser, H. O. Smith, J. C. Venter. 1995. Whole-genome random sequencing and assembly of Haemophilus influenza rd. Science 269:496-512.
Fraser, C. M., J. A. Eisen, and S. L. Salzberg. 2000. Microbial genome sequencing. Nature 406: 799-803.
Fraser, C. M., J. A. Eisen, K. E. Nelson, I. T. Paulsen, and S. L. Salzberg. 2002. The value of complete microbial genome sequencing (you get what you pay for). Journal of Bacteriology 184(23):6403-6405.
Fraser-Ligett, C. M. 2005. Insights on biology and evolution from microbial genome sequencing. Genome Research 15:1603-1610.
Fredericks, D. N., and D. A. Relman. 1996. Sequence-based identification of microbial pathogens: A reconsideration of Koch’s postulates. Clinical Microbiology Reviews 9:18-33.
Frerichs, R. R., P. S. Keim, R. Barrais, and R. Piarroux. 2012. Nepalese origin of cholera epidemic in Haiti. Clinical Microbiology and Infection 18(6):E158-E163.
Gage, K. L., and M. Y. Kosoy. 2005. Natural history of plague: Perspectives from more than a century of research. Annual Review of Entomology 50:505-528.
Gardy, J. L., J. C. Johnston, S. J. Ho Sui, V. J. Cook, L. Shah, E. Brodkin, S. Rempel, R. Moore, Y. Zhao, R. Holt, R. Varhol, I. Birol, M. Lem, M. K. Sharma, K. Elwood, S. J. Jones, F. S. Brinkman, R. C. Brunham, and P. Tang. 2011. Whole-genome sequencing and social-network analysis of a tuberculosis outbreak. New England Journal of Medicine 364(8):730-739.
Gevers, D., F. M. Cohan, J. G. Lawrence, B. G. Spratt, T. Coenye, E. J. Feil, E. Stackebrandt, Y. Van de Peer, P. Vandamme, F. L. Thompson, and J. Swings. 2005. Opinion: Re-evaluating prokaryotic species. Nature Reviews Microbiology 3(9):733-739.
Ghedin, E. 2012. Session II: Characterizing Intra-host Influenza Virus Populations to Predict Emergence. Paper presented at the Forum on Microbial Threats Workshop, The Science and Applications of Microbial Genomics, Washington, DC, Institute of Medicine, Forum on Microbial Threats, June 12.
Ghedin, E., J. Laplante, J. DePasse, D. E. Wentworth, R. P. Santos, M. L. Lepow, J. Porter, K. Stellrecht, X. Lin, D. Operario, S. Griesemer, A. Fitch, R. A. Halpin, T. B. Stockwell, D. J. Spiro, E. C. Holmes, K. St George. 2011. Deep sequencing reveals mixed infection with 2009 pandemic influenza A (H1N1) virus strains and the emergence of oseltamivir resistance. Journal of Infectious Diseases 203(2):168-174.
Ghedin, E., E. C. Holmes, J. V. Depasse, L. T. Pinilla, A. Fitch, M. E. Hamelin, J. Papenburg, G. Boivin. 2012. Presence of oseltamivir-resistant pandemic A/H1N1 minor variants before drug therapy with subsequent selection and transmission. Journal of Infectious Diseases 206(10): 1504-1511.
Gilbert, J. 2012. Session I: The Earth Microbiome Project: Modeling the Earth’s Microbiome. Paper presented at the Forum on Microbial Threats Workshop, The Science and Applications of Microbial Genomics, Washington, DC, Institute of Medicine, Forum on Microbial Threats, June 12.
Gilbert, J. A., and C. L. DuPont. 2011. Microbial metagenomics: Beyond the genome. Annual Review of Marine Science 3:347-371.
Gilbert, J. A., R. O’Dor, N. King, and T. Vogel. 2011. The importance of metagenomic surveys to microbial ecology: Or why Darwin would have been a metagenomic scientist. Microbial Informatics and Experimentation 1:5.
Girard, J. M., D. M. Wagner, A. J. Vogler, C. Keys, C. J. Allender, L. C. Drickamer, and P. Keim. 2004. Differential plague-transmission dynamics determine Yersinia pestis population genetic structure on local, regional, and global scales. Proceedings of the National Academy of Science USA 101(22):8408-8413.
Girguis, P. 2012. Session I: Population Diversity in Deep-Sea Microbial Communities. Paper presented at the Forum on Microbial Threats Workshop, The Science and Applications of Microbial Genomics, Washington, DC, Institute of Medicine, Forum on Microbial Threats, June 12.
Hacker, J., and J. D. Kaper. 2000. Pathogenicity islands and the evolution of microbes. Annual Review of Microbiology 54:641-679.
Haensch, S., R. Bianucci, M. Signoli, M. Rajerison, M. Schultz, S. Kacki, M. Vermunt, D. A. Weston, D. Hurst, M. Achtman, E. Carniel, and B. Bramanti. 2010. Distinct clones of Yersinia pestis caused the Black Death. PLoS Pathogens 6(10):e1001134.
Handelsman, J. 2004. Metagenomics. Microbiology and Molecular Biology Reviews 68(4):669-685.
Harris, S. R., E. J. Feil, M. T. G. Holden, M. A. Quail, E. K. Nickerson, N. Chantratita, S. Gardete, A. Tavares, N. Day, J. Lindsay, J. D. Edgeworth, H. de Lencastre, J. Parkhill, S. J. Peacock, and S. D. Bentley. 2010. Evolution of MRSA during hospital transmission and intercontinental spread. Science 327:467-474.
Harris, S. R., E. J. P. Cartwright, M. E. Török, M. T. G. Holden, N. M. Brown, A. L. Ogilvy-Stuart, M. J. Ellington, M. A. Quail, S. D. Bentley, J. Parkhill, S. J. Peacock. 2012. Using whole genome sequencing to dissect the cause and effect of a meticillin-resistant Staphylococcus aureus outbreak: A descriptive study. Lancet Infectious Diseases (Epub ahead of print).
Hehemann, J. H., G. Correc, T. Barbeyron, W. Helbert, M. Czjzek, and G. Michel. 2010. Transfer of carbohydrate-active enzymes from marine bacteria to Japanese gut microbiota. Nature 464(7290):908-912.
Hendriksen, R. S., L. B. Price, J. M. Shupp, J. D. Gillece, R. S. Kaas, D. M. Engelthaler, V. Bortolaia, T. Pearson, A. E. Waters, B. P. Upadhyay, S. D. Shrestha, S. Adhikari, G. Shakya, P. S. Keim, and F. M. Aarestrup. 2011. Population genetics of Vibrio cholerae from Nepal in 2010: Evidence on the origin of the Haitian outbreak. mBio 2(4):e00157-11.
Hornsey, M., N. Loman, D. W. Wareham, M. J. Ellington, M. J. Pallen, J. F. Turton, A. Underwood, T. Gaulton, T. P. Thomas, M. Doumith, D. M. Livermore, N. Woodford. 2011. Whole-genome comparison of two Acinetobacter baumannii isolates from a single patient, where resistance developed during tigecycline therapy. Journal of Antimicrobial Chemotherapy 66(7):1499-1503.
Hugenholtz, P. 2002. Exploring prokaryotic diversity in the genomic era. Genome Biology 3(2): 0003.1-0003.8.
Hugenholtz, P., and G. W. Tyson. 2008. Metagenomics. Nature 455:481-483.
The Human Microbiome Jumpstart Reference Strains Consortium. 2010. A catalog of reference genomes from the human microbiome. Science 328(5981):994-999.
Hunt, D. E., L. A. David, D. Gevers, S. P. Preheim, E. J. Alm, and M. F. Polz. 2008. Resource partitioning and sympatric differentiation among closely related bacterioplankton. Science 320(5879):1081-1085.
Huse, S. 2012. Session III: The Impact of Sequencing Errors on Estimates of Diversity in the Rare Biosphere (and Potential Solutions). Paper presented at the Forum on Microbial Threats Workshop, The Science and Applications of Microbial Genomics, Washington, DC, Institute of Medicine, Forum on Microbial Threats, June 13.
Huse, S. M., D. M. Welch, H. G. Morrison, and M. L. Sogin. 2010. Ironing out the wrinkles in the rare biosphere through improved OTU clustering. Environmental Microbiology 12(7):1889-1898.
Huse, S. M., Y. Ye, Y. Zhou, and A. A. Fodor. 2012. A core human microbiome as viewed through 16S rRNA sequence clusters. PLoS One 7(6):e34242.
Isenberg, H. D. 1988. Pathogenicity and virulence: Another view. Clinical Microbiology Reviews 1(1):40-53.
JASON. 2009. Microbial forensics. JSR-08-512.
———. 2010. The $100 genome: Implications for the DoD. JSR-11-320.
Joneson, S., J. E. Stajich, S. H. Shiu, E. B. Rosenblum. 2011. Genomic transition to pathogenicity in chytrid fungi. PLoS Pathogens 7(11):e1002338.
Kanehisa, M. 2002. The KEGG database. Novartis Foundation symposium 247:91-101; discussion 101-103, 119-128, 244-252.
Kaper, J. B., J. P. Nataro, and H. L. Mobley. 2004 Pathogenic Escherichia coli. Nature Reviews Microbiology 2(2):123-140.
Keegan, K. P., W. L. Trimble, J. Wilkening, A. Wilke, T. Harrison, M. D’Souza, and F. Meyer. 2012. A platform-independent method for detecting errors in metagenomic sequencing data: DRISEE. PLoS Computational Biology 8(6):e1002541.
Keim, P., A. Kalif, J. Schupp, K. Hill, S. E. Travis, K. Richmond, D. M. Adair, M. Hugh-Jones, C. R. Kuske, and P. Jackson. 1997. Molecular evolution and diversity in Bacillus anthracis as detected by amplified fragment length polymorphism markers. Journal of Bacteriology 179:818-824.
Knight, R., J. Jansson, D. Field, N. Fierer, N. Desai, J. Fuhrman, P. Hugenholtz, F. Meyer, R. Stevens, M. Bailey, J. I. Gordon, G. Kowalchuk, and J. A. Gilbert. 2012. Unlocking the potential of metagenomics through replicated experimental design. Nature Biotechnology 30(6):513-520.
Koch, R. 1891. Uber bakteriologische Forschung Verhandlung des X Internationalen Medichinischen Congresses, Berlin, 1890, 1, 35. August Hirschwald, Berlin. (In German.) Xth International Congress of Medicine, Berlin.
Kondrashov, A. S., and M. V. Mina. 1986. Sympatric speciation: When is it possible? Biological Journal of the Linnean Society 27:201-223.
Krediet, C., K. B. Ritchie, A. Alagely, M. Teplitski. (In Press). Coral commensal bacterial interference of metabolism and surface motility in a white pox pathogen during early colonization of coral surfaces. ISME Journal.
Kumarasamy, K. K., M. A. Toleman, T. R. Walsh, J. Bagaria, F. Butt, R. Balakrishnan, U. Chaudhary, M. Doumith, C. G. Giske, S. Irfan, P. Krishnan, A. V. Kumar, S. Maharjan, S. Mushtaq, T. Noorie, D. L. Paterson, A. Pearson, C. Perry, R. Pike, B. Rao, U. Ray, J. B. Sarma, M. Sharma, E. Sheridan, M. A. Thirunarayan, J. Turton, S. Upadhyay, M. Warner, W. Welfare, D. M. Livermore, N. Woodford. 2010. Emergence of a new antibiotic resistance mechanism in India, Pakistan, and the UK: A molecular, biological, and epidemiological study. Lancet Infectious Diseases 10(9):597-602.
La Rosa, P. S., J. P. Brooks, E. Deych, E. L. Boone, D. J. Edwards, Q. Wang, E. Sodergren, G. Weinstock, and W. D. Shannon. 2012. Hypothesis testing and power calculations for taxonomic-based human microbiome data. PLoS ONE 7(12):e52078.
Larsen, P. E., F. R. Collart, D. Field, F. Meyer, K. P. Keegan, C. S. Henry, J. McGrath, J. Quinn, and J. A. Gilbert. 2011. Predicted relative metabolomic turnover (PRMT): Determining metabolic turnover from a coastal marine metagenomic dataset. Microbial Informatics and Experimentation 1(1):4.
Larsen, P. E., D. Field, and J. A. Gilbert. 2012. Predicting bacterial community assemblages using an artificial neural network approach. Nature Methods 9(6):621-625.
Lederberg. J. 2000. Infectious history. Science 288(5464):287-293.
Lewis, K. 2011. Presentation given at the March 14-15, 2011, public workshop, “Synthetic and Systems Biology,” Forum on Microbial Threats, Institute of Medicine, Washington, DC.
Lewis, T., N. J. Loman, L. Bingle, P. Jumaa, G. M. Weinstock, D. Mortiboy, and M. J. Pallen. 2010. High-throughput whole-genome sequencing to dissect the epidemiology of Acinetobacter baumannii isolates from a hospital outbreak. Journal of Hospital Infection 75(1):37-41.
Ley, R. E., C. A. Lozupone, M. Hamady, R. Knight, and J. I. Gordon. 2008. Worlds within worlds: Evolution of the vertebrate gut microbiota. Nature Reviews Microbiology 6:776-788.
Link, V. B. 1955. A history of plague in United States of America. Public Health Monographs 26:1-120.
Lipkin, W. I. 2010. Microbe hunting. Microbiology and Molecular Biology Reviews 74(3): 363-377.
Lips, K. R., F. Brem, R. Brenes, J. D. Reeve, R. A. Alford, J. Voyles, C. Carey, L. Livo, A. P. Pessier, and J. P. Collins. 2006. Emerging infectious disease and the loss of biodiversity in a neotropical amphibian community. Proceedings of the National Academy of Sciences USA 103:3165-3170.
Lipsitch, M., C. Colijn, T. Cohen, W. P. Hanage, and C. Fraser. 2009. No coexistence for free: Neutral null models for multistrain pathogens. Epidemics 1(1):2-13.
Loman, N. J., C. Constantinidou, J. Z. Chan, M. Halachev, M. Sergeant, C. W. Penn, E. R. Robinson, and M. J. Pallen. 2012a. High-throughput bacterial genome sequencing: An embarrassment of choice, a world of opportunity. Nature Reviews Microbiology 10(9):599-606.
Loman, N. J., R. V. Misra, T. J. Dallman, C. Constantinidou, S. E. Gharbia, J. Wain, and M. J. Pallen. 2012b. Performance comparison of benchtop high-throughput sequencing platforms. Nature Biotechnology 30(5):434-439.
MacLean, D. J., D. G. Jones, and D. J. Studholme. 2009. Application of “next-generation” sequencing technologies to microbial genetics. Nature Reviews Microbiology 7:287-296.
Mao-Jones, J., K. B. Ritchie, L. E. Jones, and S. P. Ellner. 2010. How microbial community composition regulates coral disease development. PLoS Biology 8(3):e1000345.
Mardis, E. R. 2008. Next-generation sequencing methods. Annual Reviews Genomics and Human Genetics 9:387-402.
———. 2011. A decade’s perspective on DNA sequencing technology. Nature 470:198-203.
McClelland, E. E., P. Bernhardt, and A. Casadevall. 2006. Estimating the relative contributions of virulence factors for pathogenic microbes. Infection and Immunity 74(3):1500-1504.
McDaniel, L. D., E. Young, J. Delaney, F. Ruhnau, K. B. Ritchie, and J. H. Paul. 2010. High frequency of horizontal gene transfer in the oceans. Science 330(6000):50.
McDaniel, L. D., E. C. Young, K. B. Ritchie, and J. H. Paul. 2012. Environmental factors influencing gene transfer agent (GTA) mediated transduction in the subtropical ocean. PLoS ONE 7(8):e43506.
McFall-Ngai, M., E. A. Heath-Heckman, A. A. Gillette, S. M. Peyer, and E. A. Harvie. 2012. The secret languages of coevolved symbioses: Insights from the Euprymna scolopes-Vibrio fischeri symbiosis. Seminars in Immunology 24:3-8.
Medini, D., C. Donati, H. Tettelin, V. Masignani, and R. Rappuoli. 2005. The microbial pan-genome. Current Opinion in Genetics and Development 15:589-594.
Medini, D., D. Serruto, J. Parkhill, D. A. Relman, C. Donati, R. Moxon, S. Falkow, and R. Rappuoli. 2008. Microbiology in the post-genomic era. Nature Reviews Microbiology 6:419-430.
Merrell, D. S., and S. Falkow. 2004. Frontal and stealth attack strategies in microbial pathogenesis. Nature 430(6996):250-256.
Metzker, M. L. 2010. Sequencing technologies—The next generation. Nature Reviews Genetics 11:31-45.
Meyer, F. 2012. Session IV: Analyzing Metagenomic Data: Inferring Microbial Community Function with MG-RAST. Paper presented at the Forum on Microbial Threats Workshop, The Science and Applications of Microbial Genomics, Washington, DC, Institute of Medicine, Forum on Microbial Threats, June 13.
Meyer, F., D. Paarmann, M. D’Souza, R. Olson, E. M. Glass, M. Kubal, T. Paczian, A. Rodriguez, R. Stevens, A. Wilke, J. Wilkening, and R. A. Edwards. 2008. The metagenomics RAST server—A public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinformatics 9:386.
Morelli, G., Y. Song, C. J. Mazzoni, M. Eppinger, P. Roumagnac, D. M. Wagner, M. Feldkamp, B. Kusecek, A. J. Vogler, Y. Li, Y. Cui, N. R. Thomson, T. Jombart, R. Leblois, P. Lichtner, L. Rahalison, J. M. Petersen, F. Balloux, P. Keim, T. Wirth, J. Ravel, R. Yang, E. Carniel, and M. Achtman. 2010. Yersinia pestis genome sequencing identifies patterns of global phylogenetic diversity. Nature Genetics 42(12):1140-1143.
Mutreja, A., D. W. Kim, N. R. Thomson, T. R. Connor, J. H. Lee, S. Kariuki, N. J. Croucher, S. Y. Choi, S. R. Harris, M. Lebens, S. K. Niyogi, E. J. Kim, T. Ramamurthy, J. Chun, J. L. Wood, J. D. Clemens, C. Czerkinsky, G. B. Nair, J. Holmgren, J. Parkhill, and G. Dougan. 2011. Evidence for several waves of global transmission in the seventh cholera pandemic. Nature 477(7365):462-465.
Nee, S. 2004. More than meets the eye. Nature 429:804-805.
Nichol, S. T., C. F. Spiropoulou, S. Morzunov, P. E. Rollin, T. G. Ksiazek, H. Feldmann, A. Sanchez, J. Childs, S. Zaki, and C. J. Peters. 1993. Genetic identification of a hantavirus associated with an outbreak of acute respiratory illness. Science 262:914-917.
Otto, T. D. 2011. Real-time sequencing. Nature Reviews Microbiology 9:633.
Overbeek, R., T. Begley, R. M. Butler, J. V. Choudhuri, N. Diaz, H. Y. Chuang, M. Cohoon, V. de Crécy-Lagard, T. Disz, R. Edwards, et al. 2005. The subsystems approach to genome annotation and its use in the project to annotate 1000 genomes. Nucleic Acids Research 33(17):5691-5702.
Pace, N. R. 1997. A molecular view of microbial diversity and the biosphere. Science 276:734-740.
Pace, N. R., D. A. Stahl, D. J. Lane, and G. J. Olsen. 1985. The analysis of natural microbial populations by ribosomal RNA sequences. American Society for Microbiology News 51:4-12.
Pallen, M. J., and B. W. Wren. 2007. Bacterial pathogenomics. Nature 449:835-842.
Paul, J. H., E. Young, L. McDaniel, K. B. Ritchie, C. Voolstra. (In Review). Gene transfer agent mediated horizontal gene transfer in the marine environment. Proceedings of the National Academy of Sciences USA.
Pennisi, E. 2012. Search for pore-fection. Science 336:534-537.
Perry, R. D., and J. D. Fetherston. 1997. Yersinia pestis—Etiologic agent of plague. Clinical Microbiology Reviews 10(1):35-66.
Piarroux, R., R. Barrais, B. Faucher, R. Haus, M. Piarroux, J. Gaudart, R. Magloire, and D. Raoult. 2011. Understanding the cholera epidemic, Haiti. Emerging Infectious Diseases 17(7):1161-1168.
Pollitzer, R. 1951. Plague studies. 1. A summary of the history and survey of the present distribution of the disease. Bulletin of the World Health Organization 4(4):475-533.
Qin, J., R. Li, J. Raes, M. Arumugam, K. S. Burgdorf, C. Manichanh, T. Nielsen, N. Pons, F. Levenez, T. Yamada, D. R. Mende, J. Li, J. Xu, S. Li, D. Li, J. Cao, B. Wang, H. Liang, H. Zheng, Y. Xie, J. Tap, P. Lepage, M. Bertalan, J. M. Batto, T. Hansen, D. Le Paslier, A. Linneberg, H. B. Nielsen, E. Pelletier, P. Renault, T. Sicheritz-Ponten, K. Turner, H. Zhu, C. Yu, S. Li, M. Jian, Y. Zhou, Y. Li, X. Zhang, S. Li, N. Qin, H. Yang, J. Wang, S. Brunak, J. Doré, F. Guarner, K. Kristiansen, O. Pedersen, J. Parkhill, J. Weissenbach, MetaHIT Consortium, P. Bork, S. D. Ehrlich, and J. Wang J. 2010. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464(7285):59-65.
Rasko, D. 2012. Session II: Comparative Genomics of E. coli and Shigella: Identification and Characterization of Pathogenic Variants Based on Whole Genome Sequence Analysis. Paper presented at the Forum on Microbial Threats Workshop, The Science and Applications of Microbial Genomics, Washington, DC, Institute of Medicine, Forum on Microbial Threats, June 12.
Rasko, D. A., P. L. Worshamb, T. G. Abshireb, S. T. Stanley, J. D. Bannand, M. R. Wilsond, R. J. Langhamc, R. S. Deckerc, L. Jianga,, T. D. Reade, A. M. Phillippy, S. L. Salzberg, M. Popf, M. N. Van Ertg, L. J. Kenefic, P. S. Keim, C. M. Fraser-Liggett, and J. Ravel. 2011. Bacillus anthracis comparative genome analysis in support of the Amerithrax investigation. Proceedings of the National Academy of Sciences USA 108(12):5027-5032.
Read, T. D., S. L. Salzberg, M. Pop, M. Shumway, L. Umayam, L. Jiang, E. Holtzapple, J. D. Busch, K. L. Smith, J. M. Schupp, D. Solomon, P. Keim, and C. M. Fraser. 2002. Comparative genome sequencing for discovery of novel polymorphisms in Bacillus anthracis. Science 296:2028-2033.
Relman, D. A. 1993. The identification of uncultured microbial pathogens. Journal of Infectious Diseases 168:1-8.
______. 1999. The search for unrecognized pathogens. Science 284:1308-1310.
______. 2011. Microbial genomics and infectious diseases. New England Journal of Medicine 365(4):347-357.
______. 2012. Day 2 Welcoming Remarks and Summary of Day One. Forum on Microbial Threats Workshop, The Science and Applications of Microbial Genomics, Washington, DC, Institute of Medicine, Forum on Microbial Threats, June 13.
Relman, D. A., J. S. Loutit, T. M. Schmidt, S. Falkow, and L. S. Tompkins. 1990. The agent of bacillary angiomatosis: An approach to the identification of uncultured pathogens. New England Journal of Medicine 323:1573-1580.
Relman, D. A., T. M. Schmidt, R. P. MacDermott, and S. Falkow. 1992. Identification of the uncultured bacillus of Whipple’s disease. New England Journal of Medicine 327:293-301.
Ritchie, K. B. 2006. Regulation of microbial populations by coral surface mucus and mucus-associated bacteria. Marine Ecology Progress Series 322:1-14.
______. 2011. Bacterial symbionts of corals and symbiodinium. In: Beneficial microorganisms in multicellular life forms, E. Rosenberg and U. Gophna (Eds.). Berlin: Springer-Verlag Chapter 9, pp. 139-150.
Rivers, T. M. 1937. Viruses and Koch’s postulates. Journal of Bacteriology 33:1-12.
Rohde, H., J. Qin, Y. Cui, D. Li, N. J. Loman, M. Hentschke, W. Chen, F. Pu, Y. Peng, J. Li, F. Xi, S. Li, Y. Li, Z. Zhang, X. Yang, M. Zhao, P. Wang, Y. Guan, Z. Cen, X. Zhao, M. Christner, R. Kobbe, S. Loos, J. Oh, L. Yang, A. Danchin, G. F. Gao, Y. Song, Y. Li, H. Yang, J. Wang, J. Xu, M. J. Pallen, J. Wang, M. Aepfelbacher, R. Yang. 2011. Open-source genomic analysis of Shiga-toxin-producing E. coli O104:H4. New England Journal of Medicine 365(8):718-724.
Rosenblum, E. B. 2012. Session II: Evolution and Pathogenicity in the Deadly Chytrid Pathogen of Amphibians. Paper presented at the Forum on Microbial Threats Workshop, The Science and Applications of Microbial Genomics, Washington, DC, Institute of Medicine, Forum on Microbial Threats, June 12.
Rosenblum, E. B., J. E. Stajich, N. Maddox, and M. B. Eisen. 2008. Global gene expression profiles for life stages of the deadly amphibian pathogen Batrachochytrium dendrobatidis. Proceedings of the National Academy of Sciences USA 105(44):17034-17039.
Rosenblum, E. B., T. J. Poorten, M. Settles, G. K. Murdoch, J. Robert, N. Maddox, and M. B. Eisen. 2009. Genome-wide transcriptional response of Silurana (Xenopus) tropicalis to infection with the deadly chytrid fungus. PLoS ONE 4(8):e6494.
Rosenblum, E. B., T. J. Poorten, S. Joneson, and M. Settles. 2012. Substrate-Specific Gene Expression in Batrachochytrium dendrobatidis, the Chytrid Pathogen of Amphibians. PLoS One 7(11): e49924.
Rosenblum, E. B., T. J. Poorten, M. Settles, and G. K. Murdoch. 2012. Only skin deep: Shared genetic response to the deadly chytrid fungus in susceptible frog species. Molecular Ecology 21(13):3110-3120.
Rusch, D. B., A. L. Halpern, G. Sutton, K. B. Heidelberg, S. Williamson, S. Yooseph, D. Wu, J. A. Eisen, J. M. Hoffman, K. Remington, K. Beeson, B. Tran, H. Smith, H. Baden-Tillson, C. Stewart, J. Thorpe, J. Freeman, C. Andrews-Pfannkoch, J. E. Venter, K. Li, S. Kravitz, J. F. Heidelberg, T. Utterback, Y. H. Rogers, L. I. Falcón, V. Souza, G. Bonilla-Rosso, L. E. Eguiarte, D. M. Karl, S. Sathyendranath, T. Platt, E. Bermingham, V. Gallardo, G. Tamayo-Castillo, M. R. Ferrari, R. L. Strausberg, K. Nealson, R. Friedman, M. Frazier, and J. C. Venter. 2007. The Sorcerer II global ocean sampling expedition: Northwest Atlantic through Eastern Tropical Pacific. PLoS Biology 5:e77.
Santamaria-Fries, M., L. F. Fajardo, M. L. Sogin, P. Olson, and D. A. Relman. 1996. Lethal infection by a previously unclassified metazoan parasite. Lancet 347:1797-1801.
Scheutz, F., E. M. Nielsen, J. Frimodt-Møller, N. Boisen, S. Morabito, R. Tozzoli, J. P. Nataro, and A. Caprioli. 2011. Characteristics of the enteroaggregtive Shiga toxin/verotoxin-producing Escherichia coli O104:H4 strain causing the outbreak of haemolytic uraemic syndrome in Germany, May to June 2011. Eurosurveillance 16(24):pii=19889.
Schloissnig, S., M. Arumugam, S. Sunagawa, M. Mitreva, J. Tap, A. Zhu, A. Waller, D. R. Mende, J. R. Kultima, J. Martin, K. Kota, S. R. Sunyaev, G. M. Weinstock, and P. Bork. 2013. Genomic variation landscape of the human gut microbiome. Nature 493(7430):45-50.
Schrenk, M. O., D. S. Kelley, J. R. Delaney, and J. A. Baross. 2003. Incidence and diversity of microorganisms within the walls of an active deep-sea sulfide chimney. Applied and Environmental Microbiology 69(9):3580-3592.
Shapiro, B. J., J. Friedman, O. X. Cordero, S. P. Preheim, S. C. Timberlake, G. Szabó, M. F. Polz, and E. J. Alm. 2012. Population genomics of early events in the ecological differentiation of bacteria. Science 336(6077):48-51.
Sharp, K. H., and K. B. Ritchie. 2012. Multi-partner interactions in corals in the face of climate change. Biological Bulletin 223:66-77.
Sharp, K. H., K. B. Ritchie, P. J. Schupp, R. Ritson-Williams, and V. J. Paul. 2010. Bacterial acquisition in juveniles of several broadcast spawning coral species. PLoS One 5(5):e10898.
Smillie, C. S., M. B. Smith, J. Friedman, O. X. Cordero, L. A. David, and E. J. Alm. 2011. Ecology drives a global network of gene exchange connecting the human microbiome. Nature 480(7376):241-244.
Sylvan, J. B., B. M. Toner, and K. J. Edwards. 2012. Life and death of deep-sea vents: Bacterial diversity and ecosystem succession on inactive hydrothermal sulfides. mBio 3(1):e00279-11.
Takai, K., F. Inagaki, S. Nakagawa, H. Hirayama, T. Nunoura, Y. Sako, K. H. Nealson, and K. Horikoshi. 2003. Isolation and phylogenetic diversity of members of previously uncultivated e-proteobacteria in deep-sea hydrothermal fields. FEMS Microbiology Letters 218(1):167-174.
Tatusov, R. L., N. D. Fedorova, J. D. Jackson, A. R. Jacobs, B. Kiryutin, E. V. Koonin, D. M. Krylov, R. Mazumder, S. L. Mekhedov, A. N. Nikolskaya, et al. 2003. The COG database: An updated version includes eukaryotes. BMC Bioinformatics 4:41, Epub.
Teplitski, M., and K. B. Ritchie. 2009. How feasible is the biological control of coral disease? Trends in Ecology and Evolution 24(7):378-385
Thomas, P. D., V. Wood, C. J. Mungall, S. E. Lewis, J. A. Blake; Gene Ontology Consortium. 2012. On the use of gene ontology annotations to assess functional similarity among orthologs and paralogs: A short report. PLoS Computational Biology8(2):e1002386
Trimble, W. L., K. P. Keegan, M. D’Souza, A. Wilke, J. Wilkening, J. Gilbert, and F. Meyer. 2012. Short-read reading-frame predictors are not created equal: Sequence error causes loss of signal. BMC Bioinformatics 13(1):183.
Tringe, S. G., C. von Mering, A. Kobayashi, A. A. Salamov, K. Chen, H. W. Chang, M. Podar, J. M. Short, E. J. Mathur, J. C. Detter, P. Bork, P. Hugenholtz, and E. M. Rubin. 2008. Comparative metagenomics of microbial communities. Science 308:554-557.
Turnbaugh, P. J., R. E. Ley, M. Hamady, C. M. Fraser-Liggett, R. Knight, and J. I. Gordon. 2007. The human microbiome project. Nature 449:804-810.
Tyson, G. W., J. Chapman, P. Hugenholtz, E. E. Allen, R. J. Ram, P. R. Richardson, V. V. Solovyev, E. M. Rubin, D. S. Rokhsar, and J. F. Banfield. 2004. Community structure and metabolism through reconstruction of microbial genomes from the environment. Nature 428:37-43.
VandeWalle, J. L., G. W. Goetz, S. M. Huse, H. G. Morrison, M. L. Sogin, R. G. Hoffmann, K. Yan, and S. L., McLellan. 2012. Acinetobacter, Aeromonas and Trichococcus populations dominate the microbial community within urban sewer infrastructure. Environmental Microbiology 14(9): 2538-2552.
Venter, J. C., K. Remington, J. F. Heidelberg, A. L. Halpern, D. Rusch, J. A. Eisen, D. Wu, I. Paulsen, K. E. Nelson, W. Nelson, D. E. Fouts, S. Levy, A. H. Knap, M. W. Lomas, K. Nealson, O. White, J. Peterson, J. Hoffman, R. Parson, H. Baden-Tillson, C. Pfannkoch, Y. H. Rogers, and H. O. Smith. 2004. Environmental genome shotgun sequencing of the Sargasso Sea. Science 304:66-74.
Vogler, A. J., F. Chan, D. M. Wagner, P. Roumagnac, L. Lee, R. Nera, M. Eppinger, J. Ravel, L. Rahalison, B. W. Rasoamanana, S. M. Beckstrom-Sternberg, M. Achtman, S. Chanteau, and P. Keim. 2011. Phylogeography and molecular epidemiology of Yersinia pestis in Madagascar. PLoS Neglected Tropical Diseases 5(9):e1319.
Weinstock, G. 2012a. Genomic approaches to studying the human microbiota. Nature 489:13.
______. 2012b. Session I: Variation in Microbial Communities and Genomes. Paper presented at the Forum on Microbial Threats Workshop, The Science and Applications of Microbial Genomics, Washington, DC, Institute of Medicine, Forum on Microbial Threats, June 12.
White, B. J., C. Cheng, F. Simard, C. Costantini, and N. J. Besansky. 2010. Genetic association of physically unlinked islands of genomic divergence in incipient species of Anopheles gambiae. Molecular Ecology 19(5):925-939.
Whitman, W. B., D. C. Coleman, and W. J. Wiebe. 1998. Prokaryotes: The unseen majority. Proceedings of the National Academy of Sciences USA 95(12):6578-6583.
Woyke, T., D. Tighe, K. Mavromatis, A. Clum, A. Copeland, W. Schackwitz, A. Lapidus, D. Wu, J. P. McCutcheon, B. R. McDonald, N. A. Moran, J. Bristow, J-F. Cheng. 2010. One bacterial cell, one complete genome. PLoS One 5(4):e10314.
Wu, D., P. Hugenhotz, K. Mavromatis, R. Pukall, E. Dalin, N. N. Ivanova, V. Kunin, L. Goodwin, M. Wu, B. J. Tindall, S. D. Hooper, A. Pati, A. Lykidis, S. Spring, I. J. Anderson, P. D’haeseleer, A. Zemla, M. Singer, A. Lapidus, M. Nolan, A. Copeland, C. Han, F. Chen, J. F. Cheng, S. Lucas, C. Kerfeld, E. Lang, S. Gronow, P. Chain, D. Bruce, E. M. Rubin, N. C. Kyrpides, H. P. Klenk, and J. A. Eisen. 2009. A phylogeny-driven genomic encyclopaedia of Bacteria and Archaea. Nature 462:1056-1060.
Wylie, K. M., R. M. Truty, T. J. Sharpton, K. A. Mihindukulasuriya, Y. Zho, H. Gao, E. Sodergren, G. M. Weinstock, and K. S. Pollard. 2012a. Novel bacterial taxa in the human microbiome. PLoS One 7(6):e35294.
Wylie, K. M., K. A. Mihindukulasuriya, E. Sodergren, G. M. Weinstock, and G. A. Storch. 2012b.
Sequence analysis of the human virome in febrile and afebrile children. PLoS ONE 7(6):e27735.
Zhou, J., L. Wu, Y. Deng, X. Zhi, Y. H. Jiang, Q. Tu, J. Xie, J. D. Van Nostrand, Z. He, and Y. Yang. 2011. Reproducibility and quantitation of amplicon sequencing-based detection. ISME Journal 5(8):1303-1313.
Zhou, Y., H. Gao, K. A. Mihindukulasuriya, P. S. Rosa, K. M. Wylie, T. Vishnivetskaya, M. Podar, B. Warner, P. I. Tarr, D. E. Nelson, J. D. Fortenberry, M. J. Holland, S. E. Burr, W. D. Shannon, E. Sodergren, and G. M. Weinstock. 2013. Biogeography of the ecosystems of the healthy human body. Genome Biology 14(1):R1.




















































































































