
Page 193
5
DEVELOPMENT OF CANNABINOIDS DRUGS
Medicines today are expected to be of known composition and quality. Even in cases where marijuana can provide relief of symptoms, the crude plant mixture does not meet this modern expectation. The future of medical marijuana lies in classical pharmacological drug development, and indeed there has been a resurgence of scientific, as well as public, interest in the therapeutic applications of cannabinoids. After an initial burst of scientific activity in the 1970s, today's renewed interest has been fueled by major scientific discoveries discussed in previous chapters: the identification and cloning of endogenous cannabinoid receptors, the discovery of endogenous substances that bind to these receptors, and the emergence of synthetic cannabinoids that also bind to cannabinoid receptors. These scientific accomplishments have propelled interest in developing new drugs that can treat more effectively or more safely the constellation of symptoms for which cannabinoids might have therapeutic benefit (see chapter 4). Through the process of what is referred to as ''rational drug design,'' scientists manipulate the chemical structures of known cannabinoids to design better therapeutic agents. Several new cannabinoids are being developed for human use, but none has reached the stage of human testing in the United States.
The purpose of this chapter is to describe the process of and analyze the prospects for development of cannabinoid drugs. It first discusses the regulatory hurdles that every new drug encounters en route to market. It then proceeds to describe the regulatory and market experiences of
Page 194
dronabinol (tetrahydrocannabinol, or THC, in sesame oil), the only approved cannabinoid in the United States. These sections serve as a road map to determine whether the therapeutic potential of cannabinoids is likely to be exploited commercially to meet patient needs. Finally, the chapter describes what would be needed to bring marijuana to market as a medicinal plant.
The term cannabinoids is used in this chapter to refer to a group of substances that are structurally related to THC—by virtue of a tricyclic chemical structure—or that bind to cannabinoid receptors, such as the natural ligand anandamide. From a chemist's point of view, this definition encompasses a variety of distinct chemical classes. But because the purpose of this chapter is to explore prospects for drug development, both chemical structure and pharmacological activity are important; therefore, the broader definition of cannabinoids is used.
Federal Drug Development Policy
Like controlled substances, cannabinoids developed for medical use encounter a gauntlet of public health regulatory controls administered by two federal agencies: the Food and Drug Administration (FDA) of the U.S. Department of Health and Human Services (DHHS) and the Drug Enforcement Administration (DEA) of the U.S. Department of Justice. The FDA regulates human testing and the introduction of new drugs into the marketplace, whereas the DEA determines the schedule of and establishes production quotas for drugs with potential for abuse to prevent their diversion to illicit channels. The DEA also authorizes registered physicians to prescribe controlled substances. Some drugs, such as marijuana, are labeled Schedule I in the Controlled Substance Act, and this adds considerable complexity and expense to their clinical evaluation. It is important to point out that Schedule I status does not necessarily apply to all cannabinoids.
Food and Drug Administration
Under the Federal Food, Drug, and Cosmetic (FD&C) Act, the FDA approves new drugs for entry into the marketplace after their safety and efficacy are established through controlled clinical trials conducted by the drugs' sponsors.23 FDA approval of a drug is the culmination of a long, research intensive process of drug development, which often takes well over a decade.19,44 Drug development is performed largely by pharmaceutical companies, but some targeted drug development programs are sponsored by the National Institutes of Health (NIH) to stimulate further development and marketing by the private sector. The NIH's drug devel-
Page 195
opment programs—including those for AIDS, cancer, addiction, and epilepsy—have been instrumental in ushering new drugs to market in collaboration with pharmaceutical companies.33 In fact, as noted later, most of the preclinical and clinical research on dronabinol was supported by NIH.
Drug development begins with discovery, that is, the synthesis and purification of a new compound with expected biological activity and therapeutic value. The next major step is the testing of the compound in animals to learn more about its safety and efficacy and to predict its utility for humans. Those early activities are collectively referred to as the preclinical phase. When evidence from the preclinical phase suggests a promising role in humans, the manufacturer submits an Investigational New Drug (IND) application to the FDA. The IND submission contains a plan for human clinical trials and includes the results of preclinical testing and other information.20 Absent FDA objection, the IND becomes effective after 30 days, allowing the manufacturer to conduct clinical testing (testing in humans), which generally involves three phases (see Figure 5.1). The three stages of clinical testing are usually the most time-consuming phases of drug development, lasting five years on average.22 The actual time depends on the complexity of the drug, availability of patients, duration of use, difficulty of measuring clinical end points, therapeutic class, and indication (the disease or condition for which the drug has purported benefits).31
Drug development is a long and financially risky process. For every drug that ultimately reaches clinical testing through an IND, thousands of drugs are synthesized and tested in the laboratory. And only about one in five drugs initially tested in humans successfully secures FDA approval for marketing through a new drug application (NDA).19
The manufacturer submits an NDA to the FDA to gain approval for marketing when clinical testing is complete. An NDA is a massive document, the largest portion of which contains the clinical data from Phase III testing. The other technical sections of an NDA include chemistry, manufacturing, and controls; nonclinical pharmacology and toxicology; and human pharmacokinetics and bioavailability.23 In the case of a new cannabinoid, an abuse liability assessment would also probably be part of an NDA submission. In 1996 the median time for FDA review of an NDA, from submission to approval, was 15.1 months, a review period considerably shorter than that in 1990, when the figure was 24.3 months.22 The shortening of approval time is an outgrowth of the Prescription Drug User Fee Act of 1992, which authorized the FDA to hire additional review staff with so-called user fees paid by industry and imposed clear deadlines for FDA action on an NDA. With respect to the cost of a single drug's development, a number of recent studies have provided a range of estimates of
Page 196
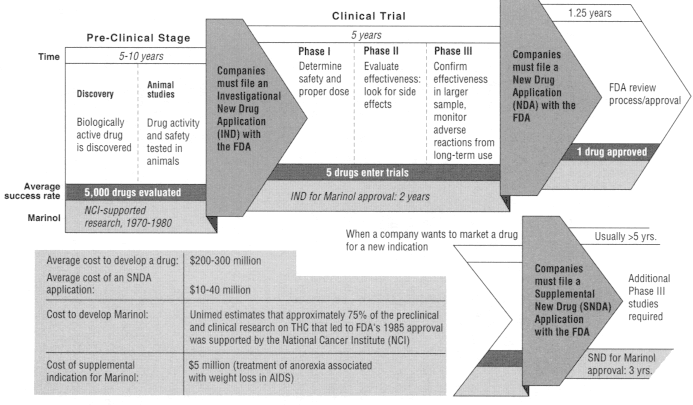
Figure 5.1
Stages of clinical testing.
Page 197
about $200-$300 million, depending on the method and year of calculation.33,44
With FDA approval of an NDA, the manufacturer is permitted to market the drug for the approved indication. At that point, although any physician is at liberty to prescribe the approved drug for another indication (an "off-label use"), the manufacturer cannot promote it for that indication unless the new indication is granted separate marketing approval by the FDA.* To obtain such approval, the manufacturer is required to compile another application to the FDA for what is known variously as an "efficacy supplement," a ''supplemental application," or a "supplemental new drug application.'' Those terms connote that the application is supplemental to the NDA. In general, collecting new data for FDA approval of an efficacy supplement is not as intensive a process as that for an NDA; it generally requires the firm to conduct two additional Phase III studies, although under some circumstances only one additional study of the drug's efficacy is needed.24 The preclinical studies, for example, ordinarily need not be replicated. The average cost to the manufacturer for obtaining approval for the new indication is typically about $10-$40 million.33 The review time to obtain FDA approval for the new indication can be considerable; a recent study of supplemental indications approved by the FDA in 1989-1994 found the approval time to exceed that for the original NDA,18 a reflection, in part, of the lower priority that the FDA accords to the review of efficacy supplements as opposed to new drugs.23
The manufacturer also must apply to the FDA to receive marketing approval for a new formulation of a previously approved drug. A new formulation is a new dosage form, including a new route of administration. An example of such a new formulation is an inhaled version of Marinol, which is currently approved only in capsule form. The manufacturer is required to establish bioequivalence, safety, and efficacy of the new formulation. The amount of evidence required for approval is highly variable, depending on the similarities between the new formulation and the approved formulation. New formulations are evaluated case by case by the FDA. In the case of Marinol, for example, an inhaled version is likely to require not only new studies of efficacy but also new studies of abuse liability. There appear to be no published peer-reviewed studies of the average cost and time for approval of a new formulation.
Two other FDA programs might be relevant to the potential availabil-
*FDA policies for off-label use are being transformed as a result of the Food and Drug Administration Modernization Act of 1997. The FDA recently promulgated new rules to give manufacturers greater flexibility to disseminate information about off-label uses (FDA, 1998b24a). As of this writing, however, court decisions have left the status of the new rules somewhat unclear.
Page 198
ity of new cannabinoids. One program is authorized under the Orphan Drug Act of 1983, which provides incentives to manufacturers to develop drugs to treat "orphan diseases." An orphan disease, as defined in an amendment to the act, is one that affects 200,000 or fewer people in the United States.* The act's most important incentive is a period of exclusive marketing protection of seven years, during which time the FDA is prohibited from approving the same drug for the same indication.5,6 Some of the medical conditions for which cannabinoids have been advocated—Huntington's disease, multiple sclerosis, and spinal cord injury (see chapter 4)—might meet the definition of an orphan disease and thus enable manufacturers to take advantage of the act's financial incentives to bring products to market. If a disease affects more than 200,000 people, the manufacturer sometimes subdivides the patient population into smaller units to qualify. For example, a drug for the treatment of Parkinson's disease is not likely to receive an orphan designation because its prevalence exceeds 200,000, but orphan designation has been accorded to drugs for subsets of Parkinson's patients, such as those suffering from early-morning motor dysfunction in the late stages of the disease.25
The other program is the Treatment-IND program, which was established by regulation in 1987 (and codified into law in 1997) to allow patients with serious and life-threatening diseases to obtain experimental medications, such as marijuana, before their general marketing.t Treatment INDs may be issued during Phase III studies to patients who are not enrolled in clinical trials, provided among other requirements that no comparable alternative drug is available.22,32,33 Thus, the treatment IND program can provide a mechanism for some patients to obtain a promising new cannabinoid before its widespread commercial availability if it reached the late stages of clinical testing for a serious or life-threatening disease.
Drug Enforcement Administration
The DEA is responsible for scheduling controlled substances, that is, drugs and other agents that possess a potential for abuse. Abuse is generally defined as nonmedical use that leads to health and safety hazards, diversion from legitimate channels, self-administration, and other untoward results. '15,21 The legislation that gives DEA the authority to regulate
*The FDA can grant orphan designation to a drug intended for a condition that affects a larger population if the manufacturer's estimated expenses are unlikely to be recovered by sales in the United States (Public Law 98-551).
tMarijuana cigarettes were available under a special FDA-sponsored Compassionate Investigational New Drug Program for desperately ill patients until March 1992, when the program was closed to new participants.48
Page 199
drugs of abuse is the Controlled Substances Act, which was passed in 1970 and amended several times. The overall purpose of the CSA is to restrict or control the availability of drugs to prevent their abuse.
Under the CSA, the DEA places each drug that has abuse potential into one of five categories. The five categories, referred to as Schedules IV, carry different degrees of restriction. Schedule I is the most restrictive, covering drugs that have "no accepted medical use" in the United States and that have high abuse potential. The definitions of the categories and examples of drugs in each are listed in Appendix C. Each schedule is associated with a distinct set of controls that affect manufacturers, investigators, pharmacists, practitioners, patients, and recreational users. The controls include registration with the DEA, labeling and packaging, production quotas, security, recordkeeping, and dispensing.15 For instance, patients with a legitimate medical need for drugs in Schedule II, the most restrictive schedule for drugs "currently with accepted medical use," can neither refill their prescriptions nor have them telephoned to a pharmacy (except in an emergency).
The scheduling of substances under the CSA is handled case by case. It may be initiated by DEA, by DHHS, or by petition from an interested party, including the drug's manufacturer or a public-interest group.15 The final decision for scheduling rests with the DEA, but for this purpose the secretary of DHHS is mandated to provide a recommendation. The secretary's recommendation* to DEA is based in part on results from abuse liability testing that the FDA requires of the manufacturer seeking approval of a new drug. Abuse liability testing is not a single test; it is a compilation of several in vitro human and animal studies, of which some of the best known are drug self-administration and drug discrimination studies.21,34 The secretary's recommendation for scheduling is formally guided by eight legal criteria, including the drug's actual or relative potential for abuse, scientific evidence of its pharmacological effect, risk to public health, and its psychic or physiological dependence liability (21 U.S.C. § 811 (b), (c)). Once the DEA receives a scheduling recommendation, its scheduling decision, including the requirement for obtaining public comment, usually takes weeks to months.33 In practice, the DEA usually adheres to the recommendation of the secretary.t Beyond the DEA,
*The FDA and the National Institute of Drug Abuse, two agencies of DHHS, work jointly to develop the medical and scientific analysis that is forwarded to the secretary, who makes a recommendation to the administrator of the DEA (DEA, 199815).
tUnder the CSA, "the recommendations of the Secretary to the Attorney General shall be binding on the Attorney General as to such scientific and medical matters, and if the Secretary recommends that a drug or other substance not be controlled, the Attorney General shall not control the drug or other substance" (21 U.S.C.§ 811 (b)).
Page 200
various state scheduling laws also affect the manufacture and distribution of controlled substances.33, 50
Under the CSA, marijuana and THC* are in Schedule I, the most restrictive schedule. The scheduling of any other cannabinoid under this act first hinges on whether it is found in the plant. All cannabinoids in the plant are automatically in Schedule I because they fall under the act's definition of marijuana (21 U.S.C. § 802 (16)). In addition, under DEA's regulations, synthetic equivalents of the substances contained in the plant and "synthetic substances, derivatives, and their isomers" whose "chemical structure and pharmacological activity" are "similar" to THC also are automatically in Schedule I (21 CFR § 1308.11(d)(27). Based on the examples listed in the regulations, the word similar probably limits the applicability of the regulation to isomers of THC, but DEA's interpretation of its own regulations would carry significant weight in any specific situation.
Prompted by a 1995 petition from Jon Gettman, a former president of the National Organization for the Reform of Marijuana Laws (NORML), to remove marijuana and THC from Schedule I, DEA gathered information which was then submitted to DHHS for a medical and scientific recommendation and scheduling recommendation, as required by the CSA. For the reasons noted above, any changes in scheduling of marijuana and THC would also affect other plant cannabinoids. For the present, however, any cannabinoid found in the plant is automatically controlled in Schedule I.
Investigators are affected by Schedule I requirements even if their research is being conducted in vitro or on animals. For example, researchers studying cannabinoids found in the plant are required under the CSA to submit their research protocol to DEA, which issues a registration that is contingent on FDA's evaluation and approval of the protocol (21 CFR § 1301.18). DEA also inspects the researcher's security arrangements. However, the regulatory implications are quite different for cannabinoids not found in the plant. Such cannabinoids appear to be unscheduled unless the FDA or DEA decides that they are sufficiently similar to THC to be placed automatically into Schedule I under the regulatory definition outlined above or the FDA or the manufacturer deems them to have potential for abuse, thereby triggering de novo the scheduling process noted above. Thus far, the cannabinoids most commonly used in preclinical research (Table 5.1) appear to be sufficiently distinct from THC that they are not currently considered controlled substances by definition (F. Sapienza, DEA, personal communication, 1998). No new cannabinoids other than
*Technically, the CSA and the regulations use the term "tetrahydrocannabinols."
Page 201
| TABLE 5.1 Cannabinoids and Related |
| Compounds Commonly Used in Research |
| Agonists |
| THC |
| WIN 55,212-2 |
| CP-55940 |
| HU-210 |
| Anandamide (natural ligand) |
| 2-Arachidonylglycerol (natural ligand) |
| Antagonists |
| SR 141716A |
| SR 144528 |
| SOURCES: Felder and Glass (1998)26 and Mechoulam et al. (1998).36 |
THC have yet been clinically tested in the United States, so scheduling experience is limited. The unscheduled status of some cannabinoids might change as research progresses. Results of early clinical research could lead a manufacturer to proceed with or lead the FDA to require abuse liability testing. Depending on the results of such studies, DHHS might or might not recommend scheduling de novo to DEA, which makes the final decision case by case.
Will newly discovered cannabinoids be subject to scheduling? That is a complex question that has no simple answer. The answer depends entirely on each new cannabinoid—whether it is found in the plant, its chemical and pharmacological relationship to THC, and its potential for abuse. Novel cannabinoids with strong similarity to THC are likely to be scheduled at some point before marketing, whereas those with weak similarity might not be. The manufacturer's submission to FDA, which contains its own studies and its request for a particular schedule, can also shape the outcome. Cannabinoids found in the plant are automatically in Schedule I until the manufacturer requests and provides justification for rescheduling. The CSA does permit DEA to reschedule a substance (move it to a different schedule) and to deschedule a substance (remove it from control under the CSA) according to the scheduling criteria (see Appendix F) and the process outlined above.
The possibility of scheduling is a major determinant of whether a manufacturer proceeds with drug development.33 In general, pharmaceutical firms perceive scheduling to be a deterrent because it limits their ability to achieve market share for the following reasons: restricted access,
Page 202
physician disinclination to prescribe scheduled substances, stigma, the additional expense for abuse liability studies, and expensive delays in reaching the market due to federal and state scheduling processes.33 Empirical evidence to support that widely held perception is difficult to find, but at least one large survey of physicians found them to have moderate concerns about prescribing opioids because of actual or perceived pressure from regulatory agencies, such as DEA.57 On the basis of a legal analysis and widespread complaints from researchers and pharmaceutical executives, the Institute of Medicine (IOM, 1995)33 recommended changes in the CSA to eliminate the act's barriers to undertaking clinical research and development of controlled substances; this position was supported in a later report on marijuana.40
Development and Marketing Of Marinol
The following material is based on the published literature (where cited), workshops sponsored by the IOM, and an interview with Robert Dudley, senior vice president of Unimed Pharmaceuticals, Inc., the manufacturer of Marinol and he holder of the NDA. Unimed markets Marinol jointly with Roxane Laboratories, Inc.
Marinol (dronabinol) is the only cannabinoid with approval for marketing in the United States.* The following description covers its development, regulatory history, pharmacokinetics, adverse effects, abuse liability, and market growth. The experience with Marinol can serve as a possible bellwether for the regulatory and commercial fate of new cannabinoids being considered for development.
Development and Regulatory History
Marinol is manufactured as a capsule containing THC in sesame oil; it is taken orally. It was approved by the FDA in 1985 for the treatment of nausea and vomiting associated with cancer chemotherapy. In 1992, the FDA approved marketing of dronabinol for the treatment of anorexia associated with weight loss in patients with AIDS.45 The preclinical and clinical research on THC that culminated in the FDA's 1985 approval was supported primarily by the National Cancer Institute (NCI), whose research support goes back to the 1970s. NCI's contribution appears pivotal, considering that Unimed, the pharmaceutical company that holds
*The only cannabinoid licensed outside the United States is nabilone (Cesamet), which is an analogue of THC available in the United Kingdom for the management of nausea and vomiting associated with cancer chemotherapy (Pertwee, 1997).46
Page 203
the NDA, estimates its contribution to have been only about 25% of the total research effort. The FDA's review and approval of Marinol took about two years after submission of the NDA, according to Unimed. To obtain approval for Marinol's second indication (through an efficacy supplement), the FDA required two more relatively small Phase III studies. The studies lasted three years and cost $5 million to complete.
Physical Properties, Pharmacokinetics, and Adverse Events
Marinol is synthesized in the laboratory rather than extracted from the plant. Its manufacture is complex and expensive because of the numerous steps needed for purification. The poor solubility of Marinol in aqueous solutions and its high first-pass metabolism in the liver account for its poor bioavailability; only 10-20% of an oral dose reaches the systemic circulation.45,60 The onset of action is slow; peak plasma concentrations are not attained until two to four hours after dosing.45,56 In contrast, inhaled marijuana is rapidly absorbed. In a study comparing THC administered orally, by inhalation, and intravenously, plasma concentration peaked almost instantaneously after both inhalation and intravenous administration; most participants' peak plasma concentrations after oral administration occurred at 60 or 90 minutes. Variation in individual responses is highest for oral THC and bioavailability is lowest.42
Marinol's most common adverse events are associated with the central nervous system (CNS): anxiety, confusion, depersonalization, dizziness, euphoria, dysphoria, somnolence, and thinking abnormality.8,9,45,59In two recent clinical trials, CNS adverse events occurred in about one-third of patients, but only a small percentage discontinued the drug because of adverse effects.8,9 Lowering the dose of dronabinol can minimize side effects, especially dysphoria (disquiet or malaise).47
Abuse Potential and Scheduling
On commercial introduction in 1985, Marinol was placed in Schedule II. This schedule, the second most restrictive, is reserved for medically approved substances that have "high potential for abuse" (21 U.S.C. § 812 (b) (2)). Unimed did not encounter any delays in marketing as a result of the scheduling process because the scheduling decision was made by the DEA before FDA's approval for marketing. Nor did Unimed encounter any marketing delays as a result of state scheduling laws. Unimed was not specifically asked by the FDA to perform abuse liability studies for the first approval, presumably because such studies had been conducted earlier.
Unimed later petitioned the DEA to reschedule Marinol from Sched-
Page 204
ule II to Schedule III, which is reserved for medically approved substances that have some potential for abuse (21 U.S.C. § 812 (b) (3)). To buttress its request for rescheduling, Unimed supported an analysis of Marinol's abuse liability by researchers at the Haight Ashbury Free Clinic of San Francisco, which treats many cannabis-dependent patients and people who have HIV/AIDS. The analysis found no evidence of abuse or diversion of Marinol after a literature review and surveys and interviews of medical specialists in addiction, oncology, cancer research, and treatment of HIV, and people in law enforcement. The authors attribute Marinol's low abuse potential to its slow onset of action, its dysphoric effects, and other factors.12 On November 5, 1998, the DEA announced a proposal to reschedule Marinol to Schedule III.17 As of this writing, no formal action on that proposal had been taken.
The rescheduling of a drug from Schedule II to Schedule III is considered important because it lifts some of the restrictions on availability. For example, Unimed expects a sales increase of about 15-20% as a result of rescheduling. In its judgment and that of many other pharmaceutical companies,33 scheduling limits market penetration; the more restrictive the schedules, the greater the limitation. The reasons are that physicians and other providers are reluctant to prescribe Schedule II drugs; patients are deterred from seeking prescriptions because of Schedule II prohibition of refills, as opposed to other commercially available scheduled substances; additional restrictions are imposed by several states, such as quantity restrictions (for example, 30-day supply limits) and triplicate prescriptions;50 and some Schedule II drugs are excluded from hospital formularies because of onerous security and paperwork requirements under federal and state controlled substances laws.
Market Growth and Transformation
Annual sales of Marinol are estimated at $20 million, according to Unimed. Of Marinol's patient population 80%/ use it for HIV, 10% for cancer chemotherapy, and about 5-10% for other reasons. The latter group is thought to consist of Alzheimer's patients drawn to the drug by a recently published clinical study indicating Marinol's promise for the treatment of their anorexia and disturbed behavior.58 As noted earlier, Unimed cannot promote Marinol for this unlabeled indication, but physicians are free to prescribe it for such an indication. Unimed is conducting additional research in pursuit of FDA approval of a new indication for Marinol in the treatment of Alzheimer disease.
The 1992 approval of Marinol for the treatment of anorexia in AIDS patients marked a major transformation in the composition of the patient population. Marinol's use had been restricted to oncology patients. The
Page 205
oncology market for Marinol gradually receded as a result of the introduction of newer medications, including such serotonin antagonists as ondansetron, which are more effective (see chapter 4, "Nausea and Vomiting) and are not scheduled. Much of the recent growth of the market for Marinol (which is about 10% per year) is attributed to its increasing use by HIV patients being treated with combination antiretroviral therapy. Marinol appears to have a dual effect, not only stimulating appetite but also combating the nausea and vomiting associated with combination therapy. Unimed is supporting a Phase II study to examine this combined effect and, with promising results, plans to seek FDA approval for this new indication.
Unimed has two forms of market protection for Marinol. In December 1992, the FDA granted Marinol seven years of exclusive marketing under the Orphan Drug Act. The market exclusivity is related to Marinol's use in anorexia associated with AIDS. Because of the designated orphan indication, the active ingredient, THC, cannot be marketed by another manufacturer for the same indication until December 1999. Other pharmaceutical manufacturers are not constrained from manufacturing and marketing THC for its other indication, antiemesis for cancer chemotherapy, but none appears to be interested in what is, by pharmaceutical company standards, a small market. In addition to market exclusivity, Unimed secured in June 1998 a "use patent" for dronabinol for the treatment of disturbed patients with dementia; this confers patent protection to Unimed for this use for 20 years from the date of filing of the application,* assuming that this indication eventually gains FDA approval.
The rate-limiting factors in the growth of the market for Marinol, according to Unimed are the lack of physician awareness of the drug's efficacy, its adverse effects, and its restricted availability as a result of placement in Schedule II. Unimed perceives only a small percentage of its market to be lost to "competition" from marijuana itself, but there are, admittedly, no reliable statistics on the number of people who have chosen to treat their symptoms with illegally obtained marijuana, despite their ability to obtain Marinol.
New Routes of Administration
It is well recognized that Marinol's oral route of administration hampers its effectiveness because of slow absorption and patients' desire for
*A use patent—also known as a process patent—accords protection for a method of using a composition or compound. A use patent is not considered as strong as a product patent, which prohibits others from manufacturing, using, or selling the product for all uses, rather than for the specific use defined in a use patent.
Page 206
more control over dosing. A drug delivered orally is first absorbed from the stomach or small intestine and then passed through the liver, where it undergoes some metabolism before being introduced into the circulation. To overcome the deficiencies of oral administration, Unimed activated an IND in 1998 as a step toward developing new formulations for Marinol. Four new formulations—deep lung aerosol, nasal spray, nasal gel, and sublingual preparation—are under study in Phase I clinical studies being conducted in conjunction with Roxane Laboratories. These formulations seek to deliver Marinol to the circulation more rapidly and directly. The first two fall under inhalation as a route of administration. Inhalation is considered the most promising method, owing to the rapidity of onset of its effects and potential for better titration of the dose by the patient, but it might also carry an increased potential for abuse. The abuse of a drug correlates with its rapidity of onset (G. Koob, IOM workshop). Sublingual route (under the tongue) administration also affords rapid absorption into the circulation, in this case from the oral mucosa. Other researchers are pursuing the delivery of THC through rectal suppositories, but this slower route might not be acceptable to many patients. Transdermal (skin patches) administration, which is best suited to hydrophilic drugs, is precluded by the lipophilicity of THC. Thus, the choice of routes of administration depends heavily on the physicochemical characteristics of the drug and on its safety, abuse liability, and tolerability.
Unimed expects the FDA to require it to conduct studies of the bioavailability, efficacy, and possibly abuse liability of any new formulation it seeks to market. Any formulation that expedites Marinol's onset of action, as suggested above, is thought to carry greater possibility of abuse. The cost of developing each new formulation is estimated by Unimed at $7-$10 million.
Unimed and Roxane are developing, or considering development of, five new indications for Marinol: disturbed behavior in Alzheimer's disease, nausea and vomiting in HIV patients who are receiving combination therapy, spasticity in multiple sclerosis, intractable pain, and anorexia in cancer and renal disease.
Costs of Marinol and Marijuana
During the IOM public workshops held during the course of this study, many people commented that an important advantage of using marijuana for medical purposes is that it is much less expensive than Marinol. But this comparison is deceptive. While the direct costs of marijuana are relatively low, the indirect costs can be prohibitive. Individuals who violate federal or state marijuana laws risk a variety of costs associated with engaging in criminal activity, ranging from increased vulner-
Page 207
ability to theft and personal injury legal fees to long prison terms. In addition, when purchasing illicit drugs there is no guarantee that the product purchased is what the seller claims it is or that it is not contaminated.
The price of Marinol for its most commonly used indication, anorexia in AIDS, is estimated at $200 per month. The less common indication—nausea and vomiting with cancer chemotherapy—is not as expensive because it is not chronic. Regardless of indication, patients' out-of-pocket expenses tend to be much less—often minimal—because of reimbursement through public or private health insurance. For indigent patients who are uninsured, Roxane sponsors a patient assistance program to defray the cost.
The street value of marijuana, according to the DEA's most recent figures, is about $5-$10 per bag of loose plant.16* At the California buyers' clubs, the price is $2-$16 per gram, depending on the grade of marijuana. The cost to a patient using marijuana depends on the number of cigarettes smoked each day, their THC content, and the duration of use. Insurance does not cover the cost of marijuana. In addition, it is possible for a person to cultivate marijuana privately with little financial investment.
Thus, Marinol appears to be less expensive than marijuana for patients with health insurance or with financial assistance from Roxane. But if the full cost of Marinol is borne out of pocket by the patient, the cost comparison is not so unambiguous. In this case the daily cost in relation to marijuana varies according to the number of cigarettes smoked: If the patient smokes two or more marijuana cigarettes per day, Marinol might be less expensive than marijuana; if the patient smokes only one marijuana cigarette per day, Marinol might be more expensive than marijuana, according to an analysis submitted to the DEA by Unimed. The cost comparisons will depend on fluctuations in the retail price and street value of Marinol and marijuana, respectively, and will vary if marijuana becomes commercially available.
In summary, Marinol has been on the U.S. market since 1985. Its commercial development depended heavily on research supported by the NIH. Marinol's market has grown to $20 million in annual sales. Further market growth is expected but is still constrained by lack of awareness, adverse effects, the oral route of administration, and restrictions imposed by drug scheduling. The manufacturer is proceeding with research on new forms of delivery to overcome the problems associated with oral administration. The manufacturer also is proceeding with research on an array of new indications for Marinol.
*The DEA did not provide an estimate of the weight of marijuana per bag.
Page 208
Market Outlook For Cannabinoids
The potential therapeutic value of cannabinoids is extremely broad. It extends well beyond antiemesis for chemotherapy and appetite stimulation for AIDS, the two indications for which the FDA has approved dronabinol (Marinol). Chapter 4 of this report assesses the possible wider therapeutic potential of marijuana and THC in neurological disorders, glaucoma, and analgesia—all conditions for which clinical research has been under way to fulfill unmet patient needs. New therapeutic uses are being explored in preclinical research. For any of these therapeutic indications, will novel cannabinoids reach the market to satisfy the medical needs of patients?
Economic Factors in Development
The outcomes of preclinical and clinical research determine whether a drug is sufficiently safe and effective to warrant FDA approval for marketing. But the decisions to launch preclinical research and to proceed to clinical trials if early results are promising are dictated largely by economic factors. A pharmaceutical company must decide whether to invest in what is universally regarded as a long and risky research path. For any given drug the question is, Will there be an adequate return on investment? The investment in this case is the high cost of developing a drug. The expectation of high financial returns on investment is what drives drug development.44,53
Market analyses are undertaken to forecast whether a drug will reap a substantial return on investment. The market analysis for a cannabinoid is likely to be shaped by various factors. The average cost of developing a cannabinoid is likely to be higher than that of developing other drugs if its clinical indication is in the therapeutic categories of neuropharmaceutical or nonsteroidal antiinflammatory drug, the two therapeutic categories associated with the highest research and development costs.19 One reason for higher costs is the need to satisfy the DEA's regulatory requirements related to drug scheduling.
On the ''market return'' side are multiple factors. A market analysis examines the expected returns from the possible markets for which a cannabinoid could be clinically pursued. The financial size of each market is calculated mostly on the basis of the current and projected patient prevalence (for a given clinical indication), sales data (if available), and competition from other products. The duration of use is also factored in—a drug needed for long-term use in a condition with an early age of onset is desirable from a marketing perspective. Factors that can augment or diminish market return include patentability and other forms of market protection,
Page 209
| TABLE 5.2 Cannabinoids Under Development for Human Use | |||||
| Name of | Investigator | Stage of | Pharmacology | U.S. FDA | Possible |
| HU-211 | Pharmos Corp. | Clinical | NMDA | None | Neuroprotection |
| CT-3 | Atlantic | Preclinical | Nonpsycho- | None | Antiinflammatory |
| THC | Unimed Roxane | Clinical | Cannabinoid | IND | (See text) |
| Marijuana | HortaPharm | Clinical | Cannabinoid | None | Multiple sclerosis |
| Donald Abrams, | Clinical | Cannabinoid | IND | HIV-related | |
| Ethan Russo, | Cannabinoid | IND | Migraine | ||
| aClinical trials are to proceed in the next few years under a license from the British Home Office.10 | |||||
| SOURCES: Glain, 199827; Atlantic Pharmaceuticals, 19977; Striem et al., 199755; Nainggolan, 199737; Zurier et al., 199861; D. Abrams and E. Russo, personal communications, 1998; R. Dudley, personal communication, 1998; Pharmaprojects Database, 1998. | |||||
reimbursement climate, restrictions in access due to drug scheduling, social attitudes, adverse effect profile, and drug interactions.33,53 New cannabinoids generally can receive product patents, giving the patent holder 20 years of protection from others seeking to manufacture or sell the same product. According to U.S. patent law, the product must be novel and "nonobvious" in relation to prior patents.28
Cannabinoids under Development
From publicly available sources, the IOM was able to learn of several cannabinoids being developed for human use (Table 5.2). With the exception of Marinol and marijuana, all are in the preclinical phase of testing in the United States. This list might not be comprehensive, inasmuch as other compounds could be under development, but that information is
Page 210
proprietary.* The table does not list the full complement of cannabinoids, both agonists and antagonists, being used in research as tools to understand the pharmacology of cannabinoids (for more comprehensive lists of cannabinoids, see Felder and Glass, 199826; Mechoulam et al., 199836; Howlett, 199530; Pertwee 199746). Nor does it list cannabinoids once considered for development but later discontinued. An 18-year survey of analgesics in development in 1980-1998 found that six of the nine cannabinoids under development for analgesia were discontinued or undeveloped,49,+ but work on most of these was halted before 1988, when the first endogenous cannabinoid receptor was discovered (chapter 3).
Three points can be made on the basis of Table 5.2. First, virtually all of the listed cannabinoids are being developed by small pharmaceutical companies or by individuals. In general, that implies that their development is considered especially risky from a commercial standpoint in that small companies are often willing to assume greater development risks than larger more established firms (W. Schmidt, personal communication, 1998). Without the benefit of sales revenues, small companies are able to fund their research through financing from venture capital, stock offerings, and relationships with established pharmaceutical companies.43
Second, with the exception of THC, no constituents of the marijuana plant appear to be undergoing development by pharmaceutical companies. A number of plant compounds have been tested in experimental models and humans. For example, the antiemetic properties and negligible side effects of D8-THC were demonstrated in a clinical trial in children who were undergoing cancer chemotherapy,' but no sponsor was interested in developing D8-THC for commercial purposes (R. Mechoulam, Hebrew University, personal communication, 1998). The absence of plant cannabinoids under development implies that the specter of automatic placement in Schedule I under the CSA is an important deterrent, even though rescheduling would occur before marketing.++ The point from the earlier discussion is that automatic, as opposed to de novo, scheduling appears to cast a pall over development of a cannabinoid found in the plant. Another impediment is that a cannabinoid extracted
*Information about the existence of an IND is proprietary; it can be confirmed only by the manufacturer, not the FDA.
+Discontinued: levonantradol, nabitan, nantradol, and pravadoline. Undeveloped: CP47497 and CP-55244.
++As a result of the FDA's approval of an NDA, the drug would be, at a minimum, rescheduled in Schedule II. Depending on abuse liability data supplied by the manufacturer and the FDA's recommendation, the drug could be moved to a less restrictive schedule or be descheduled.
Page 211
from the plant is not likely to fulfill the criteria for a product patent, although other forms of market protection are possible. Marinol, for example, was accorded orphan drug status and its manufacturer obtained a use patent.
Third, cannabinoids are being developed for therapeutic applications beyond those discussed earlier in this chapter and in chapter 4. One of the most prominent new applications of cannabinoids is for "neuroprotection," the rescue of neurons from cell death associated with trauma, ischemia, and neurological diseases.29,36 Cannabinoids are thought to be neuroprotective—through receptor-dependent51 as well as receptor-independent pathways; both THC, which binds to CB, receptors, and CBD, which does not, are potent antioxidants, effective neuroprotectants because of their ability to reduce the toxic forms of oxygen (free radicals) that are formed during cellular stress.29 The synthetic cannabinoid HU-211 (dexanabinol) is an antioxidant and an antagonist of the NMDA receptor, rather than an agonist at the cannabinoid receptor.52 Earlier research demonstrated that HU-211 protects neurons from neurotoxicity induced by excess concentrations of the excitatory neurotransmitter glutamate. Excess release of glutamate, which acts by binding to the NMDA receptor, is associated with trauma and disease.54 As an NMDA antagonist, HU-211 blocks the damaging action of glutamate and other endogenous neurotoxic agents.52,55 After having been studied in the United Kingdom in Phase I clinical trials, HU-211 progressed to Phase II clinical trials in Israel for treatment of severe closed-head trauma (Knoller et al., 1998).35
Market Prospects
It is difficult to gauge the market prospects for new cannabinoids. There certainly appears to be scientific interest, particularly for the discovery of new cannabinoids, but whether this interest can be sustained commercially through the arduous course of drug development is an open question. Research and development experience is limited; only one cannabinoid, dronabinol, is commercially available, and most of its research and development costs were shouldered by the federal government. Furthermore, the size of dronabinol's market (at about $20 million) is modest by pharmaceutical company standards. None of the other cannabinoids in development has reached clinical testing in the United States. Their scientific, regulatory, and commercial fates are likely to be very important in shaping future investment patterns. Experience with the drug scheduling process also is likely to be watched very carefully. If the early products are heavily regulated in the absence of strong abuse liability, future development might be deterred. For the present, what seems to be clear
Page 212
from the dearth of products in development and the small size of the companies sponsoring them is that cannabinoid development is seen as especially risky.
One scenario is that cannabinoids will be pursued for lucrative markets that reflect large unmet medical needs. Of the therapeutic needs for which cannabinoid receptor agonists have been tested, analgesia is by far the largest. The annual U.S. prescription and over-the-counter analgesic market in 1997 was $4.4 billion.49 Given the long-standing need for less addictive, safer, easier to use, and more effective drugs for acute and chronic pain, it would not be surprising to see cannabinoids developed to treat some segments of the current analgesic market, if their safety and effectiveness were clearly established in clinical trials.
In addition to cannabinoid receptor agonists, two classes of cannabinoid-related drugs might prove therapeutically useful: cannabinoid antagonists and inverse agonists, compounds that bind to receptors but produce effects opposite those of agonists. Neither would be subject to the same scheduling concerns as cannabinoid agonists because they are not found in marijuana and would be highly unlikely to have any abuse potential. Another set of cannabinoid-related drugs, such as those that affect the synthesis, uptake, or inactivation of endogenous cannabinoids might, however, have abuse potential because they would influence the signal strength of endogenous cannabinoids.
The development of specific cannabinoid antagonists, like SR141716A for CB1 receptors and SR144528 for CB2 receptors, has provided a substantial impetus to understand cannabinoid actions. Those compounds block many of the effects of THC in animals, and their testing in humans has just begun. Cannabinoid antagonists have physiological effects on their own, in the absence of THC. They might have important therapeutic potential in a variety of clinical situations. For example, THC reduces short-term memory, so it is possible that a CB1 antagonist like SR141716A could act as a memory-enhancing agent. Similarly, for conditions in which cannabinoids decrease immune function (presumably by binding to CB2 receptors in immune cells), a CB2 antagonist might be useful as an immune stimulant.
Cannabinoid inverse agonists would exert effects opposite those of THC and might thus cause appetite loss, short-term memory enhancement, nausea, or anxiety. Those effects could possibly be separated by molecular design, in which case inverse agonists might have some therapeutic value. One report has been published suggesting that the CB1 receptor antagonist, SR141617A,l1 is an inverse agonist, and there will likely be others.
Page 213
Regulation of and Market Outlook For Marijuana
Marijuana is not legally marketed in the United States.* No sponsor has ever sought marketing approval from the FDA for medical use of marijuana. One sponsor has an IND for a clinical safety study on HIV anorexia (D. Abrams, University of California at San Francisco, personal communication, 1998). Another has an IND pending for the treatment of migraine headaches (E. Russo, Western Montana Clinic, personal communication, 1998). Since 1970, marijuana's manufacture and distribution have been tightly restricted under the CSA, which places marijuana in Schedule I, which is reserved for drugs or other substances with "a high potential for abuse," "no currently accepted medical use," and "lack of accepted safety for use . . . under medical supervision'' (21 U.S.C. § 812 (b)(1)).
Marijuana has remained in Schedule I despite persistent efforts at rescheduling since the 1970s by advocacy groups, such as NORML. Through petitions to the DEA, advocacy groups contend that marijuana does not fit the legal criteria for a Schedule I substance, owing to its purported medical uses and lack of high abuse liability.3,4,48 Another rescheduling petition, which was filed in 1995, is being evaluated by the FDA and DEA.
Availability for Research
To use marijuana for research purposes, researchers must register with the DEA, as well as adhere to other relevant requirements of the CSA and other federal statutes, such as the FD&C act. The National Institute on Drug Abuse (NIDA), one of the institutes of NIH, is the only organization in the United States licensed by the DEA to manufacture and distribute marijuana for research purposes. NIDA performs this function under its Drug Supply Program.+ Through this program, NIDA arranges for marijuana, to be grown and processed through contracts with two organizations: the University of Mississippi and the Research Triangle Institute. The University of Mississippi grows, harvests, and dries marijuana; and the institute processes it into cigarettes. A researcher can obtain marijuana free of charge from NIDA through an NIH-approved research grant to investigate marijuana, or through a separate protocol review.39 Research grant approvals are handled through the conventional NIH peer review
*Under the CSA, its only legal use is in research under strictly defined conditions.
tThis is also the program through which several patients receive marijuana under a compassionate use program monitored by the FDA. For history and information on this effort, see Randall (1993).48
Page 214
process for extramural research, a highly competitive process with a success rate in 1997 of 32% of approved NIDA grants.41 Through the separate protocol review, in which a researcher funds research independently of an NIH grant, NIDA submits the researcher's protocol to several external reviewers who evaluate the protocol on the basis of scientific merit and relevance to the mission of NIDA and NIH.
Through those two avenues marijuana has been supplied to several research groups—most of those that apply. While there has been much discussion of NIDA's alleged failure to supply marijuana for research purposes, we are unaware of recent cases in which they failed to supply marijuana to an investigator with an NIH-approved grant for research on marijuana. Donald Abrams's difficulty in obtaining research funding and marijuana from NIDA has been much discussed,2 but the case of a single individual should not be presumed to be representative of the community of marijuana researchers. Failure of investigators who apply to NIH for marijuana research grants to receive funding is hardly exceptional: in 1998 less than 25% of all first-time investigator-initiated grant applications (known as ROls) to the NIH were funded.38
To import marijuana under the CSA for research purposes, the procedures are more complex. Under DEA regulations, marijuana can be imported, provided that the researcher is registered with the DEA, has approval for marijuana research (21 CFR § 1301.11, .13, and .18), and has a DEA-approved permit for importation (21 CFR § 1312.11, .12, and .13), and that the exporter in the foreign country has appropriate authorization by the country of exportation. Importation would enable U.S. researchers to conduct research on marijuana grown by HortaPharm, a company that has developed unique strains of marijuana. However, no U.S. researcher has imported HortaPharm's marijuana because Dutch authorities have refused to issue an export permit, despite the issuance of an import permit by the DEA (D. Pate, HortaPharm, personal communication, 1998).*
HortaPharm, which is in the Netherlands, grows marijuana as a raw material for the manufacture of pharmaceuticals. Through selective breeding and controlled production, HortaPharm has developed marijuana strains that feature single cannabinoids, such as THC or cannabidiol. The plants contain a consistently "clean" phytochemical profile and a higher
*It might eventually be possible to import HortaPharm's marijuana from England, where HortaPharm is growing its marijuana strains for research use in clinical trials for multiple sclerosis (Boseley, 1998).0 England, as the country of origin, would have to provide appropriate authorization for export of the strains to the United States. Permission to export for research purposes is part of the basis for HortaPharm's participation in this project with GW Pharmaceuticals through a special set of licenses with the British Home Office (David Pate, HortaPharm, personal communication, 1998).
Page 215
concentration of THC (16%) or other desired cannabinoids than seized marijuana. Marijuana seized in the United States in 1996 had a THC content averaging about 5%.16 Consistency of THC content is desirable because it overcomes the natural variability due to latitude, weather, and soil conditions. Product consistency is a basic tenet of pharmacology because it enables standardized dosing for regulatory and treatment purposes.
The difficulties of conducting research on marijuana were noted in the 1997 NIH report40 that recommended that NIH facilitate clinical research by developing a centralized mechanism to promote design, approval, and conduct of clinical trials.
Regulatory Hurdles to Market
For marijuana to be marketed legally in the United States, a sponsor with sufficient resources would be obliged to satisfy the regulatory requirements of both the FD&C act and the CSA.
Under the FD&C act, a botanical product like marijuana theoretically might be marketed in oral form as a dietary supplement;* however, as a practical matter, only a new drug approval is likely to satisfy the provisions of the CSA, which require prescribing and distribution controls on drugs of abuse that also have an "accepted medical use." (The final paragraphs of this section clarify the criteria for "accepted medical use.")
Bringing marijuana to market as a new drug is uncharted terrain. The route is fraught with uncertainty for at least three pharmacological reasons: marijuana is a botanical product, it is smoked, and it is a drug with abuse potential. In general, botanical products are inherently more difficult to bring to market than are single chemical entities because they are complex mixtures of active and inactive ingredients. Concerns arise about product consistency, potency of the active ingredients, contamination, and stability of both active and inactive ingredients over time. These are among the concerns that a sponsor would have to overcome to meet the requirements for an NDA, especially those related to safety and to chemistry, manufacturing, and control.
A handful of botanical preparations are on the market, but none received formal approval as a new drug by today's standards of safety and efficacy (FDA, Center for Drug Evaluation and Research, personal communication, 1998). The three marketed botanical preparations are older drugs that came to market years before safety and efficacy studies were required by legislative amendments in 1938 and 1962, respectively.
*Inhaled products may not lawfully be marketed as dietary supplements.
Page 216
One of the botanical preparations is the prescription product digitalis. Because it came to market before 1938, it is available today, having been "grandfathered" under the law; but it does not necessarily meet contemporary standards for safety and effectiveness.20 Two other botanical preparations, psyllium and senna, came to market between 1938 and 1962. Drugs entering the market during that period were later required to be evaluated by the FDA in what is known as the over-the-counter drug review process,20 through which psyllium and senna were found to be generally recognized as safe and effective and so were allowed to remain on the market as over-the-counter drugs.* Although no botanical preparations have been approved as new drugs, it is important to point out that a number of individual plant constituents, either extracted or synthesized de novo, have been approved (for example, taxol and morphine). But these drug approvals were for single constituents rather than botanical preparations themselves. The FDA is developing guidance for industry to explain how botanicals are reviewed as new drugs, but the final document might not be available before 1999.
That marijuana is smoked might pose an even greater regulatory challenge. The risks associated with smoking marijuana are described in chapter 2. The FDA would have to weigh those risks with marijuana's therapeutic benefits to arrive at a judgment about whether a sponsor's NDA for marijuana met the requirements for safety and efficacy under the FD&C act. Marijuana delivered in a novel way that avoids smoking would overcome some, but not all, of the regulatory concerns. Vaporization devices that permit inhalation of plant cannabinoids without the carcinogenic combustion products found in smoke are under development by several groups; such devices would also require regulatory review by the FDA.
The regulatory hurdles to market posed by the CSA are formidable but not insurmountable. If marijuana received market approval as a drug by the FDA, it would most likely be rescheduled under the CSA, as was the case for dronabinol. That is because a new drug approval satisfies the "accepted medical use" requirement under the CSA for manufacture and distribution in commerce.13 But a new drug approval is not the only means to reschedule marijuana under the CSA.14 For years advocates for rescheduling have argued that marijuana does enjoy "accepted medical use," even in the absence of a new drug approval. Although advocates have been unsuccessful in rescheduling efforts, their actions prompted
*Over-the-counter monographs for these products have been issued as tentative final monographs (proposed rules) but have not yet been issued in final form as final rules (FDA, Center for Drug Evaluation and Research, personal communication, 1998).
Page 217
the DEA to specify the criteria by which it would determine whether a substance had "accepted medical use." In the DEA's 1992 denial of a rescheduling petition, it listed these elements as constituting "accepted medical use": the drug's chemistry must be known and reproducible, there must be adequate safety studies, there must be adequate and well-controlled studies proving efficacy, the drug must be accepted by qualified experts, and the scientific evidence must be widely available.14
Assuming that all of those criteria were satisfied, marijuana could be rescheduled—but into which schedule? The level of scheduling would be dictated primarily by a medical and scientific recommendation to the DEA made by the secretary of DHHS.* As noted earlier, this recommendation is determined by the five scheduling criteria listed in the CSA. However, scheduling in a category less restrictive than Schedule II might be prohibited by international treaty obligations. The Single Convention on Narcotic Drugs, a treaty ratified by the United States in 1967, restricts scheduling of the plant and its resin to at least Schedule II (the more restrictive Schedule I is another option).13
Market Outlook
The market outlook for the development of marijuana as a new drug, on the basis of the foregoing analysis, is not favorable, for a host of scientific, regulatory, and commercial reasons. From a scientific point of view, research is difficult because of the rigors of obtaining an adequate supply of legal, standardized marijuana for study. Further scientific hurdles are related to satisfying the exacting requirements for FDA approval of a new drug. The hurdles are even more exacting for a botanical product because of the inherent problems with, for example, purity and consistency. Finally, the health risks associated with smoking pose another barrier to FDA approval unless a new smoke-free route of administration is demonstrated to be safe. Depending on the route of administration, an additional overlay of regulatory requirements might have to be satisfied.
From a commercial point of view, uncertainties abound. The often-cited cost of new drug development, about $200-$300 million, might not apply, but there are probably additional costs needed to satisfy the FDA's requirements for a botanical product. As noted above, no botanical products have ever been approved as new drugs by the FDA under today's stringent standards for safety and efficacy. Satisfying the legal require-
*At present, there is no practical mechanism for generating such a recommendation outside the new drug approval process, although such a mechanism could, theoretically, be developed.33
Page 218
ments of the CSA also will add substantially to the cost of development. On the positive side, so much research already has been done that some development costs will be lower. The cost of bringing dronabinol to market, for example, was reduced dramatically as a result of clinical trials supported with government funding. Nevertheless, it is impossible to estimate the cost of developing marijuana as a new drug. Estimating return on investment is similarly difficult. A full-fledged market analysis would be required for the indication being sought. Such an analysis would take into account the market limitations resulting from drug scheduling restrictions, stigma, and patentability.
The plant does not constitute patentable subject matter under U.S. patent law because it is unaltered from what is found in nature. So-called products of nature are not generally patentable.28 New marijuana strains, however, could be patentable in the United States under a product patent or a plant patent because they are altered from what is found in nature. (A product patent prohibits others from manufacturing, using, or selling each strain for 20 years; a plant patent carries somewhat less protection.) HortaPharm has not yet sought any type of patent for its marijuana strains in the United States, but it has received approval for a plant registration in Europe (David Watson, HortaPharm, personal communication, 1998).
In short, development of the marijuana plant is beset by substantial scientific, regulatory, and commercial obstacles and uncertainties. The prospects for its development as a new drug are unfavorable unless return on investment is not a driving force. It is noteworthy that no pharmaceutical firm has sought to bring it to market in the United States. The only interest in its development appears to be in England in a small pharmaceutical firm (see Boseley, 199810) and in the United States among physicians without formal ties to pharmaceutical firms (D. Abrams, University of California at San Francisco, and E. Russo, Western Montana Clinic, personal communications, 1998).
Conclusions
Cannabinoids are an interesting group of compounds with potentially far-reaching therapeutic applications. There is a surge of scientific interest in their development as new drugs, but the road to market for any new drug is expensive, long, risky, and studded with scientific, regulatory, and commercial obstacles. Experience with the only approved cannabinoid, dronabinol, might not illuminate the pathway because of the government's heavy contribution to research and development, dronabinol's scheduling history, and its small market.
There appear to be only two novel cannabinoids actively being developed for human use, but they have yet to be tested in humans in the
Page 219
United States. Their experience is likely to be more predictive of the marketing prospects for other cannabinoids. It is too early to forecast the prospects for cannabinoids, other than to note that their development at this point is considered to be especially risky, to judge by the paucity of products in development and the small size of the pharmaceutical firms sponsoring them.
The market outlook in the United States is distinctly unfavorable for the marijuana plant and for cannabinoids found in the plant. Commercial interest in bringing them to market appears nonexistent. Cannabinoids in the plant are automatically placed in the most restrictive schedule of the Controlled Substances Act, and this is a substantial deterrent to development. Not only is the plant itself subject to the same scheduling strictures as are individual plant cannabinoids, but development of marijuana also is encumbered by a constellation of scientific, regulatory, and commercial impediments to availability.
References
1. Abrahamov A, Abrahamov A, Mechoulam R. 1995. An efficient new cannabinoid antiemetic in pediatric oncology. Life Sciences 56:2097-2102.
2. Abrams DI. 1998. Medical marijuana: Tribulations and trials. Journal of Psyclhoactive Drugs 30:163-169.
3. AMA (American Medical Association Council on Scientific Affairs). 1997. Report to the AMA House of Delegates. Chicago: AMA.
4. Annas GJ. 1997. Reefer madness—the federal response to California's medical-marijuana law. The New England Journal of Medicine 337:435-439.
5. Arno PS, Bonuck K, Davis M. 1995. Rare diseases, drug development, and AIDS: The impact of the Orphan Drug Act. Milbank Quarterly 73:231-252.
6. Asbury C. 1991. The Orphan Drug Act: The first seven years. Journal of the American Medical Association 265:893-897.
7. Atlantic Pharmaceuticals. 1997. Atlantic Pharmaceuticals' proprietary compound shows promising anti-inflammatory effects in pre-clinical trials [WWW document]. URL http://www.atlan.com/p-ll-10-97ct3zurier.htm (accessed September 1998).
8. Beal JE, Olson RLL, Morales JO, Bellman P, Yangco B, Lefkowitz L, Plasse TF, Shepard KV. 1995. Dronabinol as a treatment for anorexia associated with weight loss in patients with AIDS. Journal of Pain and Symptom Management 10:89-97.
9. Beal JE, Olson R, Lefkowitz L, Laubenstein L, Bellman P, Yangco B, Morales JO, Murphy R, Powderly W, Plasse TF, Mosdell KW, Shepard KV. 1997. Long-term efficacy and safety of dronabinol for acquired immunodeficiency syndrome-associated anorexia. Journal of Pain and Symptom Management 14:7-14.
10. Boseley S. 1998. Multiple sclerosis victims to test medicinal effects of marijuana [WWW document].URL http://www.anomalous-images/news/news/227.HTML (accessed September 8, 1998).
Page 220
11. Bouaboula M, Perrachon S, Milligan L, Canat X, Rinaldi-Carmona M, Portier MB, Calandra B, Pecceu F, Lupker J, Maffrand JP, Le Fur G, Casellas P. 1997. A selective inverse agonist for central cannabinoid receptor inhibits mitogen-activated protein kinase activation stimulated by insulin or insulin-like growth factor 1. Evidence for a new model of receptor/ligand interactions. Journal of Biological Chemistry 272:2233022339.
12. Calhoun, SR, Galloway GP, Smith DE. 1998. Abuse potential of dronabinol (Marinol). Journal of Psychoactive Drugs 30:187-196.
13. Cooper RM. 1980. Therapeutic use of marijuana and heroin: The legal framework. Food Drug Cosmetic Law Journal 35:68-82.
14. DEA (Drug Enforcement Administration). 1992. Marijuana scheduling petition; denial of petition; remand. Federal Register 57:10499-10508.
15. DEA. 1998. Drugs of abuse [WWW document].URL http://www.usdoj.gov/dea/ pubs/abuse/contents.htm (accessed September 1998).
16. DEA. 1996. The National Narcotics Intelligence Consumers Committee (NNICC) report [WWW document].URL www.usdoj.gov/dea/pubs/intel/nnicc97.htm (accessed September 1998).
17. DEA. 1998b. Rescheduling of synthetic dronabinol from Schedule II to Schedule III. Federal Register 63:59751-59753.
18. DiMasi JA, Brown JS, Lasagna L. 1996. An analysis of regulatory review times of supplemental indications for already-approved drugs: 1989-1994. Drug Information Journal 30:315-337.
19. DiMasi JA, Hanson RW, Grabowski HG, Lasagna L. 1995. Research and development costs for new drugs by therapeutic category: A study of the U.S. pharmaceutical industry. Pharmaco Economics 7:152-169.
20. FDA (Food and Drug Administration). 1990. From Test Tube to Patient: New Drug Development in the United States. Rockville, MD: U.S. Department of Health and Human Services.
21. FDA. 1997b. Draft Guidelines for Research Involving the Abuse Liability Assessment of New Drugs. Rockville, MD: U.S. Department of Health and Human Services. Division of Anesthetic, Critical Care and Addiction Drug Products.
22. FDA. 1997a. Center for Drug Evaluation and Research Fact Book [WWW document]. URL http://www.fda.gov/cder/homepage (accessed September 1998).
23. FDA. 1998a. Center for Drug Evaluation and Research Handbook [WWW document]. URL http://www.fda/cder/handbook.htm (accessed September 1998).
24a. FDA. 1998b. FDA proposes rules for dissemination information on off label uses (press release, June 5). Washington, DC: U.S. Department of Health and Human Services.
24. FDA. 1998c. Guidance for industry: Providing clinical evidence of effectiveness for human drugs and biological products. Center for Drug Evaluation and Research, Center for Biologics Evaluation and Research. May 1998 [WWW document].URL http://www.fda.gov/cder/guidance/1397fnl.pdf (accessed September 1998).
25. FDA. 1998d. Office of Orphan Products Development Program Overview [WWW document].URL http://www.fda.gov/orphan/DESIGNAT/recent.htm (accessed October 14, 1998).
26. Felder CC, Glass M. 1998. Cannabinoid receptors and their endogenous agonists. Annual Reviews of Pharmacology and Toxicology 38:179-200.
27. Glain SJ. 1998. 1. Wall Street Journal.
28. Gollin MA. 1994. Patenting recipes from nature's kitchen: How can a naturally occurring chemical like taxol be patented? Biotechnology (NY) 12:406-407.
Page 221
29. Hampson AJ, Grimaldi M, Axelrod J, Wink D. 1998. Cannabidiol and (-)delta-9-tetrahydrocannabinol are neuroprotective antioxidants. Proceedings of the National Academy of Sciences USA 95:8268-8273.
30. Howlett AC. 1995. Pharmacology of cannabinoid receptors. Annual Review of Pharmacology and Toxicology 35:607-634.
31. IOM (Institute of Medicine). 1990. Modern Methods of Clinical Investigtion. Washington, DC: National Academy Press.
32. IOM. 1991. Expanding Access to Investigational Therapies for HIV Infection and AIDS. Washington, DC: National Academy Press.
33. IOM. 1995. The Development of Medications for the Treatment of Opiate and Cocaine Addictions: Issues for the Government and Private Sector. Washington, DC: National Academy Press.
34. IOM. 1996. Pathways of Addiction: Opportunities in Drug Abuse Research. Washington, DC: National Academy Press.
35. Knoller N, Levi L, Israel Z, Razon N, Reichental E, Rappaport Z, Ehrenfreund N, Biegon A. Safety and outcome in a Phase II clinical trail of dexanabinol in severe head trauma. Congress of Neurological Surgeons Annual Meeting. Seattle, WA, Oct. 7, 1998.
36. Mechoulam R, Hanus L, Fride E. 1998. Towards cannabinoid drugs-revisited. In: Ellis GP, Luscombe DK, Oxford AW, Editors, Progress in Medicinal Chemistry. vol. 35. Amsterdam: Elsevier Science. Pp. 199-243.
37. Nainggolan L. 1997. Marijuana—a missed market opportunity? Scrip Magazine.
38. National Institutes of Health (NIH). 1999. http://www.nih.gov/grants/award/ award.htm.
39. NIDA (National Institute on Drug Abuse). 1996. Research Resources: Drug Supply Systen, 10th Edition. Rockville, MD.
40. NIH (National Institutes of Health). 1997. Workshop on the Medical Utility of Marijuana. Report to the Director, National Institutes of Health by the Ad Hoc Group of Experts. Bethesda, MD, February 19-20, 1997. Bethesda, MD: National Institutes of Health.
41. NIH. 1998. FY (1970-1997 NIH (Preliminary) competing research project applications [WWW document].URL http:/silk.nih.gov/public/cbz2rfm.@www.comic.dsncc (accessed October 1998).
42. Ohlsson A, Lindgren JE, Wahlen A, Agurell S, Hollister LE, Gillespie HK. 1980. Plasma delta-9-tetrahydrocannabinol concentrations and clinical effects after oral and intravenous administration and smoking. Clinical Pharmacology and Therapeutics 28:409416.
43. OTA (Office of Technology Assessment). 1991. Biotechnology in a Global Economy. OTTAWA-494. Washington, DC: U.S. Government Printing Office.
44. OTA. 1993. Pharmaceutical R&D: Costs, Risks and Rewards. OTA-H-522. Washington, DC: U.S. Government Printing Office.
45. PDR (Physicians' Desk Reference). 1996. Physicians' Desk Reference. 50th ed. Montvale, NJ: Medical Economics Co.
46. Pertwee RG. 1997. Cannabis and cannabinoids: Pharmacology and rationale for clinical use. Pharmaceutical Science 3:539-545.
47. Plasse TF, Gorter RW, Krasnow SH, Lane M, Shepard KV, Wadleigh RG. 1991. Recent clinical experience with dronabinol. Pharmacology Biochemistry and Behavior 40:695700.
48. Randall IV B. 1993. Medical Use of Marijuana: Policy and Regulatory Issues. 93-308 SPR. Washington, DC: Congressional Research Service.
Page 222
49. Schmidt WK. 1998. Overview of current investigational drugs for the treatment of chronic pain. National Managed Health Care Congress, Second Annual Conference on Therapeutic Developments in Chronic Pain. Annapolis, MD, May 18, 1998.
50. Shapiro RS. 1994. Legal bases for the control of analgesic drugs. Journal of Pain and Symptom Managemnent 9:153-159.
51. Shen M, Piser TM, Seybold VS, Thayer SA. 1996. Cannabinoid receptor agonists inhibit glutamatergic synaptic transmission in rat hippocampal cultures. Journal of Neroscience 16:4322-4334.
52. Shohami E, Weidenfeld J, Ovadia H, Vogel Z, Hanus L, Fride E, Breuer A, Ben-Shabat S, Sheskin T, Mechoulam R. 1996. Endogenous and synthetic cannabinoids: Recent advances. CNS Drtug Reviews 2:429-451.
53. Spilker B. 1989. Multinational Drug Companies: Issues in Drug Discovertl and Developmnent. New York: Raven Press.
54. Standaert DG, Young AB. 1996. Treatment of central nervous system degenerative disorders. In: Hardman JG, Limbird LE, Molinoff PB, Ruddon RR, Gilman AG, Editors, Goodmnan & Gilman's: The Pharinacological Basis of Therapeutics. 9th ed. New York: McGraw-Hill. Pp. 503-519.
55. Striem S, Bar-Joseph A, Berkovitch Y, Biegon A. 1997. Interaction of dexanabinol (HU211), a novel NMDA receptor antagonist, with the dopaminergic system. European lournlal of Pharmacology 388:205-213.
56. Timpone JG, Wright DJ, Li N, Egorin MJ, Enama ME, Mayers J, Galetto G, DATRI 004 Study Group. 1997. The safety and pharmacokinetics of single-agent and combination therapy with megestrol acetate and dronabinol for the treatment of HIV wasting syndrome. The DATRI 004 study group. AIDS Researchi andl HwumanR Retroviruses 13:305315.
57. Turk DC, Brody MC, Akiko OE. 1994. Physicians' attitudes and practices regarding the long-term prescribing of opioids for non-cancer pain. Pain 59:201-208.
58. Volicer L, Stelly M, Morris J, McLaughlin J, Volicer BJ. 1997. Effects of dronabinol on anorexia and disturbed behavior in patients with Alzheimer's disease. International Journal of Geriatric Psychiatry 12:913-919.
59. Voth EA, Schwartz RH. 1997. Medicinal applications of delta-9-tetrahydrocannabinol and marijuana. Annals of Internal Medicine 126:791-798.
60. Wall ME, Sadler BM, Brine D, Taylor H, Perez-Reyes M. 1983. Metabolism, disposition, and kinetics of delta-9-tetrahydrocannabinol in men and women. Clinical Phartmacology and Therapeutics 34:352-363.
61. Zurier RB, Rossetti RG, Lane JH, Goldberg JM, Hunter SA, Burstein SH. 1998. Dimethylheptyl-THC-11 oic acid: A non-psychoactive antiinflammatory agent with a cannabinoid template structure. Arthritis and Rlheumatism 41:163-170.






























