

























































Below is the uncorrected machine-read text of this chapter, intended to provide our own search engines and external engines with highly rich, chapter-representative searchable text of each book. Because it is UNCORRECTED material, please consider the following text as a useful but insufficient proxy for the authoritative book pages.
11 Exposure and Biomarker Assessment in Humans he evaluation of potential reduced-exposure agents (PREPs) has to T be defined in the context of the outcome of interest (e.g., individual or population reduction in risk and disease type) and compared to an appropriate baseline (i.e., nonsmokers, former smokers, current smok- ers in the context of host susceptibility and previous level of smoke expo- sure). Tobacco exposure can be measured in the aggregate at the level of the entire population (e.g., through the measurement of tobacco sales or reported consumption in population-based surveys) and related to dis- ease incidence or change in mortality rates. These methodologies are con- sidered descriptive epidemiological tools that are useful in generating hypotheses and/or validating public health strategies, marketing pro- grams, and so forth. Exposure can also be measured at the level of the individual through biomarker measurements. This type of assessment within epidemiological studies can be used for hypothesis generation or testing. A range of methodologies and assays can be used for assessing exposure, as well as a range of assays for assessing host susceptibilities to exposure. The evaluation of a PREP can include four components: (1) external exposure measurements, (2) internal exposure measurements, (3) biomarkers estimating the biologically effective dose (Perera, 1987), and (4) biomarkers of potential harm (see Figure 11-1). The definitions of each are provided in Table 11-1 and explained further herein. There have been different definitions of types of exposure assessments used previously, but more recent understandings of biomarker uses and limitations, as 309
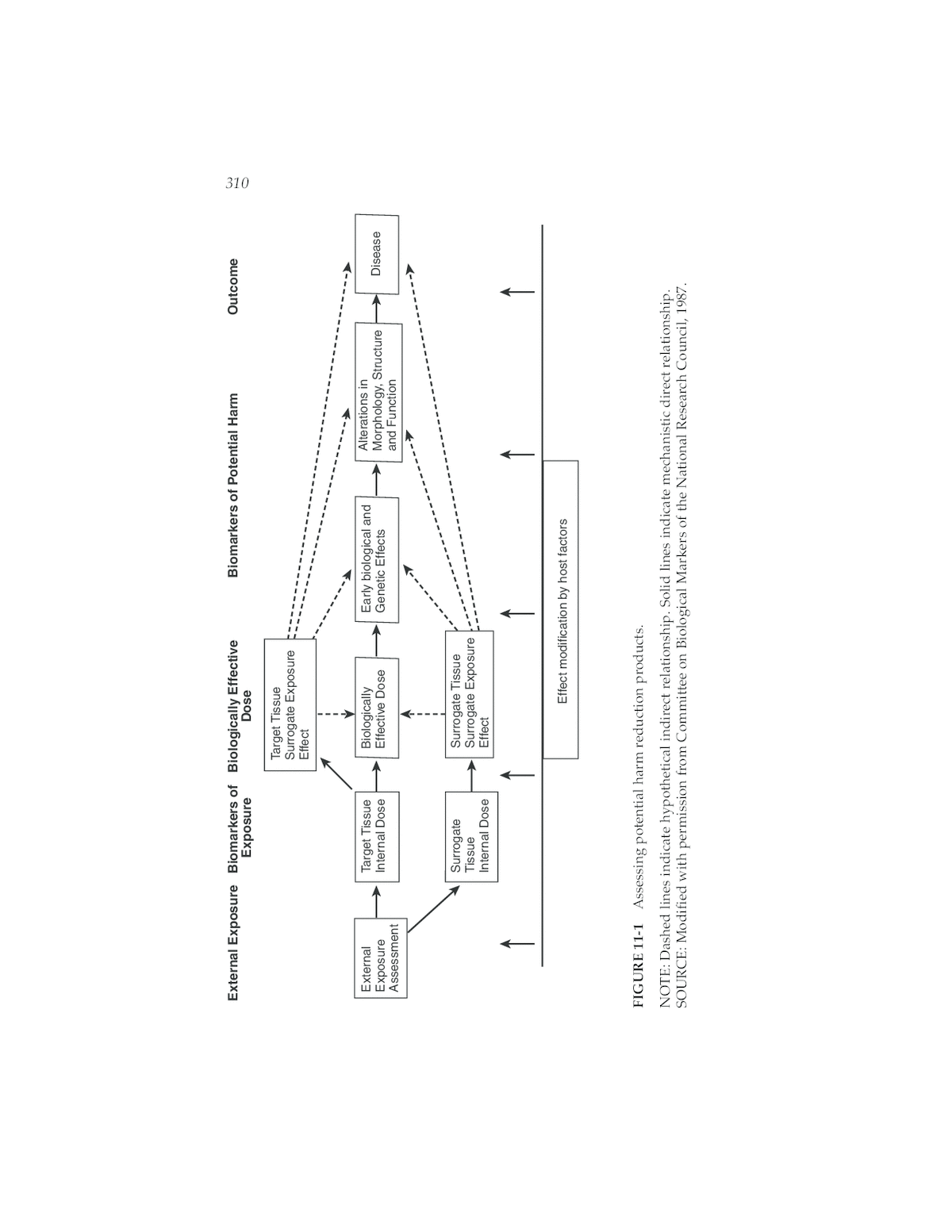
External Exposure Biomarkers of Biologically Effective Biomarkers of Potential Harm Outcome 310 Exposure Dose Target Tissue Surrogate Exposure Effect External Target Tissue Biologically Early biological and Alterations in Exposure Internal Dose Effective Dose Genetic Effects Morphology, Structure Disease Assessment and Function Surrogate Surrogate Tissue Tissue Surrogate Exposure Internal Dose Effect Effect modification by host factors FIGURE 11-1 Assessing potential harm reduction products. NOTE: Dashed lines indicate hypothetical indirect relationship. Solid lines indicate mechanistic direct relationship. SOURCE: Modified with permission from Committee on Biological Markers of the National Research Council, 1987.
EXPOSURE AND BIOMARKER ASSESSMENT IN HUMANS 311 TABLE 11-1 Exposure and Biomarker Assessment Definitions Exposure or Biomarker Assessmenta Definition External exposure A tobacco constituent or product that may reach or is at the marker portal of entry to the body Biomarker of A tobacco constituent or metabolite that is measured in a exposure biological fluid or tissue that has the potential to interact with a biological macromolecule; sometimes considered a measure of internal dose Biologically The amount that a tobacco constituent or metabolite binds to Effective Dose or alters a macromolecule; estimates of the BED might be (BED) performed in surrogate tissues Biomarker of A measurement of an effect due to exposure; these include potential harm early biological effects, alterations in morphology, structure, or function, and clinical symptoms consistent with harm; also includes âpreclinical changesâ aCategories and definitions reflect concept that the critical exposure is at the level of a biological macromolecule, so that exposure for this discussion is not limited to a measure- ment at the portal of entry to the body. well as different approaches needed for PREP evaluation lead to a need for clarification and redefinition. The latter three measurements improve upon the first by quantifying exposure at the cellular level to characterize low-dose exposures or low-risk populations, providing a relative contri- bution of individual chemical carcinogens from complex mixtures and estimating total burden of a particular exposure where there are many sources (Vineis and Porta, 1996). In assessing PREPs through biomarkers, understanding the biological effects of a wide range of exposures will be important. Within the context of this chapter, exposure at the level of the cell and critical macromolecules is considered with greater weight, rather than the traditional view of exposure at the portal of entry into a person. Biomarkers are intuitively more informative and better disease risk markers when measured in the target tissue through biopsies (e.g., oral mucosa, lung, bladder). However, biomarker assays are technically lim- ited, and target tissue can be difficult to obtain, especially in nondiseased smokers. Therefore, biomarker assays have been developed for surrogate tissues and fluids (e.g., expired breath, saliva, blood, urine). While these are technically simpler to use and easier to collect, the ability to prove a
312 CLEARING THE SMOKE TABLE 11-2 Measurements Used For Assessing Harm Reduction Products Factor Comment Type of measurement Types of measurements that can be used include external exposure assessment, biomarkers of exposure, biomarkers that represent the biologically effective dose, and biomarkers of potential harm. Depending on the context, the PREP, and the outcome of interest, different measurements might be more appropriate, although it is likely that a combination will be needed Target tissue and Is the measurement used for detecting effects in target or outcome effect surrogate tissues, and is this a measurement of pathogenesis? Dose-response data Measurements must have a dose-response relationship that is understood on a mechanistic basis. Biomarker should be able to demonstrate effects from exposure over the range of human experience, so that it can show exposure reduction from a PREP Harm reduction in Biomarker should be able to predict a decrease in disease dose-response data incidence after exposure is reduced Specificity Is the measurement specific for a tobacco product constituent, or does is also measure exposures from nontobacco products? Sensitivity Is the measurement sensitive enough to measure what it is supposed to measure in humans within the possible exposure ranges? Validation Are there sufficient data to show that the assay is reproducible? predictive value for the potential harm reduction is more difficult. It should be noted that the biomarkers discussed in this chapter refer to either target or surrogate tissue or fluid assays, but that the biologically effective dose refers to the assessment of a mechanistically relevant bio- marker only in the target organ. The following factors should be considered when evaluating mea- surements for predicting or determining the effects of a PREP. Table 11-2 summarizes these factors and Table 11-3 provides an overview of avail- able measures to predict the effects. 1. Type of measurement. Measurements are defined within four general categoriesânamely, external exposure, biomarkers of exposure, biomarkers estimating the biologically effective dose, and biomarkers of
EXPOSURE AND BIOMARKER ASSESSMENT IN HUMANS 313 potential harm. Placing a measurement solely within one category may not be possible. The external exposure assessment category is limited to those methods that are not detected by an assay using a body fluid or part. Although some external exposure methods might be poor predictors of disease risk and hence also poor measures for assessing a new product (e.g., the Federal Trade Commission [FTC] method described below), oth- ers might be strongly associated with disease risk and might therefore be better (e.g., cigarettes per day). While some external exposure assess- ments might be useful for harm reduction risk assessments (e.g., smoking history), they should not be used alone in assessing harm reduction be- cause the predictive power for disease is not sufficient without corrobora- tive biomarker data. Biomarkers of exposure are assayed in a body fluid (including exhaled air) or tissue that measures a constituent of tobacco smoke, tobacco-related products, or metabolites, where the constituent is not bound to a biomolecule. These biomarkers include unmetabolized compounds (e.g., carbon monoxide [CO], serum nicotine, carcinogen levels in serum or internal organs), biomarkers of exposure to individual cigarettes (e.g., incremental increases in exhaled CO or serum nicotine), and metabolites in any body fluid (e.g., cotinine in serum or urine, car- cinogen metabolites in urine). Biomarkers assessing the biologically effec- tive dose are those considered mechanistically related to disease outcomes (e.g., carcinogen-DNA adducts in the target tissue). Surrogate biologically effective doses, once validated, estimate a biological effect in a target organ (e.g., hemoglobin adducts or white blood cell carcinogen-DNA adducts). These biomarkers are in theory best able to link exposure (exter- nal and internal) to disease outcomes. Biomarkers of potential harm can reflect early or late damage (e.g., loss of heterozygosity in sputum, back- ground mutations in nondiseased tissues, reactive airway disease, arrhythmias, premalignant lesions, mutations in premalignant lesions, chromosomal aberrations in smoking-damaged epithelium, hypermethy- lation of genes, atherosclerosis). In this context, potential harm implies that the assay might or might not reflect actual harm and that some change in physiological function, for example, might not represent a harmful effect. 2. Target tissue and outcome relationship. A biomarker assay should be shown to be relevant to the outcome of interest. Besides having a mecha- nistic relationship to pathogenesis, data should be available to determine the predictive capacity for disease and disease reduction. This validation includes supportive evidence that the assay reflects harm reduction, such as might be done in an experimental cell culture or animal study. Assays that measure the effects in target tissue would generally have the greatest weight to support the use of a PREP. Sometimes, the target tissue effect
314 CLEARING THE SMOKE TABLE 11-3 Methods for PREP Assessment Type of Target vs. Category Measurement Surrogate Examples Strengths External External Neither Questionnaire data, FTC yield Inexpensi exposure exposure assessment Biomarker of Internal dose Target Polycyclic aromatic hydrocarbon in lung Provides i exposure tissue tissue exposur Surrogate Urinary measurement of tobacco Easily acc tissue constituent or metabolite, exhaled CO, measure carboxyhemoglobin, urinary smoking mutagenicity host cap clearanc Biologically Biologically Target Carcinogen-DNA adducts in human Reflects in effective effective dose tissue lung tissue, exfoliated bladder cells, exposur dose or oral mucosa activatio cycle co Surrogate Carcinogen-DNA or hemoglobin Does not tissue adducts; DNA adducts; lipid greater peroxidation availabl epidem Biomarker of Early biological Target Changes in RNA or protein expression, Assessme potential and genetic tissue somatic mutations, and LOH in leading harm effects normally or abnormally appearing tissue; change in methylation or gene control; mitochondrial mutations, mRNA expression arrays, or proteomics Alterations in Target Osteoporosis, hypertension, cough, Greater ab morphology, tissue hyperplasia, dysplasia, lipids, blood with ma structure, or coagulant pathways, mRNA function expression arrays, or proteomics Surrogate assays Surrogate tissue Leukocytosis; HPRT mutations; Easily acc chromosomal aberrations; circulating measure lymphocytes; mRNA or protein smoking expression via microarrays in cultured host cap blood cells clearanc Effect Measures of Neither Genetic polymorphisms for genes Reflects li modifiers interindividual involved in disease pathways high thr variation Target Enzyme induction of metabolizing Integrated enzymes exposur exposur NOTE: HPRT=hypoxanthine phosphoribosyltransferase; LOH=loss of heterozygosity.
EXPOSURE AND BIOMARKER ASSESSMENT IN HUMANS 315 Strengths Limitations Inexpensive Does not reflect actual internal doses on in lung Provides integrated measure of external Expensive; may not be specific for exposure and smoking behavior tobacco products; does not necessarily reflect biologically effective dose; tissue may be difficult to access; may be difficult to validate as a risk marker for disease co Easily accessible; provides integrated May not be specific for tobacco products; haled CO, measure of external exposure and does not necessarily reflect biologically smoking behavior; metabolites reflect effective dose; may be difficult to host capacity for metabolism and validate as a risk marker for disease clearance uman Reflects integrated measure of external Difficult to measure and validate as a er cells, exposure, smoking behavior, metabolic disease risk marker, predictive value activation, DNA repair capacity, cell- for disease risk is insufficiently studied, cycle control, and capacity for apoptosis more commonly reflects internal dose to a target macromolecule rather than disease risk in Does not require invasive procedures, Relationship to disease risk is not fully d greater amount of tissue is generally established available; more likely to be used in an epidemiological setting pression, Assessment of mechanistic pathway Tissue difficult to obtain; technically H in leading to disease difficult; relationship to disease risk earing difficult to establish; harmful effects n or gene may already be present; bioinformatics tions, with which to process information not yet available ugh, Greater ability to identify risk for disease Tissue difficult to obtain; late effects s, blood with marker where harm has already occurred; bioinformatics with which to process mics information not yet available ; Easily accessible; provides integrated Relationship to target organ effect is rculating measure of external exposure and difficult to prove; specificity for ein smoking behavior; metabolites reflect tobacco product needs to be proved; n cultured host capacity for metabolism and bioinformatics not yet available clearance nes Reflects lifetime response to exposure; Candidate gene approach will typically s high throughput possible study many polymorphisms that are not related to disease risk zing Integrated assessment of how prior Tissue technically difficult to obtain; exposures or genetic traits affect laboratory validation difficult exposures and harm osity.
316 CLEARING THE SMOKE might also be a surrogate for an effect in other tissues, and a surrogate tissue assay might reflect effects in multiple organs. 3. Dose-response data for harm. Assays that have a demonstrable dose- response relationship to actual disease outcomes is important for assess- ing a PREP and, if they do not, it should be shown to have a dose-response relationship to a biomarker of potential harm relevant to a disease path- way. The mechanistic basis for the relationship should be well under- stood in order to make meaningful interpretations of data used to assess a PREP. For example, assays that demonstrate a dose-response relationship between smoking and DNA damage in epithelial cells of a target organ could be useful. Methods assessing tobacco exposure as a complex mix- ture would have greater weight than a single component exposure. 4. Dose-response data for harm reduction. Assays that show a reduction in harm after reducing exposure to tobacco smoke or a tobacco product constituent would have the greatest weight, where the experimental de- sign uses an initial dose level for a specific duration of time followed by exposure to a lower level at a later time. The intent is to simulate the effects of a personâs switching from one level of exposure to another level of exposure. Importantly, the effects of the biomarker should be measur- ably different over the range of human exposures, so that the assessment can predictably measure the effects of exposure reduction from a PREP. Currently, there are some biomarker assays that have been assessed in former smokers or smoking cessation trials. These biomarker studies that indicate measurable decreases in effect can provide some information about the utility of markers for assessing exposure reduction. Included in this are half-life data, which must be measured and taken into account when evaluating a tobacco-related PREP. Methods assessing tobacco ex- posure as a complex mixture would have greater weight than a single component exposure. 5. Specificity. Consideration should be given to whether the effect is specific to a constituent of tobacco smoke or a tobacco product, or whether the method also measures exposure from other sources. Higher degrees of specificity are useful, although in some cases the method might be useful for assessing exposures from multiple sources other than tobacco in order to provide an understanding about relative contributions. Assays that are specific for tobaccoâs complex chemical mixture and those that are specific for a chemical or chemical class both have utility, but the former would have greater weight if appropriately validated, because persons are exposed simultaneously to all of the constituents. Validating assays for complex effects is more difficult because they may have less specificity for tobacco.
EXPOSURE AND BIOMARKER ASSESSMENT IN HUMANS 317 6. Sensitivity. The assay must be sufficiently sensitive to measure what it is supposed to measure in the human tissue of interest. This is espe- cially problematic in measuring low-level effects, for example, in assess- ing the effects of environmental tobacco smoke (ETS) exposure. 7. Validation. It is critical that biomarkers for assessing PREPs be well validated in the laboratory. Validation includes proof that the assay mea- sures what it claims to measure and that it is reproducible. Sensitivity, specificity, and predictive value are all important to consider. EXTERNAL EXPOSURE ASSESSMENT: THE FTC METHOD AND QUESTIONNAIRE DATA External exposure markers attempt to measure the amount of a to- bacco smoke or tobacco product constituent that may enter at a portal to the body. However, these predictors generally do so without regard to most interindividual differences in smoking behavior and cellular pro- cesses. There are several types of external exposure assessment, some of which are listed in Table 11-4. A common way to assess potential exposure to tobacco smoke is by measuring the yield of tobacco smoke constituents. One attempt to esti- mate delivered doses is the method adopted by the Federal Trade Com- mission in 1967. It was intended to provide a standardized estimate of tar and nicotine yield by cigarette brand, simulating a cursory observation of human smoking behavior. A cigarette is inserted into a smoking machine and lit, puffs are taken through a syringe (35 ml over 2 seconds, every 60 seconds) until the cigarette is âsmokedâ to a fixed length. Particulates are collected on a filter and weighed. Nicotine is assayed separately. Tar is measured as total particulate matter less nicotine, other alkaloids, and water. Although the machine provides yield data that can be used to compare one cigarette to another, this information has limited usefulness for understanding human exposure because people do not smoke ciga- rettes as the machine does due to different smoking behaviors. Smokers also can affect cigarette filter performance by covering ventilation holes in the filter with their lips or fingers, which would increase yields in vivo. Although FTC yields might define a comparative range of actual expo- sures, there is a wide overlap of actual to predicted yields among types of cigarettes (i.e., low, medium, and high yields), where smokers of low- nicotine cigarettes might have higher nicotine levels than those who smoke brands with higher FTC yields (Byrd et al., 1998, 1995). Altering the FTC method to simulate puffs and times for actual smokers results in
TABLE 11-4 External Exposure Assessmenta 318 Related to Variables Used a Disease Category in Literature Outcomeb Strengths Limitations FTC machine Tar yield Yes Standardized method for yields Little relationship to actual human method Nicotine yield experience Individual smoke constituent yield Subject Cigarettes per day Yes Inexpensive assessment; generally Recall is subject to self-perceptions of smoking Years of smoking considered reliable, except in some risk. Reporting is variable depending history Age of initiation circumstances listed in limitations on context, such as smoking cessation Recall of inhalation program or where recall bias might depth exist in epidemiology studies. Known Usual type of limitations for persons who are cigarette smoked switching brands or altering smoking Quitting attempts behavior; also not sufficiently reliable Cumulative tar in smoking cessation studies. Thus, not exposure sufficiently reliable in harm reduction studies Smoking Puff duration No Direct measure of inhalation exposure Measurement performed in artificial Topography Puffs per cigarette per cigarette. Can be used to assess environment Interpuff interval effects of cigarette brand switching Puff volume aReferences are not provided in this table but can be found in the text of this and disease-related chapters. bAny report related to a disease outcome where the report is plausible but has not necessarily been replicated.
EXPOSURE AND BIOMARKER ASSESSMENT IN HUMANS 319 higher exposures to tar and specific carcinogens (e.g., tobacco-specific nitrosamines and benzo[a]pyrene) (Djordjevic et al., 2000; Fischer et al., 1989; Hoffman and Hoffman, 1997). For example, using modified proto- cols to stimulate human smoking behavior, the medium-yield (0.9-1.2 mg nicotine per cigarette) and low-yield (0.8 mg nicotine per cigarette) ciga- rettes deliver similar amounts of tar per day, although by FTC method measured per cigarette yields of tar, benzo[a]pyrene (BaP), and tobacco- specific nitrosamines (TSNAs) were higher in the former (Djordjevic et al., 2000). As cigarettes with different designs are developed and mar- keted, an assumption that the FTC method of estimating yields will be comparable to existing products is premature. Over the last 30 years, data from surveys have been an important tool in the assessment of tobacco exposure among individuals and the popula- tion. They have been an effective means of tracking patterns of tobacco use and the societal perceptions that ultimately influence consumption. Individual exposure can be assessed through the measurement of the number of cigarettes smoked per day, duration of smoking, types or brands of cigarettes smoked (e.g., âtarâ delivery, filter type, type of to- bacco, mentholation), and age at initiation (IARC, 1986; Kaufman et al., 1989; La Vecchia et al., 1990; Lubin et al., 1984; Stellman and Garfinkel, 1989; U.S. DHHS, 1988; Vutuc and Kunze, 1983; Wilcox et al., 1988; Zang and Wynder, 1992). Lifetime exposures can be estimated by calculating pack-years (average packs per day multiplied by number of years smoked) or cumulative tar exposure (Zang and Wynder, 1992). A more detailed description of the most common surveys in use is presented in Table 11-5. Most analyses indicate that self-report validity among adults is good (Patrick et al., 1994). Certain limitations, however, are evident in this type of exposure assessment (Giovino, 1999; U.S.DHHS, 1994). First, sampling errors may occur in any study in which generalizations are made from a selected population sample. One example is the over- or underrepre- sentation of certain groups, especially those that exhibit significant to- bacco use or have differing smoking behavior. In fact, there is a built-in exclusion in many of the major surveillance tools of various segments of the population, such as the institutionalized mentally ill, prisoners, and those in areas of inadequate telephone coverage. Errors in response must be considered including memory errors, nonresponse errors, and mis- classifications and inconsistencies in reporting. The validity of self- reported responses can be influenced by many factors (Velicer et al., 1992), particularly the respondentâs perception of privacy (Giovino, 1999). This is especially a concern among adolescents in the home setting and among groups that have increased pressure to abstain or to quit, including preg- nant women, adolescents, and patients with heart or lung disease. One
320 CLEARING THE SMOKE TABLE 11-5 Major Tobacco Use Surveys Survey Sponsor Population Size National Health Interview National Center for Health Civilian, noninstitution- More than Survey (NHIS) Statistics, Centers for alized adults over age families Disease Control and 18; children by proxy Prevention (CDC) Behavioral Risk Factor CDC and individual states Noninstitutionalized adults Surveillance System over age 18 (BRFSS) National Health and CDC Age 2 and over Approxim Nutrition Examination 40,000 p Survey (NHANES) between 1994 National Household National Institute on Drug Noninstitutionalized Approxim Survey on Drug Abuse Abuse and Substance civilian population over 25,500 p (NHSDA) Abuse and Mental age 12 in 1998 Health Services Administration American Legacy Sixth to twelfth grade Foundation Survey students Monitoring the Future University of Michigan Eighth, tenth, and twelfth Approxim Survey (previously, the Survey Research Center grade students 50,000 s National High School from pu Senior Survey) private schools Youth Risk Behavior CDC Ninth to twelfth grade Surveillance System students (YRBSS) effort to validate self-report measures and to reveal any ETS exposure can be found in the National Health and Nutrition Examination Survey (NHANES; see Table 11-5), which collects serum cotinine levels of re- spondents (CDC, 2000; Giovino, 1999; Giovino et al., 1995; SAMHSA, 1998). Population surveys have limited practicality in evaluating the conse- quence of tobacco exposure because of the relatively long time frame required. However, in context, population assessments have been studied extensively in relation to disease outcomes and thus can be considered a
EXPOSURE AND BIOMARKER ASSESSMENT IN HUMANS 321 Size Setting Comments tution- More than 38,000 Household interview Excludes homeless not in shelters, ver age families in 1998 with responses military personnel, prisoners, proxy typed directly in hospital patients laptop computer; Data: cigarette, chewing tobacco, annual cigar, and pipe use since 1965 Oversampling of black American and Hispanic populations zed adults Computer-assisted Added smokeless tobacco use telephone questions in 1987 interviews; annual State level Approximately Personal interview Serum cotinine measurements 40,000 participants with physical exam Oversampling of children 1-5years, between 1988 and and blood tests; adults over age 60, black 1994 periodic Americans, and Mexican Americans zed Approximately Household interview; State level tion over 25,500 participants self-administered Oversampling of black Americans, in 1998 through a computer; Hispanic Americans, and youth annual rade School based Evaluates knowledge of and attitudes towards all forms of tobacco, including bidis and Kreteks d twelfth Approximately Classroom based; self- Random sample from each senior 50,000 students administered; annual class is followed after graduation from public and for longitudinal data private high schools grade Classroom based; Oversampling of black and Hispanic- self-administered; American students. Combination of biennial national, state, and local surveys crude measurement of individual risk and a better measure of population risk. These surveys do provide insight into trends of tobacco product use within and across a variety of sociodemographic groups, including age, sex, race or ethnicity, educational status, and economic status. The data can be compared to morbidity and mortality registries to understand new or changing consequences of use patterns or specific products. In addi- tion, these trends in prevalence, initiation, and cessation in turn aid in the evaluation of the effects of tobacco-related activities, policies, and inter- ventions within the general population and its subgroups.
322 CLEARING THE SMOKE Methods for assessing external exposure (e.g., number of cigarettes per day) are widely used and relatively inexpensive but do not provide an assessment of how someone smokes cigarettes and how the body re- sponds to exposure. Thus, these measures approximate the level of actual exposure and, as described below, become less reliable in assessing expo- sure reduction. Smoking topography is an additional method of assessing external exposure (e.g., how much smoke enters the lung, estimated by measuring puff volume, number of puffs per cigarette, puff duration, total inhalation time, and interpuff interval) (Bridges et al., 1990; Gritz et al., 1983; Herning et al., 1983; Hofer et al., 1992; Kolonen et al., 1992b). In the laboratory, if subjects smoke their own cigarettes, then it is presumed that the measurement reflects their usual smoking behavior. A limitation of smoking topography studies is that cigarettes are typically smoked via cigarette holders, which may influence puffing behaviors and prevent vent hole blocking that might normally occur when the cigarettes are smoked without the holder. Smoking topography studies have contrib- uted to the findings that persons who switch from high-tar and nicotine to low-tar and nicotine cigarettes increase their intake of smoke per cigarette to compensate for a lower yield of nicotine (Benowitz et al., 1986a, b). It is well established that smokers self-titrate their blood nicotine levels, such that smokers of lower-nicotine cigarettes inhale more (Benowitz et al., 1983; Benowitz et al., 1986b; Benowitz et al., 1998; Ebert et al., 1983; Gritz et al., 1983; Hill and Marquardt, 1980), and altering topography leads to differences in nicotine absorption and CO boosts (Hofer et al., 1992; Kolonen et al., 1992a). Smoking lower-nicotine delivery cigarettes in- creases puff volume (Battig et al., 1982; Bridges et. al., 1986; Kolonen et al., 1992b) and, to a lesser extent, puff duration (Bridges et al., 1990). Using a multiple regression model for prediction of nicotine blood levels, the best- fit model incorporates interpuff interval, number of puffs per cigarette, puff volume, puff duration, inhaled volume, and inhalation duration (Herning et al, 1983). These studies are difficult to interpret, however, because cigarette and topographic parameters are interrelated (Bridges et al., 1990; Kolonen et al., 1992a; Nemeth-Coslett and Griffiths, 1984). Different methods have been developed for the study of exposure to environmental tobacco smoke (EPA, 1992). Stationary and personal air monitors can be used to measure total particulates or individual constitu- ents. Some measurements, such as nicotine, are more specific for ETS. Ambient air concentrations and personal exposures to polycyclic aromatic hydrocarbons (PAHs) and other tobacco constituents can be measured, but their relationship to disease risk has not been adequately studied.
EXPOSURE AND BIOMARKER ASSESSMENT IN HUMANS 323 BIOMARKERS OF EXPOSURE Biomarkers of exposure, measured in a body fluid, tissue, or exhaled air, represent an internal dose of tobacco smoke or a tobacco product constituent that is either the parent compound or its metabolite. They are not measurements of how the constituents interact with body functions or macromolecules to cause harm. Some of these markers have been re- searched extensively, and they are more representative of actual human exposures to tobacco products than external measures of exposure. They are generally technically feasible and provide information about short- term (e.g., from a single cigarette) and long-term exposures. Examples are listed in Table 11-6, which gives a range of assays available but is not intended to be all inclusive. Because it has such a short half-life, carbon monoxide is best used for assessing recent exposures, although CO mea- surements also have been used to improve long-term exposure estimates of cigarette consumption (Law et al., 1997). The limitations of CO are that there are other sources of carbon monoxide, such as automobile exhaust and endogenous metabolism, and there is some variation with differences in physical activity, gender, and the presence of lung disease or other disease states. Nicotine blood levels are used and are helpful for assessing internal exposure primarily because it has a very short half-life. Serum, urinary, or salivary cotinine, which is a metabolite of nicotine with a longer half-life, however, has been extensively studied for confirmation of exposure in smokers, quitters, and persons exposed to ETS (Bono et al., 1996; Benowitz, 1999; Crawford et al., 1994). Cotinine levels are depen- dent on both the extent of formation from nicotine by cytochrome P450 (CYP) 2A6 and the rates of oxidation and glucuronidation of cotinine to 3- hydroxy-cotinine and glucuronide conjugates, respectively, which vary widely among individuals. Therefore, cotinine levels are only approxi- mately correlated with the daily intake of nicotine. Carbon monoxide and nicotine boosts (i.e., the difference between levels before and after a single cigarette) reflect smoking topography and exposures from an individual cigarette. Technologies exist for directly measuring internal exposure to tobacco smoke constituents in target organs through biopsies (e.g., PAHs in the lung) (Lodovici et al., 1998) and for measuring levels of metabolites of compounds (e.g., those from TSNAs in the urine) (Atawodi et al., 1998; Carmella et al., 1990, 1995). Tobacco smokers have higher levels of mu- tagens circulating in the body, which can be measured by using extracts of urine in the Ames Salmonella mutation assay (Jaffe et al., 1983; Mohtashamipur et al., 1985; Yamasaki and Ames, 1977). Levels have been found to decrease with some test cigarettes that heat, rather than burn, tobacco (Smith et al., 1996).
324 CLEARING THE SMOKE TABLE 11-6 Biomarkers of Exposurea,b Associated Variables Used Dose-Response with Cessation Chemical Specific to Category in Literature Data Available or Half-life Specificity Tobacco Nicotine-related Nicotine Yes 2 hr Yes Yes (excep biomarkers using n replacem therapy Nicotine boost (pre- Yes NA Yes Yes and post-cigarette nicotine levels) Cotinine Yes 17 hr Yes Yes (excep using N Other nicotine Yes Depends on Yes Yes (excep metabolites metabolite using N Minor tobacco Anatabine NDA 10-16 hr Yes Yes alkaloids Anabasine Carbon Exhaled CO Yes 4-6 hr Yes No monoxide CO boost (pre- and Yes NA Yes Yes post-cigarette levels) Carboxyhemoglobin Yes Hours Yes No Hydrogen Thiocyanate Yes 1-2 weeks No No cyanide
EXPOSURE AND BIOMARKER ASSESSMENT IN HUMANS 325 hemical Specific to Related to a ecificity Tobacco Disease Riskc Strengths Limitations s Yes (except when Yes Direct measure of Short half-life depen- using nicotine (addiction exposure dent on a personâs replacement only) ability to metabolize therapy [NRT]) nicotine and time of sampling. Not useful with concurrent use of NRT s Yes NDA Measures exposure to Requires two blood single cigarette draws. Short-term marker only s Yes (except when Yes Well validated; can be Short-term marker only. using NRT) (addiction measured easily in At higher levels of only) urine, plasma saliva, smoking, dose- or hair. Useful for response relationship environmental is less clear and there tobacco smoke is wide overlap among smokers s Yes (except when NDA Allows for assessment Low levels. No benefit using NRT) of nicotine metabolism over cotinine. Short term marker only s Yes NDA Useful when individuals Short-term marker only are using NRT; may be precursors to nitrosamines s No Yes Easy to measure in Other sources exist, exhaled air including endogenous processes. Short-term marker only. s Yes NDA Measures exposure to Short term marker only. single cigarette Levels vary over the day s No Yes Measures cumulative, Requires blood draw although short-term and special handling. exposure to several Benefit above that for cigarettes using exhaled CO not shown o No NDA Long-term marker. Can Many dietary sources. be measured in urine, Dose-response curve saliva, and blood. flattens at higher Saliva easy to obtain smoking levels so cannot distinguish among heavy smokers continues
326 CLEARING THE SMOKE TABLE 11-6 Continued Associated Variables Used Dose-Response with Cessation Chemical Specific to Category in Literature Data Available or Half-life Specificity Tobacco Tobacco- Urinary metabolites Yes 45 d Yes Yes specific nitrosamines Polycyclic Parent compounds NDA NDA Yes No aromatic hydrocarbons Urinary Yes NDA Yes No 3-hydroxypyrene and 1-hydroxypyrene Complex Urinary Yes Yes No No mixture assay mutagenicity NOTE: NA=not applicable; NDA=No data available. The assessment of smoking exposure using nicotine or cotinine can- not be done in smokers who are concomitantly using nicotine replace- ment products. An alternative is to determine the levels of other tobacco alkaloids, such as anatabine or anabasine in the urine (Jacob et al., 1999). BIOMARKERS ESTIMATING THE BIOLOGICALLY EFFECTIVE DOSE The biologically effective dose (Perera, 1987) is the amount of a to- bacco smoke or tobacco toxin that measurably binds to, or alters, a macro- molecule (e.g., protein or DNA) in a cell. In some cases, the macromol- ecule may be a surrogate for a target molecule. The biologically effective dose represents the net effect of metabolic activation, decreased rate of detoxification, decreased repair capacity, loss of cell-cycle checkpoint con- trol, and decreased rates of cell death. It should be noted that not all binding to, or alteration of, a macromolecule leads to an adverse health effect; so, often, what is really measured is the dose to a target macromol- ecule that estimates the biologically effective dose. Table 11-7 provides
EXPOSURE AND BIOMARKER ASSESSMENT IN HUMANS 327 hemical Specific to Related to a ecificity Tobacco Disease Riskc Strengths Limitations s Yes NDA May reflect biologically Technically difficult to effective dose measure s No Yes Measured in organs Technically difficult to where effect might obtain tissue and occur perform assay s No NDA Assay simple to perform Other exposures can be substantial o No NDA May be related to in Lack of specificity vivo mutagen exposure aSelected examples; list is not all-inclusive. bReferences are not provided in this table but can be found in the text of this and disease- related chapters. cAny report related to a disease outcome associated with tobacco where the report is plausible but has not necessarily been replicated. examples of biomarkers that estimate the biologically effective dose, but is not intended to be all inclusive. Many tobacco-related toxins and chemical carcinogens are biologi- cally inactive until transformed by cellular enzymes such as cytochrome- P450s into reactive intermediates. These reactive intermediates bind to macromolecules such as DNA and protein and disrupt their normal pro- cesses. For cancer, a common assessment of the biologically effective dose is the measurement of carcinogen-DNA adduct levels. These are formed when carcinogen metabolites are alkylated to nucleotides, creating a promutagenic lesion. There are strong laboratory animal data and some human studies that indicate a relationship between tobacco smoke con- stituents, carcinogenâDNA adduct formation, and cancer (La and Swenberg, 1996). Laboratory animal studies have shown a correlation between cancer and increased adducts in target organs (Ashurst et. al., 1983; Nakayama et al., 1984; Pelkonen et al., 1980). In humans, tobacco smoking leads to increased adduct formation in target tissues such as the lung (Phillips et al., 1988; Schoket et al., 1998; Wiencke et al., 1995) and in
328 CLEARING THE SMOKE TABLE 11-7 Biomarkers Estimating the Biologically Effective Dosea,b Target Dose- Tissue Variables Used Response Associated with Assay Chemical Category in Literature Data Cessation or Half-life Available Specificity Carcinogen- Nonidentified Yes Yes Yes No DNA adducts adducts/ 32 P-postlabeling PAH-DNA Yes 9-13 weeks Yes Yes adducts (blood) 4-Aminobiphenyl- Yes Yes Yes Yes DNA adducts NNK-DNA Yes NDA Yes Yes adducts 8-hydroxydeoxy- No Yes Yes Yes guanosine 5-(Hydroxy- No NDA No Yes methyl)uracil N-Nitrosamine- NDA 26 hr (blood; O6- Yes Yes related-DNA methyldeoxy- adducts guanosine) and 60 hr (blood; 7- methyldeoxy- guanosine)
EXPOSURE AND BIOMARKER ASSESSMENT IN HUMANS 329 Dosea,b Target Related Tissue Specific to a Assay Chemical to Disease Available Specificity Tobacco Riskc Strengths Limitations Yes No No Yes Facile assay; does not Cannot identify adducts so require knowledge mechanistic studies are of specific adducts; problematic blood may be surrogate for lung tissue. Adducts found in all tissues, including heart and blood vessels Yes Yes No Yes Can be measured in Low sensitivity and any tissue and technical difficulties make assays are available assay use limited that are sufficientlyin large-scale studies. sensitive Diet might be greater contributor than smoking Yes Yes No NDA Can be measured in Low sensitivity makes assay any tissue; has some use limited in large-scale specificity for studies smoking if no known occupational exposure Yes Yes Yes NDA Can be measured in Low sensitivity makes assay any tissue, although use limited in large-scale methodology has studies low sensitivity. Highly specific for smoking Yes Yes No NDA Can be measured in Assay has large any tissue interlaboratory variation; it is easy to introduce oxidative damage into laboratory assay; low sensitivity makes assay use limited in large-scale studies No Yes No Not Sufficient sensitivity Technically difficult avail- to use for ETS able Yes Yes No NDA Can be measured Low sensitivity makes assay in any tissue use limited in large-scale studies. Diet a common source continues
330 CLEARING THE SMOKE TABLE 11-7 Continued Target Dose- Tissue Variables Used Response Associated with Assay Chemical Category in Literature Data Cessation or Half-life Available Specificity Carcinogen- PAH-Hgb adducts Yes NDA No Yes hemoglobin (Hgb) adducts 4-Aminobiphenyl- Yes 7-9 weeks No Yes Hgb adducts Carcinogen- PAHâalbumen Yes NDA No Yes protein adducts adducts Carcinogen- Anti-BPDE serum NDA NDA No No DNA adduct antibodies antibodies Adducts Yes Yes Yes Yes Carbon Carboxy- Yes Yes No Yes monoxide hemoglobin Lipid F2-Isoprostanes No Yes No Yes peroxidation NOTE: NA=not applicable; NDA=no data available; NNK=nitrosonornicotine ketone; BPDE=benzo(a)pyrene-diol-epoxide surrogate tissues such as blood (Tang et al., 1995; Vineis et al., 1994; Wiencke et al., 1995). Evidence exists that carcinogen-DNA adduct levels in target and nontarget organs are modulated by interindividual differ- ences (Badawi et al., 1995; Grinberg-Funes et al., 1994; Kato et al., 1995;
EXPOSURE AND BIOMARKER ASSESSMENT IN HUMANS 331 Target Related Tissue Specific to a Assay Chemical to Disease Available Specificity Tobacco Riskc Strengths Limitations No Yes No NDA Large amount of Surrogate assay not yet adducts available validated against target in blood so method organ damage is facile No Yes No NDA Large amount of Surrogate assay not yet adducts available validated against target in blood so method organ damage is facile No Yes No NDA Large amount of Surrogate assay not yet adducts available validated against target in blood so method organ damage is facile No No NDA NDA May provide long-term Doubtful that a dose- marker of exposure response relationship can be established due to complexity of immune response in individuals Yes Yes No NDA Measured in organs Technically difficult to where effect might obtain tissue and perform occur assay No Yes No Yes Might also reflect a Logistical problems in surrogate measure sample handling of biologically effective dose No Yes No NDA Corroborative end Technically difficult point for oxidative damage without artifactual introduction of oxidative damage aSelected examples; list is not all-inclusive. bReferences are not provided in this table but can be found in the text of this and disease- related chapters. cAny report related to a disease outcome associated with tobacco where the report is plausible but has not necessarily been replicated. Pastorelli et al., 1998; Rojas et al., 1998; Ryberg et al., 1997; Stern et al., 1993). Interestingly, in former smokers, age of initiation may influence lung adduct levels (Wiencke et al., 1999). In humans, only a few studies have investigated a link between carcinogen-DNA adducts and cancer
332 CLEARING THE SMOKE risk. All data come from case-control studies of the lung and bladder, and almost all show a positive relationship (Dunn et al., 1991; Peluso et al., 1998; Tang et al., 1995; van Schooten et al., 1990). However, since no published prospective studies of tobacco smoking show a relationship of adducts to cancer, the case-control studies must be interpreted cautiously because there may be an effect due to differential metabolism or DNA repair. The utility of carcinogen-DNA adduct measurements in assessing harm reduction is suggested by studies showing that lung adduct levels are lower in persons who smoked filter cigarettes (van Schooten et al., 1990). Hemoglobin adducts, an estimate of the biologically effective dose, are higher in smokers than in nonsmokers (Bryant et al., 1987), and in those who smoke black rather than blond tobacco (Bryant et al, 1988). Snuff dipping may lead to even higher levels of some types of adduct than to smoking (Carmella et al., 1990). A variety of assays are available to determine carcinogenâmacro- molecular adducts in human tissues (Farmer and Shuker, 1999; Hecht, 1999; La and Swenberg, 1996; Lee et al., 1993; Wang et al., 2000). Although DNA adduct analysis is most commonly studied in relation to carcino- genesis, adducts also have been found in atherosclerotic lesions (Izzotti et al., 1995). Assay techniques include the phosphorus-32 (32P)-postlabeling assay-nucleotide chromatography (Phillips, 1997; Randerath et al., 1981), immunoassays (Lee et al., 1993), fluorescence spectroscopy (Izzotti et al., 1991), gas chromatography-mass spectroscopy (GC-MS) (Farmer and Shuker, 1999; Hecht, 1999), and electrochemical detection (Helbock et al., 1998; Park et al., 1989). Each has its strengths and limitations, and almost all are challenged by low sensitivity and/or specificity. The less specific methods, such as the 32P-postlabeling assay-nucleotide chromatography, when used as originally described (Randerath et al., 1981) or with modifi- cations (Reddy and Randerath, 1986), offer the benefit of assessing expo- sure to complex mixtures because multiple adducts are measured at the same time. However, because the assay does not identify the types of adducts, any interpretations of the results are limited. Chemical specificity is helpful in assessing harm reduction products when the adducts are specific for tobacco (e.g., TSNAs or 4-aminobiphenyl in the absence of occupational exposure), whereas adduct assays that determine levels from endogenous sources (e.g., oxidative damage, methylation) are more diffi- cult to use and interpret. The study of carcinogen-DNA adducts presents other challenges in interpretation; for example, carcinogen-DNA adduct levels are higher in the heart than in the lung (Randerath et al., 1989) while cancer is rare in the former. For the future, newer adduct methods may provide increased specificity and sensitivity, along with higher throughput. The use of target organ biomarkers can provide specific information about potentially carcinogenic effects and will best represent the biologi-
EXPOSURE AND BIOMARKER ASSESSMENT IN HUMANS 333 cally effective dose. Target organs include lung for lung diseases, oral mucosa for oral cavity diseases, bladder mucosa for bladder disease, and so forth. Surrogate markers that estimate levels in target organs, such as carcinogen-DNA adducts in blood, have been partially studied, indicat- ing that blood levels might reflect target organ levels (Mustonen and Hemminki, 1992; Mustonen et al., 1993; Tang et al., 1995; Wiencke et al., 1995), but this is not yet firmly established. Protein (Meyer and Bechtold, 1996) and hemoglobin (Wang et al., 2000) adducts also may estimate levels of exposure at the target organ and thus be surrogates. Such assays offer technological advantages because these macromolecules are more abun- dant in blood than DNA, but the relationship of these other macro- molecular adducts to DNA levels has been insufficiently studied. A few studies show the decline of adducts following short-term and long-term smoking cessation. Most studies will necessarily rely on blood levels, and the half-life of adducts in blood will depend on the life span of various blood cell types. In humans, the half-life for 4-aminobiphenyl- hemoglobin adducts is 7-9 weeks, which is shorter than the life span of a red blood cell (Jahnke et al., 1990). PAH-DNA adducts in white blood cells have a half-life of 9 to 13 weeks (Mooney et al., 1995). In human lungs, it was reported that adducts persist in the lungs of ex-smokers (Randerath et al., 1989), but it is not known whether this is truly persis- tence or the formation of new adducts from the continuing presence of tobacco constituents such as PAHs or from other exposures such as diet or air pollution (Rothman et al., 1990). Carcinogen-DNA adduct data have essentially not been used for population risk assessments. In one example, it was considered that a doubling of PAH-DNA adduct levels would result in an additional 2,400 cancer cases per million persons (van Delft et al., 1998), but the model assumed linear dose-responses; was not adjusted for age, gender, or race; and was too simplistic. BIOMARKERS OF POTENTIAL HARM These biomarkers reflect changes in a cell and its macromolecules that result from tobacco. These can range from isolated changes, with or without effects on function, to events that clearly lead to illness or are symptoms of the illness (i.e., cough). Examples of biomarkers of effect are provided in Table 11-8, which gives the reader a range of assays available but is not intended to be all inclusive. Among the most promising biomarkers of effect for assessing harm reduction claims for cancer are those that measure DNA damage or alter- ations of genetic function (mutations, gross chromosomal changes, DNA methylation of promoter regions, etc.). While these biomarkers are envi-
334 CLEARING THE SMOKE TABLE 11-8 Biomarkers of Potential Harmful Effectsa,b Associated Target Dose- with Tissue Specific Variables Used Response Cessation Assay Chemical to Category in Literature Data or Half-life Available Specificity Tobacco Enzymatic Aryl hydrocarbon No >30 d Yes Yes No induction hydroxylase CYP1A2 No NDA Yes Yes No DNA repair NDA Yes Yes NA No enzymes Microarray assays NDA NDA Yes NA No for mRNA expression and proteomics Chromosomal Chromosomal Yes Yes Yes No No alterations aberrations Micronuclei Yes Yes Yes No No Sister chromatid Yes Yes No No No exchanges Loss of Yes Yes Yes No No heterozygosity Mutations in Yes Yes No No No reporter genes (HPRT, GPA)
EXPOSURE AND BIOMARKER ASSESSMENT IN HUMANS 335 Related Specific to a Chemical to Disease Specificity Tobacco Riskc Strengths Limitations Yes No Yes Indicates acquired changes in Technically difficult to assess in susceptibility; related to large epidemiological studies DNA-adduct levels Yes No Yes Indicates acquired changes in Technically difficult to assess in susceptibility; related to large epidemiological studies DNA-adduct levels NA No NDA Indicates acquired changes in Technically difficult susceptibility; provides analysis of what is likely to be critical part of carcinogenesis NA No NDA Reflects integrated measure of Difficult to perform; multiple genotypes, provides relationship to disease risk is complex data potentially technically difficult to prove; usable for rapid identification requires extensive laboratory of important risk factors validation; RNA and protein microarray assays are expensive; large-scale studies are needed; refined bioinformatic analysis required No No Yes Can be done in blood as Very nonspecific; relationship surrogate tissue. Similar to target organ is not lesions observed in cancer. established; significant lack of Can be measured in persons specificity and wide overlap without cancer between smokers and nonsmokers No No NDA Facile assay Lack of specificity No No No Easy to do in blood as surrogate Very nonspecific; relationship tissue. Can be measured in to target organ is not persons without cancer established; predictivity for disease risk not established. Association with cancer in case-control studies may have case bias. Significant lack of specificity and wide overlap between smokers and nonsmokers No No NDA Similar lesions observed in Technically complex; cancer relationship to cancer risk unknown No No NDA Facile assay in blood Relationship to target tissue or blood unknown continues
336 CLEARING THE SMOKE TABLE 11-8 Continued Associated Target Dose- with Tissue Specific Variables Used Response Cessation Assay Chemical to Category in Literature Data or Half-life Available Specificity Tobacco Mutational load NA NDA Yes No No in target genes (p53, K-ras) Mitochondrial Deletions, NDA NDA Yes No No mutations insertions Epigenetic Whole genome NDA NDA Yes No No cancer methylation effects Hypermethylation NDA NDA Yes No No of promoter regions Lipids Blood lipids: Yes NDA Yes Yes No HDL, LDL, oxidized LDL, triglycerides Cardiovascular Heart rate, blood No Yes Yes NA No response pressure Thrombosis Bleeding time No NDA Yes No No Fibrinogen NDA NDA Yes Yes No Prothrombin time, Yes NDA Yes Yes No partial thromboplastin time, plasminogen activator inhibitor, C- reactive protein Urinary Yes No No Yes No thromboxane and prostacyclins
EXPOSURE AND BIOMARKER ASSESSMENT IN HUMANS 337 Related Specific to a Chemical to Disease Specificity Tobacco Riskc Strengths Limitations No No NDA Target gene specificity Very difficult to do in normal tissues No No NDA Provides corroborative marker Relationship to disease not established No No No Facile assay Relationship to disease unknown No No No Similar lesions observed in Technically difficult; cancers relationship to risk unknown Yes No Yes May be directly related to Levels among heavy smokers disease risk cannot be distinguished. Wide interindividual variation. Many individuals under medication therapy. Significant confounders exist NA No Yes Easy to measure; intraindividual Both interindividual and differences may be important intraindividual differences are for the individual significant. Substantial confounders exist, and many persons are on medications No No No Minimally invasive Very nonspecific Yes No NDA Pathogenically related to Does not distinguish levels of disease smoking. Nicotine might separately affect these parameters so limited use in persons using NRT Yes No NDA Leave a fingerprint at the site of their formation Yes No Yes May be markers of platelet- Technically difficult. Wide vascular interactions; reflect overlap of values due to chronic exposure individual differences in response continues
338 CLEARING THE SMOKE TABLE 11-8 Continued Associated Target Dose- with Tissue Specific Variables Used Response Cessation Assay Chemical to Category in Literature Data or Half-life Available Specificity Tobacco Platelet activation Yes NDA Yes No No and survival Blood cell White blood cell Yes Yes Yes Yes No parameters counts (i.e., lymphocytes, neutrophils, total counts) Hematocrit, Yes Yes Yes No No hemoglobin, red blood cell mass Bronchio- Inflammatory Yes Yes Yes No No alveolar cells, protein, lavage cytokines response Neutrophil Yes Yes Yes No No elastase a1- antiprotease complex α1-antitrypsin No No Yes Yes Yes Inflammatory Leukotrienes Yes NDA No Yes No mediators of response Pulmonary FEV1, FVC Yes Yes Yes No No function tests Periodontal Periodontal Yes Yes Yes No No disease height Gum bleeding Yes Yes Yes No No Osteoporosis Fractures Yes NDA NA No No Bone density NDA NDA Yes No No Skin Premature Yes NDA NA No No wrinkling
EXPOSURE AND BIOMARKER ASSESSMENT IN HUMANS 339 Related Specific to a Chemical to Disease Specificity Tobacco Riskc Strengths Limitations No No No Platelet activation in vivo might Technically difficult to use for be pathophysiologically large numbers of subjects. related to cardiac artery Significant number of thrombosis confounding variables. Smoking increases platelet counts Yes No Yes Can be a surrogate marker for Relationship to disease several processes including uncertain, although atherosclerosis and thrombosis alterations in levels are linked epidemiologically to disease. Wide interindividual and intraindividual variation and large number of confounders No No No Can reflect both cardiac and Insensitive; wide interindividual respiratory disease risk differences No No NDA Provides different types of data Bronchoscopy is too invasive for with single procedure large epidemiological studies No No NDA Provides different types of data Bronchoscopy is too invasive for with single procedure large epidemiological studies Yes Yes NDA May be specific to tobacco Requires invasive test; short smoke half-life Yes No NDA May be measured in urine, Substantial number of bronchioalveolar lavage, and confounders serum No No Yes Widely available Low sensitivity for mild disease. Decrease in function with aging. Large interindividual variation No No Yes No No Yes No No Yes Easily measured Numerous confounders No No Yes No No NA Lack of specificity; involves subjective evaluation continues
340 CLEARING THE SMOKE TABLE 11-8 Continued Associated Target Dose- with Tissue Specific Variables Used Response Cessation Assay Chemical to Category in Literature Data or Half-life Available Specificity Tobacco Fetal and Birth weight Yes Yes Yes No No neonatal effects Weight Weight loss and Yes Yes Yes No No gain NOTE: NA=not applicable; NDA=no data available; FVC=Forced vital capacity; FEV1=forced expiratory volume in 1 sec; HDL=high-density lipoprotein; LDL=low-density lipoprotein. sioned for use in developing a molecular fingerprint reflecting a particu- lar exposure, this has not occurred for tobacco carcinogens, and measur- able effects thus far are relatively nonspecific. Nonetheless, a reduction in the level of genetic damage would logically be required if a tobacco- related PREP were to be successful in reducing cancer risk, although how much reduction of genetic damage would be needed to derive a benefit in terms of disease risk is unknown. Several types of assays are available. The main limitation today is that no assays have been shown convinc- ingly to be sufficiently predictive of cancer risk. Chromosomal damage can be measured through classical cytogenetic alterations (Bender et al., 1988; Obe et al., 1982; Ramsey et al., 1995), micronuclei formation (Thorne et al., 1998), COMET (Poli et al., 1999; Speit and Hartmann, 1999), fluores- cent in situ hybridization (FISH) (Pressl et al., 1999; Ramsey et al., 1995; van Diemen et al., 1995), or polymerase chain reaction (PCR) methods assessing loss of heterozygosity (using tandem repeats or comparative genomic hybridization) (Mao et al., 1997), where the latter two methods can be used for morphologically normal-appearing cells. Mutations in reporter genes, such as hypoxanthine phosphoribosyltransferase (HPRT) (Ammenheuser et al., 1997; Hou et al., 1999; Jones et al., 1993) or glycophorin A (GPA), have been used in blood cells, but it is better to
EXPOSURE AND BIOMARKER ASSESSMENT IN HUMANS 341 Related Specific to a Chemical to Disease Specificity Tobacco Riskc Strengths Limitations No No Yes Data collection is easy Nonspecific; numerous confounders No No Yes Both a biomarker for Some people perceive weight metabolism and an important loss as a benefit of smoking, outcome for some people despite significant adverse effects associated with smoking aSelected examples; list is not all-inclusive. bReferences are not provided in this table but can be found in the text of this and disease- related chapters. cAny report related to a disease outcome associated with tobacco where the report is plausible but has not necessarily been replicated. identify mutation rates for cancer genes in biopsies from target organs or in surrogate tissues, and for genes such as p53 (Greenblatt et al., 1994) or KRAS (Lehman et al., 1996; Mills et al., 1995; Scott et al., 1997; Yakubovskaya et al., 1995). Although these assays are available, current technology limits their use in large-scale epidemiological studies. The role of mitochondrial DNA lesions is receiving greater attention for cancer risk (Fliss et al., 2000), and the lesions associated with smoking might be useful (Liu et al., 1997). Among all the assays that have potential application to assessing harm reduction claims, only two studies have assessed prospectively the cancer predictive value of chromosomal aberrations (Bonassi et al., 1995; Hagmar et al., 1994), but they consisted of pooled heterogenous popula- tions and were not focused on tobacco. Further studies are needed to indicate the value of these assays for determining harm reduction. Thus, none of these assays can be used today to allow claims of risk reduction, although in the proper setting they can suggest that such might occur. Biomarkers of pathobiological effect include morphological markers of preneoplastic lesions (e.g., dysplasia), altered phenotypic expression of normal cellular functions (e.g., overexpression of the proto-oncogene Erb- B2), and mutations in cancer-related genes such as the p53 tumor sup- pressor gene. Some of these may be considered preclinical effects that are
342 CLEARING THE SMOKE occurring before diagnosis. The lesions demonstrate a personâs pheno- type for exposure and predisposition that persist following DNA dam- age. Recent advances have made it possible to measure background mu- tations in cancer-associated genes of noncancerous tissues (Aguilar et al., 1994; Mao et al., 1997; Sidransky, 1997), which presumably are related to future cancer risk. The study of mutations in the p53 tumor suppressor gene is uniquely suited for studying cancer etiology, exposure, and susceptibility (Harris and Hollstein, 1993), because p53 is involved in many cellular processes including maintenance of genomic stability, programmed cell death, DNA repair, and others (Attardi and Jacks, 1999; Hollstein et al., 1999; Shimoda et al., 1994; Soussi et al., 2000). The p53 gene, in particular, has a more frequent spectrum of mutations in tobacco-associated lung cancers (Bennett et al., 1999). An interactive effect of alcohol drinking and ciga- rette use in oral cavity and lung cancers leads to different types of p53 mutations (Ahrendt et al., 1999, 2000; Brennan et al., 1995). Interestingly, given that the p53 mutational spectrum for lung cancer is similar world- wide (Hartmann et al., 1997), it is likely that tobacco smoke is the major determinant of lung p53 mutations worldwide. Evidence for a relation- ship of gene-environment interactions and mutation risk in the p53 gene can be found from a Japanese study of CYP1A1 (Kawajiri et al., 1996), where a fivefold increase in risk of p53 mutations was found for smokers with lung cancer and the âat-riskâ genetic variant. This risk increased further for persons who also lacked the glutathione S-transferase (GSTM1) gene. In one study from Norway, smokers with lung cancer who lacked GSTM1 also had more p53 mutations, especially transversions (Ryberg et al., 1994). For oropharyngeal tumors, the frequency of p53 mutations was increased for the same CYP1A1 variant allele (Lazarus et al., 1998). An increased risk for p53 mutations in lung cancer also has been found in Japanese persons with less common variants of CYP2E1 (Oyama et al., 1997). Newly developed technologies allow for the detection of loss of het- erozygosity (LOH) in small amounts of tissue. Losses at chromosome 3p14, 9p21, and 17p13 have been seen in the lungs of both smokers and former smokers, where the first is less frequent in former smokers than current smokers (Mao et al., 1997). An important area that has not been well studied is the effect of tobacco toxicants on the induction of enzymes that might affect cancer risk. For example, cytochrome P-450 enzymes are induced with tobacco smoking (e.g., arylhydrocarbon hydroxylases [AHHs]) (Bartsch et al., 1995; Guengerich, 2000; McLemore et al., 1990; Nakajima et al., 1991, 1995; Rojas et al., 1992). Induction is related to greater amounts of DNA damage (Bartsch et al., 1991; Geneste et al., 1991). It remains to be tested whether a
EXPOSURE AND BIOMARKER ASSESSMENT IN HUMANS 343 tobacco-related PREP can reduce AHH exposure so that other carcino- genic exposures will be less harmful. Many proteins are induced in rela- tion to DNA damage (e.g., p53) (Bjelogrlic et al., 1994). Whether higher levels of these proteins increase or decrease the risk of disease remains unknown. Several biomarkers can be studied in relation to cardiovascular dis- ease risk, but none of these are specific to tobacco smoking, such as blood lipid level (Cullen et al., 1997; Freeman et al., 1993; Hellerstein et al., 1994; Ludviksdottir et al., 1999; Stubbe et al., 1982; Wald et al., 1989), which changes with cessation (Green and Harari, 1995), or urinary excretion of thromboxane A2 metabolites (Nowak et al., 1987; Lassila et al., 1988; Rangemark et al., 1992; Wennmalm et al., 1991). F2-Isoprostanes in blood have a dose-response relationship to smoking (Morrow et al., 1995). To- bacco smoking is associated with decreased weight (Green and Harari, 1995) and therefore modifies the relationship of weight gain to increased risk of heart disease (Fulton and Shekelle, 1997). Blood pressure has been studied but is not clearly associated with smoking (Green and Harari, 1995). Other biomarkers that have been suggested to reflect an increased cardiac disease risk include reduced platelet survival (Fuster et al., 1981). Newer imaging methods such as electron-beam computed tomography (OâMalley et al., 2000; Raggi et al., 2000) are being used to assess heart disease risk, and these methods might be used to assess the decreasing rate of formation of atherosclerosis or calcium when using a PREP. Biomarkers of developing respiratory illness have been assessed in different ways, and several studies have specifically assessed the effects of smoking reduction separately from cessation. Symptoms, albeit late effects, such as cough, chronic phlegm production, wheezing, and short- ness of breath have been used and improve with smoking cessation (Buist et al., 1976; Kanner et al., 1999). Reducing smoking, without quitting, also is associated with a reduction in symptoms (Buist et al., 1976). There are many studies that explore decrements of pulmonary function related to cigarette smoking. While such decrements occur with aging independent of smoking, further decrements are induced by smoking (Lange et al., 1989; McCarthy et al., 1976). Declines in the forced expiratory volume at 1 second (FEV1) are associated with increased disease and mortality, in- cluding nonpulmonary diseases (James et al., 1999). The decline in pul- monary function tests slows with complete cessation (Buist et al., 1976; Kanner et al., 1999; Lange et al., 1989; McCarthy et al., 1976) and with greater than 25% reduction in the number of cigarettes smoked per day (Buist et al., 1976; Lange et al., 1989; McCarthy et al., 1976). Smoking reduction in the elderly apparently showed no effect in slowing the rate of decline (Lange et al., 1989). Bronchioalveolar lavage has been used, although it is invasive, and different types of assays can assess inflamma-
344 CLEARING THE SMOKE tion, neutrophil elastase α1-antiprotease complex, and α1-antitrypsin (Rennard et al., 1990). Induction of these components reverses with smok- ing reduction (Rennard et al., 1990), and some markers such as alveolar neutrophils, neutrophil elastase α1-antiprotease complexes, and alveolar macrophages decrease in smokers who reduce their amount of smoking when provided with nicotine replacement therapy (Rennard et al., 1990). Several nonspecific biomarkers of effect are related to smoking, such as leukocyte count (Parry et al., 1997; Phillips et al., 1992; Sunyer et al., 1996; Wald et al., 1989), which reverses with cessation (Green and Harari, 1995; Sunyer et al., 1996) and then increases again with resumption of smoking (Sunyer et al., 1996). Levels remain increased to some extent in former smokers compared to never smokers (Parry et al., 1997). Whether these findings, however, are independent predictors of disease risk has had limited study (e.g., mortality) (James et al., 1999), and the differences that can be found may be due to disease unrelated to smoking (Wald et al., 1989). Some of these parameters are covariates (James et al., 1999). Thus, such markers would be less useful for assessing harm reduction claims but might be useful for assessing exposure reduction claims. There are several short-term effects on the body that can be consid- ered both from the perspective of disease and as a biomarker of effect. Examples include periodontal disease, abnormal glucose tolerance tests, and decreased birthweight of infants born to mothers who smoke. Also, changes in adult body weight can be measured in the context of harm reduction. It is well known that smoking increases metabolism and de- creases appetite, while stopping smoking is associated with weight gain (OâHara et al., 1998). This can be a very important marker of smoking effects since the consideration of weight is often a factor in personsâ begin- ning smoking or resisting cessation. HOST SUSCEPTIBILITY Host susceptibility could modify the risk of tobacco-related disease and, therefore, the effects of PREPs. Host susceptibility can be influenced by genetic susceptibility, age, gender, ethnicity, health status, and so forth. These will not be discussed in detail except for genetic susceptibility, but any relevant potential modifying factor should be considered in the as- sessment of a PREP. The study of genetic susceptibilities can improve the accuracy of esti- mates of disease associations (Khoury and Wagener, 1995). Tobacco toxi- cants affect people to variable degrees. It is therefore reasonable to as- sume that harm reduction strategies would affect people differently. There is large interindividual variation in cellular responsesâfor example, in metabolism and detoxification of toxicants and DNA repair. As other
EXPOSURE AND BIOMARKER ASSESSMENT IN HUMANS 345 cellular responses to DNA damage are identified (e.g., cell-cycle delays, heat shock), interindividual variation in risk is likely to be discovered for these as well. Interindividual effects in cellular responses could be due to genetically determined enzyme expression, kinetics, or stability. Also, in- duction of enzymes from previous exposures or comorbidity also may contribute to cancer risk, and induction has a genetic component. Susceptibility to disease from genetic variability can range from small to large, depending on the genetic penetrance. Highly-penetrant cancer susceptibility genes cause familial cancers but account for less than 1% of all cancers (Fearon, 1997). Low-penetrant genes cause common sporadic cancers and can have great public health consequences (Shields and Har- ris, 2000). Genetic susceptibility can be assessed either phenotypically (measur- ing the resultant enzymatic function) or genotypically (determining the genetic code). Examples are provided in Table 11-9. Phenotypic assays may include determining enzymatic activity by administering probe TABLE 11-9 Assays for Assessing Effect Modification by Heritable Traits Assay Type Example Used in Literature Strengths Limitations Gene-based Genetic polymorph- Inexpensive, simple Functional relation- assays isms for carcinogen to perform, specific ship of genotype to metabolism and gene effect when phenotype difficult induction or DNA exists, high to prove; disease repair, smoking throughput risk for low- behavior available penetrant genes difficult to prove Phenotypic Mutagen sensitivity Reflects integrated Difficult to perform; assays for DNA repair; measure of multiple relationship to host-reactivation genotypes; provides disease risk assay for DNA complex data technically difficult repair; CYP450 potentially usable to prove; requires metabolism and for rapid identifica- extensive labora- induction studies; tion of important tory validation; RNA expression of risk factors RNA and protein specific genes; microarray assays microarray RNA are expensive; expression; large-scale studies proteomics are needed; bioinformatics not available
346 CLEARING THE SMOKE drugs to individuals and measuring blood levels or urinary metabolites, assessing carcinogen metabolic capacity in cultured lymphocytes, or es- tablishing the ratios of endogenously produced substances such as estro- gen metabolite ratios. One extensively studied phenotype in relation to smoking risk is AHH activity (Kellermann et al., 1973; Kouri et al., 1982). In general, it is preferable to use a gene-based assay to assess disease risk because DNA is easier to obtain and the assays are technically simpler. However, phenotypes usually represent a multigenic trait, which may not be adequately characterized by only one genetic assay. Therefore, there is a role for both gene- and phenotype-based assays in research studies and PREP assessments. Examples of frequently studied genetic polymorphisms in tobacco-related cancers that have been shown in some studies to modify smoking-related disease risk include the N- acetyltransferase 2 (NAT2) (Brockmoller et al., 1996, 1998; Henning et al., 1999), glutathione S-transferase M1 (GSTM1) (Bell et al., 1993; Brockmoller et al., 1996, 1998; Cullen et al., 1997; Jourenkova et al., 1998; Jourenkova- Mironova et al., 1999; Rebbeck, 1997), cytochrome P-450 1A1 (CYP1A1) genes (Bishop, 1987; Ishibe et al., 1997), glutathione S-transferase Pi (Ryberg et al., 1997), and others (Jourenkova-Mironova et al., 1999; Rosvold et al., 1995; Wiencke et al., 1997). These and other genetic polymorphisms are believed to affect levels of biomarkers, such as DNA adducts (Kato et al., 1995; Pastorelli et al., 1998; Ryberg et al., 1997; Yu et al., 1995). In the general population, DNA repair capacity decreases in humans with aging (Liu et al., 1994; Wei et al., 1993), which would make this an acquired risk factor for cancer and might explain a portion of the in- creased cancer risk in the elderly (Simpson, 1997). Both genotyping and phenotyping assays for DNA repair or cell-cycle control that affects DNA repair might be useful in identifying individuals who might benefit from harm reduction strategies. Tobacco toxicants can affect DNA repair (Grafstrom et al., 1994), so that the effects of both tobacco toxicants and heritable capacity on DNA repair can be considered in assessing harm reduction products. It should be noted that cigarette smoking induces levels of some repair enzymes (Drin et al., 1994; Hall et al., 1993; Slupphaug et al., 1992), so caution must be used for some phenotyping assays. Inherited susceptibilities via specific genetic polymorphisms that af- fect the efficiency of DNA repair (e.g., for base excision repair) have been identified recently (Mohrenweiser and Jones, 1998). Studies now being completed indicate an effect of these genetic variants on tobacco-related cancer risk (Sumida et al., 1998), some of which have functional effects on DNA repair (Lunn et al., 1999, 2000). A nonspecific DNA repair assay, which measures chromosomal aberrations in human cultured lympho- cytes after an in vitro challenge with a mutagen, has shown initial prom- ise. In this case, an increased mutagen-related aberration rate has been
EXPOSURE AND BIOMARKER ASSESSMENT IN HUMANS 347 observed in persons with primary and secondary upper aerodigestive tract cancers (Cloos et al., 1996), multiple primary cancers (Cloos et al., 1994), and lung cancer (Li et al., 1996; Spitz et al., 1995; Wei et al., 1996). Genetic susceptibilities for genes other than those involved in car- cinogen metabolism and DNA repair are also being investigated (Jin et al., 1995; Sjalander et al., 1996). There has been less study of genetic sus- ceptibilities for coronary artery disease (Gealy et al, 1999). It is likely that these genes also will play a role in modifying disease risk (see Chapter 13). GENETIC PREDISPOSITIONS TO SMOKING ADDICTION The greatest contributors to smoking addiction are the availability of tobacco and cultural acceptance of tobacco smoking. Genetics plays a lesser role. The tobacco smoking epidemic has occurred only over the last 50 to 70 years, and it is unlikely that human genetics have evolved in that amount of time. Nonetheless, twin studies indicate a genetic role for both smoking initiation and smoking persistence (Carmelli et al., 1992; Heath et al., 1993a, b). People smoke in ways that will maintain a desired blood nicotine level. Nicotine in turn stimulates reward mechanisms in the brain. Presynaptic nicotinic acetylcholine receptors stimulate the secretion of dopamine into neuronal synapses. There also are effects on other pathways, such as those that involve serotonin. For dopamine, synaptic dopamine stimulates dopamine receptors; five subtypes have been identified, which are con- sidered to be D1- or D2-like. Synaptic dopamine levels are governed by presynaptic release and the presynaptic dopamine transporter protein. In humans, there are different types of data supporting the link between nicotine and dopamine. Nicotine self-administration through tobacco smoking may reduce the adverse consequences of Parkinsonâs disease, attention deficit disorder, and schizophrenia (Bannon et al., 1995; Olincy et al., 1997; Seeman, 1995), diseases thought to be related to dopamine abnormalities. Also, smoking probably relieves depression (Gilbert and Gilbert, 1995), and the dopamine transporter inhibitor antidepressants (e.g., bupropion SR) are now used to treat nicotine addiction (Hurt et al., 1997; Jorenby et al., 1999). The genes that code for dopamine receptors (e.g., DRD2, DRD4), dopamine transporter reuptake (SL6A3), and dopamine synthesis (e.g., dopamine hydroxylase, tyrosine hydroxylase, tryptophan hydroxylase, catechol-O-methyltransferase, monoamine oxidase) are polymorphic. Some of the polymorphisms result in altered protein function. Persons with higher levels of synaptic dopamine, or âmore stimulationâ of dopamine receptors may have less rewarding effects of nicotine and so would be
348 CLEARING THE SMOKE less likely to become smokers and would more easily quit. For example, in a study of 500 smokers and nonsmokers, several candidate genes have been implicated (Lerman et al., 1998, 1999; Shields et al., 1998), whereas other studies of candidate genes have yielded null results (Lerman et al., 1997). Other investigators also have reported supporting evidence (Comings et al., 1996; Noble et al., 1994; Spitz et al., 1998). Thus, it is likely that there is a genetic contribution to smoking addiction and behavior and there may also be a genetic influence on who benefits from PREPs. BIOMARKER ASSESSMENT FOR ENVIRONMENTAL TOBACCO SMOKE EXPOSURE Biomarker assessments in persons exposed to environmental tobacco smoke are problematic because exposures occur at much lower levels than in smokers, and therefore the level of detection is limiting (Benowitz, 1999). The most consistently used biomarkers are those that reflect expo- sures, namely cotinine (serum, plasma, or urine), rather than biologically effective doses or biomarkers of effect. Such biomarkers, for example, can show that adolescents are exposed to tobacco smoke through household smoking (Bono et al., 1996). Urinary metabolites of tobacco-specific nitrosamines also have been found in persons exposed passively to smoke (Atawodi et al, 1998; Hecht et al., 1993; Parsons et al., 1998). DNA adducts in the lung are also detected in persons who are thought to be nonsmokers (Kato et al., 1995). Children exposed to modest levels of ETS have been found to have increased concentrates of 4-aminobiohenyl adducts of PAH- albumin adducts (Tang et al., 1999). Although it may follow that proven methods to reduce harm in smokers would apply to nonsmokers with passive exposure, there are circumstances in which passive smoke expo- sure might be substantial (e.g., cigar smoking). DEVELOPMENT AND VALIDATION OF BIOMARKER ASSAYS, INCLUDING QUALITY CONTROL The use of biomarkers in assessing harm reduction can be helpful only when the assays have undergone rigorous development and valida- tion. Reliance on insufficiently validated biomarkers becomes problem- atic because they are of uncertain value and so should not be used to support a claim of exposure or risk reduction. The design and develop- ment of a biomarker assay must conform to the original goalsâthat is, the assay should have sufficient specificity, it should be quantitatively repro- ducible in humans at the levels that occur when exposure reduction is achieved, and other assays should be available to corroborate the qualita- tive and quantitative results. Many pitfalls have already been found in
EXPOSURE AND BIOMARKER ASSESSMENT IN HUMANS 349 biomarker development. There are examples of biomarker assays that are more difficult to perform at levels observed in humans compared to the use of higher-level laboratory chemical standards (e.g., immunoassays) (Santella et al., 1988). Some methodologies can artifactually affect assay results (e.g., introduction of oxidative damage) (Farmer and Shuker, 1999). In some cases, measurements of in vivo formation can be skewed by exogenous exposure to the biomarker (e.g., dietary ingestion of 3- alkyladenine) (Prevost and Shuker, 1996). Validation of a biomarker assay includes a determination of repli- cability (e.g., coefficient of variation), interobserver and interlaboratory variability, intraindividual variation, and interindividual variation. These validation steps must be done using known controls that simulate human exposure levels and harm. Thus, the assay should be validated in light and heavy smokers, former smokers, and never smokers. Caution must be used in interpreting assay results in the context of certain study de- signs. For example, the reliability of biomarkers thought to be related to disease risk in case-control studies is problematic for markers that might be affected by disease status (differential case bias) (Wald et al., 1989). Research laboratories providing data that can impact individual or public health should have adequate quality control and quality assurance procedures in place. The definition of adequate will depend on the popu- lation under study and the number of subjects. In clinical pathology labo- ratories, standards and protocols have been established by organizations such as the College of American Pathologists and the National Committee for Clinical Laboratory Standards. In a research laboratory that performs biomarkers studies assessing PREPs, there should be standards for profi- ciency testing, quality improvement, quality control, use of standards, methods for interpretation, specimen handling, specimen labeling, speci- men processing, and reporting of results. There also should be criteria for facility and equipment maintenance. CONCLUSIONS The assessment of a PREP will have to consider external exposure and markers of internal exposure, estimates of the biologically effective dose, and biomarkers of potential harm. A risk reduction claim should be based on disease reduction, but time limitations mandate the use of biomarkers for both exposure and risk reduction assessments. Measure- ments of the number of cigarettes per day, smoking duration, estimated lifetime exposure, smoking topography, and so forth, provide an effective indicator of exposure that has been associated with risk. However, these measures may be insensitive to small changes in risk, are difficult to as- sess accurately over time, and have not been tested in the context of harm
350 CLEARING THE SMOKE reduction. Also, because there is interindividual variation in how the body responds to these exposures, such measures might not be suffi- ciently accurate for new products intended to decrease exposure. The relationship of external exposure markers to disease risk might be less predictable for new products. Currently, there is sufficient evidence to show that biomarkers can provide better estimates of risk in the context of exposure, and therefore they will likely be able to provide improved as- sessments for harm reduction products. However, no single biomarker has been sufficiently validated and related to disease risk to be recom- mended as an intermediate biomarker of cancer risk. Thus, different types of biomarkers along the pathway from internal exposure to biologically effective dose, and to potential harm are needed, and additional research is necessary to identify the best combination of markers to be used. Ex- perimental toxicity testing (in vitro and animal models) are not sufficient to support a tobacco-related PREP claim because only biomarkers can show that the PREP reduces exposure adequately enough to imply risk reduction. However, the use of intermediate biomarkers as surrogate risk factors for disease may overestimate the number of persons who actually develop disease because not all early changes in morphology or function progress to disease. On the other hand, it may underestimate if, as ex- pected, other mechanisms are involved in the disease process that are not reflected by the biomarkers. Therefore, the implication of a potential ben- efit in a harm reduction strategy could also be an overestimate, but this limitation in the scientific methodology for identifying sufficiently spe- cific biomarkers of risk requires acceptance at the current time. Previously, the most common way in which exposure reduction has been inferred is through the use of methods that simulate human smok- ing behavior, such as the FTC method. Although they provide a standard- ized way to assess cigarettes, it is clear that these methods have limited usefulness because people smoke their cigarettes differently than the ma- chine, with resultant differences in the types and amounts of exposure. The use of biomarkers improves exposure assessments (e.g., charac- terizing low-dose exposures or low-risk populations), provides a relative contribution of individual chemical carcinogens from complex mixtures (e.g., TSNAs and PAHs in cigarette smoke), and estimates total burden of a particular exposure where there are numerous sources (e.g., BaP from air, tobacco, diet, and occupation) (Vineis and Porta, 1996). Biomarkers also can establish differences in individual susceptibilities and whether there are differences in response depending on dose. Thus, biomarkers that measure both complex exposures and single tobacco product con- stituents are needed and should be assessed for the range of possible human exposures and those that assess complex exposures should carry a greater weight. Also, some biomarkers should be used that are less spe-
EXPOSURE AND BIOMARKER ASSESSMENT IN HUMANS 351 cific for individual tobacco constituents in order to monitor for the intro- duction of new hazards from tobacco-related PREPs. Today, there remain technical limitations to the use of biomarkers. Depending on the harmful effect, surrogate assays in nontarget fluids or organs that represent effects in target organs may be easier to perform in humans because the target tissues might not be easily accessible. How- ever, if such is the case, the relevance of the surrogate biomarker to the effect in the target organ should be demonstrated. The use of a biomarker for harm reduction assessment should include several considerations, including where it is along the pathway from ex- posure to disease, its specificity and sensitivity, available harm dose- response data, available reduction in harm dose-response data, target tissue effect, and how it is validated. The need for validation cannot be overemphasized. Each biomarker should be validated for its relationship to exposure and harm and also as a laboratory assay that provides reliable and reproducible data. Separately, the way a biomarker is affected by interindividual variation in response and by behavior should also be con- sidered. Assessment of harm and harm reduction should be made through direct human experience, as the products are used by the general popula- tion. Most of what is known about harmful tobacco products has resulted from epidemiology, supported by in vitro studies, laboratory animal stud- ies, and human experiments. However, while epidemiological studies can provide the most definitive data about tobacco harm and harm reduction products, the study of diseases with long latency (e.g., cancer, heart dis- ease, chronic obstructive pulmonary disease) is problematic because such studies require many years before they provide useful data. Thus, be- cause definitive evidence for a new risk reduction product is not available short-term markers that reflect long-term outcomes are needed. If an ap- proach for assessing risk reduction products required only epidemiologi- cal data measuring disease outcome prior to use by the public, then an opportunity to reduce morbidity and early mortality might be missed. However, the use of intermediate markers does not replace long-term follow-up and epidemiological surveillance, but allows judgments to be made until such data are forthcoming. Biomarkers of internal exposure, biologically effective dose, or poten- tial harm have been validated to different degrees. It is typically easier to show a direct relationship of external exposure to biomarkers in the fol- lowing order: internal exposure, biologically effective dose, and potential harm. Conversely, it is typically easier to show a direct relationship of disease outcome to biomarkers in the following order: potential harm, biologically effective dose, and internal exposure. It might be acceptable to rely on external exposure measurements for considering risk and dose-
352 CLEARING THE SMOKE response, but only with substantial corroborative biomarker data. The best strategy for assessing the claims for risk reduction methods is with several markers that range from exposure to outcome, one being linked to another, and at least one with which a dose-response risk assessment can be made. The recommendation that harm reduction products should be as- sessed with the use of biomarkers reflects sufficient available data to show that the public is composed of individuals with different cultural and heritable traits that affect how people use tobacco products and respond to them. To achieve the best confidence that a PREP will reduce risks for persons who cannot stop smoking, both well-validated methods for pre- dicting risk, including external exposure indicators, and the best available biomarker assays should be used. RESEARCH AGENDA There are currently different methodologies for assessing PREPs, but substantial research is needed to increase confidence in the application of these methods. Although it may be possible to improve external methods for assessing exposures, such as through modification of the FTC method or improving questionnaire assessments, there is so much variability in human smoking behavior that it is believed these methods could never be much more helpful than they already are. This recommendation does not imply that questionnaires and topography instruments are not helpful in assessing smoking behavior, because they are, but it is unlikely that the methodology can be improved substantially. Indeed, clinical epidemio- logical studies generally have to integrate more variables for smoking behavior (e.g., accurately documenting changes in smoking, brand switch- ing). The development and validation of biomarkers for assessing harm reduction must be accelerated for all diseases, especially for cardiovascu- lar and respiratory diseases because less research has been conducted compared to cancer. The use of a biomarker for assessing harm reduction should be con- sidered using the criteria provided in this chapter. Dose-response rela- tionships should be established, and the biomarker should be assessed for reversibility in smoking cessation trials. In all studies of biomarker vali- dation, consideration should be made of what nontobacco exposures, if any, would influence the biomarker study results. Also, biomarkers have to be tested and validated in different populations, to determine whether they are affected by susceptible subpopulations, and within genders, races, or ethnicities. Research efforts should focus on biomarkers that
EXPOSURE AND BIOMARKER ASSESSMENT IN HUMANS 353 might be used for existing cohort studies, where disease outcome already is known. For example, markers are needed that can be used in serum or small amounts of DNA from stored samples. This is the best way to identify a relationship between exposure, a biomarker, and disease risk. Substantial research is needed to identify the relationships between biomarkers to exposure, biologically effective doses, and biomarkers of harm. Study designs that can provide these linkages are needed, and the best evidence will come from cohort studies. Internal biomarkers of exposure such as cotinine, nicotine boosts, CO, and CO boosts provide good information about exposure, including to environmental tobacco smoke, but additional markers, such as urinary anabasine and anatabine levels, have to be developed for use in persons who are concurrently using nicotine replacement therapy. Increased efforts to measure urinary excretion of carcinogen metabolites, which are currently showing promise for use in risk assessment of active smoking and ETS, are needed. Examples include urinary excretion of tobacco- specific nitrosamines and polycyclic aromatic hydrocarbons and urinary mutagenicity, where these reflect both single and complex markers of exposure, respectively. Also, markers with longer half-lives would be useful to avoid confounding by recent changes in smoking behavior. Biomarkers that reflect the biologically effective doses of exposure to carcinogens must be improved and validated. Newer technologies are now available that are more sensitive (e.g., mass spectroscopy) and can provide more information, and these should be applied in experimental systems and human studies that were developed long before such meth- ods were available. For example, the determination of carcinogen-DNA adducts might be useful where small amounts of tissue are available (e.g., buccal swabs, sputum, blood). More biomarkers of potential harm are currently being developed than any other types. This is because pathobiological pathways are well understood and newer technologies are available to explore them. How- ever, along with better technologies will come limitations in the interpre- tation of new data (e.g., mRNA expression assays, proteomics). As re- searchers explore greater numbers of gene-smoking interactions and accumulate data for numerous genes expressed in response to exposures, it is clear that there are insufficient methods to analyze data where there are a substantial number of predictor variables. Also, some data will have to be reduced to clusters or other smaller units that are understandable in the context of biological hypotheses. Increased research is needed in methodologies to interpret these types of data, to validate the new models in the context of disease outcome. For cancer, increased efforts are needed to assess target organ assays, such as genetic damage in lung cells in sputum and exfoliated bladder
354 CLEARING THE SMOKE cells in urine, in persons years before they have a clinically detectable cancer. Given that genetic damage is only one part of the carcinogenic process, additional efforts are necessary to develop biomarkers for other pathways, such as gene silencing through hypermethylation of promoter regions. For cardiac disease, additional studies are needed to validate biomarkers of platelet function, endothelial function, endothelial thicken- ing, and plaque formation and thrombosis. For respiratory disease, better markers are needed to assess changes in lung function that predict chronic obstructive pulmonary disease and asthma, and to assess immunological changes that will increase risk of respiratory infections. It would be optimal to identify biomarkers that can be used to assess risk for several diseases. For example, biomarkers of oxidative damage might identify risk for cardiac disease, cancer, and respiratory illness. However, because the relationship of oxidative damage to these diseases remains mostly an unproved hypothesis, research is needed in this area. Biomarkers will have to assess PREPs for single tobacco constituents and complex mixtures. The use of biomarkers that can assess multiple exposures from complex mixtures is critical because new tobacco-related PREPs might include compounds that are not present in existing tobacco constituents, or the ratio of exposures to individual constituents might change. A committee of experts should be convened to consider and iden- tify those biomarkers that have the most promise and to determine what combination of biomarkers should be part of a panel for assessing PREPs. To identify those biomarkers most useful for assessing harm reduc- tion products, current efforts have to be focused on clinical trials that assess the effects of switching brands, using new products, and reducing daily consumption of tobacco through the concomitant use of nicotine replace therapy or other aids used for smoking cessation. There are unique opportunities in epidemiological studies to validate biomarkers for use in assessing harm reduction strategies. Specifically, cohorts of participants in smoking cessation programs and former smokers should be established because these individuals represent the best possible reduction in the risk due to smoking. The collection of tissues and fluids from persons who have quit smoking and comparisons of persons who do and do not develop disease would be very helpful in determining which biomarkers have the most predictive value. This should be done in the context of previous smoking history to identify which persons would obtain the greatest benefit from cessation and how biomarkers might be able to identify individuals at greatest risk within these groups. Some diseases, such as cardiovascular disease, have a rela- tively rapid decline in risk following cessation so it would be quicker to validate cardiac disease risk factors. For cancer, the studies will take much longer. Monitoring populations that are at the highest risk of cancer, such
EXPOSURE AND BIOMARKER ASSESSMENT IN HUMANS 355 as persons with resected early-stage lung cancer or bladder cancer, might be useful in this context. If a biomarker cannot predict increased risk in former smokers, it is unlikely to be useful in assessing PREPs. REFERENCES Aguilar F, Harris CC, Sun T, Hollstein M, Cerutti P. 1994. Geographic variation of p53 mutational profile in nonmalignant human liver. Science 264(5163):1317-1319. Ahrendt SA, Chow JT, Yang SC, et al. 2000. Alcohol consumption and cigarette smoking increase the frequency of p53 mutations in non-small cell lung cancer. Cancer Res 60(12):3155-3159. Ahrendt SA, Halachmi S, Chow JT, et al. 1999. Rapid p53 sequence analysis in primary lung cancer using an oligonucleotide probe array. Proc Natl Acad Sci U S A 96(13):7382-7387. Ammenheuser MM, Hastings DA, Whorton EB, Ward JB. 1997. Frequencies of hprt mutant lymphocytes in smokers, non-smokers, and former smokers. Environ Mol Mutagen 30(2):131-138. Ashurst SW, Cohen GM, Nesnow S, DiGiovanni J, Slaga TJ. 1983. Formation of benzo(a)- pyrene/DNA adducts and their relationship to tumor initiation in mouse epidermis. Cancer Res 43(3):1024-1029. Atawodi SE, Lea S, Nyberg F, et al. 1998. 4-Hydroxy-1-(3-pyridyl)-1-butanone-hemoglobin adducts as biomarkers of exposure to tobacco smoke: validation of a method to be used in multicenter studies. Cancer Epidemiol Biomarkers Prev 7(9):817-821. Attardi LD, Jacks T. 1999. The role of p53 in tumour suppression: lessons from mouse models. Cell Mol Life Sci 55(1):48-63. Badawi AF, Hirvonen A, Bell DA, Lang NP, Kadlubar FF. 1995. Role of aromatic amine acetyltransferases, NAT1 and NAT2, in carcinogen-DNA adduct formation in the hu- man urinary bladder. Cancer Res 55(22):5230-5237. Bannon MJ, Granneman JG, Kapatos G. 1995. The dopamine transporter. Bloom FE, Kupfer DL, ed. Psychopharmacology: The 4th Generation of Progress. New York: Raven Press. Pp. 179-188. Bartsch H, Petruzzelli S, De Flora S, et al. 1991. Carcinogen metabolism and DNA adducts in human lung tissues as affected by tobacco smoking or metabolic phenotype: a case- control study on lung cancer patients. Mutat Res 250(1-2):103-114. Bartsch H, Rojas M, Alexandrov K, et al. 1995. Metabolic polymorphism affecting DNA binding and excretion of carcinogens in humans. Pharmacogenetics 5 Spec No:S84-90. Battig K, Buzzi R, Nil R. 1982. Smoke yield of cigarettes and puffing behavior in men and women. Psychopharmacology (Berl) 76(2):139-148. Bell DA, Taylor JA, Paulson DF, Robertson CN, Mohler JL, Lucier GW. 1993. Genetic risk and carcinogen exposure: a common inherited defect of the carcinogen-metabolism gene glutathione S-transferase M1 (GSTM1) that increases susceptibility to bladder cancer. J Natl Cancer Inst 85(14):1159-1164. Bender MA, Awa AA, Brooks AL, et al. 1988. Current status of cytogenetic procedures to detect and quantify previous exposures to radiation. Mutat Res 196(2):103-159. Bennett WP, Hussain SP, Vahakangas KH, Khan MA, Shields PG, Harris CC. 1999. Molecu- lar epidemiology of human cancer risk: gene-environment interactions and p53 muta- tion spectrum in human lung cancer. J Pathol 187(1):8-18. Benowitz NL. 1999. Biomarkers of environmental tobacco smoke exposure. Environ Health Perspect 107 Suppl 2:349-355. Benowitz NL, Hall SM, Herning RI, Jacob P 3d, Jones RT, Osman AL. 1983. Smokers of low- yield cigarettes do not consume less nicotine. N Engl J Med 309(3):139-142.
356 CLEARING THE SMOKE Benowitz NL, Jacob P 3d, Kozlowski LT, Yu L. 1986a. Influence of smoking fewer cigarettes on exposure to tar, nicotine, and carbon monoxide. N Engl J Med 315(21):1310-1313. Benowitz NL, Jacob P 3d, Yu L, Talcott R, Hall S, Jones RT. 1986b. Reduced tar, nicotine, and carbon monoxide exposure while smoking ultralow- but not low-yield cigarettes. JAMA 256(2):241-246. Benowitz NL, Zevin S, Jacob P 3rd. 1998. Suppression of nicotine intake during ad libitum cigarette smoking by high-dose transdermal nicotine. J Pharmacol Exp Ther 287(3):958- 962. Bishop JM. 1987. The molecular genetics of cancer. Science 235:305-311. Bjelogrlic NM, Makinen M, Stenback F, Vahakangas K. 1994. Benzo[a]pyrene-7,8-diol-9,10- epoxide-DNA adducts and increased p53 protein in mouse skin. Carcinogenesis 15(4):771-774. Bonassi S, Abbondandolo A, Camurri L, et al. 1995. Are chromosome aberrations in circu- lating lymphocytes predictive of future cancer onset in humans? Preliminary results of an Italian cohort study. Cancer Genet Cytogenet 79(2):133-135. Bono R, Russo R, Arossa W, Scursatone E, Gilli G. 1996. Involuntary exposure to tobacco smoke in adolescents: urinary cotinine and environmental factors. Arch Environ Health 51(2):127-131. Brennan JA, Boyle JO, Koch WM, et al. 1995. Association between cigarette smoking and mutation of the p53 gene in squamous-cell carcinoma of the head and neck. N Engl J Med 332(11):712-717. Bridges RB, Combs JG, Humble JW, Turbek JA, Rehm SR, Haley NJ. 1990. Puffing topogra- phy as a determinant of smoke exposure. Pharmacol Biochem Behav 37(1):29-39. Bridges RB, Humble JW, Turbek JA, Rehm SR. 1986. Smoking history, cigarette yield and smoking behavior as determinants of smoke exposure. Eur J Respir Dis Suppl 146:129- 137. Brockmoller J, Cascorbi I, Kerb R, Roots I. 1996. Combined analysis of inherited polymor- phisms in arylamine N-acetyltransferase 2, glutathione S-transferases M1 and T1, mi- crosomal epoxide hydrolase, and cytochrome P450 enzymes as modulators of bladder cancer risk. Cancer Res 56(17):3915-3925. Brockmoller J, Cascorbi I, Kerb R, Sachse C, Roots I. 1998. Polymorphisms in xenobiotic conjugation and disease predisposition. Toxicol Lett 102-103:173-183. Bryant MS, Skipper PL, Tannenbaum SR, Maclure M. 1987. Hemoglobin adducts of 4- aminobiphenyl in smokers and nonsmokers. Cancer Res 47(2):602-608. Bryant MS, Vineis P, Skipper PL, Tannenbaum SR. 1988. Hemoglobin adducts of aromatic amines: associations with smoking status and type of tobacco. Proc Natl Acad Sci U S A 85(24):9788-9791. Buist AS, Sexton GJ, Nagy JM, Ross BB. 1976. The effect of smoking cessation and modifica- tion on lung function. Am Rev Respir Dis 114(1):115-122. Byrd GD, Davis RA, Caldwell WS, Robinson JH, deBethizy JD. 1998. A further study of FTC yield and nicotine absorption in smokers. Psychopharmacology (Berl) 139(4):291-299. Byrd GD, Robinson JH, Caldwell WS, deBethizy JD. 1995. Comparison of measured and FTC-predicted nicotine uptake in smokers. Psychopharmacology (Berl) 122(2):95-103. Carmella SG, Akerkar SA, Richie JP, Hecht SS. 1995. Intraindividual and interindividual differences in metabolites of the tobacco-specific lung carcinogen 4-(methylnitros- amino)-1-(3-pyridyl)-1-butanone (NNK) in smokersâ urine. Cancer Epidemiol Biomarkers Prev 4(6):635-642. Carmella SG, Kagan SS, Kagan M, et al. 1990. Mass spectrometric analysis of tobacco-spe- cific nitrosamine hemoglobin adducts in snuff dippers, smokers, and nonsmokers. Cancer Res 50(17):5438-5445. Carmelli D, Swan GE, Robinette D, Fabsitz R. 1992. Genetic influence on smokingâa study of male twins. N Engl J Med 327(12):829-833.
EXPOSURE AND BIOMARKER ASSESSMENT IN HUMANS 357 CDC (Centers for Disease Control and Prevention). 2000. National Health and Nutrition Examination Survey. [Online]. Available: http://www.cdc.gov/nchs/nhanes.htm [accessed 2001]. Cloos J, Braakhuis BJ, Steen I, et al. 1994. Increased mutagen sensitivity in head-and-neck squamous-cell carcinoma patients, particularly those with multiple primary tumors. Int J Cancer 56(6):816-819. Cloos J, Spitz MR, Schantz SP, et al. 1996. Genetic susceptibility to head and neck squamous cell carcinoma. J Natl Cancer Inst 88(8):530-535. Comings DE, Ferry L, Bradshaw-Robinson S, Burchette R, Chiu C, Muhleman D. 1996. The dopamine D2 receptor (DRD2) gene: a genetic risk factor in smoking. Pharmacogenetics 6(1):73-79. Committee on Biological Markers of the National Research Council. 1987. Biological mark- ers in environmental health research. Environ Health Perspect 74:3-9. Crawford FG, Mayer J, Santella RM, et al. 1994. Biomarkers of environmental tobacco smoke in preschool children and their mothers. J Natl Cancer Inst 86(18):1398-1402. Cullen P, Schulte H, Assmann G. 1997. The Munster Heart Study (PROCAM): total mortal- ity in middle-aged men is increased at low total and LDL cholesterol concentrations in smokers but not in nonsmokers. Circulation 96(7):2128-2136. Djordjevic MV, Stellman SD, Zang E. 2000. Doses of nicotine and lung carcinogens deliv- ered to cigarette smokers. J Natl Cancer Inst 92(2):106-111. Drin I, Schoket B, Kostic S, Vincze I. 1994. Smoking-related increase in O6-alkylguanine- DNA alkyltransferase activity in human lung tissue. Carcinogenesis 15(8):1535-1539. Dunn BP, Vedal S, San RH, et al. 1991. DNA adducts in bronchial biopsies. Int J Cancer 48(4):485-492. Ebert RV, McNabb ME, McCusker KT, Snow SL. 1983. Amount of nicotine and carbon monoxide inhaled by smokers of low-tar, low-nicotine cigarettes. JAMA 250(20):2840- 2842. EPA (Environmental Protection Agency). 1992. Respiratory Health Effects of Passive Smoking; Lung Cancer and Other Disorders. Washington, DC: EPA. Farmer PB, Shuker DE. 1999. What is the significance of increases in background levels of carcinogen-derived protein and DNA adducts? Some considerations for incremental risk assessment. Mutat Res 424(1-2):275-286. Fearon ER. 1997. Human cancer syndromes: clues to the origin and nature of cancer. Science 278(5340):1043-1050. Fischer S, Spiegelhalder B, Preussmann R. 1989. Influence of smoking parameters on the delivery of tobacco-specific nitrosamines in cigarette smokeâa contribution to relative risk evaluation. Carcinogenesis 10(6):1059-1066. Fliss MS, Usadel H, Caballero OL, et al. 2000. Facile detection of mitochondrial DNA muta- tions in tumors and bodily fluids. Science 287(5460):2017-2019. Freeman DJ, Griffin BA, Murray E, et al. 1993. Smoking and plasma lipoproteins in man: effects on low-density lipoprotein cholesterol levels and high-density lipoprotein subfraction distribution. Eur J Clin Invest 23(10):630-640. Fulton JE, Shekelle RB. 1997. Cigarette smoking, weight gain, and coronary mortality: re- sults from the Chicago Western Electric Study. Circulation 96(5):1438-1444. Fuster V, Chesebro JH, Frye RL, Elveback LR. 1981. Platelet survival and the development of coronary artery disease in the young adult: effects of cigarette smoking, strong family history and medical therapy. Circulation 63(3):546-551. Gealy R, Zhang L, Siegfried JM, Luketich JD, Keohavong P. 1999. Comparison of mutations in the p53 and K-ras genes in lung carcinomas from smoking and nonsmoking women. Cancer Epidemiol Biomarkers Prev 8(4 Pt 1):297-302.
358 CLEARING THE SMOKE Geneste O, Camus AM, Castegnaro M, et al. 1991. Comparison of pulmonary DNA adduct levels, measured by 32P-postlabelling and aryl hydrocarbon hydroxylase activity in lung parenchyma of smokers and ex-smokers. Carcinogenesis 12(7):1301-1305. Gilbert DG, Gilbert BO. 1995. Personality, psychopathology, and nicotine response as me- diators of the genetics of smoking. Behav Genet 25(2):133-147. Giovino GA. 1999. Epidemiology of tobacco use among US adolescents. Nicotine Tob Res 1(Suppl 1):S31-40. Giovino GA, Henningfield JE, Tomar SL, Escobedo LG, Slade J. 1995. Epidemiology of tobacco use and dependence. Epidemiol Rev 17(1):48-65. Grafstrom RC, Dypbukt JM, Sundqvist K, et al. 1994. Pathobiological effects of acetalde- hyde in cultured human epithelial cells and fibroblasts. Carcinogenesis 15(5):985-990. Green MS, Harari G. 1995. A prospective study of the effects of changes in smoking habits on blood count, serum lipids and lipoproteins, body weight and blood pressure in occupationally active men. The Israeli CORDIS Study. J Clin Epidemiol 48(9):1159-1166. Greenblatt MS, Bennett WP, Hollstein M, Harris CC. 1994. Mutations in the p53 tumor suppressor gene: clues to cancer etiology and molecular pathogenesis. Cancer Res 54(18):4855-4878. Grinberg-Funes RA, Singh VN, Perera FP, et al. 1994. Polycyclic aromatic hydrocarbon- DNA adducts in smokers and their relationship to micronutrient levels and the glu- tathione-S-transferase M1 genotype. Carcinogenesis 15(11):2449-2454. Gritz ER, Rose JE, Jarvik ME. 1983. Regulation of tobacco smoke intake with paced cigarette presentation. Pharmacol Biochem Behav 18(3):457-462. Guengerich FP. 2000. Metabolism of chemical carcinogens. Carcinogenesis 21(3):345-351. Hagmar L, Brogger A, Hansteen IL, et al. 1994. Cancer risk in humans predicted by in- creased levels of chromosomal aberrations in lymphocytes: Nordic study group on the health risk of chromosome damage. Cancer Res 54(11):2919-2922. Hall J, Bresil H, Donato F, et al. 1993. Alkylation and oxidative-DNA damage repair activity in blood leukocytes of smokers and non-smokers. Int J Cancer 54(5):728-733. Harris CC, Hollstein M. 1993. Clinical implications of the p53 tumor-suppressor gene. N Engl J Med 329(18):1318-1327. Hartmann A, Blaszyk H, Kovach JS, Sommer SS. 1997. The molecular epidemiology of p53 gene mutations in human breast cancer. Trends Genet 13(1):27-33. Heath AC, Cates R, Martin NG, et al. 1993a. Genetic contribution to risk of smoking initia- tion: comparisons across birth cohorts and across cultures. J Subst Abuse 5(3):221-246. Heath AC, Martin NG. 1993b. Genetic models for the natural history of smoking: evidence for a genetic influence on smoking persistence. Addict Behav 18(1):19-34. Hecht SS. 1999. DNA adduct formation from tobacco-specific N-nitrosamines. Mutat Res 424(1-2):127-142. Hecht SS, Carmella SG, Murphy SE, Akerkar S, Brunnemann KD, Hoffmann D. 1993. A tobacco-specific lung carcinogen in the urine of men exposed to cigarette smoke. N Engl J Med 329(21):1543-1546. Helbock HJ, Beckman KB, Shigenaga MK, et al. 1998. DNA oxidation matters: the HPLC- electrochemical detection assay of 8-oxo-deoxyguanosine and 8-oxo-guanine. Proc Natl Acad Sci U S A 95(1):288-293. Hellerstein MK, Benowitz NL, Neese RA, et al. 1994. Effects of cigarette smoking and its cessation on lipid metabolism and energy expenditure in heavy smokers. J Clin Invest 93(1):265-272. Henning S, Cascorbi I, Munchow B, Jahnke V, Roots I. 1999. Association of arylamine N- acetyltransferases NAT1 and NAT2 genotypes to laryngeal cancer risk. Pharmacogenet- ics 9(1):103-111.
EXPOSURE AND BIOMARKER ASSESSMENT IN HUMANS 359 Herning RI, Jones RT, Benowitz NL, Mines AH. 1983. How a cigarette is smoked deter- mines blood nicotine levels. Clin Pharmacol Ther 33(1):84-90. Hill P, Marquardt H. 1980. Plasma and urine changes after smoking different brands of cigarettes. Clin Pharmacol Ther 27(5):652-658. Hofer I, Nil R, Wyss F, Battig K. 1992. The contributions of cigarette yield, consumption, inhalation and puffing behaviour to the prediction of smoke exposure. Clin Investig 70(3-4):343-351. Hoffmann D, Hoffmann I. 1997. The changing cigarette, 1950-1995. J Toxicol Environ Health 50(4):307-364. Hollstein M, Hergenhahn M, Yang Q, Bartsch H, Wang ZQ, Hainaut P. 1999. New ap- proaches to understanding p53 gene tumor mutation spectra. Mutat Res 431(2):199- 209. Hou SM, Yang K, Nyberg F, Hemminki K, Pershagen G, Lambert B. 1999. Hprt mutant frequency and aromatic DNA adduct level in non-smoking and smoking lung cancer patients and population controls. Carcinogenesis 20(3):437-444. Hurt RD, Sachs DP, Glover ED, et al. 1997. A comparison of sustained-release bupropion and placebo for smoking cessation. N Engl J Med 337(17):1195-1202. IARC (International Agency on the Research of Cancer). 1986. IARC Monographs on the Evaluation of the Carcinogenic Risk of Chemicals to Humans: Tobacco Smoking. Vol. 38 ed. Lyon, France: IARC. Ishibe N, Wiencke JK, Zuo ZF, McMillan A, Spitz M, Kelsey KT. 1997. Susceptibility to lung cancer in light smokers associated with CYP1A1 polymorphisms in Mexican- and Af- rican-Americans. Cancer Epidemiol Biomarkers Prev 6(12):1075-1080. Izzotti A, De Flora S, Petrilli GL, et al. 1995. Cancer biomarkers in human atherosclerotic lesions: detection of DNA adducts. Cancer Epidemiol Biomarkers Prev 4(2):105-110. Izzotti A, Rossi GA, Bagnasco M, De Flora S. 1991. Benzo[a]pyrene diolepoxide-DNA ad- ducts in alveolar macrophages of smokers. Carcinogenesis 12(7):1281-1285. Jacob P, Yu L, Shulgin AT, Benowitz NL. 1999. Minor tobacco alkaloids as biomarkers for tobacco use: comparison of users of cigarettes, smokeless tobacco, cigars, and pipes. Am J Public Health 89(5):731-736. Jaffe RL, Nicholson WJ, Garro AJ. 1983. Urinary mutagen levels in smokers. Cancer Lett 20(1):37-42. Jahnke GD, Thompson CL, Walker MP, Gallagher JE, Lucier GW, DiAugustine RP. 1990. Multiple DNA adducts in lymphocytes of smokers and nonsmokers determined by 32P-postlabeling analysis. Carcinogenesis 11(2):205-211. James AL, Knuiman MW, Divitini ML, Musk AW, Ryan G, Bartholomew HC. 1999. Asso- ciations between white blood cell count, lung function, respiratory illness and mortal- ity: the Busselton Health Study. Eur Respir J 13(5):1115-1119. Jin X, Wu X, Roth JA, et al. 1995. Higher lung cancer risk for younger African-Americans with the Pro/Pro p53 genotype. Carcinogenesis 16(9):2205-2208. Jones IM, Moore DH, Thomas CB, Thompson CL, Strout CL, Burkhart-Schultz K. 1993. Factors affecting HPRT mutant frequency in T-lymphocytes of smokers and nonsmok- ers. Cancer Epidemiol Biomarkers Prev 2(3):249-260. Jorenby DE, Leischow SJ, Nides MA, et al. 1999. A controlled trial of sustained-release bupropion, a nicotine patch, or both for smoking cessation. N Engl J Med 340(9):685-691. Jourenkova-Mironova N, Voho A, Bouchardy C, et al. 1999. Glutathione S-transferase GSTM1, GSTM3, GSTP1 and GSTT1 genotypes and the risk of smoking-related oral and pharyngeal cancers. Int J Cancer 81(1):44-48. Jourenkova N, Reinikainen M, Bouchardy C, Dayer P, Benhamou S, Hirvonen A. 1998. Larynx cancer risk in relation to glutathione S-transferase M1 and T1 genotypes and tobacco smoking. Cancer Epidemiol Biomarkers Prev 7(1):19-23.
360 CLEARING THE SMOKE Kanner RE, Connett JE, Williams DE, Buist AS. 1999. Effects of randomized assignment to a smoking cessation intervention and changes in smoking habits on respiratory symp- toms in smokers with early chronic obstructive pulmonary disease: the Lung Health Study. Am J Med 106(4):410-416. Kato S, Bowman ED, Harrington AM, Blomeke B, Shields PG. 1995. Human lung carcino- gen-DNA adduct levels mediated by genetic polymorphisms in vivo. J Natl Cancer Inst 87(12):902-907. Kaufman DW, Palmer JR, Rosenberg L, Stolley P, Warshauer E, Shapiro S. 1989. Tar content of cigarettes in relation to lung cancer. Am J Epidemiol 129(4):703-711. Kawajiri K, Eguchi H, Nakachi K, Sekiya T, Yamamoto M. 1996. Association of CYP1A1 germ line polymorphisms with mutations of the p53 gene in lung cancer. Cancer Res 56(1):72-76. Kellermann G, Shaw CR, Luyten-Kellerman M. 1973. Aryl hydrocarbon hydroxylase induc- ibility and bronchogenic carcinoma. N Engl J Med 289(18):934-937. Khoury MJ, Wagener DK. 1995. Epidemiological evaluation of the use of genetics to im- prove the predictive value of disease risk factors. Am J Hum Genet 56(4):835-844. Kolonen S, Tuomisto J, Puustinen P, Airaksinen MM. 1992a. Effects of smoking abstinence and chain-smoking on puffing topography and diurnal nicotine exposure. Pharmacol Biochem Behav 42(2):327-332. Kolonen S, Tuomisto J, Puustinen P, Airaksinen MM. 1992b. Puffing behavior during the smoking of a single cigarette in a naturalistic environment. Pharmacol Biochem Behav 41(4):701-706. Kouri RE, McKinney CE, Slomiany DJ, Snodgrass DR, Wray NP, McLemore TL. 1982. Posi- tive correlation between high aryl hydrocarbon hydroxylase activity and primary lung cancer as analyzed in cryopreserved lymphocytes. Cancer Res 42(12):5030-5037. La DK, Swenberg JA. 1996. DNA adducts: biological markers of exposure and potential applications to risk assessment. Mutat Res 365(1-3):129-146. La Vecchia C, Bidoli E, Barra S, et al. 1990. Type of cigarettes and cancers of the upper digestive and respiratory tract. Cancer Causes Control 1(1):69-74. Lange P, Groth S, Nyboe GJ, et al. 1989. Effects of smoking and changes in smoking habits on the decline of FEV1. Eur Respir J 2(9):811-816. Lassila R, Seyberth HW, Haapanen A, Schweer H, Koskenvuo M, Laustiola KE. 1988. Vaso- active and atherogenic effects of cigarette smoking: a study of monozygotic twins discordant for smoking. BMJ 297(6654):955-957. Law MR, Morris JK, Watt HC, Wald NJ. 1997. The dose-response relationship between cigarette consumption, biochemical markers and risk of lung cancer. Br J Cancer 75(11):1690-1693. Lazarus P, Sheikh SN, Ren Q, et al. 1998. p53, but not p16 mutations in oral squamous cell carcinomas are associated with specific CYP1A1 and GSTM1 polymorphic genotypes and patient tobacco use. Carcinogenesis 19(3):509-514. Lee CK, Brown BG, Reed EA, Coggins CR, Doolittle DJ, Hayes AW. 1993. Ninety-day inha- lation study in rats, using aged and diluted sidestream smoke from a reference ciga- rette: DNA adducts and alveolar macrophage cytogenetics. Fundam Appl Toxicol 20(4):393-401. Lehman TA, Scott F, Seddon M, et al. 1996. Detection of K-ras oncogene mutations by polymerase chain reaction-based ligase chain reaction. Anal Biochem 239(2):153-159. Lerman C, Caporaso N, Audrain J, Main D, Bowman ED, Lockshin B, Boyd NR, Shields PG. 1999. Evidence suggesting the role of specific genetic factors in cigarette smoking. Health Psychol 18(1):14-20. Lerman C, Caporaso N, Main D, Audrain J, Boyd NR, Bowman ED, Shields PG. 1998. Depression and self-medication with nicotine: the modifying influence of the dopa receptor gene. Health Psychol 17(1):56-62.
EXPOSURE AND BIOMARKER ASSESSMENT IN HUMANS 361 Lerman C, Shields PG, Main D, et al. 1997. Lack of association of tyrosine hydroxylase genetic polymorphism with cigarette smoking. Pharmacogenetics 7(6):521-524. Li D, Wang M, Cheng L, Spitz MR, Hittelman WN, Wei Q. 1996. In vitro induction of benzo(a)pyrene diol epoxide-DNA adducts in peripheral lymphocytes as a suscepti- bility marker for human lung cancer. Cancer Res 56(16):3638-3641. Liu CS, Kao SH, Wei YH. 1997. Smoking-associated mitochondrial DNA mutations in hu- man hair follicles. Environ Mol Mutagen 30(1):47-55. Liu Y, Hernandez AM, Shibata D, Cortopassi GA. 1994. BCL2 translocation frequency rises with age in humans. Proc Natl Acad Sci U S A 91(19):8910-8914. Lodovici M, Akpan V, Giovannini L, Migliani F, Dolara P. 1998. Benzo[a]pyrene diol-ep- oxide DNA adducts and levels of polycyclic aromatic hydrocarbons in autoptic samples from human lungs. Chem Biol Interact 116(3):199-212. Lubin JH, Blot WJ, Berrino F, et al. 1984. Patterns of lung cancer risk according to type of cigarette smoked. Int J Cancer 33(5):569-576. Ludviksdottir D, Blondal T, Franzon M, Gudmundsson TV, Sawe U. 1999. Effects of nico- tine nasal spray on atherogenic and thrombogenic factors during smoking cessation. J Intern Med 246(1):61-66. Lunn RM, Helzlsouer KJ, Parshad R, et al. 2000. XPD polymorphisms: effects on DNA repair proficiency. Carcinogenesis 21(4):551-555. Lunn RM, Langlois RG, Hsieh LL, Thompson CL, Bell DA. 1999. XRCC1 polymorphisms: effects on aflatoxin B1-DNA adducts and glycophorin A variant frequency. Cancer Res 59(11):2557-2561. Mao L, Lee JS, Kurie JM, et al. 1997. Clonal genetic alterations in the lungs of current and former smokers. J Natl Cancer Inst 89(12):857-862. McCarthy DS, Craig DB, Cherniack RM. 1976. Effect of modification of the smoking habit on lung function. Am Rev Respir Dis 114(1):103-113. McLemore TL, Adelberg S, Liu MC, et al. 1990. Expression of CYP1A1 gene in patients with lung cancer: evidence for cigarette smoke-induced gene expression in normal lung tissue and for altered gene regulation in primary pulmonary carcinomas. J Natl Cancer Inst 82(16):1333-1339. Meyer MJ, Bechtold WE. 1996. Protein adduct biomarkers: state of the art. Environ Health Perspect 104 Suppl 5:879-882. Mills NE, Fishman CL, Scholes J, Anderson SE, Rom WN, Jacobson DR. 1995. Detection of K-ras oncogene mutations in bronchoalveolar lavage fluid for lung cancer diagnosis. J Natl Cancer Inst 87(14):1056-1060. Mohrenweiser HW, Jones IM. 1998. Variation in DNA repair is a factor in cancer suscepti- bility: a paradigm for the promises and perils of individual and population risk esti- mation? Mutat Res 400(1-2):15-24. Mohtashamipur E, Norpoth K, Lieder F. 1985. Isolation of frameshift mutagens from smok- ersâ urine: experiences with three concentration methods. Carcinogenesis 6(5):783-788. Mooney LA, Santella RM, Covey L, et al. 1995. Decline of DNA damage and other biomarkers in peripheral blood following smoking cessation. Cancer Epidemiol Biomarkers Prev 4(6):627-634. Morrow JD, Frei B, Longmire AW, et al. 1995. Increase in circulating products of lipid peroxidation (F2-isoprostanes) in smokers. Smoking as a cause of oxidative damage. N Engl J Med 332(18):1198-1203. Mustonen R, Hemminki K. 1992. 7-Methylguanine levels in DNA of smokersâ and non- smokersâ total white blood cells, granulocytes and lymphocytes. Carcinogenesis 13(11):1951-1955. Mustonen R, Schoket B, Hemminki K. 1993. Smoking-related DNA adducts: 32P- postlabeling analysis of 7-methylguanine in human bronchial and lymphocyte DNA. Carcinogenesis 14(1):151-154.
362 CLEARING THE SMOKE Nakajima T, Elovaara E, Anttila S, et al. 1995. Expression and polymorphism of glutathione S-transferase in human lungs: risk factors in smoking-related lung cancer. Carcinogen- esis 16(4):707-711. Nakayama J, Yuspa SH, Poirier MC. 1984. Benzo(a)pyrene-DNA adduct formation and removal in mouse epidermis in vivo and in vitro: relationship of DNA binding to initiation of skin carcinogenesis. Cancer Res 44(9):4087-4095. Nemeth-Coslett R, Griffiths RR. 1984. Determinants of puff duration in cigarette smokers: I. Pharmacol Biochem Behav 20(6):965-971. Noble EP, St Jeor ST, Ritchie T, et al. 1994. D2 dopamine receptor gene and cigarette smok- ing: a reward gene? Med Hypotheses 42(4):257-260. Nowak J, Murray JJ, Oates JA, FitzGerald GA. 1987. Biochemical evidence of a chronic abnormality in platelet and vascular function in healthy individuals who smoke ciga- rettes. Circulation 76(1):6-14. OâHara P, Connett JE, Lee WW, Nides M, Murray R, Wise R. 1998. Early and late weight gain following smoking cessation in the Lung Health Study. Am J Epidemiol 148(9):821- 830. OâMalley PG, Taylor AJ, Jackson JL, Doherty TM, Detrano RC. 2000. Prognostic value of coronary electron-beam computed tomography for coronary heart disease events in asymptomatic populations. Am J Cardiol 85(8):945-948. Obe G, Vogt HJ, Madle S, Fahning A, Heller WD. 1982. Double-blind study on the effect of cigarette smoking on the chromosomes of human peripheral blood lymphocytes in vivo. Mutat Res 92(1-2):309-319. Olincy A, Young DA, Freedman R. 1997. Increased levels of the nicotine metabolite cotinine in schizophrenic smokers compared to other smokers. Biol Psychiatry 42(1):1-5. Oyama T, Kawamoto T, Mizoue T, et al. 1997. Cytochrome P450 2E1 polymorphism as a risk factor for lung cancer: in relation to p53 gene mutation. Anticancer Res 17(1B):583- 587. Park JW, Cundy KC, Ames BN. 1989. Detection of DNA adducts by high-performance liquid chromatography with electrochemical detection. Carcinogenesis 10(5):827-832. Parry H, Cohen S, Schlarb JE, et al. 1997. Smoking, alcohol consumption, and leukocyte counts. Am J Clin Pathol 107(1):64-67. Parsons WD, Carmella SG, Akerkar S, Bonilla LE, Hecht SS. 1998. A metabolite of the tobacco-specific lung carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone in the urine of hospital workers exposed to environmental tobacco smoke. Cancer Epidemiol Biomarkers Prev 7(3):257-260. Pastorelli R, Guanci M, Cerri A, et al. 1998. Impact of inherited polymorphisms in glu- tathione S-transferase M1, microsomal epoxide hydrolase, cytochrome P450 enzymes on DNA, and blood protein adducts of benzo(a)pyrene-diolepoxide. Cancer Epidemiol Biomarkers Prev 7(8):703-709. Patrick DL, Cheadle A, Thompson DC, Diehr P, Koepsell T, Kinne S. 1994. Validity of self- reported smoking: a review and meta-analysis. American Journal of Public Health 84(7): 1086-1093. Pelkonen O, Vahakangas K, Nebert DW. 1980. Binding of polycyclic aromatic hydrocarbons to DNA: comparison with mutagenesis and tumorigenesis. J Toxicol Environ Health 6(5- 6):1009-1020. Peluso M, Airoldi L, Armelle M, et al. 1998. White blood cell DNA adducts, smoking, and NAT2 and GSTM1 genotypes in bladder cancer: a case-control study. Cancer Epidemiol Biomarkers Prev 7(4):341-346. Perera FP. 1987. Molecular cancer epidemiology: a new tool in cancer prevention. J Natl Cancer Inst 78(5):887-898.
EXPOSURE AND BIOMARKER ASSESSMENT IN HUMANS 363 Phillips AN, Neaton JD, Cook DG, Grimm RH, Shaper AG. 1992. The leukocyte count and risk of lung cancer. Cancer 69(3):680-684. Phillips DH. 1997. Detection of DNA modifications by the 32P-postlabelling assay. Mutat Res 378(1-2):1-12. Phillips DH, Hewer A, Martin CN, Garner RC, King MM. 1988. Correlation of DNA adduct levels in human lung with cigarette smoking. Nature 336(6201):790-792. Poli P, Buschini A, Spaggiari A, Rizzoli V, Carlo-Stella C, Rossi C. 1999. DNA damage by tobacco smoke and some antiblastic drugs evaluated using the Comet assay. Toxicol Lett 108(2-3):267-276. Pressl S, Edwards A, Stephan G. 1999. The influence of age, sex and smoking habits on the background level of fish-detected translocations. Mutat Res 442(2):89-95. Prevost V, Shuker DE. 1996. Cigarette smoking and urinary 3-alkyladenine excretion in man. Chem Res Toxicol 9(2):439-444. Raggi P, Callister TQ, Cooil B, et al. 2000. Identification of patients at increased risk of first unheralded acute myocardial infarction by electron-beam computed tomography. Cir- culation 101(8):850-855. Ramsey MJ, Moore DH, Briner JF, et al. 1995. The effects of age and lifestyle factors on the accumulation of cytogenetic damage as measured by chromosome painting. Mutat Res 338(1-6):95-106. Randerath E, Miller RH, Mittal D, Avitts TA, Dunsford HA, Randerath K. 1989. Covalent DNA damage in tissues of cigarette smokers as determined by 32P-postlabeling assay. J Natl Cancer Inst 81(5):341-347. Randerath K, Reddy MV, Gupta RC. 1981. 32P-labeling test for DNA damage. Proc Natl Acad Sci U S A 78(10):6126-6129. Rangemark C, Benthin G, Granstrom EF, Persson L, Winell S, Wennmalm A. 1992. Tobacco use and urinary excretion of thromboxane A2 and prostacyclin metabolites in women stratified by age. Circulation 86(5):1495-1500. Rebbeck TR. 1997. Molecular epidemiology of the human glutathione S-transferase geno- types GSTM1 and GSTT1 in cancer susceptibility. Cancer Epidemiol Biomarkers Prev 6(9):733-743. Reddy MV, Randerath K. 1986. Nuclease P1-mediated enhancement of sensitivity of 32P- postlabeling test for structurally diverse DNA adducts. Carcinogenesis 7(9):1543-1551. Rennard SI, Daughton D, Fujita J, et al. 1990. Short-term smoking reduction is associated with reduction in measures of lower respiratory tract inflammation in heavy smokers. Eur Respir J 3(7):752-759. Rojas M, Alexandrov K, Cascorbi I, et al. 1998. High benzo[a]pyrene diol-epoxide DNA adduct levels in lung and blood cells from individuals with combined CYP1A1 MspI/ Msp-GSTM1*0/*0 genotypes. Pharmacogenetics 8(2):109-118. Rojas M, Camus AM, Alexandrov K, et al. 1992. Stereoselective metabolism of (-)- benzo[a]pyrene-7,8-diol by human lung microsomes and peripheral blood lympho- cytes: effect of smoking. Carcinogenesis 13(6):929-933. Rosvold EA, McGlynn KA, Lustbader ED, Buetow KH. 1995. Identification of an NAD(P)H:quinone oxidoreductase polymorphism and its association with lung cancer and smoking. Pharmacogenetics 5(4):199-206. Rothman N, Poirier MC, Baser ME, et al. 1990. Formation of polycyclic aromatic hydrocar- bon-DNA adducts in peripheral white blood cells during consumption of charcoal- broiled beef. Carcinogenesis 11(7):1241-1243. Ryberg D, Kure E, Lystad S, et al. 1994. p53 mutations in lung tumors: relationship to putative susceptibility markers for cancer. Cancer Res 54(6):1551-1555.
364 CLEARING THE SMOKE Ryberg D, Skaug V, Hewer A, et al. 1997. Genotypes of glutathione transferase M1 and P1 and their significance for lung DNA adduct levels and cancer risk. Carcinogenesis 18(7):1285-1289. SAMHSA (Substance Abuse and Mental Health Services Administration). 1998. National Household Survey on Drug Abuse: Population Estimates 1998. [Online]. Available: http//:www.samhsa.gov/oasNHSDA/Pe1998/Pop98web1.htm#TopOfPage [accessed 2001]. Santella RM, Weston A, Perera FP, et al. 1988. Interlaboratory comparison of antisera and immunoassays for benzo[a]pyrene-diol-epoxide-I-modified DNA. Carcinogenesis 9(7):1265-1269. Schoket B, Phillips DH, Kostic S, Vincze I. 1998. Smoking-associated bulky DNA adducts in bronchial tissue related to CYP1A1 MspI and GSTM1 genotypes in lung patients. Car- cinogenesis 19(5):841-846. Scott FM, Modali R, Lehman TA, et al. 1997. High frequency of K-ras codon 12 mutations in bronchoalveolar lavage fluid of patients at high risk for second primary lung cancer. Clin Cancer Res 3(3):479-482. Seeman P. 1995. Dopamine receptors. Bloom FE, Lupfer DJ, ed. Psycopharmacology; 4th Generation of Progress. New York: Raven Press. Pp. 295-302. Shields PG, Harris CC. 2000. Cancer risk and low-penetrance susceptibility genes in gene- environment interactions. J Clin Oncol 18(11):2309-2315. Shields PG, Lerman C, Audrain J, et al. 1998. Dopamine D4 receptors and the risk of ciga- rette smoking in African-Americans and Caucasians. Cancer Epidemiol Biomarkers Prev 7(6):453-458. Shimoda R, Nagashima M, Sakamoto M, et al. 1994. Increased formation of oxidative DNA damage, 8-hydroxydeoxyguanosine, in human livers with chronic hepatitis. Cancer Res 54(12):3171-3172. Sidransky D. 1997. Nucleic acid-based methods for the detection of cancer. Science 278(5340):1054-1059. Simpson AJ. 1997. The natural somatic mutation frequency and human carcinogenesis. Adv Cancer Res 71:209-240. Sjalander A, Birgander R, Rannug A, Alexandrie AK, Tornling G, Beckman G. 1996. Asso- ciation between the p21 codon 31 A1 (arg) allele and lung cancer. Hum Hered 46(4):221- 225. Slupphaug G, Lettrem I, Myrnes B, Krokan HE. 1992. Expression of O6-methylguanine- DNA methyltransferase and uracil-DNA glycosylase in human placentae from smokers and non-smokers. Carcinogenesis 13(10):1769-1773. Smith CJ, McKarns SC, Davis RA, et al. 1996. Human urine mutagenicity study comparing cigarettes which burn or primarily heat tobacco. Mutat Res 361(1):1-9. Soussi T, Dehouche K, Beroud C. 2000. p53 website and analysis of p53 gene mutations in human cancer: forging a link between epidemiology and carcinogenesis. Hum Mutat 15(1):105-113. Speit G, Hartmann A. 1999. The comet assay (single-cell gel test). A sensitive genotoxicity test for the detection of DNA damage and repair. Methods Mol Biol 113:203-212. Spitz MR, Hsu TC, Wu X, Fueger JJ, Amos CI, Roth JA. 1995. Mutagen sensitivity as a biological marker of lung cancer risk in African Americans. Cancer Epidemiol Biomarkers Prev 4(2):99-103. Spitz MR, Shi H, Yang F, et al. 1998. Case-control study of the D2 dopamine receptor gene and smoking status in lung cancer patients. J Natl Cancer Inst 90(5):358-363. Stellman SD, Garfinkel L. 1989. Lung cancer risk is proportional to cigarette tar yield: evi- dence from a prospective study. Prev Med 18(4):518-525.
EXPOSURE AND BIOMARKER ASSESSMENT IN HUMANS 365 Stern SJ, Degawa M, Martin MV, et al. 1993. Metabolic activation, DNA adducts, and H-ras mutations in human neoplastic and non-neoplastic laryngeal tissue. J Cell Biochem Suppl 17F:129-137. Stubbe I, Eskilsson J, Nilsson-Ehle P. 1982. High-density lipoprotein concentrations increase after stopping smoking. Br Med J (Clin Res Ed) 284(6328):1511-1513. Sumida H, Watanabe H, Kugiyama K, Ohgushi M, Matsumura T, Yasue H. 1998. Does passive smoking impair endothelium-dependent coronary artery dilation in women? J Am Coll Cardiol 31(4):811-815. Sunyer J, Munoz A, Peng Y, et al. 1996. Longitudinal relation between smoking and white blood cells. Am J Epidemiol 144(8):734-741. Tang D, Santella RM, Blackwood AM, et al. 1995. A molecular epidemiological case-control study of lung cancer. Cancer Epidemiol Biomarkers Prev 4(4):341-346. Tang D, Warburton D, Tannenbaum SR, Skipper P, Santella RM, Cereijido GS, Crawford FG, Perera FP. 1999. Molecular and genetic damage from environmental tobacco smoke in young children. Cancer Epidemiol Biomarkers Prev 8(5)427-431. Thorne S, Mullen MJ, Clarkson P, Donald AE, Deanfield JE. 1998. Early endothelial dys- function in adults at risk from atherosclerosis: different responses to L-arginine. J Am Coll Cardiol 32(1):110-116. U.S. DHHS (U.S. Department of Health and Human Services). 1988. Smoking and Health; A Report of the Surgeon General. Washington, DC: U.S. Department of Health and Human Services, Centers for Disease Control and Prevention. U.S. DHHS (U.S. Department of Health and Human Services). 1994. Preventing Tobacco Use Among Young People: A Report of the Surgeon General. Washington, DC: U.S. Department of Health and Human Services, Centers for Disease Control and Prevention. van Delft JH, Baan RA, Roza L. 1998. Biological effect markers for exposure to carcinogenic compound and their relevance for risk assessment. Crit Rev Toxicol 28(5):477-510. van Diemen PC, Maasdam D, Vermeulen S, Darroudi F, Natarajan AT. 1995. Influence of smoking habits on the frequencies of structural and numerical chromosomal aberra- tions in human peripheral blood lymphocytes using the fluorescence in situ hybridiza- tion (FISH) technique. Mutagenesis 10(6):487-495. van Schooten FJ, Hillebrand MJ, van Leeuwen FE, et al. 1990. Polycyclic aromatic hydrocar- bon-DNA adducts in lung tissue from lung cancer patients. Carcinogenesis 11(9):1677- 1681. Velicer WF, Prochaska, JO, Rossi, JS, Snow, MG. 1992. Assessing outcome in smoking cessa- tion studies. Psychological Bulletin 111(1): 23-41. Vineis P, Bartsch H, Caporaso N, et al. 1994. Genetically based N-acetyltransferase meta- bolic polymorphism and low-level environmental exposure to carcinogens. Nature 369(6476):154-156. Vineis P, Porta M. 1996. Causal thinking, biomarkers, and mechanisms of carcinogenesis. J Clin Epidemiol 49(9):951-956. Vutuc C, Kunze M. 1983. Tar yields of cigarettes and male lung cancer risk. J Natl Cancer Inst 71(3):435-437. Wald NJ, Thompson SG, Law MR, Densem JW, Bailey A. 1989. Serum cholesterol and subsequent risk of cancer: results from the BUPA study. Br J Cancer 59(6):936-938. Wang LE, Bondy ML, de Andrade M, et al. 2000. Gender difference in smoking effect on chromosome sensitivity to gamma radiation in a healthy population. Radiat Res 154(1):20-27. Wei Q, Gu J, Cheng L, et al. 1996. Benzo(a)pyrene diol epoxide-induced chromosomal aber- rations and risk of lung cancer. Cancer Res 56(17):3975-3979.
366 CLEARING THE SMOKE Wei Q, Matanoski GM, Farmer ER, Hedayati MA, Grossman L. 1993. DNA repair and aging in basal cell carcinoma: a molecular epidemiology study. Proc Natl Acad Sci U S A 90(4):1614-1618. Wennmalm A, Benthin G, Granstrom EF, Persson L, Petersson AS, Winell S. 1991. Relation between tobacco use and urinary excretion of thromboxane A2 and prostacyclin me- tabolites in young men. Circulation 83(5):1698-1704. Wiencke JK, Kelsey KT, Varkonyi A, et al. 1995. Correlation of DNA adducts in blood mononuclear cells with tobacco carcinogen-induced damage in human lung. Cancer Res 55(21):4910-4914. Wiencke JK, Spitz MR, McMillan A, Kelsey KT. 1997. Lung cancer in Mexican-Americans and African-Americans is associated with the wild-type genotype of the NAD(P)H: quinone oxidoreductase polymorphism. Cancer Epidemiol Biomarkers Prev 6(2):87-92. Wiencke JK, Thurston SW, Kelsey KT, et al. 1999. Early age at smoking initiation and to- bacco carcinogen DNA damage in the lung. J Natl Cancer Inst 91(7):614-619. Wilcox HB, Schoenberg JB, Mason TJ, Bill JS, Stemhagen A. 1988. Smoking and lung cancer: risk as a function of cigarette tar content. Prev Med 17(3):263-272. Yakubovskaya MS, Spiegelman V, Luo FC, et al. 1995. High frequency of K-ras mutations in normal appearing lung tissues and sputum of patients with lung cancer. Int J Cancer 63(6):810-814. Yamasaki E, Ames BN. 1977. Concentration of mutagens from urine by absorption with the nonpolar resin XAD-2: cigarette smokers have mutagenic urine. Proc Natl Acad Sci U S A 74(8):3555-3559. Yu MC, Ross RK, Chan KK, et al. 1995. Glutathione S-transferase M1 genotype affects aminobiphenyl-hemoglobin adduct levels in white, black and Asian smokers and non- smokers. Cancer Epidemiol Biomarkers Prev 4(8):861-864. Zang EA, Wynder EL. 1992. Cumulative tar exposure. A new index for estimating lung cancer risk among cigarette smokers. Cancer 70(1):69-76.
