
1
The Story of Influenza
OVERVIEW
In the early 20th century, science was sufficiently sophisticated to anticipate that influenza, which had twice reached pandemic proportions in the late 19th century, would recur, but was largely powerless to blunt the devastating impact of the 1918 (H1N1) pandemic. Since then, mankind has gained several advantages against the disease: experience of three better characterized pandemics (1918, 1957, and 1968); knowledge of influenza viruses; capacity to design and manufacture vaccines and antiviral drugs to forestall (if not prevent) infection; and molecular technology that may one day pinpoint the viral components that produce virulence, and thereby identify targets for more effective vaccines and drugs.
Yet the world is vulnerable to the next pandemic, perhaps even more than in 1918, when the pace and frequency of global travel was considerably less than today. As the contributors to this chapter demonstrate, there is still much to be learned from past pandemics that can strengthen defenses against future threats. The chapter begins with a review of the events of 1918, the lessons they offer, and the historical and scientific questions they raise. It describes the epidemiology and symptomology of that deadly viral strain, limited efforts toward prevention and treatment, and the resulting social disruption and its exacerbation by the actions of public officials and the media.
The chapter continues with an account of molecular studies underway to determine the origin of the 1918 virus and the source(s) of its exceptional virulence. Clues are being sought by examining viruses preserved in frozen
and fixed tissues of victims of the 1918 flu. Characterization of five of the eight RNA segments of the 1918 influenza virus indicates that it was the common ancestor of both subsequent human and swine H1N1 lineages, and experiments testing models of virulence using reverse genetics approaches with 1918 influenza genes have begun in hopes of identifying genetic features that confer virulence in humans.
In a parallel effort, subsequently described, epidemiologists are analyzing death records and serological data to better understand patterns of transmission, morbidity, and mortality in past influenza pandemics. Such findings could inform planning for public health interventions to reduce the incidence of severe outcomes in future pandemics. In particular, these studies reveal a signature change in excess mortality from the elderly to younger age groups, a “pandemic age shift,” that occurred with each of the three pandemics of the 20th century. If such a shift could be recognized in incipient pandemics, it might allow sufficient time for the production and distribution of vaccine and antiviral drugs before the worst pandemic impact occurs.
1918 REVISITED: LESSONS AND SUGGESTIONS FOR FURTHER INQUIRY
John M. Barry
Distinguished Visiting Scholar
Center for Bioenvironmental Research at Tulane and Xavier Universities
The 1918–1919 influenza pandemic killed more people in absolute numbers than any other disease outbreak in history. A contemporary estimate put the death toll at 21 million, a figure that persists in the media today, but understates the real number. Epidemiologists and scientists have revised that figure several times since then. Each and every revision has been upward. Frank Macfarlane Burnet, who won his Nobel Prize for immunology but who spent most of his life studying influenza, estimated the death toll as probably 50 million, and possibly as high as 100 million. A 2002 epidemiologic study also estimates the deaths at between 50 and 100 million (Johnson and Mueller, 2002).
The world population in 1918 was only 28 percent of today’s population. Adjusting for population, a comparable toll today would be 175 to 350 million. By comparison, at this writing AIDS has killed approximately 24 million, and an estimated 40 million more people are infected with the virus.
A letter from a physician at one U.S. Army camp to a colleague puts a more human face on those numbers:
These men start with what appears to be an ordinary attack of LaGrippe or Influenza, and when brought to the Hosp. they very rapidly develop the most vicious type of Pneumonia that has ever been seen … and a few hours later you can begin to see the Cyanosis extending from their ears and spreading all over the face, until it is hard to distinguish the colored men from the white. It is only a matter of a few hours then until death comes…. It is horrible. One can stand it to see one, two or twenty men die, but to see these poor devils dropping like flies…. We have been averaging about 100 deaths per day…. Pneumonia means in about all cases death…. We have lost an outrageous number of Nurses and Drs. It takes special trains to carry away the dead. For several days there were no coffins and the bodies piled up something fierce…. It beats any sight they ever had in France after a battle. An extra long barracks has been vacated for the use of the Morgue, and it would make any man sit up and take notice to walk down the long lines of dead soldiers all dressed and laid out in double rows…. Good By old Pal, God be with you till we meet again (Grist, 1979).
That letter reflected a typical experience in American Army cantonments. The civilian experience was not much better.
In preparing for another pandemic, it is useful to examine events of 1918 for lessons, warnings, and areas for further inquiry.
The Virus Itself
The pandemic in 1918 was hardly the first influenza pandemic, nor was it the only lethal one. Throughout history, there have been influenza pandemics, some of which may have rivaled 1918’s lethality. A partial listing of particularly violent outbreaks likely to have been influenza include one in 1510 when a pandemic believed to come from Africa “attacked at once and raged all over Europe not missing a family and scarce a person” (Beveridge, 1977). In 1580, another pandemic started in Asia, then spread to Africa, Europe, and even America (despite the fact that it took 6 weeks to cross the ocean). It was so fierce “that in the space of six weeks it afflicted almost all the nations of Europe, of whom hardly the twentieth person was free of the disease” and some Spanish cities were “nearly entirely depopulated by the disease” (Beveridge, 1977). In 1688, influenza struck England, Ireland, and Virginia; in all these places “the people dyed … as in a plague” (Duffy, 1953). A mutated or new virus continued to plague Europe and America again in 1693 and Massachusetts in 1699. “The sickness extended to almost all families. Few or none escaped, and many dyed especially in Boston, and some dyed in a strange or unusual
manner, in some families all were sick together, in some towns almost all were sick so that it was a time of disease” (Pettit, 1976). In London in 1847 and 1848, more people died from influenza than from the terrible cholera epidemic of 1832. In 1889 and 1890, a great and violent worldwide pandemic struck again (Beveridge, 1977).
But 1918 seems to have been particularly violent. It began mildly, with a spring wave. In fact, it was so mild that some physicians wonder if this disease actually was influenza. Typically, several Italian doctors argued in separate journal articles that this “febrile disease now widely prevalent in Italy [is] not influenza” (Policlinico, 1918). British doctors echoed that conclusion; a Lancet article in July 1918 argued that the spring epidemic was not influenza because the symptoms, though similar to influenza, were “of very short duration and so far absent of relapses or complications” (Little et al., 1918).
Within a few weeks of that Lancet article appearing, a second pandemic wave swept around the world. It also initially caused investigators to doubt that the disease was influenza—but this time because it was so virulent. It was followed by a third wave in 1919, and significant disease also struck in 1920. (Victims of the first wave enjoyed significant resistance to the second and third waves, offering compelling evidence that all were caused by the same virus. It is worth noting that the 1889–1890 pandemic also came in waves, but the third wave seemed to be the most lethal.)
The 1918 virus, especially in its second wave, was not only virulent and lethal, but extraordinarily violent. It created a range of symptoms rarely seen with the disease. After H5N1 first appeared in 1997, pathologists reported some findings “not previously described with influenza” (To et al., 2001). In fact, investigators in 1918 described every pathological change seen with H5N1 and more (Jordon, 1927:266–268).
Symptoms in 1918 were so unusual that initially influenza was misdiagnosed as dengue, cholera, or typhoid. One observer wrote, “One of the most striking of the complications was hemorrhage from mucous membranes, especially from the nose, stomach, and intestine. Bleeding from the ears and petechial hemorrhages in the skin also occurred” (Ireland, 1928:57). A German investigator recorded “hemorrhages occurring in different parts of the interior of the eye” with great frequency (Thomson and Thomson, 1934b). An American pathologist noted: “Fifty cases of subconjunctival hemorrhage were counted. Twelve had a true hemotypsis, bright red blood with no admixture of mucus…. Three cases had intestinal hemorrhage” (Ireland, 1928:13). The New York City Health Department’s chief pathologist said, “Cases with intense pain look and act like cases of dengue … hemorrhage from nose or bronchi … paresis or paralysis of either cerebral or spinal origin … impairment of motion may be severe or mild, permanent or temporary … physical and mental depression. Intense
and protracted prostration led to hysteria, melancholia, and insanity with suicidal intent” (Jordon, 1927:265).
The 1918 virus also targeted young adults. In South African cities, those between the ages of 20 and 40 accounted for 60 percent of the deaths (Katzenellenbogen, 1988). In Chicago the deaths among those aged 20 to 40 nearly quintupled deaths of those aged 41 to 60 (Van Hartesveldt, 1992). A Swiss physician “saw no severe case in anyone over 50.”1 In the “registration area” of the United States—those states and cities that kept reliable statistics—the single greatest number of deaths occurred in the cohort aged 25 to 29, the second greatest in those aged 30 to 34, and the third in those aged 20 to 24. More people died in each one of those 5-year groups than the total deaths among all those over age 60, and the combined deaths of those aged 20 to 34 more than doubled the deaths of all those over 50 (U.S. Bureau of the Census, 1921). The single group most likely to die if infected were pregnant women. In 13 studies of hospitalized pregnant women during the 1918 pandemic, the death rate ranged from 23 to 71 percent (Jordon, 1927:273). Of the pregnant women who survived, 26 percent lost the child (Harris, 1919). (As far back as 1557, people connected influenza with miscarriage and the death of pregnant women.)
The case mortality rate varied widely. An overall figure is impossible to obtain, or even estimate reliably, because no solid information about total cases exists. In U.S. Army camps where reasonably reliable statistics were kept, case mortality often exceeded 5 percent, and in some circumstances exceeded 10 percent. In the British Army in India, case mortality for white troops was 9.6 percent, for Indian troops 21.9 percent.
In isolated human populations, the virus killed at even higher rates. In the Fiji islands, it killed 14 percent of the entire population in 16 days. In Labrador and Alaska, it killed at least one-third of the entire native population (Jordan, 1927; Rice, 1988).
But perhaps most disturbing and most relevant for today is the fact that a significant minority—and in some subgroups of the population a majority—of deaths came directly from the virus, not from secondary bacterial pneumonias.
In 1918, pathologists were intimately familiar with the condition of lungs of victims of bacterial pneumonia at autopsy. But the viral pneumonias caused by the influenza pandemic were so violent that many investigators said the only lungs they had seen that resembled them were from victims of poison gas.
Then, the Army called them “atypical pneumonias.” Today we would call this atypical pneumonia Acute Respiratory Distress Syndrome (ARDS). The Army’s pneumonia board judged that “more than half” of all the deaths among soldiers came from this atypical pneumonia (Ireland, 1928).
One cannot extrapolate from this directly to the civilian population. Army figures represent a special case both in terms of demographics and environment, including overcrowded barracks.
Even so, the fact that ARDS likely caused more than half the deaths among young adults sends a warning. ARDS mortality rates today range from 40 to 60 percent, even with support in modern intensive care units (ICUs). In a pandemic, ICUs would be quickly overwhelmed, representing a major challenge for public health planners.
Treatment and Prevention in 1918
Physicians tried everything they knew, everything they had ever heard of, from the ancient art of bleeding patients, to administering oxygen, to developing new vaccines and sera (chiefly against what we now call Hemophilus influenzae—a name derived from the fact that it was originally considered the etiological agent—and several types of pneumococci). Only one therapeutic measure, transfusing blood from recovered patients to new victims, showed any hint of success.
George Whipple, later a Nobel laureate, studied numerous vaccines and sera and found them “without therapeutic benefit.” But of some vaccines he said, “The statistical evidence, so far as it goes, indicates a probability … [of] some prophylactic value.”2 Some bacterial vaccines may have prevented particular secondary pneumonias.
Meanwhile, the public used home remedies of every description. None showed any evidence of effect.
Some nonmedical interventions did succeed. Total isolation, cutting a community off from the outside world, did work if done early enough. Gunnison, Colorado, a town that was a rail center and was large enough to have a college, succeeded in isolating itself. So did Fairbanks, Alaska. American Samoa escaped without a single case, while a few miles away in Western Samoa, 22 percent of the entire population died.
More interestingly—and perhaps importantly—an Army study found that isolating both individual victims and entire commands that contained infected soldiers “failed when and where [these measures] were carelessly
applied,” but “did some good … when and where they were rigidly carried out” (Soper, undated draft report).
Even if isolation only slowed the virus, it had some value. One of the more interesting epidemiologic findings in 1918 was that the later in the second wave someone got sick, the less likely he or she was to die, and the more mild the illness was likely to be.
This was true in terms of how late in the second wave the virus struck a given area, and, more curiously, it was also true within an area. That is, cities struck later tended to suffer less, and individuals in a given city struck later also tended to suffer less. Thus west coast American cities, hit later, had lower death rates than east coast cities, and Australia, which was not hit by the second wave until 1919, had the lowest death rate of any developed country.
Again, more curiously, someone who got sick 4 days into an outbreak in one place was more likely to develop a viral pneumonia that progressed to ARDS than someone who got sick 4 weeks into the outbreak in the same place. They were also more likely to develop a secondary bacterial pneumonia, and to die from it.
The best data on this comes from the U.S. Army. Of the Army’s 20 largest cantonments, in the first five affected, roughly 20 percent of all soldiers with influenza developed pneumonia. Of those, 37.3 percent died (Soper, 1918; undated draft report).
In the last five camps affected—on average 3 weeks later—only 7.1 percent of influenza victims developed pneumonia. Only 17.8 percent of the soldiers who developed pneumonia died (Soper, 1918).
Inside each camp the same trend held true. Soldiers struck down early died at much higher rates than soldiers in the same camp struck down late.
Similarly, the first cities struck—Boston, Baltimore, Pittsburgh, Philadelphia, Louisville, New York, New Orleans, and smaller cities hit at the same time—all suffered grievously. But in those same places, the people struck by influenza later in the epidemic were not becoming as ill, and were not dying at the same rate, as those struck in the first 2 to 3 weeks.
Cities struck later in the epidemic also usually had lower mortality rates. One of the most careful epidemiologic studies of the epidemic was conducted in Connecticut. The investigator noted that “one factor that appeared to affect the mortality rate was proximity in time to the original outbreak at New London, the point at which the disease was first introduced into Connecticut…. The virus was most virulent or most readily communicable when it first reached the state, and thereafter became generally attenuated” (Thompson and Thompson, 1934a: 215).
The same pattern held true throughout the country and the world. It was not a rigid predictor. The virus was never completely consistent. But places hit later tended to suffer less.
One obvious hypothesis that might explain this phenomenon is that medical care improved as health care workers learned how to cope with the disease. But this hypothesis collapses upon examination. In a given city, as the epidemic proceeded, medical care disintegrated. Doctors and nurses were overworked and sick themselves, and victims—possibly even a majority of victims—received no care at all late in an epidemic.
Even in Army camps, where one could expect communication between physicians from one camp to the next, there seemed to be no improvements in medical care that could account for the different mortality rates. A distinguished investigator specifically looked for evidence of improved care or better preventive measures in Army camps and found none.
A second obvious explanatory hypothesis, that the most vulnerable people were struck first, also fails. For that hypothesis to be true, Americans on the east coast had to have been more vulnerable than those on the west coast, and Americans and western Europeans had to have been more vulnerable than Australians.
But another hypothesis, although entirely speculative, may be worth exploring. If one steps back and looks at the entire United States, it seems that people across the country infected with the virus in September and early to mid-October suffered the most severe attacks. Those infected later, in whatever part of the country they were, suffered less.
At the peak of the pandemic, then, the virus seemed to still be mutating rapidly, virtually with each passage through humans, and it was mutating toward a less lethal form.
We do know that after a mild spring wave, after a certain number of passages through humans, a lethal virus evolved. Possibly after additional passages it became less virulent. This makes sense particularly if the virus was immature when it erupted in September, if it entered the human population only a few months before the lethal wave.
This hypothesis may suggest some areas for investigation.
Social Disruption and Public Health Lessons
In the United States, national and local government and public health authorities badly mishandled the epidemic, offering a useful case study.
The context is important. Every country engaged in World War I tried to control public perception. To avoid hurting morale, even in the nonlethal first wave the press in countries fighting in the war did not mention the outbreak. (But Spain was not at war and its press wrote about it, so the pandemic became known as the Spanish flu).
The United States was no different. In 1917 California Senator Hiram Johnson made the since-famous observation that “The first casualty when war comes is truth.” The U.S. government passed a law that made it pun-
ishable by 20 years in jail to “utter, print, write or publish any disloyal, profane, scurrilous, or abusive language about the government of the United States.”
One could go to jail for cursing or criticizing the government, even if what one said was true. A Congressman was jailed. Simultaneously, the government mounted a massive propaganda effort. An architect of that effort said, “Truth and falsehood are arbitrary terms…. There is nothing in experience to tell us that one is always preferable to the other…. The force of an idea lies in its inspirational value. It matters very little if it is true or false” (Vaughn, 1980).
The combination of rigid control and disregard for truth had dangerous consequences. Focusing on the shortest term, local officials almost universally told half-truths or outright lies to avoid damaging morale and the war effort. They were assisted—not challenged—by the press, which although not censored in a technical sense cooperated fully with the government’s propaganda machine.
Routinely, as influenza approached a city or town—one could watch it march from place to place—local officials initially told the public not to worry, that public health officials would prevent the disease from striking them. When influenza first appeared, officials routinely insisted at first it was only ordinary influenza, not the Spanish flu. As the epidemic exploded, officials almost daily assured the public that the worst was over.
This pattern repeated itself again and again. Chicago offers one example: Its public health commissioner said he’d do “nothing to interfere with the morale of the community…. It is our duty to keep the people from fear. Worry kills more people than the epidemic” (Robertson, 1918).
That idea—“Fear kills more than the disease”—became a mantra nationally and in city after city. As Literary Digest, one of the largest circulation periodicals in the country, advised, “Fear is our first enemy” (Van Hartesveldt, 1992).
In Philadelphia, when the public health commissioner closed all schools, houses of worship, theaters, and other public gathering places, one newspaper went so far as to say that this order was “not a public health measure” and reiterated that “there is no cause for panic or alarm.”
But as people heard these reassurances, they could see neighbors, friends, and spouses dying horrible deaths.
In Chicago, the Cook County Hospital mortality rate of all influenza admissions—not just those who developed pneumonia—was 39.8 percent (Keeton and Cusman, 1918). In Philadelphia, bodies remained uncollected in homes for days, until eventually open trucks and even horse-drawn carts were sent down city streets and people were told to bring out the dead. The bodies were stacked without coffins and buried in cemeteries in mass graves dug by steam shovels.
This horrific disconnect between reassurances and reality destroyed the credibility of those in authority. People felt they had no one to turn to, no one to rely on, no one to trust.
Ultimately society depends on trust. Without it, society began to come apart. Normally in 1918 America, when someone was ill, neighbors helped. That did not happen during the pandemic. Typically, the head of one city’s volunteer effort, frustrated after repeated pleas for help yielded nothing, turned bitter and contemptuous:
Hundreds of women who are content to sit back had delightful dreams of themselves in the roles of angels of mercy, had the unfathomable vanity to imagine that they were capable of great sacrifice. Nothing seems to rouse them now. They have been told that there are families in which every member is ill, in which the children are actually starving because there is no one to give them food. The death rate is so high and they still hold back.3
That attitude persisted outside of cities as well. In rural Kentucky, the Red Cross reported “people starving to death not from lack of food but because the well were panic stricken and would not go near the sick” (An Account of the Influenza Epidemic, 1919).
As the pressure from the virus continued, an internal Red Cross report concluded, “A fear and panic of the influenza, akin to the terror of the Middle Ages regarding the Black Plague, [has] been prevalent in many parts of the country” (The Mobilization of the American National Red Cross, 1920). Similarly, Victor Vaughan, a sober scientist not given to overstatement, worried, “If the epidemic continues its mathematical rate of acceleration, civilization could easily … disappear … from the face of the earth within a matter of a few more weeks” (Collier, 1974).
Of course, the disease generated fear independent of anything officials did or did not do, but the false reassurances given by the authorities and the media systematically destroyed trust. That magnified the fear and turned it into panic and terror.
It is worth noting that this terror, at least in paralyzing form, did not seem to materialize in the few places where authorities told the truth.
One lesson is clear from this experience: In handling any crisis, it is absolutely crucial to retain credibility. Giving false reassurance is the worst thing one can do. If I may speculate, let me suggest that almost as bad as outright lying is holding information so closely that people think officials know more than they say.
The Site of Origin
It is very possible that we will never know with certainty where the 1918 virus crossed into man. In the 1920s and 1930s, outstanding investigators in several countries launched massive reviews of evidence searching for the site of origin. They could not definitively answer the question. But they were unanimous in believing that no known outbreak in China could, as one investigator said, “be reasonably regarded as the true forerunner” of the epidemic.
They considered the most likely sites of origin to be France and the United States, and most agreed with Macfarlane Burnet, who concluded that the evidence was “strongly suggestive” that the 1918 influenza pandemic began in the United States, and that its spread was “intimately related to war conditions and especially the arrival of American troops in France” (Burnet and Clark, 1942).
My own research also makes me think that the United States was the most likely site of origin. The unearthing of previously unknown epidemiologic evidence has led me to advance my own hypothesis that the pandemic began in rural Kansas and traveled with draftees to what is now Fort Riley.
But whether the pandemic began in France or the United States is not really important. What does matter is that the pandemic most likely did not begin in Asia.
This has important implications for modern surveillance efforts. Although Asia’s population density and the close proximity of humans and animals there makes the region particularly dangerous, the evidence of 1918—confirmed by the H7N7 outbreak in Europe of 2003—demonstrates the need for surveillance worldwide.
Something else should be addressed regarding surveillance. A physician now active in public health who received his medical degree in Honduras in 1986 says that he and his colleagues were taught that there was no difference between a cold and influenza. He believes physicians in Central America and possibly elsewhere in the world routinely ignore influenza. Clearly, if we are to have an adequate surveillance system, physicians need to be alert to the disease.
Data
Outstanding laboratory investigators have made enormous progress over the years in understanding the virus and developing effective antiviral drugs as well as new technologies to make vaccines. But one area remains in which investigators have lagged behind—in applying modern insights and statistical methods to old data.
To use an analogy, a similar situation is found in the flooding of the Sacramento River, one of the few rivers in the country where flood control
is the direct responsibility of the U.S. Army Corps of Engineers. The Corps has as powerful computers as anyone, but in a recent 10-year period, the Sacramento River experienced a 100-year flood three times, devastating parts of California—despite the fact that each time the Corps raised the standard for a 100-year flood. The point is not that the river exceeded the 100-year level three times in 10 years. Random chance could account for that. The point is that it did so even though the Corps changed the definition of a 100-year flood. A senior Corps official confessed that the Corps simply did not have enough data to know what a 100-year flood was.
We may be in a similar situation with influenza. We have had only three pandemics in the 20th century. That is not a good base on which to build models. Indeed, the Centers for Disease Control and Prevention’s model of what would happen in the United States should another pandemic strike predicts that the most likely death toll would fall between 89,000 and 207,000. Yet the actual death tolls of two of the three pandemics fell well outside the predicted range. Adjusted for population, 1968 deaths were somewhat fewer than the best case scenario, and 1918 nearly 800 percent worse than the worst case. (In 1918, antibiotics would likely have lessened this gap, but the increased population of those with impaired immune systems would somewhat balance that benefit, and increase deaths.)
In addition, we have not taken advantage of the data that we do have. Several presentations at this conference demonstrate that fact—some on the plus side, by deriving findings of value by reviewing records from 1918, but also on the negative side, by making certain assumptions about 1918 that conflict with actual data.
A careful review of old data would also prove valuable. Studying 1889 (and enough data can be found, possibly from earlier pandemics as well), 1918, 1957, and 1968 might tell us whether each followed the same patterns, which in turn could help us to devise strategies for the use of antivirals and vaccines.
The Next Pandemic
Virtually every expert on influenza believes another pandemic is nearly inevitable, that it will kill millions of people, and that it could kill tens of millions—and a virus like 1918, or H5N1, might kill a hundred million or more—and that it could cause economic and social disruption on a massive scale. This disruption itself could kill as well.
Given those facts, every laboratory investigator and every public health official involved with the disease has two tasks: first, to do his or her work, and second, to make political leaders aware of the risk. The preparedness effort needs resources. Only the political process can allocate them.
CHASING THE ELUSIVE 1918 VIRUS: PREPARING FOR THE FUTURE BY EXAMINING THE PAST
Jeffery K. Taubenberger4
Department of Molecular Pathology
Armed Forces Institute of Pathology
Introduction
Influenza A viruses are negative strand RNA viruses of the genus Orthomyxoviridae. They continually circulate in humans in yearly epidemics (mainly in the winter in temperate climates) and antigenically novel virus strains emerge sporadically as pandemic viruses (Cox and Subbarao, 2000). In the United States, influenza is estimated to kill 30,000 people in an average year (Simonsen et al., 2000; Thompson et al., 2003). Every few years, a more severe influenza epidemic occurs, causing a boost in the annual number of deaths past the average, with 10,000 to 15,000 additional deaths. Occasionally, and unpredictably, influenza sweeps the world, infecting 20 to 40 percent of the population in a single year. In these pandemic years, the numbers of deaths can be dramatically above average. In 1957–1958, a pandemic was estimated to cause 66,000 excess deaths in the United States (Simonsen et al., 1998). In 1918, the worst pandemic in recorded history was associated with approximately 675,000 total deaths in the United States (U.S. Department of Commerce, 1976) and killed at least 40 million people worldwide (Crosby, 1989; Patterson and Pyle, 1991; Johnson and Mueller, 2002).
Influenza A viruses constantly evolve by the mechanisms of antigenic drift and shift (Webster et al., 1992). Consequently they should be considered emerging infectious disease agents, perhaps “continually” emerging pathogens. The importance of predicting the emergence of new circulating influenza virus strains for subsequent annual vaccine development cannot be underestimated (Gensheimer et al., 1999). Pandemic influenza viruses have emerged three times in this century: in 1918 (“Spanish” influenza, H1N1), in 1957 (“Asian” influenza, H2N2), and in 1968 (“Hong Kong” influenza, H3N2) (Cox and Subbarao, 2000; Webby and Webster, 2003). Recent circulation of highly pathogenic avian H5N1 viruses in Asia from 1997 to 2004 has caused a small number of human deaths (Claas et al., 1998; Subbarao et al., 1998; Tran et al., 2004; Peiris et al., 2004). How and
when novel influenza viruses emerge as pandemic virus strains and how they cause disease is still not understood.
Studying the extent to which the 1918 influenza was like other pandemics may help us to understand how pandemic influenzas emerge and cause disease in general. On the other hand, if we determine what made the 1918 influenza different from other pandemics, we may use the lessons of 1918 to predict the magnitude of public health risks a new pandemic virus might pose.
Origin of Pandemic Influenza Viruses
The predominant natural reservoir of influenza viruses is thought to be wild waterfowl (Webster et al., 1992). Periodically, genetic material from avian virus strains is transferred to virus strains infectious to humans by a process called reassortment. Human influenza virus strains with recently acquired avian surface and internal protein-encoding RNA segments were responsible for the pandemic influenza outbreaks in 1957 and 1968 (Scholtissek et al., 1978a; Kawaoka et al., 1989). The change in the hemagglutinin subtype or the hemagglutinin (HA) and the neuraminidase (NA) subtype is referred to as antigenic shift. Because pigs can be infected with both avian and human virus strains, and various reassortants have been isolated from pigs, they have been proposed as an intermediary in this process (Scholtissek, 1994; Ludwig et al., 1995). Until recently there was only limited evidence that a wholly avian influenza virus could directly infect humans, but in 1997 18 people were infected with avian H5N1 influenza viruses in Hong Kong, and 6 died of complications after infection (Claas et al., 1998; Subbarao et al., 1998; Scholtissek, 1994; Ludwig et al., 1995). Although these viruses were very poorly transmissible or non-transmissible (Claas et al., 1998; Subbarao et al., 1998; Scholtissek, 1994; Ludwig et al., 1995; Katz et al., 1999), their isolation from infected patients indicates that humans can be infected with wholly avian influenza virus strains. In 2003–2004, H5N1 outbreaks in poultry have become widespread in Asia (Tran et al., 2004), and at least 32 people have died of complications of infection in Vietnam and Thailand (World Health Organization, 2004). In 2003, a highly pathogenic H7N7 outbreak occurred in poultry farms in The Netherlands. This virus caused infections (predominantly conjunctivitis) in 86 poultry handlers and 3 secondary contacts. One of the infected individuals died of pneumonia (Fouchier et al., 2004; Koopmans et al., 2004; World Health Organization, 2004). In 2004, an H7N3 influenza outbreak in poultry in Canada also resulted in the infection of a single individual (World Health Organization, 2004), and a patient in New York was reported to be sick following infection with an H7N2 virus (Lipsman, 2004). Therefore, it may not be necessary to invoke swine
as the intermediary in the formation of a pandemic virus strain because reassortment between an avian and a human influenza virus could take place directly in humans.
While reassortment involving genes encoding surface proteins appears to be a critical event for the production of a pandemic virus, a significant amount of data exists to suggest that influenza viruses must also acquire specific adaptations to spread and replicate efficiently in a new host. Among other features, there must be functional HA receptor binding and interaction between viral and host proteins (Weis et al., 1988). Defining the minimal adaptive changes needed to allow a reassortant virus to function in humans is essential to understanding how pandemic viruses emerge.
Once a new virus strain has acquired the changes that allow it to spread in humans, virulence is affected by the presence of novel surface protein(s) that allow the virus to infect an immunologically naïve population (Kilbourne, 1977). This was the case in 1957 and 1968 and was almost certainly the case in 1918. While immunological novelty may explain much of the virulence of the 1918 influenza, it is likely that additional genetic features contributed to its exceptional lethality. Unfortunately not enough is known about how genetic features of influenza viruses affect virulence. The degree of illness caused by a particular virus strain, or virulence, is complex and involves host factors like immune status, and viral factors like host adaptation, transmissibility, tissue tropism, or viral replication efficiency. The genetic basis for each of these features is not yet fully characterized, but is most likely polygenic in nature (Kilbourne, 1977).
Prior to the analyses on the 1918 virus described in this review, only two pandemic influenza virus strains were available for molecular analysis: the H2N2 virus strain from 1957 and the H3N2 virus strain from 1968. The 1957 pandemic resulted from the emergence of a reassortant influenza virus in which both HA and NA had been replaced by gene segment closely related to those in avian virus strains (Scholtissek et al., 1978b; Schafer et al., 1993; Webster et al., 1995). The 1968 pandemic followed with the emergence of a virus strain in which the H2 subtype HA gene was exchanged with an avian-derived H3 HA RNA segment (Scholtissek et al., 1978b; Webster et al., 1995), while retaining the N2 gene derived in 1957. More recently it has been shown that the PB1 gene was replaced in both the 1957 and the 1968 pandemic virus strains, also with a likely avian derivation in both cases (Kawaoka et al., 1989). The remaining five RNA segments encoding the PA, PB2, nucleoprotein, matrix and non-structural proteins, all were preserved from the H1N1 virus strains circulating before 1957. These segments were likely the direct descendants of the genes present in the 1918 virus. Because only the 1957 and 1968 influenza pandemic virus strains have been available for sequence analysis, it is not clear what changes are necessary for the emergence of a virus strain with pandemic
potential. Sequence analysis of the 1918 influenza virus allows us potentially to address the genetic basis of virulence and human adaptation.
Historical Background
The influenza pandemic of 1918 was exceptional in both breadth and depth. Outbreaks of the disease swept not only North America and Europe, but also spread as far as the Alaskan wilderness and the most remote islands of the Pacific. It has been estimated that one-third of the world’s population may have been clinically infected during the pandemic (Frost, 1920; Burnet and Clark, 1942). The disease was also exceptionally severe, with mortality rates among the infected of more than 2.5 percent, compared to less than 0.1 percent in other influenza epidemics (Marks and Beatty, 1976; Rosenau and Last, 1980). Total mortality attributable to the 1918 pandemic was probably around 40 million (Crosby, 1989; Johnson and Mueller, 2002; Patterson and Pyle, 1991).
Unlike most subsequent influenza virus strains that have developed in Asia, the “first wave” or “spring wave” of the 1918 pandemic seemingly arose in the United States in March 1918 (Barry, 2004; Crosby, 1989; Jordan, 1927). However, the near simultaneous appearance of influenza in March–April 1918 in North America, Europe, and Asia makes definitive assignment of a geographic point of origin difficult (Jordan, 1927). It is possible that a mutation or reassortment occurred in the late summer of 1918, resulting in significantly enhanced virulence. The main wave of the global pandemic, the “fall wave” or “second wave,” occurred in September–November 1918. In many places, there was yet another severe wave of influenza in early 1919 (Jordan, 1927).
Three extensive outbreaks of influenza within 1 year is unusual, and may point to unique features of the 1918 virus that could be revealed in its sequence. Interpandemic influenza outbreaks generally occur in a single annual wave in the late winter. The severity of annual outbreaks is affected by antigenic drift, with an antigenically modified virus strain emerging every 2 to 3 years. Even in pandemic influenza, while the normal late winter seasonality may be violated, the successive occurrence of distinct waves within a year is unusual. The 1890 pandemic began in the late spring of 1889 and took several months to spread throughout the world, peaking in northern Europe and the United States late in 1889 or early 1890. The second wave peaked in spring 1891 (over a year after the first wave) and the third wave in early 1892 (Jordan, 1927). As in 1918, subsequent waves seemed to produce more severe illness so that the peak mortality was reached in the third wave of the pandemic. The three waves, however, were spread over more than 3 years, in contrast to less than 1 year in 1918. It is unclear what gave the 1918 virus this unusual ability to generate repeated
waves of illness. Perhaps the surface proteins of the virus drifted more rapidly than other influenza virus strains, or perhaps the virus had an unusually effective mechanism for evading the human immune system.
The influenza epidemic of 1918 killed an estimated 675,000 Americans, including 43,000 servicemen mobilized for World War I (Crosby, 1989). The impact was so profound as to depress average life expectancy in the United States by more than 10 years (Grove and Hetzel, 1968) (Figure 1-1) and may have played a significant role in ending the World War I conflict (Crosby, 1989; Ludendorff, 1919).
Many individuals who died during the pandemic succumbed to secondary bacterial pneumonia (Jordan, 1927; LeCount, 1919; Wolbach, 1919) because no antibiotics were available in 1918. However, a subset died rapidly after the onset of symptoms often with either massive acute pulmonary hemorrhage or pulmonary edema, often in less than 5 days (LeCount, 1919; Winternitz et al., 1920; Wolbach, 1919). In the hundreds of autopsies performed in 1918, the primary pathologic findings were confined to the respiratory tree and death was due to pneumonia and respiratory failure (Winternitz et al., 1920). These findings are consistent with infection by a well-adapted influenza virus capable of rapid replication throughout the entire respiratory tree (Reid and Taubenberger, 1999; Taubenberger et al.,
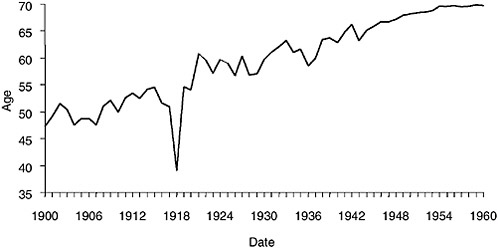
FIGURE 1-1 Life expectancy in the United States, 1900–1960, showing the impact of the 1918 influenza pandemic.
SOURCES: U.S. Department of Commerce (1976); Grove and Hetzel (1968); Linder and Grove (1943).
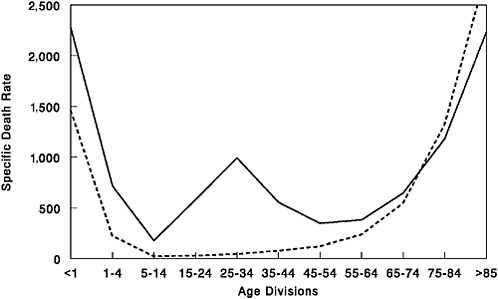
FIGURE 1-2 Influenza and pneumonia mortality by age, United States. Influenza and pneumonia specific mortality by age, including an average of the interpandemic years 1911–1915 (dashed line), and the pandemic year 1918 (solid line). Specific death rate is per 100,000 of the population in each age division.
SOURCES: U.S. Department of Commerce (1976); Grove and Hetzel (1968); Linder and Grove (1943).
2000). There was no clinical or pathological evidence for systemic circulation of the virus (Winternitz et al., 1920).
Furthermore, in the 1918 pandemic most deaths occurred among young adults, a group that usually has a very low death rate from influenza. Influenza and pneumonia death rates for 15- to 34-year-olds were more than 20 times higher in 1918 than in previous years (Linder and Grove, 1943; Simonsen et al., 1998) (Figure 1-2). The 1918 pandemic is also unique among influenza pandemics in that absolute risk of influenza mortality was higher in those younger than age 65 than in those older than 65. Strikingly, persons less than 65 years old accounted for more than 99 percent of all excess influenza-related deaths in 1918–1919 (Simonsen et al., 1998). In contrast, the less-than-65 age group accounted for only 36 percent of all excess influenza-related mortality in the 1957 H2N2 pandemic and 48 percent in the 1968 H3N2 pandemic. Overall, nearly half of the influenza-related deaths in the 1918 influenza pandemic were young adults aged 20 to 40 (Simonsen et al., 1998) (Figure 1-2). Why this particular age group suffered such extreme mortality is not fully understood (see below).
The 1918 influenza had another unique feature: the simultaneous infec-
tion of both humans and swine. Interestingly, swine influenza was first recognized as a clinical entity in that species in the fall of 1918 (Koen, 1919) concurrently with the spread of the second wave of the pandemic in humans (Dorset et al., 1922–1923). Investigators were impressed by clinical and pathological similarities of human and swine influenza in 1918 (Koen, 1919; Murray and Biester, 1930). An extensive review by the veterinarian W.W. Dimoch of the diseases of swine published in August 1918 makes no mention of any swine disease resembling influenza (Dimoch, 1918–1919). Thus, contemporary investigators were convinced that influenza virus had not circulated as an epizootic disease in swine before 1918 and that the virus spread from humans to pigs because of the appearance of illness in pigs after the first wave of the 1918 influenza in humans (Shope and Lewis, 1931).
Thereafter the disease became widespread among swine herds in the U.S. midwest. The epizootic of 1919–1920 was as extensive as in 1918–1919. The disease then appeared among swine in the midwest every year, leading to Shope’s isolation of the first influenza virus in 1930, A/swine/ Iowa/30 (Shope and Lewis, 1931), 3 years before the isolation of the first human influenza virus, A/WS/33 by Smith, Andrewes, and Laidlaw (Smith et al., 1933). Classical swine viruses have continued to circulate not only in North American pigs, but also in swine populations in Europe and Asia (Brown et al., 1995; Kupradinun et al., 1991; Nerome et al., 1982).
During the fall and winter of 1918–1919, severe influenza-like outbreaks were noted not only in swine in the United States, but also in Europe and China (Beveridge, 1977; Chun, 1919; Koen, 1919). Since 1918 there have been many examples of both H1N1 and H3N2 human influenza A virus strains becoming established in swine (Brown et al., 1998; Castrucci et al., 1993; Zhou et al., 2000), while swine influenza A virus strains have been isolated only sporadically from humans (Gaydos et al., 1977; Woods et al., 1981).
The unusual severity of the 1918 pandemic and the exceptionally high mortality it caused among young adults have stimulated great interest in the influenza virus strain responsible for the 1918 outbreak (Crosby, 1989; Kolata, 1999; Monto et al., 1997). Because the first human and swine influenza A viruses were not isolated until the early 1930s (Shope and Lewis, 1931; Smith et al., 1933), characterization of the 1918 virus strain previously has had to rely on indirect evidence (Kanegae et al., 1994; Shope, 1958).
Serology and Epidemiology of the 1918 Influenza Virus
Analyses of antibody titers of 1918 influenza survivors from the late 1930s suggested correctly that the 1918 virus strain was an H1N1-subtype
influenza A virus, closely related to what is now known as “classic swine” influenza virus (Dowdle, 1999; Philip and Lackman, 1962; Shope, 1936). The relationship to swine influenza is also reflected in the simultaneous influenza outbreaks in humans and pigs around the world (Beveridge, 1977; Chun, 1919; Koen, 1919). Although historical accounts described above suggest that the virus spread from humans to pigs in the fall of 1918, the relationship of these two species in the development of the 1918 influenza has not been resolved.
Which influenza A subtype(s) circulated before the 1918 pandemic is not known for certain. In a recent review of the existing archaeoserologic and epidemiologic data, Walter Dowdle concluded that an H3-subtype influenza A virus strain circulated from the 1889–1891 pandemic to 1918, when it was replaced by the novel H1N1 virus strain of the 1918 pandemic (Dowdle, 1999).
It is reasonable to conclude that the 1918 virus strain must have contained a hemagglutinin gene encoding a novel subtype such that large portions of the population did not have protective immunity (Kilbourne, 1977; Reid and Taubenberger, 1999). In fact, epidemiological data collected between 1900 and 1918 on influenza prevalence by age in the population provide good evidence for the emergence of an antigenically novel influenza virus in 1918 (Jordan, 1927). Jordan showed that from 1900 to 1917, the 5 to 15 age group accounted for 11 percent of total influenza cases in this series while the >65 age group similarly accounted for 6 percent of influenza cases. In 1918 the 5- to 15-year-old group jumped to 25 percent of influenza cases, compatible with exposure to an antigenically novel virus strain. The >65 age group only accounted for 0.6 percent of the influenza cases in 1918. It is likely that this age group accounted for a significantly lower percentage of influenza cases because younger people were so susceptible to the novel virus strain (as seen in the 1957 pandemic [Ministry of Health, 1960; Simonsen et al., 1998]), but it is also possible that this age group had pre-existing H1 antibodies. Further evidence for pre-existing H1 immunity can be derived from the age-adjusted mortality data in Figure 1-2. Those individuals >75 years had a lower influenza and pneumonia case mortality rate in 1918 than they had for the prepandemic period of 1911–1917.
When 1918 influenza case rates by age (Jordan, 1927) are superimposed on the familiar “W”-shaped mortality curve (seen in Figure 1-2), a different perspective emerges (Figure 1-3). As shown, those <35 years of age in 1918 accounted for a disproportionately high influenza incidence by age. Interestingly, the 5 to 14 age group accounted for a large fraction of 1918 influenza cases, but had an extremely low case mortality rate compared to other age groups (Figure 1-3). Why this age group had such a low case fatality rate cannot yet be fully explained. Conversely, why the 25 to 34 age
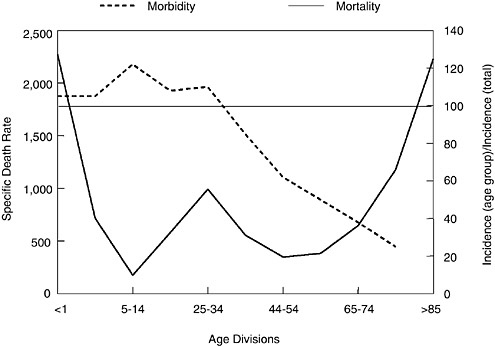
FIGURE 1-3 Influenza and pneumonia mortality by age (solid line), with influenza morbidity by age (dashed line) superimposed. Influenza and pneumonia mortality by age as in Figure 1-2. Specific death rate per age group, left ordinal axis. Influenza morbidity presented as ratio of incidence in persons of each group to incidence in persons of all ages (=100), right ordinal axis. Horizontal line at 100 (right ordinal axis) represents average influenza incidence in the total population.
SOURCES: Taubenberger et al. (2001); adapted from Jordan (1927).
group had such a high influenza and pneumonia mortality rate in 1918 remains enigmatic, but it is one of the truly unique features of the 1918 influenza pandemic.
One theory that may explain these data concerns the possibility that the virus had an intrinsically high virulence that was only tempered in those patients who had been born before 1889. It can be speculated that the virus circulating prior to 1889 was an H1-like virus strain that provided partial protection against the 1918 virus strain (Ministry of Health, 1960; Simonsen et al., 1998; Taubenberger et al., 2001). Short of this cross-protection in patients older than 29 years of age, the pandemic of 1918 might have been even more devastating (Zamarin and Palese, 2004). A second possibility remains that the high mortality of young adults in the 20 to 40 age group may have been a consequence of immune enhancement in this age group. Currently, however, the absence of pre-1918 human influenza samples and
the lack of pre-1918 sera samples for analysis makes it impossible to test this hypothesis.
Thus, it seems clear that the H1N1 virus of the 1918 pandemic contained an antigenically novel hemagglutinin to which most humans and swine were susceptible in 1918. Given the severity of the pandemic, it is also reasonable to suggest that the other dominant surface protein, NA, also would have been replaced by antigenic shift before the start of the pandemic (Reid and Taubenberger, 1999; Taubenberger et al., 2000). In fact, sequence and phylogenetic analyses suggest that the genes encoding these two surface proteins were derived from an avian-like influenza virus shortly before the start of the 1918 pandemic and that the precursor virus did not circulate widely in either humans or swine before 1918 (Fanning et al., 2002; Reid et al., 1999, 2000) (Figure 1-4). It is currently unclear what other influenza gene segments were novel in the 1918 pandemic virus in comparison to the previously circulating virus strain. It is possible that sequence and phylogenetic analyses of the gene segments of the 1918 virus may help elucidate this question.
Genetic Characterization of the 1918 Virus
Sequence and Functional Analysis of the Hemagglutinin and Neuraminidase Gene Segments
Samples of frozen and fixed lung tissue from five second-wave influenza victims (dating from September 1918 to February 1919) have been used to examine directly the genetic structure of the 1918 influenza virus. Two of the cases analyzed were U.S. Army soldiers who died in September 1918, one in Camp Upton, New York, and the other in Fort Jackson, South Carolina. The available material consists of formalin-fixed, paraffin-embedded autopsy tissue, hematoxylin and eosin-stained microscopic sections, and the clinical histories of these patients. A third sample was obtained from an Alaskan Inuit woman who had been interred in permafrost in Brevig Mission, Alaska, since her death from influenza in November 1918. The influenza virus sequences derived from these three cases have been called A/ South Carolina/1/18 (H1N1), A/New York/1/18 (H1N1), and A/Brevig
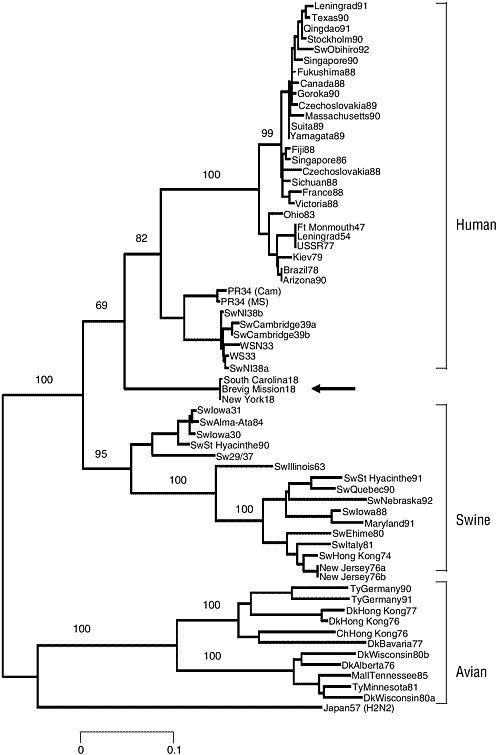
FIGURE 1-4 Phylogenetic tree of the influenza virus hemagglutinin gene segment. Amino acid changes in three lineages of the influenza virus hemagglutinin protein segment, HA1. The tree shows the numbers of unambiguous changes between these sequences, with branch lengths being proportional to number of changes.
SOURCE: Reid et al. (1999).
Mission/1/18 (H1N1), respectively. To date, five RNA segment sequences have been published (Basler et al., 2001; Reid et al., 1999, 2000, 2002, 2004). More recently, the HA sequences of two additional fixed autopsy cases of 1918 influenza victims from the Royal London Hospital were determined (Reid et al., 2003). The HA sequences from these five cases show >99 percent sequence identity, but differ at amino acid residue 225 (see below).
The sequence of the 1918 HA is most closely related to that of the A/ swine/Iowa/30 virus. However, despite this similarity the sequence has many avian features. Of the 41 amino acids that have been shown to be targets of the immune system and subject to antigenic drift pressure in humans, 37 match the avian sequence consensus, suggesting there was little immunologic pressure on the HA protein before the fall of 1918 (Reid et al., 1999). Another mechanism by which influenza viruses evade the human immune system is the acquisition of glycosylation sites to mask antigenic epitopes. The HAs from modern H1N1 viruses have up to five glycosylation sites in addition to the four found in all avian HAs. The HA of the 1918 virus has only the four conserved avian sites (Reid et al., 1999).
Influenza virus infection requires binding of the HA protein to sialic acid receptors on the host cell surface. The HA receptor binding site consists of a subset of amino acids that are invariant in all avian HAs, but vary in mammalian-adapted HAs. Human-adapted influenza viruses preferentially bind sialic acid receptors with α(2-6) linkages. Those viral strains adapted to birds preferentially bind α(2-3) linked sugars (Gambaryan et al., 1997; Matrosovich et al., 1997; Weis et al., 1988). To shift from the proposed avian-adapted receptor-binding site configuration (with a preference for α(2-3) sialic acids) to that of swine H1s (which can bind both α(2-3) and α(2-6)) requires only one amino acid change, E190D. The HA sequences of all five 1918 cases have the E190D change (Reid et al., 2003). In fact, the critical amino acids in the receptor-binding site of two of the 1918 cases are identical to that of the A/swine/Iowa/30 HA. The other three 1918 cases have an additional change from the avian consensus, G225D. Because swine viruses with the same receptor site as A/swine/Iowa/30 bind both avian- and mammalian-type receptors (Gambaryan et al., 1997), A/ New York/1/18 virus probably also had the capacity to bind both. The change at residue 190 may represent the minimal change necessary to allow an avian H1-subtype HA to bind mammalian-type receptors (Reid et al., 1999, 2003; Stevens et al., 2004; Gamblin et al., 2004; Glaser et al., 2004), a critical step in host adaptation.
The crystal structure analysis of the 1918 HA (Stevens et al., 2004; Gamblin et al., 2004) suggests that the overall structure of the receptor binding site is akin to that of an avian H5 HA in terms of its having a narrower pocket than that identified for the human H3 HA (Wilson et al.,
1981). This provides an additional clue for the avian derivation of the 1918 HA. The four antigenic sites that have been identified for another H1 HA, the A/PR/8/34 virus HA (Caton et al., 1982), also appear to be the major antigenic determinants on the 1918 HA. The X-ray analyses suggest that these sites are exposed on the 1918 HA and thus they could be readily recognized by the human immune system.
The principal biological role of NA is the cleavage of the terminal sialic acid residues that are receptors for the virus’s HA protein (Palese and Compans, 1976). The active site of the enzyme consists of 15 invariant amino acids that are conserved in the 1918 NA. The functional NA protein is configured as a homotetramer in which the active sites are found on a terminal knob carried on a thin stalk (Colman et al., 1983). Some early human virus strains have short (11-16 amino acids) deletions in the stalk region, as do many virus strains isolated from chickens. The 1918 NA has a full-length stalk and has only the glycosylation sites shared by avian N1 virus strains (Schulze, 1997). Although the antigenic sites on human-adapted N1 neuraminidases have not been definitively mapped, it is possible to align the N1 sequences with N2 subtype NAs and examine the N2 antigenic sites for evidence of drift in N1. There are 22 amino acids on the N2 protein that may function in antigenic epitopes (Colman et al., 1983). The 1918 NA matches the avian consensus at 21 of these sites (Reid et al., 2000). This finding suggests that the 1918 NA, like the 1918 HA, had not circulated long in humans before the pandemic and very possibly had an avian origin (Reid and Taubenberger, 2003).
Neither the 1918 HA nor NA genes have obvious genetic features that can be related directly to virulence. Two known mutations that can dramatically affect the virulence of influenza virus strains have been described. For viral activation, HA must be cleaved into two pieces, HA1 and HA2, by a host protease (Lazarowitz and Choppin, 1975; Rott et al., 1995). Some avian H5 and H7 subtype viruses acquire a mutation that involves the addition of one or more basic amino acids to the cleavage site, allowing HA activation by ubiquitous proteases (Kawaoka and Webster, 1988; Webster and Rott, 1987). Infection with such a pantropic virus strain can cause systemic disease in birds with high mortality. This mutation was not observed in the 1918 virus (Reid et al., 1999; Taubenberger et al., 1997).
The second mutation with a significant effect on virulence through pantropism has been identified in the NA gene of two mouse-adapted influenza virus strains, A/WSN/33 and A/NWS/33. Mutations at a single codon (N146R or N146Y, leading to the loss of a glycosylation site) appear, like the HA cleavage site mutation, to allow the virus to replicate in many tissues outside the respiratory tract (Li et al., 1993). This mutation was also not observed in the NA of the 1918 virus (Reid et al., 2000).
Therefore, neither surface protein-encoding gene has known mutations
that would allow the 1918 virus to become pantropic. Because clinical and pathological findings in 1918 showed no evidence of replication outside the respiratory system (Winternitz et al., 1920; Wolbach, 1919), mutations allowing the 1918 virus to replicate systemically would not have been expected. However, the relationship of other structural features of these proteins (aside from their presumed antigenic novelty) to virulence remains unknown. In their overall structural and functional characteristics, the 1918 HA and NA are avian-like, but they also have mammalian-adapted characteristics.
Interestingly, recombinant influenza viruses containing the 1918 HA and NA and up to three additional genes derived from the 1918 virus (the other genes being derived from the A/WSN/33 virus) were all highly virulent in mice (Tumpey et al., 2004). Furthermore, expression microarray analysis performed on whole lung tissue of mice infected with the 1918 HA/ NA recombinant showed increased upregulation of genes involved in apoptosis, tissue injury, and oxidative damage (Kash et al., 2004). These findings were unusual because the viruses with the 1918 genes had not been adapted to mice. The completion of the sequence of the entire genome of the 1918 virus and the reconstruction and characterization of viruses with 1918 genes under appropriate biosafety conditions will shed more light on these findings and should allow a definitive examination of this explanation.
Antigenic analysis of recombinant viruses possessing the 1918 HA and NA by hemagglutination inhibition tests using ferret and chicken antisera suggested a close relationship with the A/swine/Iowa/30 virus and H1N1 viruses isolated in the 1930s (Tumpey et al., 2004), further supporting data of Shope from the 1930s (Shope, 1936). Interestingly, when mice were immunized with different H1N1 virus strains, challenge studies using the 1918-like viruses revealed partial protection by this treatment, suggesting that current vaccination strategies are adequate against a 1918-like virus (Tumpey et al., 2004). In fact, the data may even allow us to suggest that the human population, having experienced a long period of exposure to H1N1 viruses, may be partially protected against a 1918-like virus (Tumpey et al., 2004).
Because virulence (in the immunologically naïve person) has not yet been mapped to particular sequence motifs of the 1918 HA and NA genes, what can gene sequencing tell us about the origin of the 1918 virus? The best approach to analyzing the relationships among influenza viruses is phylogenetics, whereby hypothetical family trees are constructed that take available sequence data and use them to make assumptions about the ancestral relationships between current and historical influenza virus strains (Fitch et al., 1991; Gammelin et al., 1990; Scholtissek et al., 1993) (Figure 1-5). Because influenza viruses possess eight discrete RNA segments that can move independently between virus strains by the process of
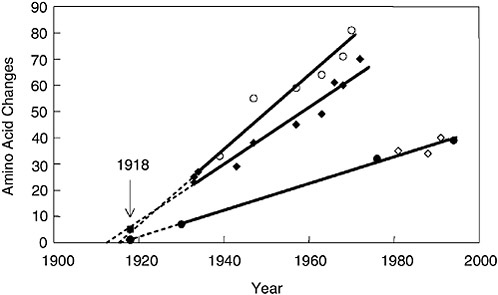
FIGURE 1-5 Change in hemagglutinin (HA) and neuraminidase (NA) proteins over time. The number of amino acid changes from a hypothetical ancestor was plotted versus the date of viral isolation for viruses isolated from 1930 to 1993. Open circles, human HA; closed diamonds, human NA; closed circles, swine HA; open diamonds, swine NA. Regression lines were drawn, extrapolated to the x-intercept and then the 1918 data points, closed square, 1918 HA; closed circle, 1918 NA were added to the graph (arrow).
SOURCES: Reid et al. (1999, 2000); Taubenberger et al. (2000).
reassortment, these evolutionary studies must be performed independently for each gene segment.
A comparison of the complete 1918 HA (Figure 1-5) and NA genes with those of numerous human, swine, and avian sequences demonstrates the following: Phylogenetic analyses based on HA nucleotide changes (either total or synonymous) or HA amino acid changes always place the 1918 HA with the mammalian viruses, not with the avian viruses (Reid et al., 1999). In fact, both synonymous and nonsynonymous changes place the 1918 HA in the human clade. Phylogenetic analyses of total or synonymous NA nucleotide changes also place the 1918 NA sequence with the mammalian viruses, but analysis of nonsynonymous changes or amino acid changes places the 1918 NA with the avian viruses (Reid et al., 2000). Because the 1918 HA and NA have avian features and most analyses place HA and NA near the root of the mammalian clade (close to an ancestor of the avian genes), it is likely that both genes emerged from an avian-like influenza reservoir just prior to 1918 (Reid et al., 1999, 2000, 2003; Fanning and
Taubenberger, 1999; Fanning et al., 2000) (Figure 1-4). Clearly, by 1918 the virus had acquired enough mammalian-adaptive changes to function as a human pandemic virus and to form a stable lineage in swine.
Sequence and Functional Analysis of the Non-Structural Gene Segment
The complete coding sequence of the 1918 non-structural (NS) segment was completed (Basler et al., 2001). The functions of the two proteins, NS1 and NS2 (NEP), encoded by overlapping reading frames (Lamb and Lai, 1980) of the NS segment, are still being elucidated (O’Neill et al., 1998; Li et al., 1998; Garcia-Sastre et al., 1998; Garcia-Sastre, 2002; Krug et al., 2003). The NS1 protein has been shown to prevent type I interferon (IFN) production by preventing activation of the latent transcription factors IRF-3 (Talon et al., 2000) and NF-κB (Wang et al., 2000). One of the distinctive clinical characteristics of the 1918 influenza was its ability to produce rapid and extensive damage to both the upper and lower respiratory epithelium (Winternitz et al., 1920). Such a clinical course suggests a virus that replicated to a high titer and spread quickly from cell to cell. Thus, an NS1 protein that was especially effective at blocking the type I IFN system might have contributed to the exceptional virulence of the 1918 virus strain (Garcia-Sastre et al., 1998; Talon et al., 2000; Wang et al., 2000). To address this possibility, transfectant A/WSN/33 influenza viruses were constructed with the 1918 NS1 gene or with the entire 1918 NS segment (coding for both NS1 and NS2 [NEP] proteins) (Basler et al., 2001). In both cases, viruses containing 1918 NS genes were attenuated in mice compared to wild-type A/WSN/33 controls. The attenuation demonstrates that NS1 is critical for the virulence of A/WSN/33 in mice. On the other hand, transcriptional profiling (microarray analysis) of infected human lung epithelial cells showed that a virus with the 1918 NS1 gene was more effective at blocking the expression of IFN-regulated genes than the isogenic parental mouse-adapted A/WSN/33 virus (Geiss et al., 2002), suggesting that the 1918 NS1 contributes virulence characteristics in human cells, but not murine ones. The 1918 NS1 protein varies from that of the WSN virus at 10 amino acid positions. The amino acid differences between the 1918 and A/WSN/33 NS segments may be important in the adaptation of the latter virus strain to mice and likely account for the observed differences in virulence in these experiments. Recently, a single amino acid change (D92E) in the NS1 protein was associated with increased virulence of the 1997 Hong Kong H5N1 viruses in a swine model (Seo et al., 2002). This amino acid change was not found in the 1918 NS1 protein.
Sequence and Functional Analysis of the Matrix Gene Segment
The coding region of influenza A RNA segment 7 from the 1918 pandemic virus, consisting of the open reading frames of the two matrix genes, M1 and M2, has been sequenced (Reid et al., 2002). Although this segment is highly conserved among influenza virus strains, the 1918 sequence does not match any previously sequenced influenza virus strains. The 1918 sequence matches the consensus over the M1 RNA-binding domains and nuclear localization signal and the highly conserved transmembrane domain of M2. Amino acid changes that correlate with high yield and pathogenicity in animal models were not found in the 1918 virus strain.
Influenza A virus RNA segment 7 encodes two proteins, the matrix proteins M1 and M2. The M1 mRNA is colinear with the viral RNA, while the M2 mRNA is encoded by a spliced transcript (Lamb and Krug, 2001). The proteins encoded by these mRNAs share their initial 9 amino acids and also have a stretch of 14 amino acids in overlapping reading frames. The M1 protein is a highly conserved 252-amino-acid protein. It is the most abundant protein in the viral particle, lining the inner layer of the viral membrane and contacting the ribonucleoprotein (RNP) core. M1 has been shown to have several functions (Lamb and Krug, 2001), including regulation of nuclear export of vRNPs, both permitting the transport of vRNP particles into the nucleus upon infection and preventing newly exported vRNP particles from reentering the nucleus. The 97-amino-acid M2 protein is a homotetrameric integral membrane protein that exhibits ion-channel activity and is the target of the drug amantadine (Hay et al., 1985). The ion-channel activity of M2 is important both during virion uncoating and during viral budding (Lamb and Krug, 2001).
Five amino acid sites have been identified in the transmembrane region of the M2 protein that are involved in resistance to the antiviral drug amantadine: sites 26, 27, 30, 31, and 34 (Holsinger et al., 1994). The 1918 influenza M2 sequence is identical at these positions to that of the amantadine-sensitive influenza virus strains. Thus, it was predicted that the M2 protein of the 1918 influenza virus would be sensitive to amantadine. This was recently demonstrated experimentally. A recombinant virus possessing the 1918 matrix segment was inhibited effectively both in tissue culture and in vivo by the M2 ion-channel inhibitors amantadine and rimantadine (Tumpey et al., 2002).
The phylogenetic analyses suggest that the 1918 matrix genes, while more avian-like than those of other mammalian influenza viruses, were mammalian adapted (Reid et al., 2002). For example, the extracellular domain of the M2 protein contains four amino acids that differ consistently between the avian and mammalian clades (M2 residues #14, 16, 18, and 20). The 1918 sequence matches the mammalian sequence at all four of
these residues (Reid et al., 2002), suggesting that the matrix segment may have been circulating in human virus strains for at least several years before 1918.
Sequence and Functional Analysis of the Nucleoprotein Gene Segment
The nucleoprotein gene (NP) of the 1918 pandemic influenza A virus has been amplified and sequenced from archival material (Reid et al., 2004). The NP gene is known to be involved in many aspects of viral function and to interact with host proteins, thereby playing a role in host specificity (Portela and Digard, 2002). NP is highly conserved, with a maximum amino acid difference of 11 percent among virus strains, probably because it must bind to multiple proteins, both viral and cellular. Numerous studies suggest that NP is a major determinant of host specificity (Scholtissek et al., 1978a, 1985). The 1918 NP amino acid sequence differs at only six amino acids from avian consensus sequences, consistent with reassortment from an avian source shortly before 1918. However, the 1918 NP nucleotide sequence has more than 170 differences from avian consensus sequences, suggesting substantial evolutionary distance from known avian sequences. Both the 1918 NP gene and protein sequences fall within the mammalian clade upon phylogenetic analysis.
Phylogenetic analyses of NP sequences from many virus strains result in trees with two main branches, one consisting of mammalian-adapted virus strains and one of avian-adapted virus strains (Gammelin et al., 1990; Gorman et al., 1991; Shu et al., 1993). The NP gene segment was not replaced in the pandemics of 1957 and 1968, so it is likely that the sequences in the mammalian clade are descended from the 1918 NP segment. The mammalian branches, unlike the avian branch, show a slow but steady accumulation of changes over time. Extrapolation of the rate of change along the human branch back to a putative common ancestor suggests that this NP entered the mammalian lineage sometime after 1900 (Gammelin et al., 1990; Gorman et al., 1991; Shu et al., 1993). Separate analyses of synonymous and nonsynonymous substitutions also placed the 1918 virus NP gene in the mammalian clade (Reid et al., 2004). When synonymous substitutions were analyzed, the 1918 virus gene was placed within and near the root of swine viruses. When nonsynonymous viruses were analyzed, the 1918 virus gene was placed within and near the root of the human viruses.
The evolutionary distance of the 1918 NP from avian and mammalian sequences was examined using several different parameters. There are at least three possibilities for the origin of the 1918 NP gene segment (Reid et al., 2004). First, it could have been retained from the previously circulating human virus, as was the case with the 1957 and 1968 pandemic virus
strains, whose NP segments are descendants of the 1918 NP. The large number of nucleotide changes from the avian consensus and the placement of the 1918 sequence in the mammalian clade are consistent with this hypothesis. Neighbor-joining analyses of nonsynonymous nucleotide sequences or of amino acid sequences place the 1918 sequence within and near the root of the human clade. The 1918 NP has only a few amino acid differences from most bird virus strains, but this consistent group of amino acid changes is shared by the 1918 NP and its subsequent mammalian descendants and is not found in any birds, resulting in the 1918 sequence being placed outside the avian clade (Reid et al., 2004). One or more of these amino acid substitutions may be important for adaptation of the protein to humans. However, the very small number of amino acid differences from the avian consensus argues for recent introduction from birds—80 years after 1918, the NP genes of human influenza virus strains have accumulated more than 30 additional amino acid differences from the avian consensus (a rate of 2.3 amino acid changes per year). Thus it seems unlikely that the 1918 NP, with only six amino acid differences from the avian consensus, could have been in humans for many years before 1918. This conclusion is supported by the regression analysis that suggests that the progenitor of the 1918 virus probably entered the human population around 1915 (Reid et al., 2004).
A second possible origin for the 1918 NP segment is direct reassortment from an avian virus. The small number of amino acid differences between 1918 and the avian consensus supports this hypothesis. While 1918 varies at many nucleotides from the nearest avian virus strain, avian virus strains are quite diverse at the nucleotide level. Synonymous/nonsynonymous ratios between 1918 and avian virus strains are similar to the ratios between avian virus strains, opening the possibility that avian virus strains may exist that are more closely related to 1918. The great evolutionary distance between the 1918 sequence and the avian consensus suggests that no avian virus strain similar to those in the currently identified clades could have provided the 1918 virus strain with its NP segment.
A final possibility is that the 1918 gene segment was acquired shortly before 1918 from a source not currently represented in the database of influenza sequences. There may be a currently unknown influenza host that, while similar to currently characterized avian virus strains at the amino acid level, is quite different at the nucleotide level. It is possible that such a host was the source of the 1918 NP segment (Reid et al., 2004).
Future Work
Five of the eight RNA segments of the 1918 influenza virus have been sequenced and analyzed. Their characterization has shed light on the origin
of the virus and strongly supports the hypothesis that the 1918 virus was the common ancestor of both subsequent human and swine H1N1 lineages. Sequence analysis of the genes to date offers no definitive clue as to the exceptional virulence of the 1918 virus strain. Thus, experiments testing models of virulence using reverse genetics approaches with 1918 influenza genes have begun.
In future work it is hoped that the 1918 pandemic virus strain can be placed in the context of influenza virus strains that preceded it and followed it. The direct precursor of the pandemic virus, the first or “spring” wave virus strain, lacked the exceptional virulence of the fall wave virus strain. Identification of an influenza RNA-positive case from the first wave would have tremendous value in deciphering the genetic basis for virulence by allowing differences in the sequences to be highlighted. Identification of pre-1918 human influenza RNA samples would clarify which gene segments were novel in the 1918 virus.
In many respects, the 1918 influenza pandemic was similar to other influenza pandemics. In its epidemiology, disease course, and pathology, the pandemic generally was different in degree but not in kind from previous and subsequent pandemics. Furthermore, laboratory experiments using recombinant influenza viruses containing genes from the 1918 virus suggest that the 1918 and 1918-like viruses would be as sensitive to the Food and Drug Administration-approved anti-influenza drugs rimantadine and oseltamivir as other virus strains (Tumpey et al., 2002). However, there are some characteristics of the pandemic that appear to be unique: Mortality was exceptionally high, ranging from 5 to 20 times higher than normal. Clinically and pathologically, the high mortality appears to be the result of a higher proportion of severe and complicated infections of the respiratory tract, not with systemic infection or involvement of organ systems outside the influenza virus’s normal targets. The mortality was concentrated in an unusually young age group. Finally, the waves of influenza activity followed each other unusually rapidly, resulting in three major outbreaks within a year’s time. Each of these unique characteristics may find their explanation in genetic features of the 1918 virus. The challenge will be in determining the links between the biological capabilities of the virus and the known history of the pandemic.
Research Agenda for the Future
The work on the 1918 influenza virus, especially its origin, has led to the support of more comprehensive influenza virus surveillance and genomics initiatives for both human and animal influenza A viruses. We believe significant advancement in the understanding of influenza biology and ecology can be made by the generation of full genomic sequences of a
large number of influenza viruses from different hosts. In conclusion, some of the questions that need to be addressed in pandemic influenza include the following:
-
Can an entire avian influenza virus adapt directly in a human, or is reassortment necessary to generate a pandemic strain?
-
Does adaptation of an avian influenza virus to humans require an intermediate host?
-
Can all possible subtypes of avian influenza virus reassort to form functional human pandemic strains, or are there biological limitations to particular HA and NA subtypes?
-
A novel HA seems to be required for a pandemic strain; what about the other gene segments?
-
Can genetic changes be mapped to “virulence”?
-
Can features of virulence be separated from the host in question? Can the viral genetic component of human virulence be modeled in experimental animal or in vitro systems?
-
What molecular changes are necessary for avian strains to adapt to mammals, and to humans in particular?
-
Can host-adaptive changes (genetic fingerprints) be used to trace the evolution of a pandemic strain through intermediate hosts?
Unless we make progress in understanding these and other issues involving the complex ecology and biology of influenza viruses, we will face the risk of revisiting the past in our future.
PANDEMIC INFLUENZA AND MORTALITY: PAST EVIDENCE AND PROJECTIONS FOR THE FUTURE5
L. Simonsen,6 D.R. Olson,7 C. Viboud,8 E. Heiman,6 R.J. Taylor,9 M.A. Miller,8 and T.A. Reichert10
SUMMARY
Pandemic influenza is often thought of as a tornado—a sudden disaster that arrives with little warning and does its worst in a relatively short time. Only three of these calamities occurred in the twentieth century. Their mortality impact ranged from devastating (the 1918 “Spanish” A(H1N1) influenza) to moderate (the 1957 Asian A(H2N2) pandemic) to mild (the 1968 “Hong Kong” A(H3N2) virus). In this paper we review the “pandemic age shift,” a signature change of mortality impact from the elderly to younger age groups that has occurred during each of these pandemics. We also suggest that the “tornado” paradigm may not be completely apt, in that past pandemics have given epidemiologic warning signs of their arrival, and generally play out over several years.
For the 1918 Spanish influenza pandemic, a new study by Olson et al. documents substantial mortality impact during a pandemic “herald wave” in early spring of 1918 in New York City, and a general lack of increased pandemic mortality in those over 45 years of age. For the 1957 pandemic, a classic study documented that the emerging H2N2 influenza virus caused substantial excess mortality during the first three seasons it was in circulation. The 1968 pandemic mortality impact was only “smoldering” in Europe during the first season and did not break into open flame until the next season, during which the majority of mortality impact occurred. Although mortality caused by the 1968 pandemic virus was unimpressive relative to surrounding severe epidemics, the age shift signature sets it apart. Furthermore, antibodies to H3-like antigens—the result of exposure to these antigens in childhood prior to 1892—relatively protected people aged 77 years and older.
Because our experience with pandemic influenza is so limited, it is difficult to predict the mortality impact of future pandemics except to say that the likely range is wide (from ~20 to ~500 deaths per 100,000 population) and that those under 65 years of age will account for a high proportion of the deaths. It may be helpful to think of pandemic mortality impact as the sum of influenza-related deaths that occur over several seasons dominated by the emerging virus until the pandemic age shift pattern gives way to “business as usual,” which typically occurs after a decade or less.
The good news from epidemiological studies for pandemic preparedness planning is that past pandemics gave significant warning signs of their arrival. In 1918, a pandemic herald wave occurred 6 months or more before the majority of mortality impact the following fall. The Asian H2N2 influenza virus was characterized by early summer, 1957, but significant mortality in the United States did not occur until October. In 1968, the pandemic wave of mortality in Europe crested a full year after the pandemic strain first arrived. Furthermore, in both the 1957 and 1968 pandemics,
much of the total impact occurred as a series of smaller “twisters” in the first several seasons after its emergence, before the total population had been affected. These facts suggest that there may well be sufficient time for production and distribution of vaccine and antiviral drugs to prevent much of the mortality impact of the next pandemic, and that these medical interventions will continue to play an important role in limiting “pandemic” mortality for years after the pandemic season. Finally, the pandemic age shift documented for all pandemics studied begs the crucial question of who should be given first priority for vaccine and antivirals, should these be in short supply in the early phase of a pandemic.
The Charge: Using Lessons from Past Pandemics to Help Project the Impact of Future Pandemics
In the case of pandemics, we are planning for the equivalent of a tornado … rare and completely unpredictable until the last minute, when a “weather watch” (e.g., pandemic alert) appears on the TV screen (Kilbourne, 1997).
The lesson of the history of pandemics appears to be that at least the initial attack may sometimes occur with gentleness and thus may afford a substantial breathing space for the preparation and use of specific vaccine (Stuart-Harris, 1970).
Why worry about pandemics of the past? Three influenza pandemics occurred in the twentieth century, and the patterns and magnitude of pandemic mortality are the only impact data available for all three of these events (Table 1-1). We believe, therefore, that continued epidemiological
TABLE 1-1 Mortality Impact in the Three Pandemics of the Twentieth Century in the United States
|
|
Antigenic Shift (Pandemic Event) |
Number of Excess Deaths in the Pandemic Season (All-Cause Deaths) |
Total Excess Mortality Rate per 100,000 Population (Crude) (All-Cause Deaths) |
|
1918–1919 A(H1N1) |
H + N (All novel virus?) |
~ 500,000 |
530 |
|
1957–1958 A(H2N2) |
H + N |
~ 60,000 |
40 |
|
1968–1969 A(H3N2) |
H only |
~ 40,000 |
18 |
analyses of historic mortality data and sero-archaeology—the study of stored serum samples to uncover when specific influenza antigens were circulating—can expand our understanding of pandemic mortality patterns and severity, and that such studies will greatly aid public health planning for pandemic influenza.
In this review, we present the story of pandemic influenza as seen through the lens of epidemiology. For the 1968 pandemic we present data comparing the mortality impact internationally—and highlight the still unexplained finding of “smoldering” pandemic activity in Europe. We review more recent efforts to characterize the signature “age shift” of pandemic influenza and highlight the value of sero-archaeology as a tool to understand what we call “the virtues of antigenic sin”—protection derived from exposure in childhood to influenza H-antigens that are recycled in later pandemic viruses. We revisit a classic study of the 1957 pandemic that analyzes age-specific mortality data from the United States, which shows that most of the mortality was spread over three seasons; we also compare the age-specific mortality impact in the United States to that in Japan. For the 1918 pandemic, we present a study of newly uncovered mortality data from New York City that tells a fetching story about a herald wave and the sparing of the elderly (Olson et al., 2004).
These efforts to study the epidemiology of past pandemic impact on mortality are akin to the efforts by virologists who, in the interest of predicting the future, are hard at work identifying and studying preserved human and animal virus specimens from the 1918 Spanish flu pandemic (Reid and Taubenberger, 2003). But instead of molecular clues to viral pathogenicity and recombination, mortality data provides some important insights into how the pandemic evolves over time, and which age groups are at highest risk for severe outcomes. First, a future pandemic may not appear as a completely unpredictable “tornado” that hits hard the first season and leaves little time for production and distribution of vaccines and antivirals. It is certainly true that, like a tornado, no one can predict precisely when a new pandemic strain might emerge. However, studies of past pandemics show that the next pandemic may well not do its worst in the first season. Instead, historical evidence shows that there can be herald waves or smoldering activity during the first season in which the pandemic influenza virus emerges, suggesting that the preparation time for pandemic vaccines and antivirals might be longer than a few months. Second, pandemic impact cannot be discussed without speaking of age. During a pandemic, the younger population is at substantially increased risk relative to non-pandemic influenza seasons; in some pandemics, this sparing of the elderly may occur as a consequence of antigen recycling. The pandemic age shift has important consequences for thinking about how best to protect the population and minimize years of life lost to future pandemic influenza.
The Pandemic Age Shift: A Signature of Pandemic Mortality Impact
It has previously been demonstrated that all three pandemics of this century were characterized by a shift in the age distribution of deaths (Simonsen et al., 1998). The younger population (in that study, persons under 65 years of age) experienced a sharply elevated mortality risk and accounted for a markedly increased fraction of all influenza-related deaths. As we will discuss below, the 1968 pandemic age shift pattern was exacerbated by the protection of the very elderly by virtue of their experience with H3 antigens as children (Simonsen et al., 2003). For the 1968 pandemic, then, the observed age shift was due to a combination of increased risk among the young and decreased risk among the elderly (Simonsen et al., 2003).
During the 1957 A(H2N2) Asian pandemic in the United States, nearly 40 percent of all influenza-related deaths occurred in the younger population under 65 years of age. The proportion of deaths among people under age 65 that occurred during A(H2N2) epidemics dropped to 5 percent by 1968, when circulation of this virus ceased. In the 1968 A(H3N2) pandemic, this proportion was approximately 50 percent, but declined to less than 10 percent over the next decade (Figure 1-6) (Simonsen et al., 2003).
FIGURE 1-6 A pandemic “decade”: Seasonal excess pneumonia and influenza (P&I) mortality rates for A(H3N2)-dominated seasons remained elevated for a decade after 1968 in persons aged 45–64 years, United States, 1968–1999.
The age shift in mortality was even more pronounced in the 1918 A(H1N1) Spanish influenza pandemic (Collins, 1931; Simonsen, 1998; Olson et al., 2004).
The 1968 Hong Kong Pandemic: Some New Observations
The Unimpressive Impact of the 1968 Pandemic on Mortality in the United States
The 1968 pandemic is the only one known in which a shift in the hemagglutinin antigen was not accompanied by a shift in the neuraminidase antigen. Perhaps for that reason, the 1968 pandemic mortality impact was not particularly severe compared to the severe epidemic in 1967–1968 (the last A(H2N2) epidemic), as well as two severe H3N2 epidemics in 1975–1976 and 1980–1981 (Table 1-2). People aged 75 years and older were far less likely to die of influenza during the pandemic than during these three surrounding epidemics, whereas people aged 45–64 years were at nearly three-fold elevated risk. Despite the differences in relative risk among age groups in different years, the absolute risk of dying of influenza during the pandemic was about 3 times higher for the elderly than for the younger age group (Table 1-2).
TABLE 1-2 The Age-Specific Impact of the 1968 Pandemic in the United States*: Comparison to Surrounding Severe Epidemics
Virtues of Antigenic Sin: The Sparing of the Elderly
Sero-archaeological studies have demonstrated that the majority of the very elderly had H3 antibodies before they were exposed to the 1968 A(H3N2) pandemic virus (Dowdle, 1999; Marine and Workman, 1969). These antibodies were remnants of the immune response to exposure to H3N? viruses that circulated before 1891 (Marine and Workman, 1969); thus, the 1968 pandemic virus apparently contained an H3 antigen “recycled” after 77 years of absence. Marine and Workman hypothesized that the pre-existing anti-H3 antibodies were the result of “original antigenic sin” (Davenport et al., 1953)—childhood exposure to H3 antigens—and that these antibodies might have protected the elderly during the 1968 A(H3N2) pandemic (Marine and Workman, 1969). We recently confirmed this hypothesis when we used U.S. national mortality data to demonstrate that people over the age of 77 were, in fact, protected from influenza-related mortality during the 1968 pandemic, compared to surrounding severe non-pandemic seasons (Simonsen et al., 2003). Even so, the absolute risk of dying from 1968 pandemic influenza was always highest among the very elderly, although this risk was likely significantly lower than it would have been without the protection provided by the anti-H3 antibodies still present in this age group. Figure 1-7 shows the several-fold increase in
FIGURE 1-7 Evidence of protection of the very elderly by “virtues of antigenic sin”: Age-specific excess P&I mortality rates for the 1968 pandemic compared to the 1980–1981 season.
pneumonia and influenza (P&I) mortality rates during the 1968 pandemic among younger age groups in the United States compared to the 1980–1981 season, and the absence of any such increase among the very elderly.
These findings have significant implications for both pandemic planning and the prioritization of high-risk groups for vaccination in the scenario of vaccine shortage. Indeed, if one wishes to minimize the number of years-of-life-lost should vaccine be in short supply, then it would be more effective to immunize the middle aged and younger elderly than the very elderly.
The European 1968 Experience: A “Smoldering” Pattern
The 1968 pandemic experience in Europe was different from that of the United States. It began with the rapid spread of a new virus, which reached Europe about 2 months after its emergence in Hong Kong (Cockburn et al., 1969). But influenza activity remained curiously weak in the wave that occurred during the 1968–1969 winter in Europe (Assaad et al., 1973; Stuart-Harris, 1970). At the same time, influenza-related mortality and morbidity increased substantially in the United States, especially among the young (Housworth and Spoon, 1971). More surprising, a much more severe wave occurred in the United Kingdom during the winter of 1969–1970, although no change in the circulating strain had been identified (Miller et al., 1971). We revisited the pandemic experience in the United States and the United Kingdom by extending the analysis of mortality data from both countries (Figure 1-8) to better describe and possibly explain the geographical differences.
FIGURE 1-8 Monthly pneumonia and influenza (P&I) mortality rate during the first two waves of the 1968 pandemic (A/H3N2) in the United Kingdom. Epidemic threshold determined by a spline-Serfling regression model.
TABLE 1-3 Comparison of the Relative Impact* of the First Two Waves of A/H3N2 Viruses and the Age Distribution of Influenza Deaths in the United States and United Kingdom, 1968–1970
In both countries, we studied the age distribution of mortality rates associated with the first and second pandemic waves of A/H3N2, which occurred during the winters of 1968–1969 and 1969–1970 (Table 1-3). Consistent with the epidemiologic signature of a pandemic (Simonsen et al., 1998), a mortality shift towards younger age groups was observed simultaneously in both the United States and United Kingdom. The shift in the first pandemic wave (1968/1969) in the United Kingdom was not quite as definitive, but was nonetheless above the background of preceding epidemic seasons. In both countries, the proportion of deaths in younger age groups was highly elevated in the second wave. This age shift is consistent with the fact that virus surveillance systems reported widespread circulation of A(H3N2) in both countries during both seasons (Miller et al., 1971; Housworth and Spoon, 1971).
Because pneumonia mortality rates throughout the year were more than two-fold higher in the United Kingdom than in the United States during the 1950s and 1960s (Langmuir and Housworth, 1969; WHO, 1971), direct comparison of the absolute excess P&I mortality impact of the two pandemic waves is less revealing than their relative impact. In the United States, the first wave (1968–1969) accounted for 70 percent of the pandemic deaths, and the second season accounted for the remaining 30 percent. In the United Kingdom, however, the proportions were reversed: the first wave accounted for only 22 percent of UK pandemic deaths, whereas the remaining 78 percent occurred in the second (Table 1-3). Given that circulation of the pandemic virus is well-documented, we use the term “smoldering pandemic” to characterize the first wave in the United Kingdom—the pandemic started slowly, but built to a more destructive conflagration in the second season. We are currently studying other countries, and it thus far appears that the UK pattern describes the typical “European” 1968 pandemic experience (Stuart-Harris, 1970), while the U.S. pattern appears to represent the North American experience (Viboud et al., 2004).
The impact of a novel influenza virus is thought to decrease over time as immunity increases in the population (Cox and Subbarao, 2000; Miller et al., 1971). The European 1968 pandemic pattern, with its smoldering delay, did not fit this pattern, however. More than 30 years later, the reasons for the “smoldering waves” and the differences between North America and Europe are not clear (Nguyen-Van-Tam and Hampson, 2003). It is possible that in Europe only, a high level of immunity to neuraminidase N2 protected the population during the first A/H3N2 wave (Stuart-Harris, 1970). Such immunity would have been acquired through past exposure to A/H2N2 viruses. The “immunity” hypothesis is supported by a high rate of asymptomatic illnesses reported during the first pandemic wave in Europe (Miller et al., 1971; Sohier and Henry, 1969). Alternatively, the different patterns may be due to minor genetic differences in the A/H3N2 viruses that circulated on the two continents. This hypothesis is difficult to address
because only a very limited number of influenza A(H3N2) genetic sequences from the 1968–1970 period are available in the public domain. A new influenza genomics initiative recently funded by the National Institute of Allergy and Infectious Diseases (NIAID) should help change this situation, however. Under this program the Institute for Genomic Research (TIGR) will sequence qualified and properly prepared influenza virus samples for investigators (http://www.niaid.nih.gov/dmid/genomes/mscs/projects.htm). This initiative should encourage scientists everywhere to dig out old isolates sitting quietly in laboratory freezers, so that their complete sequences can be placed in GenBank for all to use.
From the perspective of pandemic planning, the smoldering European 1968 pandemic experience is encouraging, in that a repeat of this pattern in a future pandemic might allow the production and distribution of pandemic vaccines to occur in time to prevent a great many deaths. Indeed, had an effective pandemic vaccine become available in Europe even a full year after the emergence of A(H3N2) viruses in 1968, the majority of deaths associated with this pandemic might have been prevented. Whether smoldering patterns will occur in future pandemics is, of course, not known.
The 1957 Asian Pandemic: Impact Over Several Seasons
The 1957 influenza pandemic, which claimed the lives of more than one million people worldwide, has long been an unofficial model scenario for a future pandemic in the United States. In order to increase the utility of this model for pandemic planners, we have recently begun to compare the well-characterized mortality patterns observed in the United States during the pandemic (Serfling et al., 1967) with those of other countries.
The U.S. Experience: Three Waves Between 1957 and 1963
The Asian H2N2 influenza virus is thought to have first emerged in China in February or March 1957. It reached the United States in early summer, at which time it caused sporadic outbreaks (Jordan, 1958a; Dunn, 1958). However, a measurable impact on U.S. mortality did not occur until October (Serfling et al., 1967). Moreover, each of the first three seasons dominated by the emerging A(H2N2) virus—1957–1958, 1959–1960 and 1962–1963—resulted in roughly equivalent spikes in excess P&I mortality rates in the U.S. population (Serfling et al., 1967) (Table 1-4). These observations suggest that the second twentieth century pandemic did not strike in a sudden, overwhelming onslaught. Instead, measurable mortality impact occurred 4 to 6 months after the virus had begun to circulate and was isolated.
Serfling’s analyses of the U.S. data also revealed that of all the deaths that occurred among age groups 45 years or younger during the first three
serious H2N2 seasons, the majority occurred in the pandemic season of 1957–1958. Conversely, those 45 years and older felt the majority of the mortality impact in the next two A(H2N2)-dominated seasons, 1959–1960 and 1962–1963. For example, 71 percent of the excess deaths among 15–19 year olds in all three seasons occurred during the pandemic 1957–1958 season, while for people aged 75 and older only 33 percent of excess deaths occurred in the pandemic season (Table 1-4). These differences in age-related mortality patterns closely reflected differences in age-specific attack rates of the influenza virus, which were available from the Cleveland Family Study (Jordan et al., 1958a) and Tecumseh, Michigan (Hennessy et al., 1964). Indeed, the attack rate among school children was very high (72.9 percent) in 1957–1958 (Jordan et al., 1958b), but far lower in 1959–1960 and 1962–1963 (Hennessy et al., 1964). It was considered possible that the lower 1957–1958 attack rates left a larger proportion of susceptible people among older cohorts during the subsequent seasons (Hennessy et al., 1964). In summary, the bulk of A(H2N2) infection and mortality among younger age groups occurred in 1957/1958; much less occurred in the subsequent seasons, during which these younger age groups were probably protected by immunity gained through their first encounter with A(H2N2) viruses. However, for the middle aged and elderly, the “pandemic” impact was almost evenly divided between the first three seasons dominated by A(H2N2) viruses (Serfling et al., 1967).
To investigate whether this age-specific mortality pattern also describes the international experience, we set out to develop a methodology for
TABLE 1-4 Relative Impact of First Three “Waves” of A(H2N2) Influenza in the United States, 1957–1963 (modified from data in Serfling et al., 1967)
|
Pandemic |
Excess P&I Mortality Rate/100,000 Population in the United States and the [Proportional Contribution to the Total for Three Seasons] |
||
|
All Ages |
School Children 5–14 Years of Age |
Elderly 75+ Years of Age |
|
|
1957/1958 |
10.6 [44%] |
1.5 [75%] |
97 [33%] |
|
1959/1960 |
7.4 [30%] |
0.3 [15%] |
102 [35%] |
|
1962/1963 |
6.3 [26%] |
0.2 [10%] |
94 [32%] |
|
Total pandemic impact (sum of 3 seasons) |
24.3 [100%] |
2.0 [100%] |
293 [100%] |
TABLE 1-5 Relative impact of first 3 “waves” of A(H2N2) influenza in JAPAN, 1957–1963 (data from Heiman et al., unpublished)
|
Pandemic |
Excess P&I Mortality Rate/100,000 Population in Japan and the [Proportional Contribution to the Total for Three Seasons] |
||
|
All Ages |
School Children 5–14 Years of Age |
Elderly 75+ Years of Age |
|
|
1957/1958 |
1.8 [44%] |
1.9 [79%] |
29 [36%] |
|
1959/1960 |
0.9 [30%] |
0.2 [8%] |
18 [22%] |
|
1962/1963 |
1.3 [26%] |
0.3 [13%] |
34 [42%] |
|
Total pandemic impact (sum of 3 seasons) |
4.0 [100%] |
2.4 [100%] |
81 [100%] |
measuring pandemic mortality burden based on annual mortality data (Heiman et al., unpublished). Annual age-specific P&I mortality data were provided by WHO for the United States and Japan. We estimated the pandemic excess mortality in 1957 and 1958 by subtracting as background the number of deaths in surrounding years when there was little or no influenza A activity. We validated this approach by comparing these U.S. age-specific excess mortality estimates with those generated using actual seasonal data (Serfling et al., 1967). We found that for Japan, the age pattern of relative impact over the first seasons was very similar to that observed in the United States (Table 1-5). Also, in contrast to the United States where there was no measurable increase in influenza-related mortality until October, P&I mortality in Japan was elevated in the early summer of 1957 (Reichert et al., 2001).
The 1918 Spanish Influenza Pandemic Revisited: Evidence for a Severe Herald Wave and Protection of the Elderly in New York City
The exact time and place that the 1918 pandemic virus originated has never been conclusively determined. Public health investigators recognized almost immediately that the so-called Spanish influenza, which spread across Europe in the late spring and summer of 1918 and exploded globally in the autumn and winter of 1918/1919, probably did not originate in Spain (Low, 1920). Reports of influenza epidemics in U.S. military training camps in spring 1918, however, led some to identify the central United States as the “presumptive primary focus” of the pandemic (Vaughn, 1921).
Although this hypothesis has been cited many times over the years, it has never been subject to rigorous reexamination. Moreover, an old analysis of regional urban U.S. mortality statistics showed that excess mortality increased in several Atlantic seaboard cities in the spring of 1918, especially in New York City, suggesting that perhaps the pandemic strain might already have been spreading in this region (Frost, 1919).
To investigate whether and when the characteristic age shift occurred in influenza seasons preceding the 1918–1919 pandemic season in the United States, age-stratified excess deaths in New York City were analyzed (Olson et al., 2004). Comparison of all-cause mortality data for people over and under age 45 indicated that a shift in age-specific excess mortality happened very early in 1918, in the midst of an ongoing influenza season. This pattern is consistent with the arrival of the pandemic virus in New York City at about this time, and the subsequent occurrence of a pandemic herald wave from February to April 1918 (Figure 1-9, Table 1-6).
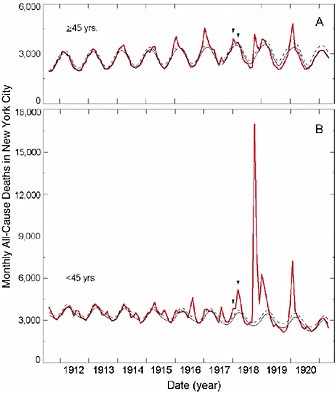
FIGURE 1-9 Monthly all-cause deaths in New York City by age. Classic Serfling model estimates of baseline mortality and 95 percent confidence limits were calculated for under-45 [B] and 45-and-over [A] all-cause deaths. The seasonal pattern indicates that the 1917–1918 influenza season (arrowheads) showed two periods with distinct age-specific peaks. (Figure modified from Olson et al., 2004.)
TABLE 1-6 Age-Specific Mortality Impact of the 1918–1919 A(H1N1) Pandemic in New York City (Population: 5 million) and During the “Herald Wave” in Early 1918 (modified from Olson et al., 2004)
|
Age Groups (years of age) |
Pre-Herald Wave January 1918 All-Cause Excess Mortality Rate/100,000 Population |
Herald Wave February–April 1918 All-Cause Excess Mortality Rate/100,000 Population |
Major Pandemic September 1918–March 1919 All-Cause Excess Mortality Rate/100,000 Population |
|
<5 |
84 |
230 |
720 |
|
5–14 |
2 |
21 |
190 |
|
15–24 |
5 |
64 |
580 |
|
25–44 |
7 |
38 |
760 |
|
45–64 |
18 |
14 |
210 |
|
65+ |
190 |
95 |
150 |
|
All ages |
21 |
56 |
530 |
The New York City data also demonstrate that mortality among people aged 45 and older during the 1918–1919 pandemic influenza season was no worse than in surrounding years. For people under age 45, however, the 1918–1919 influenza season was very bad—people in this age group were far more likely to die of influenza than in previous years. Indeed, the age groups at highest absolute risk of dying during the 1918–1919 A(H1N1) pandemic were young children and young and middle-aged adults (Table 1-6).
These findings suggest that the early 1918 pandemic herald wave was spreading as early as February 1918, 6–7 months before the beginning of the explosive 1918–1919 pandemic. Relative to preceding influenza epidemic seasons, both the herald and pandemic waves caused proportionally more mortality in younger age groups but less mortality among those over 45 years of age, possibly as the result of recycling of an H1-like antigen from half a century earlier (Olson et al., 2004).
Conclusion: Lessons from Pandemics Past for Pandemics Still to Come
Epidemiologic studies of past pandemics offer at least three important insights into what we can expect when the next influenza pandemic occurs. We believe these observations can help to guide pandemic detection and preparedness planning.
1. Mortality impact is difficult to predict, but a shift to younger ages is highly likely. Because our experience with pandemic influenza is so limited (N = 3), it is difficult to predict the mortality impact of a future pandemic. One can say, however, that the likely range is wide (from ~20 to ~500 deaths per 100,000 people) and that people under 65 years of age will account for a high proportion of these death.
2. Pandemic mortality impact is not always “tornado-like.” Pandemic influenza is not always like a sudden storm, followed by a return to clear skies. Instead, mortality rates can remain elevated for several years—during which time an effective vaccine would be in high demand. For example, in the 1957 pandemic worldwide, and in the 1968 pandemic in North America, much of the pandemic mortality impact occurred in a series of smaller but still severe twisters in subsequent years. This seems well explained by attack rates: The pattern of cumulative age-specific mortality impact during the first waves mirrored the age-specific attack rates, at least for the 1957 pandemic (Serfling et al., 1967). Thus, the majority of middle-aged and elderly people—age groups that account for most of the cumulative pandemic mortality—were only affected by the emerging strain during the second or third season after its emergence.
3. Often there is a warning. For the 1918 pandemic, a herald wave that caused substantial mortality occurred at least 6 months before the
major force of the pandemic hit in September. The 1957 pandemic virus had been characterized in Asia by the spring and was known to be circulating in the United States as early as June—months before the pandemic mortality impact began. For the 1968 pandemic, the majority of European deaths occurred after a 1-year delay. Thus, in all three pandemics, some form of warning was available.
Although mortality data are useful to characterize the patterns and impact of past pandemics, in most countries such data would not be available to allow timely detection of mortality age shifts to reveal pandemic activity. Instead, influenza virus surveillance efforts are most likely to provide the first warning of a future pandemic. And because younger people are disproportionately infected by pandemic strains when these first emerge, focusing pandemic virus surveillance efforts on isolates from children and young adults with severe outcomes of upper respiratory diseases would help to ensure that pandemic activity is detected as quickly as possible.
The idea that the next pandemic may not do all or even most of its damage in the first season is certainly good news for preparedness planning. In all three pandemics in the twentieth century, the majority of associated deaths occurred 6 months to a year after the pandemic virus first emerged. This suggests that intense and timely surveillance of both age-specific mortality and new influenza viruses could provide sufficient time for production and distribution of vaccines and antivirals to prevent much, if not most, of the mortality impact. Moreover, these medical interventions are likely to continue to play an important role for many years after the pandemic season. One should also note that the 1957 and 1968 pandemics tended to respect normal seasonality patterns, giving one hemisphere of the world an extra 6 months to prepare.
Finally, the existence of the pandemic age shift documented for all pandemics studied raises a crucial question: Who should get vaccines and antivirals first if these are in short supply, younger people or the elderly? If a future pandemic were to be like the severe 1918/1919 pandemic, in which young and middle-aged adults were the age groups at highest absolute risk of dying, then younger people should clearly get priority. But if a future pandemic were like the 1957 or the 1968 pandemics, the answer would not be so obvious. In those years, young and middle-aged adults were facing the most dramatic risk increase relative to non-pandemic influenza, yet they remained at a lower absolute risk than the elderly. The situation would be made more complex if the pandemic virus were to contain a recycled influenza antigen. In this instance, elderly age groups with prior exposure to similar antigens might be at less risk than in preceding non-pandemic seasons.
Of course, with or without recycling, a pattern like those seen in 1957 or 1968 would result in the elderly having a higher absolute risk of death.
If the metric used to measure effectiveness of vaccination were “numbers of deaths prevented,” then perhaps the elderly should be given priority—assuming they can produce an adequate antibody response to the pandemic vaccine. But if the concern is to minimize the years-of-life-lost, then the vaccine may be better used in young and middle-aged adults. This point was illustrated in a paper that sought to determine vaccination priorities by age and risk status; when basing priority on “returns due to vaccination,” an endpoint that is heavily influenced by years of life lost, the young and middle aged rose to the top of the priority list (Meltzer et al., 1999). Other authors have proposed that children be given priority to receive pandemic vaccine (Stuart-Harris, 1970; Longini et al., 1978; Reichert et al., 2001) and antivirals (Longini et al., 2004) in order to reduce transmission in the community and thereby indirectly reduce influenza impact among the elderly. The 2004 U.S. Pandemic Influenza Preparedness and Response Plan developed by the National Vaccine Program Office has not yet defined such priority groups (DHHS, 2004:24).
Given the very different proposals for how to best employ pandemic vaccines and antivirals should a shortage occur, we urge that a framework for determining priority groups be developed immediately. Such a scheme should be agreed on beforehand and be flexible enough to adapt to the likely level of disaster at hand. Any such an assessment would depend on rapid interpretation of early data on transmissibility and case fatality in the pandemic epicenter.
Will the next pandemic be 1918-like or 1957/1968-like? That is the question.
REFERENCES
An Account of the Influenza Epidemic in Perry County, Kentucky. 1919. 8/14/19, NA, RG 200, Box 689.
Assaad F, Cockburn WC, Sundaresan TK. 1973. Use of excess mortality from respiratory diseases in the study of influenza. Bull World Health Organ 49(3):219–233.
Barry JM. 2004. The Great Influenza: The Epic Story of the Deadliest Plague in History. New York: Viking Press. P. 560.
Basler CF, Reid AH, Dybing JK, Janczewski TA, Fanning TG, Zheng H, Salvatore M, Perdue ML, Swayne DE, Garcia-Sastre A, Palese P, Taubenberger JK. 2001. Sequence of the 1918 pandemic influenza virus nonstructural gene (NS) segment and characterization of recombinant viruses bearing the 1918 NS genes. Proc Natl Acad Sci USA 98:2746–2751.
Beveridge W. 1977. Influenza: The Last Great Plague, an Unfinished Story of Discovery. New York: Prodist.
Brown IH, Chakraverty P, Harris PA, Alexander DJ. 1995. Disease outbreaks in pigs in Great Britain due to an influenza A virus of H1N2 subtype. Vet Rec 136:328–329.
Brown IH, Harris PA, McCauley JW, Alexander DJ. 1998. Multiple genetic reassortment of avian and human influenza A viruses in European pigs, resulting in the emergence of an H1N2 virus of novel genotype. J Gen Virol 79:2947–2955.
Burnet F, Clark E. 1942. Influenza: A Survey of the Last 50 Years in the Light of Modern Work on the Virus of Epidemic Influenza. Melbourne, Australia: Macmillan.
Castrucci MR, Donatelli I, Sidoli L, Barigazzi G, Kawaoka Y, Webster RG. 1993. Genetic reassortment between avian and human influenza A viruses in Italian pigs. Virology 193:503–506.
Caton AJ, Brownlee GG, Yewdell JW, Gerhard W. 1982. The antigenic structure of the influenza virus A/PR/8/34 hemagglutinin (H1 subtype). Cell 31:417–427.
Chun J. 1919. Influenza: Including its infection among pigs. National Medical Journal (of China) 5:34–44.
Claas EC, Osterhaus AD, van Beek R, De Jong JC, Rimmelzwaan GF, Senne DA, Krauss S, Shortridge KF, Webster RG. 1998. Human influenza A H5N1 virus related to a highly pathogenic avian influenza virus. Lancet 351:472–477.
Cockburn WC, Delon PJ, Ferreira W. 1969. Origin and progress of the 1968-69 Hong Kong influenza epidemic. Bull World Health Organ 41(3):345–348.
Collier R. 1974. The Plague of the Spanish Lady. London, England: Macmillan. P. 266.
Collins S. 1931. Age and sex incidence of influenza and pneumonia morbidity and mortality in the epidemic of 1928-29 with comparative data for the epidemic of 1918-19. Pub Health Rep 46:1909–1937.
Colman PM, Varghese JN, Laver WG. 1983. Structure of the catalytic and antigenic sites in influenza virus neuraminidase. Nature 303:41–44.
Cox NJ, Subbarao K. 2000. Global epidemiology of influenza: Past and present. Annu Rev Med 51:407–421.
Crosby A. 1989. America’s Forgotten Pandemic. Cambridge, England: Cambridge University Press.
Davenport FM, Hennesey AV, Francis T. 1953. Epidemiologic and immunologic significance of age distribution of antibody to antigenic variants of influenza virus. J Exp Med 99:641–656.
DHHS (Department of Health and Human Services). 2004. Pandemic Influenza Response and Preparedness Plan. [Online]. Available: http://www.hhs.gov/nvpo/pandemicplan/ [accessed December 17, 2004].
Dimoch WW. 1918–1919. Diseases of swine. J Am Vet Med Assn 54:321–340.
Dorset M, McBryde CN, Niles WB. 1922–1923. Remarks on “Hog” flu. J Am Vet Med Assn 62:162–171.
Dowdle WR. 1999. Influenza A virus recycling revisited. Bull World Health Organ 77:820–828.
Duffy J. 1953. Epidemics in Colonial America. Baton Rouge, LA: LSU Press. Pp. 187–188.
Dunn FL. 1958. Pandemic influenza in 1957: Review of international spread of new Asian strain. JAMA 166(10):1140–1148.
Fanning TG, Taubenberger JK. 1999. Phylogenetically important regions of the influenza A H1 hemagglutinin protein. Virus Res 65:33–42.
Fanning TG, Reid AH, Taubenberger JK. 2000. Influenza A virus neuraminidase: Regions of the protein potentially involved in virus-host interactions. Virology 276:417–423.
Fanning TG, Slemons RD, Reid AH, Janczewski TA, Dean J, Taubenberger JK. 2002. 1917 avian influenza virus sequences suggest that the 1918 pandemic virus did not acquire its hemagglutinin directly from birds. J Virol 76:7860–7862.
Fitch W, Leiter J, Li X, Palese P. 1991. Positive Darwinian evolution in human influenza A viruses. Proc Natl Acad Sci USA 88:4270–4274.
Fouchier RA, Schneeberger PM, Rozendaal FW, Broekman JM, Kemink SA, Munster V, Kuiken T, Rimmelzwaan GF, Schutten M, Van Doornum GJ, Koch G, Bosman A, Koopmans M, Osterhaus AD. 2004. Avian influenza A virus (H7N7) associated with human conjunctivitis and a fatal case of acute respiratory distress syndrome. Proc Natl Acad Sci USA 101:1356–1361.
Frost W. 1920. Statistics of influenza morbidity. Pub Health Rep 35:584–597.
Frost WH. 1919. The epidemiology of influenza. JAMA 70:313–318.
Gambaryan AS, Tuzikov AB, Piskarev VE, Yamnikova SS, Lvov DK, Robertson JS, Bovin NV, Matrosovich MN. 1997. Specification of receptor-binding phenotypes of influenza virus isolates from different hosts using synthetic sialylglycopolymers: Non-egg-adapted human H1 and H3 influenza A and influenza B viruses share a common high binding affinity for 6'-Sialyl(N-acetyllactosamine). Virology 232:345–350.
Gamblin SJ, Haire LF, Russell RJ, Stevens DJ, Xiao B, Ha Y, Vasisht N, Steinhauer DA, Daniels RS, Elliot A, Wiley DC, Skehel JJ. 2004. The structure and receptor binding properties of the 1918 influenza hemagglutinin. Science 303:1838–1842.
Gammelin M, Altmuller A, Reinhardt U, Mandler J, Harley VR, Hudson PJ, Fitch WM, Scholtissek C. 1990. Phylogenetic analysis of nucleoproteins suggests that human influenza A viruses emerged from a 19th-century avian ancestor. Mol Biol Evol 7:194–200.
Garcia-Sastre A. 2002. Mechanisms of inhibition of the host interferon alpha/beta-mediated antiviral responses by viruses. Microbes Infect 4:647–655.
Garcia-Sastre A, Egorov A, Matassov D, Brandt S, Levy DE, Durbin JE, Palese P, Muster T. 1998. Influenza A virus lacking the NS1 gene replicates in interferon-deficient systems. Virology 252:324–330.
Gaydos JC, Hodder RA, Top FH Jr, Soden VJ, Allen RG, Bartley JD, Zabkar JH, Nowosiwsky T, Russell PK. Swine influenza A at Fort Dix, New Jersey (January–February 1976). Case finding and clinical study of cases. J Infect Dis 136:S356–S362.
Geiss GK, Salvatore M, Tumpey TM, Carter VS, Wang X, Basler CF, Taubenberger JK, Bumgarner RE, Palese P, Katze MG, Garcia-Sastre A. 2002. Cellular transcriptional profiling in influenza A virus-infected lung epithelial cells: The role of the nonstructural NS1 protein in the evasion of the host innate defense and its potential contribution to pandemic influenza. Proc Natl Acad Sci USA 99:10736–10741.
Gensheimer KF, Fukuda K, Brammer L, Cox N, Patriarca PA, Strikas RA. 1999. Preparing for pandemic influenza: The need for enhanced surveillance. Emerg Infect Dis 5:297–299.
Glaser L, Zamarin D, Taubenberger JK, Palese P. 2004 (submitted). A Single Amino Acid Substitution in the 1918 Influenza Virus Hemagglutinin Changes the Receptor Binding Specificity.
Gorman OT, Bean WJ, Kawaoka Y, Donatelli I, Guo YJ, Webster RG. 1991. Evolution of influenza A virus nucleoprotein genes: Implications for the origins of H1N1 human and classical swine viruses. J Virol 65:3704–3714.
Grist NR. 1979. Pandemic influenza 1918. Brit Med J 2(6203):1632–1633.
Grove RD, Hetzel AM. 1968. Vital Statistics Rates in the United States: 1940–1960. Washington, DC: National Center for Health Statistics. Government Printing Office.
Harris J. 1919. Influenza occurring in pregnant women: A statistical study of 130 cases. JAMA 72(14):978–980.
Hay A, Wolstenholme A, Skehel J, Smith M. 1985. The molecular basis of the specific anti-influenza action of amantadine. EMBO J 4:3021–3024.
Hennessy AV, Davenport FM, Horton RJ, Napier JA, Francis T Jr. 1964. Asian influenza: Occurrence and recurrence, a community and family study. Mil Med 129:38–50.
Holsinger LJ, Nichani D, Pinto LH, Lamb RA. 1994. Influenza A virus M2 ion channel protein: A structure-function analysis. J Virol 68:1551–1563.
Housworth WJ, Spoon MM. 1971. The age distribution of excess mortality during A2 Hong Kong influenza epidemics compared with earlier A2 outbreaks. Am J Epidemiol 94:348–350.
Ireland MW, ed. 1928. Medical Department of the United States Army in the World War. Commun Dis 9:61.
Johnson NP, Mueller J. 2002. Updating the accounts: Global mortality of the 1918–1920 “Spanish” influenza pandemic. Bull Hist Med 76:105–115.
Jordan E. 1927. Epidemic Influenza: A survey. Chicago, IL: American Medical Association.
Jordan WS, Badger GF, Dingle JH. 1958a. A study of illness in a group of Cleveland families. XVI. The epidemiology of influenza, 1948-1953. Am J Hyg 68:169–189.
Jordan WS, Denny FW, Badger GF, Curtiss C, Dingle JH, Oseasohn R, Stevens DA. 1958b. A study of illness in a group of Cleveland families. XVII. The occurrence of Asian influenza. Am J Hyg 68:190–212.
Kanegae Y, Sugita S, Sortridge K, Yoshioka Y, Nerome K. 1994. Origin and evolutionary pathways of the H1 hemagglutinin gene of avian, swine and human influenza viruses: Cocirculation of two distinct lineages of swine viruses. Arch Virol 134:17–28.
Kash JC, Basler CF, Garcia-Sastre A, Carter V, Billharz R, Swayne DE, Przygodzki RM, Taubenberger JK, Katze MG, Tumpey TM. 2004. Global host immune response: Pathogenesis and transcriptional profiling of type A influenza viruses expressing the hemagglutinin and neuraminidase genes from the 1918 pandemic virus. J Virol 78(17):9499–9511.
Katz JM, Lim W, Bridges CB, Rowe T, Hu-Primmer J, Lu X, Abernathy RA, Clarke M, Conn L, Kwong H, Lee M, Au G, Ho YY, Mak KH, Cox NJ, Fukuda K. 1999. Antibody response in individuals infected with avian influenza A (H5N1) viruses and detection of anti-H5 antibody among household and social contacts. J Infect Dis 180:1763–1770.
Katzenellenbogen JM. 1988. The 1918 influenza epidemic in Mamre. S Afr Med J 74(7): 362–364.
Kawaoka Y, Webster RG. 1988. Molecular mechanism of acquisition of virulence in influenza virus in nature. Microb Pathog 5:311–318.
Kawaoka Y, Krauss S, Webster RG. 1989. Avian-to-human transmission of the PB1 gene of influenza A viruses in the 1957 and 1968 pandemics. J Virol 63:4603–4608.
Keeton R, Cusman AB. 1918. The influenza epidemic in Chicago. JAMA 71(24):1963. (Note: The 39.8 percent corrects an earlier report in JAMA by Nuzum on 11/9/18, p. 1562.)
Kilbourne E. 1977. Influenza pandemics in perspective. JAMA 237:1225–1228.
Kilbourne ED. 1997. Perspectives on pandemics: A research agenda. J Infect Dis 176(Suppl 1):S29–S31.
Koen JS. 1919. A practical method for field diagnoses of swine diseases. Am J Vet Med 14:468–470.
Kolata GB. 1999. Flu: The Story of the Great Influenza Pandemic of 1918 and the Search for the Virus That Caused It. New York: Farrar Straus & Giroux.
Koopmans M, Wilbrink B, Conyn M, Natrop G, van der Nat H, Vennema H, Meijer A, van Steenbergen J, Fouchier R, Osterhaus A, Bosman A. 2004. Transmission of H7N7 avian influenza A virus to human beings during a large outbreak in commercial poultry farms in the Netherlands. Lancet 363:587–593.
Krug RM, Yuan W, Noah DL, Latham AG. 2003. Intracellular warfare between human influenza viruses and human cells: The roles of the viral NS1 protein. Virology 309:181–189.
Kupradinun S, Peanpijit P, Bhodhikosoom C, Yoshioka Y, Endo A, Nerome K. 1991. The first isolation of swine H1N1 influenza viruses from pigs in Thailand. Arch Virol 118:289–297.
Lamb R, Krug R. 2001. Orthomyxoviridae: The viruses and their replication. In: Knipe D, Howley P, eds. Fields Virology. Vol. 1. Philadelphia, PA: Lippincott Williams & Wilkins. Pp. 1487–1531.
Lamb RA, Lai CJ. 1980. Sequence of interrupted and uninterrupted mRNAs and cloned DNA coding for the two overlapping nonstructural proteins of influenza virus. Cell 21:475–485.
Langmuir AD, Housworth J. 1969. A critical evaluation of influenza surveillance. Bull World Health Organ 41:393–398.
Lazarowitz SG, Choppin PW. 1975. Enhancement of the infectivity of influenza A and B viruses by proteolytic cleavage of the hemagglutinin polypeptide. Virology 68:440–454.
LeCount ER. 1919. The pathologic anatomy of influenza bronchopneumonia. JAMA 72: 650–652.
Li S, Schulman J, Itamura S, Palese P. 1993. Glycosylation of neuraminidase determines the neurovirulence of influenza A/WSN/33 virus. J Virol 67:6667–6673.
Li Y, Yamakita Y, Krug R. 1998. Regulation of a nuclear export signal by an adjacent inhibitory sequence: The effector domain of the influenza virus NS1 protein. Proc Natl Acad Sci USA 95:4864–4869.
Linder FE, Grove RD. 1943. Vital Statistics Rates in the United States: 1900–1940. Washington, DC: National Office of Vital Statistics. Government Printing Office.
Lipsman J. 2004. H7N2 Avian Influenza Identified in Westchester Resident. New York: Westchester County Department of Health.
Little TR, Garofalo CJ, Williams PA. 1918. B. influenzae and present epidemic. Lancet 2:4950.
Longini IM, Ackerman E, Elveback LR. 1978. An optimization model for influenza A epidemics. Math Biosci 38:141–157.
Longini IM, Halloran ME, Nizam A, Yang Y. 2004. Containing pandemic influenza with antiviral agents. Am J Epidemiol 159:623–633.
Low RB. 1920. In: Reports on Public Health and Medical Subjects. No. 4. London, England: Her Majesty’s Stationery Office.
Ludendorff E. 1919. Meine Kriegserinnerungen 1914–1918. Berlin, Germany: Ernst Siegfried Mittler und Sohn Verlagsbuchhandlung. P. 514.
Ludwig S, Stitz L, Planz O, Van H, Fitch WM, Scholtissek C. 1995. European swine virus as a possible source for the next influenza pandemic? Virology 212:551–561.
Marine WM, Workman WM. 1969. Hong Kong influenza immunologic recapitulation. Am J Epidemiol 90:406–415.
Marks G, Beatty WK. 1976. Epidemics. New York: Scribner.
Matrosovich MN, Gambaryan AS, Teneberg S, Piskarev VE, Yamnikova SS, Lvov DK, Robertson JS, Karlsson KA. 1997. Avian influenza A viruses differ from human viruses by recognition of sialyloigosaccharides and gangliosides and by a higher conservation of the HA receptor-binding site. Virology 233:224–234.
Meltzer MI, Cox NJ, Fukuda K. 1999. The economic impact of pandemic influenza in the United States: Priorities for intervention. Emerg Infect Dis 5:659–671.
Miller DL, Pereira MS, Clarke M. 1971. Epidemiology of the Hong Kong-68 variant of influenza A2 in Britain. Br Med J 1:475–479.
Ministry of Health, United Kingdom. 1960. The influenza epidemic in England and Wales, 1957–1958. In: Reports on Public Health and Medical Subjects. Vol. 100. London, England: Ministry of Health.
Monto AS, Iacuzio DA, La Montagne JR. 1997. Pandemic influenza: Confronting a reemergent threat. J Infect Dis 176:S1–S3.
Murray C, Biester HE. 1930. Swine influenza. J Am Vet Med Assn 76:349–355.
Nerome K, Ishida M, Oya A, Oda K. 1982. The possible origin H1N1 (Hsw1N1) virus in the swine population of Japan and antigenic analysis of the isolates. J Gen Virol 62:171–175.
Nguyen-Van-Tam JS, Hampson AW. 2003. The epidemiology and clinical impact of pandemic influenza. Vaccine 21:1762–1768.
Olson DR, Simonsen L, Edleson PJ, Morse SS. 2004. In: 4th International Conference on Emerging Infectious Diseases. Atlanta, GA.
O’Neill RE, Talon J, Palese P. 1998. The influenza virus NEP (NS2 protein) mediates the nuclear export of viral ribonucleoproteins. EMBO 17:288–296.
Palese P, Compans RW. 1976. Inhibition of influenza virus replication in tissue culture by 2-deoxy-2,3-dehydro-N-trifluoroacetylneuraminic acid (FANA): Mechanism of action. J Gen Virol 33:159–163.
Patterson KD, Pyle GF. 1991. The geography and mortality of the 1918 influenza pandemic. Bull Hist Med 65:4–21.
Peiris JS, Yu WC, Leung CW, Cheung CY, Ng WF, Nicholls JM, Ng TK, Chan KH, Lai ST, Lim WL, Yuen KY, Guan Y. 2004. Re-emergence of fatal human influenza A subtype H5N1 disease. Lancet 363:617–619.
Pettit DA. 1976. A Cruel Wind: America Experiences the Pandemic Influenza, 1918–1920. P. 32. PhD dissertation, University of New Hampshire.
Philip RN, Lackman DB. 1962. Observations on the present distribution of influenza A/swine antibodies among Alaskan natives relative to the occurrence of influenza in 1918–1919. Am J Hyg 75:322–334.
Policlinico. 1918, 6/30/18, 25(26), quoted in JAMA 71(9):780.
Portela A, Digard P. 2002. The influenza virus nucleoprotein: A multifunctional RNA-binding protein pivotal to virus replication. J Gen Virol 83:723–734.
Reichert TA, Sugaya N, Fedson DS, Glezen WP, Simonsen L, Tashiro M. 2001. The Japanese experience of vaccinating school children against influenza. N Engl J Med 344:889–896.
Reid AH, Taubenberger JK. 1999. The 1918 flu and other influenza pandemics: “Over there” and back again. Lab Invest 79:95–101.
Reid AH, Taubenberger JK. 2003. The origin of the 1918 pandemic influenza virus: A continuing enigma. J Gen Virol 84:2285–2292.
Reid AH, Fanning TG, Hultin JV, Taubenberger JK. 1999. Origin and evolution of the 1918 “Spanish” influenza virus hemagglutinin gene. Proc Natl Acad Sci USA 96:1651–1656.
Reid AH, Fanning TG, Janczewski TA, Taubenberger JK. 2000. Characterization of the 1918 “Spanish” influenza virus neuraminidase gene. Proc Natl Acad Sci USA 97:6785–6790.
Reid AH, Fanning TG, Janczewski TA, McCall S, Taubenberger JK. 2002. Characterization of the 1918 “Spanish” influenza virus matrix gene segment. J Virol 76:10717–10723.
Reid AH, Janczewski TA, Lourens RM, Elliot AJ, Daniels RS, Berry CL, Oxford JS, Taubenberger JK. 2003. 1918 influenza pandemic caused by highly conserved viruses with two receptor-binding variants. Emerg Infect Dis 9(10):1249–1253.
Reid AH, Fanning TG, Janczewski TA, Lourens R, Taubenberger JK. 2004. Novel origin of the 1918 pandemic influenza virus nucleoprotein gene segment. J Virol 78(22):12462–12470.
Rice G. 1988. Black November. Wellington, New Zealand: Allen and Unwin. P. 140.
Robertson JD. 1918. Report of an Epidemic of Influenza in Chicago Occurring During the Fall of 1918. Chicago, IL: Department of Health.
Rosenau MJ, Last JM. 1980. Maxcy-Rosenau Preventative Medicine and Public Health. New York: Appleton-Century-Crofts.
Rott R, Klenk HD, Nagai Y, Tashiro M. 1995. Influenza viruses, cell enzymes, and pathogenicity. Am J Respir Crit Care Med 152:S16–S19.
Schafer JR, Kawaoka Y, Bean WJ, Suss J, Senne D, Webster RG. 1993. Origin of the pandemic 1957 H2 influenza A virus and the persistence of its possible progenitors in the avian reservoir. Virology 194:781–788.
Scholtissek C. 1994. Source for influenza pandemics. Eur J Epidemiol 10:455–458.
Scholtissek C, Koennecke I, Rott R. 1978a. Host range recombinants of fowl plague (influenza A) virus. Virology 91:79–85.
Scholtissek C, Rohde W, Von Hoyningen V, Rott R. 1978b. On the origin of the human influenza virus subtypes H2N2 and H3N2. Virology 87:13–20.
Scholtissek C, Burger H, Kistner O, Shortridge KF. 1985. The nucleoprotein as a possible major factor in determining host specificity of influenza H3N2 viruses. Virology 147:287–294.
Scholtissek C, Ludwig S, Fitch W. 1993. Analysis of influenza A virus nucleoproteins for the assessment of molecular genetic mechanisms leading to new phylogenetic virus lineages. Arch Virol 131:237–250.
Schulze IT. 1997. Effects of glycosylation on the properties and functions of influenza virus hemagglutinin. J Infect Dis 176(Suppl 1):S24–S28.
Seo SH, Hoffmann E, Webster RG. 2002. Lethal H5N1 influenza viruses escape host antiviral cytokine responses. Nat Med 8:950–954.
Serfling R. 1963. Methods for current statistical analysis of excess pneumonia-influenza deaths. Pub Health Rep 78:494–506.
Serfling RE, Sherman IL, Houseworth WJ. 1967. Excess pneumonia-influenza mortality by age and sex in three major influenza A2 epidemics, United States, 1957-58, 1960 and 1963. Am J Epidemiol 86:433–441.
Shope R. 1958. Influenza: History, epidemiology, and speculation. Pub Health Rep 73: 165–178.
Shope RE. 1936. The incidence of neutralizing antibodies for swine influenza virus in the sera of human beings of different ages. J Exp Med 63:669–684.
Shope RE, Lewis PA. 1931. Swine influenza: Experimental transmission and pathology. J Exp Med 54:349–359.
Shu L, Bean W, Webster R. 1993. Analysis of the evolution and variation of the human influenza A virus nucleoprotein gene from 1933 to 1990. J Virol 67:2723–2729.
Simonsen L, Clarke MJ, Schonberger LB, Arden NH, Cox NJ, Fukuda K. 1998. Pandemic versus epidemic influenza mortality: A pattern of changing age distribution. J Infect Dis 178:53–60.
Simonsen L, Fukuda K, Schonberger LB, Cox NJ. 2000. The impact of influenza epidemics on hospitalizations. J Infect Dis 181:831–837.
Simonsen L, Reichert TA, Miller M. 2003. In: Kawaoka Y, ed. Options for the Control of Influenza V. International Congress Series 1263. Vol. ICS 1265. Okinawa, Japan: Elsevier. Pp. 791–794.
Smith W, Andrewes C, Laidlaw P. 1933. A virus obtained from influenza patients. Lancet 225:66–68.
Sohier R, Henry M. 1969. Epidemiological data on Hong Kong influenza in France. Bull World Health Organ 41:402–404.
Soper G. 1918, November 8. The influenza-pneumonia pandemic in the American Army camps, September and October 1918. Science 454.
Soper G. undated. The Influenza Pandemic in the Camps. Undated draft report. Sanitation Corps, NA, RG 112, Box 394.
Stevens J, Corper AL, Basler CF, Taubenberger JK, Palese P, Wilson IA. 2004. Structure of the uncleaved human H1 hemagglutinin from the extinct 1918 influenza virus. Science 303:1866–1870.
Stuart-Harris CH. 1970. Pandemic influenza: An unresolved problem in prevention. J Infect Dis 122:108–115.
Subbarao K, Klimov A, Katz J, Regnery H, Lim W, Hall H, Perdue M, Swayne D, Bender C, Huang J, Hemphill M, Rowe T, Shaw M, Xu X, Fukuda K, Cox N. 1998. Characterization of an avian influenza A (H5N1) virus isolated from a child with a fatal respiratory illness. Science 279:393–396.
Talon J, Horvath CM, Polley R, Basler CF, Muster T, Palese P, Garcia-Sastre A. 2000. Activation of interferon regulatory factor 3 is inhibited by the influenza A virus NS1 protein. J Virol 74:7989–7996.
Taubenberger JK, Reid AH, Krafft AE, Bijwaard KE, Fanning TG. 1997. Initial genetic characterization of the 1918 “Spanish” influenza virus. Science 275:1793–1796.
Taubenberger J, Reid A, Fanning T. 2000. The 1918 influenza virus: A killer comes into view. Virology 274:241–245.
Taubenberger JK, Reid AH, Janczewski TA, Fanning TG. 2001. Integrating historical, clinical and molecular genetic data in order to explain the origin and virulence of the 1918 Spanish influenza virus. Philos Trans R Soc Lond B Biol Sci 356:1829–1839.
The Mobilization of the American National Red Cross During the Influenza Pandemic 1918–1919. 1920. Geneva, Switzerland. P. 24.
Thomson D, Thomson R. 1934a. Annals of the Pickett-Thomson Research Laboratory. Volume IX, Influenza. Baltimore, MD: Williams and Wilkens.
Thomson D, Thomson R. 1934b. Annals of the Pickett-Thomson Research Laboratory. Volume X, Influenza. Baltimore, MD: Williams and Wilkens.
Thompson WW, Shay DK, Weintraub E, Brammer L, Cox N, Anderson LJ, Fukuda K. 2003. Mortality associated with influenza and respiratory syncytial virus in the United States. JAMA 289:179–186.
To KF, Chan PK, Chan KF, Lee WK, Lam WY, Wong KF, Tang NL, Tsang DN, Sung RY, Buckley TA, Tam JS, Cheng AF. 2001. Pathology of fatal human infection associated with avian influenza A H5N1 virus. J Med Virol 63(3):242–246.
Tran TH, Nguyen TL, Nguyen TD, Luong TS, Pham PM, Nguyen VC, Pham TS, Vo CD, Le TQ, Ngo TT, Dao BK, Le PP, Nguyen TT, Hoang TL, Cao VT, Le TG, Nguyen DT, Le HN, Nguyen KT, Le HS, Le VT, Christiane D, Tran TT, Menno de J, Schultsz C, Cheng P, Lim W, Horby P, Farrar J; World Health Organization International Avian Influenza Investigative Team. 2004. Avian influenza A (H5N1) in 10 patients in Vietnam. N Engl J Med 350:1179–1188.
Tumpey TM, Garcia-Sastre A, Mikulasova A, Taubenberger JK, Swayne DE, Palese P, Basler CF. 2002. Existing antivirals are effective against influenza viruses with genes from the 1918 pandemic virus. Proc Natl Acad Sci USA 99:13849–13854.
Tumpey TM, Garcia-Sastre A, Taubenberger JK, Palese P, Swayne DE, Basler CF. 2004. Pathogenicity and immunogenicity of influenza viruses with genes from the 1918 pandemic virus. Proc Natl Acad Sci USA 101(9):3166–3171.
U.S. Bureau of the Census. 1921. Mortality Statistics. Washington, DC: Government Printing Office. P. 30.
U.S. Department of Commerce. 1976. Historical Statistics of the United States: Colonial Times to 1970. Washington, DC: Government Printing Office.
Van Hartesveldt FR. 1992. The 1918–1919 Pandemic of Influenza: The Urban Impact in the Western World. Lewiston, NY: Edwin Mellen Press. Pp. 121, 144.
Vaughn S. 1980. Holding Fast the Line: Democracy, Nationalism, and the Committee on Public Information. Chapel Hill, NC: University of North Carolina Press.
Vaughn WT. 1921. Influenza: An epidemiological study. Am J Hyg Monograph No. 1.
Viboud C, Grais RF, Lafont BAP, Miller MA, Simonsen L. 2004. Multi-national impact of the 1968 Hong-Kong influenza pandemic: Evidence for a smoldering pandemic. Submitted.
Wang X, Li M, Zheng H, Muster T, Palese P, Beg AA, Garcia-Sastre A. 2000. Influenza A virus NS1 protein prevents activation of NF-kappaB and induction of alpha/beta interferon. J Virol 74:11566–11573.
Webby RJ, Webster RG. 2003. Are we ready for pandemic influenza? Science 302:1519–1522.
Webster R, Rott R. 1987. Influenza virus A pathogenicity: The pivotal role of hemagglutinin. Cell 50:665–666.
Webster RG, Bean WJ, Gorman OT, Chambers TM, Kawaoka Y. 1992. Evolution and ecology of influenza A viruses. Microbiol Rev 56:152–179.
Webster RG, Sharp GB, Claas EC. 1995. Interspecies transmission of influenza viruses. Am J Respir Crit Care Med 152:S25–S30.
Weis W, Brown JH, Cusack S, Paulson JC, Skehel JJ, Wiley DC. 1988. Structure of the influenza virus haemagglutinin complexed with its receptor, sialic acid. Nature 333:426–431.
WHO (World Health Organization). 1971. Stat Bull 52:8–11.
WHO. 2004. Avian Influenza A(H7) Human Infections in Canada. [Online]. Available: http://www.who.int/csr/don/2004_04_05/en/ [accessed December 17, 2004].
Wilson IA, Skehel JJ, Wiley DC. 1981. Structure of the haemagglutinin membrane glycoprotein of influenza virus at 3 A resolution. Nature 289:366–373.
Winternitz MC, Wason IM, McNamara FP. 1920. The Pathology of Influenza. New Haven, CT: Yale University Press.
Wolbach SB. 1919. Comments on the pathology and bacteriology of fatal influenza cases, as observed at Camp Devens, Mass. Johns Hopkins Hospital Bulletin 30:104.
Woods GT, Schnurrenberger PR, Martin RJ, Tompkins WA. 1981. Swine influenza virus in swine and man in Illinois. J Occup Med 23:263–267.
Zamarin D, Palese P. 2004 (in press). Influenza virus: Lessons learned. In: Kowalski JB, Morissey JB, eds. International Kilmer Conference Proceedings. Champlain, NY: Polyscience Publications.
Zhou NN, Senne DA, Landgraf JS, Swenson SL, Erickson G, Rossow K, Liu L, Yoon KJ, Krauss S, Webster RG. 2000. Emergence of H3N2 reassortant influenza A viruses in North American pigs. Vet Microbiol 74:47–58.


























































