
Workshop discussions during Session III were organized around four case study regulatory science applications that served as focal lenses to discuss how enhanced approaches to obtaining, accessing, and integrating information could advance regulatory science.
IDENTIFICATION AND DEVELOPMENT OF MEANINGFUL BIOMARKERS
John Wagner, senior vice president, head of clinical and translational sciences, Takeda Pharmaceuticals, observed that although biomarkers have been a subject of regulatory interest since 1999, progress in their development, standardization, and regulation has not met expectations. Indeed, biomarkers remain a priority, and improvements are being offered through a number of policy initiatives and programs, such as the U.S. House of Representatives’ “21st Century Cures” bill, the Foundation for the National Institutes of Health (FNIH) Biomarkers Consortium, and the FDA Biomarker Qualification Program. With this confluence of activities, it will be important to harmonize ongoing efforts in biomarker development to avoid duplication and ensure the endeavors are separately and collectively worthwhile, Wagner said.
FDA Biomarker Qualification Pathways
Shashi Amur, scientific lead, FDA Center for Drug Evaluation and Research’s (CDER’s) Biomarker Qualification Program, discussed FDA’s objective-dependent biomarker qualification pathways (see Figure 3-1). If the intent is to only use the biomarker for a single drug development application, the sponsor would combine biomarker qualification with a regulatory submission, such as an Investigational New Drug (IND) or New
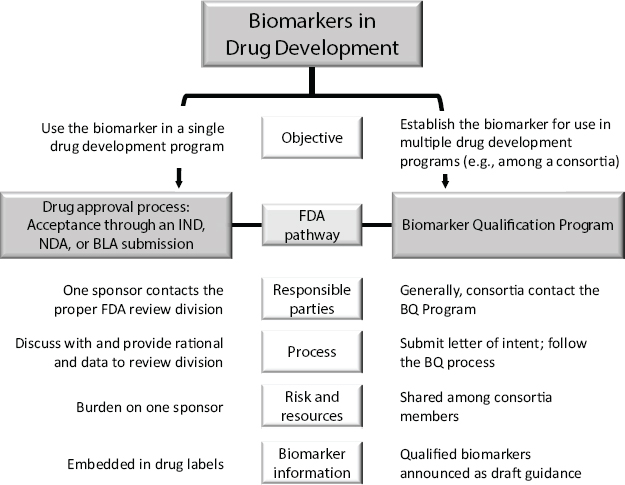
NOTES: BLA = Biological License Application; BQ = Biomarker Qualification; IND = Investigational New Drug Application; NDA = New Drug Application.
SOURCE: Amur et al., Clinical Pharmacology & Therapeutics 98 (1):34–46, 2015 (presented by Amur on October 20, 2015).
Drug Application (NDA). The biomarker can therefore only be used by that sponsor, and the information is often included in the drug label after approval.
However, Amur said, if the biomarker is intended to be used for development of multiple drugs, it goes through a separate biomarker qualification process. Often, this approach is used when consortia identify a biomarker that each member later intends to use in a separate drug development application. After approval, the biomarker is publicized on FDA’s website as draft guidance and it becomes public information.
When a biomarker is qualified, it is qualified for a specific context of use (COU), Amur said. The COU determines what level of evidence is needed, and that level of evidence then drives the qualification process. Each type of biomarker can have multiple COUs. FDA is currently involved in many ongoing biomarker projects, including efforts to establish a common taxonomy for types of biomarkers.
Amur used the example of a recently qualified biomarker, total kidney volume in autosomal dominant polycystic kidney disease (ADPKD), to illustrate several principles and best practices relating to biomarker qualification:
- Collaborative meetings (including cross-agency) facilitate scientific exchange.
- Early consultation with FDA Biomarker Qualification Review Teams (BQRTs) is critical.
- Data standardization including harmonized terminology, facilitates data aggregation and ensures that data are usable.
- The biomarker qualification process requires significant reviewer effort, including, for example, conducting additional analyses or developing an external cross-validation model.
Amur also cited initiatives to streamline biomarker qualification, including the Critical Path Innovation Meeting (CPIM) and FDA’s Limited COU Qualification. CPIM was developed by CDER to address issues in drug development identified in the 2004 FDA publication Innovation or Stagnation: Challenge and Opportunity on the Critical Path to New Medical Products. CPIMs provide a means for CDER and investigators across industry, academia, patient advocacy groups, and government to communicate to improve efficiency and success in drug development.1 FDA’s CDER provides an avenue to qualify a biomarker for a “limited” COU in order to expedite the integration of the biomarker in drug development and to possibly generate additional data that can help in qualifying the biomarker for an “expanded” context of use.
Advancing Science and Infrastructure for Biomarker Development
Gabriela Lavezzari, assistant vice president, science and regulatory advocacy, Pharmaceutical Research and Manufacturers of America (PhRMA), characterized FDA’s biomarker qualification process as having a lack of predictability, suggesting that FDA further outline what a qualification package should look like (i.e., what evidence, data standards, and assay validations are needed). By providing additional clarity on these parameters, Lavezzari said, the amount of time spent on the biomarker development process could decrease and the return on investment of researchers or consortia members could be maximized. She also encouraged conversations among stakeholders, including academia, industry, and consortia, around the development of a defined set of required evidence for qualification, commonly called “evidentiary standards,” that could aid in streamlining and providing more predictability in the qualification process.
John Michael Sauer, executive director, Predictive Safety Testing Consortium (PSTC), C-Path, highlighted C-Path’s consortium model for bio-
___________________
1 For more information, see http://www.fda.gov/Drugs/DevelopmentApprovalProcess/DrugInnovation/ucm395888.htm (accessed December 23, 2015).
marker qualification. C-Path’s consortia create partnerships that function as neutral, precompetitive spaces for pharmaceutical companies, academics, and regulatory bodies (e.g., FDA, European Medicines Agency [EMA]) to have discussions to move qualification forward. Sauer noted two areas that could use further attention to aid consortia through the biomarker qualification process: (1) better define the qualification process, specifically evidentiary standards; and (2) identify ways to better collect and share data, possibly in a repository, to use with prospective analyses.
Sauer noted that one potential way to foster a collaborative environment that encourages data sharing could be to establish a means for masking shared data so that it cannot be traced back to the originating organization and impart undesired risk. Several workshop participants also noted a need for the establishment of a common definition of “precompetitive space” to foster more collaboration in the development of biomarkers.
INTEGRATING CLINICAL TRIAL DATA
The advent of technological advances such as electronic health records (EHRs), patient registries, and social media has ushered in an era of “big data” that holds promise for driving innovation in clinical research and application. “Big data” could take on different meanings depending on the user or situation—and, as noted by several workshop participants, use of the terminology is in and of itself divisive—but can be generally characterized by the “five Vs”: volume, velocity, variety, veracity, and value, said Sam Shekar, chief medical officer, Northrup Grumman.
Developing Capabilities to Integrate and Use Big Data in Clinical Research
Martin Landray, professor of medicine and epidemiology and deputy director, Big Data Institute, University of Oxford, noted that big data can permit researchers to more effectively collect information on traditional clinical outcomes and provide greater insight into patients’ symptoms and quality of life. Big data can also allow for novel assessment of both traditional disease features, such as exercise capacity or cognitive function, and of new disease features, such as keystroke speed, that are not currently incorporated into regulatory decision making because the symptom cannot be quantified. Finally, big data can promote thinking about the economic and social consequences of disease and treatment.
Despite the current hype surrounding big data and their potential positive impact on clinical medicine, “the fundamental principles of large-scale randomized trials remain unaltered,” said Landray. When applying big data to the design of clinical trials, he noted, it is critical to focus on three areas:
the hypothesis being assessed, the intended interpretation, and the errors that could develop as a result of the analysis.
Landray outlined his key considerations in achieving reliable assessments of treatment effects in aggregated clinical trials:
- Scale—the number of participants and the number of outcomes; allows for good statistical power in the face of moderate treatment effects
- Breadth—the diversity of the populations under observation (e.g., co-morbidities, concomitant treatments), and assessments of safety and efficacy
- Length—the frequency and duration of the clinical trial or assessment
- Depth—the careful and detailed characterization of trial participants’ outcomes
In light of these principles, Landray also cautioned that accurate data do not necessarily imply that results are reliable; they must be analyzed for errors. Results generated from large enough datasets are remarkably resilient to changes in outcome due to random errors, which do not add bias and can be overcome by adhering to the principles described above, he noted. He gave the example of a large randomized dataset, in which introducing 10 percent more false-positive events does not alter the conclusions or the statistical significance of the results. Introducing even 20 percent more false events will not alter the conclusions enough to change any regulatory or clinical decisions made from the data, he said. The same pattern holds true if the calculations are performed instead for similar rates of missing events. He added:
You don’t have to (have) perfect (data) to get reliable conclusions. You do have to understand in what way you are imperfect and to what extent that is going to matter to the type of conclusion you are trying to draw. It is the avoidance of (some) errors that matter to the decision making, not the avoidance of all errors.
The data must also be analyzed for systematic errors, Landray said, which cannot be corrected after the trial has been completed; if those exist, the trial will not generate reliable results in any analyses.
Postapproval Applications for Big Data
In addition to priorities in the preclinical and early clinical stages, participants discussed needs in the postapproval space. Derek Angus, Distinguished Professor and Mitchell P. Fink Endowed Chair, Department of Critical Care Medicine, University of Pittsburgh, discussed the need for a more defined structure in postapproval safety evaluations. He explained,
The postapproval world can be characterized as a data-poor, opinion-rich environment. At the time a therapy arrives with approval, the randomized controlled trials (RCTs) evidence that led to that approval is both too broad—in that the overall treatment effect is considered average, not personalized—and too narrow, as the trial population is not considered representative of the general population.
To address these potential concerns, Angus noted, it could prove valuable to consider innovative ways in which appropriate information can be generated during the postapproval phase. He cited an example that blended a point-of-care trial, where randomized observational studies are conducted directly in a clinical setting, with a large platform trial, which uses broad inclusion criteria for admittance to the trial and relatively simple protocol design. As patients with severe pneumonia were admitted to hospitals in Europe, they were randomly assigned to 1 of 48 possible treatment regimens. Enrollment was triggered by entering admitting data into a patient’s EHR. The trial took into account both causal inference and real-world effectiveness. It considered multiple therapies and generated treatment options in real time as more data were added to the algorithms.
Angus remarked that this model is not without problems, including concerns about how to report and disseminate results from an ongoing trial. Most importantly, Angus cautioned, having complex data for a large group such as this one does not obviate the need for randomization.
Adaptive Clinical Trials
Adaptive trials allow a researcher, in a prespecified manner, to harness accumulating data to decide when and how to modify a clinical trial. This modification could encompass, for example, moving patients to the most effective treatment arm or dropping less effective arms of the study as data accumulate. Brian Alexander, assistant professor of radiation oncology, Harvard Medical School, highlighted the use of Bayesian trial design to conduct randomized adaptive clinical trials. Importantly, the statistical method employed in Bayesian trial design automatically reflects uncertainty because it is a measure of probabilities that is continuously updated by new information.
Alexander and colleagues used Bayesian trial design to model how relationships between various endpoints could be evaluated during an ongoing trial. This method allows both for incorporation of immediately useful information in randomized assignments to designated treatment arms and for evaluation of how auxiliary endpoints are associated with survival. In addition, he described how incorporating data generated outside of the clinical trial itself, such as overall health, disease progression, or imaging
results, could be used to generate a “longitudinal model” that adds “shades of gray” to an otherwise binary survival endpoint. “Death is binary, but the probability of dying is not,” he said.
Bayesian techniques also offer the potential to more formally include information generated prior to a particular clinical trial, and to apply trial results to making future decisions and evaluations. In this manner, barriers between preclinical, early phase, and late phase clinical trials can be traversed or broken down, potentially addressing some of the concerns previously described by Russ Altman and FDA, Alexander stated.
Although developing software and conducting simulations for models such as this are time-intensive, their implementation may be a worthwhile investment and enable better preparation of an overall plan for evidentiary development. More flexible clinical trial designs such as adaptive trial design could provide efficiencies by capturing data that are potentially lost during the extended process of a trial and allow clinical trial researchers to enroll new patients without the time constraints from predetermined clinical trial phase timeframes. These steps could potentially result in a better outcome from the trial and a more successful trial enterprise overall, said Alexander.
Nontraditional Approaches to Big Data Analyses: A Case Study in Rare Disease
Random errors could have a disproportionate effect on smaller clinical trials. This is because as the sample size becomes larger in any trial, outliers and missed events will have less of an effect and the model will still be accurate, as illustrated by Landray. Susan Ward, founder and executive director, the Collaborative Trajectory Analysis Program (cTAP), pointed out that in smaller trials, however, small variations in data can have an enormous impact on the model. This effect is particularly seen in trials involving rare diseases like Duchenne muscular dystrophy (DMD). In a DMD trial, Ward and colleagues noted large variations in the trial’s primary endpoint (6-minute walk distance). To address the variance, Ward and her collaborators at cTAP applied latent class trajectory analysis. This methodology was developed in social sciences and health care economics to handle variance due to heterogeneity. The method assumes that a single mean exists for a “class,” finds the optimal number of classes by minimizing variance, and allows visualization of multiple clusters of data.2 Ward pointed out that this technique used for rare disease could also be applied to more common diseases. Common diseases are increasingly recognized as groupings of heterogeneous diseases with a set of common symptoms.
___________________
2 Ward provided additional resources for the workshop participants: Leoutsakos et al., 2012; Muthen and Asparouhov, 2014; Muthen and Brown, 2009.
Techniques such as the one used by cTAP could help to tailor treatment to a particular subset of patients based on covariance or other factors, or to clarify a more significant effect for a treatment under evaluation.
Considerations for Use of Big Data
Big data can confer many benefits in such endeavors as measurement of novel outcomes, postapproval safety monitoring, and adaptive trial design. To realize these benefits, however, data need to be accessible and of sufficient format and quality for the researchers who wish to use them. Several workshop participants noted that the value of data depends on having a clear understanding of which ways the data may be of poor quality—that is, what errors, random or systematic, may have been introduced during their collection. Additionally, discussants noted the importance of understanding, when curating and analyzing data, the assays—i.e., the technologies and methodologies—that were applied in collecting the original data. Without proper understanding of the original purpose and collection methods, incorporating the data into trial design may prove challenging, if not impossible. For example, confounding factors in assays might not be obvious when looking at a database alone, but upon further review such factors could be revealed, precluding aggregation of the results.
Understanding data quality and variability also influences how confident the scientist can be in any conclusions drawn, whether those conclusions are used to interpret disease features, the social impact of treatment, or identification of new biomarkers. In any of those cases or many other applications of aggregated data, it would be detrimental and costly to focus on a perceived signal that is actually an artifact caused by failure to understand fully the variability inherent in the data and their subsequent aggregation, making high confidence in conclusions a key component of any application for big data. Marc Salit, leader, Genome-Scale Measurements, National Institute of Standards and Technology (NIST), shared his framework for understanding data, their sources of variability, and how to identify an artifact (see Box 3-1). He termed this framework a “three-legged stool.” Confidence in a measurement can only be achieved when the units of measurement are understood (metrological traceability), the likely dispersion around the result is known (measurement uncertainty), and evidence establishes that the methodology used to obtain a result has been rigorously considered (method validation3; e.g., existing benchmark data and reproducible results from previous studies).
___________________
3 With respect to method validation, Salit noted that analytic validation—the accuracy, precision, and reproducibility of a test—is distinct from clinical validation—the relevance of the test in an actual clinical condition—and is a key factor in moving forward with biomarker qualification.
Data Collection, Curation, and Harmonization
Throughout the workshop, many participants emphasized the importance of key principles in data collection, curation, and harmonization. Original data are typically usable only for the purpose for which they were originally collected; repurposing data for other analyses, observed Richard Platt, professor and chair, Harvard Medical School Department of Population Medicine, Harvard Pilgrim Health Care Institute, usually necessitates a great deal of curation. Data curation, “the active and ongoing management of data through [their] life cycle of interest and usefulness to scholarship, science, and education,” includes activities that enable “data discovery and retrieval, maintain quality, add value, and provide for re-use over time” (Cragin et al., 2007).
When data are ready to be repurposed, millions of dollars are spent on data curation and countless hours of work are dedicated to make the data fit for novel purposes, said Brian Corrigan, senior director, Pfizer Inc. He
stressed the importance of having trained professionals collect and curate the data acquired during the course of a study so that those data conform to established, acceptable data standards.
Another obstacle that can arise during the process of aggregating and repurposing previously collected data is incongruous terminology and units of measurement. Landray provided an example encountered during aggregation of intergenerational data from trials on thrombolytic therapy for treating myocardial infarction: “myocardial infarction” is now on its third universal definition. In describing his work with FDA Sentinel (see section in this chapter on Next-Generation Surveillance), Platt noted that there are 67 different units of measure for recording blood platelet counts alone in laboratory results from Sentinel data partners. To perform analytical work of value from such data, it is necessary to harmonize the data through careful curation, Platt said.
Sharing Clinical Trial Data
Aggregating data from clinical trials to create bigger datasets is of increasing interest. Kyle Myers, Center for Devices and Radiological Health (CDRH), FDA, pointed out that funders of research are more commonly requiring data sharing as a condition for receiving support. Robert Califf, deputy commissioner for medical products and tobacco, FDA (at the time of the workshop), mentioned legislative initiatives intended to incentivize broad consent from patient volunteers so that data could be more easily shared and used by multiple researchers. However, making data available for such purposes raises a number of concerns, from the need to protect patient privacy and safeguard intellectual property, to the costs of sharing and curating data (see Box 3-2).
Although numerous concerns are associated with making data available and fit for use by a wider audience, several participants offered potential solutions. Charles Jaffe, chief executive officer, Health Level Seven International (HL7), noting that data quality depends on the accuracy of data collection and storage, stressed the importance of interoperability4 in harmonizing data and making exchange more seamless. “It is becoming increasingly clear,” he said, “that we cannot continue to silo the data (in separate databases). . . . [I]t is expensive, error prone, and hard to manage.” Jaffe argued that the future of health care interoperability will be
___________________
4 Interoperability is “the ability of different information technology systems and software applications to communicate, exchange data, and use the information that has been exchanged. Data exchange schema and standards should permit data to be shared across clinicians, labs, hospitals, pharmacies, and patients regardless of the application or application vendor.” Source: http://www.himss.org/library/interoperability-standards/what-is-interoperability (accessed February 19, 2016).
in application program interfaces (APIs) such as HL7’s Fast Healthcare Interoperability Resources (FHIR) specification.
In the environment of a consortium, Enrique Avilés, chief technology officer, C-Path, found in his experience at C-Path that consortium members are willing to share data if each understands the rights of each member, the agreement, and the final use and dissemination of the end product. To accomplish this, Avilés recommended structuring collaboration around specific goals and governance criteria. Every data contribution received is handled based on predetermined agreements for the data: what can be done, how it can be shared, how to ensure appropriate anonymization and privacy, and how to integrate various data sources. All the data integrated are evaluated according to C-Path’s key objectives and formatted to the Clinical Data Interchange Standards Consortium (CDISC) standards, already accepted by FDA, EMA, and the Pharmaceuticals and Medical Devices Agency (PMDA), for continuity and consistency. Sauer suggested that data sharing could be important to develop treatments for complex diseases. Participating in an organized consortium may be the most successful way of accessing data from multiple sources, Sauer said. Successful data sharing in the future will depend on common privacy standards, common data standards, and incentives to share data.
NEXT-GENERATION SURVEILLANCE
Several workshop presentations and discussions explored new tools, methodologies, and paradigms for collection, aggregation, and analysis of surveillance data. In the session Next-Generation Surveillance, panelists discussed new systems for data aggregation and efforts to leverage community search logs, discussion forums, and other Web-based platforms such as Twitter or Facebook. Panelists also examined how data analysis methodologies could be brought to bear to apply these Web-based data toward epidemiological studies, diagnosing and tracking illness over time, tracking adverse drug events, and even for gauging the black market for pharmaceuticals.
Although some workshop participants expressed enthusiasm for the possibilities of these types of developing tools, it was noted that all of them face further refinement before they would be suitable for use by FDA to identify risks. Brian Strom, chancellor of Biomedical and Health Sciences, Rutgers, the State University of New Jersey, and Platt observed that Sentinel is currently designed to strengthen existing hypotheses rather than generate new ones, as these other tools presented do, because of the difficulty in determining how much importance to assign to unanticipated effects. It would be a burden on FDA to follow up on a potentially vast number of false-positive events, they said.
Systematic Methods for Medical Product Reporting
Patients and clinicians do not always know that current adverse event reporting tools exist or how to use them, noted John Brownstein, associate professor, Harvard Medical School. MedWatcher5 is one FDA-sponsored tool designed to allow patients to provide more detailed information about adverse events. Brownstein highlighted the power of these reporting platforms not only to inform adverse drug events, but also to illuminate the illegal use of drugs on the black market and to track the street value of medical products. These black market data, he said, could help inform a greater understanding of the public health impact of certain medical products. FDA has launched an ongoing informatics initiative, FDA Sentinel, which will incorporate electronic health data from at least 100 million people to assess the safety of marketed medical products. A description and update of FDA Sentinel given by Platt is summarized in Box 3-3.
___________________
5 MedWatcher is a mobile application (app) that allows individuals to submit voluntary reports of serious medical device problems to FDA using a smart phone or tablet. For more information, see http://www.fda.gov/MedicalDevices/Safety/ReportaProblem/ucm385880.htm (accessed December 23, 2015).
Because some medical devices may not undergo classical clinical trials, they are typically continuously assessed after they reach the market, said Danica Marinac-Dabic, director of epidemiology, CDRH, FDA. Registries linked to EHRs and unique device identifications will be valuable for continuous surveillance of medical devices. MDEpiNet6 (the Medical Device Epidemiology Network Initiative) is a public–private partnership whose mission is “to bridge evidentiary gaps, to develop datasets and innovative methodological approaches for conducting robust analytic studies, and to improve medical device safety and effectiveness understanding throughout the device life cycle.”
___________________
6 MDEpiNet is part of the Epidemiology Research Program at FDA’s CDRH. The initiative is a collaborative program through which CDRH and external partners share information and resources to enhance our understanding of the safety and effectiveness of medical devices after they are marketed. For more information see http://www.fda.gov/MedicalDevices/ScienceandResearch/EpidemiologyMedicalDevices/MedicalDeviceEpidemiologyNetworkMDEpiNet/default.htm (accessed December 23, 2015).
Web-Based Surveillance Data
Individuals are creating a new and continuous stream of data today via search engines, social media, and wearable fitness trackers, which offer unique vantage points for acquiring and aggregating health data compared to traditional clinical data sources. These new tools could be used on a global scale for tracking everything from emerging infectious disease to adverse drug events.
Search Engines and Web-Search Logs
Eric Horvitz, distinguished scientist and managing director, Microsoft Research, noted that approximately 72 percent of adult Internet users reported performing health-related inquiries online (Pew Center for Research, 2015). Bing and Google reported that about 10 percent of Web inquiries were health related. These search logs could serve as an immense source of health data, such as for surveillance of adverse reactions, he said. Horvitz highlighted that as a result of prolific use of cell phones and personal computers, individuals continuously self-report health data via Web searches. He observed that Web-based search, unlike more communicative forms of social media, may be less influenced by broader societal or communal attitudes and opinions. Combining these new techniques with traditional health care and claims data could prove informative on how to better harness the Internet for pharmaceutical surveillance and integrate nontraditional sources of information, he said.
Building off work from Altman and colleagues, who found through analysis of the FDA Adverse Event Reporting System (FAERS) that patient-reported hyperglycemia was higher when patients were taking two medications in combination, Paxil and Pravachol, than either drug alone, Horvitz and colleagues sought to confirm the real-world presence of this effect through analysis of Web-search logs. Using a generated set of terms obtained from BioPortal7 and consumer-oriented search terms in Microsoft Bing, they analyzed 1 year of Web-search logs, which revealed that this drug interaction can be accurately identified in Web searches.
Microsoft Research has harnessed this concept by establishing a tool termed BLAERS (Behavioral Log-Based Adverse Event Reporting System). BLAERS provides ongoing monitoring of Web-search logs for adverse drug events. This and similar Web search–based tools provide an opportunity to complement traditional sources of adverse drug event reporting.
___________________
7 BioPortal is a repository of biomedical ontologies developed by The National Center for Biomedical Ontology; see http://bioportal.bioontology.org (accessed December 23, 2015).
Online Discussion Forums
Online discussion forums could also serve as a profound source of nontraditional data to capture adverse drug events. John Holmes, professor of medical informatics, University of Pennsylvania, defined a discussion forum as “an online resource and a social media resource where people participate in actual conversations.” People generally participate in a chat room to talk about a particular topic, meaning the population automatically tends to segregate; for example, forums on breast cancer might typically consist of women between the ages of 40 to 70. Holmes and colleagues analyzed discussion forums using a Web crawler and a set of controlled vocabulary to extract information and develop findings. In one study, Holmes and colleagues investigated the side effects of an aromatase inhibitor (used to treat some estrogen receptor–positive breast cancers) by looking at 1,000 randomly selected messages in the discussion threads for identified side effects. In reviewing the messages, they found that 18 percent of participants mentioned at least one side effect, with some individuals reporting side effects more frequently than stated on drug warning labels. Holmes cautioned that these data would only be valuable for hypothesis generation, however, as the data are limited by the fact that there is no denominator to the analysis and thus cannot be viewed as rates or proportions.
Challenges and Limitations to Web-Based Surveillance
Several participants discussed the challenges and limitations inherent in analyzing discussion forum chats, social media, and Web search (see Box 3-4). Many speakers emphasized the importance of having a standardized vocabulary to avoid complications in data analysis related to variations in syntax. On the other hand, it was noted that certain syntactical elements could help to personalize surveillance data by elucidating the sentiments and attitudes of the individual that are not currently captured in traditional methods, and thus could be of subjective value. It was also noted that researchers will need to develop and adhere to anonymization mechanisms when publishing and presenting data to address the lack of consent inherent in Web-based surveillance mechanisms. Holmes stated that the influence of current events on user searches and comments could have a profound influence on reporting patterns. For example, news reports on H1N1 resulted in over-reporting on Web-search logs by Google Flu Trends. Where these alterations in Internet searches and postings arise, conclusions will need to be adjusted to account for this, he said.
INNOVATIVE MODELING FOR INTEGRATING DATA
Modeling can reveal effects and patterns in the data that are not apparent when only examining the raw results. Strategies and techniques for accurately modeling how a treatment is performing in either a clinical trial or a clinical application are rapidly evolving. Speakers discussed how incorporation of data collected outside of a clinical trial setting, such as medical record data, and better qualification and understanding of variability and its sources could lead to development of models that might be more effective at answering questions that arise in the postapproval space.
Quantifying and Addressing Uncertainty
Assumptions and uncertainties each increase variability in datasets, and variability affects the overall predictive power of a model. To accurately predict and build models of treatment response for clinical trials, both uncertainties and underlying assumptions should be taken into account, said Sandy Allerheiligen, vice president, Modeling and Simulation, Merck. Uncertainties in data can result from limited dataset size, bias in the sample population, heterogeneous response to treatment, or any number of effects that cannot be predicted or measured.
Statistical approaches known as value of information techniques could aid in quantifying uncertainty, said Katherine von Stackelberg, research scientist, Harvard Center for Risk Analysis. These techniques use Bayesian statistics to quantify the “opportunity loss from a decision made under uncertainty,” determining whether more data should be collected in clinical trials before bringing a product to market and helping to calculate the expected benefit of further data collection. The techniques can also serve to estimate the number of patients needed in a clinical trial to observe a treatment effect or approximate the cost of bringing more patients into a trial. Therefore, these techniques could be powerful tools for evaluating and directing research priorities. “[Value of information techniques] quantify the value of the information and allow the regulator to prioritize where additional investment is really going to lead to maximal benefit and identify those areas that have the greatest likelihood of influencing clinical practice,” said von Stackelberg.
Modeling the Placebo Effect
The placebo effect, or the measurable change in a patient’s health status that cannot be attributed to the treatment being tested, is another source of variability.8 Ariana Anderson, assistant research statistician, University of California, Los Angeles, noted the importance of modeling the placebo response in clinical trials. Modeling holds particular value in the case of rare diseases, where patient numbers are small and statistical power is low, and it may be unethical to assign patients to the placebo group, she said. If the placebo response could be predicted, it could help the clinical trial researcher to determine the magnitude of its effect in future trials. One such method for predicting and mapping the placebo response is through functional magnetic resonance imaging (fMRI) techniques. After a baseline fMRI is established in patients, this model can be used to predict treatment response over placebo for an individual patient, or potentially predict patients who would not respond to treatment because of increased susceptibility to placebo effect.
Modeling Within a Consortium Setting
One way to address variability is to approach clinical trial design and implementation via consortia. By having multiple organizations participate in a consortium, each organization can benefit from the expertise of the others and in turn can better address biases and heuristics inherent in data collection, said Corrigan. Consortia can expand the depth of data inputs and
___________________
8 For further information on this technique, see Anderson and Cohen (2013).
aid in creating model constructs that are potentially more reliable, which can, in turn, promote better understanding of disease natural histories and therapeutics. However, the actual amount of data available, data standards, data-sharing policies, and clinical trial or modeling approaches can vary among the data-collecting organizations. For a consortium to be successful, these discrepancies should be proactively addressed, Corrigan said.
As an example of a successful consortium effort, the Coalition Against Major Diseases, a public–private partnership formed by C-Path, created a disease progression model of Alzheimer’s disease (AD) that addresses critical concerns surrounding AD and the performance of treatments in clinical trials for AD, including dropout rates, placebo effect, covariants with the disease, and variability both in patient response and in the methodology used by different data collectors within the consortium. The model allows for users to quantitatively design clinical trials before they begin.
Corrigan outlined key components that lead to the successful generation of the AD model. He noted that, most importantly, success is a function of time, and consortia members should be prepared for potentially lengthy investment in constructing an effective model. Technical support, including enhancements and updates to the model, and infrastructure must be planned for in advance. Finally, establishment of data standards is critical, as is partnering with regulators early to qualify the tool and establish a context of use.
This page intentionally left blank.




















