
1
Natural Selection in Action During Speciation
SARA VIA
The role of natural selection in speciation, first described by Darwin, has finally been widely accepted. Yet, the nature and time course of the genetic changes that result in speciation remain mysterious. To date, genetic analyses of speciation have focused almost exclusively on retrospective analyses of reproductive isolation between species or subspecies and on hybrid sterility or inviability rather than on ecologically based barriers to gene flow. However, if we are to fully understand the origin of species, we must analyze the process from additional vantage points. By studying the genetic causes of partial reproductive isolation between specialized ecological races, early barriers to gene flow can be identified before they become confounded with other species differences. This population-level approach can reveal patterns that become invisible over time, such as the mosaic nature of the genome early in speciation. Under divergent selection in sympatry, the genomes of incipient species become temporary genetic mosaics in which ecologically important genomic regions resist gene exchange, even as gene flow continues over most of the genome. Analysis of such mosaic genomes suggests that surprisingly large genomic regions around divergently selected quantitative trait loci can be protected from interrace recombination by “divergence hitchhiking.” Here, I describe the formation of the genetic mosaic during early ecological speciation, consider
Departments of Biology and Entomology, University of Maryland, College Park, MD 20742.
the establishment, effects, and transitory nature of divergence hitchhiking around key ecologically important genes, and describe a 2-stage model for genetic divergence during ecological speciation with gene flow.
The origin of species is only slightly less mysterious now than it was 150 years ago when Darwin published his famous book (Darwin, 1859). Although Darwin’s idea that natural selection drives speciation has finally been widely accepted (Coyne and Orr, 2004), we still have much to learn about the nature and time course of the genetic changes that cause speciation under natural selection (Schluter, 2001, 2009; Via and West, 2008).
THE SPYGLASS
Pivotal ideas developed during the modern synthesis of the 1930s–1940s have largely determined the course of modern speciation research. Ernst Mayr (1942) developed the biological species concept, putting reproductive isolation at the center of speciation and making analysis of the evolution of reproductive isolation a clear target for speciation research. Mayr also stressed that the evolution of reproductive isolation is a fragile process that can only proceed if geographical separation renders gene flow impossible, firmly establishing allopatric speciation as the norm. Theodosius Dobzhansky (1937) identified a wide array of traits that could cause reproductive isolation, but focused much of his own research into speciation on postzygotic genetic incompatibilities (Dobzhansky, 1934), as did H. J. Muller (1942). At the time, hybrid sterility was a particularly problematic aspect of speciation because it had been unclear since Darwin (1859) how such a disadvantageous trait could evolve under natural selection. By providing a clear mechanism by which hybrid sterility could evolve (Coyne and Orr, 2004, pp. 269 and 270), Dobzhansky-Muller genetic incompatibilities (DMIs) took center stage in the genetic analysis of speciation, where they have remained ever since. Collectively, the architects of the synthesis outlined a retrospective approach to speciation that I call “the spyglass,” because it starts late in the process (or after it is complete) and looks back in time to infer the causes of speciation (Fig. 1.1A).
Unquestionably, this approach has been a rich source of information about the kinds of barriers to gene flow that can isolate species [e.g., Otte and Endler (1989); Howard and Berlocher (2003)], but alternative ideas about speciation and how to study it have met with considerable resistance during the past 70 years. Even today, allopatric speciation remains the null model against which all other mechanisms for speciation must be tested (Coyne and Orr, 2004, p. 158), and DMIs are widely regarded as
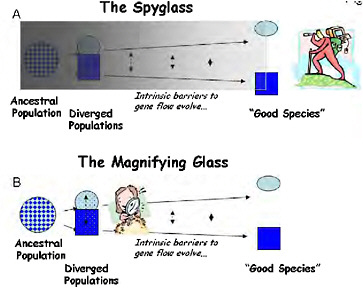
FIGURE 1-1 Two ways to study the process of speciation, which is visualized here as a continuum of divergence from a variable population to a divergent pair of populations, and through the evolution of intrinsic barriers to gene flow to the recognition of “good species.” (A) Using “the spyglass,” the process is studied by attempting to look back to see the details of speciation from the vantage point of the present. (B) Using “the magnifying glass,” the mechanisms of reproductive isolation are studied in partially isolated divergent ecotypes, which are used as models of an early stage of speciation.
the appropriate focus of research in speciation genetics (Ting et al., 2000; Masly and Presgraves, 2007; Mihola et al., 2009; Phadnis and Orr, 2009; Willis, 2009).
THE MAGNIFYING GLASS
A different view of speciation genetics is now gaining in popularity: the population-level analysis of how ecology and genetics interact in various situations to cause the evolution of barriers to gene flow (Schemske, 2000; Schluter, 2001; Via, 2001). By analyzing partially reproductively isolated ecotypes or races, the genetic changes contributing to reproductive isolation can be studied before they become confounded by additional genetic differences between species that accumulate after speciation is complete. Indeed, studying barriers to gene flow in populations that are not yet completely reproductively isolated may reveal important aspects of the process that have never been seen clearly before. This approach is particularly suitable for the analysis of speciation under divergent
selection, now called “ecological speciation” (Schluter, 2001). To contrast this approach with the more classic retrospective analyses, I call the population-level analysis of the ecological and genetic causes of reproductive isolation the “magnifying glass” (Fig. 1.1B).
The validity of population-level analyses of the ecology and genetics of the partial reproductive isolation has occasionally been questioned. Because there is no guarantee that ecotypes or races will ever attain species status, some argue that barriers to gene flow between them caused by divergent selection are irrelevant to the study of speciation (Futuyma, 1987; Coyne and Orr, 2004). Others disregard ecologically based reproductive isolation between ecotypes because it lacks permanence and could be reversed if the pattern of divergent selection changes (Coyne and Orr, 2004). However, many valid species concepts do not require or even consider permanence, and doing so simply underscores the observation that different species concepts apply at different points along a continuum of divergence from populations to well-established and permanent species (Harrison, 1998).
To supporters of the population-level approach, divergent populations are an early stage on that continuum. They argue that barriers to gene flow in partially isolated ecological races or ecotypes must be similar to those that would have been seen long ago between a pair of present-day sister species in a similar ecological situation. Although it is certainly true that not all divergent races will go on to become full species, many contemporary species must have passed through the stage of population divergence typified by ecotypes. Because the particular ecological conditions associated with divergence under selection cannot be seen clearly through the spyglass, that approach is unlikely to reveal much about the initial causes of reproductive isolation during speciation under divergent selection.
Speciation research is plagued by this awkward gulf between population-level and species-level analyses. To fully understand the mechanisms of speciation, we must cross that space, integrating magnifying glass analyses of the early parts of the process with the view of the end products through the spyglass.
HOW DOES NATURAL SELECTION CAUSE SPECIATION?
With the possible exception of reinforcement, natural selection does not directly favor phenotypic traits or genetic incompatibilities simply because they block gene flow. Instead, it is generally thought that reproductive isolation occurs indirectly, as a “by-product” of the genetic changes that increase adaptation (Coyne and Orr, 2004, p. 385). However, pooling ecologically based reproductive isolation with that caused by the accumulation
of genetic incompatibilities under the term by-product does not do much to advance our mechanistic understanding of speciation. To fully connect the views through the magnifying glass and the spyglass, we must understand how different ecological situations lead to particular types of barriers to gene flow and on what timetable these different forms of reproductive isolation evolve. It is particularly important to ask under what circumstances and how often ecologically based reproductive isolation produce virtually complete reproductive isolation before appreciable genetic incompatibility has evolved (Ramsey et al., 2003; Coyne and Orr, 2004, pp. 57–59).
THE EFFECT OF GEOGRAPHY ON SPECIATION
Ecological speciation can occur in either geographically isolated populations (allopatry) or in settings with no physical barriers to gene flow (sympatry or parapatry). When gene exchange is physically impossible, the conditions under which reproductive isolation can evolve are nonrestrictive: allopatric speciation can be driven by strong or weak divergent selection, sexual selection, uniform selection, or even stabilizing selection. It may occur quickly under divergent selection or extremely slowly under uniform or balancing selection.
In contrast, the conditions under which sympatric or parapatric speciation with gene flow can occur are not so forgiving: genetically based phenotypic divergence requires much stronger selection to occur and be maintained when gene flow is possible than when geography makes it an impossibility [e.g., Rice and Hostert (1993); Via (2001)]. In the presence of migration, the establishment of genomic regions that resist gene flow sufficiently to maintain phenotypic differentiation is only likely if divergent (or possibly sexual) selection is strong, and so the initial barriers to gene flow in sympatry are likely to evolve quickly (Rice and Hostert, 1993; Via, 2001; Hendry et al., 2007). Speciation with gene flow (Rice and Hostert, 1993) is thus unlikely to occur under weak divergent selection, and it is certainly not expected under uniform or balancing selection (except perhaps by polyploidy). One fortuitous effect of the strong selection required for speciation with gene flow is that the genomic regions that cause reproductive isolation become particularly distinctive relative to the rest of the genome. This facilitates their discovery in empirical analyses.
Until recently, genetic models of speciation with gene flow have been extremely simplified, and they have suggested quite a restrictive set of conditions for sympatric speciation. In particular, critics cite the difficulty of evolving assortative mating in the face of free recombination (Coyne and Orr, 2004, pp. 127–141). Although Felsenstein (1981) showed that this constraint is significantly reduced under linkage between genes affecting performance and mating, that result has been largely ignored (Via, 2001).
A variety of conditions that facilitate ecological speciation with gene flow are now well described (Rice and Hostert, 1993; Via, 2001). They include strong divergent selection on multiple traits associated with resource or habitat use and ecologically based selection against migrants and/or hybrids. Recent work suggests that assortative mating can evolve rather easily if habitat choice determines the choice of mates (Rice and Hostert, 1993; Via, 2001), if mate choice is a correlate of the traits under divergent selection (Schluter, 2001), or if recombination is reduced by physical linkage or pleiotropy (Felsenstein, 1981; Hawthorne and Via, 2001). Some of the best-studied divergent races in the wild, including the ecologically specialized host races of the pea aphid [Acyrthosiphon pisum (Harris)] satisfy these conditions (Via, 2001), providing strong empirical support for the argument that reproductive isolation can evolve, or at least persist, in the face of gene flow.
The first step in using the magnifying glass to study speciation involves estimating the magnitude of gene flow between a pair of partially reproductively isolated taxa. That process is not as straightforward as it may seem.
POTENTIAL VS. REALIZED GENE FLOW DURING ECOLOGICAL SPECIATION UNDER DIVERGENT SELECTION
Empirical estimates of gene flow assume neutrality of markers and a balance between migration and random drift [e.g., Hartl and Clark (1997)]. This emphasis on gene flow under neutrality implies that there is just 1 “true” estimate of gene exchange between 2 taxa. Moreover, the minimal gene flow required to counter drift under Wright’s island model (Hartl and Clark, 1997, pp. 194 and 195) conjures up a picture of gene flow as a force that will easily homogenize adjacent populations (Fig. 1.2A). Yet, one can easily find phenotypically divergent and ecologically specialized populations living in close adjacency with no physical barrier to gene flow. Is this a contradiction? Not necessarily, because the degree to which a genomic region affecting a given trait is homogenized by migration depends not on the estimated gene flow under neutrality, but on the realized gene flow at that region after selection.
It is a maxim of population genetics that migration and genetic drift affect the entire genome, whereas the effects of natural selection are limited to genomic regions harboring loci that affect the selected phenotypic traits. In ecologically specialized populations, divergent selection on traits associated with the use of resources or habitats is strong enough to maintain divergence in the parts of the genome that affect those traits, while gene flow continues in other genomic regions (Figs. 1.2B and 1.3). Between such populations, the estimated gene flow under neutrality may
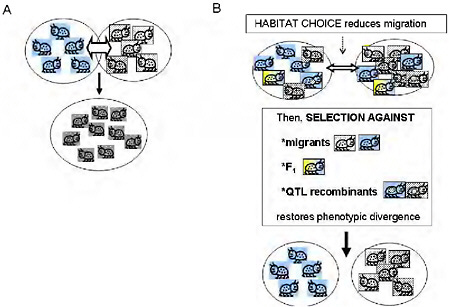
FIGURE 1-2 (A) The simple vision of gene flow as a homogenizing force in populations. Free migration and random mating between locally adapted populations is simplistically visualized as homogenizing populations and eradicating local adaptation. (B) Ecologically based barriers to gene flow under divergent selection permit genetically based phenotypic divergence under selection to be maintained in the face of the possibility of gene flow: habitat choice reduces migration, then selection against migrants and F1, and selection against QTL recombinants reduce introgression of locally adapted alleles, maintaining divergence.
grossly overestimate the realized gene exchange experienced by divergently selected genomic regions.
Because of the localized genomic effects of divergent selection on realized gene exchange, divergence early in ecological speciation with gene flow is expected to be greater in genomic regions that harbor key quantitative trait loci (QTL) than it is in regions that have no effect on the phenotypic divergence of the populations (Fig. 1.3). If, as usually thought, speciation with gene flow involves only a handful of characters (Rice and Hostert, 1993), these divergent regions may comprise a relatively small fraction of the genome, leaving the genomes of incipient species largely homogenized by ongoing gene flow and “profoundly genetically similar” [Fig. 1.3 and Kondrashov et al. (1998)]. We call the resulting pattern of genomic heterogeneity in divergence and gene exchange early in ecological speciation with gene flow “the genetic mosaic of speciation” (Via and West, 2008).
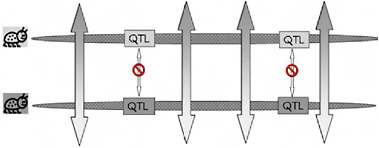
FIGURE 1-3 A cartoon version of genomic heterogeneity of gene exchange and genetic divergence under divergent selection. One chromosome from each specialized parental population is shown, with the genomic regions that contain locally adapted genes (QTL) indicated in boxes shaded to correspond to the specialized parent from which it came. Bidirectional arrows indicate gene exchange, with the pattern in the arrow tips showing that alleles from the other population are introgressing. Gene exchange can occur outside the regions containing the specialized genes, but is blocked from occurring within those regions for the reasons shown in Fig. 1.2B. This heterogeneous pattern of gene exchange under selection establishes the “genetic mosaic of speciation,” in which genetic divergence is restricted to divergently selected portions of the genomes, while other regions share polymorphisms freely through ongoing gene exchange.
USING FstOUTLIER ANALYSIS TO IDENTIFY SELECTED GENOMIC REGIONS IN THE GENETIC MOSAIC
Wright’s Fst is a widely used measure of genetic divergence between populations (Hartl and Clark, 1997). Lewontin and Krakauer (1973) proposed that genetic markers with aberrantly high Fst values (“outliers”) could be inferred to be affected by divergent selection. Recently, outlier analyses have enjoyed a renaissance because of the development of new methods based on coalescent simulation (Beaumont and Nichols, 1996; Beaumont and Balding, 2004). Fst variation is still used to eliminate selected markers from population genetic analyses that require neutrality (Luikart et al., 2003), but it is now of primary interest as a signature of divergent selection that permits detection of genomic regions involved in adaptive divergence (Beaumont, 2005; Storz, 2005).
Significant heterogeneity in marker Fst has been documented between ecologically divergent races (Wilding et al., 2001; Emelianov et al., 2004; Rogers and Bernatchez, 2005; Bonin et al., 2006; Oetjen and Reusch, 2007), with the interpretation that Fst outliers must be linked to loci affecting the phenotypic traits known to distinguish the divergent ecotypes, races, or subspecies. However, outlier analysis alone cannot reveal the cause of deviant Fst values, and the conclusion that Fst outliers must be associ-
ated with genes causing the most obvious differences between ecotypes is premature.
Using a linkage map that shows locations of the QTL affecting phenotypic traits known to be under divergent selection, the hypothesis that outliers are linked to key phenotypic traits under divergent selection can be tested: Are Fst outliers scattered randomly on the map, or are they clustered near divergently selected QTL? This approach requires a system in which the phenotypic traits involved in divergence and reproductive isolation are well known, the strength of selection on these traits has been measured, and QTL affecting the key traits have been localized on a linkage map, so that mapped markers can be used in an Fst analysis of field-collected samples. The pea aphid host races on alfalfa and red clover [A. pisum pisum (Harris)] are such a system.
PEA APHIDS ON ALFALFA AND RED CLOVER: ECOLOGICAL SPECIATION IN ACTION?
The pea aphid complex is a worldwide group of phloem-feeding insects, found primarily on legumes (Eastop, 1971). Although the pea aphid host races on alfalfa and red clover are in the same subspecies, A. pisum pisum, sympatric pea aphid populations on alfalfa, red clover, and other legumes are highly genetically divergent and ecologically specialized in the eastern United States (Via, 1991b), Europe (Sandstrom, 1996; Simon et al., 2003; Ferrari et al., 2007, 2008), and South America (Peccoud et al., 2008).
Experimental studies in both the field and laboratory have documented extensive ecologically based reproductive isolation between the pea aphid host races in eastern North America because of strong selection against migrants to the alternate host (Via, 1989, 1991a,b, 1999), environmentally mediated selection against hybrids (Via et al., 2000), and habitat choice (Via et al., 2000). It is unknown whether divergence between the pea aphid host races began in sympatry [e.g., Coyne and Orr (2004, pp. 163 and 164)]. However, conditions of the initial split are far less relevant to the study of speciation than is the fact that divergent selection currently maintains genetically based phenotypic differentiation and significant ecologically based reproductive isolation between sympatric populations.
Via and West (2008) estimated Fst between the pea aphid host races for 45 markers with known locations on a QTL map of genomic regions affecting early fecundity and behavioral acceptance of each plant. They then estimated the map distance from each marker to the nearest QTL for one of the key host use traits and found that Fst outliers were significantly clustered around the QTL involved in reproductive isolation (P < 0.05; Fig. 1.4).
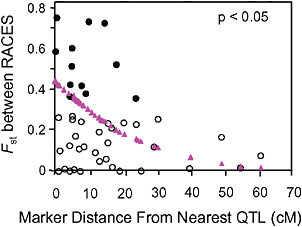
FIGURE 1-4 The relationship between the Fst values of AFLP markers, and their map distance from the nearest QTL involved in reproductive isolation between pea aphids on alfalfa and red clover. Fst outliers are shown with solid circles, and the dotted line at 10.6 cM marks the average distance from an outlier to the nearest QTL. The triangles are the predicted values from a logistic regression of the probability that a marker was an outlier on its distance to the nearest QTL (modified from Via and West, 2008).
Surprisingly, the spatial distribution of the mapped Fst outliers suggests that the signature left by divergent phenotypic selection on neutral markers can extend far from major QTL: the average outlier was 10.6 cM from the nearest QTL. A similar result was found for traits involved in the divergence of whitefish morphs in postglacial lakes (Rogers and Bernatchez, 2007), where the average outlier was 16.2 cM from the nearest QTL. In both systems, the hitchhiking regions around divergently selected QTL are far larger than expected. After a selective sweep through a large panmictic population, linkage disequilibrium rapidly erodes except in areas of reduced recombination (Begun and Aquadro, 1992). Although reduced recombination is, in fact, the explanation for the large hitchhiking regions we observed, they arise not from suppression of recombination per se, but from a reduction in the “effective recombination” between locally adapted QTL alleles in divergent populations that are subdivided (i.e., no longer randomly mating).
Charlesworth et al. (1997) analyzed the potential for hitchhiking when populations are subdivided by divergent (local) selection. Using a simulation analysis, they found that regions of linkage disequilibrium of the size we observed (Via and West, 2008) can be maintained around a divergently selected locus. This unexpected result occurs because subdivision reduces the opportunity for recombination between locally adapted QTL
alleles. In other words, divergent selection leads to fewer than expected interpopulation matings, which reduces the effective recombination of locally adapted QTL alleles below what is expected based on a map from a controlled cross.
Because speciation is, by definition, a process in which populations become increasingly subdivided, reduced effective recombination under subdivision is an important aspect of speciation with gene flow, although it has been largely unappreciated. As populations become subdivided by divergent selection and ecological specialization increases, the opportunity for interrace recombination is increasingly reduced by selection against migrants, extrinsic selection against F1 hybrids, and/or habitat choice. When recombination between QTL alleles does occur, the local disadvantage of the recombinant QTL allele further reduces the frequency of migrant alleles in advanced generation hybrids or backcrosses.
So, despite free recombination within subpopulations, the effective recombination between genotypes with different QTL alleles begins to decline as soon as divergent selection foils panmixia in early ecological speciation with gene flow, and it may also be a significant factor maintaining divergence after secondary contact. To emphasize that this form of hitchhiking is maintained only in divergent populations, and that it differs from hitchhiking after a selective sweep, I call it “divergence hitchhiking” (Via and West, 2008).
The reduction in effective recombination between divergent races means that the nominal map distance estimated from Fst outliers to the nearest QTL (Fig. 1.4) overestimates the actual probability that recombination will separate a divergent outlier allele from a locally adapted allele at a nearby QTL. In other words, the “effective map distance” between a locally adapted QTL and a nearby marker is much less than that estimated from a linkage map made using controlled matings.
We can empirically approximate the effective marker-QTL distance in pea aphids by estimating the extent to which habitat choice and divergent selection limit the opportunity for interrace mating and recombination (e.g., Fig. 1.2B). We used field sampling to estimate that habitat choice reduces migration to the alternate host to ≈11% (suggesting that 89% of possible migrants reject the alternate host plant; Via et al., 2000). The fitness of migrants from alfalfa to clover is ≈30% of that of nonmigrants, whereas the fitness of migrants from clover to alfalfa is only ≈5% of that expected for nonmigrants, for an average relative fitness of migrants of ≈17% (estimated selection against migrants of s = 0.83; Via et al., 2000). F1 hybrids do not feed as effectively on either host as the parental specialist, which reduces their fecundity (and the realized number of recombinations) by ≈50% (Via et al., 2000). So, without accounting for sexual selection against migrants, the probability of recombination between races for
a marker 10.6 cM from a QTL is the original recombination probability (0.106) discounted by the probability that a migrant will choose the alternate habitat and survive there (0.11 × 0.17), and the relative fecundity of F1 hybrids (0.5), making the effective recombination rate for that marker (0.106) × (0.11) × (0.17) × (0.5) = 0.001.
This calculation suggests that the average outlier, at a nominal distance of 10.6 cM from the nearest QTL on the linkage map (Via and West, 2008), has an effective map distance to that QTL of only ≈0.1 cM. In these populations, even a marker 50 cM from a divergently selected QTL has an effective map distance of only 0.5 cM as a result of the large decrease in effective migration caused by extensive ecologically based reproductive isolation. This estimated difference between nominal recombination and effective recombination between subdivided populations shows why such a large genomic region is expected to remain in linkage disequilibrium with a QTL under divergent selection during ecological speciation with gene flow.
It is of interest that the magnitude of effective recombination in a given genomic region changes over time as ecological specialization evolves, because greater specialization increases the magnitude of resource-based selection against migrants and hybrids. This can be illustrated with a simple example. Imagine that early in divergence only a few QTL have differentiated such that extent of habitat choice and the disadvantage of migrants or F1 is only 25% as strong as at present. Then, only 22% of potential migrants would refuse the alternate host (78% accept), selection against a migrant would be s = 0.21 (relative fitness of migrants = 0.79), and the relative fecundity of an F1 hybrid would be 0.875, making the effective rate of recombination of the average outlier with the nearest QTL (0.106) × (0.78) × (0.79) × (0.875) = 0.057. Thus, at this earlier point in divergence, the average outlier would have an effective map distance of 5.7 cM from the nearest QTL. Although smaller than the nominal map distance of 10.6 cM, it is far from the tight effective linkage seen at present. The size of each region of divergence hitchhiking therefore depends not only on the strength of divergent selection directly on that genomic region, but also on the extent to which effective migration is reduced by the earlier divergence of other QTL alleles throughout the genome.
Divergence hitchhiking has the same general effect on interrace recombination and speciation as a chromosomal inversion that happens to contain 1 or more key QTL [e.g., Noor et al. (2001), Rieseberg (2001), Machado et al. (2007)]. However, unlike inversions, which must occur relatively infrequently at the site of key QTL, divergence hitchhiking appears automatically around any QTL under strong divergent selection. More-
over, regions of divergence hitchhiking are dynamic. They increase in size as the evolution of specialization reduces the effective migration between diverging races, and regions of divergence hitchhiking around loosely linked QTL may overlap and merge. Perhaps most importantly, however, regions of divergence hitchhiking leave no permanent signature because they do not involve physical alterations to chromosomes. These regions of reduced interrace recombination can only be detected while divergence elsewhere in the genome is low. They will not be seen in retrospective analyses of good species, because as speciation progresses they become assimilated into the overall genomewide pattern of genetic divergence and by the time speciation is complete, they have disappeared.
HOW MANY QTL ARE THERE WITHIN A REGION OF DIVERGENCE HITCHHIKING?
Hawthorne and Via (2001) found that QTL for different traits under divergent selection in the pea aphid host races tended to colocalize on the linkage map. Colocalization of QTL increases selection experienced by that genomic region, thereby increasing the size of the region of divergence hitchhiking, and facilitating both QTL detection and the accumulation of additional QTL. In addition, any given QTL may actually be a cluster of several genes. Thus, it seems likely that most regions of divergence hitchhiking will contain multiple genes that affect 1 or more traits under divergent selection.
How Are Regions of Divergence Hitchhiking Delineated?
Determining the size of a given region of divergence hitchhiking is not entirely straightforward. There are 2 contrasting views:
I.
A Single Region of Divergence Hitchhiking Extends Across a Cluster of FstOutliers Around a Given QTL or Group of QTL (Fig. 1.5A).
Via and West (2008) proposed that a cluster of outliers defines a single region of divergence hitchhiking, which may include 1 or more QTL. They suggested that the boundaries of a given region of divergence hitchhiking be estimated by curve fitting in a genome scan of Fst values at various map distances around individual QTL under divergent selection (Fig. 1.5A). In this view, markers with low Fst values that lie within hitchhiking regions are interpreted as polymorphisms that predate divergence at the QTL. They are thus uninformative about population divergence and should not be used to mark the boundaries of divergence hitchhiking.
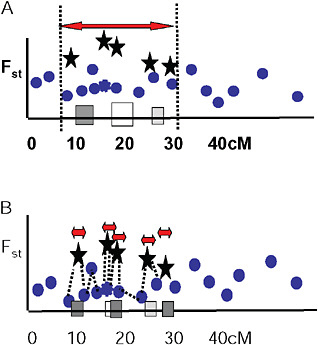
FIGURE 1-5 Two views of how to delineate regions of divergence hitchhiking. (A) A single region of divergence hitchhiking around QTL affecting one or more traits (indicated by differently shaded squares) is defined by a cluster of Fst outliers. (B) Each outlier defines a a separate hitchhiking region, bounded by a low Fst marker. In this view, outliers are thought to be either under direct divergent selection, or tightly linked to a selected gene.
II.
Each Outlier Corresponds to a Gene or QTL Under Selection or Is Itself Under Selection (Fig. 1.5B).
Ting et al. (2000) found that a DNA sequence just 1,100 bp away from the hybrid sterility gene Odysseus (Ody) was not divergent between the 2 parental species, and from this single observation they concluded that the hitchhiking region around Ody must be extremely small. Wood et al. (2008) and Smadja et al. (2008) extend this idea by suggesting that the nearest genomic region of low genetic divergence to an Fst outlier marks the boundary of its hitchhiking region (Fig. 1.5B).
Several observations are inconsistent with this hypothesis. First, outliers are easy to find. Even in studies with just a few markers (Wilding et al., 2001; Emelianov et al., 2004; Rogers and Bernatchez, 2007; Via and West, 2008), 5% or more of tested markers are generally Fst outliers. Taking 5% as a minimal estimate, this observation implies either that hitchhiking
regions are large enough that they capture 5% of randomly chosen markers (Via and West, 2008), or that so many genes are under divergent selection that 5 of them can be found with only 100 markers (Wood et al., 2008). If the latter were true, we would expect that in an average-sized genome of ≈25,000 genes, ≈1,250 of them (5%) would be involved in ecotypic differentiation and early speciation. This seems unlikely, given the prevailing view that speciation with gene flow typically involves just a handful of traits, each influenced by just a few major genes (Rice and Hostert, 1993). Second, even with very strong phenotypic selection during population divergence, the selection coefficients on each of 1,250 genes would be far too small to generate either a detectable QTL or an Fst outlier.
THE TWO STAGES OF ECOLOGICAL SPECIATION WITH GENE FLOW
Consideration of the mosaic nature of the genome during early ecological speciation with gene flow suggests that genetic change in this form of speciation occurs in 2 distinct stages:
During the first stage of ecological speciation with gene flow, genomic regions containing major QTL for key traits quickly diverge under selection and become resistant to gene exchange. This establishes the commonly observed pattern of genomic heterogeneity in divergence between incipient species, which we call the genetic mosaic.
As divergent selection proceeds, ecologically based reproductive isolation increases because of resource-based selection against migrants and hybrids and the evolution of habitat choice. These factors limit “effective migration,” i.e., the joint probability that a migrant will choose the alternate resource, survive to mate with a resident, and then that a subsequent F1 will produce a recombinant gamete. This reduction in migration increases the size of hitchhiking regions genomewide and tips the migration-selection balance, which permits QTL of smaller effect to diverge between the populations. By the end of stage 1, ecologically based reproductive isolation may be nearly complete between the new lineages, and genetic divergence is expected to be concentrated in just a handful of genomic regions.
Although this first stage of divergence may involve relatively few traits and a small fraction of the genome, the divergence of QTL for key phenotypic traits under selection defines the branching pattern with which other loci will eventually become phylogenetically concordant. The phenotypic traits that diverge under selection are those that are likely
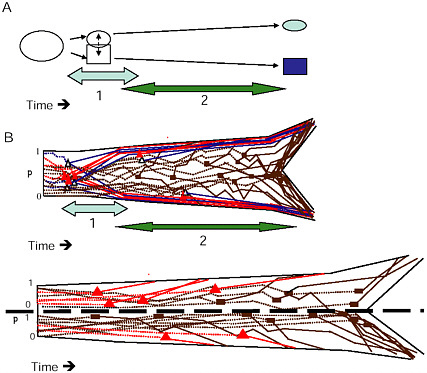
FIGURE 1-6 The two stages of ecological speciation-with-gene-flow. (A) The duration of Stage 1 is noted by the light gray, Stage 2 by the darker arrow. Events during each stage are as described in the text. (B) Cartoon of gene trees within a species tree for ecological speciation-with-gene-flow. Each gene is polymorphic in the original population, with a frequency (p) as noted on the axis at the left of the drawing. As time goes on, some loci quickly diverge at about the same time under divergent selection. Solid triangles denote selected QTL, open triangles are divergence hitchhikers. This divergence is mostly complete by the end of Stage 1. During Stage 2, a handful of additional loci diverge under selection, and loci that were unaffected by divergent selection diverge by independent responses to uniform or balancing selection, or by drift (solid rectangles). (C) Gene trees in a species tree under allopatry, symbols are as in (B) . If an allopatric population enters a new environment, there may be a period of rapid response to divergent selection similar to Stage 1 in (A) . Otherwise, genes diverge over a long time period under any combination of divergent selection, independent responses to uniform or balancing selection, or drift, eventually all coming into concordance to produce the branching pattern of the new species.
to distinguish the eventual species, and genetic divergence at the major QTL for these traits defines the branches of what will ultimately become the species tree. Because selection accelerates progression to reciprocal monophyly (Avise, 2000), loci within these genomic regions are expected to become reciprocally monophyletic long before the rest of the genome, on approximately the same timescale as the divergence of the quantitative traits that these genes affect. During this phase, few other genetic differences are expected between the incipient species. This restriction of genetic
differentiation to the genomic regions affecting key divergently selected QTL is what makes it possible to use outlier analysis during stage 1 to distinguish genomic regions harboring these branch-defining loci.
At the end of stage 1, the gene trees for loci unaffected by divergent selection will still resemble a discordant collection of “tangled twigs” (Avise, 2000, p. 307), which reflects a combination of unresolved ancestral polymorphism, recent gene flow, and stochastic effects of the coalescent process (Maddison, 1997). At this point, any phylogeographic or phylogenetic analysis based on a set of randomly chosen neutral markers is likely to yield discordant results (Beltran et al., 2002; Machado and Hey, 2003; Mallarino et al., 2004; Dopman et al., 2005; Pollard et al., 2006). If Fst outliers can be identified in such analyses, their gene trees will reflect a clear branching pattern consistent with the phenotypic divergence, but most randomly chosen markers will produce noisy and uninformative phylogenetic patterns [examples in Wilding et al. (2001), Campbell and Bernatchez (2004), Emelianov et al. (2004), Dopman et al. (2005), and Via and West (2008)].
An important prediction of this model of speciation is that phylogenetic discordance will persist through speciation and possibly well beyond. Therefore, to detect the evolutionary story of lineage branching that will be reflected in the future species tree, it is necessary to focus on analyses of Fst outliers that are in linkage disequilibrium with QTL under divergent selection [see examples in Wilding et al. (2001), Emelianov et al. (2004), Dopman et al. (2005), and Via and West (2008)]. Early in speciation, these outliers will reveal the history of adaptive divergence. The truly neutral markers in other genomic regions will still be useful, however, for analyses of demographic events such as bottlenecks or range expansion.
Preferentially using markers affected by divergent selection for phylogeographic or phylogenetic analysis of populations conflicts with the clear preference for neutral markers in species-level phylogenetics. This apparent contradiction results from the gene tree-species tree mismatch, which persists until phylogenetic concordance is reached. Until that time, variability among gene trees leads will inevitably produce highly variable “cloudograms,” instead of the simple species-level cladograms that will eventually be visible (Maddison, 1997).
In sum, there are 2 key ideas in this model: first, the branching pattern of the QTL and their linked outliers reveals the branching pattern with which all of the discordant gene trees will eventually fall in line, and second, this pattern can be seen even during the period of rampant gene tree discordance if one analyzes Fst outliers rather than a random sample of markers.
As the portions of the genome affected by divergent selection become increasingly resistant to interrace gene exchange during stage 1, genes and
sequences within regions of divergence hitchhiking can begin to diverge by genetic drift or independent responses to directional and stabilizing selection within each race. The free recombination enjoyed within races can accelerate divergence at these loci by allowing beneficial mutations to spread within races, while divergence hitchhiking blocks their export to the other race. Some allelic substitutions within regions of divergence hitchhiking may produce genetic incompatibilities between the new species. Thus, an additional prediction of this model is that genes for hybrid sterility or inviability that are found close to “branch-defining” QTL will have, on average, a greater time to most recent common ancestor than will those found in other genomic regions.
Stage 2:
Divergence by Genetic Drift and Independent Responses to Directional or Balancing Selection (Fig. 1.6A and B)
By the end of stage 1, most gene exchange is likely to have already been blocked by the ecologically based reproductive isolation that reduces effective migration. The incipient species are now essentially “ecologically allopatric.” Thus begins stage 2, in which the parts of the genome outside regions of divergence hitchhiking begin to differentiate by genetic drift or independent responses to selection within the new lineages. This secondary divergence will eventually bring all of the variation in polymorphic gene trees into widespread phylogenetic concordance with the branching pattern determined earlier by divergent selection on QTL affecting the key ecologically important traits.
Given that only a small fraction of the genome may be affected by divergent selection during stage 1, most of the eventual genetic divergence between new ecological species is likely to occur during stage 2. Genetic analyses of hybrid sterility and inviability reveal that genetic incompatibilities are numerous and scattered throughout the genome (Masly and Presgraves, 2007). It is thus probable that by the end of stage 2, the number of genetic incompatibilities that have accumulated could far outnumber, and potentially obscure, the adaptive genetic changes that were actually involved in the initial evolution of reproductive isolation under divergent selection during stage 1. This is one of the major drawbacks of the exclusive use of the spyglass in the study of speciation genetics.
In many cases of speciation by divergent selection, enough ecologically based reproductive isolation will evolve to isolate a pair of new species before many DMIs can accumulate. Even so, the genetic incompatibilities that lead to hybrid sterility and inviability play a very important role in ecological speciation because they make the ecologically based reproductive isolation that evolved earlier permanent and irreversible. By the end of stage 2, it is likely that enough genetic incompatibilities will
have accumulated that the new species and their diverse adaptations will persist even if the pattern of natural selection changes.
This 2-stage model reverses the roles that are sometimes assumed for different forms of reproductive isolation during speciation (Coyne and Orr, 1989, 1997). In allopatric speciation, postzygotic genetic incompatibilities accumulate while populations are geographically isolated. These cause reproductive isolation when populations come back into contact. It is then that ecologically based prezygotic isolation plays its secondary role, evolving under selection to “reinforce” the existing postzygotic isolation and prevent the production of sterile or inviable hybrids. In contrast, in ecological speciation with gene flow, various ecologically based barriers to gene flow evolve first, as a result of adaptation under divergent selection. Once migration between the incipient species is essentially eliminated, they become ecologically allopatric, and postzygotic incompatibilities can accumulate. These DMIs then play the secondary reinforcing role by rendering the earlier evolution of ecologically based reproductive isolation difficult to reverse.
WHY HASN’T DIVERGENCE HITCHHIKING BEEN SEEN BEFORE?
One hazard of the retrospective spyglass approach to speciation is that patterns of genetic divergence early in the process of speciation can become obscured or even invisible over time as additional divergence between the new species accumulates. This is likely to be the fate of divergence hitchhiking during the process of ecological speciation.
Early in ecological speciation with gene flow, populations diverge under selection at genomic regions that affect key ecologically important traits, while gene flow continues across the rest of the genome (Fig. 1.3). Fst outliers found during this period (stage 1) will tend to map to these selected regions, as suggested by our analyses (Via and West, 2008). However, by the time most of the genome is phylogenetically concordant and retrospective analyses begin, the genomic signature of the original divergent selection will have faded. Outliers might be found, but they should not be expected to mark the genomic regions under early divergent selection.
During stage 2 of ecological speciation with gene flow, genetic divergence occurs mostly in genomic areas that were not originally affected by divergent selection. As overall genomic divergence between the new species increases through drift or independent responses to selection, the distinctive genetic signature of divergence hitchhiking (excessive divergence in regions near divergently selected QTL) becomes assimilated into a more widespread pattern of genetic divergence between the new species. So, it is perhaps not surprising that decades of retrospective analyses
have not seen these important, but transient, regions of interrace linkage disequilibrium around divergent QTL.
Fst outliers may still be found late in stage 2 or in hybrid zones between “good species” [e.g., Yatabe et al. (2007)], but interpreting their cause becomes increasingly difficult as the overall level of genetic divergence between the new species grows. A high Fst marker seen late in speciation or in a hybrid zone is more likely to be the result of genetic drift or a recent selective sweep within 1 species than it is to be a signature of the divergent selection that caused speciation. Therefore, outliers found between new species or in hybrid zones, or between incipient species in secondary contact after a period of allopatric divergence, should not necessarily be expected to mark genomic regions affected by divergent phenotypic selection during the initial phases of ecological speciation.
Outlier analyses of races or morphs that have become partially reproductively isolated under divergent selection thus offer a privileged, but transient, view of the genetic mechanisms involved in early ecological speciation. It is in regions of divergence hitchhiking that ecological speciation with gene flow begins, and the divergence of the ecologically important QTL at their center determines the eventual pattern of branching seen later in the species tree. However, by the time that good species are recognized, the distinctive pattern of divergence hitchhiking around key QTL is gone, and the opportunity to analyze the genomic regions pivotal to ecological speciation with gene flow has been lost.
THE REAL DIFFERENCE BETWEEN SYMPATRIC AND ALLOPATRIC SPECIATION
In allopatric populations, where there is no possibility for gene exchange, virtually any type or strength of selection will eventually lead to reproductive isolation, and barriers to gene flow may be of virtually any kind. In their classic survey of reproductive isolation between Drosophila species, Coyne and Orr (1989, 1997) found that in allopatry, prezygotic (ecologically based) and postzygotic reproductive isolation (from DMIs) appeared to evolve at about the same rate.
In contrast, for speciation to occur without physical barriers to gene flow, divergent selection must be strong and “multifarious,” i.e., affecting several different traits, which causes ecologically based isolation to evolve relatively rapidly (Rice and Hostert, 1993; Schluter, 2001; Via, 2001; Hendry et al., 2007). Consistent with this, Coyne and Orr’s comparative analyses (Coyne and Orr, 1989, 1997) suggest that in sympatric populations, prezygotic isolation precedes the evolution of postzygotic isolation. The primacy of ecologically based isolation in speciation with gene flow is supported by empirical analyses of taxa in which divergent selection is
thought to have been involved in speciation. They reveal extensive prezygotic ecologically based isolation, with little or no isolation attributable to postzygotic genetic incompatibilities (Schluter, 2001; Via, 2001).
In the 2-stage model of speciation described here, allopatric speciation can occur without stage 1, but sympatric speciation cannot. There is essentially only 1 path for purely sympatric speciation: rapid divergence at genomic regions harboring QTL for traits under divergent selection, leading to significant ecologically based reduction of successful interbreeding between incipient species and ecological allopatry by the end of stage 1. Then, during stage 2, genetic incompatibilities can accumulate to reinforce the ecologically based isolation and make it permanent.
In contrast, allopatric speciation cannot be divided into distinct stages, because the accumulation of DMIs by independent responses to uniform or balancing selection can occur at the same time as the evolution of ecologically based isolation driven by divergent selection. In allopatry, any combination of divergent selection, uniform selection, and genetic drift could produce speciation (Fig. 1.6C). Because the rapid divergence under selection that characterizes ecological speciation with gene flow is not required when populations are geographically isolated (although it can happen), allopatric speciation will often take much longer than speciation with gene flow.
DIVERGENCE HITCHHIKING MAKES SYMPATRIC SPECIATION MUCH MORE LIKELY THAN COMMONLY BELIEVED
Divergence hitchhiking neutralizes the most long-standing criticism of sympatric speciation, the difficulty of maintaining linkage disequilibrium between genes involved in resource use and those that produce assortative mating [e.g., Coyne and Orr (2004, pp. 127–137); Felsenstein (1981); Hawthorne and Via (2001); Via (2001)]. Although it has been clear for some time that this problem is mitigated if the traits under divergent selection for resource use also affect mate choice (Schluter, 2001), or if there is pleiotropy or physical linkage between the 2 classes of genes (Felsenstein, 1981; Hawthorne and Via, 2001; Via, 2001), these observations have done little to quell the controversy.
By providing a simple mechanism by which combinations of genes that produce assortative mating can accumulate and be protected from recombination, divergence hitchhiking removes the major constraint on sympatric speciation that prevented its acceptance for so long. The controversy over sympatric speciation has occupied a tremendous number of researchers over the past 50 years. If additional studies in other taxa show that divergence hitchhiking is a general phenomenon, we may finally be able to put this issue behind us.
CONCLUSIONS
The genetic changes that produce speciation have fascinated researchers for many years. To date, most research on this crucial aspect of evolution has taken a retrospective approach that I call the spyglass. However, population-level analysis of the ecological and genetic mechanisms that produce reproductive isolation between partially isolated ecotypes or races (the magnifying glass) can provide a very different perspective on the problem of speciation. Both the spyglass and the magnifying glass are useful tools in the genetic analysis of speciation; any truly general theory of how speciation occurs must be consistent not only with observations from fully differentiated species, but also with mechanisms seen at the population level in partially isolated ecological races. Speciation is a multidimensional problem, and we will not solve Darwin’s mystery unless we scrutinize it from every possible vantage point.
ACKNOWLEDGMENTS
I thank John Avise and Francisco Ayala for the invitation to discuss these ideas at the Sackler Colloquium; Dolph Schluter and an anonymous referee for exceptionally thoughtful comments on the manuscript; Gina Conte for developing the multiplexed microsatellites for the new linkage map; Justin Malin for some clever computational assistance; and Casey Mason-Foley, Kelly Mills, and Jeffrey Lew for PCRs without end. My work on speciation is supported by National Science Foundation Grants DEB9796222, DEB0221221, and DEB0528288 and the U.S. Department of Agriculture National Research Initiative, Gateways to Genomics Program.






















