
Development of the Mouse Model Dramatype for Human Clinical Benefit
Kazunori Nakajima
Department Head and Assistant Professor, Department of Molecular Neurobiology, Institute of DNA Medicine, Jikei University School of Medicine, Tokyo, Japan.
PRESTO, Japan Science and Technology Corporation (JST), Saitama, Japan.
Human patients suffer from various diseases that are affected by multiple factors. In Figure 1, this “dramatype” is depicted from a physician's point of view. The dramatype not only includes “phenotype” but is also affected by several extrinsic factors represented by the proximate environment (further explained in Dr. Nomura's discussion in this volume). Here the dramatype is the patients' symptoms, which are caused by a combination of several factors including genetic background, developmental environment, and proximate environment such as current social environment and medical care. In many cases, the proximate environment plays the major role after birth.
Physicians should see each patient as an individual. Simply looking at the disease itself is not enough, because each patient has a different genetic background, different developmental environment, and different proximate environment, which are not described in a textbook. Physicians should also see the patient as a whole person. Because the final goal of biomedical research is to treat and prevent human diseases, we must always take all of these factors into account. Thus, defining the phenotype of the mouse model should be done in the context of defining dramatype.
In patients with so-called “smooth brain,” or classical lissencephaly, the brain surface is basically smooth and without the usual cortical folds, which are important in normal brains to increase the surface area dramatically. Several mouse models share some of the characteristics of this disease. For example, in the reeler (Falconer 1951; reviewed in de Rouvroit and Goffinet 1998) and yotari (Yoneshima and others 1997) mutant mice, the cerebellum is much smaller than normal and lacks the foliated structure (“smooth cerebellum”). Although these
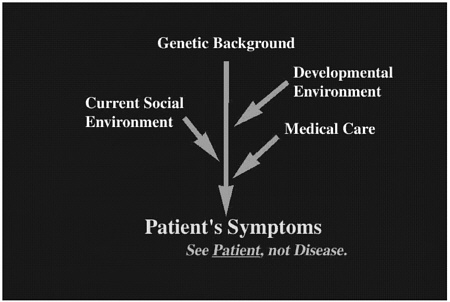
FIGURE 1 Clinical symptoms comprise human patients' “dramatype,” which includes genetic background, developmental environment, and proximate environment.
mutants do not exactly correspond to the human lissencephaly, the mechanism of the “fold” formation is nevertheless likely to be shared in some way among them. Our ultimate goals in conducting experiments with these animal models are to prevent and find a cure for this terrible disease.
The reeler is a well-known mutant mouse found approximately half a century ago. In the reeler, the neocortical structure is basically inverted because of abnormal neuronal migration and the mouse exhibits ataxic gait and tremor. The yotari, a novel mutant mouse we found unexpectedly a few years ago, has a phenotype almost identical to that of the reeler. As with humans, we started from the phenotype and took an unusual approach.
We immunized reeler mutants with homogenates of normal embryonic brains (Ogawa and others 1995). We expected the immunized animal to produce an antibody against a molecule present only in the normal brain but not in the mutant brain. This approach worked well, and we did obtain an antibody that recognized cells in the margin of the brain in the normal mouse but not in the reeler mouse. This antibody, CR-50, became a very useful tool because it inhibits the function of the antigen (del Rio and others 1997; Miyata and others 1997; Nakajima and others 1997; Ogawa and others 1995). The antigen recognized by this antibody was identified by a couple of groups and was named Reelin (D'Arcangelo and others 1995; Hirotsune and others 1995). It instructs the migrating neuroblasts to
be aligned appropriately. However, the yotari mouse has a mutation in the dab1 gene (Kojima and others 2000), which is expressed in the migrating neuroblasts (Sheldon and others 1997). We learned that the reelin and dab1 genes act on a common signaling pathway, in which the reelin signal is mediated by Dab1 to control neuronal migration (Howell and others 1999; Rice and others 1998).
A completely unexpected breakthrough occurred recently in this field when Joachim Herz, who was working on lipoprotein metabolism, made double knockout (KO) mice of the very low density lipoprotein receptor (VLDLR) gene and apolipoprotein E receptor 2 (ApoER2) gene and found that the mice's phenotype was similar to that of the reeler mouse (Trommsdorff and others 1999). It was then learned that Reelin binds to these receptors to tranduce the signal to the intracellular Dab1 protein (D'Arcangelo and others 1999; Hiesberger and others 1999). This information was surprising not only for the developmental neurobiologists but also for those working on lipid metabolism, and it reflects the importance of interaction between completely different fields. To define the phenotype of only one mouse (“my” mouse) is not enough. New discoveries most likely depend on combining data of various mice in different fields, although practically it is not always easy.
Now that I have explained some of the genetic background of a particular diseased state in mice caused by abnormal neuronal migration, I would like to focus on the extrinsic (environmental) factor. Schizophrenia is a severe disorder that affects about 1% of the whole population. Recent reports have indicated abnormal neuronal alignment in schizophrenic brains, which hints at neurodevelopmental causes for this disease. Also, reelin expression has been shown to be significantly reduced (approximately 50%) in schizophrenic patients (Impagnatiello and others 1998). The neurodevelopmental etiology of this disease has also been suggested by epidemiologic reports that indicate an association between maternal second trimester infection of influenza virus and increased risk of later development of schizophrenia (Wright and others 1995; Figure 2). Recently, Dr. S. H. Fatemi (University of Minnesota) infected pregnant mice with human influenza virus at midgestation and analyzed the neonatal brains. Interestingly, he found abnormal neuronal migration associated with reduced reelin expression (Fatemi and others 1999; Figure 2). Although the abnormal pattern of neuronal migration was not identical to that observed in the reeler mouse, this study implicates the role of an extrinsic factor for affecting the Reelin-mediated neuronal alignment.
We do not yet know whether and how Reelin is involved in the development of schizophrenia; however, one possibility is that the infected mice have produced an antibody against the viral antigen, which cross-reacts with a molecule essential for the cortical development. In addition, it is possible that multiple genetic factors (such as reelin, dab1, VLDLR, and ApoER2) and environmental factors affect the final pattern of neuronal alignment through a common molecular pathway (as seen in Figure 1).
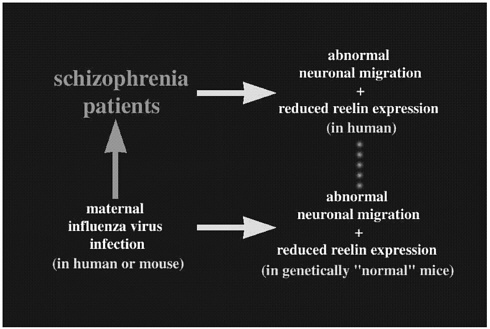
FIGURE 2 An abnormal neuronal migration pattern is hypothetically combined with reduced reelin expression to produce an antibody against the viral antigen, resulting in the development of schizophrenia.
In the context of dramatype development, it will be important to know at which step (genetic background, developmental environment, or proximate environment) each factor (or gene of interest) is involved because our ultimate goal is to overcome the dramatype (patient symptoms) by controlling all of these factors.
REFERENCES
D'Arcangelo, G., G.G. Miao, S.-C. Chen, H.D. Soares, J.I. Morgan, and T. Curran. 1995. A protein related to extracellular matrix proteins deleted in the mouse mutant reeler. Nature 374:719-723.
D'Arcangelo, G., R. Homayouni, L. Keshvara, D.S. Rice, M. Sheldon, and T. Curran. 1999. Reelin is a ligand for lipoprotein receptors. Neuron 24:471-479.
de Rouvroit, C.L., and A.M. Goffinet. 1998. The reeler mouse as a model of brain development. Adv. Anat. Embryol. Cell Biol. 150:1-106.
del Rio, J.A., B. Heimrich, V. Borrell, E. Foerster, A. Drakew, S. Alcantara, K. Nakajima, T. Miyata, M. Ogawa, K. Mikoshiba, P. Derer, M. Frotscher, and E. Soriano. 1997. A role for Cajal-Retzius cells and reelin in the development of hippocampal connections. Nature 385:70-74.
Falconer, D.S. 1951. Two new mutants, “trembler” and “reeler,” with neurological actions in the house mouse (Mus musculus L.). J. Genet. 50:192-201.
Fatemi, S.H., E.S. Emamian, D. Kist, R.W. Sidwell, K. Nakajima, P. Akhter, A. Shier, S. Sheikh, and K. Bailey. 1999. Defective corticogenesis and reduction in reelin immunoreactivity in cortex and hippocampus of prenatally infected neonatal mice. Mol. Psychiatr. 4:145-154.
Hiesberger, T., M. Trommsdorff, B. Howell, A.M. Goffinet, M.C. Mumby, J.A. Cooper, and J. Herz. 1999. Direct binding of Reelin to VLDL receptor and ApoE receptor 2 induces tyrosine phosphorylation of disabled-1 and modulates Tau phosphorylation . Neuron 24:481-489.
Hirotsune, S., T. Takahara, N. Sasaki, K. Hirose, A. Toshiki, T. Ohashi, M. Kusakabe, Y. Murakami, M. Muramatsu, S. Watanabe, K. Nakao, M. Katsuki, and Y. Hayashizaki. 1995. The reeler gene encodes a protein with an EGF-like motif expressed by pioneer neuron. Nature Genet. 10:77-83.
Howell, B.W., T.M. Herrick, and J.A. Cooper. 1999. Reelin induced tyrosine phosphorylation of disabled 1 during neuronal positioning . Genes Dev. 13:643-648.
Impagnatiello, F., A.R. Guidotti, C. Pesold, Y. Dwivedi, H. Caruncho, M.G. Pisu, D.P. Uzunov, N.R. Smalheiser, J.M. Davis, G.N. Pandey, G.D. Pappas, P. Tueting, R.P. Sharma, and E. Costa. 1998. A decrease of reelin expression as a putative vulnerability factor in schizophrenia. Proc. Natl. Acad. Sci. U S A 95:15718-15723.
Kojima T., K. Nakajima, and K. Mikoshiba. 2000. The disabled 1 gene is disrupted by a replacement with L1 fragment in yotari mice. Mol. Brain Res. 75:121-127.
Miyata, T., K. Nakajima, K. Mikoshiba, and M. Ogawa. 1997. Regulation of Purkinje cell alignment by reelin as revealed with CR-50 antibody. J. Neurosci. 17:3599-3609.
Nakajima, K., K. Mikoshiba, T. Miyata, C. Kudo, and M. Ogawa. 1997. Disruption of hippocampal development in vivo by CR-50 mAb against reelin. Proc. Natl. Acad. Sci. U S A 94:8196-8201.
Ogawa, M., T. Miyata, K. Nakajima, K. Yagyu, M. Seike, H. Yamamoto, and K. Mikoshiba. 1995. The reeler gene-associated antigen on Cajal-Retzius neuron is crucial molecule for laminar organization of cortical neuron. Neuron 14:899-912.
Rice, D.S., G. Sheldon, G. D'Arcangelo, K. Nakajima, D. Goldorwitz, and T. Curran. 1998. Disabled-1 acts down stream of reelin in a signaling pathway that controls laminar organization in the mammalian brain. Development 125:3719-1729.
Sheldon, M., D.S. Rice, G. D'Arcangelo, K. Yoneshima, K. Nakajima, K. Mikoshiba, B. Howell, J.A. Cooper, D. Goldowitz, and T. Curran. 1997. Scrambler and yotari disrupt the disabled gene and produce a reeler-like phenotype in mice. Nature 389:730-733.
Trommsdorff, M., M. Gotthardt, T. Hiesberger, J. Shelton, W. Stockinger, J. Nimph, R.E. Hammer, J. A. Richerdson, and J. Herz. 1999. Reeler/disabled-like disruption of neuronal migration in knockout mice lacking the VLDL receptor and ApoE receptor 2. Cell 97:698-701.
Wright, P., N. Takei, L. Rifkins, and R.M. Murray. 1995. Maternal influenza, obstetric complications and schizophrenia. Am. J. Psychiatr. 152:1714-1720.
Yoneshima, H., E. Nagata, M. Matsumoto, M. Yamada, K. Nakajima, T. Miyata, M. Ogawa, and K. Mikoshiba. 1997. A novel neurological mutant mouse, yotari, which exhibits reeler-like phenotype but expresses CR-50 antigen/reelin. Neurosci. Res. 29:217-223.





