
4
Information Related to Biologic Plausibility
The committee reviewed all the relevant experimental studies of 2,4-di-chlorophenoxyacetic acid (2,4-D), 2,4,5-trichlorophenoxyacetic acid (2,4,5-T), 4-amino-3,5,6-trichloropicolinic acid (picloram), dimethylarsinic acid (DMA, also called cacodylic acid), and 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) that have been published since Update 2012 (IOM, 2014), and it has incorporated the findings into this chapter when it is appropriate and into the biologic-plausibility sections of Chapters 7–13 when they are of consequence for particular health outcomes. For each substance, this chapter includes a review of toxicokinetic properties, a brief summary of the toxic outcomes investigated in animal experiments, and a discussion of underlying mechanisms of action as illuminated by in vitro studies. The final section of this chapter discusses complicating factors in extrapolating findings from laboratory experimentation to humans and two emerging subjects in molecular and biologic science that provide novel insights into the potential mechanisms of xenobiotic-induced disease, which may thereby establish greater biologic plausibility for various toxic responses being associated with exposure to the herbicides sprayed in Vietnam.
The establishment of biologic plausibility through laboratory studies strengthens the evidence of a cause–effect relationship between herbicide exposure and health effects reported in epidemiologic studies, and thus supports the existence of the less stringent relationship of association, which is the target of this committee’s work. Experimental studies of laboratory animals or cultured cells make it possible to observe the effects of herbicide exposure under highly controlled conditions, which is difficult or impossible to do in epidemiologic studies. The conditions that are controlled include the genetic differences among people, the frequency and magnitude of exposure, exposure to other chemicals,
and preexisting health conditions, all of which can be controlled in a laboratory animal study.
Once a chemical contacts the body, it becomes subject to the processes of absorption, distribution, metabolism, and excretion. The combination of those four biologic processes determines the concentration of the chemicals in the various tissues and organs in the body and how long each organ or tissue is exposed to it and thus influences its pharmacologic and possibly toxic activity.
The absorption of a substance in an organism normally takes place by uptake into the bloodstream from mucous surfaces, such as the intestinal walls of the digestive tract during ingestion. Low solubility, chemical instability in the stomach, and an inability of the substance to permeate the intestinal wall can all reduce the extent to which the substance is absorbed after being ingested. The solubility of a chemical in fat and its hydrophobicity influence the pathways by which it is absorbed, its relative potential to be metabolized (structurally transformed), and ultimately whether it persists in the body or is excreted. Absorption is a critical determinant of a chemical’s bioavailability, that is, the fraction of it that reaches the systemic circulation. In addition to ingestion, the routes of exposure experienced by humans are inhalation (entry via the airways) and dermal exposure (entry via the skin). Animal studies may involve additional routes of exposure that are not ordinarily encountered by humans, such as intravenous or intraperitoneal injection, in which a chemical is injected into, respectively, the bloodstream or the abdominal cavity.
Distribution refers to the movement of a substance from the site of entry to the tissues and organs where it may have its ultimate effect or be sequestered. Distribution takes place most commonly via the bloodstream.
Metabolism is the process by which a foreign substance is chemically modified when it enters an organism. For many environmental toxicants, this process takes place largely in the liver via the action of enzymes, including cytochrome P450s, which catalyze the oxidative metabolism of many chemicals. As metabolism occurs, the parent chemical is converted into new chemicals called metabolites, which are often more water-soluble (polar) and thus more readily excreted. When the resulting metabolites are pharmacologically or toxicologically inert, metabolism has deactivated the administered dose of the parent chemical and thus reduced its effects on the body. Metabolism may, however, generate a chemical that is more potent or more toxic than the parent compound. Excretion is the removal of substances or their metabolites from the body, most commonly in urine or feces, whereas elimination applies to the disappearance of the parent molecule from the bloodstream. The rate of excretion of a chemical from the body is often limited by the rate of metabolism of the parent chemical into more water-soluble, readily excreted metabolites. Excretion is often incomplete, especially in the case of chemicals that resist biotransformation, and incomplete excretion results in the accumulation of foreign substances that can adversely affect biologic functions. Elimination is referred to as “first-order” when its rate is directly proportional to
TABLE 4-1 Estimates of TCDD Half-Life in Humans and Animals
| Reference | Half-Lifea | Confidence Interval | Comment |
|---|---|---|---|
| Human studies: | |||
| Leung et al., 2006 | 0.4 year | Breastfed infants, 0–1 year after exposure | |
| Aylward et al., 2005a | Toxicokenetic model estimates for exposures: | ||
| < 3 years | > 10,000 pg/g of serum lipid | ||
| > 10 years | < 50 pg/g of serum lipid | ||
| Emond et al., 2005 | PBPK model based on 10 Ranch Hand veterans: | ||
| Weeks | 40,000 pg/g of serum lipid | ||
| >10 years | 138 pg/g of serum lipid | ||
| Flesch-Janys et al., 1996 | 7.2 years | Adult males, Boehringer cohort | |
| Geusau et al., 2002 | 1.7 yearsb | 0–3 years after exposure: | |
| Adult female 1, 144,000 pg/g of serum lipid | |||
| 3.4 yearsb | Adult female 2, 26,000 pg/g of serum lipid | ||
| Kumagai and Koda, 2005 | 1.1–2.3 years | Adult male, incinerator workers, 0–1.3 years after exposure | |
| Michalek et al., 2002 | 0.34 yearb | Adult males, Seveso cohort, 0–3 months after exposure | |
| 6.9 years | 3–16 years after exposure | ||
| 9.8 years | Adult females, Seveso cohort, 3–16 years after exposure | ||
| 7.5 years | Adult males, Ranch Hands, 9–33 years after exposure | ||
| Needham et al., 1994 | 7.8 years | 7.2–9.7 years | Adults, Seveso cohort |
| Pirkle et al., 1989 | 7.1 years | 5.8–9.6 years | Adult males, Ranch Hands, 9–23 years after exposure |
| Milbrath et al., 2009 | 7.2 years | Reference half-life for 48.7-year-old | |
| Sorg et al., 2009 | 15.4 months | Victor Yushchenko: TCDD at 108,000 ppt lipid | |
| Animal studies: | Monkeys | ||
| Neubert et al., 1990 | 73.7 days | 60.9–93.8 days | single injection |
| Mice | |||
| DeVito and Birnbaum, 1995 | 15 days | female B6C3F1 | |
| Gasiewicz et al., 1983 | 11 daysc | C5BL/6J | |
| 24.4 daysc | DBA/2J | ||
| 12.6 daysc | B6D2F1/J | ||
| Koshakji et al., 1984 | 20 days | male ICR/Ha Swiss |
| Reference | Half-Lifea | Confidence Interval | Comment |
|---|---|---|---|
| Rats | |||
| Emond et al., 2006 | Inducible elimination PBPK model estimates: | ||
| 10 days | 103 μg/kg acute treatment | ||
| 75 days | 10-3 μg/kg acute treatment | ||
| Hurst et al., 1998 | 8 days | Pregnant female Long-Evans, excretion from liver | |
| Pohjanvirta and Tuomisto, 1990 | 21.9 days | Male Han/Wistar, resistant strain | |
| Viluksela et al., 1996 | 20.2 days | Long-Evans, TurkuAB strain | |
| 28.9 daysd | Long-Evans, Charles River strain | ||
| Weber et al., 1993 | 16.3 ± 3.0 days | Male Sprague-Dawley |
aHalf-lives of TCDD in humans based on measurement of TCDD in serum samples.
bShorter half-lives measured in humans during first months after exposure or in severely contaminated persons consistent with nonlinear elimination predicted by physiologically based pharmacokinetic (PBPK) models (for example, Carrier et al., 1995). Greater half-life in females attributed to greater BMI index.
cTotal cumulative excretion of 3H-TCDD–derived radioactivity.
dAttributed to differences in dilution due to different growth rates.
the amount of chemical then in the body, which also means that the chemical’s half-life is independent of dose. A half-life is defined as the time required for the plasma concentration or the amount of a chemical in the body to be reduced by half. The half-life of TCDD in humans varies with body mass index (BMI), age, sex, and concentration in the body and has been found to vary from 0.4 to more than 10 years (see Table 4-1).
Collectively, the routes and rates of absorption, distribution, biotransformation or metabolism, and excretion of a toxic substance make up the toxicokinetics (or pharmacokinetics for chemicals used as pharmaceutical agents) of the substance. Those processes determine the amount of a particular substance or metabolite that reaches specific organs or cells and that persists in the body. Understanding the toxicokinetics of a chemical is useful for assembling a valid reconstruction of a human exposure, but it is most important in assessing the risk of effects from exposure to a chemical by determining the concentration of the active chemical in target tissues. The principles involved in toxicokinetics are similar from chemical to chemical, although the degree to which different processes influence distribution depends on the structure and other inherent properties of a particular chemical. Thus, the lipophilicity or hydrophobicity of a chemical and its structure influence the pathways by which it is metabolized and whether it persists in the body or is excreted. The degree to which different toxicokinetic processes influence the toxic potential of a chemical depends on
metabolic pathways, which often differ among species. For that reason, attempts at extrapolation from experimental animal studies to human exposures must be done extremely carefully.
Many chemicals were used by the US armed forces in Vietnam. The nature of the substances themselves was discussed in detail in Chapter 4 of the original Veterans and Agent Orange: Health Effects of Herbicides Used in Vietnam (VAO) report (IOM, 1994). Four herbicides documented in military records were of particular concern and are examined here: 2,4-D, 2,4,5-T, picloram, and cacodylic acid. This chapter also examines TCDD, the most toxic congener of the tetrachlorodibenzo-p-dioxins (tetraCDDs), also commonly referred to as dioxin, which is a contaminant of 2,4,5-T. Considerably more information is available on TCDD than on the herbicides themselves. Other contaminants present in 2,4-D and 2,4,5-T are of less concern. Except as noted, the laboratory studies of the chemicals of concern used pure compounds or formulations; the epidemiologic studies discussed in later chapters often tracked exposures to mixtures.
PICLORAM
Chemistry
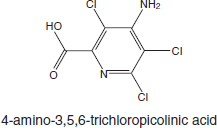
Picloram (Chemical Abstracts Service Number [CAS No.] 1918-02-1; see chemical structure in Figure 4-1) was used with 2,4-D in the herbicide formulation Agent White, which was sprayed in Vietnam. It is also used commonly in Australia in a formulation that has the trade name Tordon 75D®. Tordon 75D contains several chemicals, including 2,4-D, picloram; a surfactant, diethyleneglycolmonoethyl ether; and a silicone defoamer. A number of studies of picloram used such mixtures as Tordon formulations or other mixtures of 2,4-D and picloram that are similar to Agent White.
Toxicokinetics
The original VAO committee reviewed studies of the toxicokinetics of picloram. Studies of animals showed a rapid absorption through the gastrointestinal
tract and a rapid elimination of picloram as the unaltered parent chemical in urine. Nolan et al. (1984) examined the toxicokinetics of picloram in six healthy male volunteers who were given a single oral dose of 0.5 or 5.0 mg/kg or a dermal dose of 2.0 mg/kg. Picloram was rapidly absorbed in the oral study and rapidly excreted unchanged in urine. More than 75 percent of the dose was excreted within 6 hours, and the remainder with an average half-life of 27 hours. On the basis of the quantity of picloram excreted in urine in the dermal study, the authors noted that only 0.2 percent of the picloram applied to the skin was absorbed. Because of its rapid excretion, picloram has low potential to accumulate in humans.
In general, the literature on picloram toxicity continues to be sparse. Studies of humans and animals indicate that picloram is rapidly eliminated as the parent chemical. Studies of animals indicate that picloram is sparingly toxic at high doses.
Toxicity Profile
The original VAO committee reviewed studies of the carcinogenicity, genotoxicity, acute toxicity, chronic systemic toxicity, reproductive and developmental toxicity, and immunotoxicity of picloram. In general, there is some evidence of carcinogenicity in some rodent models but not in other species (NCI, 1978). Because of some concern that contaminants in the picloram (in particular, hexachlorobenzene) might be responsible for the carcinogenicity, picloram itself has not been established as a chemical carcinogen.
Studies conducted by the Environmental Protection Agency (EPA) (1988) yielded no evidence that picloram is a genotoxic agent. Picloram is considered a mild irritant; it has produced erythema in rabbits only at high doses. The available information on the acute toxicity of picloram is paltry. Some neurologic effects—including hyperactivity, ataxia, and tremors—were reported in pregnant rats exposed to picloram at 750 or 1,000 mg/kg (Thompson et al., 1972).
Chronic Systemic Toxicity
Several studies have reported various effects of technical-grade picloram on the livers of rats. In the carcinogenicity bioassay conducted by Stott et al. (1990), treatment-related hepatomegaly, hepatocellular swelling, and altered tinctorial properties were noted in the central regions of the liver lobules in the groups exposed at 60 and 200 mg/kg per day. Males and females exposed at the 200 mg/kg dose had higher liver weights than controls. The no-observed-effect level (NOEL) was 20 mg/kg per day, and the lowest observed-effect level was 60 mg/kg per day for histologic changes in centrilobular hepatocellular tissues. According to EPA (1988), hexachlorobenzene (a contaminant of technical-grade picloram at 197 ppm) was probably not responsible for the hepatic effects. Gorzinski and
colleagues (1987) also reported a dose-related increase in liver weights, hepatocellular hypertrophy, and changes in centrilobular tinctorial properties in male and female F344 rats exposed to picloram at 150 mg/kg per day and higher in the diet for 13 weeks. In a 90-day study, cloudy swelling in the liver cells and bile duct epithelium occurred in male and female F344 rats given 0.3 percent or 1.0 percent technical picloram in the diet (EPA, 1988). Hepatic effects have also been reported in dogs exposed to picloram: Increased liver weights were reported in beagles that received 35 mg/kg per day or more in the diet for 6 months (EPA, 1988). No other effects of chronic exposure to picloram have been reported.
Reproductive and Developmental Toxicity
The reproductive toxicity of picloram was evaluated in a two-generation study; however, few animals were evaluated, and no toxicity was detected at the highest dose tested, 150 mg/kg per day (EPA, 1988). Some developmental toxicity was produced in rabbits exposed to picloram by gavage at 400 mg/kg per day on gestation days (GDs) 6–18. Fetal abnormalities were forelimb flexure, fused ribs, hypoplastic tail, and omphalocele, each occurring in a single litter (John-Greene et al., 1985). Some maternal toxicity was observed at that dose, however, and EPA concluded on the basis of the sporadic nature of the findings that the malformations were not treatment related (EPA, 1988). No teratogenic effects were produced in the offspring of rats given picloram by gavage at up to 1,000 mg/kg per day on GDs 6–15, but the occurrence of bilateral accessory ribs was significantly increased (Thompson et al., 1972).
Immunotoxicity
Studies of the potential immunotoxicity of picloram have included dermal sensitization in humans and rodent immunoassays. In one study, 53 volunteers received nine 24-hour applications of 0.5 mL of a 2 percent potassium picloram solution on the skin of both upper arms. Each volunteer received challenge doses 17–24 days later. The formulation of picloram (its potassium salt) was not a skin sensitizer or an irritant (EPA, 1988). In a similar study, a 5 percent solution of picloram (M-2439, Tordon 101 formulation) produced a slight dermal irritation and a sensitization response in six of the 69 volunteers exposed. When the individual components of M-2439—picloram, triisopropanolamine (TIPA) salt, and 2,4-D TIPA salt—were tested separately, no sensitization reaction occurred (EPA, 1988). Tordon K+, but not technical-grade picloram, was also found to be a skin sensitizer in guinea pigs (EPA, 1988). CD1 mice exposed to Tordon 202C (94 percent 2,4-D and 6 percent picloram) had no consistent adverse effects on antibody responses (Blakley, 1997), but the lack of a consistent response may be due to the fact that CD1 mice are outbred.
Mechanisms
No well-characterized mechanisms of toxicity for picloram are known.
CACODYLIC ACID
Chemistry
Arsenic (As) is a naturally occurring element that exists in a trivalent form (As+3 or AsIII) and a pentavalent form (As+5 or AsV). See Figure 4-2 for the chemical structures of selected arsenic-containing compounds; sodium arsenite, which contains AsIII, is generally considered to be the most toxic of these arsenic compounds. Arsenic is commonly present in drinking-water sources that are associated with volcanic soils and can reach high concentrations (over 50 ppb). Numerous human health effects have been attributed to drinking-water exposure, particularly bladder, skin, and lung cancers and vascular diseases. Arsenic exists
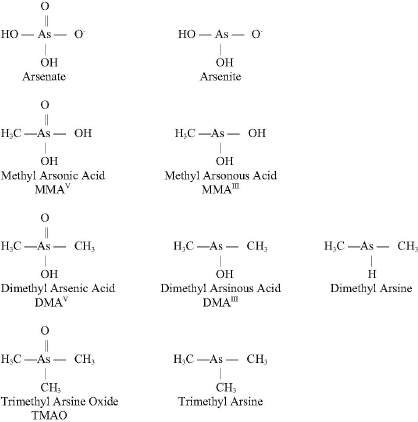
in both inorganic and organic (including methylated) forms and is readily metabolized in humans and other species, and inorganic arsenic can be converted to organic forms. Although organic forms can be converted into inorganic forms by microorganisms in the soil, there is no evidence that this can occur in humans or other vertebrate species (Cohen et al., 2006).
The arsenic in cacodylic acid (CAS No. 75-60-5) has a valence of +5. Cacodylic acid (also known as dimethylarsinic acid [DMAV] by its more standard chemical name) was the form of arsenic used in Agent Blue, one of the mixtures used for defoliation in Vietnam. DMAV made up about 30 percent of Agent Blue. Agent Blue was chemically and toxicologically unrelated to Agent Orange, which consisted of phenoxy herbicides contaminated with dioxin-like compounds.
Potential cacodylic acid exposure of Vietnam veterans would have involved direct exposure to exogenous DMAV, rather than exposure to inorganic arsenic, which would have led to endogenous formation of MMAV and MMAVIII and then DMAV, as shown in Figure 4-3. The old hypothesis that methylation of inorganic arsenic was a detoxifying mechanism has been dispelled by newer studies. Direct treatment of laboratory animals with these metabolic products has demonstrated them to be linked to increased incidence of cancers and non-cancer health outcomes, but there are no studies of health effects in humans following direct exposure to DMA that could provide epidemiologic evidence of association for DMA as required by the Agent Orange Act. It cannot be assumed, particularly given the
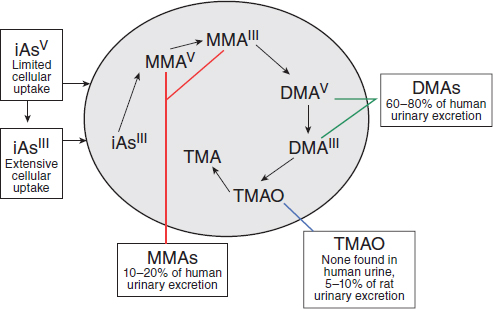
SOURCE: Adapted with permission from Cohen et al., 2006.
apparent lack of endogenous demethylation, that direct exposure to DMA would be equivalent to exposure to inorganic arsenic. Consequently, VAO committees have considered and reviewed toxicologic studies in which animals were directly exposed to DMA, but the extensive literature on the health effects of exposure to inorganic arsenic (including the epidemiologic research, animal experiments, and mechanistic studies) has been regarded as not primarily pertinent to DMA exposure and has not been considered. For information about effects of inorganic arsenic exposure, the reader is referred to recent reviews (IARC, 2012a; NRC, 2013).
Toxicokinetics
Investigations of its metabolism and disposition have generally found DMAV to be rapidly excreted and mostly unchanged in the urine of most animal species after systemic exposure (Cohen et al., 2006; Suzuki et al., 2010). However, rats differ from most other mammals (including humans) in that a larger percentage (10 percent) of DMAV binds to hemoglobin in red blood cells, which leads to a considerably longer half-life in blood (Cui et al., 2004; Suzuki et al., 2004). The binding of DMAV to hemoglobin is 10 times higher in rats than in humans (Lu et al., 2004). Chronic exposure of normal rat hepatocytes to DMAV results in decreased uptake and increased excretion, so that over time they developed resistance to its cytotoxic effects (Kojima et al., 2006); the tolerance was mediated by the induction of glutathione-S-transferase activity and of multiple-drug–resistant protein expression. Adair et al. (2007) examined the tissue distribution of DMA in F344 rats after drinking-water exposure to DMA for 14 days and found that it was extensively metabolized to trimethylated forms that may play a role in toxicity. In a study of DMA treatment of Wistar rats for 10 weeks, the metabolism to trimethylated forms was far less apparent, and the tissue distribution of DMA and trimethylated metabolites was strikingly different (Liu et al., 2015). Thus, there may be differential effects of exposure duration or rat strains, or both, in DMA distribution and metabolism.
A physiologically based pharmacokinetic (PBPK) model of intravenous and ingested DMAV has been developed on the basis of mouse data (Evans et al., 2008). Similar models have been developed for humans on the basis of exposure to inorganic arsenic (El-Masri and Kenyon, 2008), but these models have limited relevance for assessing potential harm to Vietnam veterans who are presumed to have been directly exposed to DMAV.
Although epidemiologic studies of direct exposure to DMAV are not available, investigations into the relationship between health outcomes and the metabolic profiles of humans exposed to inorganic arsenic provide some insight into the roles of the individual metabolites in producing adverse outcome. An increased incidence of urothelial cancer (bladder, kidney, renal pelvis, ureter, and urethra combined) among people exposed to high levels of inorganic arsenic
in drinking water was found in those who generate more MMAV and less DMAV endogenously (Huang SK et al., 2008). Also, lower risk of arsenical skin lesions was associated with evidence of higher arsenic methylation capacity in people in areas of high arsenic exposure via the drinking water (Zhang et al., 2014a,b) and smelter workers (Wen et al., 2012). These results could suggest that elevated cumulative levels of urinary MMAV may be causally associated with increased risk of inorganic arsenic-induced adverse health outcomes, but they could also imply that complete methylation of inorganic arsenic to DMAV and resulting enhanced excretion are relatively protective.
Toxicity Profile
This section discusses the toxicity associated with organic forms of arsenic, most notably DMAV because it is the active ingredient in Agent Blue. The toxicity of inorganic arsenic is not considered relevant to veteran exposures to Agent Blue.
Neurotoxicity
Kruger et al. (2006) found that both DMAIII and DMAV significantly attenuated neuronal ion currents through N-methyl-D–aspartate receptor ion channels, whereas only DMAV inhibited ion currents through α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptors. The data suggest that those methylated forms of arsenic may have neurotoxic potential.
Immunotoxicity
Previous studies have shown that a low concentration of DMAV (10–7 M) could increase the proliferation of human peripheral blood monocytes after their stimulation with phytohemagglutinin, whereas it took a high concentration (10–4 M) to inhibit release of interferon-γ. This suggested that various immunomodulatory effects of DMAV have their own concentration specificity (Di Giampaolo et al., 2004).
Skin Toxicity
In a recent evaluation of the effects of topical exposure of pregnant mice to DMA (valence not stated) on the skin of the dams and offspring (Kim E et al., 2012), no effects were observed in offspring, but the exposure did increase skin thickness in the area of application and alter the expression of apoptosis-related genes (Bcl-2, Bad, caspase-12). The results suggested that transient DMA exposure can be a skin irritant and produce dermatitis.
Genotoxicity and Carcinogenicity
DMAIII and DMAV are genotoxic and increase oxidative stress and cause DNA damage, particularly aneuploidy, but they are, such as inorganic arsenic species, poor mutagens (Rossman and Klein, 2011). Gómez et al. (2005) demonstrated that DMAIII induces a dose-related increase in DNA damage and oxidative stress in Jurkat cells. DMAIII was considerably more potent than DMAV in inducing DNA damage in Chinese hamster ovary cells (Dopp et al., 2004), which was associated with a greater uptake of DMAIII into the cells. An additional study showed that DMAV permeates membranes poorly, but when forced into cells by electroporation it can induce DNA damage (Dopp et al., 2005). Similarly, an analysis of arsenical dimethylated metabolites in human bladder cancer cells found dimethylmonothioarsinic acid (DMMTAV) and DMAIII to be the most toxic and DMAV to be less toxic in terms of DNA damage (Naranmandura et al., 2011). DNA damage from DMMTAV was shown to be related to the accumulation of reactive oxygen species and down-regulation of p53 and p21 (DNA repair proteins); these processes were mediated in part through intracellular conversion of DMMTAV to DMAV and DMAIII (Naranmandura et al., 2011). Thus, although extracellular DMAV has little toxic effect in cells because of its low uptake, intracellular DMAV can be highly toxic. Gene-expression profiling of bladder urothelium after chronic exposure to DMAV in drinking water showed significant increases in genes that regulate oxidative stress (Sen et al., 2005), whereas hepatic gene-expression profiling showed that DMAV exposure induced changes consistent with oxidative stress (Xie et al., 2004). In vivo, DMAV-induced proliferation of the urinary bladder epithelium could be attenuated with the antioxidant N-acetylcysteine (Wei et al., 2005). Arsenicals, including DMA, also interfere with certain DNA repair mechanisms, both base- and nucleotide-excision repair, and may thereby act as co-carcinogens enhancing the effect of other genotoxic carcinogens (Rossman and Klein, 2011). In fact, DMA is a stronger inhibitor of nucleotide-excision repair than inorganic arsenic (Shen et al., 2008).
DMAIII and DMAV are carcinogenic. Cancers have been induced in the urinary bladder, kidneys, liver, thyroid glands, and lungs of laboratory animals exposed to high concentrations of DMA. In a 2-year bioassay of F344/Crl rats fed a diet containing 40 or 100 ppm DMAV, the females consuming the highest dose (100 ppm) developed urothelial carcinomas and papillomas in the bladder, and males and females at both dose levels (40 and 100 ppm) developed hyperplastic nonneoplastic changes in the bladder (Arnold et al., 2006). Wei et al. (2002) exposed male F344/DuCrj rats to DMA via the drinking water and found statistically significant incidences of bladder hyperplasia and transitional cell papillomas and carcinomas at doses of 50 and 100 ppm. Similarly, Wang A et al. (2009) found that exposure of F344 rats to DMAV in drinking water at 1, 4, 40, or 100 ppm resulted in a change in the urinary bladder epithelium, but there were no changes in DNA repair capacity. In another study, Cohen et al. (2007b) exposed
F344 rats to DMAV in the diet for 2 years and found an increase in bladder tumors in those receiving 100 ppm; the researchers postulated that trimethylated forms of arsenic may be responsible for bladder cancer in rats. Direct intravesical administration of 90 mg/kg DMAV to female adult rats resulted in increased bromodeoxyuridine labeling in urothelial cells, indicating DNA damage, weak neutrophil infiltration, and the proliferation of urothelial epithelium mediated through modest increases in oxidative-stress indexes (Takahashi et al., 2011). Increased urothelial cell proliferation was also found following DMA exposure via the drinking water (Wei et al., 2002). It is noteworthy that co-treatment with an antioxidant, N-acetylcysteine, worsened the DMAV-induced bladder injury rather than ameliorating it as expected, suggesting that the carcinogenic mechanism of DMAV is more complicated than simple production of oxidative stress. In the mouse lung, DMAV acts as a tumor initiator (Yamanaka et al., 2009) and as a tumor promoter (Mizoi et al., 2005). DMAV can also act as a complete carcinogen, inducing lung tumors in susceptible strains of mice, including those with deficient DNA-repair activity (Hayashi et al., 1998; Kinoshita et al., 2007). In F344/DuCrj rats treated with a mixture of carcinogens for 4 weeks, subsequent exposure to DMA (not indicated whether this was DMAIII or V) via the drinking water for 24 weeks caused tumor promotion in the urinary bladder, kidney liver, and thyroid gland but inhibited the induction of tumors of the nasal passages (Yamamoto et al., 1995). In a similarly designed experiment, DMA (not indicated whether this was DMAIII or V) was found to be a bladder tumor promotor after treatment with the bladder carcinogen N′-butyl-N′-(4-hydroxybutyl) nitrosamine (Wanibuchi et al., 1996). Yamanaka et al. (2009) suggested that DMAIII can act as a tumor promoter through the formation of a DMAIII radical after the reduction of DMAV. Recent studies have also found that oral exposure of adult mice to 200 ppm DMAV in addition to fetal arsenic exposure can act as a promoter of renal and hepatocellular carcinoma, markedly increasing tumor incidence beyond that produced by the fetal arsenic exposure alone (Tokar et al., 2012). These findings emphasize how multiple life events can contribute to an adverse health outcome in which adult DMAV exposure triggered an otherwise dormant disease.
Mechanisms
Oxidative stress is a common theme that runs through the literature on the mechanisms of action of arsenic, particularly with regard to cancers in animals, although some studies have suggested that methylated arsenicals (MMAIII and DMAIII) can induce aneuploidy in mammalian cells at concentrations below those required to produce oxidative stress after in vitro exposure (Kligerman and Tennant, 2007) and that oxidative stress can be induced at non-cytotoxic concentrations (Rossman and Klein, 2011). Other studies have shown that mice that are deficient in enzymes associated with repair of oxidative DNA damage are highly susceptible to induction of tumors, particularly lung tumors, by DMAV (Kinoshita
et al., 2007). The chemical reaction of arsenicals with thiol groups in sensitive target tissues, such as red blood cells and kidneys, may also be a mechanism of action of organic arsenicals (Naranmandura and Suzuki, 2008).
Cohen et al. (2007b) postulated that cytotoxicity-induced regenerative cell proliferation in the urothelium is a major factor in the carcinogenicity of DMA to the rat bladder, and indeed urothelial cell proliferation is increased following DMA exposure (Takahashi et al., 2011; Wei et al., 2002). However, whether this can be taken to indicate that there is a dose threshold for DMA carcinogenicity remains uncertain in view of the above mentioned in vitro data from Kligerman and Tennant (2007) and Rossman and Klein (2011). There may also be epigenetic effects involved in DMA carcinogenicity, as suggested by a study in humans in which there was a significant association between global DNA methylation and urinary DMA levels (Tellez-Plaza et al., 2014).
The variation in the susceptibility of various animal species to tumor formation caused by inorganic and organic arsenic is thought to depend heavily on differences in metabolism and distribution. Thus, genetic differences may play an important role. Numerous investigators have examined potential human susceptibility factors and gene polymorphisms that may increase a person’s risk of cancer and other diseases induced by arsenicals (Aposhian and Aposhian, 2006; Hernandez et al., 2008; Huang SK et al., 2008; Huang YK et al., 2008; McCarty et al., 2007; Meza et al., 2007; Steinmaus et al., 2007, 2010), but as yet polymorphisms that may contribute to a person’s susceptibility to DMA-induced cancers or tissue injury have not been identified.
Summary
DMA is genotoxic and carcinogenic in certain animal models and in vitro assays, including studies in human cells, and it interferes with DNA repair mechanisms and has epigenetic effects that may be involved in gene damage and carcinogenesis. However, it is not clear whether these effects would also occur in humans directly exposed to DMA.
PHENOXY HERBICIDES: 2,4-DICHLOROPHENOXY ACID
AND 2,4,5-TRICHLOROPHENOXYACETIC ACID
Chemistry
2,4-D (CAS No. 94-75-7) is an odorless crystalline powder that, when pure, is white in color (see Figure 4-4); it may appear yellow when phenolic impurities are present. The melting point of 2,4-D is 138°C, and the free acid is corrosive to metals. It is soluble in water and in a variety of organic solvents (such as acetone, alcohols, ketones, ether, and toluene). 2,4,5-T (CAS No. 93-76-5) is an odorless, white to light-tan solid with a melting point of 158°C. 2,4,5-T is noncorrosive

and is soluble in alcohol and water. It reacts with organic and inorganic bases to form salts and with alcohols to form esters.
Uses of 2,4-D and 2,4,5-T
2,4-D has been used commercially in the United States since World War II to control the growth of broadleaf plants and weeds on range lands, lawns, golf courses, forests, roadways, parks, and agricultural land; it remains a widely used herbicide approved for use by the European Union and EPA. Formulations include 2,4-D amine and alkali salts and esters, which are mobile in soil and readily absorbed through the leaves and roots of many plants. Like 2,4-D, 2,4,5-T was developed and marketed as a herbicide during World War II. However, the registration for 2,4,5-T was canceled by EPA in 1978 when it became clear that it was contaminated with TCDD during the manufacturing process. It is recognized that the production of 2,4-D also involves the generation of some dioxin contaminants, even some with dioxin-like activity, but the fraction of TCDD is comparatively very small, as discussed in Chapter 3 in conjunction with Figure 3-1, which describes the chemistry leading to TCDD contamination during the manufacture of 2,4,5-T from tricholophenol.
The herbicidal properties of 2,4-D and 2,4,5-T are related to the chemical’s ability to mimic the plant growth hormone indole acetic acid. They are selective herbicides in that they affect the growth of only broadleaf dicots (which include most weeds) and do not affect monocots, such as wheat, corn, and rice.
Toxicokinetics
Several studies have examined the absorption, distribution, metabolism, and excretion of 2,4-D and 2,4,5-T in animals and humans. Data on both compounds are consistent among species and support the conclusion that the absorption of oral or inhaled doses is rapid and complete. A recent study indicates that 2,4-D can bind to innate intestinal, intracellular lipid-binding proteins, which may be how these compounds move through columnar absorptive epithelial cells from the intestines to systemic distribution (Carbone and Velkov, 2013). Absorption
through the skin is much lower but may be increased with the use of sunscreens or alcohol (Brand et al., 2002; Pont et al., 2004). After absorption, 2,4-D and 2,4,5-T are distributed widely in the body but are eliminated quickly, predominantly in unmetabolized form in urine (Sauerhoff et al., 1977), but 2,4,5-trichlorophenol and 2,4-dichlorophenol have been identified as trace metabolites in urine. The half-life of single doses of 2,4-D or 2,4,5-T in humans has been estimated to be about 18–23 hours and is highly dependent on urinary pH (Gehring et al., 1973; Kohli et al., 1974; Sauerhoff et al., 1977; WHO, 1984). Hines et al. (2003) found that concentrations of 2,4-D and its metabolites in the urine of herbicide applicators—that is, those who apply the herbicides—were consistent with 2,4-D urinary half-life estimates of 13–40 hours in humans.
Toxicity Profile
The toxicity database on 2,4-D is extensive,1 whereas the available data on the toxicity of purified 2,4,5-T, independent of its contamination by TCDD, are sparse. TCDD is much more toxic than 2,4,5-T, and much of the toxicity attributed to 2,4,5-T in early studies was later shown to be caused by the TCDD contaminant. The following summary therefore focuses on 2,4-D toxicity, and information on pure 2,4,5-T is added when it is available.
After a single oral dose, 2,4-D is considered to produce moderate acute toxicity with an LD50 (dose lethal to 50 percent of exposed animals) of 375 mg/kg in rats, 370 mg/kg in mice, and from less than 320 to 1,000 mg/kg in guinea pigs. Rats and rabbits have dermal LD50s of 1,500 mg/kg and 1,400 mg/kg, respectively. 2,4,5-T itself also produces moderate acute toxicity, with oral LD50s of 389 mg/kg in mice and 500 mg/kg in rats. Death from acute poisoning with 2,4-D or 2,4,5-T has been attributed to the ability of the chemicals to uncouple oxidative phosphorylation, a vital process used by almost all cells in the body as the primary means of generating energy. After exposure to a high dose, death due to multiple organ failure can occur rapidly. Studies in rats, cats, and dogs indicate that the central nervous system is the principal target organ for acute 2,4-D toxicity in mammals and suggest that the primary site of action is the cerebral cortex or the reticular formation (Arnold et al., 1991; Dési et al., 1962a,b). Based on case reports, neurotoxicity in humans is the predominant effect of acute inhalation and oral exposure to 2,4-D; symptoms include stiffness of the arms and legs, lack of coordination, lethargy, anorexia, stupor, and coma. 2,4-D is also an irritant of the gastrointestinal tract, causing nausea, vomiting, and diarrhea.
Chronic exposure to 2,4-D at relatively high concentrations has been shown to produce a variety of toxic effects, including hepatic and renal toxicity, neurotoxicity, and hematologic changes. A NOEL of 2,4-D of 1 mg/kg was identified
________________
1See http://toxnet.nlm.nih.gov, search on “2,4-D” or “2,4,5-T,” accessed July 24, 2015.
for renal toxicity in rats (Hazleton Laboratories America, 1986). Exposure to 2,4-D was associated with reduced survival and decreased growth rates of offspring of mothers fed high doses during pregnancy, which were associated with maternal toxicity (Munro et al., 1992). Charles et al. (2001) found 2,4-D did not affect fertility or produce teratogenic effects in the offspring of rats or rabbits gavaged with doses lower than 90 mg/kg/day, which caused overt maternal toxicity. A recent one-generation study in which rats were fed diets containing 2,4-D (females: up to 40 mg/kg/day; males: up to 45 mg/kg/day) from 4 weeks before breeding through 3 weeks of lactation confirmed these results, and furthermore found that even at the highest exposure there is no evidence of interaction with the androgen, estrogen, or steroidogenesis pathways in the pups (Marty et al., 2013). Other studies, however, suggest that exposure to 2,4-D does have an impact on the male reproductive system (Alves et al., 2013; Joshi et al., 2012). Exposure of adult male rats to 2,4-D at doses as low as 150 mg/kg for 30 days resulted in a reduction in the weight of the testes, prostate, epididymis, and seminal vesicles and a reduction in sperm density (Joshi et al., 2012). Alves et al. (2013) show that the exposure of rat Serotoli cells in culture at 10 μM 2,4-D resulted in alterations in cellular metabolism linked to effective spermatogenesis, which could be a mechanism that would reduce sperm density. Mazhar et al. (2014) treated pregnant rats by gavage on GDs 1–19 with 100 mg/kg/day 2,4-D alone or administered with 100 mg/kg/day of the antioxidant vitamin E. Morphological and skeletal defects and low birth weight were observed in the fetuses of dams treated only with 2,4-D, but not in those whose mothers were also treated with vitamin E, thereby suggesting that 2,4-D exposure elicits fetotoxicty through inducing oxidative stress. In vitro exposure of human erythrocytes to 2,4-D caused changes in antioxidant enzyme activity as well as increased protein carbonyls, also indicating induction of oxidative stress (Bukowska, 2003). Similar changes were observed in the liver of exposed rats (Tayeb et al., 2010, 2013). Immunotoxicity of 2,4-D has been reported in a small number of studies, including a few studies of 2,4-D applicators showing both immunosuppression (Faustini et al., 1996) and immuno-stimulation (Figgs et al., 2000; Holland et al., 2002). At high doses that produced clinical toxicity in experimental animals, a suppression of the antibody response was observed, whereas other measures of immune function were normal. The immunotoxicity of 2,4,5-T has not been evaluated in laboratory animals.
The carcinogenicity of 2,4-D and 2,4,5-T has been studied in rats, mice, and dogs after exposure in their food, direct placement in their stomachs, or exposure of their skin. Early studies in mice (NTIS, 1968) and rats (Hansen et al., 1971) found little to suggest tumor induction when animals were treated with 2,4-D by gavage or subcutaneously. Hazelton Laboratories of America (1986, 1987) conducted a series of studies in rats and mice, which all had negative results except one that found an increased incidence of brain tumors in male rats—but not female rats—that received the highest dose of 45 mg/kg/day 2,4-D in their feed.
The occurrence of malignant lymphomas in dogs kept as pets was reported to be higher when owners reported that they used 2,4-D on their lawns than when they did not (Hayes et al., 1991, 1995), but detailed reanalysis did not confirm this finding (Kaneene and Miller, 1999). A controlled study that used dogs exposed to 2,4-D in the laboratory had negative results. Timchalk (2004) suggested that dogs are not relevant for comparative evaluation of human health risk attributable to 2,4-D exposure, because they excrete 2,4-D less efficiently than rats or humans. Given the degree of variability observed in humans (Hines et al., 2003), however, the canine information might be applicable for some people.
2,4-D is not metabolized to reactive intermediates capable of interacting with DNA, and the evidence supports the conclusion that 2,4-D is not a genotoxic carcinogen. However, Sandal and Yilmaz (2011) found that lymphocytes from smokers show genotoxic damage after exposure to 2,4-D, whereas lymphocytes from non-smokers do not, which suggests that although 2,4-D may not be a carcinogen, it may influence the activity of known carcinogens.
2,3,7,8-TETRACHLORODIBENZO-P-DIOXIN
Chemistry
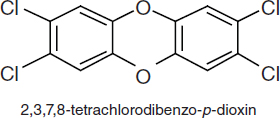
TCDDs are polychlorinated dibenzo-p-dioxins that have a triple-ring structure consisting of two benzene rings connected by an oxygenated ring with four attached chlorine atoms; in the case of the dioxin congener of greatest concern, 2,3,7,8-TCDD (commonly called simply TCDD), the chlorine atoms are attached at the 2, 3, 7, and 8 positions of the benzene rings (see Figure 4-5). The chemical properties of TCDD include a molecular weight of 322, a melting point of 305–306°C, a boiling point of 445.5°C, and a log octanol–water partition coefficient of 6.8 (National Toxicology Program substance profile). It is very lipophilic or fat soluble, is virtually insoluble in water (19.3 ng/L), and is soluble in organic solvents, such as benzene and acetone. It has been suggested that volatilization of dioxin from water may be an important mechanism of transfer from the aqueous to the atmospheric phase (EPA, 2004); however, because of its very low water solubility, most TCDD is bound to sediments and particulate matter.
Toxicokinetics
The disposition of TCDD (which includes its absorption, distribution, biotransformation, and excretion) have been extensively studied in humans and a number of other animal models in the past 30 years. Given the plethora of data, this section highlights and summarizes only key findings.2
TCDD is absorbed into the body rapidly but is eliminated slowly. Because it is very lipophilic, resistant to biotransformation, and slowly eliminated, the concentration of TCDD in the lipid fraction of blood serum is thought to be in dynamic equilibrium with that in the lipid fraction in other tissue compartments. Thus, the lipid-adjusted blood serum concentration of TCDD is used to estimate total body burdens; at high TCDD concentrations, however, the liver sequesters some of the dioxin, so a lipid adjustment that ignores the hepatic fraction would underestimate the total body burden. The exposure of humans to TCDD is thought to occur primarily via the mouth, skin, and lungs. In laboratory animals, oral administration of TCDD has been shown to result in absorption of 50 to 93 percent of the administered dose (Nolan et al., 1979; Rose et al., 1976). Similarly, a study performed in a 42-year-old man found that 87 percent of the oral dose was absorbed (Poiger and Schlatter, 1986). Dermal absorption appears to be dose-dependent: Lower absorption occurs at higher doses (Banks and Birnbaum, 1991). Studies performed in vitro with tissues isolated from humans indicate that human skin may not be readily penetrable (Weber et al., 1991). The varied and complex environmental matrices make environmental exposures difficult to quantify. Animal studies have demonstrated that the presence of soil or lipophilic agents dramatically reduces dermal absorption of TCDD: Application in an activated carbon–water paste essentially eliminates absorption in contrast with the absorption of the pure compound dissolved in solvents. Oral bioavailability of TCDD and related compounds also depends on the matrix: Contaminated breast milk and food products have much higher bioavailability than soil-bound or sediment-bound TCDD, and activated carbon essentially blocks oral bioavailability (Olson, 2012).
After ingestion and gastrointestinal absorption, TCDD associates primarily with the lipoprotein fraction of the blood and later partitions into the cellular membranes and tissues (Henderson and Patterson, 1988). TCDD is distributed to all compartments of the body; the amounts differ from organ to organ, but most studies indicate that the primary distribution of TCDD is in the liver and adipose tissues. For example, in a human volunteer it was found that 135 days after ingestion 90 percent of TCDD was in fat (Poiger and Schlatter, 1986), and TCDD persists in adipose tissue in the rhesus monkey (Bowman et al., 1989). The
________________
2A more exhaustive review and support documents may be accessed at http://www.atsdr.cdc.gov/ToxProfiles/tp.asp?id=366&tid=63, http://www.epa.gov/ncea/pdfs/dioxin/nas-review, or http://cfpub.epa.gov/ncea/iris2/chemicalLanding.cfm?substance_nmbr=1024, accessed July 24, 2015.
distribution and elimination of TCDD depend on the tissue examined, the time that has elapsed since exposure, total exposure, and other factors. For example, the concentration of cytochrome P450 1A2 (CYP1A2) in the liver is increased by TCDD (Poland et al., 1989). Direct binding of TCDD to CYP1A2 is thought to result in the sequestration of TCDD in the liver and to inhibit its distribution to other tissues. The importance of CYP1A2 concentrations for the toxic actions of TCDD has also been demonstrated in several laboratory situations; for instance, CYP1A2-knockout mice were more susceptible than wild-type mice to TCDD immunotoxicity (Smialowicz et al., 2008), and maternal hepatic CYP1A2 was found to sequester TCDD and protect mouse fetuses against TCDD-induced teratogenesis (Dragin et al., 2006). In addition, the distribution of TCDD is age dependent, as shown by studies in which young animals displayed the highest concentration of TCDD in the liver and aged animals the highest concentrations in kidneys, skin, and muscle (Pegram et al., 1995). Finally, the rate of elimination of TCDD, particularly after low exposures, depends heavily on the amount of adipose tissue mass (Aylward et al., 2005a; Emond et al., 2005, 2006).
In laboratory animals, TCDD is metabolized slowly. It is eliminated primarily in feces as both the parent chemical and its more polar metabolites. However, elimination appears to be dose dependent; at low doses, about 35 percent of the administered dose of TCDD was detected in the feces; at higher doses, about 46 percent was observed (Diliberto et al., 2001). The dose-dependent occurrence of TCDD metabolites in the feces is thought to be due to the increased expression of metabolizing enzymes at higher doses and to hepatic sequestration, which makes dioxins more available for metabolism.
Milbrath et al. (2009) conducted a comprehensive review of studies that reported the congener-specific elimination rates of TCDD and related compounds and analyzed the relationships between the apparent half-lives of the compounds as a function of age, body fat, smoking status, and breastfeeding. In infants (under 2 years old), the compounds have a reported half-life of 0.4 year (Leung et al., 2006), and in adults, a half-life of 7.2 years (Milbrath et al., 2009). Aging results in an increase in and redistribution of body fat and lipophilic chemicals that alters their rate of elimination (Van der Molen et al., 1996). Human studies of the Ranch Hand cohort have consistently found a similar relationship between an increasing half-life of TCDD and an increasing BMI (Michalek and Tripathi, 1999; Michalek et al., 1992, 1996). Smoking and breastfeeding are associated with promoting the elimination of TCDD and, in the case of breastfeeding, exposing infants through breast milk. Polycyclic aromatic hydrocarbons (PAHs) in cigarette smoke are capable of inducing CYP1A1, 1A2, and 1B1, which in turn may increase the rate of metabolism and subsequent elimination of TCDD. A 30 percent decrease in TCDD plasma half-life has been associated with smoking (Flesch-Janys et al., 1996).
Special Case of the Poisoning of Victor Yushchenko
In 2004 Victor Yushchenko, a candidate for the presidency of the Ukraine, was poisoned with TCDD. It led to severe chloracne and a blood serum TCDD concentration of 108,000 ppt (pg/g lipid), which was about 50,000 times as great as that in the general population at the time. The incident provided an opportunity to assess the toxicokinetics of TCDD after what was apparently a single large exposure. Serum and fat analysis of TCDD supports the first-order elimination half-life of 15.4 months in Yushchenko, and the similar decay curves confirmed that TCDD was in equilibrium between serum lipids and subcutaneous fat (Sorg et al., 2009). That is much shorter than the 7.2-year reference half-life reported by Milbrath et al. (2009) and supports the dose-dependent elimination of TCDD, which is associated with the induction of potential TCDD-metabolizing enzymes (CYP1A1, 1A2, and 1B1) in very high TCDD exposures. Two metabolites of TCDD (2,3,7-trichloro-8-hydroxydibenzo-p-dioxin and 1,3,7,8-tetrachloro-2-hydroxydibenzo-p-dioxin) were detected in Yushchenko’s feces, serum, and urine but not in his fat or skin. Over a 12-month period, about 38 percent of the TCDD-derived material was eliminated as metabolites (95 percent in feces, 5 percent in urine) and 62 percent as parent chemical. The metabolite:TCDD ratio in the blood serum was about one-fiftieth of that in the feces; this supports the conclusion that the metabolites were not originally ingested with TCDD (Sorg et al., 2009). The very slow metabolism of TCDD has been previously reported in laboratory animal models (Gasiewicz et al., 1983; Olson, 1986; Olson et al., 1980; Poiger and Schlatter, 1979) and in humans (Wendling et al., 1990). It is also noteworthy that the structures of the human metabolites are the same as previously reported in the rat and dog (Poiger et al., 1982; Sawahata et al., 1982). A continued analysis of Yushchenko’s condition has revealed putative metabolomic and transcriptomic biomarkers that may prove useful for predicting health effects in populations with significant TCDD exposures (Jeanneret et al., 2014; Saurat et al., 2012).
In light of the variables discussed above and the effect of differences in physiologic states and metabolic processes, which can affect the mobilization of lipids and possibly of compounds stored in them, complex PBPK models have been developed to integrate exposure dose with organ mass, blood flow, metabolism, and lipid content in order to predict the movement of toxicants into and out of each organ. A number of modeling studies have been performed recently in an effort to understand the relevance of animal experimental studies to the exposures that occur in human populations (Aylward et al., 2005a,b; Beaudouin et al., 2010; Emond et al., 2005).
Toxicity Profile
Effects on Tissues and Organs of Laboratory Animals
The effects of TCDD in laboratory animals have been observed in a number of species (rats, mice, guinea pigs, hamsters, monkeys, cows, and rabbits) after the administration of a variety of doses and after periods that represent acute exposures (less than 24 hours), subchronic exposures (1 day–3 months), and chronic exposure (more than 3 months). Some differences have been observed between species, particularly with respect to the degree of sensitivity, but in general the effects observed are qualitatively similar. Relatively high exposures of TCDD affect a variety of organs and result in organ dysfunction and death. The lethal toxicity of TCDD waries widely among animal species; the oral LD50 of the chemical varies from 1 μg/kg in guinea pigs to 5,000 μg/kg in hamsters. The developing fetus, however, is especially vulnerable to TCDD exposure, and there is only about a 10-fold variability in fetal lethal potency among these species (Kransler et al., 2007; Peterson et al., 1993; Poland and Knutson, 1982). One characteristic of TCDD exposure is a wasting syndrome that includes the loss of adipose and muscle tissue and severe weight loss, but the specific mechanisms of lethality remain unknown. In most rodents, exposure to TCDD leads to hepatic enlargement, the presence of hepatic lesions, and impaired hepatic function. The thymus is also sensitive. Finally, in both humans and nonhuman primates, TCDD exposure results in chloracne and associated dermatologic changes. As will be discussed in more detail in Chapters 7–13, studies performed in animal models have indicated that exposure to TCDD adversely affects the heart, the skin, and the immune, endocrine, and reproductive systems and increases the incidence of cancers of the liver, skin, thyroid, adrenal cortex, hard palate, nasal turbinates, tongue, and respiratory and lymphatic systems (ATSDR, 1998; Barouki et al., 2012; Birnbaum, 1994; Huff et al., 1994; Knerr and Schrenk, 2006). When TCDD has been administered to pregnant animals, birth defects—such as cleft palate, malformations of the reproductive organs of male and female progeny, and abnormalities in the cardiovascular, pulmonary, endocrine, skeletal, and nervous systems—have been observed. Of course, effects arising from perinatal exposure are not in question for Vietnam veterans themselves, but this activity is of concern with respect to their offspring. The developmental origins of health and disease are discussed in more detail in Chapter 10.
Effects on Enzymes, Hormones, and Receptors in
Laboratory Animals and Cultured Cells
In addition to adversely affecting the ability of specific organs to fulfill their normal physiologic roles, TCDD has been found to alter the function and expression of essential proteins, particularly a number of enzymes. The enzymes
that are most affected by TCDD are ones that act on or metabolize xenobiotics and hormones, often by changing the chemicals’ polarity (water solubility), and thus promoting the elimination of the metabolites. Among the enzymes affected by TCDD, the best studied is CYP1A1, which metabolizes some xenobiotics. In laboratory animals, exposure to TCDD commonly results in an increase in CYP1A1 in most tissues; CYP1A1 therefore is often used as a marker of TCDD exposure. Related enzymes whose levels are also increased with TCDD exposure include CYP1B1 and CYP1A2, which together with CYP1A1 are capable of biotransforming some procarcinogens to potentially mutagenic and carcinogenic metabolites.
In addition to CYP1A1 and CYP1A2, TCDD can affect other enzymes that metabolize hormones, such as thyroid hormones, retinoic acid, testosterone, estrogens, and adrenal steroids. Those hormones transmit their signals by interacting with specific proteins called receptors and in this manner initiate a chain of events in many tissues of the body. For example, the binding of the primary female sex hormone, estrogen, to the estrogen receptor promotes the formation of breasts and the thickening of the endometrium, regulates the menstrual cycle, and influences brain development. Exposure to TCDD can increase the metabolism of estrogen and thus lead to a decrease in the amount of estrogen available for binding and activating the estrogen receptor. The ultimate effect of TCDD is an interference with all the bodily functions that are regulated by estrogens. Similarly, the actions of TCDD on the adrenal steroids can adversely affect their ability to regulate glucose tolerance, insulin sensitivity, lipid metabolism, body weight, vascular function, and cardiac remodeling. In addition to changing the amount of hormone present, TCDD has been found to interfere with the ability of receptors to fulfill their role in transmitting hormone signals. Those actions of TCDD on enzymes and hormone receptors are thought to underlie, in part, the observed developmental and reproductive effects and cancers that are hormone responsive.
Effects on Paths of Cellular Differentiation
The broad spectrum of TCDD effects on hormone and growth factor systems, cytokines, and other signal-transducer pathways indicates that TCDD is an extremely powerful growth dysregulator (Birnbaum, 1994). Research performed primarily in cultured cells has shown that TCDD can affect the ability of cells to undergo such processes as proliferation, differentiation, and apoptosis. During the proliferation process, cells grow and divide. When cells are differentiating, they are undergoing a change from less specialized to more specialized. Cellular differentiation is essential for an organism to mature from a fetal to an adult state. In the adult, proper differentiation is required for the normal functioning of the body, for example, in maintaining a normally responsive immune system. The processes of controlled cell death, such as apoptosis, are similarly important during development of the fetus and are necessary for normal physiologic functions
in the adult. Apoptosis is a way for the body to eliminate damaged or unnecessary cells. The ability of a cell to undergo proliferation, differentiation, and apoptosis is tightly controlled by an intricate network of signaling molecules that allows the body to maintain the appropriate size and number of all the specialized cells that form the fabric of complex tissues and organs. Any disruption of the network that alters the delicate balance of cell fate can have severe consequences, including impairment of the function of the organ because of the absence of specialized cells. Alternatively, the presence of an excess of some kinds of cells can result in the formation and development of tumors. Thus, the ability of TCDD to disrupt the normal course of a specific cell to proliferate, differentiate, or undergo apoptosis is thought to underlie (at least in part) its adverse effects on the immune system and the developing fetus and its ability to promote the formation of some cancers.
Mechanisms
TCDD binds and activates the aryl hydrocarbon receptor (AHR) in the cells of virtually every tissue in the body. The ability of TCDD to bind to the AHR with high affinity is necessary—but not sufficient—to produce most of the adverse effects associated with TCDD exposure, including those from direct TCDD binding to and activation of the AHR and later alterations in the expression of TCDD-regulated genes as well as to those signaling pathways altered through interactions with the AHR pathway (Poland and Knutson, 1982; Safe, 1990; Schmidt and Bradfield, 1996; Whitlock, 1990).
The AHR functions as a ligand-activated nuclear transcription factor. Upon binding of agonists (ligands), such as TCDD, the AHR forms a heterodimer with a structurally related protein called AHR nuclear translocator (ARNT). The dimeric complex binds to core DNA sequences called xenobiotic-responsive elements (XREs) or dioxin-responsive elements (DREs) in the promotor region of responsive genes and enhances the transcription of those genes. Many of the AHR-regulated genes encode drug-metabolizing enzymes, such as CYP1A1, CYP1A2, CYP1B1, and a variety of phase II conjugating enzymes. Although the up-regulation of these enzymes is a sensitive biomarker of exposure to TCDD and in part contributes mechanistically to some of the adverse effects of TCDD, the tissue-, species-, time-, and dose-specific modulation (increase or decrease) of many genes is thought to contribute to the wide array of toxic responses to TCDD exposure (Black et al., 2012; Boverhof et al., 2006; Ovando et al., 2006, 2010; Perdew, 2008; Puga et al., 2009; Schneider et al., 2014).
AHR Signaling Pathways
The primary and most intensely studied pathway by which TCDD elicits biologic responses is depicted in Figure 4-6. In the absence of a bound ligand,
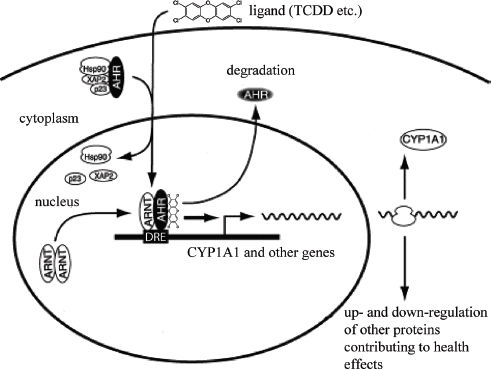
the inactive AHR is retained in the cytoplasm of the cell in a complex consisting of two molecules of the heat-shock protein Hsp90, one molecule of prostaglandin E synthase 3 (p23) (Kazlauskas et al., 1999), and one molecule of the immunophilin-like protein hepatitis B virus X-associated protein 2 (XAP2) (Petrulis et al., 2003), previously identified as either AHR-interacting protein (AIP; Ma and Whitlock, 1997) or AHR-associated protein 9 (ARA9; Carver and Bradfield, 1997). The hsp90 dimer–p23 complex plays multiple roles in the protection of the AHR from proteolysis, maintaining it in a conformation that makes it accessible to ligand binding at the same time that it prevents the premature binding of ARNT (Carver et al., 1994; Pongratz et al., 1992; Whitelaw et al., 1993). XAP2 interacts with the carboxyl terminus of hsp90 and with the AHR nuclear-localization signal (NLS), a short amino acid domain that targets the receptor for interaction with nuclear-transport proteins. The binding of XAP2 blocks such an interaction, preventing the inappropriate trafficking of the receptor into the nucleus (Petrulis et al., 2003).
The binding of ligands (such as TCDD) induces the release of XAP2 and the exposure of the NLS and leads to the binding of nuclear-import proteins and
translocation of the cytosolic complex into the nucleus (Davarinos and Pollenz, 1999; Song and Pollenz, 2002). Once in the nucleus, Hsp90, p23, and XAP2 dissociate from the AHR, which allows the binding of ARNT (Hoffman et al., 1991; Probst et al., 1993). The activated AHR–ARNT heterodimeric complex is then capable of directly or indirectly interacting with DNA by binding to recognition sequences in the regulatory region of responsive genes (Dolwick et al., 1993; Probst et al., 1993).
The canonical DNA recognition motif of the AHR–ARNT complex is referred to as the AHR-responsive element (AHRE, also referred to as the DRE or the XRE, for dioxin- or xenobiotic-response element, respectively). This element is found in the promoter region of AHR-responsive genes and contains the core sequence 5′-GCGTG-3′ (Shen and Whitlock, 1992), which is part of a more extensive consensus-binding sequence, 5′-T/GNGCGTGA/CG/CA-3′ (Lusska et al., 1993; Yao and Denison, 1992). The AHR–ARNT complex binds to the AHRE core sequence in such a manner that ARNT binds to 5′-GTG-3′ and AHR binds to 5′-TC/TGC-3′ (Bacsi et al., 1995; Swanson et al., 1995). A second type of element, termed AHRE-II, 5′-CATG(N6)C[T/A]TG-3′, has been shown to be capable of acting indirectly with the AHR–ARNT complex (Boutros et al., 2004; Sogawa et al., 2004). The end result of the process is the recruitment of the transcriptional machinery associated with RNA polymerase II and the initiation of differential changes in the expression of the genes bearing the AHR–ARNT recognition motif. Many of the genes code for proteins responsible for detoxification reactions directed at the elimination of the ligand. Research suggests that posttranslational modifications in histone proteins may modify the response (Hestermann and Brown, 2003; Schnekenburger et al., 2007).
In addition to the widely accepted view that the actions of TCDD are mediated by the binding of the activated AHR–ARNT dimer to AHREs on DNA, which results in altered gene expression (see Figure 4-6), more recent studies suggest that a “nongenomic” pathway within the cytoplasm also contributes to the toxic effects of TCDD, as reviewed by Matsumura (2009). The TCDD-mediated activation of AHR within the cytoplasm does not involve binding to ARNT or DNA and appears to contribute to rapid inflammatory responses associated with TCDD (Sciullo et al., 2008). In several cell lines, the activation of protein kinase C (PKC) and the later activation of the serine phosphorylated form of cytosolic phospholipase A2 (cPLA2) takes place within 15 min of TCDD exposure (Dong and Matsumura, 2008; Park et al., 2007). It is proposed that within the cytoplasm, the TCDD-mediated activation of AHR leads to a rapid increase in intracellular Ca2+, plus activation of cPLA2, protein kinases, and pro-inflammatory proteins, such as cyclooxygenase (COX-2) (Matsumura, 2009). This pathway and other alternative mechanisms of TCDD-mediated AHR activation have also been reviewed by Denison et al. (2011) and Perdew (2008).
AHR Physiology
The vertebrate AHR is presumed to have evolved from its counterpart in invertebrates, in which it serves a ligand-independent role in normal development processes. The ancestral function of the AHR appears to be the regulation of specific aspects of embryonic development, it having acquired the ability to bind xenobiotic compounds only during vertebrate evolution (Hahn, 2001). The invertebrate AHR also functions as a transcription factor and binds to the same dimerization partner (ARNT) and DNA-response elements as the vertebrate protein, but it does not respond to any of the environmental ligands recognized by the vertebrate receptor. Instead, it regulates diverse developmental processes that are independent of exogenous ligand exposure, such as neuronal differentiation during worm development in Caenorhabditis elegans (Huang et al., 2004; Qin and Powell-Coffman, 2004) or normal morphogenesis of legs, antennae, and bristles in Drosophila melanogaster (Adachi-Yamada et al., 2005; Céspedes et al., 2010). In developing vertebrates, the AHR seems to play a role in cellular proliferation and differentiation and, in keeping with this role in invertebrates, also has a developmental role in craniofacial, pulmonary, renal, cardiovascular, and reproductive tract morphogenesis and blood cell differentiation (Birnbaum et al., 1989; Fernandez-Salguero et al., 1997; Lahvis et al., 2005). Other potential functional roles of the AHR include reproduction, innate immunity, tumor suppression, and blood-pressure regulation (Fujii-Kuriyama and Kawajiri, 2010).
The clearest adaptive physiologic response to AHR activation is the induction of xenobiotic-metabolizing enzymes involved in detoxification of toxic ligands. Evidence of that response, which was described above, was first observed in conjunction with the induction of Cyp1a1, which resulted from exposure to PAHs or TCDD and was directly related to the activation of the AHR signaling pathway (Israel and Whitlock, 1983, 1984). Because of the presence of the AHRE motif in their gene promoters, other metabolizing genes were tested and found to be induced by AHR ligands, which led to the identification of a so-called AHR gene battery of phase I and phase II detoxification genes that code for the drug-metabolizing enzymes CYP1A1, CYP1A2, CYP1B1, NQO1, ALHD3A1, UGT1A2, and GSTA1 (Nebert et al., 2000). Presumably, vertebrates have evolved those enzymes to detect a wide array of foreign, potentially toxic chemicals, represented in the wide variety of substrates that the AHR is able to bind to and whose biotransformation and elimination it is able to facilitate.
A potential complication of the adaptive responses elicited by AHR activation is the induction of a toxic response. Toxicity may result from the adaptive response itself if the induction of metabolizing enzymes results in the production of toxic metabolites. For example, the PAH benzo[a]pyrene (B[a]P), an AHR ligand, induces its own metabolism and detoxification by the AHR-dependent signaling mechanism described earlier, but paradoxically becomes bioactivated to a toxic metabolite in several tissues by a metabolism that depends on CYP1A1 and CYP1B1
activity (Harrigan et al., 2004). A second potential source of AHR-mediated toxicity may be aberrant changes in global gene expression beyond those observed in the AHR gene battery. The global changes in gene expression may lead to deleterious changes in cellular processes and physiology. Microarray and other transcriptomic analyses have proved invaluable in understanding and characterizing that response (Boverhof et al., 2006; Martinez et al., 2002; Ovando et al., 2006, 2010; Puga et al., 2000, 2004; Takeda et al., 2012; Vezina et al., 2004).
Endogenous AHR functions likely involve interaction with endogenous ligands that activate specific physiological processes. Several chemicals and chemical classes have been identified as putative endogenous ligands including equilenin, indigoids, 2-(1′H-indole-3′-carbonyl)-thiazole-4-carboxylic acid methyl ester, leukotrienes, heme metabolites, arachidonic acid metabolites, tryptophan metabolites, and ultraviolet (UV) photoproducts of tryptophan (Guyot et al., 2013; Nguyen and Bradfield, 2008). The tryptophan catabolite and AHR ligand kynurenine has been identified as a tumor promoter that suppresses antitumour immune responses and promotes tumour-cell survival and motility (Opitz et al., 2011). The tryptophan UV photoproduct 6-formylindolo[3,2-b]carbazole (FICZ) is a high affinity AHR ligand, an inducer of CYP1A1 (Wei et al., 1998), and a substrate for CYP1A1 (Wincent et al., 2012). This autoregulatory loop maintains endogenous low levels of FICZ that influence circadian rhythms, responses to UV light, homeostasis associated with pro- and anti-inflammatory processes, and genomic stability (Wincent et al., 2012). FICZ has been shown to reduce the inflammatory response in skin inflammation models (Di Meglio et al., 2014) and enhances NK cell control of tumors (Shin et al., 2013).
It is clear that the AHR is an essential component of the toxicity of dioxin and of dioxin-like chemicals (DLCs). Homozygous deletion of the AHR in mice leads to a phenotype that is resistant to the toxic effects of TCDD and to the carcinogenic effects of B[a]P (Fernandez-Salguero et al., 1996; Lahvis and Bradfield, 1998; Schmidt et al., 1996). The AHR knockout mice, however, have other phenotypic effects, including reduced liver size, hepatic fibrosis, and cardiovascular abnormalities. AHR knockout rats also demonstrate resistance to the toxic effects of TCDD, and, in contrast to mice, display pathological alterations to the urinary tract in the absence of TCDD (Harrill et al., 2013). Hence, it is likely that dioxin has effects that are due to the disruption of endogenous AHR functions and that are unrelated to the intrinsic toxicity of some of its ligands.
Definition of Dioxin-Like Compounds, Toxic
Equivalence Factor, and Toxic Equivalents
TCDD has the highest affinity for the AHR, but many other chemicals have dioxin-like properties: They have similar chemical structures, have similar physiochemical properties, and cause a common battery of toxic responses because of their relatively high affinity for the AHR. Because of their hydrophobic nature and
| Chemical | TEF | ||||
|---|---|---|---|---|---|
| Chlorinated dibenzo-p-dioxins | |||||
| 2,3,7,8-TCDD | 1.0 | ||||
| 1,2,3,7,8-PeCDD | 1.0 | ||||
| 1,2,3,4,7,8-HxCDD | 0.1 | ||||
| 1,2,3,6,7,8-HxCDD | 0.1 | ||||
| 1,2,3,7,8,9-HxCDD | 0.1 | ||||
| 1,2,3,4,6,7,8-HpCDD | 0.01 | ||||
| OctoCDD | 0.0003 | ||||
| Chlorinated dibenzofurans | |||||
| 2,3,7,8-TCDF | 0.1 | ||||
| 1,2,3,7,8-PeCDF | 0.03 | ||||
| 2,3,4,7,8-PeCDF | 0.3 | ||||
| 1,2,3,4,7,8-HxCDF | 0.1 | ||||
| 1,2,3,6,7,8-HxCDF | 0.1 | ||||
| 1,2,3,7,8,9-HxCDF | 0.1 | ||||
| 2,3,4,7,8,9-HxCDF | 0.1 | ||||
| 1,2,3,4,6,7,8-HpCDF | 0.01 | ||||
| 1,2,3,4,7,8,9-HpCDF | 0.01 | ||||
| OctoCDF | 0.0003 | ||||
| Non-ortho-substituted PCBs | |||||
| PCB 77—3,3′,4,4′-tetraCB | 0.0001 | ||||
| PCB 81—3,4,4′,5-tetraCB | 0.0003 | ||||
| PCB 126—3,3′,4,4′,5-pentaCB | 0.1 | ||||
| PCB 169—3,3′,4,4′,5,5′-hexaCB | 0.03 | ||||
| Mono-ortho-substituted PCBs | |||||
| PCB 105—2,3,3′,4,4′-pentaCB | 0.00003 | ||||
| PCB 114—2,3,4,4′,5-pentaCB | 0.00003 | ||||
| PCB 118—2,3′,4,4′,5-pentaCB | 0.00003 | ||||
| PCB 123—2′,3,4,4′,5-pentaCB | 0.00003 | ||||
| PCB 156—2,3,3′,4,4′,5-hexaCB | 0.00003 | ||||
| PCB 157—2,3,3′,4,4′,5′-hexaCB | 0.00003 | ||||
| PCB 167—2,3′,4,4′,5,5′-hexaCB | 0.00003 | ||||
| PCB 189—2,3,3′,4,4′,5,5′-heptaCB | 0.00003 | ||||
NOTE: CB, chlorinated biphenyl; CDD, chlorinated dibenzo-p-dioxin; CDF, chlorinated dibenzofuran; PCB, polychlorinated biphenyl.
SOURCE: Adapted from van den Berg et al., 2006.
resistance to metabolism, these chemicals persist and bioaccumulate in the fatty tissues of animals and humans. Although there are several hundred polychlorinated, polybrominated, and mixed polychlorinated-polybrominated dibenzo-p-dioxins, dibenzofurans, and biphenyls, only a relatively small number of congeners of these chemical classes display dioxin-like activity. Only 17 polychlorinated dibenzo-p-dioxins and polychlorinated dibenzofurans with chlorine at the 2, 3, 7, and 8
positions and a few of the coplanar polychlorinated biphenyls that are often measured in environmental samples are recognized as being DLCs.
In the context of risk assessment, these polychlorinated–polybrominated dibenzo-p-dioxin, polychlorinated dibenzofuran, and biphenyl DLCs are commonly found as complex mixtures when detected in environmental media and biologic tissues or when measured as environmental releases from specific sources. That complicates the human health risk assessment that may be associated with exposures to varied mixtures of DLCs. To address the problem, the concept of toxic equivalence has been elaborated by the scientific community, and the toxic equivalence factor (TEF) has been developed and introduced to facilitate the risk assessment of exposures to those chemical mixtures. On the most basic level, TEFs compare the potential toxicity of each DLC found in a mixture with the toxicity of TCDD, the most toxic member of the group. The procedure involves assigning individual TEFs to the DLCs on the basis of in vivo and in vitro potency relative to TCDD, which is assigned a TEF of 1.0. The DLCs have been assigned TEFs ranging from 0.00001 to 1.0 by the World Health Organization (WHO) (van den Berg et al., 2006, as summarized in Table 4-2). Interim TEF values have been established for brominated congeners by the most recent (2011) joint WHO–UNEP (UN Environment Programme) meeting to evaluate the WHO TEF scheme. The recommendation is to use the TEF of the corresponding chlorinated congener as an interim TEF value for brominated congeners for human risk assessment (van den Berg et al., 2013).
When several chemicals are present in a mixture, the toxicity of the mixture is estimated by multiplying the TEF of each DLC in the mixture by its mass concentration and summing the products to yield the TCDD toxic equivalents (TEQs) of the mixture. In that approach to assessing the dioxin-like activity of a complex real-world mixture of DLCs, an environmental or biologic specimen with a 100-ppt (100-pg/g) TEQ is toxicologically equivalent to 100-ppt TCDD. There are two accepted specialized methods for assessing the DLCs in a complex biologic or environmental specimen: One involves analytic chemistry that quantifies specific DLCs (high-resolution gas chromatography–mass spectroscopy), and the other is a reporter-gene biologic screen that assesses dioxin-like activity due to binding to the AHR in a transformed cell line (CALUX, EPA method 4435). Epidemiologic studies discussed in this and other updates assess exposure by reporting the specific concentration of TCDD in a specimen or by expressing dioxin-like activity in a complex mixture in units of TEQs.
Carcinogenic Classification
EPA and the International Agency for Research on Cancer (IARC), a branch of WHO, have defined criteria to classify the potential carcinogenicity of chemicals on the basis of the weight of scientific evidence from animal, human, epidemiologic, mechanistic, and mode-of-action studies. EPA classified TCDD as a
“probable human carcinogen” in 1985 and as “carcinogenic to humans” in a 2003 reassessment. In 1998 the IARC panel of experts concluded that the weight of scientific evidence supported the classification of dioxin as a class I carcinogen, that is, as “carcinogenic to humans.” Four years later the US National Toxicology Program upgraded its classification to “known to be a human carcinogen.” In 2006 a panel of experts convened by the National Research Council to evaluate the EPA reassessment concluded that TCDD was “likely to be carcinogenic to humans”; this designation reflected the revised EPA Guidelines for Carcinogen Risk Assessment made public in 2005.
Genotoxicity
Genotoxicity refers to a deleterious action that affects the integrity of a cell’s DNA. Genotoxic substances are known to be potentially mutagenic or carcinogenic. Although TCDD is carcinogenic in humans and laboratory animals, it is generally classified as nongenotoxic and nonmutagenic (Wassom et al., 1977). There is no evidence of covalent binding of TCDD or its metabolites to DNA (Poland and Glover, 1979). TCDD does interact with DNA through a receptor-mediated pathway that involves the initial binding of TCDD to the AHR, binding of the activated receptor complex to DREs on DNA and later alterations in the expression of TCDD-regulated genes, and altered signaling of the biologic pathways that interact with the AHR signal-transduction mechanism (Poland and Knutson, 1982; Safe, 1990; Schmidt and Bradfield, 1996; Whitlock, 1990). TCDD, 2,4,5-T, and 2,4-D were not mutagenic in Salmonella typhimurium with or without the addition of liver metabolic-activation enzymes (Blevins, 1991; Mortelmans et al., 1984). TCDD-induced cytogenetic damage in laboratory mice showed no increase in the frequencies of sister-chromatid exchanges, chromosomal aberrations, or micronuclei in the bone marrow cells of either C57Bl/6J or DBA/2J mice after the administration of a single high dose of TCDD—up to 150 μg/kg (Meyne et al., 1985). TCDD did not alter the frequency or the spectrum of mutations in male and female Big Blue transgenic rats (Thornton et al., 2001). There is one report of a positive result with TCDD in a test that measured the induction of chromosomal deletions resulting from intrachromosomal recombination in mouse embryos in vivo (Schiestl et al., 1997).
In summary, although TCDD does have some genotoxic activity, the vast majority of studies did not detect mutagenic activity of TCDD in a variety of in vitro and in vivo short-term tests.
Epigenetic Activity
Chromosomes contain the genetic material of an organism and are composed of both DNA and proteins called histones. Interactions between DNA
and histones regulate the accessibility of DNA to binding factors that activate or suppress gene transcription. This interaction is controlled by chemical modifications of the DNA (generally methylation) and histones (such as sulfation and acetylation), which are maintained by enzymatic processes. These modifications are considered “epigenetic” because they control the function of genes without changing the coding sequence. TCDD has been demonstrated to cause changes to the epigenetic marks on the chromosomes, potentially altering the function of numerous genes. Below is a brief summary of the epigenetic effects of TCDD. More detailed information about epigenetic mechanisms in general can be found later in this chapter, particularly concerning somatic modifications in an individual, and again in Chapter 10 with respect to effects that may affect offspring of an exposed organism.
The exposure of rodents has been shown to result in the methylation of DNA in the adult rodents’ tissues—sperm, mammary tissue, and a number of other solid tissues (Papoutsis et al., 2013; Somm et al., 2013). Developmental exposure to TCDD has been shown in rodents to alter DNA methylation in preimplantation embryos (Wu et al., 2014). The effects on DNA methylation were found to persist in rats for three generations following maternal exposure to TCDD and so could essentially be considered permanent and heritable (Manikkam et al., 2012a). When Olsvik et al. (2014) fed female zebrafish TCDD in their diet prior to breeding, they found no change in the level of DNA methylation throughout the genomes of them or their embryos, but altered methylation was identified by probes for the promoter regions of a number of specific genes with corresponding marked elevations in the expression of CYP1A1 and CYP1B1 in the mothers and embryos (Olsvik et al., 2014). To date, however, there have not been publications reporting on the persistence of methylation or other epigenetic modifications in the offspring of males exposed to TCDD as adults.
Other Toxic Outcomes
Chloracne is a signature effect of high exposure to TCDD and DLCs in some species and in humans who are sensitive.
There is an extensive body of evidence from experimental studies in animal-model systems that TCDD, other dioxins, and several DLCs are immunotoxic (Kerkvliet, 2009). Although the available evidence on dioxin immunotoxicity in humans is scant, mechanistic considerations support the notion that chemical alterations of immune function would cause adverse health outcomes because of the critical role that the immune system plays in general protection—fighting off infection and eliminating cancer cells at early stages. Because of those considerations, the chemicals are potential immunotoxicants.
Similarly, reproduction and embryonic development clearly are targets of TCDD, other dioxins, and DLCs; it is found consistently that the adverse effects are more prevalent during fetal development than in the adult. Although data
on those effects in humans are practically nonexistent, some good data are now emerging on the developmental effects of DLCs in humans (Mocarelli et al., 2008). Human and animal studies have revealed other potential health outcomes, including cardiovascular disease, hepatic disease, thyroid dysfunction, lipid disorders, neurotoxicity, and metabolic disorders, such as diabetes.
A number of effects of TCDD exposure in vitro appear to be independent of AHR-mediated transcription and in at least one instance perhaps independent of AHR itself. Guo et al. (2004) showed that TCDD induced expression of transforming growth factor-α and other genes involved in extracellular matrix deposition in cells from mice that had homozygous ablation of the Ahr gene. Studies have shown that TCDD can mobilize calcium from intracellular sources and increase calcium imported from the culture medium (Puga et al., 1995). Mitochondrial oxidative stress has been shown to be induced when calcium is mobilized (Senft et al., 2002). Calcium mobilization by TCDD may have an important effect on signal-transduction mechanisms that control gene expression, inasmuch as several proto-oncogenes, such as c-fos, are activated by calcium changes.
Summary of Biologic Plausibility That TCDD
Induces Adverse Effects in Humans
Mechanistic studies in vitro and in laboratory animals have characterized the biochemical pathways and types of biologic events that contribute to the adverse effects of exposure to TCDD. For example, much evidence indicates that TCDD, acting via the AHR in partnership with ARNT, alters gene expression. Receptor binding may result in release of other cytoplasmic proteins that alter the expression or activity of other cell-regulatory proteins. Mechanistic studies also indicate that many other cellular-component proteins contribute to the gene-regulatory effect and that the response to TCDD exposure involves a complex interplay between genetic and environmental factors. Comparative data from animal and human cells in vitro and from tissues suggest a strong qualitative similarity among species in their response to TCDD, and this further supports the applicability to humans of the generalized model of initial events in response to dioxin exposure.
Biochemical and biologic responses to TCDD exposure are considered adaptive or simply reflective of exposure and not adverse in themselves if they take place within the normal homeostatic ranges of an organism. However, they may exceed normal physiologic boundaries or constitute early events in a pathway that leads to damage in sensitive members of the population. In the latter case, the response is toxic and would be expected to cause an adverse health effect. Those generalizations about dose–response and individual variability are central to establishing biologic plausibility, which in the case of TCDD largely relies on extrapolation from animal studies to human risks.
OVERARCHING TOXICOLOGIC ISSUES RELATED
TO THE CHEMICALS OF INTEREST
Limitations of Extrapolating Results of Laboratory
Studies to Human Responses
In some instances, the toxic responses identified in laboratory-animal and cell-culture studies are not detected in epidemiologic studies after human exposure to the same chemicals. Although animal and cell-culture studies provide important links to understanding the biochemical and molecular mechanisms associated with toxicity induced by xenobiotics, many factors must be considered in extrapolating their results to human disease and disease progression. The following are key factors that might limit the ability of laboratory studies to predict human responses completely and accurately.
- Magnitude and duration of exposure In many instances, animal and cell-culture studies are conducted at higher exposures and for shorter durations than are typical in human exposures. For example, the concentrations of TCDD used in animal studies can be many times higher than was typical in the TCDD exposures of Vietnam veterans during their military service. In addition, TCDD is a persistent organic pollutant, and this results in human exposure that occurs over a lifetime, whereas animal studies seldom examine chronic low-level exposure that occurs over a period of many months or years, except those that evaluate chronic toxicity or carcinogenicity. Animal studies that establish a measurement of body burden over a specific period provide the best potential for extrapolation to humans.
- Toxicokinetics The toxicokinetics—absorption, distribution, metabolism, and excretion—of xenobiotics can vary widely between laboratory animals and humans. As shown in Table 4-1, the biologic half-life of TCDD varies from 8 to 29 days in rats and mice to about 7 years in humans even though the drug-metabolizing enzymes—including cytochrome P450 1A1, 1A2, and 1B1—are up-regulated or induced via TCDD-mediated activation of the AHR in both rat and human liver (Black et al., 2012).
- Timing of exposure Many organ systems are more susceptible to xenobiotic exposure during critical stages of development, differentiation, or function—such as during gestation or in the face of another external challenge (for example, antigens, smoking, dietary salt, and fat)—than at other times. Therefore, the response of some systems (such as the immune or cardiovascular systems) may depend on the timing of exposure relative to the other challenges.
- Exposure composition Most animal and cell-culture studies involve exposure to single chemicals or a well-defined mixture, but most human exposures are to complex mixtures from multiple sources, so it is difficult
- to definitively attribute any observed effects to a particular component of the environment.
- Difference in AHR affinity The binding affinity of AHR for TCDD differs between species (discussed in Okey et al., 2005). Many of the strains of mice used for toxicologic studies harbor a high-affinity AHR allele (AHRb), and these mice exhibit greater sensitivity to hepatic CYP1A induction, immunosuppression, birth defects, and other responses than do strains that carry the low-affinity allele (AHRd). Such a simple allelic difference in AHR affinity has not been observed in humans, and the TCDD-binding affinity of the AHR found in most humans more closely resembles the low-affinity mouse AHRd allele. Nonetheless, Nebert et al. (2004) reported that some people have TCDD-binding affinity that is 12 times higher than that in others. Thus, although humans are generally considered less sensitive on the basis of an AHR that has low TCDD-binding affinity, this assumption may not apply to everyone.
- Complex disease etiology and environment The etiology of human diseases is highly influenced by genetics, environmental factors, and gene–environment interactions; these factors can be protective as well as deleterious. In addition to the chemical of interest, the environmental factors that commonly influence human responses include diet, prescription and over-the-counter pharmaceuticals, cigarette smoking, alcohol consumption, physical activity, and stress. Stress (not to be confused with oxidative stress) produced via known or unknown sources is a well-known modifier of human disease responses (for example, immune and cardiovascular responses). Furthermore, stress is an ever-present variable that is difficult to assess or control for in epidemiologic studies because there is substantial individual variation in response to it (Cohen et al., 2007a). In contrast, laboratory studies are often conducted with inbred strains of animals and under tightly controlled experimental conditions, thus possibly underestimating or overestimating the potential contribution of a single chemical exposure to disease development. On the other hand direct cause-and-effect relationships are more easily established in animal studies because of their standardization.
- Sex differences There are well-known differences in susceptibility to xenobiotic exposures between male and female animals, some of which are modified by sex steroids. For example, female Sprague Dawley rats are significantly more responsive to the hepatotoxic (neoplastic and non-neoplastic) effects of TCDD than are males of the same strain (Kociba et al., 1978).
Epigenetics
Epigenetics is the term used to describe the mechanisms that regulate gene expression and genomic stability, but that involve no changes in DNA sequence.
The epigenetic marks on DNA and bound histones are mitotically stable because they are maintained every time a cell divides. The totality of epigenetics marks in each cell, termed the line epigenome, creates and maintains the identity and function of the cell type (Christensen and Marsit, 2011; Cortessis et al., 2012; Skinner et al., 2010).
Conrad Waddington coined the term “epigenetics” in the 1940s to describe environment–gene interactions that alter biologic traits (Waddington, 1940, 1953, 1956). It was not until the 1970s, however, that the first molecular epigenetic factor was described: DNA methylation, the chemical addition of a methyl group to DNA (Holliday and Pugh, 1975). In the 1980s, the role of DNA methylation in modifying gene expression—turning genes on and off—was established (Chen and Riggs, 2005). In the 1990s, the chemical modification of histone proteins associated with DNA was shown to also modify gene expression, thus establishing a second molecular epigenetic mechanism (Turner, 1998). In the early 2000s, various small noncoding RNA molecules were shown to regulate DNA activity (Sato et al., 2011). Around 2005, the first mapping of the yeast epigenome was conducted (Pokholok et al., 2005). Since that time the mapping of cell-specific human epigenomes has accelerated under the National Institutes of Health Epigenomics Roadmap and the ENCODE projects, and more than 100 have been identified (Kundaje et al., 2015). The studies show that epigenetic marks act together in an exquisitely choreographed fashion to control cellular differentiation and the cellular ability to interact with, process, and initiate events and to respond to the signals and needs of the individual and local tissue environment.
Today, the processes recognized as epigenetic mechanisms are DNA methylation (Chen and Riggs, 2005; Holliday and Pugh, 1975), histone modification (Turner, 1998), alterations in chromatin structure (Murr, 2010), and modulation of expression by some micro RNA molecules (Valeri et al., 2009). DNA methylation is the addition of a methyl group onto specific nucleotides. In mammals it occurs mostly at cytosine nucleotides that are adjacent to guanine nucleotides, but it can also occur at cytosine nucleotides followed by other bases in embryonic cells and brain cells (Lister et al., 2013). Methylation of DNA in the promoter region of the gene can reduce the expression of the adjacent gene. Other modulations of DNA include hydroxymethylation (which is prominent in stem cells and brain cells), formylcytosine, and carboxylcytosine (Cheng et al., 2014; Song et al., 2013). Histones are the proteins that bind and form complex structures with DNA called nucleosomes in which DNA is wrapped around the histone core. The combination of DNA and histones is called chromatin. Chemical modifications of histones, such as methylation and acetylation, can alter the histone structure and modify gene expression by attracting protein complexes that can stimulate or repress transcription, in part by changing nucleosome spacing (Reid et al., 2009; Zhang and Pradhan, 2014). The most recently recognized epigenetic factor consists of small noncoding RNA molecules that can associate with mRNA and regulate gene expression.
The interaction of all those epigenetic processes creates the epigenome, which has a critical role in regulating gene expression (Christensen and Marsit, 2011; Cortessis et al., 2012; Skinner et al., 2010). The variation that is possible in the epigenome is startling: The histone proteins that control chromatin configurations have dozens of possible modifications, and there are upwards of 50 million nucleotides in the DNA where methylation can occur and participate in regulating the cellular state. That implies that trillions of configurations of the epigenome are possible, though expression of about half of all genes is common to all cells. Epigenetic marks are erased and re-established at two times during the life cycle, shortly after fertilization and during gametogenesis to allow gamete-specific epigenomes to be converted to cell-specific epigenomes and vice versa (Dean, 2014).
Environmental epigenetics is the study of how environmental factors—such as nutrition, toxicants, and stress—alter epigenetic programming. In particular, it provides a molecular mechanism—other than mutations in the DNA itself—by which environmental factors can influence disease etiology (Jirtle and Skinner, 2007; Szyf, 2007). The role of epigenetics in disease etiology has been shown for cancers and a number of other diseases (Christensen and Marsit, 2011; Cortessis et al., 2012; Skinner et al., 2010). In addition, exposure to environmental factors at critical times of development when epigenomes are shifting has the ability to alter epigenetic programming and to cause changes in gene expression because these are times when epigenomes are evolving rapidly as stems cell differentiate into more mature cell types (Skinner et al., 2010). Hence, immune responses, fetal development, and gamete formation are important examples of physiological processes whose functioning can be affected by environmentally induced epigenetic changes.
New investigative tools and a more refined understanding of the epigenetic process have given rise to active research on the nature of the relationship between environmental exposure to epigenetically active agents and the occurrence of diverse disease states, including cancers, reproductive-developmental problems, immune dysregulation, diabetes, obesity, and psychiatric illnesses (Brookes and Shi, 2014). The committee sought to review data on the potential relationship of the exposures of interest with adverse epigenetic effects in the directly exposed veterans in an attempt to find evidence linking the exposures to disease processes that might have been mediated epigenetically. We also sought to review relevant data on female veterans and male veterans separately inasmuch as the epigenetic consequences of exposures could be different, particularly in the case of adverse reproductive outcomes.
A relevant example is that the in vitro exposure of preimplantation embryos to TCDD alters the DNA methylation of imprinted genes (Wu et al., 2004). Similar results were obtained more recently when several solid tissues and sperm DNA were analyzed in adult male mice exposed to TCDD in utero (Somm et al., 2013). In utero exposure to TCDD was also shown to cause DNA methylation
and reduced expression of the BRCA1 (breast cancer) gene in mammary tissue in adult female offspring (Papoutsis et al., 2013). More generally, studies of the developmental origins of health and disease have shown that early-life exposures or environmental influences can be associated with the onset of disease much later in life (Barker et al., 2010). These early developmental alterations in the epigenome provide a molecular mechanism by which environmental exposures of female veterans can have effects on their children into adulthood.
Most epigenetic modifications occur in somatic cells and are heritable only within the altered cell line, thereby having the potential to generate effects in the exposed individual, but not in that individual’s offspring. The possibility exists, however, of epigenetic transgenerational inheritance, which involves the environment promoting a stable alteration in the germ line that is transmitted to later generations (Schmidt, 2013; Skinner, 2014; Skinner et al., 2010; Wei et al., 2015). Manikkam et al. (2012b) have shown that the exposure of pregnant female rats to TCDD at critical times of development of the germ line (when epigenetic programming is being established) can lead to abnormalities in the third-generation offspring, including kidney disease and changes in the ovaries and sperm of the offspring. There are few data on possible male-mediated heritable effects of TCDD or other environmental compounds. The sparse data include the results of studies of lead, a known developmental toxicant. Lead exposure can alter semen quality in males (Alexander et al., 1996) and has been shown to induce paternally mediated developmental toxicity in rats (Anjum et al., 2011); this demonstrates that male-mediated effects on reproduction can be induced by reproductive toxicants. It has been suggested that environmental exposures can result in reduced fertility (Guerrero-Bosagna and Skinner, 2014; Paoloni-Giacobino, 2014). However, a serious need exists for additional study of the question, particularly of the effects of the compounds of interest to this committee.
In summary, the ability of epigenetic mechanisms to regulate gene expression coupled with the interaction of the epigenome and the environment might underlie the ability of xenobiotic exposure to contribute to disease development and the potential for offspring to inherit the effects of the disrupted epigenetic processes.
Developmental Immunotoxicity
A second emerging field in the biologic sciences that may provide insight into the mechanism of xenobiotic-induced disease is developmental immunotoxicity (DIT), the study of the disruption of the developing immune system by xenobiotic exposure. The developing immune system is among the most sensitive physiologic targets of prenatal and childhood environmental insult. The sensitivity is due, in part, to the novel processes of gene rearrangement, somatic-cell selection, and immune-cell distribution that are required to produce a security system that can effectively protect not only the child but also the aging adult
against external challenges without itself producing immune-mediated chronic disease. To produce that security system, the immune system, as it matures, must coordinate steps that result in highly specialized immune cells that are capable of self-versus-non-self-recognition and that are tailored to the specialized functional environments of different tissues and organs (such as brain, lungs, skin, liver, gastrointestinal tract, and reproductive tract). A disruption of immune development can place the integrity of the organism at risk.
Among the known risk factors for DIT are various chemicals including heavy metals, some pesticides, industrial solvents such as trichloroethylene, and polychlorinated biphenyls. The adverse outcomes of DIT may become apparent soon after exposure or can emerge much later in life (Gascon et al., 2013). Often, childhood or adult infections can trigger the appearance of DIT-associated immune problems that were established earlier in life (Dietert, 2009). DIT-induced alterations can also contribute to myriad health problems related to dysfunction or pathologic conditions in virtually any tissue or organ. Chemicals, drugs, infectious agents, and physical and emotional stressors can act synergistically and increase the risk of DIT. Not everyone is at identical risk for DIT. People who have particular genotypes may be at increased risk for specific chemical-induced DIT on the basis of heritable factors that affect metabolism or immune vulnerability.
The heightened sensitivity of the developing immune system is due to the existence of critical developmental windows of vulnerability during which environmental interference with key steps of immune maturation can change the entire course of immune development and result in later-life immune dysfunction and an increased risk of disease. The events programmed for these critical developmental windows have several basic features:
- They are necessary, usually one-time events of early development, with no equivalents in adults.
- They lock in building blocks on which additional maturational events rely.
- If they do not occur both on time and efficiently, the ramifications are usually profound, prolonged, and irreversible.
Examples of critical windows of immune vulnerability and the chemicals that can cause disruptions have been described in several reviews (Dietert and Dietert, 2008; Dietert and Piepenbrink, 2006; Dietert et al., 2000; Holsapple et al., 2003; Landreth, 2002) and include
- The process of the seeding of immune cells in tissues where they grow into resident populations.
- The selection process of thymocytes in the thymus to distinguish nonself from self during the development of acquired immunity.
- The maturation of macrophage populations in the lung, in the brain, and elsewhere.
- The maturation of dendritic cells to provide balanced immune responses.
- The initial development, expansion, and seeding to the periphery of t-regulatory cell populations.
The increased sensitivity of the fetal, neonatal, and juvenile immune systems compared with the immune system of an adult can be manifested as a sensitivity to lower doses of chemical exposure than doses that affect the adult, a greater persistence of the immune problems that follow exposure than are seen in adults, a broader array of immune problems than are experienced by adults, and greater likelihood that a second later-life chemical exposure or environmental stressor will trigger an unexpected immune problem.
It is important to note that disruption of immune maturation is not the only route for DIT. Early-life chemical exposure may affect the status of genes (the epigenome) in such a way that their pattern of expression in later life is affected and thereby alter immune functional capacity. Such changes in gene status that affect immune status could occur in the exposed generation (for people exposed in utero or during childhood), or they could carry through one or more additional generations as a result of true epigenetic alterations.








































