
8
In the Light of Directed Evolution: Pathways of Adaptive Protein Evolution
JESSE D. BLOOM* and FRANCES H. ARNOLD†*
Directed evolution is a widely used engineering strategy for improving the stabilities or biochemical functions of proteins by repeated rounds of mutation and selection. These experiments offer empirical lessons about how proteins evolve in the face of clearly defined laboratory selection pressures. Directed evolution has revealed that single amino acid mutations can enhance properties such as catalytic activity or stability and that adaptation can often occur through pathways consisting of sequential beneficial mutations. When there are no single mutations that improve a particular protein property, experiments always find a wealth of mutations that are neutral with respect to the laboratory-defined measure of fitness. These neutral mutations can open new adaptive pathways by at least 2 different mechanisms. Functionally neutral mutations can enhance a protein’s stability, thereby increasing its tolerance for subsequent functionally beneficial but destabilizing mutations. They can also lead to changes in “promiscuous” functions that are not currently under selective pressure, but can subsequently become the starting points for the adaptive evolution of new functions. These lessons about the coupling between adaptive and neutral protein evolution in the laboratory offer insight into the evolution of proteins in nature.
Proteins are the molecular workhorses of biology, responsible for carrying out a tremendous range of essential biochemical functions. The existence of proteins that can perform such diverse tasks is a testament to the power of evolution, and understanding the forces that shape protein evolution has been a long-standing goal of evolutionary biology. More recently, it has also become a subject of interest among bioengineers, who seek to tailor proteins for a variety of medical and industrial applications by mimicking evolution. Although they approach the study of protein evolution from different perspectives and with different ultimate goals, evolutionary biologists and bioengineers are interested in many of the same broad questions.
In examining these questions, we begin by considering the continuing relevance of one of the earliest analyses of protein evolution, performed >40 years ago by the great chemist Linus Pauling and his colleague Emile Zuckerkandl (Zuckerkandl and Pauling, 1965). Working at the time when it was first becoming feasible to obtain amino acid sequences, Pauling and Zuckerkandl assembled the sequences of hemoglobin and myoglobin proteins from a range of species. They compared the sequences with an eye toward determining the molecular changes that accompanied the evolutionary divergence of these species. But although it was already known [in part from Pauling’s earlier work on sickle cell anemia (Pauling et al., 1949; Ingram, 1957)] that even a single mutation could alter hemoglobin’s function, the number of accumulated substitutions seemed more reflective of the amount of elapsed evolutionary time than any measure of functional alteration. Summarizing their research, Pauling and Zuckerkandl (1965) wrote:
Perhaps the most important consideration is the following. There is no reason to expect that the extent of functional change in a polypeptide chain is proportional to the number of amino acid substitutions in the chain. Many such substitutions may lead to relatively little functional change, whereas at other times the replacement of one single amino acid residue by another may lead to a radical functional change. Of course, the two aspects are not unrelated, since the functional effect of a given single substitution will frequently depend on the presence or absence of a number of other substitutions.
This passage highlights 2 key issues that continue to occupy researchers nearly a half-century later. First, natural proteins evolve through a combination of neutral genetic drift and functionally selected substitutions. Although probably every evolutionary biologist would acknowledge the existence of both types of substitutions, their relative prevalence is debated with often startling vehemence (Gillespie, 1984; Blum, 1992). The intractability of this debate is caused in large part by the difficulty
of retrospectively determining whether long-ago substitutions were the subject of selective pressures.
The second issue highlighted by Pauling and Zuckerkandl, the potential for an adaptive mutation’s effect to depend on the presence of other possibly nonadaptive mutations, has been a topic of much discussion among protein engineers (Lockless and Ranganathan, 1999; Reetz et al., 2005; Weinreich et al., 2006). The reason is that the presence of epistatic coupling between mutations has the potential to profoundly affect the success of protein optimization strategies. In the absence of epistasis, a protein can always be improved by a simple hill-climbing approach, with each successive beneficial mutation moving further up the path toward some desired objective. But such a hill-climbing approach can in principle be confounded by epistasis, because selectively favored “uphill” steps (beneficial mutations) may only be possible after several “sideways” or “downhill” steps (neutral or deleterious mutations).
Over the last decade, protein engineers have performed hundreds of directed evolution experiments to improve properties such as catalytic activity, binding affinity, or stability (Eijsink et al., 2005; Johannes and Zhao, 2006; Jackel et al., 2008). The results of these experiments offer substantial insight into the possible pathways of adaptive protein evolution and the interplay between adaptive and neutral mutations. In the next section, we describe how a typical directed evolution experiment is implemented. We then provide a specific example of how directed evolution was successfully applied to a cytochrome P450 enzyme. Drawing on this example and a wealth of other work, we then generalize to draw what we consider to be three of the main empirical lessons from directed evolution. Finally, we discuss how these lessons can help inform an understanding of natural protein evolution.
DESIGN OF A DIRECTED EVOLUTION EXPERIMENT
Although directed evolution experiments vary widely in their details, they all use the same basic evolutionary algorithm illustrated in Fig. 8.1. The experiment begins with a parent protein and an engineering goal (e.g., 10-fold improved catalytic activity on a particular substrate). The gene for the parent protein is mutagenized to produce a library of mutant genes. Proteins encoded by these mutant genes are then produced and screened (or selected) for the desired function, and the improved proteins are used as the parents for another round. Beneficial mutations are accumulated until the goal is reached or no further improvements are found. The success of the experiment obviously depends on the feasibility of the target function and whether measurable improvements can be accumulated to reach the goal.
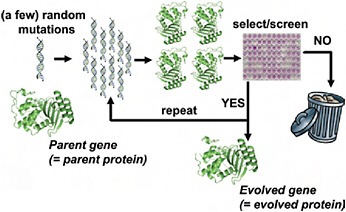
FIGURE 8.1 Schematic outline of a typical directed evolution experiment. The researcher begins with the gene for the parent protein. This parent gene is randomly mutagenized by using error-prone PCR or some similar technique. The library of mutant genes is then used to produce mutant proteins, which are screened or selected for the desired target property (e.g., improved enzymatic activity or increased stability). Mutants that fail to show improvements in the screening/selection are typically discarded, while the genes for the improved mutants are used as the parents for the next round of mutagenesis and screening. This procedure is repeated until the evolved protein exhibits the desired level of the target property (or until the student performing the experiments graduates).
There are myriad ways to implement the 3 key steps of this evolutionary algorithm: mutagenesis, screening/selection, and the decision on how to choose the parents for the next round. These choices obviously affect how successfully an experiment achieves its engineering goal and determine how closely the experiment mimics the process of natural molecular evolution. Here, we do not address how the implementation affects the experiment’s engineering success, because this issue is widely discussed in the directed evolution literature (suffice it to say that the reason that many implementations are in use is that no one approach has been decisively proven to be more effective than the others). Instead, we simply give a general overview of the most common implementations, so that a reader from outside of the field can gain a sense of the extent to which directed and natural evolution are comparable.
The 2 most common procedures for mutagenizing the parent genes are error-prone PCR and DNA shuffling (Stemmer, 1994). As its name suggests, error-prone PCR copies the parent genes while introducing a few mutations (usually 1 or 2 per gene), and therefore mimics imperfect DNA replication. DNA shuffling is a procedure that both mutagenizes and recombines homologous genes at crossover points of high sequence iden-
tity and therefore approximately imitates the natural process of homologous recombination (although the parents are often more diverged than naturally recombining proteins). A common variation on both of these techniques is to bias the creation of mutant proteins with the goal of increasing the fraction of improved mutants, for example, by targeting functionally important residues for mutagenesis (Park et al., 2005) or choosing recombination crossover points based on structural information (Voigt et al., 2002). Researchers also sometimes use techniques to specifically change several residues simultaneously with the goal of finding coupled beneficial mutations, although (as discussed below) the mutations discovered by such approaches often turn out to have been individually beneficial, and so presumably could have also been discovered separately with lower mutation rates.
The procedure used to identify improved mutants depends on the details of the particular protein and engineering goal. In some cases, the protein property of interest can be coupled to the survival of the host cell, thereby allowing for direct genetic selection of cells carrying improved mutants. Such selections can often be applied to libraries consisting of millions of different mutants. More frequently, it is not possible to design an effective selection, and mutants must be assayed directly in a high-throughput screen. In these cases, the researcher is typically able to examine libraries of a few thousand different mutants. Such screening is nonetheless sufficient to examine most of the possible individual mutations to a parent protein, because a 200-aa protein possesses only 19 × 200 = 3,800 unique single mutants (even fewer are accessible via single nucleotide changes).
The last step in the evolutionary algorithm is using the results from the screening/selection to choose the parents for the next generation. For reasons that are not entirely clear, directed evolution experiments rarely use schemes in which each mutant contributes to the next generation with a probability proportional to its measured fitness. (In contrast, fitness-proportionate selection is widely used in computational genetic algorithms.) Instead, researchers typically choose one or a few of the best mutants as parents for the next generation. Proteins undergoing directed evolution therefore experience a series of population bottlenecks in which most of the genetic variation is purged. At the same time, the adaptive-walk nature of these experiments provides little opportunity for deleterious or neutral mutations to spread (unless they hitchhike along with beneficial ones). These experiments therefore typically fail to fully recapitulate the evolutionary dynamics of either small populations (rapid genetic drift including the occasional fixation of deleterious mutations) or large populations (the maintenance of substantial levels of standing genetic variation). Directed evolution therefore probably sheds more light on the question of
how beneficial mutations arise, rather than on how these mutations would actually spread in a naturally evolving population.
AN EXAMPLE OF DIRECTED EVOLUTION: CONVERTING A CYTOCHROME P450 FATTY ACID HYDROXYLASE INTO A PROPANE HYDROXYLASE
As an example of protein adaptation via directed evolution, we describe how the substrate specificity of a cytochrome P450 enzyme was dramatically altered (Fasan et al., 2007, 2008). The cytochrome P450 monooxygenase superfamily provides a beautiful example of how nature has generated a whole spectrum of catalysts from one common framework: >7,000 P450 sequences identified from all kingdoms of life catalyze the oxidation of a vast array of organic compounds. We wanted to know how easily we could alter one of these natural enzymes (P450 BM3, a bacterial fatty acid hydroxylase) to hydroxylate small alkanes such as propane and ethane, a reaction that is catalyzed in nature by a different class of monooxygenase enzymes.
Although wild-type P450 BM3 has only weak activity on long-chain alkanes (and no measurable activity on short alkanes), we hypothesized that mutants of this enzyme displaying enhanced alkane activity might acquire measurable ability to hydroxylate slightly shorter alkanes. Further mutagenesis of active variants, with screening on progressively shorter-chained substrates, could ultimately generate activity on the smallest, gaseous alkanes. This approach of breaking down an apparently difficult problem (such as obtaining activity on a substrate very different from the native substrate) into a series of smaller problems lowers the bar for each evolutionary step and can even allow new activities to be acquired 1 mutation at a time.
Iterative rounds of random mutagenesis, recombination of beneficial mutations, and screening for activity on successively smaller alkanes, or alkane surrogates, led to the creation of P450 PMO, whose ability to hydroxylate propane is comparable with the wild-type enzyme’s activity on fatty acids (Fasan et al., 2007). This enzyme contains 23 amino acid substitutions relative to its wild-type ancestor; complete respecialization for the new substrate required changing only a little more than 2% of the amino acid sequence.
At one point during the evolution, however, no further improvements in activity on propane were found, even though the activity remained well below the native enzyme’s activity on its preferred substrates. Upon measuring the stability of the evolved enzyme and its precursors, we found that the mutations that had enhanced activity on propane had also decreased the protein’s stability to the extent that the enzyme could tolerate only a low fraction of new mutations. Intentionally selecting for several
stabilizing mutations recovered much of the lost stability and allowed for the subsequent discovery of further mutations that improved activity. Fig. 8.2 summarizes the overall changes in activity and stability that occurred during the entire directed evolution trajectory (Fasan et al., 2008).
EMPIRICAL LESSONS FROM THE DIRECTED EVOLUTION OF PROTEINS
In this section, we offer what we consider to be some of the general lessons about protein adaptation that can be drawn from directed evolution experiments.
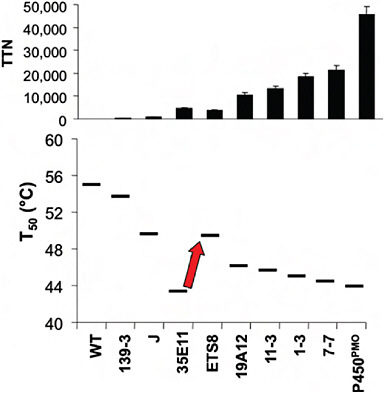
FIGURE 8.2 Activity and stability changes during the directed evolution of a cytochrome P450 enzyme for activity on short-chain alkanes. (Upper) The changes in activity on propane (total turnover number, TTN) during steps along the directed evolution trajectory. (Lower) The changes in protein stability (measured as T50 values for heat inactivation). During the steps of directed evolution, the protein was selected for activity on progressively shorter alkanes, without regard to stability. The exception is the step indicated by the arrow, where stabilizing mutations were intentionally selected to recover some of the lost stability. Data are taken from Fasan et al. (2008).
Many Desirable Protein Properties Can Be Improved Incrementally, Through Single Mutations
Perhaps the most surprising result from directed evolution experiments is simply how effectively random mutation and selection are able to enhance target protein properties. In most cases where the researcher has been able to devise a high-throughput and sensitive screening assay, it has proved possible to find mutations that improve function (usually a catalytic activity or binding affinity). Directed evolution experiments naturally classify mutations as beneficial, neutral, or deleterious, depending on how they affect the target property. These studies tend to reach remarkably similar conclusions about the fractions of mutations that fall into each of these 3 classifications, despite applying different methodologies to different proteins to optimize different properties. Typically, ≈30–50% of random mutations are strongly deleterious (Shafikhani et al., 1997; Guo et al., 2004; Drummond et al., 2005), 50–70% are approximately neutral (Shafikhani et al., 1997; Guo et al., 2004; Drummond et al., 2005), and perhaps 0.5–0.01% are beneficial (Castle et al., 2004; Garrett et al., 2004; Palackal et al., 2004; Reetz et al., 2004; Aharoni et al., 2005; Solbak et al., 2005; Bloom et al., 2006). These experiments therefore make clear that, in a laboratory context, it is almost always possible to find a substantial number of neutral mutations and usually at least a few that enhance stability or an existing function.
Most cases where directed evolution fails to immediately find beneficial mutations come when the bar is set too high, such as searching for activity on a new substrate on which the parent protein is completely inert. Such functional jumps may simply be too big for single mutations. However, these functions can usually still be generated by taking a more incremental path, as in the case described above where a cytochrome P450 became a propane hydroxylase by first becoming an octane hydroxylase (Meinhold et al., 2005; Fasan et al., 2008). A similar approach of identifying appropriate intermediate challenges was used to engineer a steroid receptor to respond to a novel ligand (Chen and Zhao, 2005). In both cases, the target activity was absent in the initial parent protein, making it refractory toward improvement by any single mutation. Selection on the intermediate substrates gave rise to low levels of the target activities, which were rapidly improved by beneficial single mutations.
These findings indicate that directed protein evolution can usually avoid being stymied by local fitness peaks, where no further incremental improvements are possible. Concern about becoming trapped on local optima probably comes from viewing evolution as occurring on a landscape created by assigning a fitness to each possible genotype. Although fitness landscapes are conceptually valid constructs, the mind effectively visualizes only 3D spaces, which are often reduced to 2 dimensions for ease
of representation on paper. However, a 300-residue protein can undergo 5,700 unique single amino acid mutations, each of which represents a different direction on the fitness landscape. For a protein to occupy a peak in such a multidimensional landscape, a step in each of these directions must lead to a decrease in fitness, meaning that all 5,700 possible mutations are deleterious. In contrast, every protein evolved in the laboratory has many possible neutral mutations, and often several beneficial ones, at least as measured by a specific biochemical assay. It may therefore be more helpful to think of protein evolution in terms of neutral networks (Smith, 1970; Huynen et al., 1996) rather than in terms of fitness peaks (see Fig. 8.3). The key difference is that fitness peaks imply a need for multiple simultaneous mutations to escape from a trap, whereas the neutral network view emphasizes the availability of many possible evolutionary pathways, which may include initially neutral and immediately beneficial mutations.
Much of the Epistatic Coupling Between Mutations Is Simply Explained in Terms of Protein Stability, Which Can Underlie Variation in Both Mutational Robustness and Evolvability
The fact that most proteins can be engineered through sequential single mutations must not be interpreted to indicate that epistatic coupling between mutations does not exist. Both directed evolution and retro-
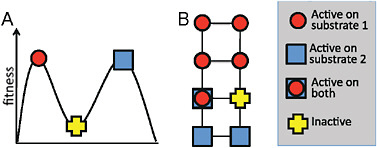
FIGURE 8.3 Fitness landscapes and neutral networks. (A) A fitness landscape in which a protein at a peak corresponding to activity on substrate 1 can only reach the peak corresponding to activity on substrate 2 by taking a downhill step corresponding to a deleterious mutation. (B) A neutral network in which a protein that is active on substrate 1 may initially be unable to achieve activity on substrate 2 with a single mutational step, but can reach activity on the latter substrate through a series of neutral steps. Although both fitness landscapes and neutral networks are conceptually valid views of evolution, fitness landscapes tend to emphasize the possibility of becoming trapped on peaks, whereas neutral networks emphasize the availability of neutral mutations and their potential coupling to adaptation. In the context of directed evolution, proteins have been found empirically to always have many possible neutral mutations.
spective analyses of natural evolution have uncovered clear examples of mutations whose beneficial effects are contingent on the existence of other initially neutral or even slightly deleterious mutations. The surprising empirical lesson is that such epistasis frequently occurs through a simple mechanism, allowing it to be both easily understood and leveraged for engineering purposes.
Early experimental clues to the common origin of much of protein mutational epistasis came from genetic studies that found that the same “global suppressor” mutations could often remedy the effects of several different deleterious mutations (Shortle and Lin, 1985; Rennell and Poteete, 1989). In many cases, the global suppressor mutations stabilized the protein’s folded structure, suggesting that they compensated for destabilization caused by the deleterious mutations (Shortle and Lin, 1985). The role of stability compensation in adaptive evolution was demonstrated in a study showing that a naturally occurring antibiotic-resistance enzyme acquired activity on new antibiotics by pairing a stabilizing mutation with one or more catalytically beneficial but destabilizing mutations (Wang et al., 2002).
The contribution of directed evolution experiments has been to demonstrate the ubiquity of such stability-mediated epistasis. Introducing just 1 stabilizing mutation into a lactamase enzyme reduced the fraction of random single amino acid mutations that inactivated the protein by one-third (Bloom et al., 2005). A cytochrome P450 enzyme that had been engineered to contain a handful of stabilizing mutations was nearly twice as tolerant to random mutations (Bloom et al., 2006). And a thermostable chorismate mutase was a remarkable 10-fold more tolerant to randomization of a large helical region than its mesostable counterpart (Besenmatter et al., 2007). The extensive stability-mediated epistasis suggested by these experiments can be visualized in terms of a protein stability threshold, as illustrated in Fig. 8.4. In a directed evolution experiment, stability is under selection only insofar as the protein must fold to its proper 3D structure with sufficient stability to perform the target biochemical function. Mutations that increase or decrease stability are therefore neutral as long as the protein remains more stable than some threshold value. But because most mutations are destabilizing, an initially neutral stabilizing mutation can increase a protein’s robustness to other, subsequent mutations.
Directed evolution has shown the crucial role that stability-based epistasis can play in adaptive evolution. One experiment directly compared the frequency with which a marginally stable and a highly stable cytochrome P450 enzyme could acquire activities on a set of new substrates upon random mutation. Libraries of mutants of both enzymes were screened, and a markedly higher fraction of mutants of the stable protein were found to exhibit the new activities (Bloom et al., 2006). This increased evolvability of the stable enzyme was caused by its ability to better toler-
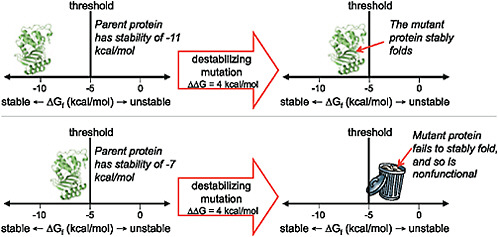
FIGURE 8.4 The effect of a mutation can depend on the stability of the protein into which it is introduced. As shown here, proteins that are more stable than the threshold can fold and function, whereas those that are less stable than the threshold fail to fold and are therefore nonfunctional. A particular functionally beneficial but destabilizing mutation may therefore only be tolerated by a protein that has previously accumulated one or more stabilizing substitutions.
ate catalytically beneficial but destabilizing mutations (Bloom et al., 2006). These results indicate that stabilizing mutations increase evolvability by the same mechanism that they increase mutational robustness.
The existence of widespread stability-mediated epistasis further explains why trapping on fitness peaks is not an important concern in directed protein evolution, although it does emphasize a role for neutral mutations. A protein that has been pushed to the margins of tolerable stability may lose access to functionally beneficial but destabilizing mutations. But this protein is still not stuck on a fitness peak, because it can regain its mutational robustness and evolvability by accumulating initially neutral but stabilizing mutations. In a nondirected context, such a process might require a time-consuming wait for stabilizing mutations to spread by neutral drift. But in a directed evolution experiment, the process can be expedited by intentional selection for stabilizing mutations, as was done in the cytochrome P450 experiment described above.
Adaptive Protein Evolution Relies Heavily on the Prevalence of Promiscuous Functions, and Protein Promiscuity in Turn Fluctuates with Neutral Mutations
Directed protein evolution experiments have demonstrated that once a biochemical function is present at even a low level, it can usually be
improved via an adaptive pathway of sequential beneficial single mutations. But how can a bioengineer induce a protein to take the first step of acquiring a trace of the target activity? One answer is, conveniently, the desired activity is often already there. Naturally occurring enzymes frequently possess low levels of various “promiscuous” activities, which can rapidly expand during laboratory evolution (O’Brien and Herschlag, 1999; Copley, 2003; Aharoni et al., 2005; Khersonsky et al., 2006).
Preexisting promiscuous activities have been used as starting points for many directed evolution experiments (Aharoni et al., 2005; Yoshikuni et al., 2006). For example, the cytochrome P450 directed evolution experiment described above used as a starting point the native enzyme’s promiscuous activity on octane. A particularly compelling example comes from phosphotriesterase and lactonase enzymes. These enzymes have been engineered for efficient activity on several new substrates through selection of mutations that enhance existing promiscuous functions (Harel et al., 2004; Aharoni et al., 2005; Amitai et al., 2006). Complementary work has shown that selection for a promiscuous activity likely explains the natural origin of a bacterial enzyme that hydrolyzes a synthetic compound only recently introduced into the environment (Afriat et al., 2006).
Laboratory evolution also indicates that promiscuous activities themselves can fluctuate substantially with neutral mutations. In separate experiments, lactonase (Amitai et al., 2007) and cytochrome P450 (Bloom et al., 2007c) enzymes were neutrally evolved by mutagenesis and selection for retention of the primary target function. Neutrally evolved variants were then examined for activity on several nontarget substrates. In many cases, the neutral mutations had led to changes in these promiscuous activities (Amitai et al., 2007; Bloom et al., 2007c). Neutral mutations can therefore set the stage for adaptation by providing a varied set of evolutionary starting points. Actually performing neutral evolution in the laboratory may be of limited engineering value because of the considerable experimental effort required to accumulate a substantial number of substitutions. But protein engineers routinely screen and recombine naturally occurring protein homologs (Crameri et al., 1998; Landwehr et al., 2007), thereby exploiting the genetic drift that underlies the divergence of their sequences and promiscuous activities. Recombination is therefore an excellent way to tap into the benefits of natural neutral evolution.
IMPLICATIONS FOR UNDERSTANDING NATURAL PROTEIN EVOLUTION
What is implied about natural protein evolution by the foregoing lessons from the laboratory? The main complication in applying these observations to natural protein evolution is determining whether similar
general selection pressures apply. Interpretation of mutational steps during directed evolution is performed in an objective way by scoring the mutant protein in a prespecified experimental assay. Therefore, it is always clear whether a mutation is beneficial, neutral, or deleterious. Of course, mutations that are scored as neutral may still affect any of an almost interminable list of other properties not measured in the assay (stability, expression level, codon usage, promiscuous activity, etc.). And herein lies the main argument against generalizing the results of directed evolution to natural evolution: experimental assays are relatively insensitive, and natural proteins evolve under pleiotropic constraints not present in the laboratory. A similar argument has been invoked against the idea of widespread neutral evolution of natural proteins, namely that apparently neutral mutations actually induce subtle, but important, alterations that do affect fitness (Blundell and Wood, 1975; Gillespie, 1991).
In the absence of an experimental method for measuring how changes in a protein affect biological fitness, these issues will always to some degree remain a matter of speculation [hence the intractability of the long-standing debate between selectionism and neutralism (Nei, 2005)]. Clearly, natural proteins are subject to additional constraints not present in most laboratory experiments, because they must function in vivo while minimizing deleterious interactions with other cellular components or pathways. In addition, laboratory evolution experiments usually impose a very strong selection for the target protein property, such that mutations that benefit the target property may be selected even to the detriment of other properties. The fact that laboratory evolution tends to impose fewer pleiotropic constraints and stronger positive selection means that it almost certainly overestimates the frequency of neutral and beneficial mutations relative to natural evolution. Therefore, whereas 50–70% of protein mutations are neutral in a laboratory assay, the percentage is surely lower in nature. The degree to which this is true will of course vary in accordance with the principles of population genetics, with proteins from viruses being more highly optimized than those from species like mammals with large genomes and small effective population sizes (Lynch, 2007). But Pauling and Zuckerkandl’s nearly half-century old observation (Zuckerkandl and Pauling, 1965) that “many substitutions may lead to relatively little functional change” seems more true than ever in an era where abundant genome sequences have revealed numerous proteins with diverged sequences that nonetheless perform largely conserved functions. This fact strongly suggests that one of the key lessons from directed evolution also applies to natural protein evolution: rather than occupying completely optimized fitness peaks, most proteins have many available evolutionary paths and often direct adaptive paths to improve potentially useful functions.
The prevalence of stability-mediated epistasis revealed by laboratory evolution also has important implications for understanding natural protein evolution. As is suggested by Fig. 8.4, whether a mutation is neutral or deleterious can be conditional on the stability of the protein in which it occurs. In contrast, most mathematical treatments of neutral evolution make the (often unspoken) assumption that a constant fraction of mutations is neutral. Several classic results commonly attributed to the neutral theory are no longer necessarily true if the fraction of neutral mutations is conditional on protein stability. In particular, in such a scenario, neutral evolution can lead to overdispersion of the molecular clock (Takahata, 1987; Bloom et al., 2007b), an influence of population size on substitution rate (Bloom et al., 2007b), and a dependence of mutational load and robustness on both population size and the structure of the underlying neutral space (van Nimwegen et al., 1999; Wilke and Adami, 2003; Bloom et al., 2007a). These results suggest the importance of continually updating theories of molecular evolution to reflect expanding knowledge about the details of the molecules in question.
The lessons of directed evolution also caution against attributing all properties of natural proteins to adaptive causes. For example, most enzymes are only marginally more stable than is required by their natural environment (Somero, 1995). This marginal stability was long argued to be an adaptive trait, providing an optimal degree of flexibility that favored high catalytic activity (Somero, 1995; Fields, 2001). This adaptive argument has been undermined by evolutionary engineering experiments demonstrating that enzyme stability can be dramatically increased without concomitant loss of catalytic activity (Serrano et al., 1993; Giver et al., 1998; Van den Burg et al., 1998). Instead, both simulations (Taverna and Goldstein, 2002) and theory (Bloom et al., 2007b; Zeldovich et al., 2007) show that the marginal stability of proteins can arise neutrally because most mutations are destabilizing. Although there are a few proteins whose marginal stability is clearly adaptive (Canadillas et al., 2006), the marginal stability of most proteins is likely the result of neutral mutation-driven processes. Other properties, such as catalytic or substrate promiscuity, that arise naturally during laboratory evolution should probably also be assigned neutral rather than adaptive origins.
Another important contribution of directed evolution has been to demonstrate 2 clear mechanisms whereby neutral mutations shape the available adaptive pathways. Selectively neutral mutations that increase stability can promote evolvability by allowing for subsequent beneficial but destabilizing mutations (Bloom et al., 2006), whereas neutral mutations that alter promiscuous activities (Amitai et al., 2007; Bloom et al., 2007c) can create the starting points for subsequent adaptive evolution. Evolutionary engineers leverage the coupling between neutral and adaptive mutations
in at least 2 ways: directly selecting for functionally neutral but stabilizing mutations and screening and recombining pools of diverged homologous sequences (Crameri et al., 1998; Landwehr et al., 2007). Natural evolution does not so deliberately exploit the potential benefits of neutral mutations, but genetic drift and preexisting diversity may play a similarly important role in natural adaptive evolution. Indeed, ancestral protein reconstruction experiments have elucidated specific adaptive events that appear to have been contingent on the initial occurrence of approximately neutral substitutions (Ortlund et al., 2007).
The overall picture that emerges from evolutionary engineering is that proteins, although clearly highly refined by evolution, retain a substantial capacity for neutral and adaptive change. In many ways, this picture is complementary to that offered by more traditional biochemical characterizations, which often focus on the exquisitely tuned interactions that can underlie a protein’s evolved function. Directed evolution does not dispute the subtlety of such interactions, nor does it usually offer such a careful description of the details of protein function. But although biochemists typically choose for their studies the most interesting examples, evolutionary engineers by necessity deal with the broader statistics of random mutations and evolutionary possibilities. These statistics suggest that proteins enjoy access to many neutral mutations, which can in turn open new adaptive avenues. Ultimately, a more detailed understanding of these evolutionary pathways will be of value in both protein engineering and evolutionary biology.
ACKNOWLEDGMENTS
F.H.A. is supported by the U.S. Department of Energy and the U.S. Army. J.D.B. is supported by a Caltech Beckman Institute Postdoctoral Fellowship and the Irvington Institute Fellowship Program of the Cancer Research Institute.
















