
6
Computational/Chemical Processes in Self-Assembly
Key Points
- There is a need to develop theory and simulation that can connect models across scales and incorporate emergent phenomena to realize functionality by design capabilities. In spite of great advances in modeling at all levels, particularly with coarse-grained models, there is still a critical lack of understanding about how long-range order on the mesoscale emerges from very short-range interactions and what one could treat less precisely.
- There are many tools in the computational chemistry toolbox that can be brought to bear on the problem of bridging the microscopic and macroscopic world, and among them are the coarse-grained models and atom-by-atom simulation of whole systems. These models can provide complementary information about mechanism and mesoscale behavior.
- Entropy is usually thought of as counteracting the enthalpic driving force for self-assembly, but in many mesoscale systems, entropy is actually helping the assembly process, though the degree to which that occurs is still unclear.
- The shape of particles can have a profound effect on self-assembly even in the absence of interparticle interactions.
- Designing proteins with specific three-dimensional structures by changing amino acid sequences so that they would then self-assemble into interesting mesoscale structures has been a longstanding engineering challenge that has still yet to be solved.
The workshop’s final session featured five presentations on how computation and chemistry can be brought to bear on the challenge of understanding self-assembly of mesoscale structures. Todd Yeates, Professor of Chemistry and Biochemistry at the University of California, Los Angeles, provided some examples of the role that symmetry plays in self-assembly and its use in creating a broad range of materials with diverse applications. William Noid, Associate Professor of Chemistry at Penn State University, described various approaches for multiscale modeling, including the use of bottom-up models for representing thermodynamic properties. Klaus Schulten, Professor of Physics and Director of the National Institutes of Health Center for Macromolecular Modeling & Bioinformatics at the University of Illinois, Urbana-Champaign, discussed how intelligent mesoscale biological systems can be resolved entirely in chemical detail and how that information can be used to mimic the robust and adaptive mesoscale molecular strategies that evolved in nature. Sharon Glotzer, the Stuart W. Churchill Collegiate Professor of Chemical Engineering, and Professor of Materials Science and Engineering, Physics, Applied Physics, and Macromolecular Science & Engineering at the University of Michigan, addressed the role that entropy, order, and function play in self-assembly processes. Gregory Voth, the Haig P. Papazian Distinguished Service Professor of Chemistry at the University of Chicago, discussed how multiscale simulations can help
reveal key features of the structure and self-assembly of large multiprotein and protein-membrane complexes such as those involved in remodeling membranes. An open discussion moderated by workshop organizing committee and Chemical Sciences Roundtable member Miguel Garcia-Garibay, from the University of California, Los Angeles, followed the five presentations.
DESIGNING NOVEL SELF-ASSEMBLING PROTEIN MATERIALS: RECENT SUCCESSES AND FUTURE PROSPECTS
The past few decades have seen spectacular success in using nucleic acids, particularly DNA molecules, to assemble into sophisticated objects, said Todd Yeates, and the reason for that success comes from the fact that the rules by which DNA molecules self-associated are simple, following the basic rules of Watson–Crick base pairing (Han et al. 2013, Ke et al. 2012, Rothemund 2006, Seeman 2010). In contrast, designing proteins with specific three-dimensional structures by changing amino acid sequences so that they would then self-assemble into interesting objects has been a long-standing engineering challenge that has still yet to be solved. However, using the underlying principles of symmetry that exist in nature, research teams such as the one headed by Yeates have recently developed design strategies that are realizing success in designing protein molecules that self-assemble into mesoscale structures.
Some 20 years ago, a graduate student of Yeates was working on the problem of trying to understand why proteins crystallize with a specific preference for different kinds of symmetries. She discovered that a key element for understanding why proteins crystallize in the arrangements that they do is a mathematical property known as the minimum contact requirements for achieving connectivity in a molecular solid (Wukovitz and Yeates 1995). Crystalline solids follow space group symmetries, Yeates explained, but what had never been examined before was the minimum different number of ways molecules had to touch each other in order to form three-dimensional structures. “This turned out to be a math problem, and the question of how many different distinct ways a molecule has to contact itself in order to form a connected network is essentially a problem of group theory,” said Yeates.
What this insight means is that the minimum contact number is a function of symmetry. For example, to create an arrangement with what is called P2 symmetry, the molecule has to touch itself in at least three different ways no matter the shape of the individual molecule. In contrast, P4 symmetry requires just two contacts. What this rather simple but powerful concept translates into is that “an astonishing range of architectures and symmetries can be generated using only two symmetric points of protein–protein interaction,” explained Yeates. That realization, he added, frames the engineering problem of how to take a molecule and engineer it so that it self-assembles.
With part one of the problem solved—coming up with a rule to guide engineering work—Yeates and his team turned to part two, figuring out how to design a protein to have the necessary two points of contact and build in the interfaces needed to create the right geometry. He noted that his group started down this path before the advent of sequence design—the idea of taking a protein molecule and redesigning its amino acid sequence in a specific way so that it would interact with itself. Instead, they used natural protein oligomers that already had natural interfaces built into them. For example, many proteins form dimers or trimers and creating a fusion product of two of these proteins, using genetic fusion to create a gene that codes for both components, is in principle creating a larger protein that has the two natural interfaces needed to create bigger structures.
The one problem with this approach is that it is necessary to control the degrees of freedom in the fusion protein so that the two halves orient themselves correctly relative to one another. “If you just fuse two protein molecules together, you’d probably expect to get some kind of gel or just some kind of glue,” said Yeates, “but since we were trying to design very specific symmetric geometries, the question is how to fuse together two protein molecules in a way that you can control or anticipate what the geometric relationship would be between the parts that are joined.”
A solution to this problem was to work with proteins that had a terminal alpha helix because that afforded the opportunity to use computational methods to predict how the rigid nature of the alpha-helical structures would dictate geometry (Padilla, Colovos, and Yeates 2001). This is not a way to control geometry, said Yeates, but a way of
anticipating it, and it triggered a combinatorial chemistry exercise of taking known proteins with known types of helices, hooking them together, sometimes with short alpha-helical linkers, and modeling how these constructs would self-assemble. As a guide for this design effort, Yeates’ team used the angles of intersection in Platonic solids to develop the geometric rules for combining different symmetry elements, and then built one protein that their calculations predicted would assemble into a single 12-unit construct with tetrahedral symmetry. In fact, the protein did self-assemble but into multiple species that had a much broader size distribution than expected.
Yeates commented that the resulting paper drew little attention, even among the members of his own group, and this effort languished for more than a decade until a new bioengineering student joined the lab and decided to revisit this earlier work. By taking advantage of the better graphics technology that had been developed during the intervening years and turning a fresh eye on the structures, this student decided that there was one amino acid in the original design that, because of its location, could interfere with the predicted self-assembly process. When the “corrected” protein was expressed, it folded neatly into four distinct species that were separable using gel electrophoresis. These species, said Yeates, corresponded to the four possible ways of taking twofold and threefold axes of symmetry to make closed symmetries (Lai, King, and Yeates 2012). An additional amino acid change produced a construct that then self-assembled into one species that could be crystallized and whose structure was determined by x-ray crystallography to be a somewhat squashed tetrahedron (Figure 6-1) (Lai, Cascio, and Yeates 2012). This student then repeated this work with another fusion protein that self-assembled into a 230-angstrom cube containing 24 subunits, and in this case the crystal structure varied from the prediction by roughly 1 angstrom (Lai et al. 2014).
A second approach that has realized success has been taken by a former student of Yeates’ who is using sequence design rather than combinatorial approaches. The idea here was to take one natural oligomer and use computational methods for sequence design to introduce the second interface at exactly the right geometry (King et al. 2012). Of the 90 different constructs that this computational approach predicted would work, two did and they have since been crystallized. The resulting crystal structures matched the predicted structures within about an angstrom, said Yeates. Further work showed that this approach could be extended to two-component assembly by defining an intended target symmetry, identifying arrangements where the molecules touch each other, and then using computational methods to introduce the right amino acids to make that happen.
In his final remarks, Yeates said that of the nearly 50 possible combinations of two symmetry point groups to create self-assembling structures, only two have been explored. Going forward, the goal will be to demonstrate that these other possibilities can be made. If successful, these designed mesoscale assemblies could have use in creating multivalent antigen displays, bi- and multispecific reagents for cell recruitment, and layers and three-dimensional materials for high-density enzyme or receptor display.
BOTTOM-UP MULTISCALE APPROACHES FOR MESOSCALE PHENOMENA: PROGRESS AND OPPORTUNITIES
In preparing his presentation for this workshop, William Noid reviewed the Department of Energy report on mesoscale science and came away with two thoughts about mesoscale phenomena. The first is that there is a need to develop theory and simulation that can connect models across scales and incorporate emergent phenomena to realize functionality by design capabilities. The second idea is that there are many tools in the computational chemistry toolbox that can be brought to bear on this problem of bridging the microscopic and macroscopic world, and among them are the coarse-grained models of the sort that he uses in his research. The challenge, he said, is that if the goal is to capture the properties that emerge when moving from the atomic to the mesoscale, it will be important to ensure that a low-resolution, coarse-grained model will incorporate the physics that are governed by atomic interactions.
Coarse-grained models, Noid explained, can simulate longer length scales and time scales than atomic-scale models, and they can reach equilibrium, something that is difficult with fine-grained models because of the computational requirements of those more detailed models. Given that coarse-grained models take so much less
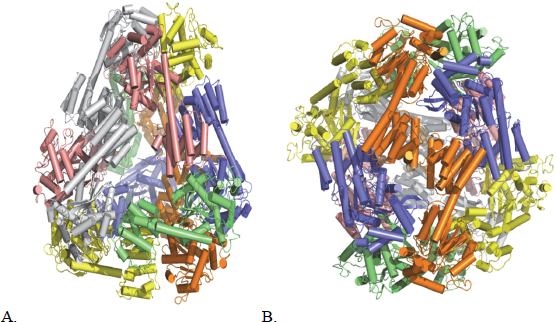
Figure 6-1 (A) The atomic structure of a designed protein cage with 12 subunits in a pseudotetrahedral symmetry. (B) The intended symmetry of the same protein cage with 12 subunits. SOURCE: Lai, Cascio, and Yeates (2012). Reprinted with permission from Science.
computational power, it is easy to run a simulation multiple times and determine the statistical confidence of simulated properties, as well as to systematically study parameter space and the impact of changing external conditions. From a conceptual point of view, coarse-grained models have advantages that go beyond their reduced computational burden. For example, it is possible to tailor coarse-grained models for particular systems and to focus efforts on essential features of a system while eliminating unnecessary details. Some coarse-grained models, for example, represent groups of water molecules or some details of some amino acids as a single entity rather than as multiple atoms. Another type might represent a protein not as a collection of atoms but as some kind of elastic network medium. Coarse-grained models provide a great deal of freedom, so it is up to the researcher to decide how best to represent a system and to create an abstract representation of the important pieces of the system and how they interact with one another.
Noid noted that while there are some theoreticians who believe that coarse-grained models are a waste of time—that the goal should be to build as detailed a model as possible and wait for computer technology to advance to the point that these models can be run in their full glory—the fact is that new things happen on different length and time scales and so describing them with the same microscopic physics is missing the point. “You want to be able to capture the physical forces driving phenomena at particular length scales,” said Noid. “That is real physics and that is what coarse-grained models allow you to do.”
There are two different broad strategies for coarse-graining, top down and bottom up. Top down entails taking an experimental observation to parameterize a coarse-grained model. Alternatively, it is possible to create a coarse-grained model to observe what type of features emerge on particular length and time scales as the properties of the model change, which is what Sharon Glotzer talked about in her presentation. The bottom-up approach to coarse-graining takes a more detailed model of the system that is believed to be an accurate representation of a particular system. Not only would one use the model to capture generic phenomena, but one would also to try to figure out the mesoscopic manifestations of the atomic interactions for that particular system. This is a powerful approach, said Noid, because it enables the development of rigorous theories that connect these different length scales. The bottom-up approach, he added, relies upon a rigorous statistical mechanics framework to go from a well-vetted fine-grained model to a coarse-grained model that captures the
phenomena of interest. The challenge in this approach, though, is to ensure that when going from a high-resolution to a lower-resolution model the correct forces and physics are captured.
Noid outlined a few of the computational approaches that people have employed to calculate wave functions and probability distributions. He described a method, known as force matching, that takes the forces generated by an atomistic model and the forces generated by a coarse-grained model and tries to minimize the difference between the two when averaging over structures or particles. This last approach is quite powerful, said Noid, because not only does it provide the desired coarse-grained energy potential, it also provides a variant framework that enables a systematic improvement of the model’s approximations. Also, this approach boils down to a direct linear least-squares problem, which means that it can be solved exactly for many different types of systems, and it makes predictions that work in practice for many systems even without force information for a structure. “You can show in practice that you can generate a model of any protein in a databank and recover the underlying potentials quantitatively,” said Noid.
He concluded his talk by noting that his group focuses on bottom-up methods and finds that they are very accurate for soft materials, but not always so accurate for modeling more complex biological materials. More work needs to be done there, he added, as there is a need to develop theories for working out the best way to represent systems. “The key idea though is that this many-body potential of mean force in statistical mechanics gives us an exact rigorous framework for any system, for any resolution, to try to make this perfect coarse-grained model, and now we just have to figure out how to make good approximations for that,” said Noid.
VIEW OF INTELLIGENT MESOSCALE BIOLOGICAL SYSTEMS AT CHEMICAL RESOLUTION: WHAT CAN BE LEARNED
To Klaus Schulten, computation serves as a microscope to provide views of biomaterials that are otherwise unavailable. He said that one of the great lessons he has learned over 40 years of studying biological materials is that “if you really want to play your cards well in the mesoscale world of materials and devices that you should think not only about how to build these materials, but that there is a lot of good chemistry that you can do with these materials. That is something that the physicists cannot do and the material scientists cannot do, but you chemists can do.”
As an example of that, Schulten discussed the cellular microtubule, which he said not only has interesting material properties, such as mechanical stability, but the ability to control chemistry, particularly the hydrolysis of guanosine triphosphate (GTP). Fresh microtubes, he explained, are loaded with GTP, and when they’re spent and fall apart they have guanosine diphosphate, but they have an intermediate state that is slightly disordered and more interesting in terms of its mechanics. More interesting, he said, is the fact that the physical properties of these materials are controlled by the molecules they bind.
He also discussed the cellulosome, which is a finger-like structure on the surface of bacteria that produce cocktails of enzymes needed to break down cellulose and other plant cell wall components. But besides producing the right enzymes with the best distribution to work on the substrates at hand, the cellulosomes themselves have polymer properties that enable them to help pull apart the cellulose in wood so that the enzymes have better access to their substrates.
The virus capsid, another example of a mesoscale biological “device,” was long thought to be simply a container of genetic material, but that notion was wrong. “The capsid is actually a communication surface: It is communicating with the cell that is being infected to make the cell help with its cellular effectors, its proteins, to guide the capsid to the right place in the cell,” said Schulten.
Returning to the example of the microtubule, Schulten said it has a number of structural properties that enable it to interact with proteins that change its properties, such as its flexibility, in response to chemistry going on at the microtubule surface. Using data from several sources, including electron microscopy and crystallographic studies, Schulten and his team have created a computational model of the microtubule at the atomic level that can reproduce the microtubule’s behavior as chemistry is happening on its surface. Similarly, his team has modeled the modular nature of the cellulosome to try to understand the changes it undergoes as it interacts with cellulose to generate strong chemical bonds that help it pull cellulose apart.
His group has also solved the structure of the HIV-1 capsid using cryoelectron microscopy and all-atom molecular dynamics simulations to make sure that the structure they deduced is stable (Zhao et al. 2013). One thing these experiments revealed is that the surface of the capsid is more heterogeneous than the typical icosahedral virus and more closely resembles Ebola or influenza virus. All told, there are some 1,300 protein molecules that pattern the capsid surface and that can move and distribute themselves and create barriers and channels through which ions can move. The capsid can also interact with cyclophilin, a protein that cells use to guide molecules to the nucleus, which the HIV-1 capsid has to do in order to inject its genetic material into the nucleus. He noted that this information should be useful for developing drugs that interfere with HIV-1 function.
In the final minutes of his presentation, Schulten discussed work aimed at understanding the mesoscale structure and function of the organelles that house the many enzymes and cofactors needed for photosynthesis. He briefly described the process by which light energy is harvested by chlorophylls and carotenoids, triggering a flow of excitons that travel to the photosynthetic reaction center, where ATP is generated, and then discussed how modeling and simulation work has enabled his group to detail the movement of molecules within the photosynthetic apparatus to enable photosynthesis to occur and provide a better understanding of the chemistry involved in this process.
EMERGENCE OF ENTROPY, ORDER, AND FUNCTION AT THE MESOSCALE
From Sharon Glotzer’s perspective of someone who is interested in entropy and order, there are three emerging trends in mesoscale science. The first is the idea of what she called patchy particles and shape entropy. A decade or so ago, when researchers began synthesizing a wide range of nanoparticles with a variety of shapes and functionalizing them with different ligands, it became clear that much of the physics and chemistry driving the self-assembly of building blocks into higher-ordered nanostructures was very similar despite the fact that these materials and the processes used to make them were quite different. To make sense of this, Glotzer and her colleagues developed a conceptual framework that was useful for understanding what kind of structures would be possible by tuning different anisotropic characteristics of the building blocks (Glotzer and Solomon 2007). This framework was developed by doing a principle component analysis that broke apart all of these different types of attributes, and it has served as a guide for further experimentation and for deciding what types of particles are likely to self-assemble into a desired structure (Figure 6-2).
Using a coarse-grained approach of the sort that Noid discussed, Glotzer’s group has been working with Christopher Murray’s group at the University of Pennsylvania to understand why the particles that Murray’s group was making were not assembling into the predicted structures. As an example, Glotzer described how lanthanum fluoride plates functionalized with oleic acids produced two very different geometries and how her group’s modeling work showed that as the aspect ratio of the plates changed, different facets became expressed more than other facets. Moreover, the model showed that the ligand binding density on the surfaces are sensitive to which facets are expressed. Thus, the aspect ratio controls the expression of facets, which controls the ligand binding density and, ultimately, the strength of the interactions between the various surfaces and the particles and shapes that emerge. This illustrates how interaction patchiness and shape control structure (Ye et al. 2013a).
The Glotzer and Murray groups were able to extend this work to understand what would happen when mixing together particles of completely different shapes, such as spheres and rods (Ye et al. 2013b). “Depending on how you tune the asymmetry of these interactions you can get anything from a fully phase-separated state to a lamellar phase, a crystal of the rods with the spheres in between to a full and proper crystal, just by subtle changes in the patchiness of the interactions around these antisymmetric particles,” said Glotzer.
Typically, when looking at this type of self-assembly, enthalpy dominates, with van der Waals interactions and hydrogen bonding coming into play at nanometer and colloidal scales. Glotzer noted, though, that there is an emerging understanding that entropy and entropic bonds also can control self-assembly, and that this appreciation could lead to the ability to make even more complex mesoscale structures through self-assembly processes. As an example, she described studies that her group
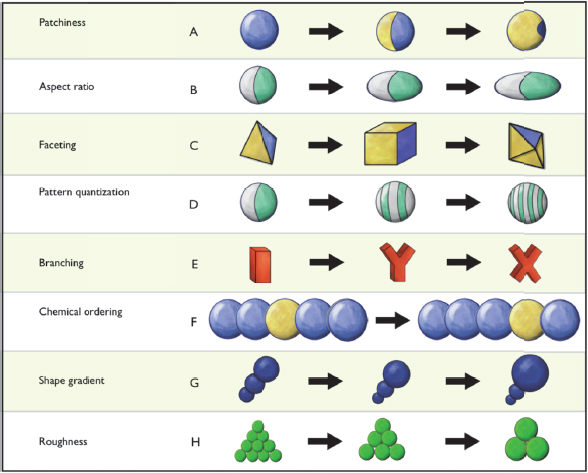
Figure 6-2 Principal component analysis reveals a nearly infinite-dimensional design space for nanoparticles. SOURCE: Glotzer and Solomon (2007). Reprinted with permission from Nature Materials.
conducted with cadmium telluride and cadmium selenide nanoparticles. These particles, which are tetrahedral in shape, are coated with ligands, are charged, and have dipole moments. Using a coarse-grained model, Glotzer and her collaborator were able to predict a dozen different types of structures, including sheets, wires, twisted chiral shapes, and superparticles, that they were then able to observe.
It has been known for decades, Glotzer explained, that entropy maximization can cause a disordered system to order, a principle upon which colloid physics depends. To see if entropy alone could drive structure formation, she and her collaborators modeled what would happen if they threw tetrahedrally shaped nanoparticles into a box. What emerged was a complete surprise—a dodecagonal quasicrystal of very complex structure with 82 particles in the unit cell and with no interactions emerging from the surrounding fluid (Haji-Akbari et al. 2009). This discovery, she said, turned out to be the tip of the iceberg in terms of the structures that could be generated simply by tuning particle shape (Damasceno, Engel, and Glotzer 2012a). Glotzer said she now believes that it is possible to generate every space group observed for crystals just by tuning particle shape and relying solely on entropy (Figure 6-3), and her group is now looking into that possibility. Already, her group has generated upward of 50 different space groups including some very complex structures such as chiral beta manganese with 20 particles in a unit cell and gamma brass, with 52 particles in a unit cell from slight truncated and augmented dodecahedra.
Entropy has usually been thought of as counteracting the enthalpic driving force for self-assembly, but Glotzer’s group has used potential mean force and torque modeling to quantify how much entropy is actually helping the assembly process. This method calculates the potential of mean force and torque between two particles in the middle of a sea of particles, and it can quantify how entropy maximization produces emergent effective entropic bonds that help align particles with one another (Damasceno, Engel, and Glotzer 2012b).

Figure 6-3 Tuning entropy by tuning shape. SOURCE: Damasceno, Engel, and Glotzer (2012a). Reprinted with permission from ACS Nano.
“We can now start to put these entropic bonds on the same footing as other kinds of chemical bonds and think about how to incorporate them into design rules,” said Glotzer.
The second major trend in mesoscale science identified by Glotzer is understanding the forces that are involved in self-assembly, meaning how long-range order develops from short-range forces. The goal of this work is to understand how giant, complex unit cells and other structures form with a periodicity and order well beyond the range of interparticle forces and to use that information to design for metastability. Already, Glotzer and her collaborators have generated the first icosahedral quasicrystal ever self-assembled from a disordered fluid in a computer simulation, and what is particularly noteworthy about this achievement, she said, is that they were able to self-assemble this structure from a one-component system of spherically symmetric particles interacting with a completely isotropic pair potential that is less than three particle diameters (Engel et al. 2015). “We’re seeing for the first time that experimental capabilities and simulation capabilities for these very large systems are starting to overlap so that we can start to understand exactly how these structures are
growing and understand nucleation and growth problems,” said Glotzer.
For example, two different types of particles, rhombic dodecahedra and spheres, both self-assemble from a fluid into a face-centered cubic, but spheres make poor crystals while rhombic dodecahedra make beautiful crystals. It would be good to know why that is so, said Glotzer, and to understand the kinetic pathway that these particles are following as they self-assemble. The goals are to begin to understand why differently shaped particles will self-assemble into the same crystal structure, predict which of those shapes will make the best crystal, and determine if an assembly pathway can be designed to force the system to make that crystal.
The third major trend that Glotzer addressed is the development of functional building blocks that will not only self-assemble but that are reconfigurable. As an example, she described the self-assembly of ellipsoid Janus particles that were gold on one side and poly(methyl methacrylate) on the other side into long braid-like fibers that were predicted by computer simulation. What is interesting about these particles is that turning an electric field on and off polarizes the gold and causes the fibers to stretch, acting as a colloidal actuator that could represent a first step toward creating artificial muscle fibers (Shah et al. 2015). She also described two other examples in which polydispersed distributions of particles and mixtures of particles and proteins that first self-organize into large superparticles then self-assemble into crystals. In one case, using cadmium telluride particles mixed with cytochrome c enzymes, the transmission electron micrographs showed that the individual particles were heterogeneously mixed within the mesoscale crystals (Park et al. 2014). She noted as an aside that these crystals are photoactive and could be used to carry out chemical reactions.
She also discussed work leading to reconfigurable colloidal clusters that she has been doing with David Pine’s group at New York University. These clusters are made by mixing spherical particles with spheres that have a dimple in them, and in a painstaking set of experiments using optical tweezers, they were able to get these colloids to transition back and forth between different configurations. “If we can assemble these into regular structures that can transition from one state to another, you could store information in this system,” said Glotzer, who estimated that it could be possible to store a terabyte of information in a teaspoon of such a colloid.
She concluded her remarks with a brief description of another project her group has started that looks at active particles and whether they will rotate with the same fixed applied torque in opposite directions when energy is pumped into the system. A constant input of energy rotates the particles and induces effective interactions among the particles that drive emergent, mesoscale phenomena.
MULTISCALE COMPUTER SIMULATION OF MESOSCALE BIOMOLECULAR COOPERATIVITY
In the workshop’s final presentation, Gregory Voth discussed the use of a full multiscale simulation hierarchy as a means of understanding membrane remodeling during endocytosis, a process that he said is not well understood at the mesoscale. There are many different pathways for endocytosis, one of which is the clathrin-mediated pathway in which a complex network of proteins cooperates to bring things into or take things out of the cell through the formation of a so-called “bud” at the membrane’s surface. This bud begins as a dent or pit in the membrane and then proceeds to encapsulate the material entering the cell and allow it to be transported within the cell. One set of proteins involved in this pathway are known as the BAR domain proteins, which resemble tiny bananas and are believed to be curvature sensors and curvature generators. As the membrane bud forms, these proteins form a scaffold upon which other proteins can build to facilitate the clipping of this bud to reform the cell wall and bring the bud into the cell.
When inserted into liposomes, BAR domain proteins such as amphiphysin and endophilin, all of which have a cluster of positively charged amino acids that interact with the negatively charged lipids of the cell membrane, can form some interesting structures that change shape (Figure 6-4) and that mesoscopic simulations can reproduce computationally (Ayton et al. 2009). The two questions of importance regarding this observation, said Voth, are whether it is possible to understand what is happening and whether they are relevant to biology. The answer to the second question, he added, is “not so relevant.”

Figure 6-4 Mesoscopic simulations (left) and electron micrographic images (right) of liposomes containing BAR-domain proteins. SOURCE: Ayton et al. (2009). Reprinted with permission from Biophysical Journal.
Several years ago, said Voth, research on the binding of endophilin to liposomes of different sizes, and thus curvatures, found that binding affinity increased significantly as the curvature of the liposome increased. The hypothesis proposed to explain this observation was that endophilin was binding to defects that occur in the membrane as it curves, and as the curvature increased, so, too, would the number and size of the defects (Bhatia et al. 2009, Drin et al. 2007, Hatzakis et al. 2009). Voth’s group calculated the free energy of binding at the molecular scale and found that there is a 5-kilocalorie-per-mole driving force for endophilin’s helical structure to fold into a defect (Cui, Lyman, and Voth 2011). “That was a critical step in our ability to model these systems, tying into atomistic scale information and building that into coarse-grained models,” said Voth. A critical element of this binding, he added, are the helical elements of these proteins.
To further explore the role that these helices play in membrane interactions and remodeling, Voth’s group ran simulations comparing BAR-domain proteins with their helices, called N-BAR, or without their helices, called BAR because the N-terminal helices are absent. These simulations showed that the helices are critical to the stability of the membrane–protein complex. With the BAR
proteins, the membranes are very disordered and will fall apart in the simulations. With the N-BAR proteins, the simulations produced structures with zigzag packing of the helices, which can be observed at poor resolution with cryoelectron microscopy. He noted that new techniques using single-electron detectors may help solve this resolution problem.
Additional simulations with flat membranes found that at high density—one protein molecule for every 300 lipid molecules—N-BAR forms a scaffold that induces the membranes to take on a uniform tubular curvature and develop small membrane invaginations. Further simulation work predicted that at high density, N-BAR will cause 200- to 300-nm-diameter liposomes to transform into reticulated tubular structures. A three-dimensional reconstruction of a cryoelectron micrograph tomogram confirmed the existence of a tubular network (Simunovic et al. 2013).
It turns out, Voth explained, that most of what these simulations produced can be described using a mesoscopic field theory that treats membranes as elastic and that allows the membrane to couple to an energy field provided by the proteins. This type of model couples the motion of the lipids with the motion of the proteins, as well as interactions between the proteins, and it produces the same results as the coarse-grained molecular-level model. He noted that “we’ve gotten pretty good at going from the atomistic level to the coarse-grained level, but what’s really missing is a direct map between the two.”
Examining the richness of these remodeled vesicles reveals a very rich phase diagram at high protein densities that predicts that different kinds of structures will form. These structures are modulated by the strength of protein binding to the surface of the liposome and its composition, density of protein at the surface, and other factors. One of Voth’s graduate students noticed that string-like features can form even at the initial phase of remodeling at high protein densities and that the formation of these strings is much less when the helices are missing. This led to simulations at much lower protein densities that produced a remarkably different behavior. Instead of forming lattices or even partial lattices, these long banana-like proteins formed strings. Moreover, these strings go into troughs in the membrane’s curvature in the opposite way from the high-density cases where tubules preform (Simunovic, Srivastava, and Voth 2013). In addition, the vesicles show large deformations that appear to be responding to string formation. What appears to be happening is that proteins binding to the surface of the membrane form linear aggregates and that two lines of protein form in side-by-side interactions. The proteins then induce local invagination or welling and curvature of the membrane, a high-energy process that is compensated for in part by the binding energy of the protein.
What is most interesting about this arrangement of proteins and lipids is that it has the effect of lowering entropy, which would make the free energy of this process go in the wrong direction, Voth explained. What seems to be happening, he said, is that membrane tension modulates protein–protein interactions in a way that disfavors linear aggregation and instead causes the proteins to form strings. Preliminary experiments using atomic force microscopy have produced images with striking similarity to those predicted by coarse-grained simulations.
In conclusion, Voth said that the low-density regime, rather than high-density conditions, is consistent with experimental observations of endocytosis, with linear aggregation of protein at the initial stages of remodeling. In the high-density region, excess bending energy breaks the membrane and as a result creates tubules.
Neil Henson, from Los Alamos National Laboratory, started the discussion and asked if a fair representation of a modeling approach for simulating a mesoscale object that does chemistry would be to “first understand the chemical reaction at the atomic level, and then the barriers, thermodynamics, and kinetics, and then abstract that information into a more coarse-grained model and perhaps to a mesoscopic theory in order to understand the effect of that chemistry on the mesoscale structure.” Voth said that this is not always the case and that many successful modeling efforts start with the coarse-grained model. Glotzer said that both atomic-scale and course-scale models are needed. “If the community had waited to study nanoparticle self-assembly by first studying nucleation and growth of the nanoparticles in solution to see how they grow, and then throwing ligands in to stop the growth and built all that up, we would never have gotten to self-assembly,” said Glotzer. She noted that starting at
the coarse-grained level can work well if there are insights from experimental data or finer-scale simulations that can be used to parameterize the model.
Schulten stated that today’s atomic-scale models can simulate a volume of about 1 cubic micron, or about 100 million to 1 billion atoms. Given that capability, the most straightforward approach would be to take the best force field parameters available and use those to simulate at the atomic scale and build from there. “My own principle is that I can only simplify what I understand and that it might be dangerous to go straight to coarse-graining before you really know the chemical details,” said Schulten. Voth added that working at the molecular scale is challenging because of limits in sample space and free energy barriers, and Glotzer wondered what the restrictions are with regard to time scale. Schulten replied that his group simulated the entire HIV-1 viral capsid for 1.5 microseconds and that there are methods for extending these simulations for multiple milliseconds, such as replica exchange molecular dynamics and transition pair sampling. “Using smart sampling methods, you can go for many milliseconds, but if you don’t know where you are heading, you are limited by a microsecond or so,” said Schulten.
Session moderator Garcia-Garibay noted that one of the things about mesoscale phenomena that the field often thinks about is emergent behavior, and he asked the panel if simulation and modeling can address emergent behavior or are there behaviors at the mesoscale that cannot be described in terms of first principles, such as geometry, topology, and long- and short-range interactions. Yeates said that, at the scales at which he works, the models generate more or less exact structures, but as he moves to larger scales he expects that there will be mesoscopic behavior where what will be important is the length scale over which order persists and those will be a challenge given what is known today. He noted, though, that Glotzer’s work with rigid materials is dealing with long-range order, but it is not clear whether these methods will work with soft, more flexible materials. “I think we’ll find out how length scales relate to flexibility as we go to extended symmetries instead of closed symmetries,” said Yeates.
Schulten said that electron microscopy is a natural tool for these longer length scales and that experimental data can inform these modeling efforts. One of the difficulties in using microscopy, said Voth, is that they require assuming that there is some sort of symmetry present and that they are done at liquid nitrogen temperatures, which limits motion and entropy considerations among other things, and so the translation to room-temperature behavior might not be straightforward.
Chris Mundy from the Pacific Northwest National Laboratory (PNNL) asked the panelists if they were considering the role of solvent in their studies given that solvent effects at the nanometer scale can be significant. Voth said that coarse-grained models of the sort that his and Glotzer’s groups run have an implicit solvent represented as hydrophobic associations that have to be estimated. “You have to build in the physics,” said Voth. Glotzer added that her approach has been to first do fully atomistic simulations with water present and gain insights from those models to feed into the coarse-grained models, an approach that Schulten agreed is a good one for including solvent effects in larger-scale models.
Patricia Thiel asked if there is a connection between these modeling studies and what is happening in biomineralization. Glotzer said there is an absolute link because these modeling efforts are asking the exact same questions: How do these assemblies grow, how does nucleation occur, and where do these processes lead once crystal growth starts? Jim De Yoreo then asked how these modeling approaches apply to biomineralization given that they are run at very high densities and natural systems operating at low volume fractions. Glotzer acknowledged that her work, for example, will not explain what is happening initially at low density, but once interactions start happening and molecules or smaller structures start aggregating, factors such as entropy can explain phenomena seen in the mesoscale.
Garcia-Garibay then asked the panel to comment on the idea that Paul Weiss put forth in the workshop’s plenary address that many things at this scale are metastable, that they are constantly in flux. Glotzer said that a major trend in mesoscale modeling now is to design for metastability, but that in fact, little is known about metastability on the mesoscale and how to guide assembly down a path to metastability instead of the globally favored stable energy state. She added that the active matter work that she mentioned briefly is all about dealing with systems that are never in thermodynamic
equilibrium and that are always driven to some steady state that is not the low-energy stable state. The big question there is whether it is possible to map that state to a thermodynamic equilibrium-like system that has a free energy function. “Is there a function that can describe your active system so that you can treat it as though it’s thermodynamic?” she asked. That is an open question that she predicted will be answered in the next 10 years.
Schulten remarked that living systems are metastable for a good reason, which is that the result is a more robust system. “Life is not stable, life is changing, and you have to adjust to the circumstances. That is why these systems are never in a very rigid state, because they have to be in different states,” said Schulten. During repair, for example, structures are being disassembled and rebuilt, usually starting from some existing surface that can guide reassembly. “Metastability is a principle of adaptability and robustness,” he added.
Bruce Garrett from PNNL asked the panel to comment on the importance of entropy in these systems, and Voth said that entropy is very important. “I think a lot of current thinking in biology is driven too much by structural biology, which is ordered, but biology is driven by free energy, not equilibrium, so why wouldn’t biology utilize entropy?” said Voth. “What we’re increasingly finding is that proteins may be driven to bind because some part of them becomes disordered and so it’s free energy that’s facilitating that behavior.” Glotzer remarked that entropy is a global concept that cannot be understood locally. By definition, it is an emergent property of a system, she said, and understanding the role of entropy is a terrific opportunity. “We don’t know yet how to design to exploit entropy to give us the structures that we want,” said Glotzer.
Noid commented that, as you change the resolution of the model, what you’re effectively doing is you’re taking configurational entropy out of the high-resolution model and you’re transferring it into the effect of interactions of the low-resolution model. And so, depending upon the level of resolution that you look at and describe your system with, your interactions become increasingly entropic and decreasingly energetic; you can show that very simply and mathematically.” He added that there are cases where there are general design principles that are well understood with regard to how entropy drives those processes, but in terms of predicting the role of entropy in driving most systems, that is still a challenging prospect. Schulten made the final comment, which was that in protein folding there is a very subtle balance between entropy and enthalpy that can be shifted by very subtle factors. As an example, he cited the case of blood clotting, which is triggered by a conformational change in protein structure triggered by small shifts in blood flow.














