
7
Enhancing the Value of Clinical Trial Registration1
|
Recommendation on Registration of Clinical Studies on ClinicalTrials.gov from the IOM Report The Future of Drug Safety: Promoting and Protecting the Health of the Public Recommendation 4.11 The committee recommends that Congress require industry sponsors to register in a timely manner at clinicaltrials.gov, at a minimum, all phase 2 through 4 clinical trials, wherever they may have been conducted, if data from the trials are intended to be submitted to the FDA as part of an NDA [New Drug Application], sNDA [supplemental New Drug Application], or to fulfill a postmarket commitment. The committee further recommends that this requirement include the posting of a structured field summary of the efficacy and safety results of the studies. |
Since the International Committee of Medical Journal Editors began requiring registration of trials in a public trials registry as a condition of consideration for publication, the number of trials registered on ClinicalTrials.gov has increased. Nevertheless, the value and the transparency of the system are not optimal. To address the weaknesses of the current system, the IOM report recommended enhancing clinical trial registration (Recommendation 4.11). Dr. Zarin discussed approaches to achieving this goal and the cost of their implementation.
A major focus of the IOM report was improved communication with the public. ClinicalTrials.gov is already a valuable resource to the public; however, the modifications proposed in Recommendation 4.11 were
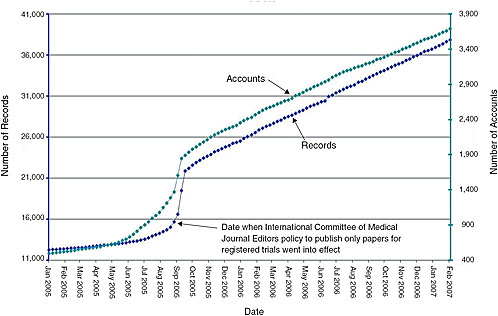
FIGURE 7-1 Monthly registration of trials on ClinicalTrials.gov. Presently, journal editors will publish papers only for registered trials. As indicated by the constant slope, even after this policy went into effect, the National Library of Medicine was capable of handling large increases in the number of trials registered.
SOURCE: Zarin, 2007.
intended to increase its value. Dr. Zarin discussed the resource challenges of what she identified as four components of Recommendation 4.11, noting that none of those components will be easy to implement:
-
Expanded scope of mandatory trial registration—The system has yet to reach a steady state, and could readily handle an increase in the number of trials registered without requiring a significant budget increase (Figure 7-1). While the current budget for ClinicalTrials.gov is just over $3 million per year, the system taps the National Library of Medicine’s (NLM) $300 million annual budget for search engine capabilities, hardware, personnel, etc. Some key functions of the registry, however, would be affected by and could benefit from policy changes:
-
Providing objective criteria for determining whether mandated trials are registered. Examples of criteria that would be easy to monitor include intervention type (e.g., drug trials), phase, and number of subjects. Examples of criteria that would be difficult to monitor include those that use subjective language (e.g., “serious conditions”).
-
Giving the National Institutes of Health (NIH) the flexibility to define “acceptable” entries and input rules so that information in the registry will be meaningful.
-
Enabling users to find the information they want. Currently, about 20 percent of industry drug records use untracked serial numbers instead of the names of drugs, thereby hindering searches for information on the drug trials. An example of how a policy change could improve this situation is Maine’s recent bill requiring drug companies to post a form indicating all previous names or aliases of each drug.
-
Addition of a results database—ClinicalTrials.gov is already linked to published results whenever possible and could be linked to drugs@fda if trial identifiers were used. For de novo results, however, quality assurance is complex, and validation of all results would be challenging since these data are more complex than other trial data, the stakes are higher, and NIH would not have access to the full results dataset. The current validation system is based on both automated and manual checks, correction of errors when found, and an archived site that tracks changes. But difficult-to-detect errors still occur. The resource needs for adding results data would depend on the number of trials. As of 2006, the system was receiving an average of about 923 new trial registrations a month (Figure 7-2), and between 160 and 500 trials in the registry are being completed each month.
-
Scientific review—Between 40 and 200 trials would need to be reviewed each week, and it is important to note that the FDA reviews for one of these trials could consist of 30+ pages of complex analyses and other information. It is unclear who would be able to review database entries for their concordance with complex FDA reviews.
-
Monitoring and enforcement—It is unclear within the various bills currently before Congress what the roles of the FDA and NIH would be, although several proposals to keep those roles simple have been put forth (e.g., using objective definitions of scope, using NCT numbers2 and incorporating them into business processes).
In summary, expanding the number of trials in the registry could probably occur at no significant increase in cost to ClinicalTrials.gov. Adding a structured results database, however, would be a complex task costing on the order of $10–20 million annually in addition to what is being spent on the registry. With respect to scientific review and monitoring and enforcement, it is unclear what would need to be done and how much it would cost.
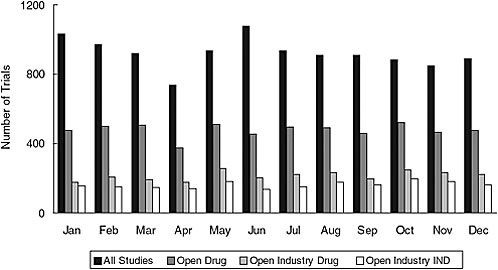
FIGURE 7-2 New trials received by ClinicalTrials.gov in 2006. The average total number of new trial registrations received per month was 923; the average number of open drug trials received per month was 478; the average number of open industry drug trials received per month was 215; and the average number of open industry IND trials received per month was 162.
NOTE: IND = Investigational New Drug.
SOURCE: Zarin, 2007.
When questioned about the history and mandate of the ClinicalTrials. gov registry, Dr. Zarin responded that the registry has been functioning since 2000, although some sponsors (NIH and some drug companies) have registered older studies. The current mandates for registration include the Food and Drug Modernization Act, Section 113, which mandates registering all Investigational New Drug (IND) studies with efficacy end points for serious and life-threatening conditions, and the above-mentioned requirement of the International Committee of Medical Journal Editors to register trials for any intervention that is clinically directed as a condition for publication. Again, one of the challenges is determining whether a trial that is mandated to be registered is actually registered. In response to another question, Dr. Zarin noted that while ClinicalTrials.gov accepts and welcomes data on nonclinical trials (i.e., observational data), it cannot enforce their inclusion.




