
1
Background Information
This report focuses on the health effects of low-dose, low-LET (low linear energy transfer) radiation. In this chapter the committee provides background information relating to the physical and chemical aspects of radiation and the interaction of radiation with the target molecule DNA. The committee discusses contributions of normal oxidative DNA damage relative to radiation-induced DNA damage and describes the DNA repair mechanisms that mammalian cells have developed to cope with such damage. Finally, this chapter introduces a special subject, the physical characteristics that determine the relative biological effectiveness (RBE) of neutrons, estimates of which are required in the derivation of low-LET radiation risk estimates from atomic bomb survivors.
PHYSICAL ASPECTS OF RADIATION
The central question that must be resolved when considering the physical and biological effects of low-dose ionizing radiation is whether the effects of ionizing radiation and the effects of the free radicals and oxidative reaction products generated in normal cellular metabolism are the same or different. Is ionizing radiation a unique insult to cells, or are its effects lost in the ocean of naturally occurring metabolic reaction products? Can cells detect and respond to low doses of ionizing radiation because of detectable qualitative and quantitative differences from endogenous reaction products?
Different Types of Ionizing Radiation
Ionizing radiation, by definition, contains enough energy to displace electrons and break chemical bonds. Charged particles, such as high-energy electrons, protons, α-particles, or fast heavy ions, are termed directly ionizing because, while they traverse the cell, they ionize numerous molecules by direct collisions with their electrons. Electromagnetic radiations, such as X- and γ-rays, consist of photons that can travel relatively large distances in tissue without interaction. Once an interaction with one of the electrons in the material occurs, part or all of the photon energy is transferred to the electron. The energetic electrons released in this way produce the bulk of ionizations. X- and γ-rays are accordingly termed “indirectly ionizing” radiation. This term is also applied to fast neutrons, because they too traverse large distances in tissue without interaction but can, in occasional collisions, transfer much of their energy to atomic nuclei that in turn produce the main part of the ionizations.
In addition to the distinction between indirectly ionizing and directly ionizing (i.e., uncharged and charged radiation) a distinction is made between sparsely ionizing, or low-LET, and densely ionizing, or high-LET, radiation. The (unrestricted) LET of an ionizing charged particle is defined as the average energy lost by the particle due to electronic interactions per unit length of its trajectory; it is expressed in kiloelectronvolts per micrometer (keV/μm).1 High-energy electromagnetic radiations, such as X-rays or γ-rays, are sparsely ionizing since, in tissue, they release fast electrons that have low LET. Neutrons are densely ionizing because in tissue they release fast protons and heavier atomic nuclei that have high LET.
Figure 1-1 gives the LET of electrons as a function of their kinetic energy and compares it to the considerably higher LET of protons. It is seen that electrons are generally sparsely ionizing while protons are, at moderate energies, densely ionizing. However it is also noted that very energetic protons, as they occur in altitudes relevant to aviation and in space, are sufficiently fast to be sparsely ionizing.
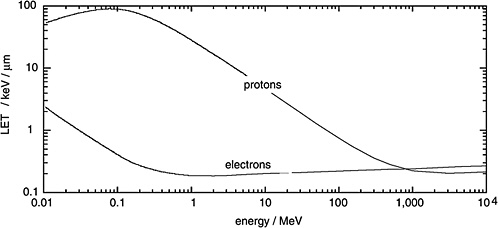
FIGURE 1-1 Linear energy transfer of protons and electrons in water. SOURCE: Data from ICRU (1970).
The effects of high-LET particles (i.e., protons and heavier ions) are outside the scope of this report. However, neutrons and their high relative biological effectiveness must be considered in the context of low-LET risk estimates derived from the observations on delayed health effects among A-bomb survivors. The reason is that a small fraction of the absorbed dose to A-bomb survivors was due not to the predominant high-energy γ-rays, but to fast neutrons. Because of the greater effectiveness of these fast neutrons, this small dose component must be taken into consideration.
Photon Spectral Distributions
The absorption and scattering of photons depends on their energy. The γ-rays from radioactive decay consist of monoenergetic photons that do not exceed several million electronvolts (MeV) in energy; γ-rays that result from the fission of uranium or plutonium have a spectrum of energies with a maximum of 2 MeV. Higher-energy γ-rays, up to 7 MeV, can be generated by inelastic scattering, as occurred in the neutron-nitrogen interaction from the atomic bomb explosions in Hiroshima and Nagasaki.
Artificially produced X-rays have a wide spectrum of energies resulting from the deceleration of electrons as they traverse high-atomic-number materials. A continuous distribution of photon energies is generated, with a mean energy of about one-third the maximal energy of the accelerated electrons. Added filtration selectively removes the “soft” (i.e., less energetic) photon component and, thus, hardens the X-rays. Discrete energy “spikes” also occur in the X-ray spectrum; these spikes originate in the ejection of electrons from atoms of the affected element, which is followed by the transition of electrons from outer shells to inner shells of the atom releasing photons of discrete energy. Conventional X-rays, used for diagnostic radiology, are commonly produced with accelerating voltages of about 200 kV. For mammography, where high contrast is sought and only a moderate thickness of tissue must be traversed by the X-rays, the low acceleration voltage of 29 kV is usually employed.
There are three different types of energy-transfer processes whereby photons of sufficient energy eject electrons from an atom, which can then interact with other atoms and molecules to produce a cascade of alterations that ultimately lead to observable biological effects. These are the photoelectric process, Compton scattering, and pair production.
At low energies (<0.1 MeV), the photoelectric process dominates in tissue. A photon interacts with and ejects an electron from one of the inner shells of an atom. The photon is extinguished, and most of its energy is imparted to the ejected electron as kinetic energy.
At medium photon energies (about 0.5–3.5 MeV), Compton scattering is the most probable event. Compton scattering occurs when an incoming photon’s energy greatly exceeds the electron-binding energy of the affected atom. In this case the energy of the incoming photon is converted into the kinetic energy of an ejected electron and a secondary “scattered” photon. The scattered photon has less energy than the primary photon and can undergo further Compton scattering until its energy is sufficiently degraded for the photoelectric process to occur.
At energies greater than 1.02 MeV, pair production can occur. A photon interacts with an atomic nucleus, and the photon energy is converted into a positron and an electron. The photon energy above 1.02 MeV is converted into the kinetic energy of the newly created particles. The electron and the positron interact with and can ionize other molecules.
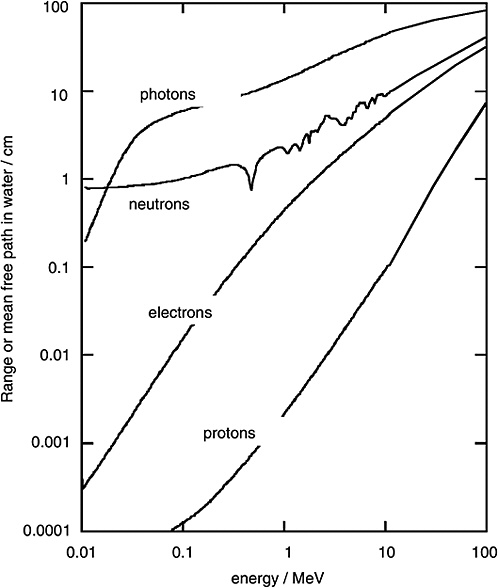
FIGURE 1-2 Mean free path of photons and neutrons in water and range of electrons and protons. SOURCE: Data from ICRU (1970).
The positron ultimately interacts with another electron, and this results in an “annihilation” event in which the mass is extinguished and two 0.51 MeV photons are emitted in opposite directions. The annihilation photons can themselves produce further ionizations.
Figure 1-2 shows the mean free path for monoenergetic photons (i.e., the average distance in water until the photon undergoes an interaction). To compare the penetration depth of photon radiation with that of electron radiation, the mean range of electrons of specified energy is given in the same diagram. It is seen that the electrons released by photons are always considerably less penetrating than the photons themselves.
Figure 1-3 compares in terms of the distributions of photon energy fluence the γ-rays from the A-bomb explosions with the distributions of photon energy for orthovoltage X-rays and low-energy mammography X-rays. These different electromagnetic radiations are all classified as low-LET (i.e., sparsely ionizing) radiation. There are, nevertheless, differences in effectiveness and possibly also differences in the risk for late effects due to these radiations.
Track Structure
The passage of fast electrons through tissue creates a track of excited and ionized molecules that are relatively far apart. X- and γ-rays produce electrons with relatively low linear energy transfer, (i.e., energy loss per unit track length) and are considered low-LET radiation. For example, the track average of unrestricted LET of the electrons liberated by cobalt-60 (60Co) gamma rays is about 0.25 keV/μm, which can be contrasted with an average LET of about 180 keV/μm for a 2 MeV α-particle, a high-LET radiation. LET is an important measure in the evaluation of relative biological effectiveness (ICRU 1970; Engels and Wambersie 1998) of a given kind of radiation.
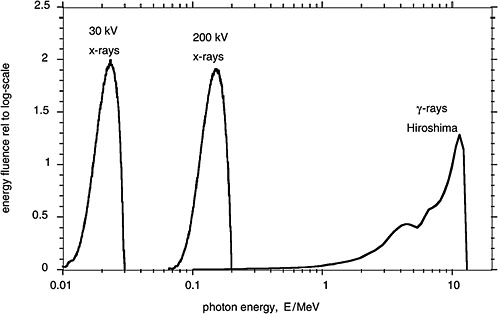
FIGURE 1-3 Distributions of photon energy fluence for mammography X-rays, orthovoltage X-rays, and γ-rays from the atomic bomb explosion in Hiroshima. The distributions of the energy fluence relative to the logarithmic scale of energy are plotted, because they represent roughly the fractional contribution of incident photons of specified energy to the dose absorbed by a person. SOURCE: Data from Seelentag and others (1979) and Roesch (1987).
Different Effectiveness of γ-Rays and X-Rays
LET and Related Parameters of Radiation Quality
While γ-rays and X-rays of various energies are all sparsely ionizing, in the body they generate electrons with somewhat different spectra of LET values (ICRU 1970). To quantify the differences, reference is usually made to the dose average LET or to the mean values of the related microdosimetric parameter dose-averaged linear energy, y.
Figure 1-4 gives the dose average LET values for the electrons released by monoenergetic photons (solid curves) and compares these values to the averages for 29 kV mammography X-rays and 200 kV X-rays (solid circles and squares, respectively; ICRP 2003). In addition to the dose average, LD, of the unrestricted LET, the diagram contains the dose averages, LD,Δ, of the restricted LET, LΔ. The restricted LET treats the Δ-rays beyond the specified cutoff energy Δ as separate tracks. This accounts in an approximate way for the increased local energies due to Δ-rays and therefore provides larger values that are more meaningful than those of unrestricted LET.
High-energy photons (e.g., 60Co γ-rays) release Compton electrons of comparatively high energy and correspondingly low LET. Photons of less energy (e.g., conventional 200 kV X-rays) produce less energetic Compton electrons with higher LET. This explains the substantial difference between the mean LET of high-energy γ-rays and conventional X-rays. For lower-energy X-rays the photon energy is further reduced, and the photo effect (i.e., the total transfer of photon energy to electrons) begins to dominate. Accordingly, the average energy of the electrons begins to increase again, which explains the relatively small difference in average LET between 200 kV X-rays and soft X-rays. At very low photon energies (i.e., less than about 20 keV) the LET values increase strongly, but these ultrasoft X-rays are of little concern in radiation protection because of their very limited penetration depth.
The dose average, LD,Δ, of the restricted LET is a parameter that correlates with the low dose effectiveness of photon or electron radiation. With a cutoff value Δ=1keV, the numerical values of LD,Δ are consistent with a low-dose RBE of about 2 for conventional X-rays versus γ-rays. A similar dependence on photon energy is seen in the related microdosimetric parameter dose lineal energy, y, which has been used as reference parameter by the liaison committee of the International Commission on Radiological Protection (ICRP) and the International Commission on Radiation Units and Measurements (ICRU) in The Quality Factor in Radiation Protection (ICRU 1986). Figure 1-5 gives values of its
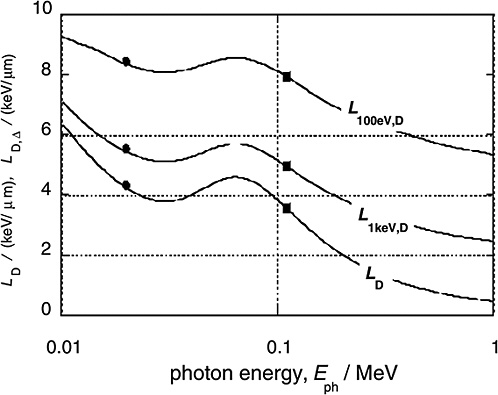
FIGURE 1-4 The dose mean restricted and unrestricted linear energy transfer for electrons liberated by monoenergetic photons of energy Eph. The dots and squares give the values for the 29 kVp and the 200 kVp X-rays. They are plotted at the weighted photon energies of the X-ray spectra. SOURCE: Data from Kellerer (2002).
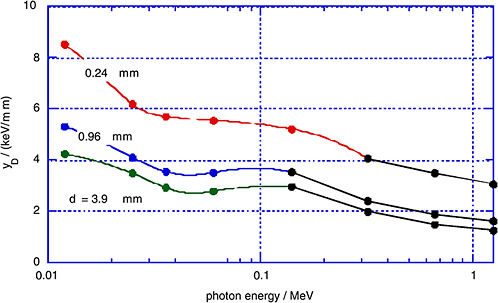
FIGURE 1-5 Measured dose average lineal energy, yD, for monoenergetic photons and for different simulated site diameters, d. SOURCE: Data from Kliauga and Dvorak (1978).
dose average, yD, as measured by Kliauga and Dvorak (1978) for various photon radiations and for different simulated site diameters, d.
The γ-rays from the atomic bomb explosions had average energies between 2 and 5 MeV at the relevant distances (Straume 1996). Figures 1-4 and 1-5 do not extend to these energies; however, it is apparent from Figures 1-4 and 1-5 that the mean values of the restricted LET or the lineal energy do not decrease substantially beyond a photon energy of 1 MeV. There is, thus, little indication that the hard γ-rays from the atomic bombs should have an RBE substantially less than unity compared to conventional 60Co γ-rays.
Information from In Vitro Studies
It has long been recognized in experimental radiobiology that low-LET radiations do not all have the same effectiveness at low doses. With regard to mutations in Tradescantia, aberrations in human lymphocytes, and killing of mouse oocytes (Bond and others 1978), conventional 200 kV X-rays have been found to be about twice as effective at low doses as high-energy γ-rays. Fast electrons may be even less effective than γ-rays. These differences are most clearly documented in cell studies and, especially, in studies on chromosome aberrations (Sinclair 1985; ICRU 1986). The most reliable and detailed data on photon RBE exist for chromosome aberrations in human lymphocytes. Edwards and others (1982) have obtained the data for dicentrics in human lymphocytes listed in Table 1-1 for 15 MeV electrons, 60Co γ-rays, and 250 kV X-rays. New data have since confirmed these substantial differences of effectiveness for different types of penetrating low-LET radiations.
Sasaki and colleagues (1989; Sasaki 1991) have determined the yields of dicentrics in human lymphocytes over a broad range of photon energies. The upper panel of Figure 1-6 gives the linear coefficients (and standard errors) from linear-quadratic fits to the dose dependencies. The closed circles relate to γ-rays and to broad X-ray spectra; the squares, to characteristic X-rays and monoenergetic photons
TABLE 1-1 Low-Dose Coefficients (and standard errors) for Induction of Chromosome Aberrations in Human Lymphocytes by Low-LET Penetrating Radiation
from synchrotron radiation. The lower panel gives analogous data obtained by Schmid and others (2002).
The diagram demonstrates that there is a substantial decrease of the yield of dicentrics from conventional X-rays to γ-rays. The photon energies below 20 keV are of special interest with regard to biophysical consideration, but are less relevant to exposure situations in radiation protection. They are included here to show the full trend of the energy dependence.
It is seen that the low-dose RBE for dicentrics for moderately filtered 200 kV X-rays is about 2–3 relative to γ-rays, while the RBE of mammographic X-rays (29 kV) relative to the moderately filtered 200 kV X-rays is somewhat in excess of 1.5.
The data for dicentrics in Figures 1-6 are reasonably consistent with the LET values in Figure 1-4 for a cutoff value in excess of 1 keV. The difference by a factor of 2–3 in the low-dose effectiveness of conventional X-rays and γ-rays has been known and, even if it should apply equally to radiation-induced late effects, would not necessarily require a departure from the current convention for radiation protection, which assigns the radiation weighting factor unity to all photon radiations. However, the difference has to be noted whenever risk estimates are derived from exposures to γ-rays and then applied to X-rays.
Apart from these considerations it is uncertain whether the marked dependence of the low-dose RBE on photon energy for chromosome aberrations also is representative for late radiation effects in man. The dependence of RBE on photon energy for dicentric chromosomes reflects the fact that the dose dependencies have large curvature for 60Co γ-rays (α/β = 0.2 Gy in the data reported by Schmid and others 2002), but little curvature for 29 kV X-rays (α/β = 1.9 Gy). If there were no curvature below 1 Gy in the dose relations for chromosome aberrations, the low-dose RBE of 29 kV X-rays would be only 1.65 compared to 60Co γ-rays. Since the dose dependence for solid tumors among A-bomb survivors indicates little curvature, the dependence of risk on photon energy may be similarly weak for tumor induction in man. It is of interest to compare the biophysical information and the experimental results to the radioepidemiologic evidence for health effects.
Information from Radioepidemiology
Numerous epidemiologic studies on medical cohorts have provided risk estimates that exhibit considerable variation. Many of these studies on patients relate to X-ray exposures, but there is no consistent epidemiologic evidence for higher risk factors from X-rays than from γ-rays. In fact, while the risk estimates from medical studies are not inconsistent with those for atomic bomb survivors, they tend to be, as a whole, somewhat lower (UNSCEAR 2000b). The radiation-related increase in breast cancer incidence can serve as an example because it has been most thoroughly studied.
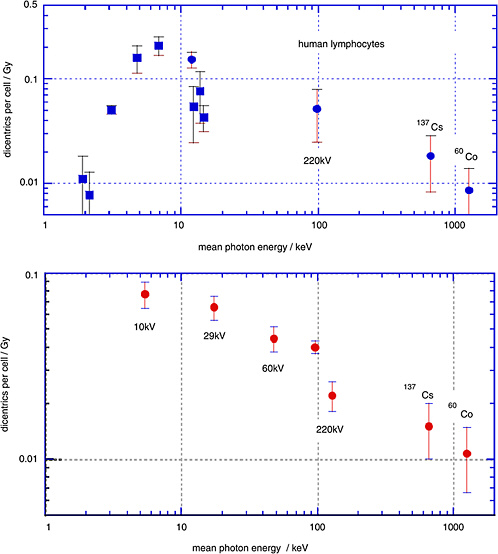
FIGURE 1-6 Data points are linear coefficients (and standard errors) of the dose dependence for dicentric chromosomes in human peripheral blood lymphocytes. Squares are for monoenergetic photons; circles are X-ray spectra or γ-rays. The two data points in the lower panel labeled 220 kV both had 220 kV generating voltage, but the filtration was different. SOURCE: Upper panel: Data from Sasaki and others (1989; Sasaki 1991). Lower panel: Data from Schmid and others (2002).
Figure 1-7 gives risk estimates from major studies on radiation-induced breast cancer. The estimated risk coefficients (and 90% confidence intervals) are expressed in terms of the excess relative risk (ERR) per gray and the excess absolute risk (EAR) per gray per 10,000 person-years (PY).
The uncertainties are large, and the risk estimates vary widely because the patient treatment regimes differed not only in the type of radiation but also in the various exposure modalities, such as acute, fractionated, or protracted exposure; whole- or partial-body exposure; exposure rate; and
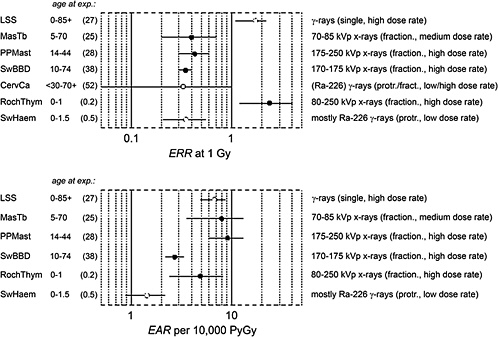
FIGURE 1-7 Excess relative risk (and 90% confidence interval) from various epidemiologic studies of breast cancer. The upper panel shows the excess relative risk per gray, the lower panel, the absolute risk per 10,000 person-years per gray. (For the description of individual studies, see UNSCEAR 2000b and Preston and others 2002a.) The confidence limit for the study of cervical carcinoma patients is recalculated. Cohorts: LSS: Life Span Study of atomic bomb survivors; MasTb: Massachusetts tuberculosis patients; PPMast: New York postpartum mastits patients; SwBBD: Swedish benign breast disease patients; CervCa: cervical cancer patients (case-control study); RochThym: Rochester infants with thymic enlargement; SwHem: Swedish infants with skin hemangioma.
magnitude of the exposure. Furthermore, there are ethnic differences, including those related to life-style, that are associated with greatly different background rates of breast cancer. Populations with low spontaneous rates tend to exhibit comparatively high ERR, while their EAR tends to be low. This complicates the comparison of risk estimates, since it remains uncertain whether relative or absolute excess incidence is the more relevant measure of risk.
The various exposed cohorts also differ considerably in the duration of follow-up and, especially, the age at exposure. The last two studies (RochThym, SwHem) relate to exposures in childhood, while the remainder refer to exposures at intermediate or higher ages. The last factor is especially critical, because both ERR and lifetime integrated EAR decrease substantially with increasing age at exposure.
The dominant influence of the various modifying factors makes it impossible on the basis of epidemiologic data to confirm the difference in effectiveness between γ-rays and X-rays or the difference between X-rays of different energies. Studies related to other types of cancer are even further removed from providing an answer. Thus, although cell studies and biophysical considerations suggest a low-dose RBE for conventional X-rays versus hard γ-rays of about 2–3, this difference cannot be confirmed at present through epidemiologic investigations.
Effects of Radiation on DNA, Genes, and Chromosomes
The probability that a low-LET primary electron will interact with a DNA molecule along its track is low, but a direct interaction of this sort is possible (Nikjoo and others 2002). Along the primary electron track, secondary electrons with lower energies are also formed, producing clusters of ionizations (see Figure 1-8, panel A). If such an ionization cluster occurs near a DNA molecule, multiple damages can occur in a very localized segment of the DNA (Figure 1-8, panel B). These clusters have been referred to as as clustered-damage or locally multiply damaged sites (LMDS) (Ward and others 1985; Goodhead 1994).
Figure 1-8 illustrates two typical structures of electron tracks produced by low-LET photons (e.g., γ-rays). The wavy lines outside the sphere represent primary and second-
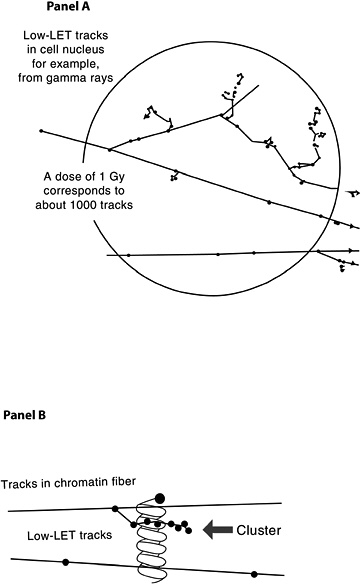
FIGURE 1-8 Panel A: Illustration of primary and secondary electron tracks producing clusters of ionization events. The calculated number of tracks is based on a nucleus with a diameter of 8 μm. The track size is enlarged relative to the nucleus to illustrate the theoretical track structure. Panel B: Illustration of clustered damage. The arrow identifies an ionization cluster near a DNA molecule to represent the possibility of locally multiply damaged sites. Only a segment of the electron track is illustrated in Panel B.
ary photons; the straight lines represent the paths of ejected electrons. For clarity of presentation, the size of the tracks is increased relative to the cell and is not drawn to scale. As the energetic electron interacts with atoms of the material, secondary electrons are produced and kinetic energy is lost. Such collisions can result in deflection of the primary electron from its original path (Figure 1-8, panel A). Important components of the track structure are the clusters of secondary ionizations that occur in a very small volume (see Figure 1-8, panel B). These clusters, acting directly or indirectly on the DNA molecule, may produce clustered damage, LMDS, that may in turn be refractory to repair. The likely site of health effects of low-dose radiation is the genetic material, which directs the structure and function of the organism. This genetic material is made up of DNA organized into genes and chromosomes (for a brief description, see Appendix A). Radiation can damage DNA as described in this chapter, and the damage can result in cell lethality, impaired cell function, or may produce damage involved in the carcinogenic process. Radiation has also been shown to produce heritable gene mutations in animals. For a basic description of gene mutations, see Appendix A.
Relative Biological Effectiveness of Neutrons
This report assesses the biological effects of low-LET radiation, that is, photons and electrons. It does not deal with densely ionizing radiation, such as heavy ions (including α-particles) and fast neutrons. Although neutrons need not be considered here on their own account, they must be accounted for in the analysis of the most important source of information on radiation risks, observations on the atomic bomb survivors of Hiroshima and Nagasaki. Such analysis requires consideration of the relative biological effectiveness of neutrons. The following remarks deal with the RBE of neutrons in general terms.
According to the 1986 dosimetry system, DS86, only a small fraction of the absorbed dose to atomic bomb survivors was due to neutrons—about 2% in Hiroshima in the most relevant dose range and 0.7% in Nagasaki (Roesch 1987). The current reevaluation of the Hiroshima and Nagasaki dosimetry, DS02, is in general agreement with these observations. However, although the absorbed dose fraction of neutrons was small in both cities, it is known from a multitude of radiobiological investigations that the RBE of small neutron doses can be large enough for even the small absorbed dose fraction to add appreciably to the late health effects among atomic bomb survivors.
Fast neutrons interact with exposed tissue predominantly by releasing recoil nuclei. At neutron energies up to a few million electronvolts, the energy transfer is predominantly to protons. On the average, a neutron transfers half its energy to a recoil proton in a collision. Neutrons of 1 MeV therefore produce recoil protons with an average initial energy of 500 keV. At a neutron energy of 0.4 MeV, the typical recoil proton energy is 200 keV, enough to allow the proton to go through its maximal LET of about 100 keV/μm, which is reached at its Bragg peak energy of 0.1 MeV. The ionization density in such proton tracks is far greater than that in an electron track, as depicted in Figure 1-1. It is evident that the resulting high local energy concentration will produce far more clusters of closely spaced ionizations than do low-LET photons and thus more LMDS (clustered damage) that may remain unrepaired or misrepaired. In addition, recoil protons have track lengths of a few micrometers, so critical damage can, with fairly high probability, be caused in neighboring chromosomal structures. The interaction of closely spaced chromosomal damage has long been noted to
be a critical factor in the production of chromosomal aberrations (Lea 1946).
Recoil protons with energy of a few hundred kiloelectronvolts appear, in line with the above biophysical considerations, to be the particles that produce maximal cellular damage per unit energy imparted. This is confirmed by various experimental studies that consistently demonstrate the maximal effectiveness of neutrons at a neutron energy of about 0.4 MeV (Kellerer and Rossi 1972b).
The dose-effect relationship, E(D), for photons can in many radiobiological investigations be described as a linear quadratic function of absorbed dose:
(1-1)
In experiments with fast neutrons, the effect is typically proportional to the absorbed dose, Dn, of neutrons over a variable dose range depending on the tissue and effect:
(1-2)
The linear dose coefficient, an, for neutrons is always substantially larger than the linear dose coefficient, a, for photon radiation. The RBE of neutrons is defined as the ratio of a γ-ray dose to the neutron dose that produces the same effect:
(1-3)
In terms of Equations (1-1) and (1-2), RBE can be expressed as a function of the neutron dose or the photon dose. The latter expression is somewhat simpler:
(1-4)
This implies that RBE assumes its maximal value, RBEmax = an/a, at low doses, whereas it decreases with increasing dose and then tends to be inversely proportional to the photon dose.
Experimental Observations
Indeed, numerous experimental investigations of chromosomal aberrations, cellular transformations, and cell killing have confirmed that maximal RBE values of neutrons occur at low doses and that, at somewhat higher doses, RBE varies inversely with increasing reference dose (i.e., the photon dose). The same has been observed for more complex effects such as opacification of the lens and, more important in the context of risk assessment, induction of tumors in animals. A synopsis of such findings was provided in the context of the microdosimetric interpretation of the neutron RBE (Kellerer and Rossi 1972b).
Although the general features of the dependence of neutron RBE on dose are brought out consistently in experimental studies, the numerical values of RBE vary, and the variation appears to be largely a matter of the different magnitude of the linear dose component for photon radiation.
Cell survival curves usually exhibit pronounced initial slopes, and the observed maximal neutron RBE rarely exceeds a factor of about 10. For dicentric chromosomal aberrations in human lymphocytes, values of about 70 are obtained for the maximal RBE of 0.5 MeV neutrons against γ-rays (Dobson and others 1991; Schmid and others 2000). This large maximal value might be seen as an indication of an exceptionally high effectiveness of neutrons at low doses. In fact the dose-effect relationship for neutrons is simply linear, and the high maximal RBE of neutrons is merely a reflection of the very shallow and imprecisely known (standard error, 30–40%) initial slope in the dose-effect relationship for γ-rays. The RBE of neutrons versus a γ-ray dose of 1 Gy is only about 12 (Bauchinger and others 1983; Schmid and others 2000).
In the context of risk estimation, the major interest is in neutron RBEs that have been evaluated in animal experiments with regard to tumor induction. A multitude of results have been reported in the literature for many tumor systems (NCRP 1990). Experiments with rodents show considerable variation, especially in female mice and rats, and this variation reflects the decisive influence of hormonal status. In experiments with female Sprague-Dawley rats, Shellabarger and others (1980) found that 4 mGy of fast neutrons produced as many mammary neoplasms as 0.4 Gy of X-rays, which implied an RBE of 100. Broerse and Gerber (1982) used female Sprague-Dawley rats, which have a much lower spontaneous incidence, and found substantially lower values of neutron RBE. However, considerable differences in neutron RBE at higher doses were observed for different tumor types. As an extreme example, one may refer to lung adenomas in female RFM mice, in which there is a clear reduction in age-adjusted incidence after γ-ray exposures up to about 2 Gy, but neutron doses of 0.2 Gy cause a substantial increase (Ullrich and others 1976). The simple assumptions made in the calculation of RBE do not seem to be applicable in such a case.
In view of this complexity, it appears best to refer to experiments with male mice or rats that determine the overall incidence of solid tumors. In an extensive series of studies of the French Commissariat a l’Energie Atomique using male Sprague-Dawley rats, a fission neutron dose of 20 mGy was consistently found to be equivalent to an acute γ-ray dose of 1 Gy with regard to both nonlethal tumors (Lafuma and others 1989) and lethal tumors (Wolf and others 2000). This comparison corresponds to a neutron RBE of 50 against a reference γ-ray dose of 1 Gy. When the experiments were evaluated in terms of life shortening as a proxy for tumor mortality, the inferred RBE was closer to 30 (Wolf and others 2000). Smaller values of the RBE—around 20 compared to a γ-ray dose of 1 Gy and about 15 compared to X-rays—are suggested by major studies with mice that were evaluated in
terms of life shortening, again as a reflection of increased mortality from tumors (Storer and others 1988; Carnes and others 1989; Covelli and others 1989).
In all experimental studies with rodents, it was difficult or impossible to determine excess tumor rates at γ-ray doses substantially less than 1 Gy. For the purpose of risk estimation, it is therefore assumed in this report that the relevant animal experiments with rodents indicate a neutron RBE for solid tumors of 20–50 compared to a reference γ-ray dose of 1 Gy. Experimental evidence suggests lower neutron RBEs for leukemia; in experiments with RFM mice (Ullrich and Preston 1987), an RBE of about 3 was seen versus a γ-ray dose of 0.5 Gy; at lower γ-ray doses, statistical uncertainty did not permit the specification of a neutron RBE.
CHEMICAL ASPECTS OF RADIATION
Electron Ionization of Water Molecules and Indirect Effects on DNA
As previously described, free electrons can be produced by X- and γ-ray interactions with atoms in tissue. These electrons can then interact with the DNA molecule and create damage in the form of strand breaks or damaged bases; these are known as direct effects. Indirect effects can occur after a photon interacts with a water molecule. Water molecules make up 70% of human tissue. Ejection of an electron from a water molecule by an incoming photon produces an ionized water molecule, H2O+. Trapping of the electron by polarizing water molecules produces a so-called hydrated electron, e-aq. When the ionized water molecule collides with another water molecule, it reacts to produce a highly reactive hydroxyl radical, OH•, according to the reaction
Other reactions produce a hydrogen radical (H•), hydrogen peroxide, and water. Thus, these reactions produce three important reactive species—e-aq, H•, and OH•, which have initial relative yields of about 45%, 10%, and 45%, respectively, in the case of γ-radiation. The reactive species can damage DNA, and such damage is termed an indirect effect.
The relatively long-lived (about 10−5 s) OH• radical is believed to be the most effective of the reactive species; as an oxidizing agent, it can extract a hydrogen atom from the deoxyribose component of DNA, creating a DNA radical. Early experiments demonstrated that about 70% of the DNA damage can be prevented by the addition of OH• scavengers (Roots and Okada 1972). Because OH• is so highly reactive, it has been estimated that only the radicals formed within about 3 nm of DNA can react with it (Ward 1994). Although DNA is deemed the most important target for biological damage that leads to health effects, other sites—such as the nuclear membrane, the DNA-membrane complex, and the outer cell membrane—may also be important for some biological effects. Signal transduction from cell membrane phospholipids damaged by free radicals and oxidizing reactions is an important natural process. This is one set of biochemical pathways by which the effects of ionizing radiation may overlap with the effects of endogenous processes, such as macrophage oxidative bursts. These processes may underlie those seen in irradiated cells that have been characterized as “bystander effects” and “adaptation” (see Chapter 2).
Nikjoo and colleagues (1997, 2002) have modeled the probability of electron and OH• radical interaction with DNA. In a 1997 publication, they modeled the spectrum of DNA damage (direct energy deposition and reactions with diffusing OH• radicals) induced by low-energy secondary electrons (0.1–4.5 keV). They note that to extrapolate available epidemiologic and experimental data from high-dose and high-dose-rate studies to the relevant low levels of single isolated tracks, it is essential to develop a more molecular and mechanistic approach based on the amounts, types, and repairability of the early molecular damage that results from the initial physical and chemical processes. Their calculations for secondary electrons show that most (about 66–74%) low-energy electron interactions in DNA “do not lead to damage in the form of strand breaks and when they do occur, they are most frequently single strand breaks” (SSBs). Although the data are complex, SSB percentages in their study range from about 22 to 27% in the electron energy range of 0.1–4.5 keV and double-strand break (DSB) percentages range from about 1.4–2.4% in the same energy range. However, more than 30% of DSBs are of a more complex form; these complex breaks are somewhat analogous to LMDS, but Nikjoo and colleagues do not include base damage in their model. Their calculations also indicate that the DNA damage tends to be along short lengths of DNA: 1–34 base pairs (bp) for 0.3 and 1.5 keV electrons. The authors conclude that the large deletions seen in radiation-induced mutations may have other mechanisms, such as nonhomologous recombination (Nikjoo and others 1997).
In the case of energetic electron interactions with DNA (0.1 eV to 100 keV electrons), Nikjoo and others (2002) estimate that more than 80% of the interactions do not cause damage in the form of DNA SSBs. Of the interactions that do cause strand breaks, the authors calculate that a small percentage (about 0.5–1.4%) produce DSBs. They note, however, that there is still a considerable contribution (>20%) to the DSB yield from complex DSBs in which a simple DSB is accompanied by at least one additional strand break within 10 bp. As in the low-energy study just described, this model does not include any contribution to the yield of strand breaks from damaged bases.
Another recent study suggests that single low-energy electrons can produce DNA SSBs and DSBs at energies below ionization thresholds (Boudaiffa and others 2000). The authors speculate that these breaks are initiated as direct damage by resonant electron attachment to DNA compo-
nents followed by bond dissociation. The breaks were produced in DNA in a vacuum, so the relevance of the resonance phenomenon to DNA breaks in the intracellular aqueous environment is open to question. Hanel and colleagues (2003) have shown that electrons at energies below the threshold for electronic excitation (<3 eV) can decompose gas-phase uracil to generate a mobile hydrogen radical. The relevance of this observation to DNA damage in vivo awaits further experimentation.
Spontaneous DNA Damage Relative to Radiation-Induced DNA Damage
DNA is an unstable chemical entity under in vitro conditions because it is the target of a variety of reactive small molecules. DNA undergoes degradative reactions caused by active hydrolysis that result in depurination and deamination, and it undergoes base adduct formation by reactions with metabolites and coenzymes, damage by reactive oxygen species generated by “leakage” from mitochondria, lipid peroxidation, and many other sources of spontaneous damage (Lindahl 1993; Marnett and Burcham 1993; Beckman and Ames 1997; Lindahl and Wood 1999; see Table 1-2).
More than 90% of naturally occurring oxidation in a cell originates in the mitochondria, and oxidative nuclear damage occurs only for reactive products that can migrate far enough to enter the nucleus and react with DNA. The cell nucleus consequently is almost anoxic (Joenje 1989), and oxidative damage is quenched about fiftyfold by histones and by suppression of Fenton oxidants. However, the nucleus is not radiobiologically hypoxic (<8 μmol/L). The superoxide radical (O2−) formed by one-electron reduction of molecular oxygen is generated in all aerobic cells. Chemical or enzymatic dismutation of O2− produces hydrogen peroxide, H2O2. Although proteins and small molecules, such as glutathione, serve as scavengers for reactive oxygen and thus protect the nucleic acids, there is a considerable amount of oxidative DNA base damage per cell per day (Saul and Ames
TABLE 1-2 Rates of Production and Steady-State Levels of Spontaneous DNA Damage in Mammalian Cellsa
|
Result of Damage |
Production Rates |
Steady-State Levelsb |
|
Depurination |
9000–10,000 per day |
<100 |
|
Deamination |
100–500 per day |
<100 |
|
3-Methyladenine |
600 per day |
<50 |
|
8-Hydroxyguaninec |
500–1000 per day |
100 (15,000) |
|
aFor comparison, background radiation of 5 mGy produces an average of about 1 electron track per cell resulting in 5–10 damaged bases, 2.5–5.0 SSBs, and 0.25 DSBs. bValues are for repair-proficient normal cells. Value in parentheses is for repair-deficient liver cells. cBest estimate of 8-hydroxyguanine values, disregarding reports of high values where chemical oxidation occurred during sample preparation. |
||
1986). However, the steady-state level of DNA damage is low, so most of the spontaneous and metabolically generated damage is apparently repaired efficiently and correctly. Although DNA in cells is basically unstable, the instability is counteracted by DNA repair processes.
Strong evidence pointing to differences between X-ray damage and oxidative damage has come from studies in the yeast Saccharomyces cerevisiae. A genome-wide collection of nearly 5000 deletion mutants in all nonessential genes is now available for this species. Using this collection, all genes that were required for resistance to the lethal effects of X-rays and hydrogen peroxide were determined (Birrell and others 2001, 2002). Of those that were resistant to either agent, few genes were in common and their rankings were different. Of the top 100 genes conferring resistance to X-rays, only 35 were in the top 100 that were sensitive to hydrogen peroxide (see Annex A-1). These rankings indicate that the types of damage caused by X-rays and hydrogen peroxide were significantly different and required different mechanisms for repair. In another study using these deletion mutants, the oxidative damage caused by five different oxidants was found to differ significantly, indicating an unexpected complexity for oxidative damage (Thorpe and others 2004). Despite these differences, all of the oxidants caused predominantly protein damage, and few of the genes involved in DNA repair were involved in resistance to damage caused by any of these oxidants. These studies indicate that DNA damage is a more significant factor in resistance to X-ray damage than to oxidative damage. These studies also showed that the genes whose expression was induced by X-rays or hydrogen peroxide were not the genes required for resistance to these agents; few of the X-ray DNA repair genes in particular were inducible by damage (Birrell and others 2002).
Background Radiation
Added to the sources of spontaneous damage and metabolically produced oxidative DNA damage is background radiation, which includes radon, cosmic rays, terrestrial γ-radiation, and natural radioisotopes in the human body. Collectively, background radiation is responsible for delivering an average effective dose per person worldwide of about 2.4 mSv per year (typical range, 1–10 mSv; UNSCEAR 2000b). This background value includes radon exposure, the health effects of which are not evaluated in this report. Medical sources of radiation (diagnostic X-rays, nuclear medicine, and so on) can substantially increase a person’s yearly radiation exposure.
Ionizing radiation produces several kinds of damage in DNA, including SSBs and DSBs in DNA chains, DNA-DNA covalent cross-links, and DNA-protein covalent cross-links and a large variety of oxidative changes in the nucleotide bases (Hutchinson 1985; Ward 1988). The identified oxidative base products of ionizing radiation are chemically iden-
tical with those produced by other oxidizing agents, such as H2O2 in the presence of iron or copper ions or those resulting from the normal metabolic production of free radicals that are by-products of the transport of electrons to oxygen in mitochondria (Dizdaroglu and others 1987, 1991; Gajewski and others 1990; Nackerdien and others 1991; Dizdaroglu 1992; Beckman and Ames 1997). It has been argued in the scientific press and the lay press that the quantity of spontaneous and metabolically generated DNA damage is many orders of magnitude greater than that resulting from low, protracted doses of radiation from environmental sources. This argument implies that the contribution from low doses of ionizing radiation is trivial and can be ignored (Billen 1990; Beckman and Ames 1997)—in other words, that the DNA damage produced by background radiation and the low doses of radiation to which some workers are exposed does not add appreciably to the extensive spontaneous and metabolic damage. However, measurement of naturally produced DNA damage generated by reactive oxygen species is difficult, and some early estimates of DNA products of spontaneous damage, such as 8-hydroxyguanine, are not likely to be accurate estimates, but rather to be overestimates due to chemical oxidation after extraction. An additional consideration is that the distribution of oxidative events produced by radiation may, in some cases, have a unique impact on DNA.
Locally Multiply Damaged Sites
Accumulated evidence shows that the products of ionizing radiation may differ from chemically generated oxidation products in their microdistribution rather than in the chemistry of individual lesions (Ward 1981, 1988, 1994). A portion of the energy of ionizing radiation, primarily that from secondary electrons, is deposited in large enough packets to produce clusters of OH• radicals. Because OH• has a very short range, owing to its high reactivity, it can produce a cluster of damage within a few base pairs of DNA if the cluster is generated within 3 nm of the DNA. Ward and colleagues (1985) have referred to such lesions as LMDS. The probability of clustered damage or LMDS increases with dose and LET but is independent of dose rate because it results from the passage of a single particle track (Prise and others 1994; Holley and Chatterjee 1996; Rydberg 1996; Nikjoo and others 2001). A DSB resulting from a single energy deposition is the most obvious example of an LMDS, but other combinations of strand breaks, cross-links, and base or sugar products can also occur (Ward 1994). Furthermore, both direct interactions of radiation with DNA and reactions of OH• contribute to the complexity of LMDS (clustered damage; Nikjoo and others 1997).
A second property of ionizing radiation that might distinguish it from chemically generated oxidation products is the extensive production of peroxyl radicals due to initial radical damage to molecules other than DNA (Floyd 1995; Milligan and others 1996). Peroxyl radicals produce oxidized bases, but not DNA strand breaks, and might account for the greater-than-expected yield of base damage, as opposed to strand breaks, observed in irradiated cells (Nackerdien and others 1992). Peroxyl radicals might also account for the production of double base lesions by single radicals that have been observed in irradiated oligonucleotides (Box and others 1995).
Ward and colleagues (1985) have calculated that 5 μM H2O2 can produce 15 Gy-equivalents of SSBs in mammalian cell DNA in 30 min through OH• generation catalyzed by iron ions bound to DNA; on the basis of these SSB yields, it takes 1000 Gy-equivalents to kill cells. At the D37 dose of cell killing, it has been calculated that each cell will have sustained 2.5 million SSBs from H2O2. In contrast, the D37 dose for low-LET ionizing radiation produces only 1000 SSBs and 40 DSBs—damage that is not characteristic of lethal doses of H2O2. Such data suggest that DSBs and other LMDS (clustered damage) produced by ionizing radiation and a few radiomimetic chemicals are the primary lethal lesions. A recent study that used the phosphorylation of the histone protein H2AX as a marker for DSBs suggests that the yield of DSBs as a function of dose is linear down to as low a dose as 3 mGy (Rothkamm and Lobrich 2003). The fraction of the energy deposited that can yield LMDS increases with LET, and LMDS are generally thought to explain the increased biological effectiveness of high-LET radiation in inducing DNA damage. Whether such LMDS are poorly repaired is still a matter of conjecture, especially in view of the multiple homologous and nonhomologous mechanisms of repair of DNA breaks. At the least, clustering will create complex DSBs within up to 10 bp or so (Ward and others 1985; Holley and Chatterjee 1996; Nikjoo and others 2001). Because of the wrapping of DNA around nucleosomes and the organization of the chromatin fiber, some clusters might include DSBs at two or more sites that are several thousand base pairs apart or even removed from each other by the distance of a chromosomal loop (about 100 kbp; Lobrich and others 1996; Rydberg 1996). For cells to survive without mutations, DNA damage must be faithfully repaired. Yet because large regions of the genome in somatic cells do not contain active genes or contain genes that are not expressed, inaccurate repair that simply restores the integrity of the DNA may be sufficient to produce viable cells that have minimal alterations in function. Conversely, it has been argued that whereas spontaneous damage is readily repaired in repair-competent cells, the DSBs and clustered lesions produced by even low-LET radiation are likely to be repaired with difficulty or incorrectly, if at all (Ward 1994). However, detailed experimental comparisons between the biological effects associated with the repair of spontaneous damage versus damage due to ionizing radiation have yet to be made.
In summary, LMDS (clustered damage) may be viewed as complex lesions associated with ionizing radiation and not with normal endogenous oxidative processes. If they are
refractory to repair, the risk to humans posed by ionizing radiation may be viewed as greater than that posed by endogenous oxidative stress.
MOLECULAR MECHANISMS OF DNA REPAIR
Ionizing radiation can cause a wide array of damage to individual DNA bases and SSBs and DSBs resulting from deoxyribose destruction (for basic biological and genetic concepts, see Appendix A). Damaged bases are repaired by mechanisms that involve excision and replacement of individual damaged bases (base-excision repair) or of larger oligonucleotide fragments (nucleotide-excision repair). SSBs are repaired in a process similar to base-excision repair with some of the same enzymatic components. DSBs potentially involve a number of repair processes, especially because organisms require the ability to distinguish between breaks caused by damage and those associated with normal processes, such as recombination, telomere maintenance, DNA replication, and processing of genes encoding antibodies. Some DSBs are simply rejoined end to end in a process called nonhomologous end joining (NHEJ). Others are repaired by a process of homologous recombination (HR) in which the broken strand is repaired by crossing over with an adjacent identical DNA sequence; this generally occurs only during or after chromosome duplication and before chromosome segregation. Damage, especially DSBs, also elicits a signal transduction process that uses a cascade of kinase and other protein modifications and changes in gene transcription, all of which contribute to a cellwide response to DNA damage.
Base-Excision Repair
Release of altered bases by base-excision repair (BER) is initiated by DNA glycosylases that hydrolytically cleave the base-deoxyribose glycosyl bond of a damaged nucleotide residue (Figure 1-9). A present estimate would be that human cell nuclei have ten to twelve different DNA glycosylases, which have varied but overlapping specificities for different base damage. BER has two main pathways that result in replacement of the damaged base with either a short or a long patch.
A common strategy for DNA glycosylases, deduced largely from structural studies, appears to be facilitated diffusion along the minor groove of DNA until a specific type of damaged nucleotide is recognized. The enzyme then kinks the DNA by compression of the flanking backbone in the same strand as the lesion, flips out the abnormal nucleoside residue to accommodate the altered base in a specific recognition pocket, and mediates cleavage (Parikh and others 1998). The DNA glycosylase then may remain clamped to the damaged site until displaced by the next enzyme in the BER pathway, APE1 (also called HAP1), which has greater affinity for the abasic site. This strategy (Parikh and others 1998; Waters and others 1999) protects the cytotoxic abasic residue and may delay the rearrangement of the base-free deoxyribose into a reactive free-aldehyde conformation that could cause cross-linking and other unwanted side effects.
The main human apurinic-apyrimidinic (AP) endonuclease, APE1, occupies a pivotal position in BER of anomalous residues, recognizing and cleaving at the 5′ side of abasic sites generated by spontaneous hydrolysis, reactive oxygen species, and DNA glycosylases. Abasic sites generated by nonenzymatic depurination probably outnumber those generated by all of the DNA glycosylases; consequently, APE1 and subsequent key proteins in the BER pathway (XRCC1 and polymerase β) are essential, whereas mice with knockouts of various DNA glycosylases so far investigated have been viable (Wilson and Thompson 1997). In a substrate recognition process similar to DNA glycosylases, APE1 flips out the base-free deoxyribose residue from the double helix before chain cleavage (Gorman and others 1997; Parikh and others 1998). When bound to DNA, the APE1 protein interacts with the next enzyme in the BER pathway, POL β, and recruits the polymerase to the site of repair (Bennett and others 1997). POL β has two distinct domains that are well suited for DNA gap filling during BER. The larger domain is the polymerase domain itself; a small basic NH2-terminal domain contains an AP lyase activity that excises the abasic sugar-phosphate residue at the strand break (Matsumoto and Kim 1995; Sobol and others 1996). POL β also interacts with the noncatalytic XRCC1 subunit of the XRCC1-DNA ligase III heterodimer. Consequently, XRCC1 acts as a scaffold protein by bringing the polymerase and ligase together at the site of repair and interacts with poly(ADP-ribose) polymerase and polynucleotide kinase (Whitehouse and others 2001); further stabilization of the complex may be achieved by direct binding of the NH2-terminal region of XRCC1 to the DNA SSB (Kubota and others 1996; Marintchev and others 1999). XRCC1 contributes to the normal X-ray resistance of mammalian cells, and mutant cells with a defective XRCC1 protein are hypersensitive to ionizing radiation.
When the terminal sugar-phosphate residue has a more complex structure that is relatively resistant to cleavage by the AP lyase function of POL β, DNA strand displacement may occur instead—involving either POL β or a larger polymerase such as POL δ—for filling in gaps a few nucleotides long (Fortini and others 1998; Dianov and others 1999). The FEN1 structure-specific nuclease removes the displaced flap, and the PCNA protein stimulates these reactions (Wu and others 1996; Klungland and Lindahl 1997), acting as a scaffold protein in this alternative pathway in a way similar to that of XRCC1 in the main pathway. Another replication factor, DNA ligase I (LIG1), then completes this longer-patch form of repair. An important property of FEN1 here, in addition to processing the 5′ ends of Okazaki fragments during lagging-strand DNA replication, is to minimize the possibility of hairpin-loop formation and slippage during
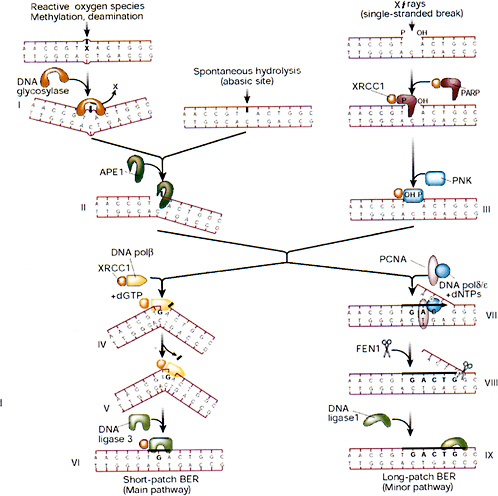
FIGURE 1-9 Base-excision repair. This pathway repairs single-base damage (from X-rays, reactive oxygen species, methylation, or deamination), apurinic sites, and SSBs (from X-rays). A damaged base is removed by glycosylases, leaving an apurinic site that is a substrate for apurinic endonuclease (APE1), which converts it into a SSB. X-ray breaks are modified by XRCC1, polynucleotide kinase (PNK), and poly(ADP-ribose) polymerase (PARP) to produce a cleaved substrate with 3′ and 5′ termini similar to those produced by APE1. The break is then patched by short- or long-patch BER. The short-patch pathway predominates in mammalian cells, and involves polymerase β, which can remove a 5′-deoxyribose moiety by its lyase activity and then insert a single base patch that is sealed by DNA ligase III. The long-patch pathway involves polymerase δ or ε, which is anchored to DNA by a PCNA collar and carries out strand displacement synthesis. The displaced flap is cleaved by the structure-specific endonuclease FEN1, and the patch is sealed by ligase I. XRCC1 is a nonenzymatic scaffold protein that interacts with many of the participants of BER and anchors them to the substrate and hands on repair intermediates through successive stages of BER. SOURCE: Reproduced with permission from J.H. Hoeijmakers (2001).
strand displacement and subsequent DNA synthesis, which might otherwise result in local expansion of sequence repeats (Tishkoff and others 1997; Freudenreich and others 1998). The temporary inefficiency of this process during early mammalian development could explain the origin of several human syndromes that are associated with expansion of triplet repeats in relevant genes.
A series of pairwise interactions between the relevant proteins in BER seem to occur in most cases without any direct strong protein-protein interactions in the absence of DNA. The XRCC1-LIG3 heterodimer is the only preformed complex, and no large preassembled multiprotein BER complex is likely to exist. Nevertheless, the consecutive ordered interactions may protect reaction intermediates and ensure efficient completion of the correction process after initial DNA damage recognition.
Nucleotide-Excision Repair of Cyclodeoxynucleosides
The great majority of endogenous DNA lesions produced by reactive oxygen species are corrected by the BER pathway, and the contributions of the different pathways of nucleotide-excision repair (NER) and mismatch repair are very minor. However, exposure of DNA or cells to ionizing radiation under hypoxic conditions causes the formation of 5′, 8-purine cyclodeoxynucleosides. This chemically stable and distorting form of DNA damage, in which the purine is attached by two covalent bonds to the sugar-phosphate backbone, can be removed only by NER (Heyer and others 2000; Kuraoka and others 2000). Similarly, a major lipid peroxidation product, malondialdehyde, reacts with G to produce an exocyclic pyrimidopurinone (M1G) that requires NER for repair. These are not the major mutagenic or cytotoxic lesions that occur as a consequence of exposure to ionizing radiation, but they could be critical in individuals with impaired ability to perform NER.
Repair of Single-Strand Breaks
Reactive oxygen species cause DNA strand breaks by destroying deoxyribose residues. Such SSBs are processed and repaired by the same enzymes responsible for the later stages of BER, sometimes with the additional steps of exonucleolytic removal of base pairs and phosphorylation of 5′ termini by DNA kinase. In contrast to the continuous protection of DNA reaction intermediates when an altered base residue is replaced however, the initial strand break is fragile and attracts unwelcome recombination events. An abundant nuclear protein, poly(ADP-ribose) polymerase-1 (PARP1), appears to have as its main role the temporary protection of DNA single-strand interruptions (Le Rhun and others 1998; Lindahl and Wood 1999). PARP1 rapidly shuttles strand breaks in DNA on and off, with NAD-dependent synthesis of poly(ADP-ribose) as its release mechanism. PARP1 knockout mice are viable but show increased numbers of spontaneous sister-chromatid exchanges and sensitivity to ionizing radiation. Extracts of cells from such mice contain low concentrations of other PARP enzymes, which may have distinct unknown roles but could also have backup functions. Crossing PARP1 knockout mice with severe combined immunodeficient disease knockout mice that lack DNA-dependent protein kinase, which is required for VDJ recombination during lymphocyte development, alleviates the DNA-processing defect in the latter and allows some low-fidelity recombination (Morrison and others 1997). PARP1 plays no clear role in the BER process itself, as POL β and LIG3 do, but it interacts with the scaffold protein XRCC1 and may in this way accelerate the recruitment of these repair enzymes for strand interruptions (Mackey and others 1999).
Repair of Double-Strand Breaks
Exposure of DNA to ionizing radiation produces about 5–7% as many DSBs as SSBs (e.g., see earlier discussion of Nikjoo and others 1997, 2000). DSBs are sites at which a surprisingly large number of proteins can bind, carry out strand-break repair, and initiate a complex series of cellular signals that regulate cell cycle progression and the induction and activation of many downstream genes. Cells often encounter DNA DSBs under natural circumstances. These include termini (e.g., telomeres at chromosome ends); recombination intermediates; and immunoglobulin rearrangement during the processing of antibody genes (which leads to increased versatility in the repertoire of immature immunocytes), during the processing of stalled or collapsed replication forks arrested by damage on the template strand and during topoisomerase action on DNA. DSB repair enzymes have been suggested as playing an essential role in telomere maintenance in normal undamaged cells (Blackburn 2000).
One critical difference between metabolically generated DSBs and those generated by ionizing radiation is that some fraction of the latter contain complex radiochemical damage that results in LMDS. LMDS (clustered damage) involve frank breaks, radiolytic fragments as termini, and base damage that is processed into breaks by cellular glycosylases (Blaisdell and Wallace 2001). DSBs thus are not inherently novel, although substantial differences between natural and radiation-induced breaks are likely. Cells contain many genes that code for DNA-binding proteins and signal transduction pathways that respond specifically to DNA double-strand breakage. Consequently, cells can distinguish between a naturally occurring end of DNA at a telomere or recombination structure, for example, and a DSB at an unusual location with atypical chemistry. This suggests that metabolic responses to DSBs and LMDS are highly evolved in most cell types and that cells are not completely unprepared and unequipped for these kinds of lesions, but are in fact able to exercise considerable discrimination in their detection and repair. Cells can also repair damage by novel chemicals, such
as cisplatinum, which was newly synthesized in the twentieth century, an indication that novelty or uniqueness is no barrier to the repair of DNA damage.
Repair of DSBs involves a number of biochemically distinct processes. Direct rejoining of the broken ends occurs by several mechanisms, generally described as NHEJ. A fast NHEJ process involves end-binding proteins (Ku70, Ku80, and DNA-PK; Baumann and West 1998; Critchlow and Jackson 1998; Zhao and others 2000), and a slower process involves the hMre11/hRad50/Nbs1 DNA-binding and exonuclease complex that appears to act on refractory, complex breaks (Haber 1998; Petrini 1999). A more complicated rejoining process—homologous recombination—depends on matching damaged DNA with its identical sequence in a sister chromatid after DNA replication or in the homologous chromosome in diploid cells. This process depends on the hRad51 protein, which facilitates homologous pairing, and accessory proteins, such as hRad52, hRad54, XRCC2, and XRCC3 (Thompson 1996). How cells coordinate these processes and determine which should be used under various circumstances is unknown. Coordination may be under the control of the Brca1 and Brca2 proteins. Brca1 binds to unusual DNA structures (Parvin 2001) and is found in a large complex that contains many repair and replication proteins (Wang and others 2000).
The proteins directly involved in DNA strand-break repair do not appear to be inducible (Tusher and others 2001) or to be strongly influenced by p53 functions, except where recombination is involved. Radiation-induced genes represent predominantly cellular signaling molecules, particularly those induced by transactivation by p53. Radiation does, however, activate a series of protein kinases, of which ATM (ataxia-telangiectasia-mutated) is the most prominent, that modify the activity of many other proteins in the repair pathways (Bakkenist and Kastan 2003).
Nonhomologous End Joining—Fast Reaction
DSBs begin to rejoin rapidly after irradiation, with half-times of about 10 min or less (Ward and others 1991). This rapid rejoining involves accumulation of the end-binding proteins Ku70 and Ku80, DNA-PK kinase, the DNA ligase IV-XRCC4 heterodimer, PARP, and others (Figure 1-10). The same factors are also an integral part of the normal process of immunologic rearrangement (Labhart 1999). Conceivably, if the LMDS contains damaged bases, the ends will also require repair steps involving glycosylases, apurinic endonuclease, and DNA polymerase β. Attempted repair by these BER enzymes can enhance DSB formation and loss of base pairs, which then must be repaired by NHEJ (Blaisdell and Wallace 2001). Attempted BER of LMDS in human lymphoblastoid cells produces lethal and mutagenic DSBs (Yang and others, 2004). Small deletions associated with NHEJ have been mapped by sequencing techniques and range up to about 10 nucleotides (Daza and others 1996).
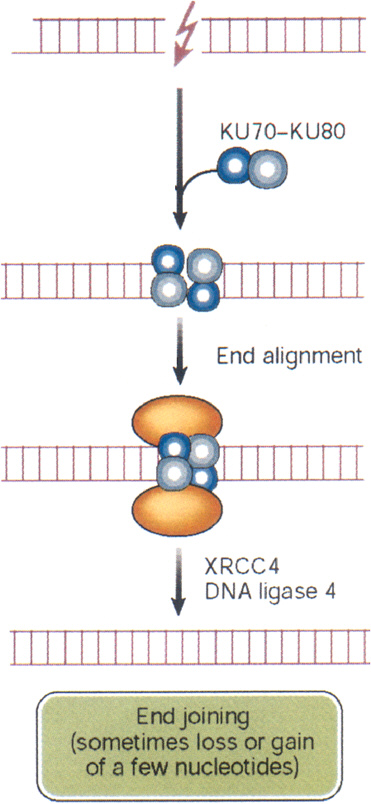
FIGURE 1-10 Nonhomologous end joining: this repair pathway re-ligates DNA DSBs by using the end-binding proteins Ku70 and Ku80 to maintain alignment, and p450 kinase acts as a binding factor. The region across the break is then sealed by ligase IV and its cofactor XRCC4. The sealed break often gains or loses a few nucleotides, especially if the break is an LMDS. In some cases, nonhomologous end joining appears to be responsible for large DNA deletions and chromosome aberrations. In these cases, considerably more than a few nucleotides can be lost. SOURCE: Reproduced with modifications and with permission from Hoeijmakers (2001).
The histone protein H2AX is phosphorylated rapidly over large regions of DNA around sites of DSBs by ATM kinase (Burma and others 2001). Loss of H2AX phosphorylation occurs rapidly with the repair of DSBs, but the biochemical details of dephosphorylation remain to be ascertained. A recent study showed that in human cells, a background level of H2AX phosphorylation occurred in about 5% of the cells. After low doses of X-rays that initially increased the level to 10%, most cells eliminated this phosphorylation, except for a small fraction in which it persisted unless the cell entered DNA synthesis (Rothkamm and Lobrich 2003). Whether this means that a small fraction of cells cannot repair some classes of LMDS or that dephosphorylation of H2AX can be slower than repair itself in a subset of cells remains to be determined.
The DNA-PK kinase is a member of a class of phosphatidyl-3-inosityl enzymes that includes ataxia-telangiectasia-mutated (ATM) and ataxia-telangiectasia-related (ATR) kinases, all of which are involved in signaling the presence of DNA damage (Shiloh 2001, 2004; Figure 1-11). Although DNA-PK kinase can phosphorylate many proteins in vitro, it is unclear which proteins it usually phosphorylates in vivo. Early cytologic evidence of X-ray damage is phosphorylation of a histone protein to create γ-H2AX foci that are visible microscopically within minutes of irradiation.
Nonhomologous End Joining—Slow Reaction
After the rapid phase of rejoining is complete, the repair of DSBs slows to a second phase with a half-time of several hours. Foci containing the hMre11/hRad50/Nbs complex form or persist and reach a maximum at about 4–6 h. Because this complex has endonuclease and DNA-binding activity, it may be involved in the slower repair of refractory DSBs that cannot be repaired by the earlier, fast mechanism. The complex is not active unless the Nbs1 protein is phosphorylated on several sites by ATM kinase (Figure 1-11), which is itself activated by DNA breaks (Shiloh 2001; Bakkenist and Kastan 2003). The precise DNA structures involved in these refractory breaks are unknown. However, one model suggests that nuclease action by the Mre11 complex resects single DNA strands and that short regions of sequence identity (microhomologies) can be used for alignment and rejoining of DNA strands (Figure 1-12).
Homologous Recombination
Repair of a DSB by HR involves matching the two broken ends of a DNA strand with identical sequences of intact DNA (Figure 1-12). The broken and intact molecules are aligned according to their sequences and encompassed by a toroid of hRad51 molecules that facilitate repair by having DNA single strands invade their homologues, producing an X-shaped four-armed structure called a Holliday junction. Resolution of this structure by specific junction nucleases produces two intact double-strand DNA molecules with or without exchanges according to the orientation of the resolution nuclease actions. The activity of hRad51 is enhanced by other factors, such as hRad52, XRCC2, and XRCC3, and suppressed by p53, which binds to both Holliday junctions and hRad51 (Buchhop and others 1997). HR is much more efficient and important for repair in yeast and somatic chick cells than in normal (nonmalignant) mammalian (human) somatic cells, where NHEJ is the dominant mechanism for DSB repair (Sonoda and others 1998). However, there are exceptions, and there may be times in the cell cycle, such as late S, when HR assumes greater importance because of the proximity of sister chromatids (Thompson 1996). The low level of sister-chromatid exchange, a form of HR, induced by X-rays and high-LET radiation indicates that, in absolute terms, HR remains a minor pathway for the repair of damage caused by ionizing radiation in somatic cells.
There is some question about the source of an identical matching sequence for repair by HR in somatic human cells. A homologous sequence may be the other allele on a chromosome of a recently replicated sister-chromatid sequence on a daughter chromatid or a similar sequence in a repetitive region along the same chromosome. In the latter case the sequences may not be identical over long regions, and the mechanism is known as “homeologous” recombination. Recombination between alleles on separate chromosomes occurs at much lower frequency than between identical sequences on sister chromatids or arranged in tandem on the same chromosome. In general, HR between sister chromatids may occur at higher frequencies late in the cell cycle (e.g., late S; Thompson and Schild 1999), and homeologous recombination is likely to result in the loss of intervening sequences with the production of deletion mutations.
The HR involving hRad51 can be visualized immuno-histochemically: foci containing hRad51, Brca1, and other proteins can be seen microscopically soon after irradiation (Scully and others 1997). Cells generally exhibit either hRad51 foci or hMre11/hRad50/Nbs foci, but not both, and the choice of which of the mutually exclusive pathways an irradiated cell follows may be determined by Brca1 (Parvin 2001).
DSB Signal Transduction and Inducible Repair
Bacteria live in a highly variable environment and have evolved efficient inducible DNA repair processes to deal with sudden challenges of DNA damage from oxygen free radicals, ionizing radiation, chemicals, and ultraviolet radiation. These inducible repair pathways are now mechanistically well understood. In Escherichia coli, the regulatory genes soxR, ada, and lex control transcription of DNA repair functions, and increased amounts of relevant DNA repair enzymes can be produced in response to environmental challenges. In mammalian cells, the same types of DNA damage are recognized by similar DNA repair enzymes. However, a
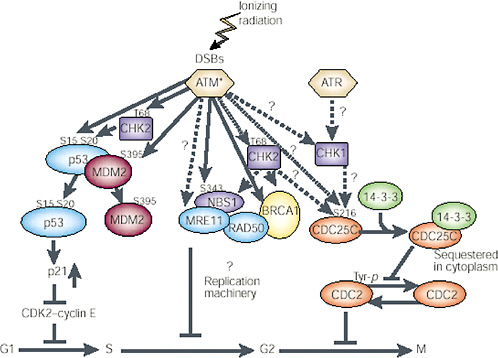
FIGURE 1-11 Network of protein kinases activated by DNA DSBs. ATM is the primary kinase that phosphorylates downstream kinases. The specific activity of ATM is increased after introduction of DSBs in DNA through ionizing radiation or other means; this then activates other proteins by phosphorylation (denoted by amino acid symbol and number) and in a cell cycle-specific manner. G1phase: Activated ATM (ATM*) directly phosphorylates three proteins involved in controlling p53 functions or levels—p53 (serine 15), CHK2 (threonine 68), and MDM2 (serine 395). CHK2 kinase may also be activated by ATM and in turn phosphorylate p53 on serine 20. This phosphorylation event and the phosphorylation of MDM2 seem to inhibit binding of MDM2 to p53 and should result in an increase in p53 protein. The increased p53 protein transcriptionally induces p21, which inhibits CDK2-cyclin E and causes arrest in the G1 phase of the cycle. S phase: Activated ATM also phosphorylates NBS1 (serine 343), and this phosphorylation event is required for the ionizing radiation-induced S-phase arrest. NBS1 exists in a complex with MRE11, RAD50, and BRCA1. The potential role of these proteins in S-phase arrest remains to be clarified; CHK2 may also be involved in this pathway, after activation by ATM, through phosphorylation of BRCA1 or NBS1. G2 phase: Details of the downstream targets of ATM at the G2 checkpoint have not been determined. CHK2 and CHK1 may be targets for ATM and ATR in the G2-M checkpoint pathway, respectively. CDC25C and 14-3-3 have been implicated in regulation of CDC2 kinase and progression through G2. Dashed arrows and question marks represent possible signaling steps; solid arrows represent reported phosphorylation events. SOURCE: Reproduced with permission from Kastan and Lim (2000).
major difference from microorganisms is that mammalian enzymes are constitutively expressed. Thus, there are no transcription control or mammalian counterparts of soxR, ada, and lex. This situation presumably reflects the much greater constancy of cellular environment in complex multi-cellular organisms. Therefore, the work on inducible DNA repair in bacteria offers no direct guidelines for the relative resistance of human cells repeatedly exposed to DNA-damaging agents.
Many reports have appeared about adaptive responses involving increased resistance or hypersensitivity in mammalian cells in response to single or multiple doses of ionizing radiation (adaptive effects). There are also reports that the effects of radiation on single cells can influence the response of adjacent nonirradiated cells (bystander effect). These reports are discussed specifically in Chapter 2, but this chapter describes the general stress response and signal transduction pathways that are known to occur after exposure to radiation.
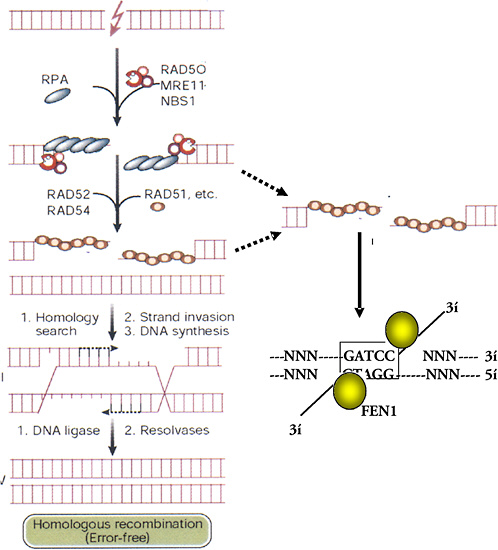
FIGURE 1-12 HR- and microhomology-mediated DSB repair. These two pathways for repair of DSBs are driven by stretches of homologous DNA. HR requires an identical sequence spanning the part of the DNA molecule containing the break and extensive remodeling of the broken DNA termini. Mre11/Rad50/Nbs1 resects individual strands by its 5′- to 3′-exonuclease activity and binds homologous double-stranded DNA by the Rad50 moiety. Exposure of single-stranded regions with only small regions of homology flanking the original break can allow microhomology-mediated strand-break rejoining coupled by cleavage of overhanging strands by FEN1 and resynthesis of any resulting gaps. The repair will, at the least, result in loss of one of the regions of microhomology. Exposure of single-stranded regions homologous to adjacent double-stranded DNA can lead to strand invasion and HR. Single-stranded regions are coated with single-strand binding protein (RPA); homology search and strand invasion are mediated by Rad52, 54, Brca 1 and 2, and Rad51. The complex structure produced forms a Holliday junction that is cleaved by junction-specific nucleases (resolvases), and associated polymerase and ligases complete an error-free exchange of DNA strands. SOURCE: Modified reproduction and reproduced with permission of J. Hoeijmakers (2001).
Damage to cells elicits increases and decreases in the expression of many genes. Recent microarray analysis has shown that these changes can involve hundreds of genes and that different stresses can invoke both a common set of genes and genes that are peculiar to particular kinds of stress (Amundson and others 1999a, 1999b). Despite the large number of affected genes, none appears to be directly involved in repair of DSBs (Tusher and others 2001). Central to most damage responses is stabilization of the tumor-suppressor gene p53, which occurs as a result of posttranslational phosphorylation or acetylation of the protein (Blattner and others 1999; Figure 1-11). Multiple potential serine and threonine residues in p53 are capable of being phosphorylated by different kinases in response to cellular stress, and several thousand combinations of modifications are possible in an irradiated cell. Resolving the functional role of any particular site can be difficult (Blattner and others 1999). The kinases include ATM, ATR, Chk1, Chk2, DNA-dependent protein kinase, and casein kinase I and II (Blattner and others 1999; Chehab and others 2000). (For the role that p53, pRb, cdc25C, chk1, chk2, 14-3-3 proteins, bub1, and the various cyclins and cyclin-dependent kinases play in radiation-induced checkpoints in G1, G2, and mitosis, see Little 1994; Jacks and Weinberg 1998; Lengauer and others 1998; Schmidt-Kastner and others 1998; Chan and others 1999; Ford and Pardee 1999; White and Prives 1999).
ATM is a centrally important kinase for X-ray damage that is activated by DNA DSBs (Bakkenist and Kastan 2003; Figure 1-11). In X-irradiated cells, phosphorylation of serine 15 and 37 interferes with the association of p53 with another protein mdm2 that also becomes phosphorylated and normally causes degradation of p53, extending its lifetime. The increased stability of p53 in irradiated cells permits it to form a tetramer and then act as a transactivating factor, increasing the expression of many other genes. Clearly, this will result in large-scale alterations of the gene expression pattern of irradiated cells that can influence their behavior. One downstream target for p53 is the cell cycle regulator protein p21; increased transcription of p21 due to p53 results in delays in the onset of DNA synthesis (the G1 checkpoint) and reduced DNA synthesis due to p21 binding the replication factor PCNA. The major response of cells to ionizing radiation is a reduction in initiation of the S phase and of replication origins during S. Another important radiation-responsive gene is GADD45; both this and p21 showed a linear dose-response relation for induction from 20 to 500 mGy with no indication of a threshold (Amundson and others 1999b).
Most of the members of the signal transduction pathways including ATM, p53, Chk1, Chk2, Brca1, and hMre11/hRad50/Nbs1 are protein products of tumor-suppressor genes. Loss of function of these members can result in genomic instability and in some instances may contribute to a series of events resulting in malignancy. They influence cell cycle checkpoints, DNA replication, DNA repair, and recombination. Thus, it is possible for a single DNA DSB to activate ATM and p53 and create a cell-wide response through this cascade of protein modifications and alterations in gene expression.
These signal transduction pathways are also activated by extracellular signals working through specific receptors on the cell membrane that then activates kinases, such as MAPKs, which phosphorylate p53. Irradiated cells also generate extracellular signals that resemble cytokines released during normal in vivo cell-cell communication processes (Herrlich and others 1992). These can, through receptors on adjacent cells or gap junctions, result in activation of the signal transduction pathways in nearby cells. These multiple intracellular and extracellular pathways of protein modification and signal transduction may constitute the mechanisms by which many of the transient alterations in cellular metabolism occur after exposure to ionizing radiation (Blattner and others 1994).
Some responses observed in particular regimes of exposure to ionizing radiation and given unique names (e.g., adaptive response, bystander effect, genomic instability) may constitute particular manifestations of these general stress responses and signal transduction pathways. These apparently distinct radiation responses have been described mainly in cell biology experiments, and in no case do they have solid biochemical support or mechanistic understanding. In addition to controversy among laboratories, some of the responses described appear to be valid only within a limited dose range and under particular experimental conditions. It is also unclear whether different types of cells, such as epithelial cells, fibroblasts, and lymphoid cells, respond similarly or differently in this regard. Some of the inducible responses appear to be complex in that they depend on participation of intercellular gap junctions in communicating radiation responses to neighboring cells. Work on this subject is in the preliminary, descriptive stage, and there is no understanding of what compounds or factors would be transferred between cells in the gap junction. Therefore, it is difficult to evaluate whether the phenomena are of any general physiologic significance.
SUMMARY
In this chapter the committee has provided background information relating to the physical and chemical aspects of radiation and the interaction of radiation with the target molecule DNA. The chapter describes the physics of electrons and beta particles, which are important contributors to direct DNA damage after ionizing radiation exposure, and introduces a special subject—the effect that neutron RBEs have on low-LET radiation risk estimates. Radical formation by ionizing radiation and its contribution to DNA damage are also described. The committee has discussed the contributions of normal oxidative DNA damage relative to radiation-induced DNA damage and described the DNA repair mechanisms that mammalian cells have developed to cope with
such damage. Modeling of electron interactions with DNA suggests that when more than one strand break occurs due to an electron interaction, approximately 30% of the breaks will be multiple events (three or more) that occur over a very small distance. These multiple events, sometimes referred to as LMDS, would be expected to occur at the same average rate per electron traversal of the DNA, whether the overall dose is high or low. It is reasonable to expect that multiple lesions of this sort would be more difficult to repair or might be prone to misrepair. This may explain the apparent inconsistency between the lethality and mutagenicity of agents that principally cause DNA single-strand breaks and ionizing radiation, which also produces double-strand breaks and LMDS. Furthermore, modeling of multiple damages in a small length of DNA suggests that the normal cellular oxidative damage of DNA may differ qualitatively from that due to ionizing radiation. Recent information is presented as an annex to this chapter, about a significant disparity in the genes that repair oxidative damage in yeast DNA and genes that repair radiation damage.
ANNEX 1A: IONIZING RADIATION AND OXIDATIVE DAMAGE—A VIEWPOINT FROM SACCHAROMYCES CEREVISIAE
Approximately 4800 deletion mutations have been made in all the nonessential genes in the yeast Saccharomyces cerevisiae. These have been used by two groups of investigators to identify the genes responsible for resistance against ionizing radiation, ultraviolet light, cisplatin, and a number of different oxidizing agents (hydrogen peroxide, diamide, linoleic acid 13-hydroperoxide, menadione, and cumene hydroperoxide; Birrell and others 2001, 2002; Game and others 2003; Thorpe and others 2004; Wu and others 2004). The set of genes required for resistance against a particular agent is an indication of the nature of the cellular biochemical pathways required to restore viability and, indirectly, of the kind of damage generated by the agent. If a common set of genes is required for several different agents, these will point to a common or overlapping chemical nature of the damage. The striking observation about the results in S. cerevisiae is that the sets of genes required for resistance against each agent differed significantly from each other. When pairwise comparisons were made between ionizing radiation and each oxidant, the overlap was low: less than half of the genes required for resistance against ionizing radiation were also required for resistance to oxidative damage (Figures 1A-1, and 1A-2).
Large numbers of genes not obviously involved in DNA repair fall within the list of sensitive mutants to ionizing radiation and oxidants. Several genes whose deletion produced sensitivity to radiation and oxidants were involved in DNA replication and recombination, suggesting that this process was vulnerable to all kinds of cellular damage in yeast. In contrast, the most important genes in human cells for repair by NHEJ were not represented among the sensitive mutants because this is a minor pathway in yeast. An additional observation is that the set of genes whose expression was induced by damage differed from the genes required for resistance against each agent, implying that repair genes were not among those induced by damage (Birrell and others 2002).
The committee carried out a detailed comparison of the genes reported by each group, using publicly available data sets. One group (Birrell and others 2001, 2002; Game and others 2003) reported the response of the complete set of 4800 genes and ranked them in sequence, from most sensitive to least sensitive. About 10% of all genes (470) showed some degree of sensitivity to ionizing radiation. The other group (Thorpe and others 2004) reported only those genes that showed sensitivity to at least one oxidant (approximately 675 genes) and ranked them in categories 1–7, with the most sensitive in category 1.
Comparison between these data sets is complicated by different methods of reporting and different technical approaches to determining sensitivity. Comparisons were therefore made in general terms rather than gene by gene, and only those genes were considered that were reported by both groups. The committee first compared the genes required for resistance against hydrogen peroxide as reported by two independent research groups, to establish the consistency of the data (Figure 1A-1). A set containing about 200 genes was common to both groups as necessary for resistance to hydrogen peroxide. Of these, 150 were also sensitive to ionizing radiation. Since different methods were used to detect sensitivity and rank the strains, some differences are not surprising. The common set of 150 genes required for resistance to both ionizing radiation and hydrogen peroxide included those involved in postreplication repair and recombination, but the genes that ranked among the most sensitive toward ionizing radiation were ranked lower on the list for hydrogen peroxide (Birrell and others 2002).
The committee then compared the genes required for resistance to different oxidizing agents with those required for resistance to X-rays (Figure 1A-2). The overlap was small in comparison to the number of genes required for resistance to ionizing radiation; conversely, more than half of the genes required for resistance to each oxidant were also required for resistance to ionizing radiation. However, the same genes were not involved for each oxidant.
The implication of these results is that each agent that is toxic to S. cerevisiae produces a unique spectrum of cellular damage, with some overlap. The relevance of these comparisons to this report lies in the attempts that have been made to explain low-dose ionizing radiation as no more than a special case of oxidative damage (Pollycove and Feinendegen 2003). If this were true, low doses of ionizing radiation would be insignificant compared to the levels of naturally occurring reactive oxygen species and could therefore be ignored as having no detrimental health effects. How-
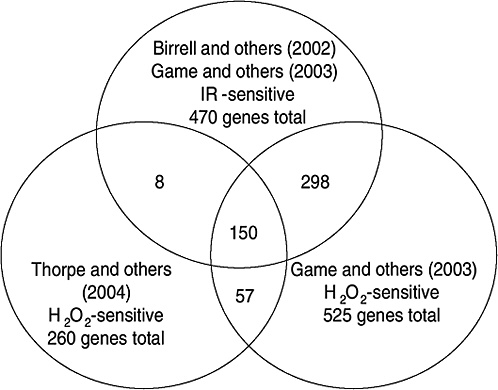
FIGURE 1A-1 Venn diagram representing the overlap among genes involved in resistance against ionizing radiation and hydrogen peroxide as indicated in the reports cited. Numbers in regions of overlap represent the number of genes responsible for resistance against two agents as reported by one or another group.
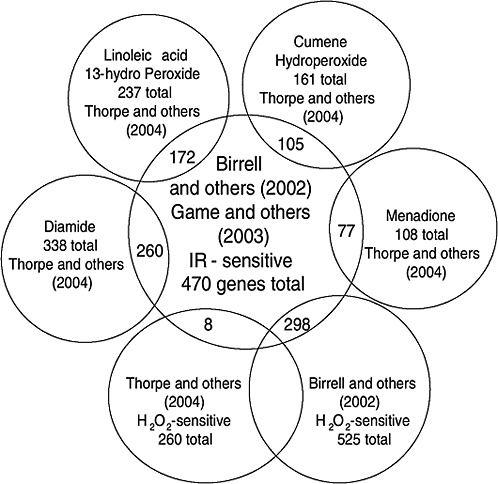
FIGURE 1A-2 Venn diagram representing the overlap among genes involved in resistance against ionizing radiation and various oxidizing agents as indicated in the reports cited. Numbers in regions of overlap represent the number of genes responsible for resistance against two agents as reported by one or another group.
ever, each oxidizing agent involved a significantly different set of genes, which also differed from those required for protection against X-rays, indicating that oxidative damage cannot be considered a single entity, but is dependent on the chemical source of the oxidation. Mutants sensitive to hydrogen peroxide included an overrepresentation of mitochondrial respiratory functions, but those sensitive to diamide encompassed genes involved in vacuolar protein sorting. This makes it especially difficult to predict what kinds of damage would result from endogenous reactive oxidative species. Endogenous damage could present its own unique spectrum of genes required for resistance, different from each of the exogenous sources as well as from ionizing radiation.
These results must be confirmed and extended to human cells, because the genes known to be involved in repair of DNA DSBs by NHEJ (Ku70, Ku80, and DNA-PK) were rarely found among those involved in resistance to ionizing radiation or oxidative damage in yeast, where they play a very minor role. The majority of genes required for resistance to oxidative damage were, however, considered by one set of authors (Thorpe and others 2004) as more representative of damage to the protein components of the cell than to DNA. These included genes required for transcription, protein trafficking, and vacuolar function.
These damage responses in S. cerevisiae are, however, dominated by the efficient homologous recombination that plays a major role in response to DNA damage (Kelley and others 2003). Homologous recombination may therefore mask some of the effects caused by loss of genes on pathways that may be minor in yeast but more important in mammalian cells (Swanson and others 1999; Gellon and others 2001; Morey and others 2003). For example, mice that are defective in apurinic endonuclease are embryonic lethals, and blastocysts derived from these nulls are radiosensitive (Xanthoudakis and others 1996; Ludwig and others 1998). RNAi ablation of a pyrimidine-specific DNA glycosylase in mice confers radiosensitivity (Rosenquist and others 2003). Although the results described in yeast do indicate differences between ionizing radiations and oxidizing agents, the extent of differences or of overlap may not be the same in mammalian cells.
These results in S. cerevisiae, however, provide no support for the attempts to equate low-dose ionizing radiation with endogenous oxidative reactions. The committee would expect even greater divergence between ionizing radiation and oxidative damage in human cells because of the higher ratio of cytoplasmic and nuclear proteins to DNA than in S. cerevisiae and the greater role of NHEJ.
























