
Proceedings of a Workshop
| IN BRIEF | |
|
December 2021 |
THE FOOD AND DRUG ADMINISTRATION’S EMERGENCY USE AUTHORIZATION: LESSONS LEARNED FROM THE PAST TO GUIDE THE FUTURE
Proceedings of a Workshop—in Brief
INTRODUCTION
The U.S. Food and Drug Administration (FDA) has responsibility for protecting public health by ensuring the safety, efficacy, and security of drugs, biological products, and medical devices. Since 2004, the Secretary of Health and Human Services has had the authority to “authorize the introduction into interstate commerce, during the effective period of a declaration…, a drug, device, or biological product intended for use in an actual or potential emergency.1 That is, in certain declared emergencies, FDA has the option to authorize use of a new product or a new use of an approved product—an authority known as Emergency Use Authorization (EUA)—if it has “reason to believe that the product may be effective and that its known benefits outweigh its known risks.” By contrast, in non-emergency situations, applicants must demonstrate a product’s safety and effectiveness through a lengthier, more extensive process.
On October 5–6, 2021, the Committee on Science, Technology, and Law of the National Academies of Sciences, Engineering, and Medicine (the National Academies) convened a virtual workshop on the EUA process. In introducing the workshop, planning committee chair William Schultz (Zuckerman Spaeder LLP) explained it was designed to examine FDA’s recent and historic use of EUAs, discuss lessons learned during the COVID-19 pandemic, and consider how those lessons might inform future efforts. The workshop also highlighted emergency mechanisms used by other health regulators (see Box 1) and considered how U.S. and global regulatory partners can strengthen cooperation in responding to global health emergencies.
__________________
1 21 USC Section 360bbb-3. The Project BioShield Act of 2004 (Public Law 108-276) amended the Federal Food, Drug and Cosmetic Act to authorize FDA to issues EUAs. 21 U.S.C. Sections 360bbb-3, 360bbb-3a, and 360bbb-3b. For more information, see “Emergency Use Authorization of Medical Products and Related Authorities,” https://www.fda.gov/regulatory-information/search-fda-guidance-documents/emergency-use-authorization-medical-products-and-related-authorities.
![]()
OVERVIEW OF EMERGENCY USE AUTHORIZATIONS AND SIMILAR AUTHORITIES
EUAs in the United States
In the opening session, former FDA Commissioner Margaret A. Hamburg (Nuclear Threat Initiative) discussed use of EUA in the United States.
“Our nation’s experience with COVID-19 has highlighted the value of the EUA mechanism,” Hamburg said. However, she noted, “it raises fundamental issues about how to balance risks and benefits, urgency and scientific rigor, and how to navigate complex questions about the intersection of scientific expertise, value judgments, individual autonomy versus collective goods, political pressures, public accountability, and societal needs.” As FDA Commissioner, Hamburg was involved with EUAs related to efforts to combat H1N1, Ebola, and Zika, but, she said, “none of those circumstances compare with COVID-19 in terms of the size, scope, urgency, or devastating impact, nor in all the other complexities swirling around.”2
The EUA process began in the early 2000s in recognition of new threats that had few FDA-approved medical countermeasures,3 Hamburg explained. The Project Bioshield Act of 2004 (PL 108-276)4 gave FDA the authority, in well-defined, declared emergencies, to authorize the use of drugs, devices, diagnostics, and other medical products not previously approved, cleared, or licensed or for off-label use of approved products. In 2005, the Public Readiness and Emergency Preparedness (PREP) Act (PL 109-148)5 added liability protection from tort liability6 to incentivize private sector development of medical countermeasures.
Initiation of the EUA process requires one of four conditions: (1) the Defense Secretary determines existence of a present or potential military emergency; (2) the Department of Homeland Security (DHS) Secretary determines existence of a present or potential domestic emergency; (3) the Health and Human Services (HHS) Secretary determines existence of a present or potential public health emergency; or (4) the DHS Secretary issues a material threat determination. If the HHS Secretary issues a declaration of emergency based upon one of these determinations, the FDA Commissioner may authorize EUAs if certain criteria are met (see Box 2).
An EUA expires when the emergency ends, Hamburg explained. Such authorizations require less safety and efficacy data than full approval. EUAs can be revoked, amended, or modified to protect public health or safety. Full approval of new products (or the new use of approved products) does require a more comprehensive submission, including additional safety and efficacy data along with the necessary administrative, manufacturing, and other information.
Hamburg stressed that careful consideration of available data and the ability to change course, as appropri-
__________________
2 Prior to COVID, FDA issued just over 20 EUAs. During the COVID-19 emergency, FDA has issued more than 500 EUAs to date, and this is the first time an EUA was approved for a novel vaccine.
3 “Medical countermeasures” are FDA-regulated products (biologics, drugs or devices) that may be used in the event of a potential public health emergency. See https://www.fda.gov/emergency-preparedness-and-response/about-mcmi/what-are-medical-countermeasures.
4 For the full text of PL 108-276, see https://www.congress.gov/108/plaws/publ276/PLAW-108publ276.pdf. The threats initially initiated by the legislation included chemical, biological, radiological and nuclear threats associated with terrorism.
5 For the full text of PL 109-148, see https://www.congress.gov/109/plaws/publ148/PLAW-109publ148.pdf. The Act provides drug manufacturers with immunity from tort claims of damages caused by vaccines, drugs, or other countermeasures.
6 A tort is an act or omission that gives rise to injury or harm to another and amounts to a civil wrong for which courts impose liability. See https://www.law.cornell.edu/wex/tort.
ate, are critical to the EUA process. She noted the importance of protecting FDA, its staff, and its regulatory processes from inappropriate political pressures. Political influence can never be fully eliminated, she acknowledged, and FDA leaders must be able to engage with political leaders to inform policy while protecting the FDA’s ability to perform its scientific and analytic tasks.
Effective preparedness and response are closely linked to public trust and confidence, Hamburg said. She urged FDA and other agencies to continually exhibit transparency, integrity, and a commitment to evidence-based decision-making. Continuous learning by public health officials and regulatory bodies can inform a better outcome for the next crisis, she concluded.
OTHER GLOBAL EFFORTS
The second workshop speaker, Emer Cooke (European Medical Agency [EMA]) described the EMA’s emergency response toolkit and emphasized the value of international collaboration. Although regulators develop different solutions in emergencies, she said, they face common problems in supporting the timely availability of and access to quality, safe, and effective vaccines, drugs, and diagnostics.
The EMA’s Conditional Marketing Authorisation (CMA) permits fast-track review and approval of medicines to fulfill unmet medical needs. CMA applicants can submit less comprehensive data than required for standard approvals, but still supportive of a positive benefit/risk profile and provide subsequent data.7 The speedy assessment is also facilitated by informal “rolling reviews.”8
Cooke emphasized the importance of preparedness to respond to public health emergencies. Building on experience from H1N1 and Ebola emergencies, a a health-threats plan was agreed to in 2018 encompassing rolling reviews; a dedicated platform to manage reviews and interact with applicants and experts; and ready-to-go legal frameworks, procedures, and plans. EMA also developed the OPEN Pilot in December 2020. This program permits the World Health Organization (WHO) and non-EU regulators to participate in EMA scientific evaluations for COVID-19 vaccines and therapeutics.9 Although each authority maintains its scientific and regulatory independence, participants share expertise and tackle common challenges. In this context, onwards reliance on WHO expedited its Emergency Use Listings (EULs) where EMA is the reference national regulatory authority also need to be considered.
__________________
7 According to Cooke, CMA has been used 60 times since its introduction in 2006, 13 in 2020 alone, including for four vaccines. For more information on the CMA process, see https://www.ema.europa.eu/en/human-regulatory/marketing-authorisation/conditional-marketing-authorisation.
8 The EMA defines a rolling review as “an ad hoc procedure used in an emergency context to allow EMA to continuously assess the data for an upcoming highly promising application as they become available, i.e. preceding the formal submission of a complete application for a new marketing authorization” See https://www.ema.europa.eu/en/documents/other/ema-initiatives-acceleration-development-support-evaluation-procedures-covid-19-treatments-vaccines_en.pdf.
9 Information on the Pilot can be found in “Questions and Answers on the Pilot Project ‘OPEN.’” Available: https://www.ema.europa.eu/en/documents/other/questions-answers-pilot-project-open_en.pdf.
Cooke chairs the International Coalition of Medicines Regulatory Authorities (ICMRA), a network of global health regulatory authorities. ICMRA enabled strategic collaboration and communication during the pandemic, with a major focus on alignment and convergence. Cooke said, without it, “we would not have achieved half of what [… was achieved as] an international community.” ICMRA is distilling best practices based upon input from 13 regulatory authorities.10 Common themes include a need for flexibility and agility in existing frameworks, challenges in how to determine whether “enough data” existed for decision-making, and maintenance of standards and public confidence.
Ongoing data generation and safety monitoring are essential, Cooke said, but warned “these are resource-intensive processes” that “take their toll on our experts and systems.” She urged continuing the unprecedented information-sharing that occurred between global health regulators and with industry during the pandemic, as well as greater use of regulatory reliance to reduce duplication of effort and expedite national decision-making.11
From EMA’s perspective, Cooke said emergency use authorization and conditional marketing authorization worked better for vaccines than for therapeutics, primarily because of the quality of clinical trials. She emphasized the need for well-designed trials and international pharmacovigilance collaboration.12 Significant challenges remain, she commented. “We have used the tools available to speed up and deliver approvals in record time, but we have not managed to address global supply and procurement,” she concluded.
USE OF THE EMERGENCY USE AUTHORIZATION DURING THE COVID-19 PANDEMIC
Three FDA centers regulate the products developed to combat COVID-19. The moderator of the first panel session, Joshua Sharfstein (Johns Hopkins Bloomberg School of Public Health), thanked the speaker representatives of the centers for their efforts during the pandemic: Patrizia Cavazzoni (Director, Center for Drug Evaluation and Research); Peter Marks (Director, Center for Biologics Evaluation and Research); and Jeffrey Shuren (Director, Center for Devices and Radiological Health).13
Center for Drug Evaluation and Research (CDER)
Communication, Cavazzoni said, has been the pillar of CDER’s pandemic response as it coordinated with other agencies, international counterparts, and the research community. In April 2020, CDER created the Coronavirus Treatment Acceleration Program (CTAP) to accelerate development of COVID-19 therapeutics. During the pandemic, CDER received unprecedented interest from researchers who had never submitted an application for an Investigational New Drug (IND). While this presented challenges, CDER found the EUA structure to be robust and flexible, Cavazzoni said. Although less data are required for EUAs, she noted the requirement for enhanced post-EUA surveillance. Based on these data collected, EUAs can be revised or revoked, as happened with hydroxychloroquine sulfate.14
The standard for an EUA issuance is applied in a dynamic environment, she explained, and its application differs across the centers due to the different qualities of the products they regulate. Despite a surge of clinical trials globally, she noted few produced sufficient, actionable data and collective resources were not used efficiently for regulatory decisions. She observed, however, that master protocols have proven valuable in advancing pandemic responses.15
Cavazzoni noted that transparency is critical for instilling public confidence. FDA makes its reviews public, but delays may occur while negotiating disclosure protection with sponsors. She suggested FDA be given broader authority to disclose information related to the safety and efficacy of the products it has approved for emergency use.
Staff across FDA rose to meet the demands, Cavazzoni commented, but an increased workload is not sustain-
__________________
10 When complete, the report will be available at www.icmra.info.
11 Regulatory reliance allows a national regulatory authority in one jurisdiction to take into account assessments performed by another authority or trusted institution, while remaining independent regarding the decisions based on those assessment. Available: https://www.raps.org/news-and-articles/news-articles/2020/6/who-drafts-recommendations-for-regulatory-reliance.
12 Pharmacovigilance is the detection, assessment, monitoring, and prevention of adverse effects or other medicine/vaccine related problems. Available: https://www.who.int/teams/regulation-prequalification/pharmacovigilance.
13 The Center for Drug Evaluation and Research (CDER) regulates therapeutics; the Center for Biologics Evaluation and Research (CBER) regulates vaccines; and the Center for Devices and Radiological Health (CDRH) regulates medical devices to include diagnostic tests, ventilators, and personal protective equipment.
14 For a timeline of EUA actions, see https://www.nationalacademies.org/event/10-04-2021/the-food-and-drug-administrations-emergency-use-authorization-lessons-learned-from-the-past-to-guide-the-future-a-workshop.
15 Cavazzoni cited to the RECOVERY trial set up in the United Kingdom; (see https://www.recoverytrial.net) and the ACTIV trial, set up by NIH in the United States (see https://www.nih.gov/research-training/medical-research-initiatives/activ.
able or advisable in the long term. CDER, for instance, saw a 43 percent increase in Investigational New Drug Applications and a 26 percent increase in formal meeting requests with no increase in staffing (see Figure 1). She suggested that FDA build enduring surge capacity in core scientific disciplines and supporting operations.
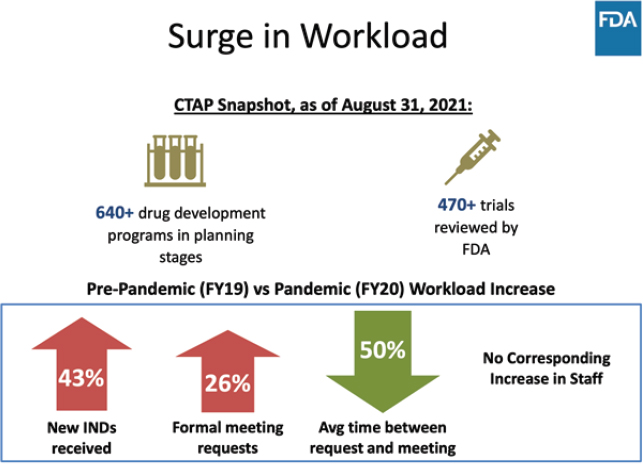
SOURCE: Patrizia Cavazzoni, Workshop Presentation, October 5, 2021.
Sharfstein asked whether CDER should establish a minimum standard for data from applicants for therapeutics, noting the criticism the agency received for its hydroxychloroquine EUA. Cavazzoni responded that the EUA should be taken in context. “There was no standard of care, patients were dying in hospitals, and no available therapies were on the horizon,” she said. Cavazzoni noted that the EUA for hydroxychloroquine was limited in scope: the drug was only authorized for the treatment of certain hospitalized patients with COVID-19 when a clinical trial was not available or participation in a clinical trial was not feasible. The EUA was based on the totality of available evidence, as the statute allows, she said, and was revoked when new data came in and the agency determined that the criteria for issuance were no longer met.
Center for Biologics Evaluation and Research (CBER)
COVID-19 vaccine development demonstrates the flexibility of the EUA, said Marks. Flexibility is important, he said, because the statute was developed for potentially more destructive disasters, such as chemical warfare.
At the time of the workshop, three vaccines had EUAs, and one of those also had full approval in the adult population. In granting EUA approval of vaccines, CBER required “clear and compelling efficacy in large well-designed phase 3 trials,” as well as quality, safety, and efficacy evaluation; public advisory meetings; and enhanced post-deployment surveillance.
Sharfstein noted that although CBER set a clear standard for guidance, data on pregnant women and children have lagged. Marks agreed, but attributed the lag in data collection to a lack of foresight in development rather than the EUA process. “As soon as we had evidence of effectiveness in November 2020, we probably should have accelerated development in pediatric settings, for the immune-compromised, and for pregnant women,” he said.
Center for Devices and Radiological Health (CDRH)
This is the seventh emergency in which EUAs were used for medical devices, said Shuren, but COVID-19 required a different playbook for a multitude of products including personal protective equipment (PPE), tests, and ventilators. To date, CDRH has received more than 7,100 EUA and pre-EUA applications and authorized 1,749 devices. Shuren concurred that stakeholder communication and engagement have been critical. For example, CDRH initiated webinars with test developers, created templates to facilitate information submittal, and issued 28 guidance documents.
As with other products, the EUA standard for the authorization of diagnostic tests is lower than full market approval, Shuren explained. CDRH seeks to be flexible and innovative, although some issues emerged in implementation. For example, a notification (self- certification) policy was published to increase test availability of molecular diagnostic and serology tests, but Shuren acknowledged its unintended consequences.16 Poor-performing molecular tests entered the marketplace and had to be revalidated, redesigned, or removed, he said, and poor performing serology tests entered the marketplace and had to be removed while being inappropriately touted by some officials as a means to open up the economy.
Shuren noted how South Korea responded to the pandemic.17 Prior to the pandemic, the country had invested in prepositioning tests for rapid development, stock-piled testing supplies, and guaranteed purchases and reimbursement that minimized manufacturers’ risks. While the South Korean and U.S. validation approaches were similar, the South Korean EUA process was more focused and open for only one month. Earlier access to patient specimens and a centralized capacity to evaluate clinical performance conserved resources and expedited test validation and authorization, Shuren said.
Tests should not enter the marketplace “sight unseen,” Shuren stated. In the future, he suggested prepositioning commercial developers, de-risking the enterprise for manufacturers, investing in centralized performance validation in both emergencies and “peacetime”, setting up a more robust mechanism for sample sharing between countries, and investing in novel technologies, especially point-of-care and at-home tests.
Noting the array of available devices and uses, Sharfstein asked whether other agencies might have helped guide FDA by defining use cases. Shuren agreed and replied that a national testing strategy could have determined which tests were needed, controlled distribution and supply of tests and testing supplies, and established mechanisms for reporting results.
Discussion
Sharfstein asked the center directors if EUA flexibility could open the door to external meddling. Marks said that transparency and data-sharing with advisory committees protect against interference. Although some errors were made, science ultimately corrected them, he added. Cavazzoni added that the agility of the EUA allows for an evolution of thinking based on evolving data, as demonstrated when an EUA is revoked.
Companies, rather than FDA, sometimes become the prime communicators with the public about EUAs, Sharfstein observed. Cavazzoni explained that laws and regulations constrain what FDA can communicate. Based on CDER’s experience, she said she would welcome broader authorities to share information, including for EUAs that are declined. Shuren suggested that a greater understanding about how and what information is communicated could increase public trust in FDA decision-making. Misinformation on social media poses a particular challenge, Marks added.
During a second session at the end of the first day of the workshop, Sharfstein asked the directors what is “high on their list” for the future. Cavazzoni supported continued work on platform trials to have equity-based infrastructure at the ready. Shuren emphasized preparedness. “If we are prepared, we do not have to use the EUA as much, he said.” Marks noted the surge in FDA workload during the pandemic. He urged thinking about how to work most efficiently with companies in the future.
In response to whether changes to the EUA are needed, Shuren acknowledged the EUA standard allows more uncertainty, but “we deal with uncertainty all the time.” Calculations of benefit and risk differ based on circumstances, he noted. Cavazzoni added, “The public needs to understand the EUA is not an end in itself. The evidence generation continues [after the EUA is granted].” Marks said that developing standards is a challenge because each emergency is unique. On the device side, setting clear expectations about performance and other characteristics is important, Shuren said.
To Cavazzoni, “the big takeaway is the critical importance of collaboration across U.S. agencies and with international counterparts, the research community, and patients.” She underscored the value of an agile and flexible framework, perhaps accompanied by more explicit explanations about decisions made within it. Marks urged, “If we are thinking about anything, think minor redecoration not renovation.” Shuren agreed. EUA flexibility, he said, has been invaluable.
__________________
16 Policy for Coronavirus Disease-2019 Tests during the Public Health Emergency (Revised). Available: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/policy-coronavirus-disease-2019-tests-during-public-health-emergency-revised.
17 FDA Center for Devices and Radiological Health. 2021, South Korea’s Response to COVID-19. Available: https://www.fda.gov/media/149334/download.
COMMUNICATION AND TRANSPARENCY FOR EUAS USED DURING THE COVID PANDEMIC
Nirav Shah (Maine Center for Disease Control), moderator of the second panel session, noted that how EUAs are understood and used on the ground has been critical to the COVID-19 response. Discussion focused on implementation before the emergency declaration, in the early stages, and in more mature stages.
Session panelist Scott Becker (American Public Health Laboratories Association) praised FDA’s professionals, whose work has atypically been in the foreground during COVID-19. From a laboratory perspective, he said, while the EUA process worked well, it could be tweaked to address lessons learned. Panelist Anne Zink (Alaska Department of Health and Human Services) echoed thanks and the need to apply lessons for the future, but appreciated that she and colleagues could assure the public that the EUA process is safe, robust, and puts science first. Panelist Mike Fraser (Association of State and Territorial Health Officials) reported similar comments from officials in other states, despite early questions about the process. WHO’s Emergency Use Listing Procedure (EUL) process meant that, within 15 days, 101 low- and middle-income countries authorized entry of vaccines into their countries, noted panelist Mariângela Simão (WHO).
When Shah asked how FDA could improve public confidence, Zink responded when information first becomes public through social media and company releases, it is “hard to message on a state level.” She urged alignment of timelines, updates, and approvals across the federal government. She also called attention to challenges, such as access to testing, in rural or remote locations. Simão observed that the world is driven by press statements, but health professionals and regulators must be driven by evidence within transparent guidelines. In advance of the next pandemic, she called for legal changes to allow FDA to share more information with WHO and other regulators.
Becker suggested that testing problems occurred because there was little experience with the EUA process for diagnostics. The first few weeks were tough, he said, but FDA quickly established pathways for applicants to work with FDA. He expressed hope that the community of practice FDA created with test developers continues, but a pathway for public health surveillance testing is still needed. Fraser commented on the larger context in which COVID-19 was politicized. While the majority of state health officials trust the EUA process, he said, explaining it to the public is complicated.
Shah asked the panelists about potential changes to the EUA process. Based on focus group feedback, Fraser suggested clearer public explanations about such concepts as emergency versus experimental use. Legal changes to enable FDA to more fully explain decisions would benefit the nation and the world, Zink added. With greater knowledge and countermeasures in place, Simão suggested more caution in assessing risks and benefits. However, Becker noted urgency remains in some lesser developed areas, such as rapid testing.
Several panelists noted that better preparation and coordination could reduce confusion. “When an EUA is posted on the FDA website, the work is just beginning for us,” Fraser said. State officials need to be prepared for questions such as racial and ethnic composition of trials or the effects of therapeutics on pregnant woman and children. “Health officials are put in a position to explain how FDA operates,” he added. “The more we can anticipate and share, the better.”
Workshop planning committee chair Schultz asked about possible testing-related changes. Becker called for a systems approach to bring stakeholders together. “We had separate lanes for everybody and, even within the laboratory community, for different labs,” he said. He suggested agencies that interact with the lab system come together. Simão observed that diagnostics regulation and quality are uneven across countries, although ICMRA is working on harmonized approaches. Zink identified low-cost rapid testing as a cornerstone for improvement.
When thinking about the next pandemic, Fraser called for anticipating communications challenges and tracking EUAs in the pipeline. Becker emphasized the value of preparedness and open dialogue with regulators. Zink urged a change in mindset: “The EUA process is fundamentally a public health process and not a company process,” she said.18 “We need to change our mindset thinking about legality, transparency, and communications.” Simão underscored the need for a global approach to regulation.
AN EVALUATION OF THE PROCESS FOR AUTHORIZING EUAS AND TRANSITIONING TO FULL APPROVAL
William Schultz moderated a discussion of EUAs and the transition from authorized emergency use to full approval.
__________________
18 Companies frequently disseminate information about FDA EUA decisions prior to FDA communication with the public.
When asked to evaluate how FDA used its EUA authority, session panelists Monica Ghandi (University of California San Francisco) and Luciana Borio (ARCH Venture Partners and Johns Hopkins University) commented that the process worked particularly well with vaccines, but, Borio added, FDA did not fully leverage lessons from previous epidemics. She noted “pain points” around virus testing, although some of the challenges lay outside the EUA context, and therapeutics that received premature EUAs. Panelist Aaron Kesselheim (Harvard Medical School and Brigham and Women’s Hospital) highlighted the mixed record for therapeutics. He noted that although problems arose with the EUA for hydroxychloroquine, the EUA for remdesivir demonstrated the usefulness of the process.
Panelist Ruth Faden (Johns Hopkins University) said, as a bioethicist, she looks at unintended consequences. Using vaccines as an example, Faden observed that the public health community is telling the public that the vaccine is safe and effective, but the regulatory authority is saying that it does not have sufficient information for full approval. She articulated a need for better explanations of what an EUA is and is not. Faden also noted that a legitimate EUA decision should be made using the best available science and independent of political conflicts interest or corruption. Ghandi noted that some patients in the safety net clinic where she works felt that the vaccine process seemed experimental and rushed. Roll-out in doctor’s offices or pharmacies, rather than mass vaccination sites, would have allowed for more one-on-one conversations regarding patient concerns, she asserted.
When asked if FDA’s EUA broad statutory authority should be revised, Borio said she does not favor restrictions, despite what she considered to be some failures in implementation. Kesselheim added that, although the intention of the statute is that EUAs are time-limited to respond to acute threats, some EUAs remain in place from previous emergencies. Time limitations or other changes can occur either statutorily or by guidance and practice, he noted.
Faden noted the conundrum in continuing trials with placebo-controlled arms19 once vaccine roll-out began. Besides ethical considerations, clinical trial participants were withdrawing from vaccine clinical trials once EUAs had been granted for the vaccine. Kesselheim suggested that well-designed trials, including several supported by NIH, could alleviate concerns about EUAs.20 Borio urged companies to “bring products to the finish line” for full approval.
INTERNATIONAL COOPERATION
The next session looked at regulatory emergency authorizations through a global lens. Agnés Saint-Raymond (EMA) moderated a session on international cooperation in the regulation of emergency use authorizations.
Session panelist Soumya Swaminathan (WHO) identified several high-level panels, including the Independent Panel for Pandemic Preparedness21 and Response and the Independent Oversight and Advisory Committee (IOAC) for the WHO Health Emergencies Programme22 that conducted reviews of pandemic preparedness and response. The reviews identified as challenges different guidelines among organizations, lack of preparedness, lack of transparency regarding the scientific basis for recommendations, and responses that exacerbated inequalities. Global health regulatory strengths include the health workforce, experience-based national responses, and vaccine development. Swaminathan noted that the WHO panel recommended the establishment of a pre-negotiated platform to facilitate access to tools and supplies.
In February 2020, WHO held its first global research and innovation forum to set a research roadmap for COVID-19, and thousands of researchers have participated in WHO expert groups, Swaminathan said. Other efforts include the Solidarity Trial for collaboration around therapeutics by researchers around the world23 and the Access to COVID-19 Tools (ACT) Accelerator to provide global solutions for diagnostics, therapeutics, and vaccines.24 Swaminathan emphasized the importance of global regulatory coordination for equitable access. “It’s important to look at not just the R&D, but the evidence that informs the policy and then granting of regulatory approval,” she said.
Panelist Nicole Lurie (Coalition for Epidemic Preparedness [CEPI]) discussed FDA’s global positioning, evidence generation, and a path forward. Although FDA’s expertise is globally recognized and international collaboration occurred, FDA’s U.S. focus means that “there is a sense that what FDA says is irrelevant for many developing countries,” she stated. Lurie suggested that FDA offer views tempered to other settings to better serve the international community.
While acknowledging many useful trials, Lurie pointed to murkiness with regard to who has responsibility for
__________________
19 A placebo arm refers to a group of participants in a clinical trial that receive a placebo.
20 The NIH trials Kesselheim identified as examples were Researching COVID to Enhance Recovery (RECOVER) initiative and Accelerating COVID-19 Therapeutic Interventions and Vaccines (ACTIV).
21 Available: https://theindependentpanel.org/mainreport/.
22 Available: https://www.who.int/news/item/25-05-2021-ioac-statement-at-the-seventy-fourth-world-health-assembly-25-may-2021.
23 Available: https://www.who.int/emergencies/diseases/novel-coronavirus-2019/global-research-on-novel-coronavirus-2019-ncov/solidarity-clinical-trial-for-covid-19-treatments.
24 Available: https://www.who.int/initiatives/act-accelerator.
generating evidence, especially in the post-EUA period. She suggested that high-income countries have the responsibility to generate high-quality evidence and support low- and middle-income countries in their efforts. While countries must make their own regulatory decisions, she called for better harmonization among regulators.
Panelist Murray Lumpkin (Gates Foundation) noted that, in working with WHO and low- and middle-income countries, the Gates Foundation found that FDA’s EUA decisions have international ramifications. For regulators in other countries to base actions on FDA’s work, they require full transparency about the reasoning behind FDA decisions. Two challenges exist: (1) U.S. confidentiality practices limit what FDA can share with other regulators; and (2) resource-constrained settings often receive different versions of the product than the versions distributed in the U.S. or other high income countries. Lumpkin said that global regulators need access to unredacted information to know why FDA authorized (or did not authorize) products and which version of the product was authorized. EMA and other agencies that share documentation have become the agencies of choice for the WHO and resource-constrained agencies, he added. “I would argue that current U.S. confidentiality practices are 20th-century relics of a pharmaceutical ecosystem that no longer exists,” he stated. He suggested examining confidentiality policies to determine if they are aligned to meet current realities, even if only during emergencies.
Panelist Analia Porras (Pan American Health Organization [PAHO]) explained that PAHO works with Latin American and Caribbean health regulatory authorities and procures medical products for those countries. PAHO developed a reliance-based process during COVID-19, starting with PPE and diagnostics. The WHO EUL provided a transparent and efficient mechanism that helped with the uptake of COVID-19 vaccines by Latin America and the Caribbean, according to Porras. She echoed Lumpkin’s remarks that the international community needed greater access to the data and information underlying FDA’s EUA decisions to increase trust and transparency. “The era of blind trust in regulatory decisions is over. It’s not enough to publish decisions; the global community needs access to the basis of those decisions,” she said.
PAHO launched its own network of COVID-19 regulatory focal points, Porras noted. She called for improved post-marketing surveillance to maintain confidence in the EUA process.
Panelist Jörg Schläpfer (Swissmedic) said that, while he was speaking from the perspective of a single national authority, international cooperation is key. Emergency authorization must take into account risk-benefit profiles in different settings, he continued. Switzerland has historically had strong reluctance to vaccines, he explained, and emergency authorization for vaccines has never been an option. Instead, Switzerland sped up the review and approval of vaccines within existing legal processes without using EUAs, while Switzerland’s COVID-19 Act foresaw emergency procedures for therapeutics to treat ill patients.25
Discussion
During discussion, Swaminathan noted the importance of academia in conducting discovery research, but said the proliferation of small clinical trials has not been the most productive. She suggested large platform trials may be needed during emergencies, as well as more global representation in these trials. Porras said academic researchers have been invaluable in post-marketing vigilance programs. Noting the success of WHO in setting an agenda that helped focus academic research, Lurie observed that the United States did not undertake this.
Schläpfer underscored the need for marketing surveillance of diagnostics to ensure suitability. Porras added in the early stages, it was difficult to advise countries about performance, use, and origin of products. Emergency use has to be coupled with validation, she said. Lumpkin said the hard part, especially early in the pandemic, was how to interpret and act on test results at the patient, community, and international levels. Swaminathan noted that the diagnostics field is less well developed than the fields of therapeutics and vaccines. She warned that sub-standard diagnostics can cause harm and waste resources.
To Schläpfer, how to communicate the notion of emergency approval, in light of public expectations, is essential. It is important to convey that no corners are cut regarding safety and quality, he said. Porras emphasized the importance of having the authority and resolve to make decisions about a product post-market. Lumpkin urged real-time availability of data behind both positive and negative FDA EUA decisions. Lurie added that FDA should require companies to agree to full transparency. Swaminathan emphasized the need to follow the science and urged FDA to play an active international role in setting benchmarks and sharing data.
__________________
25 For more information on Switzerland’s COVID-19 Act, see https://www.admin.ch/gov/en/start/documentation/votes/20210613/covid-19-act.html.
HOW SHOULD EUA AUTHORITY BE USED FOR FUTURE PANDEMICS?
Session moderator David Vladeck (Georgetown Law Center) asked panelists to consider what the COVID-19 pandemic can teach about how to deal with inevitable future pandemics.
Former FDA Commissioner Robert Califf (Verily and Google Health) said that there is a critical need for a cohesive federal plan that encompasses vaccines, diagnostics, and therapeutics; public health systems; supply chains; interaction with global regulators; and other issues. Califf’s co-panelist Dave Chokshi (New York City Department of Health and Mental Hygiene) identified two areas for improvement from a local perspective. The first, he said, relates to communications, beginning with a clear presentation of the rationale for the emergency declaration. The second relates to coordination across federal agencies.
Panelist Peter Lurie (Center for Science in the Public Interest) suggested that EUA criteria be revisited and clearer guidelines developed to strengthen FDA’s evidence standard. He questioned whether flexibility in the EUA statute is responsible for uneven performance across FDA. “As far as we can tell, the flexibility only went one way—bringing products to market,” he said, although he acknowledged that what FDA rejected is unknown. He posited that the flexibility opens the door to political pressure. Panelist Patricia Zettler (Ohio State University School of Law) noted that FDA’s EUA flexibility goes beyond whether or not to authorize a product. EUAs are temporary, and FDA has the power to revise or revoke them and to place conditions on additional data collection. “The question,” she said, “is how can FDA best use its flexibility and broad powers? Transparency could be an important tool to reduce the uncertainty created by that flexibility and perhaps reduce opportunities for inappropriate political pressure.”
Vladeck asked whether structural issues need to be fine-tuned or overhauled so the government speaks with one voice. To Califf, a cohesive national plan would alleviate some issues. Lurie agreed with the benefit of a single voice, but expressed concern if disagreements that are an expected part of scientific or regulatory processes were hidden. He lauded the FDA’s Equal Voices campaign, which allows FDA medical officers to dissent from advisory committee decisions.26
Chokshi acknowledged the inherent tension between flexibility and coordination. “To me, the task is bringing about coordination at the level of the [HHS] Secretary in a way that flows from the initial declaration of the emergency,” he said. “The Secretary should ‘remain the conductor of the symphony’ from that point forward.” Zettler characterized advisory committees as an important transparency tool. She suggested that a unified plan could help in the messaging.
Califf commented that the more pressure brought to bear on federal agencies, the greater the importance of having agency staff with the expertise and judgment to deal with that pressure. A larger crisis, he said, is many people’s deep distrust of the government, which is often influenced by misinformation. Chokshi noted the problem extends to distrust in experts. He also reminded the group that without EUAs for vaccines, the delays in securing full approval would have had a huge negative impact: hundreds of thousands of lives were saved thanks to the vaccine EUAs.
Lurie said allegations that science fell short are frustrating, given the historic success of the scientific effort. While some topics, such as the use of preprints by the media, will be judged over time, he praised FDA’s use of advisory committees and guidance documents. He urged that the agency be better prepared to respond to misleading information about products it reviews. Zettler reiterated that EUA statutory language creates communications challenges. She noted the same terminology and processes apply for drugs, devices, and products, yet they fall under different regulatory regimes in non-emergency times. She questioned whether the same language and processes for each category should be used in the EUA process.
Zettler said testing may be an area where flexibility is beneficial, and Lurie noted that the regulatory structure around therapeutics and vaccines is stronger than that for diagnostics. Chokshi said the presence of a testing strategy would have allowed for more effective EUA use for diagnostics, especially at the pandemic’s start. Califf noted that the medical device industry is fragmented with a few big companies and hundreds of thousands of start-ups, which leads to innovation as well as errors. However, he said, this situation cannot be addressed in the setting of an emergency.
Reflecting on New York City’s emergency in spring 2020, Chokshi said that an important shortcoming was the inability to use data to inform real-world decisions. Epidemiology must be both retrospective and prospective, he said. Waiting for more data or for a more complete epidemiological picture is not realistic in a pandemic.
__________________
26 For the policies and procedures of Equal Voice, see https://www.fda.gov/media/79353/download.
THE PATH FORWARD: EQUITIES AND ENHANCED PREPARATION FOR THE FUTURE
Rogério Gaspar (WHO) moderated the final workshop panel on equity and global cooperation among regulatory authorities.
Panelist Richard Hatchett (CEPI) commented that, while each threat is unique, outbreaks begin locally before expanding globally. FDA, as a leading regulator, needs to consider how to position itself to support future international response, he said. CEPI has set a goal of compressing vaccine development to 100 days, he noted, which would require regulators to determine what can be put in place beforehand.27 The speed with which COVID-19 vaccines were developed was predicated on previous work on prototype pathogens, he observed. He commented, “We need to think about adapting regulatory mechanisms to interdigitate with work happening in R&D to prepare for potential unknown future pathogens.”
Panelist Helen Rees (South African Health Products Regulatory Authority) noted that COVID-19 called attention to the importance of regulatory authorities, which are insufficiently supported in many countries. Sharing data among regulators has been difficult, she said, and called for a global discussion about what reliance and harmonization really means, and how we talk to each other in an emergency.” She encouraged applicants to be more transparent. “We should stop seeing ourselves as ring-fenced national entities and start seeing ourselves as part of a global network,” Rees said.
Regulators have a central role to play in increasing access by the world’s poorest populations to vaccines and other countermeasures, said panelist June Raine (Medicines and Healthcare Products Regulatory Authority). Efforts to expedite access to medical products have included early engagement, rolling dossier reviews, and rapid approval of trials. In addition, meaningful partnerships with the public, industry, philanthropy, and other agencies have expanded access.
Raine urged regulators to embed innovation and not return to old approaches. To strengthen collaboration, she endorsed ICMRA and EMA efforts to achieve greater harmonization and streamline regulatory processes. She also emphasized the importance of data sharing. Regulators must remove unintended barriers that slow progress, she said. “The way forward has to look like more of what we are doing,” she said, “with greater innovation, embedding the best practices that regulators have adopted, taking managed risks, systematically sharing information, and making the exceptional the everyday.”
Panelist Seth Berkley (Gavi Alliance) spoke about his organization’s role in strengthening countries’ abilities to roll out vaccines. Berkeley explained that Gavi requires WHO pre-qualification prior to vaccine purchase, as many countries do not have stringent regulations or well-functioning regulatory agencies. The large number of companies and manufacturing sites around the world have created particular challenges. Information-sharing by more well-established regulatory agencies, rolling submissions, and sharing of dossiers would be of great help, he said. He also called for building capacity because of the “evolutionary certainty” of future outbreaks. When donations come from countries with well-established regulatory agencies, he added, sharing dossiers is still necessary for local acceptance.
Discussion
During discussion, Rees noted that many countries do not have legal frameworks such as EUA or CMA and instead use more specific legislation or policy measures. Raine underscored the value of an enabling, independent framework during emergencies. Berkley called for transparency between regulatory agencies, even if information must be withheld from the public for trade reasons. Without transparency, he said, delays occur. “It is time to think about confidentiality rules and open them up,” he stated.
Raine said CEPI’s 100 Days Mission for vaccine development involves a range of activities. While highly ambitious, each of the mission’s recommendations has an owner, action, and time frame. Hatchett explained that, by analyzing each stage of COVID-19 vaccine programs, it is possible to shorten timelines.
Berkley praised FDA staff but urged FDA to see itself in a global context. Rees called for global regulatory convergence. She also emphasized the need for equity in the identification of pathogens for further vaccine research, noting many pathogens that require vaccines occur in low- and middle-income countries. Hatchett called for common
__________________
27 Pandemic Preparedness Partnership. 2021. 100 Days Mission to Respond to Future Emergency Pandemics. A Report to the G7. https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/992762/100_Days_Mission_to_respond_to_future_pandemic_threats__3_.pdf.
approaches and innovation. Raine concluded, “We must keep up the momentum. Let’s converge with the interests of patients all over the world in mind.”
CONCLUDING REMARKS FROM THE WORKSHOP PLANNING COMMITTEE
During the closing session, members of the planning committee offered concluding remarks.
Hamburg highlighted suggestions by many presenters about placing FDA’s actions into a broader context, including an international one. She noted a recurring emphasis on transparency, information-sharing, and coordination as critical, both domestically and internationally. She suggested that the U.S. National Academies could call for expanded access to information and greater transparency regarding FDA decisions, an examination of how much evidence is enough in different contexts, and a consideration of how to set research agendas. She also commented on the huge opportunities to strengthen clinical trial infrastructure and post-market surveillance.
Sharfstein emphasized the importance of international collaboration and highlighted presenters’ comments that the United States could more strongly support global efforts. In considering the domestic use of EUA, he observed that different issues emerge at different stages. Before an emergency is declared, for example, better research, public-health use cases, and pre-positioning companies could make the EUA process more effective. Sharfstein reflected on the comments from several presenters who support the EUA statute but called for more clarity about standards and what happens post-authorization.
Shah focused on comments about transparency and clear communications. Although it is easy to state that increased transparency is better, he noted that many participants raised the concern about confusing information from multiples sources. Preparedness is top of mind for the public health community, he commented, and highlighted suggestions that included soliciting input from diverse stakeholders, tracking the status of EUA applications, and streamlining communications.
Julie Gerberding (Merck) emphasized the need to make the emergency use authorization process work globally and to minimize discontinuity across regulatory environments. She cited FDA guidelines on vaccine approval as an example of clear communications that expedited industry’s ability to participate in EUA while reducing uncertainty. She urged attention to developing guidelines for future products. Public education is still necessary, she added, as is demonstrated by vaccine uptake hesitancy.
Saint-Raymond called for an end to nationalism in a global pandemic. She urged undertaking “inter-crisis work” to develop transparency and data-sharing agreements among regulators. She highlighted the need to define responsibilities around evidence generation and other issues to advance knowledge globally with stakeholders that include industry, patients, and healthy populations.
Vladeck focused on the weakness, fragmentation, and under-resourcing of the public health infrastructure domestically and internationally. This infrastructure must be modernized before the next pandemic, he said. A weakness identified during the workshop, he added, is a lack of coordination. Refinement of processes to achieve greater coordination and uniformity of communications is needed before the next crisis, he concluded.
During the current pandemic, Gaspar noted that some problems were avoided by building upon responses to previous threats. However, he added, while the science moved fast and the level of cooperation and performance was high, challenges remain in the areas of research, regulation, deployment, and especially diagnostics. He reminded the group that during a pandemic, “until all are protected, no one is protected.”
Information-sharing came up in many contexts, Schultz commented, and asked the committee about the downside of information-sharing. Hamburg said that some information, such as confidential or proprietary information, should be shared with regulators but not more broadly. Clear rules would be needed, she suggested. Gerberding said that, because the stakes are so high and public interest so intense, there have been examples of entities releasing information to the media before FDA fully reviewed their applications. She suggested that industry internal agreements or standards about disclosure might be worth exploring. Saint-Raymond highlighted the different information-sharing needs of regulators and the public. Gaspar suggested that self-regulation by pharmaceutical companies may provide a way to avoid disclosure before regulatory reviews. In his view, it is impossible to combat a global pandemic when authorities are not sharing unredacted information with each other.
Schultz said the workshop demonstrated the need to engage in appropriate planning. FDA developed a detailed vaccine plan that was well received. While this approach might also work for drugs, testing requires a different strategy because of the multitude of tests, manufacturers, and regulatory systems, he posited. He called for additional thinking about testing, perhaps internationally.
DISCLAIMER: This Proceedings of a Workshop—in Brief has been prepared by PAULA WHITACRE as a factual summary of what occurred at the workshop. The committee’s role was limited to planning the event. The statements made are those of the individual workshop participants and do not necessarily represent the views of all participants, the planning committee, the Committee on Science, Technology, and Law, or the National Academies.
REVIEWERS: To ensure that it meets institutional standards for quality and objectivity, this Proceedings of a Workshop—in Brief was reviewed by MARK MCCLELLAN, Duke University; ANALIA PORRÁS, Pan American Health Organization; and JOSHUA SHARFSTEIN, Johns Hopkins Bloomberg School of Public Health. MARILYN BAKER, National Academies of Sciences, Engineering, and Medicine, served as the review coordinator.
Planning Committee for A Workshop: WILLIAM B. SCHULTZ (Chair) (Zuckerman Spaeder LLP); ROGÉRIO GASPAR (World Health Organization); JULIE L. GERBERDING (Merck & Co., Inc.); MARGARET A. HAMBURG (Nuclear Threat Initiative); AGNÈS SAINT-RAYMOND (Former European Medicines Agency); NIRAV SHAH (Maine Center for Disease Control); JOSHUA M. SHARFSTEIN, (Johns Hopkins Bloomberg School of Public Health); DAVID C. VLADECK (Georgetown University Law Center).
National Academies of Sciences, Engineering, and Medicine Staff: REBECCA EVERLY, Acting Interim Director; JOE S. CECIL, Senior Advisor; ANITA EISENSTADT, Responsible Staff Officer; STEVEN KENDALL, Program Officer; and DOMINIC LOBUGLIO, Senior Program Assistant.
FUNDERS: Financial support for this activity was provided by the Gates Foundation and the Rockefeller Foundation.
For additional information about the Planning Committee for the Food and Drug Administration’s Emergency Use Authorization: Lessons Learned from the Past to Guide the Future, visit https://www.nationalacademies.org/event/10-04-2021/the-food-and-drug-administrations-emergency-use-authorization-lessons-learned-from-the-past-to-guide-the-future-a-workshop.
Suggested citation: National Academies of Sciences, Engineering, and Medicine. 2021. The Food and Drug Administration’s Emergency Use Authorization: Lessons Learned from the Past to Guide the Future: Proceedings of a Workshop—in Brief. Washington, DC: The National Academies Press. https://doi.org/10.17226/26441.
Policy and Global Affairs

Copyright 2021 by the National Academy of Sciences. All rights reserved.













